Postinjury Administration of Pituitary Adenylate CyclaseActivating Polypeptide (PACAP) Attenuates Traumatically
Induced Axonal Injury in Rats
ANDREA TAMÁS,1 ANDREA ZSOMBOK,2,3 ORSOLYA FARKAS,2 DÓRA REGLÖDI,1JÓZSEF PÁL,2,5 ANDRÁS BÜKI,2 ISTVÁN LENGVÁRI,1 JOHN T. POVLISHOCK,4
and TAMÁS DÓCZI2,5
ABSTRACT
Pituitary adenylate cyclase activating polypeptide (PACAP) has several different actions in the ner-vous system. Numerous studies have shown its neuroprotective effects both in vitro and in vivo. Pre-viously, it has been demonstrated that PACAP reduces brain damage in rat models of global andfocal cerebral ischemia. Based on the protective effects of PACAP in cerebral ischemia and the pres-ence of common pathogenic mechanisms in cerebral ischemia and traumatic brain injury (TBI), theaim of the present study was to investigate the possible protective effect of PACAP administered 30min or 1 h postinjury in a rat model of diffuse axonal injury. Adult Wistar male rats were subjectedto impact acceleration, and PACAP was administered intracerebroventricularly 30 min (n � 4), and1 h after the injury (n � 5). Control animals received the same volume of vehicle at both time-points(n � 5). Two hours after the injury, brains were processed for immunhistochemical localization ofdamaged axonal profiles displaying either �–amyloid precursor protein (�-APP) or RMO-14 im-munoreactivity, both considered markers of specific features of traumatic axonal injury. Our re-sults show that treatment with PACAP (100 �g) 30 min or 1 h after the induction of TBI resultedin a significant reduction of the density of �-APP–immunopositive axon profiles in the corticospinaltract (CSpT). There was no significant difference between the density of �-APP–immunopositiveaxons in the medial longitudinal fascicle (MLF). PACAP treatment did not result in significantlydifferent number of RMO-14–immunopositive axonal profiles in either brain areas 2 hours post-in-jury compared to normal animals. While the results of this study highlighted the complexity of thepathogenesis and manifestation of diffuse axonal injury, they also indicate that PACAP should beconsidered a potential therapeutic agent in TBI.
Departments of 1Anatomy (Neurohumoral Regulations Research Group of the Hungarian Academy of Sciences), 2Neurosurgeryand 3Central Laboratory of Animal Research, University of Pécs, Medical Faculty, Pécs, Hungary.
4Department of Anatomy and Neurobiology, Virginia Commonwealth University, Richmond, Virginia.5Clinical Neuroscience Research Group of the Hungarian Academy of Sciences, Department of Neurosurgery, University of
Pécs, Medical Faculty, Pécs, Hungary.
INTRODUCTION
NEUROPATHOLOGICAL STUDIES, clinical observations,and other data derived from animal models have sig-
nificantly increased the understanding of both primaryand secondary brain damage over the past decades. In up to 45% of severely head-injured patients, the neuro-pathological changes after head injury may be due to theeffects of secondary mechanisms. The early insults, suchas transient global ischemia, haematoma, or diffuse ax-onal injury, are followed and complicated by secondarymechanisms, which may be mediated by complex cas-cades of biochemical processes. Many of these secondaryposttraumatic events have been targeted as potential sitesfor pharmacological interventions to prevent or to sig-nificantly reduce secondary brain damage (Bullock,1993).
Numerous neuroprotective drugs and therapeutic in-terventions have been tested in different animal modelsof traumatic brain injury (TBI). Despite the obvious dif-ferences in their pathogenesis, TBI and ischemic braindamage do share some common pathways providing asolid basis for the application of—at least partially—sim-ilar treatment strategies. Such mechanisms include exci-totoxicity, overproduction of free radicals, inflammation,and apoptosis (Bramlett and Dietrich, 2004; Leker andShohami, 2002).
One of the most widely used therapeutic approachesis hypothermia. Postinjury hypothermia is extensivelystudied and utilized for neuroprotection in several mod-els of ischemia and traumatic injuries of the nervous sys-tem (Bethea and Dietrich, 2002; Bramlett et al., 1995;1997; Koizumi and Povlishock, 1998; Lyeth et al., 1993).Hypothermia prevents free radical production (Globus etal., 1995), nitric oxide synthesis (Sakamoto et al., 1997),intracellular calcium accumulation (Mitani et al., 1991),and the rise in excitatory amino acid secretion (Koizumiet al., 1997).
There are several other therapeutic interventions effec-tive both in cerebral ischemia and TBI. The efficacy of posttraumatic Mg2� as a neuroprotective agent has been shown in both diffuse axonal injury and brain ischemia (Heath and Vink, 1999; Tsuda et al., 1991). Sim-ilarly, treatment with NMDA channel blockers limits ex-citotoxin-induced secondary neuronal damage in modelsof TBI (Goda et al., 2002) and cerebral ischemia (Dirnaglet al., 1990). Inhibition of inflammatory mediators and cal-pain has also been proven effective in both models (Bukiet al., 1999b, 2003; Cernak et al., 2002; Hong et al., 1994;Kadoya et al., 1995; Nakayama et al, 1998; Sanderson etal., 1999; Yoshimoto and Siesjo, 1999).
Several neurotrophic factors, such as brain-derivedneurotrophic factor, nerve growth factor and fibroblast
growth factor, have been shown to play an important rolein the endogenous response following both cerebral ischemia and traumatic brain injury, and to be effectivein both brain pathologies when given exogenously (Di-etrich et al., 1996; Kawamata et al., 1997; Leker andShohami, 2002; Truettner et al., 1999). Pituitary adeny-late cyclase-activating polypeptide (PACAP) is a neu-ropeptide structurally belonging to the secretin/glucagon/VIP family. PACAP was isolated from ovine hypothala-mus based on its ability to activate adenylate cyclase inthe pituitary gland. PACAP is a bioactive peptide withdiverse activities in the nervous system. In addition to itsmore classic role as a neurotransmitter, PACAP functionsas a neurotrophic factor (Somogyvari-Vigh and Reglodi,2004). Numerous studies have shown its neuroprotectiveeffect in vitro and in vivo (Arimura, 1998; Somogyvari-Vigh and Reglodi, 2004; Vaudry et al., 2000a).
