PLK1 and HOTAIR Accelerate ProteasomalDegradation of SUZ12 and ZNF198 duringHepatitis B Virus–Induced Liver CarcinogenesisHaoZhang1, AhmedDiab1, HuitaoFan1, SaravanaKumarKailasamMani1, RonaldHullinger1,Philippe Merle2, and Ourania Andrisani1
Abstract
Elucidating mechanisms of hepatitis B virus (HBV)–mediatedhepatocarcinogenesis is needed to gain insights into the etiologyand treatment of liver cancer. Cells where HBV is replicatingexhibit increased expression of Plk1 kinase and reduced levels oftwo transcription repression factors, SUZ12 and ZNF198. SUZ12is an essential subunit of the transcription repressive complexPRC2. ZNF198 stabilizes the transcription repressive complexcomposed of LSD1, Co-REST, and HDAC1. These two transcrip-tion repressive complexes are held together by binding the longnoncodingRNAHOTAIR. In this study, we linked these regulatoryeventsmechanistically by showing that Plk1 induces proteasomaldegradation of SUZ12 and ZNF198 by site-specific phosphoryla-tion. Plk1-dependent ubiquitination of SUZ12 and ZNF198 wasenhanced by expression ofHOTAIR, significantly reducing SUZ12andZNF198 stability. In cells expressing theHBVXprotein (HBx),
downregulation of SUZ12 and ZNF198 mediated global changesinhistonemodifications. In turn,HBx-expressing cells propagatedan altered chromatin landscape after cell division, as exemplifiedby changes in histone modifications of the EpCAM promoter, atarget of PRC2 and LSD1/Co-REST/HDAC1 complexes. Notably,liver tumors from X/c-myc bitransgenic mice exhibited down-regulation of SUZ12 and ZNF198 along with elevated expressionof Plk1, HOTAIR, and EpCAM. Clinically, similar effects weredocumented in a set of HBV-related liver tumors consistent withthe likelihood that downregulation of SUZ12 and ZNF198 leadsto epigenetic reprogramming of infected hepatocytes. Becauseboth Plk1 and HOTAIR are elevated in many human cancers, wepropose that their combined effects are involved in epigeneticreprogramming associated broadly with oncogenic transforma-tion. Cancer Res; 75(11); 2363–74. �2015 AACR.
IntroductionChronic infection by the hepatitis B virus (HBV) is a major
etiologic factor in the development of hepatocellular carcinoma(HCC; ref. 1). The World Health Organization estimates that 250million people globally are chronically infected with HBV.Despite the availability of the HBV vaccine, the vaccine is notalways protective, and children born of infected mothers becomechronically infected. Current treatments include antiviral nucle-oside analogs, but eventually this treatment results in viral resis-tance (2). In advanced stage HCC, targeted therapies such assorafenib (anti-MAPK) are of modest but significant benefit(3). Thus, new and effective mechanism-based therapies areneeded, along with new prognostic markers for molecular stagingof the disease (4).
Pathogenesis of HBV-mediatedHCC involves effects of chronicinflammation of the liver (5) and effects of the HBV X protein(HBx), which acts as a weak oncogene (6) or co-factor in hepa-tocarcinogenesis (7).HBVDNA integrates into the host genomeatearly steps of clonal tumor expansion, and themajority of tumorsdisplay continued expression ofHBx (8).HBx is amultifunctionalprotein (9, 10), is required for the HBV life cycle (11), and isimplicated in HCC pathogenesis by a mechanism not yet under-stood. Regarding thismechanism, our studies (12, 13) discoveredan inverse relationship between the protein levels of two tran-scription repressive factors, SUZ12 and ZNF198, and the mitoticpolo-like kinase1 (Plk1; ref. 14). This intriguing relationshipbetween Plk1, SUZ12, and ZNF198 was observed in cellular andanimal models of HBx- and HBV-mediated oncogenic transfor-mation as well as in HBV-replicating cells (15).
SUZ12 is one of the three essential subunits of the transcriptionrepressive PRC2 complex (polycomb repressive complex 2). Theother two core subunits of PRC2 include EED protein and thehistone methyltransferase EZH2 (16). This chromatin-modifyingcomplex epigenetically regulates lineage selection during embry-onic development and stem cell differentiation (17, 18) bytrimethylating histone3 on lysine 27 (H3K27me3), a transcrip-tion silencing modification (16). PRC2 forms multiprotein com-plexes with other chromatin-modifying proteins (19, 20), andassociates with noncoding RNAs (21). PRC2 binds the longnoncoding RNA (lncRNA) HOTAIR, which also binds anothertranscription repressive complex composed of lysine demethy-lase1 (LSD1), Co-REST, and histone deacetylase 1 (HDAC1;ref. 22). Interestingly, the LSD1/Co-REST/HDAC1 complex,
1Department of Basic Medical Sciences and Purdue Center for CancerResearch, Purdue University, West Lafayette Indiana. 2Centre deRecherche en Canc�erologie de Lyon, UMR INSERM 1052, CNRS5286, Lyon Cedex, France.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
A. Diab and H. Fan contributed equally to this article.
Corresponding Author: Ourania Andrisani, Purdue University, B034 HansenBldg, 201 South University St, West Lafayette, IN 47907. Phone: 765-494-8131;Fax: 765-494-0781; E-mail: [email protected]
which enzymatically removes histone acetylations and H3K4methylations that activate transcription, is stabilized by the chro-mosomal protein ZNF198 (23). Thus, HOTAIR couples the tran-scription repressive activity of PRC2 and LSD1/Co-REST/HDAC1complexes (22). In addition to the involvement of these chro-matin-modifying complexes in cell fate determination duringembryonic development, accumulating evidence links deregu-lated expression of polycomb proteins (24) and ZNF198 (25,26) to cancer pathogenesis.
In liver tumors of X/c-myc bitransgenicmice and those inducedby chronic infection with Woodchuck hepatitis virus (WHV),increased protein levels of Plk1 correlated with reduced proteinlevels of SUZ12 and ZNF198 (15). Our studies suggested thatPlk1, activated by HBx in late G2 phase (27), is responsible fordownregulating the protein levels, but not the transcription ofSUZ12 and ZNF198 (12, 13, 27). Studies by others have shownthat Plk1phosphorylation induces proteasomal degradationof itssubstrates involved in cell cycle progression (28, 29). ElevatedPlk1 occurs in various types of human cancers, including livercancer (30). The function of Plk1 substrates ranges from proteinsinvolved in mitotic progression, chromosome segregation, DNAreplication, and signal transduction (28–30). In this study, weshow that Plk1 signals by phosphorylation the proteasomaldegradation of the chromatin-modifying proteins SUZ12 andZNF198. Clinically, elevated expression of HOTAIR and Plk1 wasquantified in aggressive liver tumors derived from chronicallyinfected HBV patients, supporting the involvement of this novelmechanism in HBV-mediated hepatocarcinogenesis.
Materials and MethodsCell culture, transfections, plasmids, siRNAs, andsynchronization protocols
Tetracycline-regulated HBx-expressing 4pX-1 cells, derivedfrom mouse hepatocyte AML12 cell line (31), was grown asdescribed (32) in the presence of tetracycline (5 mg/mL) orwithout tetracycline for 16 to 18 hours to allow HBx expression.
