17
Point Specific Alignment Methods PSI – BLAST & PHI – BLAST
| Date post: | 21-Dec-2015 |
| Category: |
Documents |
| View: | 247 times |
| Download: | 1 times |

Point Specific Alignment Methods
PSI ndash BLAST
amp
PHI ndash BLAST
In order to control the quality of the sequence matches in a BLAST search controls are placed on the E ndash value of the result
The Expect value (E) is a parameter that describes the number of hits one can expect to see just by chance when searching a database of a particular size It decreases exponentially with the Score (S) that is assigned to a match between two sequences Essentially the E value describes the random background noise that exists for matches between sequences For example an E value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance This means that the lower the E-value or the closer it is to 0 the more significant the match is However keep in mind that searches with short sequences can be virtually indentical and have relatively high E-value This is because the calculation of the E-value also takes into account the length of the Query sequence This is because shorter sequences have a high probability of occurring in the database purely by chance
One criticism of this type of control is that sequences having basically the same functionality may be missed in the search since they score over the threshold value Here is one possible cure
The Expect value can also be used as a convenient way to create a significance threshold for reporting results You can change the Expect value threshold on most main BLAST search pages When the Expect value is increased from the default value of 10 a larger list with more low-scoring hits can be reported
Another strategy is to change the rewardpenalty ratio in the scoring system
Many nucleotide searches use a simple scoring system that consists of a reward for a match and a penalty for a mismatch The (absolute) rewardpenalty ratio should be increased as one looks at more divergent sequences A ratio of 033 (1-3) is appropriate for sequences that are about 99 conserved a ratio of 05 (1-2) is best for sequences that are 95 conserved a ratio of about one (1-1) is best for sequences that are 75 conserved
On the other hand if we become too liberal in expanding these parameters or change ratios without reason we find that we can find matches for almost any sequence For example consider the amino acid sequence (V was used in place of U)
CVTTHESTEAKWITHASHARPKNIFEELSESTRINGYMEATWILLRESULT
We will use the protien ndash protein BLAST for short sequences using a non-redundant database an Expect Value of 20 and the PAM30 matrix and the Smith-Wateman algorithm
We get 37 matches for this nonsense sequence The highest scoring match has an E-value of 13
gi|121705510|ref|XP_0012710181| C6 transcription factor put 346 13
gi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv ory 337 23
gi|84625349|ref|YP_4527211| HmsF protein [Xanthomonas oryzae 337 23
gi|123445879|ref|XP_0013116951| hypothetical protein TVAG_49 329 42
gi|123469845|ref|XP_0013181321| helicase putative [Trichomo 325 56
gi|118032193|ref|ZP_015036441| conserved hypothetical protei 325 56
gtgi|121705510|ref|XP_0012710181| C6 transcription factor putative [Aspergillus clavatus NRRL 1]
gi|119399164|gb|EAW095921| C6 transcription factor putative [Aspergillus clavatus NRRL 1] Length=887 Score = 346 bits (74)
Expect = 13 Identities = 1632 (50) Positives = 1932 (59) Gaps = 932 (28)
Query 26 EE---LSESTRINGYM----EATWI--LLRES 48
EE L+ES+R GYM E TW+ L RES
Sbjct 223 EEDLNLTESSRATGYMGKNSELTWMQRLQRES 254
gtgi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv oryzae KACC10331]
gi|58428129|gb|AAW771661| HmsF protein [Xanthomonas oryzae pv oryzae KACC10331] Length=663 Score = 337 bits (72)
Expect = 23 Identities = 1641 (39) Positives = 2241 (53) Gaps = 1441 (34)
Query 18 HARP--KNIFEELSESTRINGYMEATWIL------LRESVL 50
AR K+I+E+L+ IN YME IL LR++ L
Sbjct 460 QARQIIKDIYEDLA----INSYMEG--ILFHDDGYLRDTEL 494
Even using Local Sequence Alignment Techniques and Scoring Matrices such as high powers of PAM or low values of BLOSUMn Database Searching may not find what we want
bull Many homologous sequences share only limited sequence identity
bull While they may adopt the same three-dimensional structure they may not have apparent similarity in pair wise alignments
bull Cases are known where BLAST and FASTA miss 10 ndash 20 of ldquomeaningfulrdquo hits
bull Scoring matrices do not accurately portray the similarity that may exist within a particular family of proteins They are tied to a more general database
In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

