Page 1
“PREPARATION AND EVALUATION OF LIPOSPHERE
BASED TOPICAL DRUG DELIVERY SYSTEM CONTAINING
A NSAID DRUG”
THESIS
Submitted in partial fulfillment for the degree of
MASTER OF PHARMACY
In
Industrial Pharmacy
In The Faculty of Medicine
Sant Gadge Baba Amravati University, Amravati.
RESEARCH SCHOLAR
Mr. LEELADHAR PRAJAPATI
B. PHARM
RESEARCH GUIDE
Dr. P. S. KAWTIKWAR
(M. PHARM, Ph.D.)
2012-2013
DEPARTMENT OF PHARMACEUTICS
SUDHAKARRAO NAIK INSTITUTE OF PHARMACY, PUSAD-445204
SANT GADGE BABA AMRAVATI UNIVERSITY, AMRAWATI
MAHARASHTRA. (INDIA)
Page 2
Affectionately Dedicated To, God,
Who is always with me.
My family,
Whose affection and love are infinite with me in adversity and prosperity.
The Guide,
To whom, I shall remain indebted for giving new shape and path to my life.
Page 3
(Research Guide) Dr. P.S. Kawtikwar M.Pharm. Ph.D.
Professor & H.O.D.
Department of Pharmaceutics,
S. N. Institute of Pharmacy,
Pusad- 445 204.MS (India)
CERTIFICATE
This is to certify that the investigations described in this thesis
entitled “Preparation and evaluation of liposphere based topical drug
delivery system containing a NSAID drug” was carried out by Mr.
Leeladhar Prajapati in the laboratories of Department of pharmaceutics,
S.N. Institute of pharmacy, Pusad under my supervision and guidance.
The thesis work was carried out and submitted in partial fulfillment
of the requirements for the Degree of Master of Pharmacy in
Industrial Pharmacy, in the faculty of Medicine of Sant Gadge Baba
Amravati University, Amravati.
This thesis is now ready for examination and evaluation.
Date: Dr. P. S. Kawtikwar
Place: [Research Guide]
Page 4
Dr. D. M. Sakarkar M.pharm. Ph.D. Principal, S.N. Institute of Pharmacy, Pusad -445204, MS (India)
CERTIFICATE
This is to certify that the investigations described in this thesis
entitled “Preparation and evaluation of liposphere based topical drug
delivery system containing a NSAID drug” was carried out by Mr.
Leeladhar Prajapati in the laboratories of Department of pharmaceutics,
S.N. Institute of pharmacy, Pusad under my supervision and guidance.
The thesis work was carried out and submitted in partial fulfillment of the
requirements for the Degree of Master of Pharmacy in Industrial
Pharmacy, in the faculty of Medicine of Sant Gadge Baba Amravati
University, Amravati.
The thesis is now ready for examination and evaluation.
Date: Dr. D. M. Sakarkar
Place: Principal
Page 5
Mr. Leeladhar Prajapati
B. Pharm.
Department of Pharmaceutics
S. N. Institute of Pharmacy
Pusad-445204, MS (India)
DECLARATION
It gives me great pleasure and satisfaction to declare that the thesis
entitled “Preparation and evaluation of liposphere based topical drug
delivery system containing a NSAID drug” was reviewed and submitted
in partial fulfillment of the requirements for the Degree of Master of
Pharmacy in Industrial Pharmacy, in the Faculty of Medicine of Sant
Gadge baba Amravati University, Amravati under guidance and supervision
of Dr. P.S. Kawtikwar and I hereby declare that
This thesis is now ready for examination and evaluation.
Date: Mr. Leeladhar Prajapati
Place:
Page 6
INSTITUTIONAL ANIMAL ETHICS COMMITTEE
(Reg. No. CPCSEA/729/02/a/ CPCSEA)
Prof. S.V. Tembhurne
Member Secretary (IAEC)
Ref. No. SNIOP/IAEC/2012-13/CPCSEA/IAEC/IP-PL/12-2012
Mr. Leeladhar Prajapati
(M.pharm Student)
Department of Pharmaceutics
Sudhakarrao Naik Institute of Pharmacy, pusad
Dist-Yavatmal-445204
This is to certify that the proposal of Mr.Leeladhar Prajapati
for the Study entitled “Preparation and evaluation of liposphere based
topical drug delivery system containing a NSAID drug” was approved by
Institutional Animal Ethics Committee (IAEC) of S.N. Institute of Pharmacy
Pusad in the IAEC meeting held on dated 07/01/2013 and the proposal
number CPCSEA/IAEC/IP-PL/12-2012
Hence the certificate is issued.