Previously, it has been demonstrated that PACAP hasneuroprotective effects in a rat model of global and focalcerebral ischemia treated before and after the injury(Uchida et al., 1996; Reglodi et al., 2000, 2002). Based onthe findings with PACAP in cerebral ischemia and thepresence of common pathogenetic mechanisms in cerebralischemia and TBI, we applied PACAP therapy in a ratmodel of TBI in a previous study. It was found that PACAPhad neuroprotective effects when administered immedi-ately after the injury (Farkas et al., 2004). In acute cere-bral injuries, it is of utmost importance to determine thetherapeutic time window of candidate neuroprotectiveagents, since the immediate therapy is not possible in mostcases. Of similar importance is the identification of thera-peutic interventions that could be applied at the scene ofthe accident in an attempt to prolong the time window forfurther neuroprotective interventions and measures to in-hibit progression to irreversible structural changes. Theaim of the present study was therefore, to investigate thepossible protective effect of PACAP in a rat model of TBIgiven 30 min and 1 h postinjury in order to examine thepossibility of delaying the therapeutic window.
MATERIALS AND METHODS
Induction of Traumatic Brain Injury
Adult Wistar male rats (375–400 g) were subjected toimpact acceleration TBI in accordance with previous de-scriptions (Marmarou et al., 1994; Foda and Marmarou,1994). Animals were anesthetized in a bell jar for 5 minunder 4% isoflurane (Forane, Abott, Hungary) and a 2:1mixture of N2O:O2, endotracheally intubated, and main-tained under 1.5–2% isoflurane. Rectal and temporal tem-perature were monitored and maintained at 37°C with aventral heating pad (FHC BOWDOINHAMME 04008
POSTINJURY ADMINISTRATION OF PACAP IN TBI
687
USA Temperature Control). A midline scalp incision wasmade and the skull was exposed with blunt dissection. Pari-etal bones were exposed, cleaned, and dried with bone wax.A 10-mm stainless steel disc helmet was secured with den-tal acrylic to the skull between bregma and lambda. Ratswere then fastened prone to a foam bed beneath the injurytower and injured from 2 m with a 450-g weight. After in-jury, animals were immediately ventilated with 100% O2,the helmet was removed, and the skull examined for signof fracture, which, if found, disqualified the rat from fur-ther evaluation. Selected animals underwent monitoring ofother physiological parameters including invasive bloodpressure, pulse oxymetry and arterial blood gas analysis.All procedures were performed in accordance with the eth-ical guidelines of NIH and guidelines approved by the Uni-versity of Pécs (no. BA02/2000-31/2001) to minimize painand suffering of the animals. The minimal number of ani-mals required to achieve statistically meaningful results wasused in all cases.
Drug Administration
A single bolus of 100 �g PACAP (Sigma, Hungary)dissolved in 5 �L of physiological saline was adminis-tered intracerebroventriculary 30 min after the injury inone group of rats (n � 4), and 1 h after the injury in an-other group (n � 5). Control animals received the samevolume of vehicle at both time-points (n � 5). The doseof PACAP was selected based on previous studies de-scribing dose-dependent characteristics of PACAP ad-ministration (Farkas et al., 2004). Sham-injured rats (n �4) were anesthetized, intubated and the parietal boneswere exposed without performing the impact accelera-tion injury. The animals were sacrificed for the histolog-ical assessment 2 h after brain injury.
Immunohistochemistry
At the designed survival time, animals were reanes-thetized with an overdose of sodium pentobarbital andtranscardially perfused with Zamboni’s fixative (Zam-boni and Martino, 1967). The brains were removed andimmersed in the same fixative overnight (16–18 h). Basedupon previously published observations concerning thetopography of diffusely injured axons in the rat brain-stem (Povlishock et al., 1997), a median 5-mm-wideblock of the brain was removed using a sagittal brainblocking device (Braintree Scientific Inc.) to include theregion extending from the interpeduncular fossa to thefirst cervical segment. The brainstem was blocked justlateral to the pyramids to encompass known vulnerablefiber tracts in this model of TBI: the corticospinal tract(CSpT) and the medial longitudinal fascicle (MLF). Thetissue was sectioned with Vibrotome Series 1000 (Poly-
sciences Inc., Warrington, PA) at a thickness of 40 �mand collected in 0.1 M phosphate buffer. The sectionswere collected in two groups in a semi-serial fashion,rinsed three times for 10 min in phosphate-buffered saline(PBS), and processed for immunhistochemical localiza-tion of damaged axonal profiles.
Half of the sections were single labeled for the detec-tion of RMO-14 immunoreactivity. This antibody inknown to exclusively target an epitope on the rod domainsof altered NF-M subunits, which are exposed upon mod-ification of the NF sidearms, and assumed consequence ofcalcium induced enzymatic processes (Lee et al., 1987;Okonkwo et al., 1998; Povlishock et al., 1997). Every sec-ond section was processed for immunohistochemical de-tection of the �–amyloid precursor protein (�-APP). Thisclassical marker of traumatic axonal injury (TAI) is car-ried by fast axoplasmic transport and will pool at foci af-fected by TAI. In the present study, we utilized a poly-clonal antiserum targeting the C-terminus of �-APP(Gentleman et al., 1993; Stone et al., 2000). Vibratomesections microwaved for optimizing antigenicity with theretention of excellent ultrastructural detail according toformer publications (Stone et al., 1999). Sections fromboth groups were then washed in PBS containing 1% nor-mal goat serum (Fluca), and were incubated for 35 minwith 0.2% Triton X (Sigma-Aldrich, Hungary) in 10% nor-mal goat or horse serum, respectively (NGS or NHS;Sigma Chemical Co., St. Louis, MO) in PBS. After twoquick rinses in PBS containing 1% NGS (or NHS in thecase of RMO-14) the above defined groups of sectionswere incubated overnight in rabbit anti-APP antibody at adilution of 1:3000 (Zymed Laboratories, Hartford, CT) orin mouse monoclonal RMO-14 antibody (kindly providedby Dr. John Q. Trojanowski, University of PennsylvaniaDepartment of Pathology) at the dilution of 1:500. Hav-ing been rinsed 3 � 10 min in PBS containing 1% NGS(or NHS), sections were incubated in biotinylated anti-rab-bit immunoglobuline (Sigma) derived from goat or in1:400 dilution of biotinylated, rat adsorbed anti-mouseimmunoglobulin derived from horse (Vector) for 60 min.After incubation in avidin biotin-peroxidase complex(ABC kit, Vector) and rinsing in PBS and 0.1 M phos-phate buffer 3 � 10 min and 2 � 10 min respectively, sec-tions were processed for visualization of the immunohis-tochemical complex using 0.05% diaminobenzidine(Sigma) and 0.01% hydrogen peroxide in 0.1 M PBS. Thesections were then mounted and cleared for routine lightmicroscopic examination.
