Predictive methods Predictive methods using DNA using DNA sequences sequences Unit 11 Unit 11 BIOL221T BIOL221T : Advanced : Advanced Bioinformatics for Bioinformatics for Biotechnology Biotechnology Irene Gabashvili, PhD
Transcript
Predictive methods Predictive methods using DNA using DNA sequencessequences
Unit 11Unit 11BIOL221TBIOL221T: Advanced : Advanced Bioinformatics for Bioinformatics for
BiotechnologyBiotechnology
Irene Gabashvili, PhD
Reminders from last Reminders from last weekweek
Polymorphism and mutationsPolymorphism and mutations Mapping and SequencingMapping and Sequencing Genomic Map ElementsGenomic Map Elements Types of MapsTypes of Maps ResourcesResources Practical UsePractical Use
Polymorphism - Polymorphism - Types of Types of variationvariation
SNP[snp_class], SNP[snp_class], True single nucleotide True single nucleotide polymorphism polymorphism
"coding nonsynonymous“"coding nonsynonymous“ locus region, intron, exception locus region, intron, exception mrna, utr , splice sitemrna, utr , splice site ““coding synonymous“coding synonymous“
Nonsynonymous Nonsynonymous MutationsMutations
MissenseMissense – type of nonsynonymous – type of nonsynonymous (different amino acid in the product of (different amino acid in the product of mutated genes)mutated genes)
EXAMPLE: sickle-cell disease The EXAMPLE: sickle-cell disease The replacement of A by T at the 17th nucleotide replacement of A by T at the 17th nucleotide of the gene for the beta chain of hemoglobin of the gene for the beta chain of hemoglobin changes the codon GAG (for glutamic acid) changes the codon GAG (for glutamic acid) to GTG (which encodes valine). Thus the 6th to GTG (which encodes valine). Thus the 6th amino acid in the chain becomes valine amino acid in the chain becomes valine instead of glutamic acidinstead of glutamic acid
Nonsynonymous Nonsynonymous MutationsMutations
Another example of a Another example of a missensemissense mutation: In one patient with cystic mutation: In one patient with cystic fibrosis (fibrosis (Patient BPatient B), the substitution ), the substitution of a T for a C at nucleotide 1609 of a T for a C at nucleotide 1609 converted a glutamine codon (converted a glutamine codon (CAGCAG) ) to a to a STOPSTOP codon ( codon (TAGTAG). The protein ). The protein produced by this patient had only the produced by this patient had only the first 493 amino acids of the normal first 493 amino acids of the normal chain of 1480 and could not function. chain of 1480 and could not function.
Nonsynonymous Nonsynonymous MutationsMutations
TThe new nucleotide changes a codon that he new nucleotide changes a codon that specified an amino acid to one of the specified an amino acid to one of the STOPSTOP codons (codons (TAATAA, , TAGTAG, or , or TGATGA). Therefore, ). Therefore, translationtranslation of the messenger RNA of the messenger RNA transcribedtranscribed from this mutant gene will from this mutant gene will stop prematurely. The earlier in the gene stop prematurely. The earlier in the gene that this occurs, the more truncated the that this occurs, the more truncated the protein product and the more likely that it protein product and the more likely that it will be unable to function. These type of will be unable to function. These type of mutations are called mutations are called nonsensenonsense mutations mutations
Insertions and Deletions Insertions and Deletions (Indels)(Indels)
ADRB1[gene] AND human[orgn] AND "in-ADRB1[gene] AND human[orgn] AND "in-
del"[snp_classdel"[snp_class] ] Base pairs may be added (Base pairs may be added (insertionsinsertions) or ) or removed (removed (deletionsdeletions) from the DNA of a ) from the DNA of a gene. The number can range from one to gene. The number can range from one to thousands. thousands.
As a result, translation of the gene can be As a result, translation of the gene can be "frameshifted". Indels of three nucleotides "frameshifted". Indels of three nucleotides or multiples of three may be less serious. or multiples of three may be less serious.
