Potassium(I) in water from Theoretical Calculations Maria Rudbeck U.U.D.M. Project Report 2006:9 Examensarbete i matematik, 20 poäng Handledare: Daniel Spånberg, Institutionen för materialkemi och Lars-Erik Persson, Matematiska institutionen Examinator: Lars-Erik Persson Oktober 2006 Department of Mathematics Uppsala University
Transcript
Potassium(I) in waterfrom Theoretical Calculations
Maria RudbeckU.U.D.M. Project Report 2006:9
Examensarbete i matematik, 20 poäng
Handledare: Daniel Spånberg, Institutionen för materialkemioch Lars-Erik Persson, Matematiska institutionen
Examinator: Lars-Erik Persson
Oktober 2006
Department of Mathematics
Uppsala University
Abstract The optimized geometry and energy for [K(H2O)n=1-8]+ clusters have been studied using different
basis sets and different methods (HF, MP2(FULL), BLYP and B3LYP). Moreover, two analytical potentials
have been developed for potassium in water based on MP2(FULL) and B3LYP computations using the 6-
311g* for potassium and the aug-cc-pVTZ(H2O) basis set for water. Two molecular dynamics simulations
have been performed using the potentials, separately, together with a polarizable water potential. The
simulation using both models show that the local water structure in both the first and second solvation shell
of the ion is flexible. The distribution of the coordination numbers for both the models is wide (5-12) and the
average coordination number higher for the model based on the MP2(FULL) calculations (8.1) than for the
model based on B3LYP calculations (6.5). This difference can mostly be explained by differences in K+-
(H2O)n binding energies for the two methods, as well as the fact that the water model was developed using
MP2.
“If it is true that every theory must be based upon observed facts, it is equally true that facts can not be observed without the guidance of some theory. Without such guidance, our facts would be desultory and fruitless; we could not retain them: for the most part we could not even perceive them.”
(Comte, A. (1974 reprint). The positive philosophy of Auguste Comte freely translated and condensed by Harriet Martineau. New York, NY: AMS Press. (Original work published in 1855, New York, NY: Calvin Blanchard, p. 27.)
”Every attempt to employ mathematical methods in the study of chemical questions must be considered profoundly irrational and contrary to the spirit of chemistry. If mathematical analysis would ever hold a prominent place in chemistry – an aberration which is happily almost impossible - it would occasion a rapid and widespread degeneration of that science”
Auguste Comte, 1830.
Contents
1. Introduction
2. Theory
2.1 Quantum Mechanics
2.1.1 Basis sets
2.1.2 Hartree-Fock Theory
2.1.3 Many-Body Perturbation Theory
2.1.4 Coupled Cluster Theory
2.1.5 Density Functional Theory
2.1.6 Hybrid Hartree-Fock/Density Functional Theory
2.1.7 Time versus Accuracy
2.1.8 Counterpoise Basis Set Superposition Error Correction
2.2 Molecular Dynamics
2.2.1 Ensembles
2.2.2 The Potential Energy
2.2.3 Periodic Boundary Conditions
2.2.4 Long-Range Forces
2.2.5 Equations of Motion
2.2.6 Constraint Dynamics
3. Methods
3.1 Quantum Mechanical Calculations
3.1.1 Selection of Methods and Basis Sets
3.1.2 Optimization of [K(H2O)2-8]+
3.1.2 The Potential Energy Curve
3.1.4 Fitting the Potential Energy Curve to the Analytical Potential
3.2 Molecular Dynamical Simulations
3.2.1 Simulation Protocol
3.2.2 Analysis of Molecular Dynamic Simulation Results
4. Results and Discussion
4.1 Quantum Mechanical Results
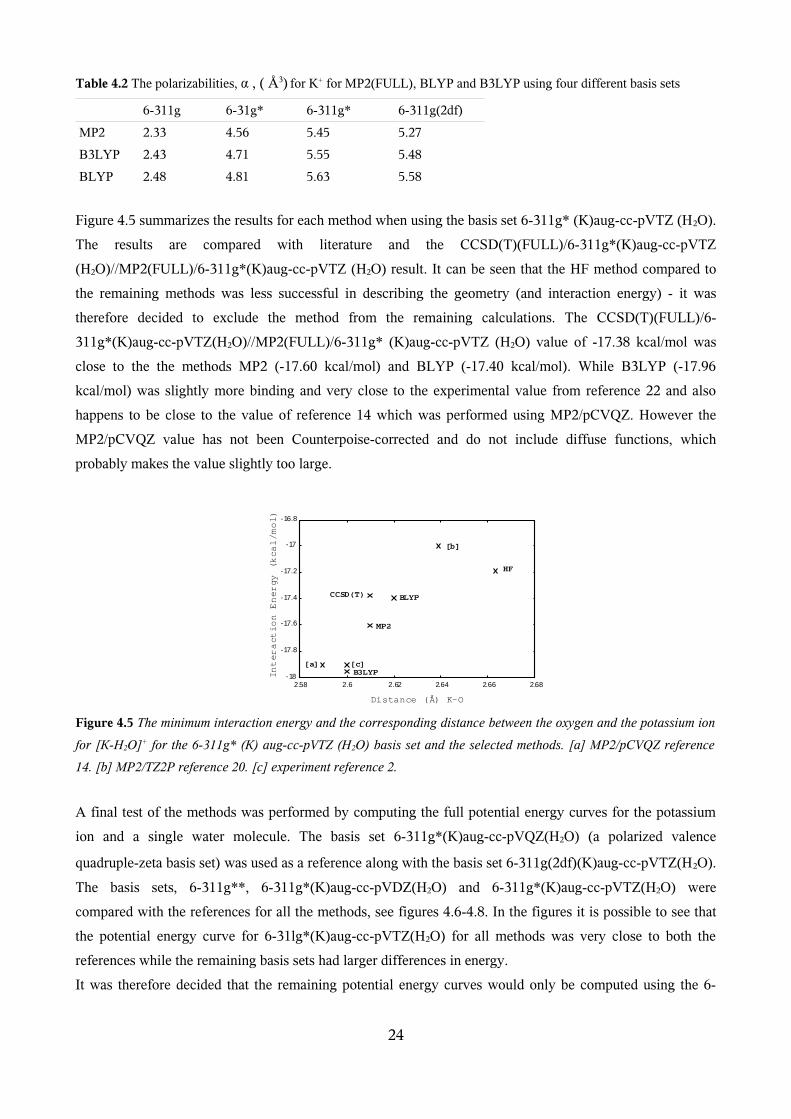
4.1.1 Selection of Methods and Basis Sets
4.1.2 Optimization of [K(H2O)2-8]+
4.1.3 The Potential Energy Curve
4.1.4 Fitting the Potential Energy Curve to the Analytical Potential
4.2 Molecular Dynamical Results
5. Conclusions
6. Acknowledgment
7. References
8. Appendix I-II
1
1. Introduction
Auguste Comte, often called the father of sociology, named himself the Pope of Positivism. If he was alive
today he would probably be even an inch happier - since mathematical methods are now employed in
chemical questions contrary to his rather sombre prediction (see the pretext). However, theoretical
descriptions need to be handled with great care since small inaccuracies, in energy for instance, can give
large differences in the calculated chemical properties. In order to be “within chemical accuracy” the energy
needs to lie within 1-2 kcal/mol of the true values. The goal of this thesis was to study the structural