Image Analysis
Semi-serial brainstem sections were examined with aNikon light microscope interfaced with a computer-as-
TAMÁS ET AL.
688
sisted image analysis system (NIH Image J software) ina blinded fashion. At the pontomedullary junction, twoadjacent grids of 40,000 �m2 were superimposed overthe CSpT and the MLF, images were captured and digi-tized at a magnification of �50 and the total number ofdamaged APP and RMO-14 immunopositive axonal pro-files within this area were marked and counted. The meandensity of immunopositive axons was computed as anumber of immunopositive axons per mm2.
Statistical Analysis
Student t-test was used to compare the density of im-munopositive profiles (expressed as mean number/mm2)between the control and PACAP-treated animals.
RESULTS
Neither the use of the above-described experimentalTBI protocol nor the administration of PACAP resultedin any significant alteration in the physiological parame-ters monitored during the experiment (temporal and rec-tal temperature, blood pressure, and blood gases), con-sistent with previous observations from our laboratoriesand others’ (Buki et al., 1999a, 2003; Otani et al., 2002;Reglodi et al., 2000, 2002; Singleton et al., 2001). Pre-vious observations with PACAP treatment found a tran-sient, slight drop in blood pressure only when the pep-tide was administered intravenously: fall in the bloodpressure was observed 5 min after intravenous injection,but it soon returned to normal (Reglodi et al., 2000). Con-sistent with the current study, no such alteration wasfound in previous studies when PACAP was given icv(Reglodi et al., 2002).
Light microscopic examination of vehicle- and drug-treated animals subjected to TBI and reacted for the vi-sualization of �-APP and RMO-14 antibodies, revealeddiscrete focal immunoreactivity within scattered axons inthe CSpT and the MLF. Sham-injured animals did notdisplay immunoreactive axonal profiles. Morphologicalcharacteristics of swollen, occasionally disconnected �-APP–immunoreactive axonal segments and the lobu-lated/vacuolated, partially or totally disconnected RMO-14–immunoreactive axonal segments appeared entirelyconsistent with previous descriptions of damaged axonalprofiles 2 h post-injury (Buki et al., 1999a,b, 2000; Stoneet al., 2000).
Treatment with PACAP (100 �g) at 30 min or 1 hafter the induction of TBI resulted in a significant re-duction of �-APP–immunopositive axon profiles in theCSpT when compared to control animals 2 h postinjury.There was no significant difference between the num-ber of �-APP–immunopositive axons in the MLF andRMO-14–immunopositive axonal profiles in both brainareas at 2 h after the injury compared to normal animals(Figs. 1,2,3).
Quantitative findings followed by statistical analysisconfirmed that postinjury administration of 100 �g ofPACAP significantly reduced the mean density of dam-aged �-APP–immunoreactive axonal profiles in theCSpT from 812.7 � 29.0 to 501.9 � 81.4 (p � 0.001) orto 459.7 � 27.7 (p � 0.0001) 30 min or 1 h after injury,respectively (Figs. 1,3). There was no significant changein the MLF, where the mean number of �-APP–im-munoreactive axon profiles varied from 89.8 � 14.9 inthe control group to 99.3 � 60.4 and to 53.0 � 5.0 ingroups treated with PACAP at 30 min and 1 h postin-jury, respectively (Fig. 1).
POSTINJURY ADMINISTRATION OF PACAP IN TBI
689
FIG. 1. Mean densities of amyloid precursor protein (APP)–immunopositive axons in the corticospinal tract (CSpT) and themedial longitudinal fascicle (MLF) in control and pituitary adenylate cyclase activating polypeptide (PACAP)–treated animals.**p � 0.01, ***p � 0.001 versus control group.
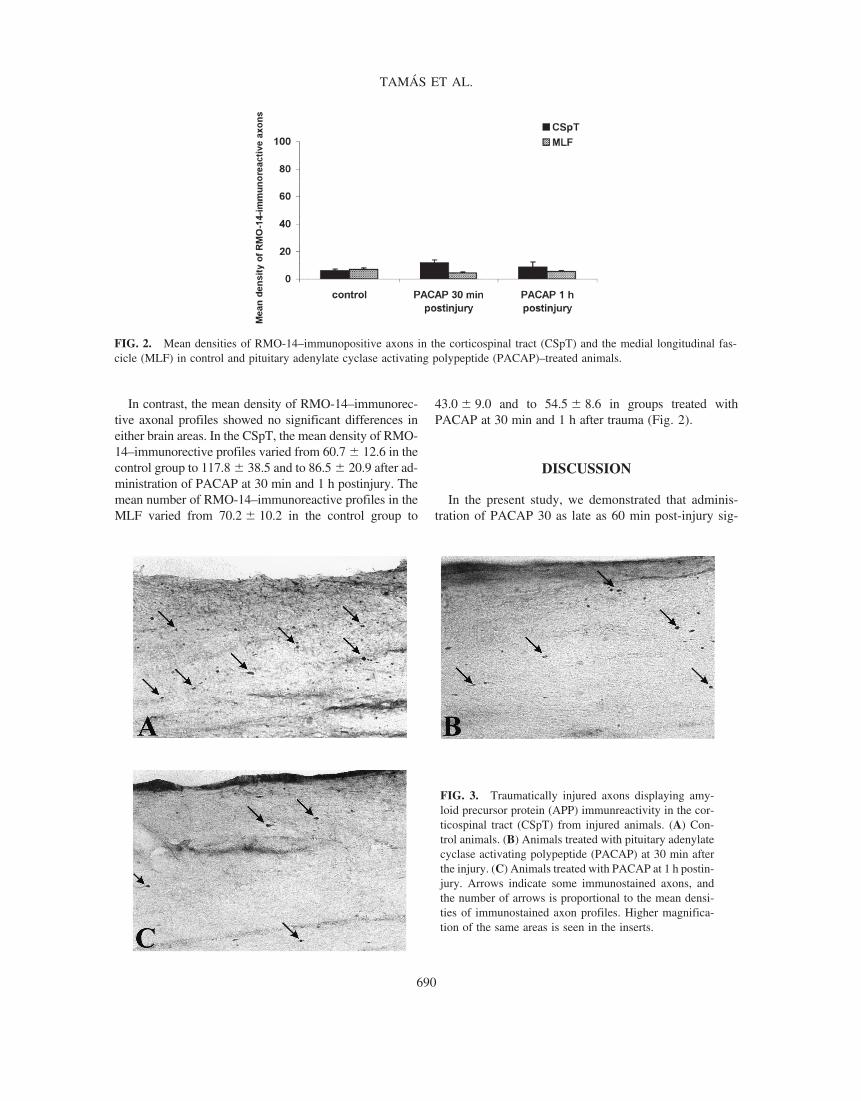
In contrast, the mean density of RMO-14–immunorec-tive axonal profiles showed no significant differences ineither brain areas. In the CSpT, the mean density of RMO-14–immunorective profiles varied from 60.7 � 12.6 in thecontrol group to 117.8 � 38.5 and to 86.5 � 20.9 after ad-ministration of PACAP at 30 min and 1 h postinjury. Themean number of RMO-14–immunoreactive profiles in theMLF varied from 70.2 � 10.2 in the control group to
43.0 � 9.0 and to 54.5 � 8.6 in groups treated withPACAP at 30 min and 1 h after trauma (Fig. 2).