Huntington's diseaseHuntington's disease and the and the fragile X fragile X syndromesyndrome are examples of are examples of trinucleotide trinucleotide repeatrepeat diseases caused by insertion diseases caused by insertion
Silent and splice-site Silent and splice-site mutationsmutations
For example, if the third base in the For example, if the third base in the TCTTCT codon for codon for serineserine is changed to any one of the is changed to any one of the other three bases, serine will still be encoded. other three bases, serine will still be encoded. Such mutations are said to be silent because Such mutations are said to be silent because they cause no change in protein (synonymous)they cause no change in protein (synonymous)
Nucleotide signals at the splice sites guide the Nucleotide signals at the splice sites guide the enzymatic machinery. If a mutation alters one enzymatic machinery. If a mutation alters one of these signals, then the intron is not of these signals, then the intron is not removed and remains as part of the final RNA removed and remains as part of the final RNA molecule. This alters the sequence of the molecule. This alters the sequence of the protein product. protein product.
Types of Maps – see Types of Maps – see MapViewerMapViewer
Predictive methods using Predictive methods using DNA sequences, B&O: DNA sequences, B&O:
chapter 5chapter 5 Gene Prediction methodsGene Prediction methods Gene Prediction ProgramsGene Prediction Programs How good the methods are?How good the methods are? Promoter AnalysisPromoter Analysis Strategies and ConsiderationsStrategies and Considerations
Markov modelsMarkov models
HMMs in Gene PredictionHMMs in Gene Prediction
Discriminant Analysis in Gene PredictionDiscriminant Analysis in Gene Prediction
What is Computational Gene Finding?What is Computational Gene Finding?
Given an uncharacterized DNA sequence, find out:Given an uncharacterized DNA sequence, find out:
Which region codes for a protein?Which region codes for a protein? Which DNA strand is used to encode the Which DNA strand is used to encode the
gene?gene? Which reading frame is used in that strand?Which reading frame is used in that strand? Where does the gene starts and ends?Where does the gene starts and ends? Where are the exon-intron boundaries in Where are the exon-intron boundaries in
eukaryotes?eukaryotes? (optionally) Where are the regulatory (optionally) Where are the regulatory
Ab initio, “intrinsic”, “template” (1Ab initio, “intrinsic”, “template” (1stst and 2and 2ndnd) vs “extrinsic”, “look-up” ) vs “extrinsic”, “look-up” (3(3rdrd and 4 and 4thth))
Eukaryotes vs Eukaryotes vs ProkaryotesProkaryotes
Genes separated by intergenic DNA, Genes separated by intergenic DNA, coding exons separated by large coding exons separated by large introns vs ORFs adjacent to one introns vs ORFs adjacent to one another another
Prokaryotic Vs. Eukaryotic Gene Prokaryotic Vs. Eukaryotic Gene FindingFinding
Prokaryotes:Prokaryotes:
small genomes 0.5 – 10small genomes 0.5 – 10·10·1066 bpbp high coding density (>90%)high coding density (>90%) no intronsno introns
– Gene identification relatively Gene identification relatively easy, with success rate ~ 99%easy, with success rate ~ 99%
Problems:Problems:
overlapping ORFsoverlapping ORFs short genesshort genes finding TSS and promotersfinding TSS and promoters
Eukaryotes:Eukaryotes:
large genomes 10large genomes 1077 – 10 – 101010 bp bp low coding density (<50%)low coding density (<50%) intron/exon structureintron/exon structure
– Gene identification a complex Gene identification a complex problem, gene level accuracy problem, gene level accuracy ~50%~50%
Problems:Problems: manymany
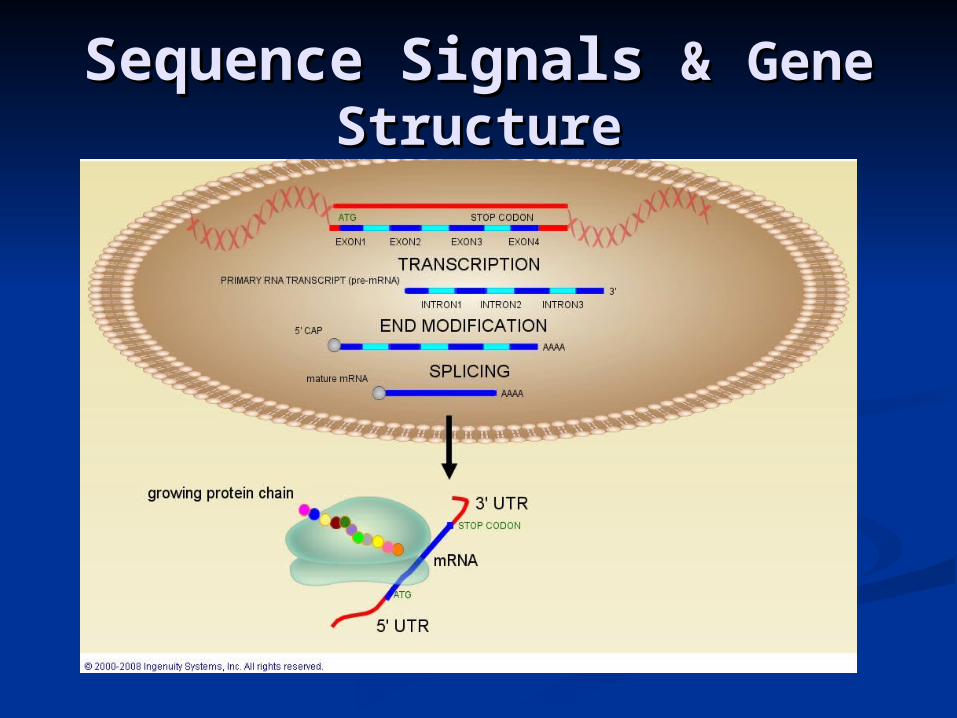
Gene StructureGene Structure
Gene Finding: Different Gene Finding: Different ApproachesApproaches
Similarity-based methods (extrinsic)Similarity-based methods (extrinsic) - use similarity to - use similarity to
annotated sequencesannotated sequences::
proteinsproteins cDNAscDNAs ESTsESTs
Comparative genomicsComparative genomics - Aligning genomic sequences from - Aligning genomic sequences from different speciesdifferent species
Ab initioAb initio gene-finding (intrinsic) gene-finding (intrinsic)
Integrated approachesIntegrated approaches
Similarity-based methodsSimilarity-based methods Based on sequence conservation due to functional Based on sequence conservation due to functional
constraintsconstraints
Use local alignment tools (Smith-Waterman algo, BLAST, Use local alignment tools (Smith-Waterman algo, BLAST, FASTA) to search protein, cDNA, and EST databasesFASTA) to search protein, cDNA, and EST databases
Will not identify genes that code for proteins not already in Will not identify genes that code for proteins not already in databases (can identify ~50% new genes)databases (can identify ~50% new genes)
Limits of the regions of similarity not well definedLimits of the regions of similarity not well defined
Comparative GenomicsComparative Genomics
Based on the assumption that coding Based on the assumption that coding sequences are more conserved than non-codingsequences are more conserved than non-coding
Alignment of homologous regionsAlignment of homologous regions
Difficult to define limits of higher similarityDifficult to define limits of higher similarity
Difficult to find optimal evolutionary distance Difficult to find optimal evolutionary distance (pattern of conservation differ between loci)(pattern of conservation differ between loci)
Summary for Extrinsic Summary for Extrinsic ApproachesApproaches
Strengths:Strengths:
Rely on accumulated pre-existing Rely on accumulated pre-existing biological data, thus should produce biological data, thus should produce biologically relevant predictionsbiologically relevant predictions
Weaknesses:Weaknesses:
Limited to pre-existing biological dataLimited to pre-existing biological data Errors in databasesErrors in databases Difficult to find limits of similarityDifficult to find limits of similarity
Signal SensorsSignal Sensors Signal – a string of DNA recognized by the cellular Signal – a string of DNA recognized by the cellular
machinerymachinery
Signal SensorsSignal Sensors
Various pattern recognition method are Various pattern recognition method are used for identification of these signals:used for identification of these signals:
Example of Consensus Example of Consensus SequenceSequence
obtained by choosing the most frequent base at each position of obtained by choosing the most frequent base at each position of the multiple alignment of subsequences of interestthe multiple alignment of subsequences of interest
Leads to loss of information and can produce Leads to loss of information and can produce many false positive or false negative predictionsmany false positive or false negative predictions
TATAAT
TATRNT
MELONMANGOHONEYSWEETCOOKY
MONEY