Place: Pusad [Prof. S.V. Tembhurne]
SSUUDDHHAAKKAARRRRAAOO NNAAIIKK IINNSSTTIITTUUTTEE OOFF PPHHAARRMMAACCYY
Nagpur road, Pusad Dist: Yavatmal (M.S) 445204
Phone (07233)-247308; Fax (07233)-247308
Web site: www.sniop.ac.in
Page 7
LIST OF TABLES
Table
No.
Title Page
No.
1. Composition and active ingredients for formulations of lipospheres 19
2. Factors influencing morphology of Lipospheres 25
3. Factors influencing entrapment efficiency 26
4. Factors influencing drug release 27
5. List of instrument used 45
6. List of chemical and reagent used 46
7. Formulations codes of lipospheres different batchs (F1-F12) 51
8. Topical gel formulation of optimized batch 55
9. Physical characters of Aceclofenac drug 60
10. Interpretation of Aceclofenac IR 62
11. UV calibration curve reading of Aceclofenac in phosphate buffer
saline pH 7.4 63
12. Evaluations of lipospehers 64
13. Mean particle size (mps) of different formulation of liposphers 66
14. Cumulative percentage drug release from various formulations of
lipospheres (F1-F4) 72
15. Cumulative percentage drug release from various formulations of
lipospheres (F5-F8) 73
16. Cumulative percentage drug release from various formulations of
lipospheres (F9-F12) 74
17. Drug release kinetics for the various formulations of lipospheres 75
18. Result of pH, Viscosity, Drug content and Spreadability 77
19. Cumulative percent (%) drug release of optimize batch (F8, LF8) 79
20. Percentage inhibition for anti-inflammatory activity 81
21. Stability studies for the formulation LF7 and LF8 82
Page 8
LIST OF FIGURES
Fig.
No.
Title Page
No.
1 Interactions of phospholipids in aqueous media. 6
2 Schematic liposphere 7
3 A cross sectional view of human skin 8
4 Epidermis with Stratum corneum including a corneocytes cluster 9
5 Skin appendages 13
6 Plasma drug concentration profile for controlled release 17
7 Schematic representation of the methods of production of LS: melt
dispersion and solvent evaporation 21
8 UV spectrum of aceclofenac 61
9 FTIR Spectra of aceclofenac drug 61
10 Calibration Curve of Aceclofenac 63
11. Entrapment efficiency (F1-F12) 64
12 Photograph of optimized liposphers batch F7 65
13 Photograph of optimized liposphers batch F8 65
14 FT-IR spectra of Aceclofenac 67
15 FT-IR Spectra of physical mixture 68
16 DSC Thermogram of Aceclofenac 69
17 DSC Thermogram of optimized formulation F8 and F6 70
18 SEM images of drug loaded lipospheres 70,71
19 In vitro drug release of batch F1,F2,F3,F4 72