DISCUSSION
In the present study, we demonstrated that adminis-tration of PACAP 30 as late as 60 min post-injury sig-
TAMÁS ET AL.
690
FIG. 2. Mean densities of RMO-14–immunopositive axons in the corticospinal tract (CSpT) and the medial longitudinal fas-cicle (MLF) in control and pituitary adenylate cyclase activating polypeptide (PACAP)–treated animals.
FIG. 3. Traumatically injured axons displaying amy-loid precursor protein (APP) immunreactivity in the cor-ticospinal tract (CSpT) from injured animals. (A) Con-trol animals. (B) Animals treated with pituitary adenylatecyclase activating polypeptide (PACAP) at 30 min afterthe injury. (C) Animals treated with PACAP at 1 h postin-jury. Arrows indicate some immunostained axons, andthe number of arrows is proportional to the mean densi-ties of immunostained axon profiles. Higher magnifica-tion of the same areas is seen in the inserts.
nificantly reduced traumatically evoked axonal injury inthe most vulnerable CSpT in the brainstem. Thus, ourstudy provides further evidence for the previously de-scribed neuroprotective effects of PACAP.
Recently, many therapeutic agents have been used inmodels of TBI, such as antiinflammatory and antiapop-totic drugs, as well as substances protective against ex-citotoxicity, to attenuate the main pathogenetic mecha-nisms present in brain injuries (Leker and Shohami,2002). Most recent results indicate that PACAP may alsobe a promising therapeutic agent in injuries of the ner-vous system. PACAP is thought to interact with growthfactors during development and following injury (Somo-gyvari-Vigh and Reglodi, 2004). In a model of TBI, in-crease of PACAP mRNA has been observed in the vul-nerable cortex and dentate gyrus concomitant with thedecrease of TUNEL-positive apoptotic cells, which mayindicate a role for PACAP in reducing the number ofapoptotic neurons (Skoglosa et al., 1999). Since its dis-covery, numerous in vitro studies have demonstrated thatPACAP has neurotrophic and neuroprotective effects.The first reports of the in vivo efficacy of PACAP showedthat the peptide exerts neuroprotection in a rat model ofglobal and focal cerebral ischemia (Ohtaki et al. 2003;Reglodi et al., 2000, 2002; Uchida et al., 1996). Sub-sequently, it has been shown that local injections ofPACAP enhance facial nerve recovery after transection,and is neuroprotective in models of optic nerve transec-tion, spinal cord injury, and 6-OHDA–induced lesion(Katahira et al., 2003; Kimura et al., 2003; Reglodi et al.,2004; Seki et al., 2003) Based on the findings withPACAP in nervous injuries and the presence of commonfeatures of pathogenetic mechanisms in cerebral ischemiaand TBI, we administered PACAP in a rat model of TBI.
The application of PACAP was considered on the ba-sis of widespread mechanisms of action. Glutamate ex-citotoxicity plays a role in the damage caused by TBI,and NMDA-antagonists have been shown to reduce theneuronal damage after impact-acceleration brain injury(Goda et al., 2002). The survival promoting effect ofPACAP has been demonstrated against glutamate-in-duced toxicity in cultured cortical and retina neurons(Frechilla et al., 2001; Morio et al., 1996; Shoge et al.,1999). A recent study has shown that PACAP upregu-lates several genes that increase resistance to neurotoxicagents, including cytochrome P450 and FGF regulatedprotein (Vaudry et al., 2002b). PACAP also increases thevelocity of glutamate uptake by cortical astroglial cellsby promoting the expression of glutamate transportersand by inducing the expression of glutamine synthetase(Figiel and Engele, 2000).
Inflammation is not restricted to infectious or autoim-mune disorders of the nervous system, but occurs in cere-
bral ischemia, trauma and neurodegenerative disorders(Leker and Shohami, 2002). Inhibition of COX-2, anenyzme responsible for the production of prostaglandinsat sites of inflammation, improves cognitive and motoroutcome following diffuse TBI in rats (Cernak et al.,2002). Interleukin-1 receptor antagonists attenuate re-gional neuronal cell death and cognitive dysfunction af-ter experimental brain injury (Sanderson et al., 1999). Inaddition, postinjury hypothermia, which is widely uti-lized for neuroprotection in TBI, also has anti-inflam-matory effect (Deng et al., 2003). PACAP has beenproven to be a potent inactivator of induced microglialrelease of proinflammatory cytokines and chemokines,such as TNF�, IL-1�, IL-6, IL-12, and NO (Delgado etal., 2002, 2003; Kim et al., 2000). Moreover, PACAPstimulates the anti-inflammatory cytokine IL-10 (Ganeaand Delgado, 2002). Recently, it has also been shownthat PACAP has inhibitory action on chemotaxis and neu-trophil migration which may play a role in its neuropro-tective effect (Kinhult et al., 2001). A few studies havedemonstrated that the anti-inflammatory actions ofPACAP exist also in vivo (Delgado et al., 1999).