properties of the potassium ion in water, an ion which is not only used in alkali batteries and as a fertilizer
[1] but is probably best known for its activities in the plasma membrane in the human cells [2] – a feature
which was acknowledged with two Nobel prizes in chemistry 1991 and 2003. It is already known that the
potassium ion's radius is greater than the sodium ion - but are more water molecules coordinated to the
potassium ion in an aqueous solution? Numerous X-ray and neutron diffraction studies have been performed
for aqueous K+-solutions at different concentrations; see for example the review article by Ohtaki and Radnai
from 1993 [3]. Since the bond length between a potassium ion and a water molecule is close to the bond
length between two water molecules, the hydration structure of the ion is very difficult to measure using the
diffraction methods. Therefore the studies have a large spread ranging from 4 to 8 in coordination number.
EXAFS studies [4] have the advantage over normal X-ray diffraction methods that it has a high selectivity of
the central elements and therefore the K+/H2O distances are completely decoupled from the H2O/H2O
distances. However, the method is also less reliable with respect to the coordination number and therefore
the large spread in the coordination number still exists. In this thesis, properties such as the coordination
number of potassium in water and how the solvated ion fits into bulk water was studied theoretically using
the theories quantum mechanics (QM) and molecular dynamics (MD).
The theory of QM was derived in the beginning of the 20th century and is based on all properties of a system
being fully obtainable from the wave function. The wave function is obtained by solving the Schrödinger
equation – an equation which can only be solved exactly for very simple problems involving no more than
two particles, but it can be solved approximately for many-body systems. The methods employed in this
thesis all assume that the solution of the wave function of the electrons and the wave function of the nuclei
can be separated, due to the large mass difference, the so-called Born-Oppenheimer approximation. Three of
the methods, the ab initio, the Density Functional Theory (DFT) and the hybrid Hartree-Fock (HF)/DFT,
assume that the location of the nuclei is fixed and that the properties of a system is obtained from the
electronic wave function. Within the three basic theories, a number of subdivisions exist - five of which were
employed in this thesis and are described in the theory section.
Due to the time consumption and computational expense, QM is not an ideal tool when studying large
systems including 100s of molecules. Classical molecular dynamics (MD) is a method which also employs
the Born-Oppenheimer approximation but follows the time evolution of the nuclei. The theory is developed
2
using classical mechanics and is solved through Newtons' equations of motion. Just like QM, MD is
branched into a range of methods - in this thesis classical MD was performed where the particle interactions
were described using an analytical potential. The analytical potential was derived through fitting a potential
expression to QM calculations, contrary to the most common approach used in the literature, namely fitting
to a small set of experimental data.
The main focus of this thesis was the optimizations and potential energy calculations of clusters
[K(H2O)n=1-8]+ using QM, the fitting of an analytical potential to the QM computations and finally the MD
simulations. Subsequently, the simulations were analyzed with respect to the coordination number and the
orientation of the water molecules surrounding the potassium in the first and second solvation shells.
3
2. Theory
Quantum Mechanics (QM) and Molecular Dynamics are two complementary techniques: In this thesis, the
QM was considered time-independent and only the electronic degrees of freedom were taken into
consideration, while the MD was time-dependent and only took the motion of the nuclei into account
explicitly. The more computational expensive but more accurate QM was used in order to study smaller
systems while MD was used for the larger systems. Below is a short summary of the two techniques. It
should be noted that the QM energy has been abbreviated with E while the energy used in the MD simulation
has been abbreviated with U in this thesis.