Example of (Positional) Example of (Positional) Weight MatrixWeight Matrix
Computed by measuring the frequency of every element of every Computed by measuring the frequency of every element of every position of the site (weight)position of the site (weight)
Score for any putative site is the sum of the matrix values (converted Score for any putative site is the sum of the matrix values (converted in probabilities) for that sequence (log-likelihood score)in probabilities) for that sequence (log-likelihood score)
Disadvantages: Disadvantages: cut-off value requiredcut-off value required assumes independence between adjacent basesassumes independence between adjacent bases
TACGAT
TATAAT
TATAAT
GATACT
TATGAT
TATGTT
1 2 3 4 5 6
A 0 6 0 3 4 0
C 0 0 1 0 1 0
G 1 0 0 3 0 0
T 5 0 5 0 1 6
Example of Decision TreeExample of Decision Tree
Markov ModelsMarkov Models
Ingredients of a Markov Ingredients of a Markov ModelModel
Collection of statesCollection of states
{S{S11, S, S22, …,S, …,SNN}}
State transition probabilities (transition State transition probabilities (transition matrix)matrix)
AAijij = P(q = P(qt+1t+1 = S = Si i | q| qtt = S = Sjj))
Initial state distributionInitial state distribution
ii = P(q = P(q11 = S = Sii))
Hidden Markov ModelsHidden Markov Models
Ingredients of a HMMIngredients of a HMM Collection of states:Collection of states: {S{S11, S, S22,…,S,…,SNN}}
State transition probabilities (transition matrix)State transition probabilities (transition matrix)
AAijij = P(q = P(qt+1t+1 = S = Si i | q| qtt = S = Sjj))
Initial state distributionInitial state distribution
ii = P(q = P(q11 = S = Sii))
Observations:Observations: {O{O11, O, O22,…,O,…,OMM}}
BBjj(k) = P(v(k) = P(vtt = O = Okk | q | qtt = S = Sjj))
Examples of Gene Examples of Gene FindersFinders
FGENESFGENES – linear DF for content and signal sensors and DP for – linear DF for content and signal sensors and DP for finding optimal combination of exonsfinding optimal combination of exons
GeneMarkGeneMark – HMMs enhanced with ribosomal binding site – HMMs enhanced with ribosomal binding site recognitionrecognition
GenieGenie – neural networks for splicing, HMMs for coding sensors, – neural networks for splicing, HMMs for coding sensors, overall structure modeled by HMMoverall structure modeled by HMM
GenscanGenscan – WM, WA and decision trees as signal sensors, HMMs for – WM, WA and decision trees as signal sensors, HMMs for content sensors, overall HMMcontent sensors, overall HMM
HMMgeneHMMgene – HMM trained using conditional maximum likelihood – HMM trained using conditional maximum likelihood
MorganMorgan – decision trees for exon classification, also Markov Models – decision trees for exon classification, also Markov Models
MZEFMZEF – quadratic DF, predict only internal exons – quadratic DF, predict only internal exons
Genscan ExampleGenscan Example
Developed by Chris Burge 1997Developed by Chris Burge 1997
One of the most accurate One of the most accurate ab initioab initio programsprograms
Uses explicit state duration HMM to Uses explicit state duration HMM to model gene structure (different length model gene structure (different length distributions for exons)distributions for exons)
Different model parameters for Different model parameters for regions with different GC contentregions with different GC content
Ab initioAb initio Gene Finding is Gene Finding is DifficultDifficult
Genes are separated by large intergenic regionsGenes are separated by large intergenic regions