Apoptotic cell death plays a role in tissue injury fol-lowing brain trauma and can be prevented by manipula-tion of the steps that lead to activation of the caspase cas-cade (Leker and Shohami, 2002). Recently, it has beendemonstrated that inhibition of the caspase cascade re-duces post-TBI (Fink et al., 1999). PACAP decreases thenaturally occurring apoptotic cell death as well as apop-tosis induced by various agents in neuronal cell lines(Canonico et al., 1996; Shioda et al., 1998; Somogyvari-Vigh and Reglodi, 2004; Tanaka et al., 1997; Vaudry etal., 2000b, 2002a). In cerebellar granule cells, PACAPreduces caspase-3 activation, which is also activated inTBI (Buki et al., 2000; Vaudry et al., 2000b).
Mitochondrial damage has long been associated withthe pathogenesis of TBI (Maxwell et al., 1997; Okonkwoet al., 1999). The mitochondrial failure purportedlycaused by calcium-induced opening of the permeabilitytransition pore is a key phenomenon in multiple neuro-logical insults, including axonal injury (Buki et al., 2000;Pettus et al., 1994). Cyclosporin A, a potent inhibitor ofCa2�-induced mitochondrial permeability transition,blunts the progression of calcium-mediated cytoskeletalchange and reduces the number of disconnected/dys-functional axons in rat TBI (Buki et al., 1999b; Okonkwoet al., 1999). Aconitase, a key mitochondrial enzyme in-fluencing the viability of neurons in response to oxida-tive stress, is inactivated by a deprivation of Ca2� influxinto neurons and PACAP attenuates this inactivation(Tabuchi et al., 2003).
In the present study we demonstrated that administra-tion of PACAP at 30 min and 1 h after the injury reduced
POSTINJURY ADMINISTRATION OF PACAP IN TBI
691
traumatically evoked axonal injury as assessed by im-munocytochemical marker �-APP in the CSpT, whilethere was no significant changes in the MLF. Similar sur-prising finding of this study was the lack of therapeuticeffect in the case of damaged RMO-14–immunoreactivefibers. These findings might be explained by several rea-sons: first, the design of this study may not have allowedenough statistical power to establish significant differ-ence in the MLF where the density of damaged axonalprofiles was found in previous studies about 10% of thatof the CSpT. Further, recent observations by Stone et al.(2000) indicated that APP and RMO-14 purportedly la-bel—at least partially—different axon populations; 2 hpost-injury, the former is more frequently localized to theCSpT while the latter is prevalent in the MLF at this timepoint. Similar results have been obtained by Suehiro etal. (2001) where the immunophilin ligand FK506 was ad-ministered following posttraumatic hypothermia associ-ated with rapid rewarming. Recently, it has been demon-strated that the ability to protect mitochondria fromundergoing mitochondrial permeability transition is veryimportant in the neuroprotection following posthy-pothermic rewarming (Okonkwo and Povlishock, 1999),and RMO-14 immunoreactivity correlates with the per-centage of calpain-mediated spectrin proteolysis and mi-tochondrial damage (Buki et al., 1999a; Povlishock et al.,1997; Povlishock and Stone, 2001). Based on the obser-vation that FK506 did not decrease the number of RMO-14–positive fibers, the authors confirmed that FK506 hasno known action on mitochondrial permeability transi-tion (Liu et al., 1991; Suehiro et al., 2001). Presently,there are no data available on the effects of PACAP oncalpain-mediated spectrin proteolysis, which should cor-relate RMO-14 immunoreactivity. The differences be-tween the structure of fibers situated in the CSpT and theMLF, and the different pathogenesis of axonal damagein these structures could also be the reason for differentprotective effects (Povlishock and Stone, 2001; Suehiroet al., 2001).
A major finding in our study is that PACAP was ef-fective even when given delayed after the trauma. Nu-merous investigations have shown the protective effectof different treatments applied before injury (Buki et al.,2003; Singleton et al., 2001) and immediately after theinjury (Koizumi and Povlishock, 1998). Clinical thera-pies on the other hand focus on delayed administrationstrategies post-injury. It has been shown that cyclosporinA has neuroprotective effect when given 30 min after TBI(Buki et al., 1999b), while MgSO4 improves motor out-come when administered up to 24 h after injury in ratTBI (Heath et al., 1999). Previously, we have demon-strated that PACAP is effective when applied immedi-ately after the injury (Farkas et al., 2004), and in this
study we have proven that PACAP has neuroprotectiveeffect also 30 min and 1 h after TBI. These findings in-dicate a considerable therapeutic window existing for theuse of PACAP in TBI, implicating PACAP to the ther-apy of traumatic axonal injury.
ACKNOWLEDGMENTS
This work was supported by the National Science Re-search Fund (OTKA T034491, T035195, T046589,T048724), Fogarty IRCA (1-RO3-TW0131302A1), NIH(grant NS20193), National Science Projects (NFKP1A/00026/2002 and ALK 000126/2002), and the HungarianAcademy of Sciences, Bolyai and Bekesy Scholarships.
REFERENCES
ARIMURA, A. (1998). Perspectives on pituitary adenylate cy-clase activating polypeptide (PACAP) in the neuroendocrine,endocrine, and nervous systems. Jpn. J. Physiol. 48, 301–331.
BETHEA, J.R., and DIETRICH, W.D. (2002). Targeting thehost inflammatory response in traumatic spinal cord injury.Curr. Opin. Neurol. 15, 355–360.
BRAMLETT, H.M., GREEN, E.J., DIETRICH, W.D., BUSTO,R., GLOBUS, M.Y., and GINSBERG, M.D. (1995). Post-traumatic brain hypothermia provides protection from sen-sorimotor and cognitive behavioral deficits. J. Neurotrauma12, 289–298.
BRAMLETT, H.M., DIETRICH, W.D., GREEN, E.J., andBUSTO, R. (1997). Chronic histopathological consequencesof fluid-percussion brain injury in rats: effects of post-trau-matic hypothermia. Acta Neuropathol. (Berl.) 93, 190–199.
BRAMLETT, H.M., and DIETRICH, W.D. (2004). Patho-physiology of cerebral ischemia and brain trauma: similari-ties and differences. J. Cereb. Blood Flow Metab. 24, 133–150.
BUKI, A., KOIZUMI, H., and POVLISHOCK, J.T. (1999a).Moderate posttraumatic hypothermia decreases early calpain-mediated proteolysis and concomitant cytoskeletal compro-mise in traumatic axonal injury. Exp. Neurol. 159, 319–328.
BUKI, A., OKONKWO, D.O., and POVLISHOCK, J.T.(1999b). Postinjury cyclosporin A administration limits ax-onal damage and disconnection in traumatic brain injury. J.Neurotrauma 16, 511–521.