2.1. Quantum Mechanics
In classical mechanics, the motion of matter is described by Newtons' three equations of motion – the total
energy is constant in the absence of external forces, the response of the particles when the forces are acting
on them and that to every action there is an equal and opposite reaction. Quantum mechanics (QM) is used
when the three equations fail to describe light particles, such as electrons, and suggests that a particle can be
described as a wave rather than a finite object traveling along a definitive path. In other words, in QM the
dynamical properties are expressed by a wavefunction which can be obtained by solving the Schrödinger
equation. The Schrödinger equation was proposed by the Austrian physicist Erwin Schrödinger 1926 and its
time-independent form is shown in equation 2.1.
H=E (2.1)
where
H=−ℏ2
2m∇2V (2.2)
and
∇ 2=2
x22
y22
z2 (2.3)
for a three-dimensional system.
Since the Schrödinger equation cannot be solved exactly for many-body systems, a first step in the treatment
is the Born-Oppenheimer approximation, which states that the nuclei can be considered stationary compared
to the electrons due to the large relative mass. This approximation allows the Schrödinger equation to be
written as a product of two wave functions – one for the electronic part and one for the nuclei (see equation
2.4). Since the wave function for the nuclei is in this case considered fixed, it can be assumed to be a
constant at arbitrary locations and be computed using classical mechanics.
tot nuclei , electrons=electronsnuclei (2.4)
4
Apart from the Born-Oppenheimer approximation other approximations need to be adopted in order to solve
the equation. Four different strategies to solve the Schrödinger equation are discussed below in sections
2.1.2-2.1.5. A more detailed description of the methods can be found in the references 5 and 6. Some of the
methods use the variation theory which says that the energy corresponding to an arbitrary wave function is
never less than the true energy - consequently the best function corresponds to the minimum in energy. Other
methods use the perturbation theory which aims to find an easier computed but related problem and then
considers how the difference between the real and simpler problems affects the solution. The perturbation
theory is very useful when the difference between the two problems is small.
2.1.1 Basis sets
Atomic orbitals describe where an electron may be found in a single atom whereas the molecular orbitals
describes where an electron may be found in a molecule. Each molecular orbital is commonly, and in the
methods employed, expressed as a linear combination of atomic orbitals (LCAO), i.e. atomic-like functions
are used as basis sets, see equation 2.5.
i=∑v=1
K
cviv (2.5)
The most natural way to describe the atomic orbitals is through Slater type orbitals, STO, since these are the
exact solutions of the Schrödinger equation for the hydrogen atom, see equation 2.5. However, when solving
the Schrödinger equation for many-electron systems, integrals of products of the basis functions are required.
Since the calculations can be very expensive, the STO:s are often replaced with Gaussian type orbitals,
GTO:s (see equation 2.7). The advantage with the GTO:s is that the product of two orbitals can be expressed
as one, see equation 2.8.
x h yk z l e− r (2.6)
x h yk z l e− r 2
(2.7)
Aa e−a ra2
⋅Ab e−b rb2
=Ac e−c rc2
(2.8)
where α is a radial extent and the sum of the Cartesian variables h, k and l determines the order of the
Gaussian.
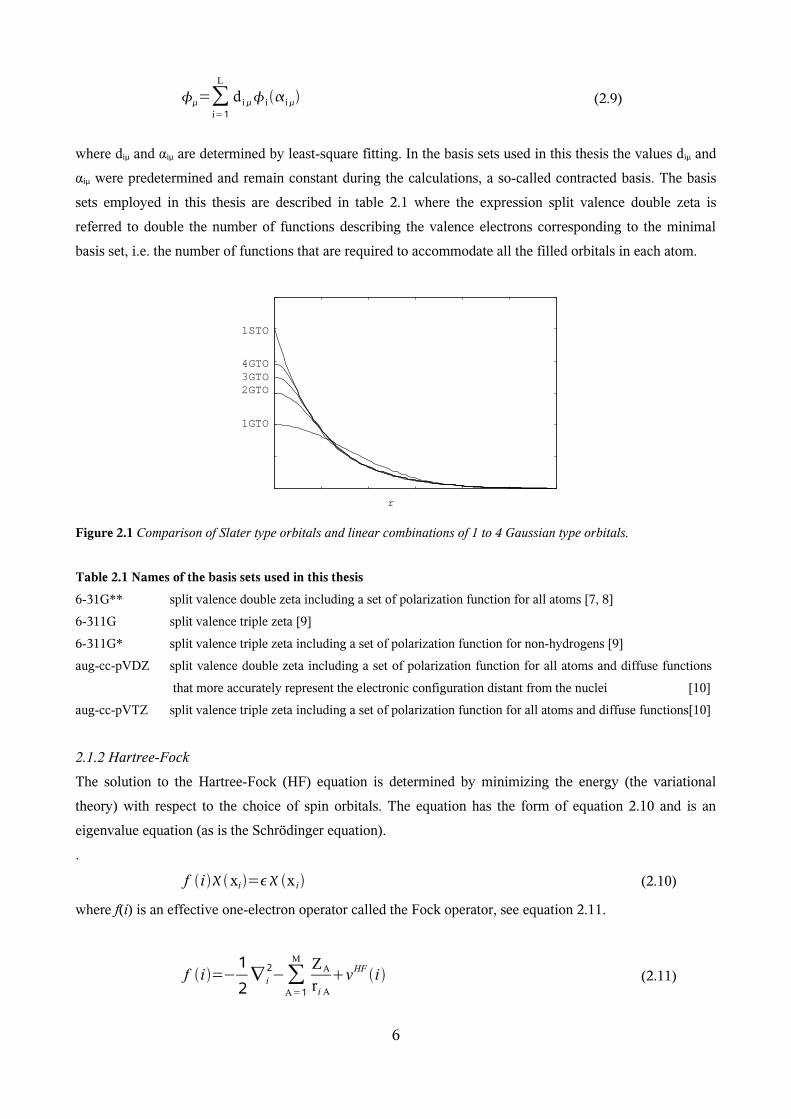
However, GTO:s do have disadvantages – for instance the STO:s do not have a cusp at the origin (see figure
2.1), i.e the values of the derivatives, at r=0, of the STO:s are negative while for the GTO:s it is zero. One
way to overcome the shortcomings of one GTO, is to express the basis sets as linear combinations of several
GTO:s, see equation 2.9, since the expansions improve the description of the atomic orbitals (see figure 2.1).