In order to control the quality of the sequence matches in a BLAST search controls are placed on the E ndash value of the result
The Expect value (E) is a parameter that describes the number of hits one can expect to see just by chance when searching a database of a particular size It decreases exponentially with the Score (S) that is assigned to a match between two sequences Essentially the E value describes the random background noise that exists for matches between sequences For example an E value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance This means that the lower the E-value or the closer it is to 0 the more significant the match is However keep in mind that searches with short sequences can be virtually indentical and have relatively high E-value This is because the calculation of the E-value also takes into account the length of the Query sequence This is because shorter sequences have a high probability of occurring in the database purely by chance
One criticism of this type of control is that sequences having basically the same functionality may be missed in the search since they score over the threshold value Here is one possible cure
The Expect value can also be used as a convenient way to create a significance threshold for reporting results You can change the Expect value threshold on most main BLAST search pages When the Expect value is increased from the default value of 10 a larger list with more low-scoring hits can be reported
Another strategy is to change the rewardpenalty ratio in the scoring system
Many nucleotide searches use a simple scoring system that consists of a reward for a match and a penalty for a mismatch The (absolute) rewardpenalty ratio should be increased as one looks at more divergent sequences A ratio of 033 (1-3) is appropriate for sequences that are about 99 conserved a ratio of 05 (1-2) is best for sequences that are 95 conserved a ratio of about one (1-1) is best for sequences that are 75 conserved
On the other hand if we become too liberal in expanding these parameters or change ratios without reason we find that we can find matches for almost any sequence For example consider the amino acid sequence (V was used in place of U)
CVTTHESTEAKWITHASHARPKNIFEELSESTRINGYMEATWILLRESULT
We will use the protien ndash protein BLAST for short sequences using a non-redundant database an Expect Value of 20 and the PAM30 matrix and the Smith-Wateman algorithm
We get 37 matches for this nonsense sequence The highest scoring match has an E-value of 13
gi|121705510|ref|XP_0012710181| C6 transcription factor put 346 13
gi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv ory 337 23
gi|84625349|ref|YP_4527211| HmsF protein [Xanthomonas oryzae 337 23
gi|123445879|ref|XP_0013116951| hypothetical protein TVAG_49 329 42
gi|123469845|ref|XP_0013181321| helicase putative [Trichomo 325 56
gi|118032193|ref|ZP_015036441| conserved hypothetical protei 325 56
gtgi|121705510|ref|XP_0012710181| C6 transcription factor putative [Aspergillus clavatus NRRL 1]
gi|119399164|gb|EAW095921| C6 transcription factor putative [Aspergillus clavatus NRRL 1] Length=887 Score = 346 bits (74)
Expect = 13 Identities = 1632 (50) Positives = 1932 (59) Gaps = 932 (28)
Query 26 EE---LSESTRINGYM----EATWI--LLRES 48
EE L+ES+R GYM E TW+ L RES
Sbjct 223 EEDLNLTESSRATGYMGKNSELTWMQRLQRES 254
gtgi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv oryzae KACC10331]
gi|58428129|gb|AAW771661| HmsF protein [Xanthomonas oryzae pv oryzae KACC10331] Length=663 Score = 337 bits (72)
Expect = 23 Identities = 1641 (39) Positives = 2241 (53) Gaps = 1441 (34)
Query 18 HARP--KNIFEELSESTRINGYMEATWIL------LRESVL 50
AR K+I+E+L+ IN YME IL LR++ L
Sbjct 460 QARQIIKDIYEDLA----INSYMEG--ILFHDDGYLRDTEL 494
Even using Local Sequence Alignment Techniques and Scoring Matrices such as high powers of PAM or low values of BLOSUMn Database Searching may not find what we want
bull Many homologous sequences share only limited sequence identity
bull While they may adopt the same three-dimensional structure they may not have apparent similarity in pair wise alignments
bull Cases are known where BLAST and FASTA miss 10 ndash 20 of ldquomeaningfulrdquo hits
bull Scoring matrices do not accurately portray the similarity that may exist within a particular family of proteins They are tied to a more general database
In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