Genes are not continuous, but split in a number of (small) Genes are not continuous, but split in a number of (small) coding exons, separated by (larger) non-coding intronscoding exons, separated by (larger) non-coding introns
in humans coding sequence comprise only a in humans coding sequence comprise only a few percents of the genome and an average few percents of the genome and an average of 5% of each geneof 5% of each gene
Sequence signals that are essential for elucidation of a gene Sequence signals that are essential for elucidation of a gene structure are degenerate and highly unspecificstructure are degenerate and highly unspecific
Alternative splicingAlternative splicing
Repeat elements (>50% in humans) – some contain coding Repeat elements (>50% in humans) – some contain coding regionsregions
Problems with Problems with Ab initioAb initio Gene FindingGene Finding
No biological evidenceNo biological evidence
In long genomic sequences many In long genomic sequences many false positive predictionsfalse positive predictions
Prediction accuracy high, but not Prediction accuracy high, but not sufficientsufficient
Evaluation of Gene Evaluation of Gene Finding ProgramsFinding Programs
Calculating accuracy of programs’ Calculating accuracy of programs’ predictionspredictions
Many evaluation studies, one of the Many evaluation studies, one of the earliest:earliest:
Burset and Guigó, 1996 (vertebrate Burset and Guigó, 1996 (vertebrate sequences)sequences)
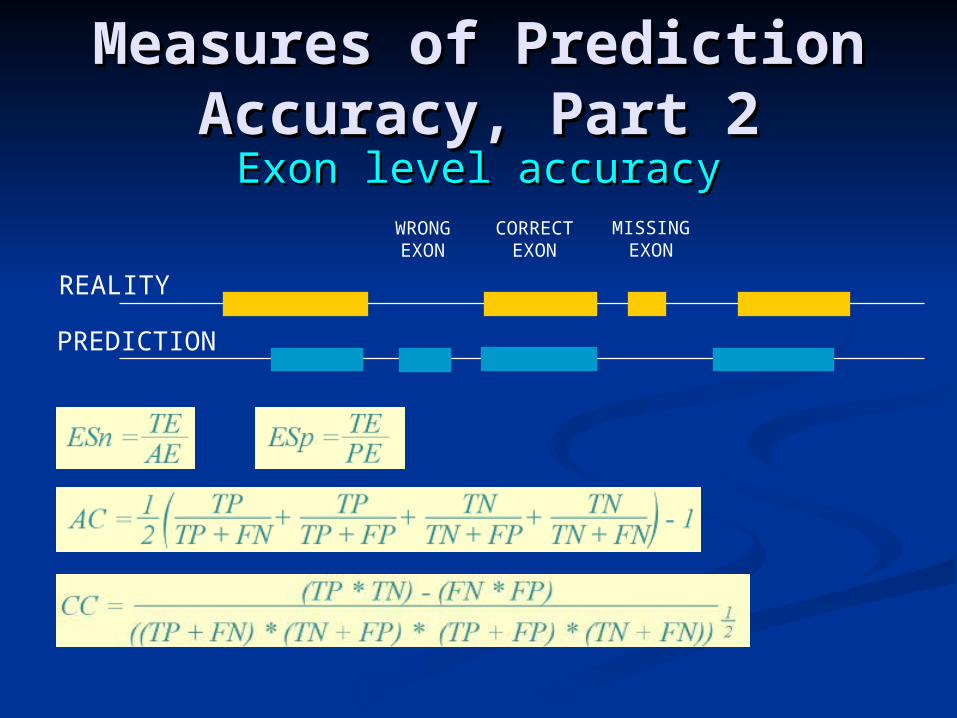
Measures of Prediction Measures of Prediction Accuracy, Part 2Accuracy, Part 2
Exon level accuracyExon level accuracy
REALITY
PREDICTION
WRONGEXON
CORRECTEXON
MISSINGEXON
41
Integrated Approaches for Integrated Approaches for Gene FindingGene Finding
Programs that integrate results of similarity Programs that integrate results of similarity searches with searches with ab initioab initio techniques techniques (GenomeScan, FGENESH+, Procrustes)(GenomeScan, FGENESH+, Procrustes)
Programs that use synteny between Programs that use synteny between organisms (ROSETTA, SLAM)organisms (ROSETTA, SLAM)
Integration of programs predicting different Integration of programs predicting different elements of a gene (EuGène)elements of a gene (EuGène)
Combining predictions from several gene Combining predictions from several gene finding programs (combination of experts)finding programs (combination of experts)
Set of methods used and they way Set of methods used and they way they are integrated differs between they are integrated differs between individual programsindividual programs
Different programs often predict Different programs often predict different elements of an actual genedifferent elements of an actual gene
they could complement each they could complement each other yielding better predictionother yielding better prediction
Gene Prediction LinksGene Prediction Links
http://genome.imim.es/geneid.html http://genes.mit.edu/GENSCAN.html FGENES, commercial, but can tryFGENES, commercial, but can try http://www.softberry.com/berry.pht
ml?topic=fgenes&group=programs&subgroup=gfind
GeneIDGeneID
Hierarchical approach: Hierarchical approach: Splice sites and stop codons predicted and Splice sites and stop codons predicted and
scored using position-specific weight scored using position-specific weight matricesmatrices