The more functions added, the better the description.
5
=∑i=1
L
d iii (2.9)
where diμ and αiμ are determined by least-square fitting. In the basis sets used in this thesis the values diμ and
αiμ were predetermined and remain constant during the calculations, a so-called contracted basis. The basis
sets employed in this thesis are described in table 2.1 where the expression split valence double zeta is
referred to double the number of functions describing the valence electrons corresponding to the minimal
basis set, i.e. the number of functions that are required to accommodate all the filled orbitals in each atom.
Figure 2.1 Comparison of Slater type orbitals and linear combinations of 1 to 4 Gaussian type orbitals.
Table 2.1 Names of the basis sets used in this thesis
6-31G** split valence double zeta including a set of polarization function for all atoms [7, 8]
6-311G split valence triple zeta [9]
6-311G* split valence triple zeta including a set of polarization function for non-hydrogens [9]
aug-cc-pVDZ split valence double zeta including a set of polarization function for all atoms and diffuse functions
that more accurately represent the electronic configuration distant from the nuclei [10]
aug-cc-pVTZ split valence triple zeta including a set of polarization function for all atoms and diffuse functions[10]
2.1.2 Hartree-Fock
The solution to the Hartree-Fock (HF) equation is determined by minimizing the energy (the variational
theory) with respect to the choice of spin orbitals. The equation has the form of equation 2.10 and is an
eigenvalue equation (as is the Schrödinger equation).
.
f ixi= x i (2.10)
where f(i) is an effective one-electron operator called the Fock operator, see equation 2.11.
f i=−1
2∇ i
2−∑A=1
M ZA
ri A
vHF i (2.11)
6
1STO
r
1GTO
2GTO3GTO4GTO
where vHF(i) is the average potential experienced by the ith electron due to the presence of the other
electrons. In order to simplify the Schrödinger equation, the many-body problem is approximated with an
one-electron problem in which the electron-electron repulsion is treated in an average way – the Hartree-
Fock Approximation. Since vHF(i) depends on the spin orbitals of the other electrons, the Hartree-Fock
equation is non-linear and must be solved iteratively. This procedure is called the self-consistent-field (SCF)
and the idea is to make an initial guess at the spin orbitals, calculate the average field (i.e. vHF(i)) and then
solve the Hartree-Fock equation. The spin orbitals obtained are subsequently used in order to calculate a new
field and a new set of spin orbitals. The procedure is repeated until self-consistency is reached.
The disadvantage with the HF method is that it fails to describe the electron correlation – the equation does
not take the correlation of the motion of the electrons into account. In other words the instantaneous
Coulomb interactions that keeps the electrons of opposite spin apart are not accounted for. Consequently the
energy from HF is too high - the difference between the HF energy and the exact energy is defined as the
correlation energy. However, it should be remembered that while HF already determines the total energy
99% correctly small energy differences are relevant in chemistry for instance in chemical reactions.
2.1.3 Many-body Perturbation Theory
One way of tackling the electron correlation is to split the total Hamiltonian, H, in two pieces: a zeroth-order
part, H0, which has known eigenfunctions and a perturbation, H' (see equation 2.12). When the zeroth order
Hamiltonian is the HF Hamiltonian, the many-body perturbation theory is referred to as MØller-Plesset
(MP).
H = H 0 +λ H' (2.12)
where λ is a parameter between 0 and 1 which describes the strength of the perturbation. The first, second,
etc order wave function can be described as a Taylor expansion in powers of the perturbation parameter, see
equation 2.13 and 2.14.
E=∑i=0
n
i E i (2.13)
=∑i=0
n
ii (2.14)
The higher the order of corrections, the more complicated types of equation need to be considered for the
energy correction. The second order corrections, MP2, accounts for about 80-90% of the electron correlation
and is the most economical wave function based method including electron correlation.
7
2.1.4 Coupled Cluster methods (CC)
Coupled Cluster theory is one of the more reliable QM methods but is also more computationally expensive
and was therefore only used as a reference in this thesis. The theory also uses the HF equation as a base but
the wave function is improved by adding a cluster function. The purpose of the functions is to correlate the
motions of any two electrons within a selected pair of occupied orbitals. The complexity of the equations and
the corresponding computer codes, as well as the cost of the computation increases sharply with the highest
level of excitation (the quantum state with the highest energy allowed). For many applications sufficient
accuracy may be obtained with the so-called CCSD, which includes single and double excitations, and the
more accurate (and more expensive) CCSD(T) is often called "the gold standard of quantum chemistry" for
its excellent compromise between the accuracy and the cost for the molecules near equilibrium geometries.
2.1.5 Density Functional Theory
The Density Functional theory (DFT) is an approach which has become very popular in chemistry since the
late 1980s and 1990s. DFT is based on the fact that the ground-state energy and other properties of a system
can be obtained solely from the electron density, ρ. This fact was shown by Hohenberg and Kohn 1965 and
can be written as in equation 2.15.