20 In vitro drug release of batch F4,F5,F6,F8 73
21 In vitro drug release of batch F9,F10,F11,F12 74
22 First order drug release plot of optimized batch F7 76
23 First order drug release plot of optimized batch F8 76
24 Higuchi’s drug release plot of optimized batch F7 76
25 Higuchi’s drug release plot of optimized batch F8 76
26 Peppas korsmeyer’s drug release plot of optimized batch F7 76
27 Peppas korsmeyer’s drug release plot of optimized batch F8 76
Page 9
28 Lipospheres based gel of optimized batch 78
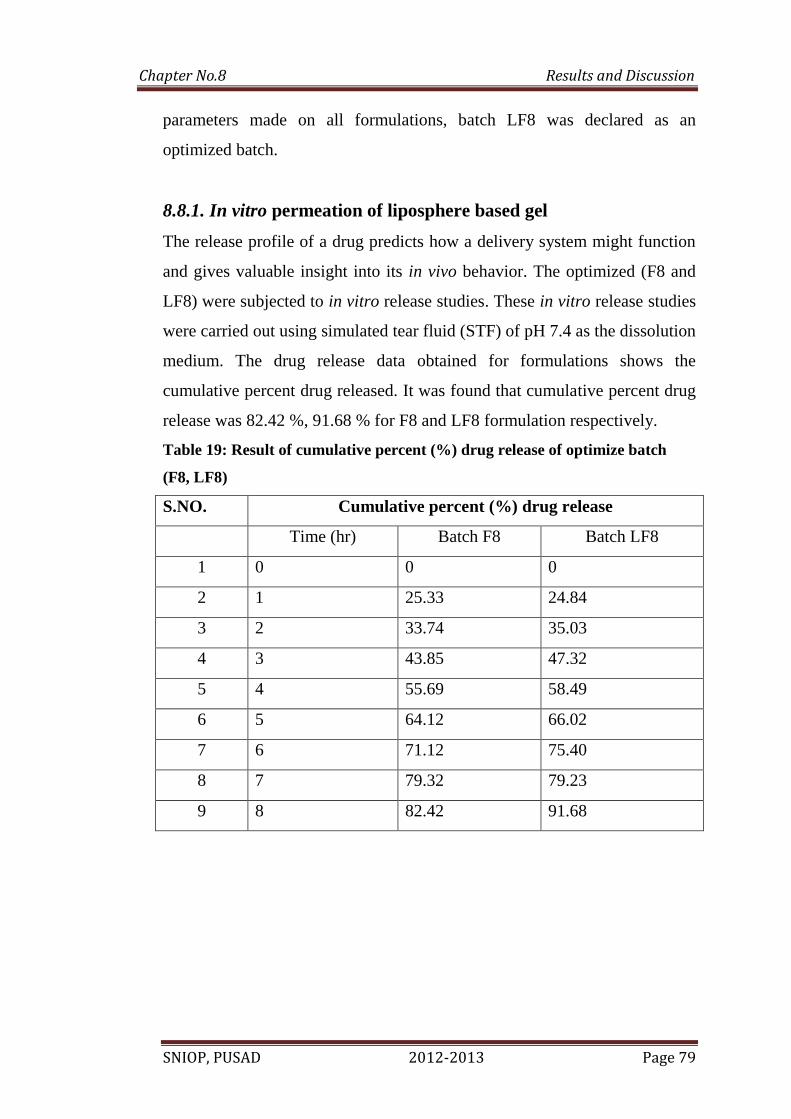
29 In vitro release profile of optimized formulation F8 and LF8 80
30 Skin-irritation testing (Draize patch test) 80
Page 10
LIST OF ABBREVATIONS
Base unit Symbol
Hour h
Second s
Minute min
Milliliter ml
Micrometer μm
Nanometer nm
Millimeter mm
Milligram mg
Gram g
Cubic meter m3
Revolutions per minute rpm
Centimeter cm
Potassium Bromide KBr
Centipoise cps
BP British Pharmacopoeia
LS Liposphere
Page 11
ACKNOWLEDGEMENT
First and foremost I would like to express my deepest pray to ALL
MIGHTY, love and thanks to my father Mr. Teerath Prasad Prajapati, my mother
Mrs. Shivkali Prajapati, for their extreme patience and support. I know this took a
long time but your sacrifice and understanding has allowed me to persevere.
Let me start by expressing my sincere and special thanks to my esteem guide
Respected Dr. P. S. Kawtikwar Sir, H.O.D. Dept. of Pharmaceutics, Sudhakarrao
Naik Institute of pharmacy, Pusad. I am thankful to him for giving me freedom to
work, timely advice and valuable suggestions. Under his constant guidance,
encouragement, and positive attitude towards work has instilled more confidence in
me. “Thank you Sir” for all you has done.
I owe a great debt of gratitude to Dr. D. M. Sakarkar Sir, Principal,
Sudhakarrao Naik Institute of pharmacy, Pusad for providing the necessary
facilities. This thesis would not have become a reality without his constant quest for
knowledge and critical evaluation.
I’m grateful to Prof. R. B. Wakade Sir, Prof. A.H.harsulkar sir, Prof.
A.M.Mahale sir, & Prof. J.K. Jadhav, Prof. A.R. Tapas sir and prof. S. V.
Tembhurne sir for their proper orientation to my work to make it possible.
I express my deepest thanks to VAV Life Science Ms. Stuti Singh mam and
Mr. Swanand Malode sir for valuable suggestions and motivation.
I express my deepest and special thanks to my elder Brothers Anand and
Yogesh & my sister for their keen interest, love and motivation in my life.