HBx expression was confirmed by RT-PCR. Synchronization of4pX-1 cells in G1–S by the double thymidine block (dTB) wasperformed as described (27). Synchronization of 4pX-1 cells inG2–M was by incubation with 100 nmol/L nocodazole for 12hours, and release from the block for 4 hours. HepAD38 cells,derived from HepG2 cell line, were grown as described (33).Transient transfections in HEK293T cells or 4pX-1 cells were per-formed employing Lipofectamine 2000 (Invitrogen) as describedby manufacturer, with 4 mg each of the following mammalianexpression plasmids: SUZ12-HA, ZNF198-GFP (34), Plk1CA-GFP,Plk1KD-GFP, Ubiquitin-FLAG (35, 36), and pcDNA3-HOTAIR(22).HOTAIRsiRNAandscrambledcontrol siRNAwere transfectedin 4pX-1 cells using Lipofectamine RNAiMAX (Invitrogen).
All cell lines used in this study are routinely tested for myco-plasma (employing commercially available kits), and retention ofdescribed properties and growth characteristics. 293HEK andAML12 cell lines were purchased from the ATCC. HepAD38 cellline (33) was obtained from Dr. C. Seeger, Fox Chase CancerCenter, Philadelphia, PA, in 2010. Expression ofHBV pregenomicRNA in HepAD38 cells is tested by PCR at regular intervals, lessthan 6 months. HBx expression in tetracycline-regulated HBx-expressing cell lines (4pX-1, 4pX-1-SUZ12kd) is confirmed by PCRat regular intervals, less than 6 months. Knockdown of SUZ12 in4pX-1-SUZ12kd cell line is confirmed by immunoblots comparedwith 4pX-1 cells. Selectionof knockdown cells is carriedout by cellsorting based on GFP fluorescence, should the expected knock-down of SUZ12 is lost. This is performed as needed. Last cellsorting for GFPþ cells was performed in 2014.
In vitro Plk1 kinase assaysIn vitro Plk1 kinase assays were performed exactly as described
(35, 36), employing affinity purified SUZ12-HA and ZNF198-GFP following transient expression in HEK293 cells. Site-directedmutagenesis of putative Plk1 phosphorylation sites in SUZ12-HAandZNF19-GFPwas performed employing theQuickChange site-directed mutagenesis Kit (Stratagene). Mutations were confirmedby DNA sequencing.
A C D HepAD38HepAD384pX-1HBx
relativeintensity
relativeintensity
kDa
150
75
50
kDa
150
75
50
kDa
150
250
75
20
7550
15
150
75
50
ZNF198-GFP
BI2636HEK293T
0.21
1.81
6.81
150
75
50
504.51
2.51
0.41
0.371
0.4
10 105 5
1 0.31 0.8
1 0.41 0.7
3.11.31 2.7
1 2.4
1SUZ12-HA
ZNF198-GFP
SUZ12-HA
Actin
Actin
relativeintensity
relativeintensity
MG132ZNF198-GFP
SUZ12-HA
Actin
Actin
relativeintensity
relativeintensity
relativeintensity
relativeintensity
Day
ZNF198
ZNF198
HB
V r
eplic
atio
nH
BV
rep
licat
ion
SUZ12
SUZ12
HBc
H3
Plk1T210
Plk1T210HBx
relativeintensity
relativeintensity
ZNF198
SUZ12
Actin
B
Figure 1.Plk1 downregulates SUZ12 and ZNF198 proteins. A, left, 4pX-1 cells, transiently transfected with SUZ12-HA and ZNF198-GFP encoding plasmids, were grownwith (þ) or without (�) HBx expression by tetracycline removal for 48 hours; whole cell extracts were immunoblotted with GFP or HA antibodies. Right,whole cell extracts from 4pX-1 cells treated with nocodazole (100 nmol/L) for 12 hours and grown with (þ) or without (�) HBx expression for 16 hours wereimmunoblottedwith ZNF198 or SUZ12 antibodies. B, HEK293T cells transiently transfectedwith SUZ12-HA and ZNF198-GFP encoding plasmidswere grownwith (þ)or without (�) BI2536 (250 nmol/L) for 12 hours or MG132 (2.5 mmol/L). Left, DMSO served as vehicle control. Right, transfected cells were synchronized in G2–M byaddition of nocodazole (100 nmol/L) for 12 hours. Lysates were immunoblotted with HA or GFP antibodies. C, immunoblots of indicated proteins usingwhole cell extracts fromHepAD38 cells grownwith (þ) orwithout (�) HBV replication by tetracycline removal for 5 and 10 days. D, immunofluorescencemicroscopyof indicated proteins, � HBV replication in HepAD38 cells.
Zhang et al.
Cancer Res; 75(11) June 1, 2015 Cancer Research2364
Immunoblots, immunoprecipitations, and immunofluores-cence microscopy were performed employing standard protocols(13). Antibodies used were as follows: HA (Sigma), FLAG(Sigma), GFP (Santa Cruz), Plk1 (Abcam), phospho-Plk1T210
Chromatin immunoprecipitation assaysChromatin immunoprecipitation (ChIP) assays were per-
formed using 4pX-1 cells synchronized by nocodazole (100ng/mL) in G2–M for 12 hours, followed by release from noco-dazole block for 4 hours, with or without HBx expression bytetracycline removal for 16 hours. The Millipore ChIP Assay Kit(Cat. No. 17-295) and the Mono-Methyl-Histone H3 (Lys4)Antibody #9723S (Cell Signaling) and Anti-Histone H3 (trimethyl K27) antibody-ChIP Grade (ab6002) were employed.
Reverse transcription and quantitative real-time PCRRNA was isolated from normal liver and liver tumors of X/c-
myc mice (6) and human HBV-related HCCs employing the
PureLink RNA Mini Kit (12183018A; Invitrogen). Liver tissuesfrom chronic HBV patients with HCC, tumor and peritumoraltissue, were obtained from the French National BiologicalResources Centre following approved consent from the FrenchLiver Tumor Network Scientific Committee. cDNA was synthe-sized from 2.0 mg total RNA isolated using the iScript cDNASynthesis Kit (170-8891; Bio-Rad). Quantitative real-time PCRreactions were performed in triplicates and normalized toGAPDH employing FastStart Essential DNA Green Master(06924204001; Roche), SYBR Green (Roche), and Roche Light-Cycler 96. The 2�DDCt method was used for analysis. Primersequences are listed in Supplementary Data.
ResultsHBx and Plk1 regulate protein levels of SUZ12 and ZNF198
To investigate whether Plk1, which becomes activated by HBxexpression (27), downregulates SUZ12 and ZNF198 proteins(15), we transfected mammalian expression vectors encodingSUZ12-HA and ZNF198-GFP in tetracycline-regulated HBx-expressing 4pX-1 cells (32). Expression of HBx decreased by>50% protein levels of transfected SUZ12-HA and ZNF198-GFPas well as endogenous SUZ12 and ZNF198 proteins (Fig. 1A). Tolink thisHBx-mediated downregulation of SUZ12 and ZNF198 toPlk1, HEK293T cells transfected with SUZ12-HA and ZNF198-GFP were treated with Plk1 inhibitor BI2536 or proteasomal
Figure 2.In vitro Plk1 kinase assays of SUZ12 andZNF198 and site-directed mutants.A, affinity purified SUZ12-HA andZNF198-GFP isolated from transfectedHEK293T cells and Plk1 (35, 36) wereused in in vitro Plk1 kinase assays.Reactions were performed with (þ) orwithout (�) Plk1 in the presence ofg-32P-ATP and analyzed by SDS-PAGEand autoradiography (35, 36).B, functional regions of SUZ12 (40) andZNF198 (23) and putative Plk1phosphorylation sites are shown. Ser toAla substitutions constructed athighlighted residues. C and D, in vitrokinase assays of WT SUZ12-HA (C) andZNF198-GFP (D) and site-directedmutants, as indicated. Commassie bluestaining shows the same gel used forautoradiography. Relative intensityquantifiedby ImageJ software is ratio ofsignal from in vitro kinase reactionversus corresponding Commassieblue-stained band, expressedrelative to WT (n ¼ 3).