BUKI, A., OKONKWO, D.O., WANG, K.K., and POVLI-SHOCK, J.T. (2000). Cytochrome c release and caspase ac-tivation in traumatic axonal injury. J. Neurosci. 20, 2825–2834.
BUKI, A., FARKAS, O., DOCZI, T., and POVLISHOCK, J.T.(2003). Preinjury administration of the calpain inhibitor
BULLOCK, R. (1993). Opportunities for neuroprotective drugsin clinical management of head injury. J. Emerg. Med. 11,Suppl 1, 23–30.
CANONICO, P.L., COPANI, A., D’AGATA, V., et al. (1996).Activation of pituitary adenylate cyclase activating polypep-tide receptors prevents apoptotic cell death in cultured cere-bellar granule cells. Ann. N.Y. Acad. Sci. 805, 470–472.
CERNAK, I., O’CONNOR, C., and VINK, R. (2002). Inhibi-tion of cyclooxygenase 2 by nimesulide improves cognitiveoutcome more than motor outcome following diffuse trau-matic brain injury in rats. Exp. Brain Res. 147, 193–199.
DELGADO, M., POZO, D., MARTINEZ, C., et al. (1999). Va-soactive intestinal peptide and pituitary adenylate cyclase-ac-tivating polypeptide inhibit endotoxin-induced TNF-� pro-duction by macrophages: in vitro and in vivo studies. J.Immunol. 162, 2358–2367.
DELGADO, M., JONAKAIT, G.M., and GANEA, D. (2002).Vasoactive intestinal peptide and pituitary adenylate cyclaseactivating polypeptide inhibit chemokine production in acti-vated microglia. Glia 39, 148–161.
DELGADO, M., LECETA, J., and GANEA, D. (2003). Va-soactive intestinal peptide and pituitary adenylate cyclase ac-tivating polypeptide inhibit the production of inflammatorymediators by activated microglia. J. Leukoc. Biol. 73,155–164.
DENG, H., HAN, H.S., CHENG, D., SUN, G.H., and YENARI,M.A. (2003). Mild hypothermia inhibits inflammation afterexperimental stroke and brain inflammation. Stroke 34,2495–2501.
DIETRICH, W.D., ALONSO, O., BUSTO, R., and FINKLE-STEIN, S.P. (1996) Posttreatment with intravenous basic fi-broblast growth factor reduces histopathological damage fol-lowing fluid-percussion brain injury in rats. J. Neurotrauma13, 309–316.
DIRNAGL, U., TANABE, J., and PULSINELLI, W. (1990).Pre- and post-treatment with MK-801 but not pretreatmentalone reduces neocortical damage after focal cerebral ischemia in the rat. Brain Res. 527, 62–68.
FARKAS, O., TAMAS, A., ZSOMBOK, A., et al. (2004). Ef-fects of pituitary adenylate cyclase activating polypeptide(PACAP) in a rat model of traumatic brain injury. Regul.Peptides 123, 69–75.
FIGIEL, M., and ENGELE, J. (2000). Pituitary adenylate cy-clase activating polypeptide (PACAP), a neuron-derived pep-tide regulating glial glutamate transport and metabolism. J.Neurosci. 20, 3596–3605.
FINK, K.B., ANDREWS, L.J., BUTLER, W.E., et al. (1999).Reduction of post-traumatic brain injury and free radical pro-duction by inhibition of the caspase-1 cascade. Neuroscience94, 1213–1218.
FODA, M.A., and MARMAROU, A. (1994). A new model ofdiffuse brain injury in rats. Part II. Morphological character-ization. J. Neurosurg. 80, 301–313.
FRECHILLA, D., GARCIA-OSTA, A., PALACIOS, S., CEN-ARRUZABEITIA, E., and DEL RIO J. (2001). BDNF me-diates the neuroprotective effect of PACAP-38 on rat corti-cal neurons. Neuroreport 12, 919–923.
GANEA, D., and DELGADO, M. (2002). Vasoactive intesti-nal peptide (VIP) and pituitary adenylate cyclase activatingpolypeptide (PACAP) as modulators of both innate and adap-tive immunity. Crit. Rev. Oral Biol. Med. 13, 229–237.
GENTLEMAN, S.M., NASH, M.J., SWEETING, C.J., GRA-HAM, D.I., and ROBERTS, G.W. (1993). Beta-amyloid pre-cursor protein (beta-APP) as a marker for axonal injury af-ter head injury. Neurosci. Lett. 160, 139–144.
GLOBUS, M.Y., ALONSO, O., DIETRICH, W.D., BUSTO,R., and GINSBERG, M.D. (1995). Glutamate release and freeradical production following brain injury: Effects of post-traumatic hypothermia. J. Neurochem. 65, 1704–1711.
GODA, M., ISONO, M., FUJIKI, M., and KOBAYASHI, H.(2002). Both MK801 and NBQX reduce the neuronal dam-age after impact-acceleration brain injury. J. Neurotrauma 19,1445–1456.
HEATH, D.L., and VINK, R. (1999). Improved motor outcomein response to magnesium therapy received up to 24 hoursafter traumatic diffuse axonal brain injury in rats. J. Neuro-surg. 90, 504–509.
HONG, S.C., GOTO, Y., LANZINO, G., SOLEAU, S., KAS-SELL, N.F., and LEE, K.S. (1994). Neuroprotection with acalpain inhibitor in a model of focal cerebral ischemia. Stroke25, 663–669.
KADOYA, C., DOMINO, E.F., YANG G., STERN J.D., andBETZ, A.L. (1995). Preischemic but not postischemic zincprotoporphirin treatment reduces infarct size and edema ac-cumulation after temporary focal cerebral ischemia in rats.Stroke 26, 1035–1038.
KATAHIRA, M., YONE, K., ARISHIMA, Y., et al. (2003).The neuroprotective effects of PACAP on spinal cord injury(SCI) in rats. Regul. Peptides 115, 49(abst).
KAWAMATA, T., DIETRICH, W.D., SCHALLERT, T., et al.(1997). Intracisternal basic fibroblast growth factor enhancesfunctional recovery and up-regulates the expression of a mo-lecular marker of neuronal sprouting following focal cere-bral infarction. Proc. Natl. Acad. Sci. USA 22, 8179–8184.