Page 12
I am thankful to all teaching and non-teaching staff of our college for valuable
suggestions and motivation, which they bestowed on me right from the inception to
the successful completion of the work.
I’m really very much grateful to Mr. M.D.Wandhare, for his tips, his
guidance and his cooperation during my dissertation work.
I express my deepest and special thanks to my colleagues,Prashant, Girish, Ketan,
Mayur, Lukesh, Anant, Pankaj, Ghanashyam, Dhanraj, Amol, Rahul, Sushil,
Nikita, Radhe, Anish, Akanksha, Nilakshi, for their keen interest, love and support
in my dissertation work.
There are many others whose names flashed across my mind when I enlist
those who have given grateful support to me. It would rather impracticable to
mention each of them separately but I am conscious my obligation and thanks them
collectively.
Leeladhar Prajapati
Page 13
Table of Contents
SR. S.NO. TITLE PAGE NO.
1.
INTRODUCTION
1-28
2.
LITERATURE REVIEW
29-35
3.
AIM AND OBJECTIVES
36
4.
PLAN OF WORK
37
5.
DRUG PROFILE
38-44
6.
LIST OF MATERIAL AND EQUIPMENT
45-47
7.
EXPERIMENTAL WORK
48-59
8.
RESULTS AND DISCUSSION
60-82
9.
SUMMARY AND CONCLUSION
83-85
10.
FUTURE SCOPE
86
11.
REFERENCES
87-97
12.
ERRATA
98
Page 14
Abstract
SNIOP, PUSAD 2012-2013
Abstract
The purpose of this study was to prepare lipospheres containing aceclofenac
intended for topical delivery with the aim of exploiting the favorable properties
of this carrier system and developing a sustained release formula to overcome
the side effects resulting from aceclofenac oral administration. Lipospheres
were prepared using different lipid cores (carnauba wax, bees wax, steryl
alcohol) and phospholipids coats (egg phosphatidylchoine and soya
phosphatidylcholine) by melt dispersion technique. Characterization of the
prepared lipospheres formulation carried out through photomicroscopy,
scanning electron microscopy (SEM), particle size analysis, diffential scanning
caorimetry (DSC), and In vitro drug release and stability study. It was
uniformly dispersed after suitably gelled by Carbopol 940 preparation. The
characterization of the prepared lipospheres was based on topical gel
rheological study, pH, spreadability, drug content, skin irritation test. No
oedema and erythema were observed after administration of lipospheres based
aceclofenac gel on rabbit skin. The anti-inflammatory effect of liposphere
systems was assessed by the rat paw edema technique and was compared to the
marketed product. Results revealed that liposphere systems were able to entrap
aceclofenac at very high levels (101.2%). The particle size of liposphere
systems was well suited for topical drug delivery. DSC revealed the molecular
dispersion of aceclofenac when incorporated in lipospheres. Lipospheres were
substantially stable after 3 months storage at 2–8 °C. Liposphere topical gel was
found to possess superior anti-inflammatory activity compared to the marketed
product.
Key words: Aceclofenac, entrapment efficiency, formulation of topical gel,
animal experiment, stability, sustained release.
Page 15
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 1
1. Introduction
One approach for increasing the beneficial action of drugs and
decreasing systemic adverse effects is to deliver the necessary amount of drugs
to the diseased sites, where they are most needed, for the appropriate period of
time1-3
. Although the drug delivery system concept is not new, great progress
has recently been made in the treatment of a variety of diseases. Particulate
carriers (e.g., polymeric nano- and Microparticles, fat emulsion, and liposomes)
possess specific advantages and disadvantages. For instance, in the case of
polymeric Microparticles, the degradation of the polymer might possibly cause
systemic toxic effects through the impairment of the reticuloendothelial system4
or by accumulation at the injection Site5, cytotoxic effects have indeed been
observed in vitro after phagocytosis of particles by human macrophages and
granulocytes6. In addition, organic solvent residues deriving from the
preparation procedures, such as the solvent evaporation technique often used
for liposome7 and polyester microparticles
8 can be present in the delivery
system and could result in severe acceptability and toxicity problems9. To solve
these adverse effects, lipid microspheres, often called lipospheres (LS), have
been proposed as a new type of fat-based encapsulation system for drug
delivery of bioactive compounds (especially lipophilic compounds). LS consist
of solid microparticles with a mean diameter usually between 0.2 and 500 μm,
composed of a solid hydrophobic fat matrix in which the bioactive compounds
are dissolved or dispersed10-12
. LS have some advantages over other delivery
systems, such as good physical stability, low cost of ingredients, ease of
preparation and scale-up, and high entrapment yields for hydrophobic drugs.