Exons built from identified “defining” sites. Exons built from identified “defining” sites. Scored as the sum of scores of defining sites Scored as the sum of scores of defining sites plus the score of their coding potentialplus the score of their coding potential
Maximization of all the score to assemble Maximization of all the score to assemble gene structuregene structure
Latesr versions of the program add sequence Latesr versions of the program add sequence similarity searchessimilarity searches
GeneSCAN - Underlying Hidden GeneSCAN - Underlying Hidden Markov Model programMarkov Model program
FGENES – linear discriminant FGENES – linear discriminant analysis to identify splice sites, analysis to identify splice sites, exons, promoter elementsexons, promoter elements
Genewise – compares a genomic Genewise – compares a genomic sequence with a protein sequence or sequence with a protein sequence or with HMMs representing protein with HMMs representing protein sequencessequences
How good the methods How good the methods are?are?
Different methods - different results. Different methods - different results. How to measure accuracy?How to measure accuracy?
SpecificitySpecificity : proportion of predicted : proportion of predicted elements, genes that are realelements, genes that are real
CorrelationCorrelation coefficient combines both coefficient combines both
Screening Test for Screening Test for Occult CancerOccult Cancer
100 patients with occult cancer: 100 patients with occult cancer: 95 have "x" in their blood95 have "x" in their blood
100 patients without occult cancer: 100 patients without occult cancer: 95 do not have "x" in their blood95 do not have "x" in their blood
5 out of every 1000 randomly selected 5 out of every 1000 randomly selected individuals will have occult cancerindividuals will have occult cancer
SENSITIVITY
SPECIFICITY
PREVALENCE
2 X 2 2 X 2 TableTable
Occult CancerPresent
Occult CancerAbsent
"x" present"x" absent
100,000
99,500
50025475
4,975 94,525
5,450 94,550
If a patient has “x” in his blood, chance of occult cancer
is 475 / 5475 = 8.7%
Standard TerminologyStandard Terminology
DiseasePresent
DiseaseAbsent
Test positive
Testnegative
EntirePopulation
FP + TN
TP + FNTrue
Positives(TP’s)
FalsePositives
(FP’s)
TrueNegatives
(TN’s)
FalseNegatives
(FN’s)
TP + FP FN + TN
DefinitionsDefinitions
PV+ = PREDICTIVE VALUE = TPTP + FP = P(D+|T+)
SPECIFICITY = TNFP + TN = P(T-|D-)
SENSITIVITY = TPTP + FN = P(T+|D+)
What is a “Positive What is a “Positive Test”?Test”?
All the analysis has assumed that it is All the analysis has assumed that it is clear whether a test is positive or clear whether a test is positive or negativenegative
In reality, many tests involve In reality, many tests involve continuous values so that one result continuous values so that one result may be “more positive” than anothermay be “more positive” than another
How should one define the cut-off at How should one define the cut-off at which a test is judged to be which a test is judged to be abnormal?abnormal?
• More false positives• Higher sensitivity• Lower specificity
More on Projects:More on Projects:
vaccine development (Ramya)vaccine development (Ramya)http://immunax.dfci.harvard.edu/PEPVAC/ HIV & the Black Plague – HarshalHIV & the Black Plague – Harshal







































































![A Memoir of Toni Wolff By Irene Irene Champernowne [part2]](https://static.documents.pub/doc/80x56/568bd6131a28ab20349ac25d/a-memoir-of-toni-wolff-by-irene-irene-champernowne-part2.jpg)