Plk1- and HOTAIR-Mediated Degradation of SUZ12 and ZNF198
www.aacrjournals.org Cancer Res; 75(11) June 1, 2015 2365
inhibitor MG132 (Fig. 1B). Treatment with BI2536 or MG132restored SUZ12-HA and ZNF198-GFP protein levels (Fig. 1B),suggesting SUZ12 and ZNF198 are downregulated via Plk1-medi-ated phosphorylation.
These effects of HBx on SUZ12 and ZNF198 proteins werealso observed in HBV-replicating HepAD38 cells (13, 15). Inthe HepAD38 cellular model of HBV replication, the integratedHBV genome is under control of the Tet-off promoter (33).HBV replication is initiated by tetracycline removal, allowingtranscription of the pregenomic RNA, the template of viralreplication. In the presence of HBV replication, using the viralcore protein HBc as marker for HBV replication, endogenous
SUZ12 and ZNF198 protein levels were reduced (Fig. 1C).Importantly, employing the phospho-T210 Plk1 (Plk1T210)antibody that detects active Plk1, we observe that in HBV-replicating cells, Plk1 was activated (Fig. 1C) and localized tothe nucleus (Fig. 1D).
Plk1 phosphorylates SUZ12 and ZNF198 in vitroBecause SUZ12 and ZNF198 are nuclear proteins and active
Plk1 was in the nucleus of HBV-replicating cells (Fig. 1D), theseresults suggest SUZ12 and ZNF198 could serve as Plk1 sub-strates. To test this hypothesis, we performed in vitro Plk1 assays(Fig. 2). SUZ12-HA and ZNF198-GFP proteins were expressed
A
100
150
75
ZNF198-GFP
IP
Inp
ut
IgG HAIgG HA
IP
Inp
ut
WB: ZNF198-GFP
WB: LSD1
WB: HDAC10.11 0.61
0.31 0.91
0.61 0.9
kDa
1
relativeintensity
relativeintensity
relativeintensity
Plk1KD
Plk1CA
100
150
75
S303A/S305A/S309A
IP
Inp
ut
IgG HAIgG HA
IP
Inp
ut
WB: ZNF198-GFP
WB: LSD1
WB: HDAC10.71 0.61
1.61 1.41
0.91 1.9
kDa
1
relativeintensity
relativeintensity
relativeintensity
Plk1KDPlk1CA
100
150
75
S1056A/S1064A
IP
Inp
ut
IgG HAIgG HA
IP
Inp
ut
WB: ZNF198-GFP
WB: LSD1
WB: HDAC10.71 0.31
1.41 0.31
1.51 1.5
kDa
1
relativeintensity
relativeintensity
relativeintensity
Plk1KDPlk1CA
C
D
SUZ12-HA
IP
Inp
ut
IgG HAIgG HA
IP
Inp
ut
WB: SUZ12-HA
WB: EZH20.31 0.71
0.41 0.5
75
100
kDa
1
relativeintensity
relativeintensity
Plk1KD
Plk1CA
IP
Inp
ut
IgG HAIgG HA
IP
Inp
ut
WB: SUZ12-HA
WB: EZH20.81 0.21
0.71 0.6
75
100
kDa
1
relativeintensity
relativeintensity
S539A/S541A/S546AS539D/S541D/S546D
B
Figure 3.Plk1-mediated phosphorylations of SUZ12 and ZNF198 disrupt interaction with respective chromatin-modifying complexes. A, transient coexpression of SUZ12-HAand Plk1CA or Plk1KD in HEK293T cells. Whole cell extracts of transfected cells immunoprecipitated with IgG or HA antibodies; immunoprecipitates (IP) wereimmunoblotted (WB) with EZH2 or HA antibodies. Relative intensity quantified versus input. B, transient coexpression of ZNF198-GFP and Plk1CA or Plk1KD inHEK293T cells.Whole cell extracts of transfected cells immunoprecipitatedwith IgG or GFP antibodies. Immunoprecipitateswere immunoblotted for GFP, HDAC1, orLSD1. C and D, transient coexpression of indicated site-directed mutants of SUZ12 (C) and ZNF198 (D) in HEK293T cells. Immunocipitates were immunoblotted withindicated antibodies (n ¼ 3).
Zhang et al.
Cancer Res; 75(11) June 1, 2015 Cancer Research2366
by transient transfection in HEK293T cells and purified byaffinity chromatography. In vitro kinase assays with affinitypurified substrates and Plk1 (36) were performed in the pres-ence of g-P32-ATP, analyzed by SDS-PAGE, and autoradiogra-phy (35, 36). Indeed, wild-type (WT) SUZ12 and ZNF198proteins were phosphorylated in vitro by Plk1 (Fig. 2A).
Next, we examined the protein sequence of SUZ12 andZNF198 for conserved, consensus Plk1 phosphorylation sites(D/E-X-S/T-�-D/E), and constructed Ser to Ala substitutions atthose sites (Fig. 2B). Phosphoproteomic studies have shownSUZ12 is phosphorylated in vivo at S546 using nocodazole-arrested Hela cells (37) and S539 in human embryonic stemcells, hESCs (38); on the other hand, ZNF198 is phosphory-lated in vivo at S305 using nocodazole-arrested Hela cells (39).However, the kinases mediating these phosphorylations areunknown.
To determine if these sites in SUZ12 and ZNF198 are phos-phorylated by Plk1, we expressed each mutant protein inHEK293T cells, purified them by affinity chromatography, andperformed in vitro Plk1 kinase assays (Fig. 2C and D). SUZ12proteins with Ser to Ala substitutions at residues 539, 541, and546 (single and triple site mutants) were used in in vitro kinaseassays. The results show that all three siteswerephosphorylated byPlk1, the triple mutant showing 70% reduction in phosphoryla-tion (Fig. 2C), and identify Plk1 as the kinase mediating thein vivo–detected phosphorylations of SUZ12 at S539 and S546(37, 38).
Similar analyses with ZNF198 identified two clusters of puta-tive Plk1 phosphorylation sites in vitro. A cluster of serine residuesat the N-terminus of ZNF198, S303, S305, and S309, and a clusterat the C-terminus, S1056 and S1064. The triple Ser to Alamutant,S303A/S305A/S309A, consistently exhibited the lowest level ofphosphorylation in vitro, in comparison with the double S1056A/S1064Amutant (Fig. 2D). Importantly, in vivophosphorylation ofS305 of ZNF198was detected by phosphoproteomic studies (39),suggesting that Plk1 is the kinase that phosphorylates S305 ofZNF198 in vivo.
Plk1 phosphorylation dissociates SUZ12- and ZNF198-containing complexes
In SUZ12, residues 539, 541, and 546 phosphorylated by Plk1in vitro (Fig. 2C) are adjacent (S539 and S541) and within (S546)the VEFS box (Fig. 2B) that interacts with EZH2 (40), suggestingthat phosphorylation at those sites will disrupt SUZ12 interactionwith EZH2. Accordingly, we examined the effect of expression ofconstitutively active (CA)Plk1CAon the interactionof SUZ12withEZH2. Expression of active Plk1CA decreased the amount of EZH2that coimmunoprecipitated with SUZ12, in comparison with thekinase dead (KD) Plk1KD (Fig. 3A). To confirm these results, weexamined endogenous EZH2 interaction with SUZ12 mutantscontaining Ser to Ala or Ser to Asp substitutions at all three sites(S539, S541, and S546). The phospho-mimetic substitutions (Serto Asp) significantly reduced interaction of SUZ12 with EZH2(Fig. 3B).