Because of their large range in particle size, LS can be administered by
different routes- such as orally, subcutaneously, intramuscularly, or topically-or
they can be used in cell encapsulation, thus allowing them to be proposed for
treatment of a number of diseases13 15
. For instance, the in vivo distribution of
LS demonstrated a high affinity to vascular wells (including capillaries),
Page 16
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 2
inflamed tissues, and granulocytes16-17
. LS have been used for the controlled
delivery of various types of drugs, including vasodilator and antiplatelet drugs,
anti-inflammatory compounds, local anesthetics, antibiotics, and anticancer
agents; they have also been used successfully as carriers of vaccines and
adjuvants18
. For a biocompatible formulation suitable for human administration,
triglycerides and monoglycerides have been chosen as the biomaterials for LS
because of their high biocompatibility, high physicochemical stability, and drug
delivery release. LS were prepared by two alternative approaches, namely, the
melt dispersion and the solvent evaporation techniques. Liposphere formulation
is an aqueous micro dispersion of solid water insoluble spherical micro the
lipospheres are made of solid hydrophobic triglycerides with a monolayer of
phospholipids embedded on the surface of the particle. Liposphere formulation
is appropriate for oral, parenteral and topical drug delivery system. The solid
core containing a drug dissolved or dispersed in a solid fat matrix and used as
carrier for hydrophobic drugs. Several techniques, such as solvent
emulsification evaporation, hot and cold homogenization and high pressure
homogenization have been used for the production of liposphere. Benefits of
liposphere drug delivery system are;
a) Improving drug stability.
b) Possibility for controlled drug release.
c) Controlled particle size.
d) High drug loading.
In addition, use of lipospheres for oral administration, it can protect the drug
from hydrolysis, as well as improve drug bioavailability. Therefore, the present
review articles focused on achievements of lipospheres formulation to deliver
the drugs in the targeted sites.
Polymers as carriers19
for “difficult to deliver” drugs, “to be targeted
drugs” and to achieve a desired release pattern is a popularly known and widely
exploited concept in formulation Technology. With the emergence of polymer
Page 17
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 3
era, delivery systems like microspheres20
nanoparticles21
found their way into
pharmaceutical market. Natural, semi synthetic and synthetic polymers are now
being widely explored in the formulation of multiparticulate systems apart from
several other applications. Multi particulate systems comprise of several small
discrete units containing drug substance entrapped or encapsulated in polymer
and offer several advantages over single unit dosage forms. They show
predictable gastric emptying resulting in less inter and intra-subject variability,
gastric emptying independent of the state of nutrition, high degree of dispersion
in the digestive tract, lower risk of dose dumping and reduced local irritation,
increased bioavailability, reduced risk of systemic toxicity. Despite these
advantages, use of polymers in drug delivery poses several safety concerns.
Polymers used in the preparation of micro/nanospheres can produce toxic
degradation products causing systemic toxic effects through the impairment of
reticuloendothelial system (RES).
Polymers precipitate toxic effects due to accumulation of products at injection
site. In vitro studies have shown cytotoxic effects after phagocytosis of polymer
particles by human macrophages and granulocytes. Eg: Pre degraded poly (L-
lactic acid) (P-PLLA) and non treated PLLA (N-PLLA) particles, both having
diameters not exceeding 38 μm, were injected intraperitoneally in mice.
Nondegradable polytetrafluoroethylene (PTFE) particles and the carrier
solution were used as control. Cells of the abdominal cavity were harvested after
1, 2, 3, 4, 5, and 7 days to study the effect on the morphology of cells and
viability due to degradation products. TEM (Transmission Electron Microscopy)
studies indicated that, upon injection of particles in the peritoneal cavity,
macrophages demonstrated signs of cell damage, cell death, and cell lysis due to
phagocytosis of a large amount of PPLLA particles.
Methods like solvent evaporation used in the preparation of liposomes and
polyester Microparticles 22
leave organic solvent residues in the product which
can result in severe acceptability and toxicity problems.
Page 18
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 4
Due to several limitations with polymeric delivery systems, extensive
attempts are being made to develop alternate carriers. Lipids23
especially, are
now being studied widely due to their attractive properties namely
physicochemical diversity, biocompatibility, biodegradability, ability to increase
the oral bioavailability of poorly water soluble drug moieties, thus making them
ideal candidates as carriers for problematic drugs.