Another strategy is to change the rewardpenalty ratio in the scoring system
Many nucleotide searches use a simple scoring system that consists of a reward for a match and a penalty for a mismatch The (absolute) rewardpenalty ratio should be increased as one looks at more divergent sequences A ratio of 033 (1-3) is appropriate for sequences that are about 99 conserved a ratio of 05 (1-2) is best for sequences that are 95 conserved a ratio of about one (1-1) is best for sequences that are 75 conserved
On the other hand if we become too liberal in expanding these parameters or change ratios without reason we find that we can find matches for almost any sequence For example consider the amino acid sequence (V was used in place of U)
CVTTHESTEAKWITHASHARPKNIFEELSESTRINGYMEATWILLRESULT
We will use the protien ndash protein BLAST for short sequences using a non-redundant database an Expect Value of 20 and the PAM30 matrix and the Smith-Wateman algorithm
We get 37 matches for this nonsense sequence The highest scoring match has an E-value of 13
gi|121705510|ref|XP_0012710181| C6 transcription factor put 346 13
gi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv ory 337 23
gi|84625349|ref|YP_4527211| HmsF protein [Xanthomonas oryzae 337 23
gi|123445879|ref|XP_0013116951| hypothetical protein TVAG_49 329 42
gi|123469845|ref|XP_0013181321| helicase putative [Trichomo 325 56
gi|118032193|ref|ZP_015036441| conserved hypothetical protei 325 56
gtgi|121705510|ref|XP_0012710181| C6 transcription factor putative [Aspergillus clavatus NRRL 1]
gi|119399164|gb|EAW095921| C6 transcription factor putative [Aspergillus clavatus NRRL 1] Length=887 Score = 346 bits (74)
Expect = 13 Identities = 1632 (50) Positives = 1932 (59) Gaps = 932 (28)
Query 26 EE---LSESTRINGYM----EATWI--LLRES 48
EE L+ES+R GYM E TW+ L RES
Sbjct 223 EEDLNLTESSRATGYMGKNSELTWMQRLQRES 254
gtgi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv oryzae KACC10331]
gi|58428129|gb|AAW771661| HmsF protein [Xanthomonas oryzae pv oryzae KACC10331] Length=663 Score = 337 bits (72)
Expect = 23 Identities = 1641 (39) Positives = 2241 (53) Gaps = 1441 (34)
Query 18 HARP--KNIFEELSESTRINGYMEATWIL------LRESVL 50
AR K+I+E+L+ IN YME IL LR++ L
Sbjct 460 QARQIIKDIYEDLA----INSYMEG--ILFHDDGYLRDTEL 494
Even using Local Sequence Alignment Techniques and Scoring Matrices such as high powers of PAM or low values of BLOSUMn Database Searching may not find what we want
bull Many homologous sequences share only limited sequence identity
bull While they may adopt the same three-dimensional structure they may not have apparent similarity in pair wise alignments
bull Cases are known where BLAST and FASTA miss 10 ndash 20 of ldquomeaningfulrdquo hits
bull Scoring matrices do not accurately portray the similarity that may exist within a particular family of proteins They are tied to a more general database
In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

gtgi|121705510|ref|XP_0012710181| C6 transcription factor putative [Aspergillus clavatus NRRL 1]
gi|119399164|gb|EAW095921| C6 transcription factor putative [Aspergillus clavatus NRRL 1] Length=887 Score = 346 bits (74)
Expect = 13 Identities = 1632 (50) Positives = 1932 (59) Gaps = 932 (28)
Query 26 EE---LSESTRINGYM----EATWI--LLRES 48
EE L+ES+R GYM E TW+ L RES
Sbjct 223 EEDLNLTESSRATGYMGKNSELTWMQRLQRES 254
gtgi|58583535|ref|YP_2025511| HmsF [Xanthomonas oryzae pv oryzae KACC10331]
gi|58428129|gb|AAW771661| HmsF protein [Xanthomonas oryzae pv oryzae KACC10331] Length=663 Score = 337 bits (72)
Expect = 23 Identities = 1641 (39) Positives = 2241 (53) Gaps = 1441 (34)
Query 18 HARP--KNIFEELSESTRINGYMEATWIL------LRESVL 50
AR K+I+E+L+ IN YME IL LR++ L
Sbjct 460 QARQIIKDIYEDLA----INSYMEG--ILFHDDGYLRDTEL 494
Even using Local Sequence Alignment Techniques and Scoring Matrices such as high powers of PAM or low values of BLOSUMn Database Searching may not find what we want
bull Many homologous sequences share only limited sequence identity
bull While they may adopt the same three-dimensional structure they may not have apparent similarity in pair wise alignments
bull Cases are known where BLAST and FASTA miss 10 ndash 20 of ldquomeaningfulrdquo hits
bull Scoring matrices do not accurately portray the similarity that may exist within a particular family of proteins They are tied to a more general database
In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

Even using Local Sequence Alignment Techniques and Scoring Matrices such as high powers of PAM or low values of BLOSUMn Database Searching may not find what we want
bull Many homologous sequences share only limited sequence identity
bull While they may adopt the same three-dimensional structure they may not have apparent similarity in pair wise alignments
bull Cases are known where BLAST and FASTA miss 10 ndash 20 of ldquomeaningfulrdquo hits
bull Scoring matrices do not accurately portray the similarity that may exist within a particular family of proteins They are tied to a more general database
In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

In an attempt to correct this the idea of a Position Specific Scoring Matrix (PSSM) was developed
In PSI-BLAST the query sequence is subjected to a normal BLAST search From this a multiple-sequence alignment is made between the query and all ldquosignificantrdquo hits
A new scoring matrix of size L rows and 20 columns is derived using the frequency of the proteins within each position of the alignment (L is the length of the query sequence)
The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