We also examined whether Plk1-mediated phosphorylationof ZNF198 disrupts interaction with LSD1/Co-REST/HDAC1complex (23). ZNF198-GFP was cotransfected with eitherPlk1CA or Plk1KD. Upon expression of inactive Plk1KD,ZNF198-GFP coimmunoprecipitated both LSD1 and HDAC1(Fig. 3C). By contrast, active Plk1CA reduced coimmunopreci-pitation of these proteins (Fig. 3C). Cotransfection of active
Plk1CA with N-terminal S303A/S305A/S309A or C-terminalS1056A/S1064A ZNF198 mutants demonstrated that only theN-terminal mutant maintained interaction with LSD1 andHDAC1 (Fig. 3D, compare left vs. right). We conclude thatPlk1 phosphorylation at the N-terminal sites dissociatesZNF198 from the LSD1/Co-REST/HDAC1 complex.
Plk1 phosphorylation promotes ubiquitin-mediateddegradation of SUZ12 and ZNF198
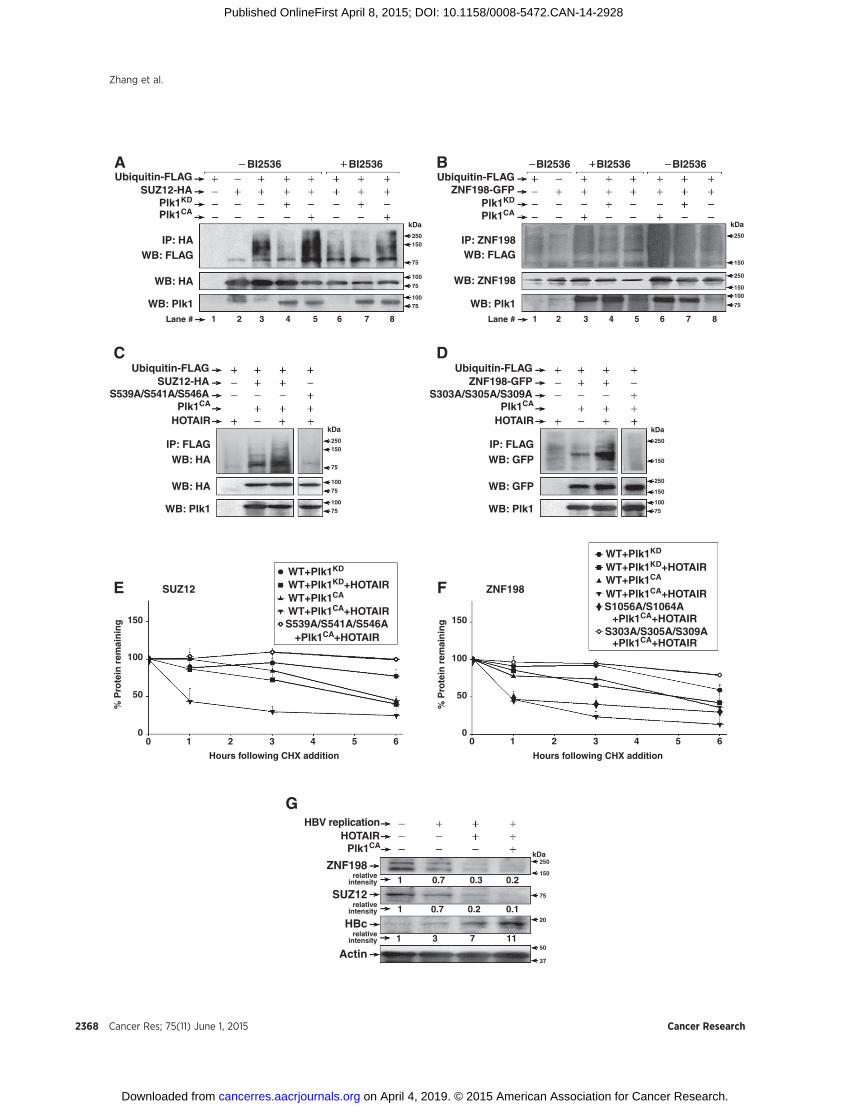
Because phosphorylation by Plk1 is often associated withubiquitin-mediated proteolysis of its substrates (29), we inves-tigated whether Plk1 phosphorylation of SUZ12 and ZNF198regulates their ubiquitination. Expression vectors encodingSUZ12-HA or ZNF198-GFP were cotransfected in HEK293Tcells with plasmids encoding ubiquitin-FLAG, and eitherPlk1CA or Plk1KD. All transfection assays were performed inthe presence of MG132, an inhibitor of proteasomal degrada-tion. Ubiquitinated WT SUZ12-HA, detected by immunoblotswith FLAG antibody, was observed upon expression of activePlk1CA (Fig. 4A, lane 5), whereas the Plk1 inhibitor BI2536suppressed ubiquitination (Fig. 4A, lane 8). Similar results wereobserved with WT ZNF198-GFP (Fig. 4B; compare lane 6without BI2536 vs. lane 3 with BI2536). Because ubiquitina-tion of SUZ12 and ZNF198 depends on active Plk1 (Fig. 4A andB), we examined the ubiquitination potential of their Plk1phosphorylation mutants in transfected HEK293T cells. Thetriple Ser to Ala mutants of SUZ12 and ZNF198 (Fig. 2) lackedubiquitination (Supplementary Fig. S1A and S1B), supportingthat Plk1-mediated phosphorylations at those sites are neces-sary for ubiquitination.
HOTAIR increases ubiquitination of SUZ12 and ZNF198,reducing their stability
HOTAIR functions as scaffold in ubiquitination by interactingwith RNA binding E3 ubiquitin ligases (41). Because HOTAIRbridges PRC2 to the LSD1/Co-REST/HDAC1 complex, stabilizedby ZNF198 (23), we investigated whether HOTAIR has a role inubiquitination of SUZ12 and ZNF198. We cotransfected inHEK293T cells expression vectors encoding HOTAIR and WTSUZ12-HA or ZNF198-GFP, together with active Plk1CA.Increased ubiquitination of SUZ12 and ZNF198 was observedwith overexpression of transfected HOTAIR (SupplementaryFig. S1C and S1D; Fig. 4C and D). By contrast, the triple mutantsof SUZ12 and ZNF198 that cannot be phosphorylated by Plk1(Fig. 2) lacked ubiquitination, even with HOTAIR overexpression(Fig. 4C and D).
To directly demonstrate the role of Plk1 and HOTAIR on thestability of SUZ12 and ZNF198 proteins, we quantified theirhalf-life (t1/2) following treatment with cyclohexamide(Fig. 4E and F; Supplementary Fig. S2). Coexpression of activePlk1CA and HOTAIR reduced the half-life of WT SUZ12 andZNF198 to 1 hour, in comparison with t1/2 > 6 hours withexpression of inactive Plk1KD and HOTAIR. Conversely, thetriple SUZ12 and ZNF198 mutants that cannot be phosphor-ylated by Plk1 (Fig. 2) exhibited enhanced stability, even in thepresence of active Plk1CA and HOTAIR. By contrast, the t1/2 ofC-terminal ZNF198 mutant (S1056A/S1064A) resembled WTZNF198 (Fig. 4F). We interpret these results to mean thatHOTAIR facilitates ubiquitination of Plk1-phosphorylatedSUZ12 and ZNF198 proteins, thereby accelerating their pro-teasomal degradation.