E 〚r 〛=∫V ext r r d rF 〚r 〛 (2.15)
where the first term describes the interaction of the electrons possessed by an external potential Vext and the
second term describes the sum of the kinetic energy of the electrons and the interelectronic interactions. F is
the complicated part of the equation – it is described by Kohn and Sham according to equation 2.16.
F 〚 r 〛=EKE 〚 r 〛EH 〚 r 〛E XC 〚r 〛 (2.16)
The kinetic energy, EKE, is thus described according to equation 2.17. Where the density is expanded in a set
of one-electron orbitals, ψi, the so-called Kohn-Sham orbitals.
E KE 〚 r 〛=∑i=1
N
∫i−∇2
2 i r d r (2.17)
The electron-electron Coulombic energy, EH, is described according to equation 2.18.
E H 〚r 〛=1 2 ∬ r 1 r2
∣r1−r 2 ∣d r1 d r2 . (2.18)
8
The Exc component is the crucial feature to the DFT theory, its exact form is not known, therefore it has been
expressed in very many alternative approximate ways. This thesis employed the BLYP expression, i.e.
Becke's (B) description for the exchange energy and Lee, Yang and Parr's definition for the correlation
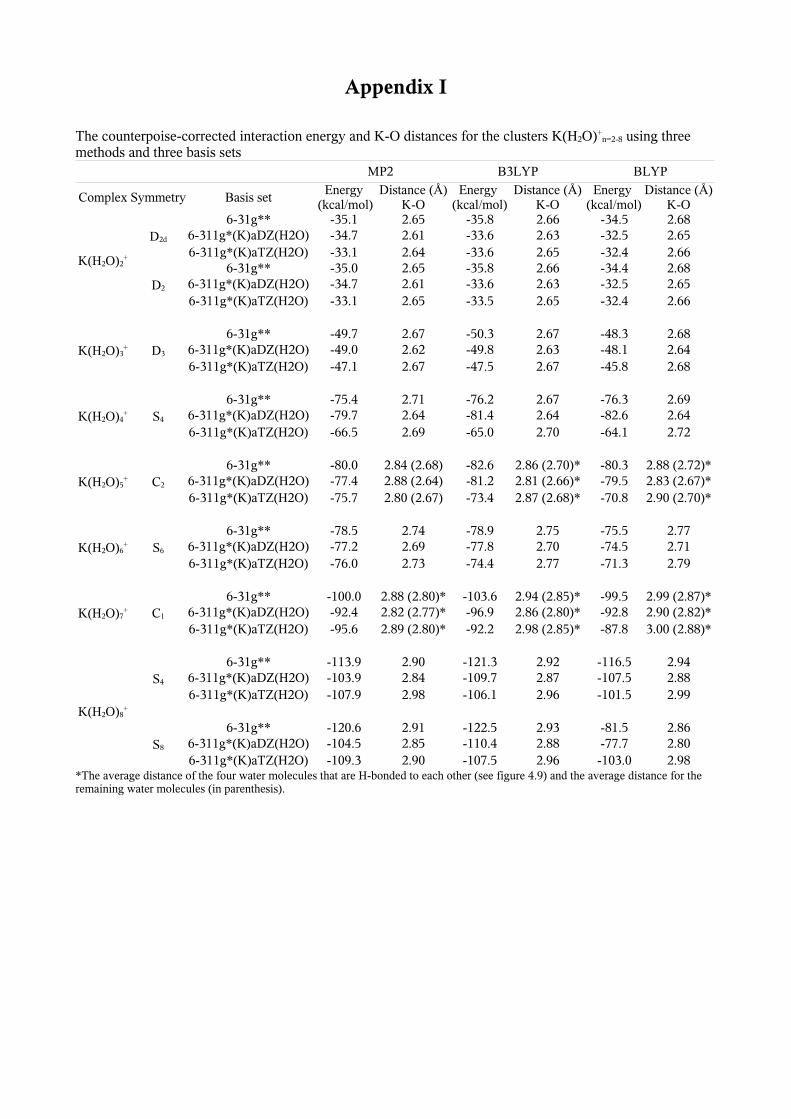
+ -109.3 2.90 -107.5 2.96 -103.0 2.98*The average distance for the four water molecules that are H-bonded to each other and the average distance for the remaining water
molecules (in parenthesis).
4.1.3 The Potential Energy Curves
The potential energy curves were computed using four different orientations for each cluster as is described
in the method section. An example of such potential energy curve for a cluster with 3 water molecules
incorporating different geometries (see figure 3.1) is shown in figure 4.10. From the figure it is possible to
see that the orientation (a) in figure 3.1 has a large attraction energy while the orientation (d) only has a
repulsive impact.
Figure 4.10 The interaction energy between the oxygen and the potassium ion when including 3 water molecules and
when computing with MP2(FULL). The letters a, b, c and d correspond to the orientations in figure 3.1.