The previous example was taken from
Pevsner J Bioinformatics and Functional Genomics
Wiley-LISS 2003 p139
And involves a search with Query sequence RBP4 (NP_006735)
Here is a portion of the PSSM generated by Pevsnerrsquos Search
Note Lines 6 11 12 14 15 16 and 42 all of which are scores for A against the 20 proteins
The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

The PSSM is then used as the query (not your original sequence) to the database and another search to the database
The statistical significance of each match is estimated and results are reported
These last three steps are repeated iteratively until no new sequences are reported that fall above the given significance level or the user chooses to terminate the search
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations
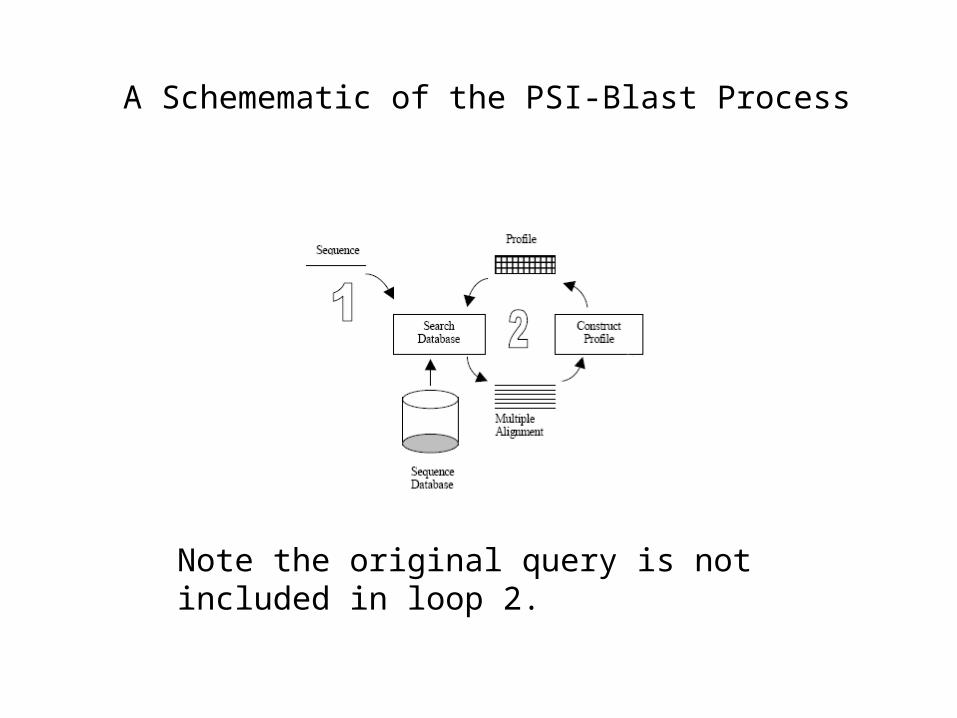
A Schemematic of the PSI-Blast Process
Note the original query is not included in loop 2
Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

Pevsner reported the following data concerning his 2002 search with original query NP_006735
At this point we will do an update of these results by going to httpwwwncbinlmnihgovblast and choosing the PSI-BLAST option with the default parameters
A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

A Dramatic Illustration of the Increased Sensitivity ot PSI-BLAST Searching
PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

PHI-BLAST stands for Pattern-Hit Initiated BLAST
Often it is the case that a protein of interest contains a signature pattern of amino acids and residues that help to define it as part of the family This ldquosignaturerdquo may be rather short in terms of its length within the sequence but it is important in defining a structural of functional domain It may even be the characteristic of an unknown function as is the case in the following example
Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

Care must be taken to choose a pattern that is not common within the database The algorithm only allows patterns that are expected to occur at most once in every 5000 residues
In the previous example the pattern is GXW where the X may be any amino acid Then we specify candidates for the following amino acids [YF] [EA] or [IVLM] These choices are based on our observation of the test sequences and our knowledge of the behavior of proteins (common protein substitutions hydrophobicity etc)
The database search is then performed looking for sequences that contain the prescribed pattern
Further iterations may be done based on this output using PSI-BLAST which no longer uses the PHI pattern but the PSSM from the first report
The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

The output from the PHI-BLAST program is the same as that of the PSI-BLAST program except that the position of the pattern is highlighted in each of the alignments
The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

The following alignment was obtained from an investigation of immunoglobulin C-Region Domains
We will investigate the conserved sequence LXCLV using PHI-BLAST
Our starting point is with the Ig 2A C region of the mouse SwissProt Accession P01865
We enter this information into the PHI-BLAST page
The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

The first iteration of this search yields 31 new statistically significant hits One of these is given below Note the rsquos over the location of the pattern LXCLV
Subsequent iterations are performed by PSI-BLAST independent of the pattern This search converged after 13 iterations

