Plk1- and HOTAIR-Mediated Degradation of SUZ12 and ZNF198
www.aacrjournals.org Cancer Res; 75(11) June 1, 2015 2367
To confirm that this mechanism is operational with endog-enous SUZ12 and ZNF198 proteins, we coexpressed Plk1CA
and HOTAIR in HBV-replicating HepAD38 cells. Expressionplasmids were transfected on day 1 after tetracycline removal,
and cells were harvested 48 hours later. Coexpression of Plk1CA
and HOTAIR significantly reduced endogenous levels ofZNF198 and SUZ12 (Fig. 4G). Interestingly, under these con-ditions, HBc levels increased by 11-fold, in agreement with
A B
C
D
4pX-1 cells
HBxBI2536
Hours afterDTB release
SUZ12
4 8 10 0 4 8 0 4 8 10
0.5 10
08
06
04
02
Control s
iRNA
siHOTA
IR
00
0.4
0.3
0.2
% o
f In
pu
t
% o
f In
pu
t
HBxHBx
HBxHBx
0.1
0.0
100
ZNF198
p-Plk1T210
Plk1
H3K27me3
HBxBI2536
BI2536
SUZ12
ZNF198
SUZ12
CHX
1 0.9 1.1 0.7 0.3 0.1 0.21
1 0.9 0.9 0.6 0.5 0.2 0.31
0 1 3 6 0 1 3 6
ZNF198
Actin
Actin
relativeintensity
relativeintensity
H3K4me3
H3K4me2
H3K4me1
Histone3
Lane # 2 3 4 5 6 7 8 9 10 11 12
0.4 2.22 1.871 1 1
0.37 1.67 1.471 1 1
kDa75
kDa75
150
50
kDaHours
75
150
50
75
75
50
50
15
20
15
20
15
20
15
20
15
20
250
150
1
EpCAM
H3K27me3 H3K4me1
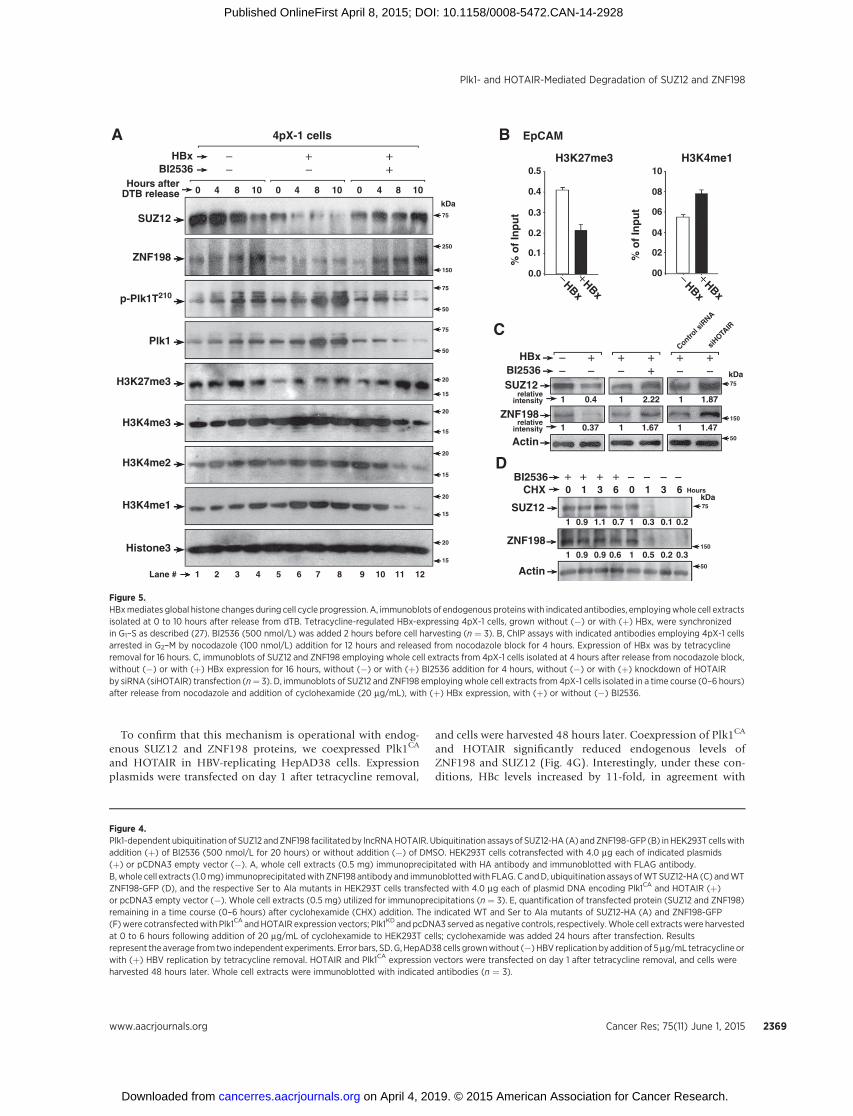
Figure 5.HBxmediates global histone changes during cell cycle progression. A, immunoblots of endogenous proteinswith indicated antibodies, employingwhole cell extractsisolated at 0 to 10 hours after release from dTB. Tetracycline-regulated HBx-expressing 4pX-1 cells, grown without (�) or with (þ) HBx, were synchronizedin G1–S as described (27). BI2536 (500 nmol/L) was added 2 hours before cell harvesting (n ¼ 3). B, ChIP assays with indicated antibodies employing 4pX-1 cellsarrested in G2–M by nocodazole (100 nmol/L) addition for 12 hours and released from nocodazole block for 4 hours. Expression of HBx was by tetracyclineremoval for 16 hours. C, immunoblots of SUZ12 and ZNF198 employing whole cell extracts from 4pX-1 cells isolated at 4 hours after release from nocodazole block,without (�) or with (þ) HBx expression for 16 hours, without (�) or with (þ) BI2536 addition for 4 hours, without (�) or with (þ) knockdown of HOTAIRby siRNA (siHOTAIR) transfection (n¼ 3). D, immunoblots of SUZ12 and ZNF198 employing whole cell extracts from 4pX-1 cells isolated in a time course (0–6 hours)after release from nocodazole and addition of cyclohexamide (20 mg/mL), with (þ) HBx expression, with (þ) or without (�) BI2536.
Figure 4.Plk1-dependent ubiquitination of SUZ12 andZNF198 facilitated by lncRNAHOTAIR. Ubiquitination assaysof SUZ12-HA (A) andZNF198-GFP (B) inHEK293T cellswithaddition (þ) of BI2536 (500 nmol/L for 20 hours) or without addition (�) of DMSO. HEK293T cells cotransfected with 4.0 mg each of indicated plasmids(þ) or pCDNA3 empty vector (�). A, whole cell extracts (0.5 mg) immunoprecipitated with HA antibody and immunoblotted with FLAG antibody.B,whole cell extracts (1.0mg) immunoprecipitatedwith ZNF198 antibody and immunoblottedwith FLAG. C andD, ubiquitination assays ofWTSUZ12-HA (C) andWTZNF198-GFP (D), and the respective Ser to Ala mutants in HEK293T cells transfected with 4.0 mg each of plasmid DNA encoding Plk1CA and HOTAIR (þ)or pcDNA3 empty vector (�). Whole cell extracts (0.5 mg) utilized for immunoprecipitations (n ¼ 3). E, quantification of transfected protein (SUZ12 and ZNF198)remaining in a time course (0–6 hours) after cyclohexamide (CHX) addition. The indicated WT and Ser to Ala mutants of SUZ12-HA (A) and ZNF198-GFP(F)were cotransfectedwith Plk1CA andHOTAIR expression vectors; Plk1KD and pcDNA3 served as negative controls, respectively.Whole cell extractswere harvestedat 0 to 6 hours following addition of 20 mg/mL of cyclohexamide to HEK293T cells; cyclohexamide was added 24 hours after transfection. Resultsrepresent the average from two independent experiments. Error bars, SD. G, HepAD38 cells grownwithout (�) HBV replication by addition of 5mg/mL tetracycline orwith (þ) HBV replication by tetracycline removal. HOTAIR and Plk1CA expression vectors were transfected on day 1 after tetracycline removal, and cells wereharvested 48 hours later. Whole cell extracts were immunoblotted with indicated antibodies (n ¼ 3).