KIM, W.K., KAN, Y., GANEA, D., HART, R.P., GOZES, I.,and JONAKAIT M. (2000). Vasoactive intestinal peptide andpituitary adenylate cyclase activating polypeptide inhibit tu-mor necrosis factor-� production in injured spinal cord andin activated microglia via cAMP-dependent pathway. J. Neu-rosci. 20, 3622–3630.
KIMURA, H., KAWATANI, M., ITO, E., and ISHIKAWA, K.(2003). Effects of pituitary adenylate cyclase activating
POSTINJURY ADMINISTRATION OF PACAP IN TBI
693
polypeptide on facial nerve recovery in the guinea pig. Laryn-goscope 113, 1000–1006.
KINHULT, J., UDDMAN, R., LAAN, M., LINDEN, A., andCARDELL, L.O. (2001). Pituitary adenylate cyclase acti-vating polypeptide inhibits neurophil chemotaxis. Peptides22, 2151–2154.
KOIZUMI, H., and POVLISHOCK, J.T. (1998). Posttraumatichypothermia in the treatment of axonal damage in an animalmodel of traumatic axonal injury. J. Neurosurg. 89, 303–309.
KOIZUMI, H., FUJISAWA, H., ITO, H., MAEKAWA, T., DI,X., and BULLOCK, R. (1997). Effects of mild hypothermiaon cerebral blood flow-independent changes in cortical ex-tracellular levels of amino acids following contusion traumain the rat. Brain Res. 747, 304–312.
LEE, V.M., CARDEN, M.J. SCHLAEPFER, W.W., and TRO-JANOWSKI, J.Q. (1987). Monoclonal antibodies distinguishseveral differentially phosphorylated states of the two largestrat neurofilament subunits (NF-H and NF-M) and demon-strate their existence in the normal nervous system of adultrats. J. Neurosci. 7, 3474–3488.
LEKER, R.R., and SHOHAMI E. (2002). Cerebral ischemiaand trauma—different etiologies yet similar mechanisms:neuroprotective opportunities. Brain Res. Rev. 39, 55–73.
LIU, J., JR., FARMER, D.J., LANE, W.S., FRIEDMAN, J.,WEISSMAN, I., and SCHREIBER, S.L. (1991). Calcineurinis a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815.
LYETH, B.G., JIANG, J.Y., and LIU, S. (1993). Behavioralprotection by moderate hypothermia initiated after experi-mental traumatic brain injury. J. Neurotrauma 10, 57–64.
MARMAROU, A., FODA, M.A., VAN DEN BRINK, W.,CAMPBELL, J., KITA, H., and DEMETRIADOU, K.(1994). A new model of diffuse brain injury in rats. Part I:Pathophysiology and biomechanics. J. Neurosurg. 80, 291–300.
MAXWELL, W.L., POVLISHOCK, J.T., and GRAHAM, D.L.(1997). A mechanistic analysis of nondisruptive axonal in-jury: a review. J. Neurotrauma 14, 419–440.
MITANI, A., KADOYA, F., and KATAOKA, K. (1991). Tem-perature dependence of hypoxia-induced calcium accumula-tion in gerbil hippocampal slices. Brain Res. 562, 159–163.
MORIO, H., TATSUNO, I., HIRAI, A., TAMURA, Y., andSAITO, Y. (1996). Pituitary adenylate cyclase activatingpolypeptide protects rat-cultured cortical neurons from glu-tamate-induced cytotoxicity. Brain Res. 741, 82–88.
NAKAYAMA, M., UCHIMURA, K., ZHU, R.L., et al. (1998).Cyclooxygenase-2 inhibition prevents delayed death of CA1hippocampal neurons following global ischemia. Proc. Natl.Acad. Sci. USA 95, 10954–10959.
OHTAKI, H., DOHI, K., YIN, T., TAKAKI, A., NAKAJO, S.,and SHIODA S. (2003). Neuroprotective effect of PACAP38
through IL-6 after focal ischemia in mouse. Regul. Peptides15, 53(abst).
OKONKWO, D.O., PETTUS, E.H., MOROI, J., and POVLI-SHOCK, J.T. (1998). Alteration of the neurofilament sidearmand its relation to neurofilament compaction occurring withtraumatic axonal injury. Brain Res. 784, 1–6.
OKONKWO, D.O., and POVLISHOCK, J.T. (1999). An in-trathecal bolus of cyclosporin A before injury preserves mi-tochondrial integrity and attenuates axonal disruption in trau-matic brain injury. J. Cereb. Blood Flow Metab. 19, 443–451.
OKONKWO, D.O., BUKI, A., SIMON, R., and POVLI-SHOCK, J.T. (1999) Cyclosporin A limits calcium-inducedaxonal damage following traumatic brain injury. Neuroreport10, 353–358.
OTANI, N., NAWASHIRO, H., FUKUI, S., et al. (2002). Tem-poral and spatial profile of phosphorylated mitogen-activatedprotein kinase pathways after lateral fluid percussion injuryin the cortex of the rat brain. J. Neurotrauma 12, 1587–1596.
PETTUS, E.H., CHRISTMAN, C.W., GIEBEL, M.L., andPOVLISHOCK, J.T. (1994). Traumatically induced alteredmembrane permeability: its relationship to traumatically in-duced reactive axonal change. J. Neurotrauma 11, 507–522.
POVLISHOCK, J.T., and STONE, J.R. (2001). Traumatic ax-onal injury, in: Head Trauma: Basic, Preclinical and Clini-cal Directions. L.P. Miller and R.L. Hayes (eds), Wiley: NewYork.
POVLISHOCK, J.T., MARMAROU, A., MCINTOSH, T.,TROJANOWSKI, J.Q., and MOROI, J. (1997). Impact ac-celeration injury in the rat: evidence for focal axolemmalchange and related neurofilament sidearm alteration. J. Neu-ropathol. Exp. Neurol. 56, 347–359.
REGLODI, D., SOMOGYVARI-VIGH, A., VIGH, S., KOZ-ICZ, T., and ARIMURA, A. (2000). Delayed systemic ad-ministration of PACAP38 is neuroprotective in transient mid-dle cerebral artery occlusion in the rat. Stroke 31, 1411–1417.
REGLODI, D., TAMAS, A., SOMOGYVARI-VIGH, A., et al.(2002). Effects of pretreatment with PACAP on the infarctsize and functional outcome in rat permanent focal cerebralischemia. Peptides 23, 2227–2234.