27
-50
-40
-30
-20
-10
0
10
20
30
40
50
1 2 3 4 5 6 7InteractionEnergy
(kcal/mol)
Distance (Å) K-O
a b
c
d
4.1.4 Fitting the Potential Energy Curve to the Analytical Potential
The result of the fitting of the ab initio points to the analytical potential can be seen in figure 4.11 and the
parameters in the Buckingham potential can be seen in table 4.4. Appendix 2 includes an example program
to perform such a fitting procedure for one ion and one water molecule, although this program was not used
when computing the parameters in table 4.4. The λ value corresponds determines the amount of shielding,
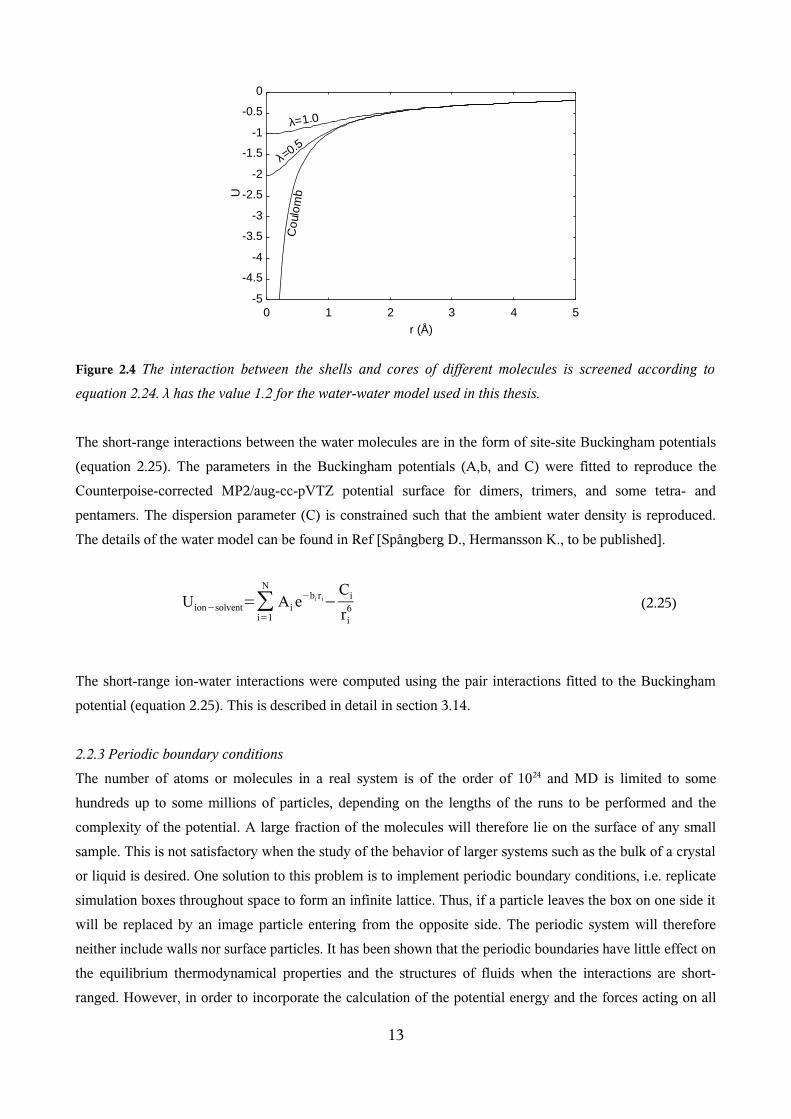
i.e. the reduction of nuclear charge to effective nuclear charge, see section 2.2.4. The figures show that the
quality of the fits are very good for both methods. However, all the parameters differ between the two
methods in table 4.4 – which is due to the potential energy differences in table 4.3.
Fig 4.11a and b. The fitting of the ab initio points to the Buckingham potential using a) MP2(FULL) and b) B3LYP.
Both of the analytical potentials are very well fitted to the Buckingham potential. All 1605 data points for all the
[K(H2O)n=1-8]+ clusters are included in these plots.
Table 4.4 The Parameters for the Buckingham Potential. λ is the shielding parameter which corresponds to a shielding
value, i.e. the reduction of nuclear charge to effective nuclear charge.
Two potassium-water radial distribution functions computed from MD-simulations using the potentials fitted
to the ab initio data from MP2(FULL) and B3LYP are presented in figure 4.12. For simplicity the two
models will be referred by QM “labels”, but it should be noted that they refer to the parametrized versions of
the QM energies. The distance and the radial distribution function values at the first peak maximum and the
first minimum for both the oxygen-ion and the hydrogen-ion radial distribution function are presented in
table 4.5. The figure and the table also include the coordination numbers of the first solvation shell of the
potassium ion. The results show a quite large difference in the coordination number (8.1 for MP2(FULL) and
6.5 for B3LYP) and the distance between the potassium ion and the oxygen (and the hydrogen). The
differences can once again be explained by the differences in the potential energies in table 4.3 and thereby
the parametric differences in table 4.4. In table 4.6 the coordination number and the distance to the first
maximum peak for the oxygen are compared to literature results – it can be concluded that the obtained
values lie at the end points of previous simulation results. It can also be concluded that most of the values are
closer to the MP2(FULL) than that of the B3LYP model. The experimental coordination number for the
potassium ion is determined with much uncertainty in the wide range of 4-8 [3]. In fact it is one of the most
difficult ions to study using X-ray and neutron diffraction methods [3] - the difficulty can be explained by
the length of the K+ - H2O bond being very close to the H2O - H2O distance in bulk water and thus it only
possible to determine the hydration number when the water structure is assumed. It should also be mentioned
that the experiments were performed at higher concentrations than the current simulation.
Figure 4.12 The Radial Distribution function for potassium in water and the integration of the coordination
29
Table 4.5 Features extracted from the ion-water radial distribution function, g(r). rmax and rmin are the distances between
the potassium (K) and the oxygen (O) (or hydrogen (H)) at the first maximum and minimum. n is the average
coordination number which is obtained by integrating the ion-oxygen g(r) to the first minimum.