Plk1- and HOTAIR-Mediated Degradation of SUZ12 and ZNF198
www.aacrjournals.org Cancer Res; 75(11) June 1, 2015 2369
earlier results showing that knockdown of SUZ12 and ZNF198enhances HBV replication (13).
HBx downregulates SUZ12 and ZNF198 during cell cycleprogression in untransformed hepatocytes and alters thechromatin landscape of daughter cells
To investigate whether this mechanism occurs in HBx-expres-sing cells, we employed tetracycline-regulated HBx-expressing4pX-1 cells (32). HBx activates Plk1 in late G2 phase (27).Employing dTB to synchronize 4pX-1 cells in G1–S, with orwithout HBx expression (27), we monitored protein levels ofSUZ12 and ZNF198 as a function of cell cycle progression.Samples were collected at 4 hours (S phase), 8 hours (G2), and10 hours (late G2–M) after release from dTB, as a function ofHBx expression and BI2536 treatment (Supplementary Fig.S3A). Immunoblots demonstrate reduction in SUZ12 andZNF198 levels with HBx expression (Fig. 5A, lanes 5–8) thatparallels activation of Plk1 (p-Plk1T210) from 4 to 10 hoursafter release from dTB (Fig. 5A, lanes 5–8 and SupplementaryTable S1). Importantly, treatment of HBx-expressing cells withBI2536 reversed this reduction (Fig. 5A, lanes 9–12). Next,we determined the effect of the reduction in SUZ12 andZNF198 proteins on global H3 modifications mediatedby the PRC2 and LSD1/Co-REST/HDAC1 complexes, respec-tively. Concomitant with the reduction in SUZ12, we observedreduction in H3K27me3 levels in HBx-expressing cells. Con-versely, the levels of activating modifications on H3K4 wereincreased. Treatment of HBx-expressing cells with BI2536reversed these effects (Supplementary Table S1 shows quanti-fication of Fig. 5A).
Because HBx, via Plk1-mediated downregulation of SUZ12and ZNF198, altered global histone modifications during cellcycle progression (Fig. 5A), we investigated whether theseepigenetic changes are propagated to daughter cells. We syn-chronized 4pX-1 cells in G2–M by nocodazole, released cellsfrom the block for 4 hours, allowing progression to G1 phase(Supplementary Fig. S3B). Employing ChIP assays, we exam-ined H3 modifications of the EpCAM promoter, a target of thePRC2 and LSD1/Co-REST/HDAC1 complexes (15). ChIP assaysshow that HBx expression decreased the silencing H3K27me3
modification mediated by PRC2, whereas the activating histonemodification H3K4me1, associated with loss of LSD1 function,was increased (Fig. 5B). This altered chromatin landscape of theEpCAM promoter in HBx-expressing cells after cell division issupported by immunoblots showing decreased SUZ12 andZNF198 levels in nocodazole-treated cells released from theblock for 4 hours (Fig. 5C). Moreover, the reduction in endog-enous SUZ12 and ZNF198 levels was restored by treatmentwith Plk1 inhibitor BI2536 and siRNA knockdown of HOTAIR(Fig. 5C), supporting involvement of both Plk1 and HOTAIR intheir downregulation. Lastly, protein levels of endogenousSUZ12 and ZNF198 were stabilized in cyclohexamide- andBI2536-treated cells (Fig. 5D), further supporting the role ofPlk1 in SUZ12 and ZNF198 degradation.
Plk1 and HOTAIR are elevated in liver tumors of X/c-myc miceand chronic HBV-infected patients
Liver tumors from X/c-myc bitransgenic mice (6) exhibitincreased levels of Plk1 and reduced levels of SUZ12 andZNF198 proteins (15). Based on the mechanism of Plk1-medi-ated degradation of SUZ12 and ZNF198 described in this study(Fig. 4), we quantified expression levels of Plk1 and HOTAIRRNAs in liver tumors from X/c-myc bitransgenic mice. Weobserved enhanced expression of Plk1 mRNA and HOTAIR inthe same tumor, ranging from 2-fold to >10-fold (Fig. 6A,right). We also quantified Plk1 and HOTAIR RNA levels inliver tumors of chronically infected HBV patients (Fig. 6B).Group B patients exhibit at least 2-fold increased expression ofboth Plk1 and HOTAIR.
Because liver tumors of X/c-myc bitransgenic mice displayincreased expression of EpCAM (Fig. 6C, left; ref. 15), we alsoquantified EpCAM expression in the HBV-related HCCs. Interest-ingly, increased EpCAM expression was observed in group BHCCs, those displaying at least 2-fold increased expression ofPlk1 and HOTAIR (Fig. 6C, right). Lastly, we determined theclinical significance of the expression of Plk1 and HOTAIR inHBV-related HCCs (Table 1). HOTAIR upregulation in group BHCCs strongly correlates with the appearance of satellite nodulesin the tumors (P ¼ 0.02), a well-recognized factor of aggres-siveness of HCC; furthermore, group B tumors are more prone
Figure 6.Plk1 and HOTAIR overexpression in liver tumors from X/c-myc mice and HBV-infected patients. PCR quantification of Plk1 and HOTAIR RNAs using total RNAisolated from liver tumors (A) of X/c-myc bitransgenic mice compared with control (Ctrl) RNA (normal mouse liver or peritumoral tissue), shown as box plots orhistograms in liver tumors from individual mice. B, PCR quantification of Plk1 and HOTAIR RNAs using RNA isolated from HBV-related HCCs compared withperitumoral tissue. Quantitative PCR reactions were performed in identical triplicates (Plk1 and HOTAIR) using GAPDH as internal control. C, quantificationof EpCAM mRNA in liver tumors from X/c-myc mice (left) and HBV-mediated HCCs (right), expressed relative to Ctrl RNA. PCR quantification of RNAsfromHBV-related HCC samples were performed in duplicates employing PCR arrays and expressed relative to normal liver (average value from 8 patients). P valuesare shown. D, model illustrates the mechanism by which HBx-activated Plk1 by phosphorylation signals ubiquitination and proteasomal degradation ofSUZ12 andZNF198. HOTAIRaccelerates ubiquitination andproteasomal degradation of SUZ12 andZNF198, likely acting as scaffold for recruitment ofRNA-binding E3ligases, as described by Yoon et al. (41).
Table 1. Correlation of HOTAIR and Plk1 upregulation between tumor and nontumor (ratio � 2) to clinicopathologic features of 13 HBV-related HCCs (group B)
Marker overexpression in tumors HOTAIR(þ) Plk1(þ) Group A versus group B
to develop satellite nodules in comparison with those of group A(P ¼ 0.05).
DiscussionLiver tumors from X/c-myc bitransgenic mice and wood-
chucks chronically infected by WHV display an inverse rela-tionship between Plk1 and chromatin-regulating proteinsSUZ12 and ZNF198 (15). In this study, we provide evidenceof a direct, causal link between Plk1 activation and reducedprotein levels of SUZ12 and ZNF198. As shown in the model(Fig. 6D), Plk1 phosphorylates SUZ12 and ZNF198 at specificsites (Fig. 2), disrupts their association with the respectivechromatin-modifying complexes (Fig. 3), and signals protea-somal degradation of SUZ12 and ZNF198 by ubiquitination(Fig. 4). Interestingly, their ubiquitination is facilitated byHOTAIR, acting as ubiquitination scaffold (41). The combinedaction of active Plk1 and HOTAIR accelerates degradation ofSUZ12 and ZNF198, decreasing their half-life by >15-fold,whereas the mutants of SUZ12 and ZNF198 that cannot bephosphorylated by Plk1 are stable (Fig. 4). The E3 ubiquitinligases involved in this mechanism are presently unknown,although the RNA-binding E3 ligases Dzip3 and Mex3b, shownto bind to HOTAIR (41), are likely candidates.