REGLODI, D., LUBICS, A., TAMAS, A., SZALONTAY, L.,and LENGVARI, I. (2004). Pituitary adenylate cyclase acti-vating polypeptide protects dopaminergic neurons and im-proves behavioral deficits in a rat model of Parkinson’s dis-ease. Behav. Brain Res. 151, 303–312.
SAKAMOTO, K.I., FUJISAWA, H., KOIZUMI, H., et al.(1997). Effects of mild hypothermia on nitric oxide synthe-sis following contusion trauma in the rat. J. Neurotrauma 14,349–353.
SANDERSON, K.L., RAGHUPATHI, R., SAATMAN, K.E.,MARTIN, D., MILLER G., and MCINTOSH, T.K. (1999).Interleukin-1 receptor antagonist attenuates regional neuronal
TAMÁS ET AL.
694
cell death and cognitive dysfunction after experimental braininjury. J. Cereb. Blood Flow Metab. 19, 1118–1125.
SEKI, T., IZUMI, S., SHIODA, S., and ARIMURA A. (2003).Pituitary adenylate cyclase activating polypeptide (PACAP)protects ganglion cell death against cutting of optic nerve inthe rat retina. Regul. Peptides 115, 55(abst).
SHIODA, S., OZAWA, H., DOHI, K., et al. (1998). PACAPprotects hippocampal neurons against apoptosis: involvementof JNK/SAPK signaling pathway. Ann. N. Y. Acad. Sci. 865,111–117.
SHOGE, K., MISHIMA, H.K., SAITOH, T., et al. (1999). At-tenuation by PACAP of glutamate-induced neurotoxicity incultured retinal neurons. Brain Res. 839, 66–73.
SINGLETON, R.H., STONE J.R., OKONKWO D.O., PELLI-CANE, A.J., and POVLISHOCK, J.T. (2001). The im-munophilin ligand FK506 attenuates axonal injury in an im-pact-acceleration model of traumatic brain injury. J.Neurotrauma 18, 607–614.
SKOGLOSA, Y., LEWEN, A., TAKEI, N., HILLERED, L.,and LINDHOLM, D. (1999). Regulation of pituitary adeny-late cyclase activating polypeptide and its receptor type 1 af-ter traumatic brain injury: comparison with brain-derivedneurotrophic factor and the induction of neuronal cell death.Neuroscience 90, 235–247.
SOMOGYVARI-VIGH, A., and REGLODI, D. (2004). Pitu-itary adenylate cyclase activating polypeptide: a potentialneuroprotective peptide. Curr. Pharm. Des. 10, 2861–2889.
STONE, J.R., WALKER, S.A., and POVLISHOCK, J.T.(1999). The visualisation of a new class of traumatically in-jured axons through the use of a modified method of mi-crowave antigen retrieval. Acta Neuropathol. 97, 335–345.
STONE, J.R., SINGLETON, R.H., and POVLISHOCK, J.T.(2000). Antibodies to the C-terminus of the beta-amyloid pre-cursor protein (APP): a site-specific marker for the detectionof traumatic axonal injury. Brain Res. 871, 288–302.
SUEHIRO, E., SINGLETON, R.H., STONE, R.J., and POVLI-SHOCK, J.T. (2001). The immunophilin ligand FK 506 at-tenuates the axonal damage associated with rapid rewarmingfollowing posttraumatic hypothermia. Exp. Neurol. 172,199–210.
TABUCHI, A., FUNAJI, K., NAKATSUBO, J., FUKUCHI,M., TSUCHIYA, T., and TSUDA, M. (2003). Inactivationof aconitase during apoptosis of mouse cerebellar granuleneurons induced by a deprivation of membrane depolariza-tion. J. Neurosci. Res. 71, 504–515.
TANAKA, J., KOSHIMURA, K., MURAKAMI, Y., SOHMIYA,M., YANAIHARA, N., and KATO, Y. (1997). Neuronal pro-
tection from apoptosis by pituitary adenylate cyclase activatingpolypeptide. Regul. Peptides 72, 1–8.
TRUETTNER, J., SCHMIDT-KASTNER, R., BUSTO, R., et al.(1999) Expression of brain-derived neurotrophic factor, nervegrowth factor, and heat shock protein HSP70 following fluidpercussion brain injury in rats. J. Neurotrauma 16, 471–486.
TSUDA, T., KOGURE, K., NISHIOKA, K., and WATAN-ABE, T. (1991). Mg2� administered up to twenty-four hoursfollowing reperfusion prevents ischemic damage of the CA1neurons in the rat hippocampus. Neuroscience 44, 335–341.
UCHIDA, D., ARIMURA, A., SOMOGYVARI-VIGH, A., SHI-ODA, S., and BANKS, W. (1996). Prevention of ischemia-induced death of hippocampal neurons by pituitary adenylate-cyclase activating polypeptide. Brain Res. 736, 280–286.
VAUDRY, D., GONZALEZ, B.J., BASILLE, M., YON, L.,FOURNIER, A., and VAUDRY, H. (2000a). Pituitary adeny-late cyclase activating polypeptide and its receptors: fromstructure to functions. Pharmacol. Rev. 52, 269–324.
VAUDRY, D., GONZALEZ, B.J., BASILLE, M., et al.(2000b). The neuroprotective effect of pituitary adenylate cy-clase activating polypeptide on cerebellar granule cells is me-diated through inhibition of the CED3-related cystein pro-tease caspase-3/CPP32. Proc. Natl. Acad. Sci. USA 97,13390–13395.
VAUDRY, D., ROUSELLE, C., BASILLE, M., et al. (2002a).Pituitary adenylate cyclase activating polypeptide protects ratcerebellar granule neurons against ethanol-induced apoptoticcell death. Proc. Natl. Acad. Sci. USA 99, 6398–6403.
VAUDRY, D., CHEN, Y., RAVNI, A., HAMALINK, C.,ELKAHLOUN, A.G., and EIDEN, L.E. (2002b). Analysisof the PC12 cell transcriptome after differentiation with pi-tuitary adenylate cyclase activating polypeptide (PACAP). J.Neurochem. 83, 1272–1284.
YOSHIMOTO, T., and SIESJO, B.K. (1999). Posttreatmentwith the immunsuppressant cyclosporin A in transient focalischemia. Brain Res. 839, 283–291.
ZAMBONI, L., and MARTINO, L. (1967). Buffered picricacid–formaldehyde: a new rapid fixative for electron mi-croscopy. J. Cell Biol. 35, 148A.