Model rmax KO g(rmax KO) rminKO g(rminKO) rmax KH g(rmax KH) n
MP2 2.80 3.17 3.78 0.80 3.34 2.22 8.11
B3LYP 2.73 2.99 3.60 0.72 3.35 2.13 6.46
Table 4.6 Structural results from the literature. Comparing the values to the results in table 4.5 show that the
coordination numbers in the literature are closer to MP2 than B3LYP.
Method Comments rmax n
MC Polarizable model. Potential derived from ab initio calculations up to quadruplets [22]2.79 7.85QM/MM-MD First solvation shell treated with HF [23] 2.78 7.8MD CHARMM22 force field [24] 2.9 7.6
*The average distance of the four water molecules that are H-bonded to each other (see figure 4.9) and the average distance for the remaining water molecules (in parenthesis).
Appendix II
A python program which develops a simple analytical potential between K+ and H2O
1. Indata.py: Reads in a file including energy and coordinates from QM-computations
2. Koordin.py: Distinguishes the potassium and the water atoms x-, y-, z-coordinates from each
other
3. Radie.py: Computes the distances between the atoms
4. v.py: Computes the Coulomb energy
5. Umod.py: Computes the total interaction energy
6. extra.py Combines data
7. f.py Computes the variance between the total interaction energy from Umod.py and the
QM energy
8. DUMOD.py Differentiates the variance between the total interaction energy from Umod.py and
the QM energy
9. rand2.py A tool for the steepest descent method
10. final.py The final computations – using the steepest descent method the variance is
minimized and the parameters A, b, C from the Buckingham potential are optimized
1. Indata.py: Reads in a file including energy and coordinates from QM-computations. Separates the energy variable and the four atoms (K, O, H and H) coordinates.
def lasin(): f=open("h2ok.out2") #Opens the file with the QM computations en=[] co=[] co1=[] co2=[] co3=[]
while 1: x=f.readline() #Reads in line by line if not x: break en.append(x) # Defines an array including the energy f.readline() co.append(f.readline()) # Defines an array including the coordinates for the potassium f.readline() co1.append(f.readline()) #Defines an array including the coordinates for oxygen co2.append(f.readline()) #Defines an array including the coordinates for the first hydrogen atom co3.append(f.readline()) #Defines an array including the coordinates for the second hydrogen atom at=f.readline() return (en, co, co1, co2, co3)
2.Koordin.py: Distinguishes the potassium and the water atoms x-, y-, z-coordinates from each other
import indatadef koord(): en,co, co1, co2, co3 = indata.lasin() #Reads in the data from the indata file n=len(en)-1 g=0 energy=[] while 1: x=float(en[g]) #Defines the energy values as decimal numbers energy.append(x) g+=1 if g > n: break
nco=len(co)-1 gco=0 cox=[] coy=[] coz=[] while 1: sp=co[gco] tp=sp.split() #Splits the potassium ions coordinates into x-, y- and z-coordinates cox.append(float(tp[0])) #Defines the coordinates as decimal numbers coy.append(float(tp[1])) coz.append(float(tp[2])) gco+=1 if gco > nco: break
nco1=len(co1)-1 gco1=0 co1x=[] co1y=[] co1z=[] co2x=[] co2y=[] co2z=[] co3x=[] co3y=[] co3z=[] while 1: sp1=co1[gco1] tp1=sp1.split() #Splits the oxygen atoms coordinates into x-, y- and z-coordinates sp2=co2[gco1] tp2=sp2.split() #Splits the first hydrogen atoms coordinates into x-, y- and z-coordinates sp3=co3[gco1] tp3=sp3.split() #Splits the second hydrogen atoms coordinates into x-, y- and z-coordinates co1x.append(float(tp1[0])) #Defines the coordinates as decimal numbers co1y.append(float(tp1[1])) co1z.append(float(tp1[2])) co2x.append(float(tp2[0])) co2y.append(float(tp2[1])) co2z.append(float(tp2[2])) co3x.append(float(tp3[0])) co3y.append(float(tp3[1])) co3z.append(float(tp3[2])) gco1+=1 if gco1 > nco1: break
for i in xrange(n): xko.append(cox[i]-co1x[i]) #Calculates the differences between the coordinates yko.append(coy[i]-co1y[i]) zko.append(coz[i]-co1z[i]) xh1k.append(cox[i]-co2x[i]) yh1k.append(coy[i]-co2y[i]) zh1k.append(coz[i]-co2z[i]) xh2k.append(cox[i]-co3x[i]) yh2k.append(coy[i]-co3y[i]) zh2k.append(coz[i]-co3z[i])
rko.append(math.sqrt(xko[i]**2+yko[i]**2+zko[i]**2)) #Computes the distances between the atoms rh1k.append(math.sqrt(xh1k[i]**2+yh1k[i]**2+zh1k[i]**2)) rh2k.append(math.sqrt(xh2k[i]**2+yh2k[i]**2+zh2k[i]**2))
n=len(energy) expo=[] r6=[] UMOD=[] f=0 for i in xrange(n): e=ako*math.exp(-bko*rko[i]) #Computes the Buckingham potential energy e2=cko/(rko[i]**6) UMOD.append(V[i]+e-e2) #Adds the Coulomb energy and the Buckingham potential return UMOD