1.1.1 Advantages of lipid based delivery systems:
Lipid based delivery systems disperse, solubilize and maintain solubility of
drug in GI fluids.
Bioavailability of most of the lipophilic drugs is altered in the presence of
lipid content in food.
Lipid carriers mimic such lipid food and thus reduce the food effect on
bioavailability of drugs and render flexibility to dosage regimen.
Transfer drug into bile-salt mixed micelle and promote lymphatic uptake of carrier-
drug particles.
Influence gut wall permeability.
Normalize and/or modify pharmacokinetic parameters. However few concerns
related to using lipids as carriers can be overcome by well characterizing
physicochemical and testing methodologies for lipid drug delivery systems and
are as follows:
Limiting solubility of drug in lipid core which determines entrapment
efficiency.
Quantity of excipient required.
Stability of drug.
Chemical stability issues like drug and carrier compatibility.
Physical stability of lipid dosage forms like polymorphic phase transitions of
drug and lipid during preparation and storage and stability of semisolids.
Page 19
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 5
Also research should be focused towards developing reliable in vitro and in vivo
testing methodologies for lipid based delivery systems and understanding the in
vivo fate of drug carried by such delivery systems and influence of co
administered drugs/lipids on the bioavailability of drugs.
Lipid based drug delivery systems like solid lipid nanoparticles (a
technology owned by Skye Pharma) and lipospheres are now being studied
widely. Solid lipid nanoparticles24
are nanosized lipid carriers in which lipidic
core contain the drug in dissolved or dispersed state. These systems were
designed to substitute polymeric carriers due to the inherent toxicity.
Lipospheres are lipid based dispersion systems in which drug is dissolved or
dispersed in lipidic core, the surface of which is embedded with emulsifier layer.
1.1.2 Advantages of lipospheres carriers:
1. Easily available, low cost, GRAS (Generally Recognized As Safe) listed raw
materials.
2. Feasible simple production techniques that do not employ high energy
process like homogenization which will otherwise compromise the stability of
labile active pharmaceutical ingredient.
3. High entrapment for lipophilic drugs.
4. Extended release of entrapped drug after a single injection from few hours to
several days
5. Good physical stability
6. Administration by several routes.
1.2. Colloidal drug delivery systems
Many of the drug substances are characterized by poor aqueous
solubility, which causes many formulation problems. Besides the use of co-
solvents, drug24
complexation and solubilization in surfactant micelles,
incorporation in colloidal carrier systems represents an alternative way to
Page 20
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 6
render poorly water soluble drugs applicable for effective therapy. Furthermore
incorporation of drugs in particulate carriers provides a possibility to
manipulate the drug release. In last few years the colloidal carriers have been
used for site specific targeting especially in cancer chemotherapy. Based on the
carrier material the conventional vehicles used as drug carriers can be divided
into 2 groups
1. Polymeric carriers.
2. Lipidic carriers.
a. Liposomes.
b. Lipoproteins.
c. Lipid O/W emulsions.
d. Lipospheres.
The lipidic carriers are more preferred than polymeric carriers to avoid
potential toxicological problems. The vehicles of all the above lipidic carriers
are composed of physiological lipids such as phospholipids, cholesterol,
cholesterol esters and triglycerides.
Phospholipid molecule
Page 21
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 7
Micelle Lipid Bilayer Lipospheres
Figure 1: Interactions of Phospholipids in Aqueous Media.
Lipospheres
Figure 2: Structure of Liposphere Figure 3: Schematic Liposphere
1.3. The structure of skin
Skin is anatomically divided into three principal and distinct layers, from
the outside of skin inward, including stratum corneum (10–20 μm thick), viable
Page 22
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 8
epidermis (50–100 μm thick), and dermis (1000-2000 μm thick). A fatty
subcutaneous layer resides beneath the dermis. It should be pointed out that all
the thickness specified here are representative only, since the actual thickness of
each layer varies several fold from place to place on the body. Adnexal
appendages, including hair follicles, associated sebaceous glands and pili
muscles, apocrine and eccrine sweat glands, can be found dispersing throughout
of the skin, varying in number and size depending on body site. The cross
section of skin structure is shown is (Figure 4)
Fig 4: A cross sectional view of human skin, Source: From Ref. (Lu and Flynn, 2009)
1.3.1 Stratum corneum (SC)
Stratum corneum is the outmost superficial layer of the skin and also the
principal barrier element of the skin. SC consists of several layers of completely
keratinized flattened dead cells, corneocytes, each of which is about 30 μm in
diameter with a hexagonal shape and 0.5-0.8 μm in thickness. These acutely
flattened corneocytes are highly organized and stacked vertically 15 to 25 cell
layers, which are embedded into a specialized and well structured intercellular
lipid matrix.