This novel mechanism that regulates SUZ12 and ZNF198stability is significant for several reasons.1. In eukaryotic cells, transcription occurs in the context of
chromatin, and often in response to extracellular signals.However, how signal transduction pathways communicatewith chromatin is not well understood. Such mechanismscould act directly to modify histones (42) or indirectly byregulating the activity of chromatin-modifying andremodeling complexes (43). For example, EZH2, themethyltransferase of PRC2, is modified byphosphorylation by AKT and CDKs, thereby regulating itsenzymatic activity (44–46) and substrate specificity (47).Recent studies have identified a series of molecular stepsinitiated by oncogenic Ras signaling that epigeneticallysilence tumor suppressor genes (48). However, in additionto silencing tumor suppressor genes, oncogenic signalingmust epigenetically modify chromatin components toactivate transcription of genes linked to proliferation andoncogenic transformation. In this study, we identified sucha mechanism, involving Plk1, overexpressed in manyhuman cancers (28), regulating the stability of epigeneticregulators.
2. This mechanism is operational upon activation of Plk1 byHBx, shown in the model of Fig. 6D, occurring during cellcycle progression, in the G2 phase (Fig. 5). Downregulationof endogenous SUZ12 and ZNF198 coincides with Plk1activation in HBx-expressing cells, reversed by inhibition ofPlk1 with BI2536. The consequence of SUZ12 and ZNF198downregulation is global changes in H3 modifications.Specifically, the EpCAM gene, silenced by PRC2 andZNF198-LSD1/Co-REST/HDAC1 complexes (15),immediately after cell division exhibits H3 modificationsassociated with gene activation. We propose that withcontinued expression of HBx, as during chronic HBVinfection, additional epigenetic alterations result in cellularreprogramming linked to oncogenic transformation.Indeed in SUZ12 knockdown cells, we have identified an
active DNA demethylation mechanism that allowsexpression of EpCAM by HBx (49). Importantly, EpCAM isexpressed in hepatic cancer stem cells (50). Thus,downregulation of SUZ12 beyond a threshold may beassociated with acquisition of an oncogenic cellularprogram.
3. The involvement of HOTAIR in significantly decreasingthe stability of SUZ12 and ZNF198 in the presence ofactivated Plk1 is another important discovery of this study(Fig. 4). The elevated expression of Plk1 and HOTAIR inliver tumors of X/c-myc bitransgenic mice as well as in agroup of human HBV-related HCCs (Fig. 6) supports therelevance of this mechanism to HBV-mediated livercarcinogenesis. The enhanced expression of HOTAIR andPlk1 disrupts PRC2 and LSD1/Co-REST-HDAC1complexes, leading to re-expression of genes silenced bythese complexes. Indeed, we detect enhanced expressionof EpCAM both in liver tumors of X/c-myc mice and ingroup B HCCs of HBV-infected patients (Fig. 6C), thosedisplaying elevated expression of both Plk1 and HOTAIR.The strong correlation between enhanced HOTAIRexpression and the aggressive HCC phenotype supportsthe significance of this mechanism to HCC pathogenesis(Table 1), and the potential use of HOTAIR expression asa biomarker for aggressive HCC. Because Plk1 andHOTAIR are overexpressed in many human cancers (51),including a group of HBV-mediated HCCs (Fig. 6), wespeculate this novel mechanism contributes to cellularreprograming observed during oncogenic transformation.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: H. Zhang, H. Fan, P. Merle, O. AndrisaniDevelopment of methodology: H. Zhang, H. FanAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): A. Diab, S.K. Kailasam Mani, P. MerleAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): H. Zhang, A. Diab, H. Fan, P. MerleWriting, review, and/or revision of the manuscript: A. Diab, H. Fan,S.K. Kailasam Mani, R. Hullinger, P. Merle, O. AndrisaniAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): H. ZhangStudy supervision: P. Merle, O. AndrisaniOther (development of figures): R. Hullinger
AcknowledgmentsThe authors thank Drs. X. Liu and J. Li for assistance and sharing of reagents
for in vitro Plk1 kinase assays, M. Gorospe and H. Chang for providing theHOTAIR expression vector, E. Tran for critical review of this article, andP. Pascuzzi for bioinformatics analyses.
Grant SupportThis work was supported by NIH grant DK044533 (O. Andrisani). Shared
Resources (flow cytometry and DNA sequencing) are supported by NIH grantP30CA023168 to Purdue Cancer Research Center.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received October 6, 2014; revisedMarch 10, 2015; accepted March 19, 2015;published OnlineFirst April 8, 2015.
Cancer Res; 75(11) June 1, 2015 Cancer Research2372
References1. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and
hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet1981;2:1129–33.
2. Zoulim F, Poynard T, Degos F, Slama A, El Hasnaoui A, Blin P, et al. Aprospective study of the evolution of lamivudine resistance mutations inpatients with chronic hepatitis B treated with lamivudine. J Viral Hepat2006;13:278–88.
3. Llovet JM, Ricci S,Mazzaferro V,Hilgard P, Gane E, Blanc JF, et al. Sorafenibin advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–90.
4. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcino-ma. Hepatology 2008;48:1312–27.
5. Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, et al.Extensive oxidative DNA damage in hepatocytes of transgenic mice withchronic active hepatitis destined to develop hepatocellular carcinoma.Proc Natl Acad Sci U S A 1994;91:12808–12.
6. Terradillos O, Billet O, Renard CA, Levy R, Molina T, Briand P, et al. Thehepatitis B virus X gene potentiates c-myc-induced liver oncogenesis intransgenic mice. Oncogene 1997;14:395–404.
7. Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as atumor promoter in development of diethylnitrosamine-induced preneo-plastic lesions. J Virol 2001;75:3851–8.
8. Su Q, Schroder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P.Expression of hepatitis B virus X protein in HBV-infected human livers andhepatocellular carcinomas. Hepatology 1998;27:1109–20.
9. Andrisani OM, Barnabas S. The transcriptional function of the hepatitis Bvirus X protein and its role in hepatocarcinogenesis (Review). Int J Oncol1999;15:373–9.
10. BouchardMJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol2004;78:12725–34.
11. Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein isrequired for viral infection in vivo. J Virol 1994;68:2026–30.
12. Studach LL, Rakotomalala L, Wang WH, Hullinger RL, Cairo S, BuendiaMA, et al. Polo-like kinase 1 inhibition suppresses hepatitis B virus Xprotein-induced transformation in an in vitro model of liver cancerprogression. Hepatology 2009;50:414–23.
13. Wang WH, Studach LL, Andrisani OM. Proteins ZNF198 and SUZ12 aredown-regulated in hepatitis B virus (HBV) X protein-mediated hepatocytetransformation and in HBV replication. Hepatology 2011;53:1137–47.
14. Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycleanalysis and chromosomal localization of human Plk1, a putative homo-logue of themitotic kinasesDrosophila polo and Saccharomyces cerevisiaeCdc5. J Cell Sci 1994;107 (Pt 6):1509–17.
15. Studach LL,Menne S, Cairo S, BuendiaMA,Hullinger RL, Lefrancois L, et al.Subset of Suz12/PRC2 target genes is activated during hepatitis B virusreplication and liver carcinogenesis associated with HBV X protein.Hepatology 2012;56:1240–51.
16. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark inlife. Nature 2011;469:343–9.
17. Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-widemapping of Polycomb target genes unravels their roles in cell fate transi-tions. Genes Dev 2006;20:1123–36.
18. Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb groupprotein Suz12 is required for embryonic stem cell differentiation. Mol CellBiol 2007;27:3769–79.
19. Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, et al.Interactions between JARID2 and noncoding RNAs regulate PRC2 recruit-ment to chromatin. Mol Cell 2014;53:290–300.
20. Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F,et al. Dnmt3L antagonizes DNA methylation at bivalent promotersand favors DNA methylation at gene bodies in ESCs. Cell 2013;155:121–34.
21. Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell2010;40:939–53.
22. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Longnoncoding RNA as modular scaffold of histone modification complexes.Science 2010;329:689–93.
23. GockeCB, YuH. ZNF198 stabilizes the LSD1-CoREST-HDAC1 complex onchromatin through its MYM-type zinc fingers. PLoS One 2008;3:e3255.
24. RichlyH, Aloia L,Di Croce L. Roles of the Polycomb group proteins in stemcells and cancer. Cell Death Dis 2011;2:e204.
25. Moinzadeh P, Breuhahn K, Stutzer H, Schirmacher P. Chromosomealterations in human hepatocellular carcinomas correlate with aetiologyand histological grade–results of an explorative CGH meta-analysis. Br JCancer 2005;92:935–41.
26. Xiao S, Nalabolu SR, Aster JC,Ma J, Abruzzo L, Jaffe ES, et al. FGFR1 is fusedwith a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lympho-ma syndrome. Nat Genet 1998;18:84–7.
27. Studach L,WangWH,WeberG, Tang J,Hullinger RL,MalbrueR, et al. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both theDNA damage checkpoint and DNA repair resulting in partial polyploidy.J Biol Chem 2010;285:30282–93.
28. Luo J, Liu X. Polo-like kinase 1, on the rise from cell cycle regulation toprostate cancer development. Protein Cell 2012;3:182–97.
29. Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, et al.Polo-like kinase-1 controls proteasome-dependent degradation of Claspinduring checkpoint recovery. Curr Biol 2006;16:1950–5.
30. Cholewa BD, Liu X, Ahmad N. The role of polo-like kinase 1 in carcino-genesis: cause or consequence? Cancer Res 2013;73:6848–55.
31. Wu JC, Merlino G, Fausto N. Establishment and characterization ofdifferentiated, nontransformed hepatocyte cell lines derived from micetransgenic for transforming growth factor alpha. Proc Natl Acad Sci U S A1994;91:674–8.
32. Tarn C, Bilodeau ML, Hullinger RL, Andrisani OM. Differential immediateearly gene expression in conditional hepatitis B virus pX-transformingversus nontransforming hepatocyte cell lines. J Biol Chem 1999;274:2327–36.
33. Ladner SK, OttoMJ, Barker CS, Zaifert K,WangGH, Guo JT, et al. Inducibleexpression of human hepatitis B virus (HBV) in stably transfected hepato-blastoma cells: a novel system for screening potential inhibitors of HBVreplication. Antimicrob Agents Chemother 1997;41:1715–20.
34. Kunapuli P, Kasyapa CS, Chin SF, Caldas C, Cowell JK. ZNF198, a zincfinger protein rearranged in myeloproliferative disease, localizes to thePML nuclear bodies and interacts with SUMO-1 and PML. Exp Cell Res2006;312:3739–51.
35. Li H, Liu XS, Yang X, Song B, Wang Y, Liu X. Polo-like kinase 1 phosphor-ylation of p150Glued facilitates nuclear envelope breakdown duringprophase. Proc Natl Acad Sci U S A 2010;107:14633–8.
36. Yang X, LiH, Zhou Z,WangWH,Deng A, Andrisani O, et al. Plk1-mediatedphosphorylation of Topors regulates p53 stability. J Biol Chem 2009;284:18588–92.
37. Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, et al.Large-scale characterization of HeLa cell nuclear phosphoproteins. ProcNatl Acad Sci U S A 2004;101:12130–5.
38. Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT,Kratchmarova I, et al. System-wide temporal characterization of the pro-teome and phosphoproteome of human embryonic stem cell differenti-ation. Sci Signal 2011;4:rs3.
39. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-basedapproach for high-throughput protein phosphorylation analysis and sitelocalization. Nat Biotechnol 2006;24:1285–92.
40. Yamamoto K, Sonoda M, Inokuchi J, Shirasawa S, Sasazuki T. Polycombgroup suppressor of zeste 12 links heterochromatin protein 1alpha andenhancer of zeste 2. J Biol Chem 2004;279:401–6.
41. Yoon JH, Abdelmohsen K, Kim J, Yang X, Martindale JL, Tominaga-Yamanaka K, et al. Scaffold function of long non-coding RNA HOTAIRin protein ubiquitination. Nat Commun 2013;4:2939.
42. Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histoneH3 is required for proper chromosome condensation and segregation. Cell1999;97:99–109.
43. Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration andstorage platform. Nat Rev Mol Cell Biol 2013;14:211–24.
44. Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, et al. Cyclin-dependentkinases regulate epigenetic gene silencing through phosphorylation ofEZH2. Nat Cell Biol 2010;12:1108–14.
45. Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, et al. Phosphor-ylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 2010;24:2615–20.
www.aacrjournals.org Cancer Res; 75(11) June 1, 2015 2373
Plk1- and HOTAIR-Mediated Degradation of SUZ12 and ZNF198
46. Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, et al. CDK1-dependentphosphorylation of EZH2 suppressesmethylation ofH3K27 andpromotesosteogenic differentiation of humanmesenchymal stem cells. Nat Cell Biol2011;13:87–94.
47. Kim E, KimM, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation ofEZH2 activates STAT3 signaling via STAT3 methylation and promotestumorigenicity of glioblastoma stem-like cells. CancerCell 2013;23:839–52.
48. Wajapeyee N, Malonia SK, Palakurthy RK, Green MR. Oncogenic RASdirects silencing of tumor suppressor genes through ordered recruitmentof transcriptional repressors. Genes Dev 2013;27:2221–6.
49. Fan H, Zhang H, Pascuzzi P, Andrisani O. Hepatitis B virus Xprotein induces EpCAM expression via active DNA demethylationdirected by RelA in complex with EZH2 and TET2. Oncogene 2015,in press.
50. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al.EpCAM-positive hepatocellular carcinoma cells are tumor-initiatingcells with stem/progenitor cell features. Gastroenterology 2009;136:1012–24.
51. Wan Y, Chang HY. HOTAIR: Flight of noncoding RNAs in cancer metas-tasis. Cell Cycle 2010;9:3391–2.
Cancer Res; 75(11) June 1, 2015 Cancer Research2374













![Reproductive Long Intergenic Noncoding RNAs Exhibit Male … · opmental patterning (HOX TRANSCRIPT ANTISENSE RNA [HOTAIR]), the induction and maintenance of pluripotency in embryonic](https://static.documents.pub/doc/80x56/5f50b0a1706f3517f829f0e1/reproductive-long-intergenic-noncoding-rnas-exhibit-male-opmental-patterning-hox.jpg)













![HOTAIR and its surrogate DNA methylation signature ... · oligonucleotide arrays, Gene Expression Omnibus acces-sion [GEO:GSE13876]). Patients gave informed consent for collection](https://static.documents.pub/doc/80x56/6120770f18fd6b3bae792c6e/hotair-and-its-surrogate-dna-methylation-signature-oligonucleotide-arrays-gene.jpg)