6. extra.py Combines data
import vimport Umodimport radie
def ex(a, b, c):
V=v.Vim() UMOD=Umod.umod(V, a, b, c) energy, rko, rh1k, rh2k = radie.radierna() return (a, b, c, V, UMOD, energy)
7. f.py Computes the variance between the total interaction energy from Umod.py and the QM energy
import extraimport math
def F(a, b, c): a, b, c, V, UMOD, energy = extra.ex(a, b, c) n=len(UMOD) f=0 for ii in xrange(n): f+=((UMOD[ii]-energy[ii])**2)*math.exp(-energy[ii]) #Computes the variance for each distance and adds the
#terms func=f/(n**2) return func
8. DUMOD.py Differentiates the variance between the total interaction energy from Umod.py and the QM energy
for i in xrange(n): dUako=math.exp(-bko*rko[i]) #Computes the inner derivative relative each parameter dUbko=-ako*rko[i]*math.exp(-bko*rko[i]) dUcko=1/(rko[i]**6)
#Computes the derivative relative to each parameter dfako+=2*(UMOD[i]-energy[i])*dUako*math.exp(-energy[i]) dfbko+=2*(UMOD[i]-energy[i])*dUbko*math.exp(-energy[i]) dfcko+=2*(UMOD[i]-energy[i])*dUcko*math.exp(-energy[i]) df=[dfako/n**2, dfbko/n**2, dfcko/n**2] #Computes the total derivative relative all parameters return (df)
9. rand2.py A tool for the steepest descent method
import DUMODimport extraimport fimport math
def RAND(a, b, c, s0, s1, s2): alpha=-1.0 #Defines three new A, b and C parameters which are used in the line search to find the best alpha a0=a+alpha*s0 a1=b+alpha*s1 a2=c+alpha*s2
while a1<-1.3: #Chooses other alpha values in case the start guess alpha value is inappropriate alpha=alpha*0.1 a0=a+alpha*s0 a1=b+alpha*s1 a2=c+alpha*s2 n=alpha fa=f.F(a0, a1, a2) #Computes the variance using the new parameters alpha-=(abs(alpha)/10) while 1: #Computes new parameters until a minimum variance is found a0=a+alpha*s0 a1=b+alpha*s1 a2=c+alpha*s2 fx=fa fa=f.F(a0, a1, a2) if fx<fa : break alpha-=abs(alpha)*0.1
fa=fx while 1: n=alpha
alpha+=(abs(alpha)/10) a0=a+alpha*s0 a1=b+alpha*s1 a2=c+alpha*s2 fx=fa fa=f.F(a0, a1, a2) if fx<fa: break if alpha>-1e-10: break n=alpha
alpha=abs(alpha) if fx>fa: while 1: n=alpha alpha+=(abs(alpha)/10) a0=a+alpha*s0 a1=b+alpha*s1 a2=c+alpha*s2 fx=fa fa=f.F(a0, a1, a2) if fx<fa: break
a0=a+n*s0 a1=b+n*s1 a2=c+n*s2 fa=f.F(a0, a1, a2) nb=n+10 while 1: a0=a+(nb)*s0 a1=b+(nb)*s1 a2=c+(nb)*s2 fb=f.F(a0, a1, a2) if fa<fb: break nb=nb+10
nc=n-abs(n)*1e-6 aa=1 while 1: a0=a+(nc)*s0 a1=b+(nc)*s1 a2=c+(nc)*s2 fc=f.F(a0, a1, a2) if fa<fc: break nc=n-abs(n)*10**aa aa+=aa
return nc, n, nb #Returns the “favorite“ alpha value and two “extra values” - one lower and one higher
10. final.py The final computations – using the steepest descent method the variance is minimized and the parameters A, b, C from the Buckingham potential are optimized
alpha, alpha2, alpha3 =rand2.RAND(a, b, c) while 1: a0=a-alpha*df[0] #Three new A, B and C values are defined by using the value which was a little
#lower than the alpha value defined in rand2.py a1=b-alpha*df[1] a2=c-alpha*df[2] fa=f.F(a0, a1, a2) #The variance is computed using these parameters
b0=a-alpha2*df[0] #Three new A, B and C values are defined by using the value the alpha value #defined in rand2.py
b1=b-alpha2*df[1] b2=c-alpha2*df[2] fb=f.F(b0, b1, b2) #The variance is computed using these parameters c0=a-alpha3*df[0] #Three new A, B and C values are defined by using the last alpha value c1=b-alpha3*df[1] c2=c-alpha3*df[2] fc=f.F(c0, c1, c2)) #The variance is computed using these parameters
t=alpha2-alpha ft=fb-fc s=alpha2-alpha3 fs=fb-fa if abs(ft-fs)<1e-10: #If the variance between the three “points” is close, the minimum variance is found break
x=alpha2 – 0.5*((((t**2)*ft)-((s**2)*fs))/((t*ft)-s*fs)) #If the three “points are not close, new alpha values are #defined
y=alpha2-x if fa>=fc: alpha=alpha2 else: alpha3=alpha2 alpha2=x
aa=a bb=b cc=c a=a-alpha2*df[0] #The new parameters b=b-alpha2*df[1] c=c-alpha2*df[2] fval=f.F(a, b, c) #The new variance print "Fval=",fval, a, b, c
if abs(fval-fold)<1e-9: #If the new variance is very close to the “old” variance, the minimum variance is #found
break fold=fval
print a, b, c, fval
a, b, c, V, UMOD, energy = extra.ex(a,b,c)energy, rko, rh1k, rh2k = radie.radierna()f=open ("h2ok.dat", "w")n=len(UMOD)-1i=0while 1: print >> f, UMOD[i], energy[i], rko[i] #The analytical potential is saved and can be visualized in the h2ok.dat file i+=1 if i>n: breakf.close()