Page 23
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 9
The most simplistic organizational description of SC is advocated by which is
the classic “brick-and-mortar” assembly (Figure 5). The intracellular space of
corneocytes is literally packed with structural protein, semi crystal line α
keratin intermixed with more amorphous β keratin. The intracellular space is
dense, offering little freedom of movement to drug molecules. Thus, the
corneocytes work as “brick” being thermodynamically impenetrable. While the
intercellular space of corneocytes is filled with a lipid “mortar” formed of
cholesterol, free fatty acids, and ceramides, which seals horny structure.
Fig.5: Brick-and-mortar model of Stratum corneum and penetration routes
through it, Source from Ref. (Elias, 1981)
However, the brick-and-mortar skin model is not enough to describe the
panorama of the SC. In fact, the cells from basal layer of epidermis, which we
describe further in the text, to the SC are built up in clusters, which represent
the basic skin permeation resistance unit.It is these clusters that are separated by
surface corrugations (wrinkle line), which often reach several micrometers into
the basal layer of the epidermis (Figure 6).
Fig.6: Epidermis with Stratum corneum including a corneocytes cluster,
Source from Ref. (Cevc and Vierl, 2010)
Page 24
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 10
In addition, the basolateral side of stratum corneum is in direct contact with the
living epidermal mass, where the corneocytes contain water at high
thermodynamic activity of the physiological milieu. On the other hand, its
external surface interfaces the environment, where air tends to have a far lower
water activity. Consequently a water gradient is established and water diffuses
out through the stratum corneum. Under such a normal hydration situation, the
stratum corneum takes up moisture to the extent of 15% to 20% of its dry
weight. It should be pointed out here that the hydration condition of SC plays
an important role on the drug molecules skin penetration, which would be
discussed further in the text.
1.3.2 Viable epidermis
The viable epidermis is underlying the stratum corneum. It is
multilayered when viewed under microscope, including, from bottom to top,
basal layer (stratum germinativum), spinous layer (stratum spinosum), granular
layer (stratum granulosum) and lucid layer (stratum lucidum). Each layer is
defined by position, shape, and morphology and also reflects the progressive
differentiation of keratinocytes which eventuates into their death and placement
as chemically and physically resistant “brick” in stratum corneum. However,
when physicochemically considered, the viable epidermis is just a group of
tightly massed live cells, which results in a singular diffusion area or resistance
in percutaneous absorption process. Water found in this live epidermis has an
activity equivalent to that of 0.9 % NaCl.
While the interface between stratum corneum and epidermis is flat, the one
between epidermis and dermis is papillose, which increases their contact
surface area and then allows for the diffusion of nutrients or other biological or
medicated molecules between dermis and epidermis.
Page 25
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 11
The epidermis itself is avascular. Besides keratinocytes, Langerhans cells also
can be found in viable epidermis. They are antigen presenting cells in the skin’s
immunological responses. Moreover, another kind of cells, melanocytes, are
strategically placed in the epidermis just above the epidermis and dermis
junction. When influenced by melanocyte-stimulating hormone or ultraviolet
radiation, melanocytes synthesize and deposit the pigment granules into skin,
which gives rise to the skin coloration.
1.3.3 Dermis
Dermis is directly adjacent to the epidermis and extends from the
epidermal-dermal junction to the subcutaneous tissue (Figure 4). Dermis
consists of a net-work of irregular connective tissue, which provides the
mechanical support for the skin.
The matrix of this connective tissue consists of structure fibers, such as
collagen, reticulum, and elastin. These fibers are embedded in an amorphous
mucopolysaccharidic gel called the ground substance.
The dermis can be arbitrarily divided in into a superficial papillary layer
and a deep reticular layer. The upper papillary layer is thin, one fifth of
thickness of the dermis, and protrudes in to the epidermis giving rise to the
dermal papilla, and also provides the support of the delicate capillary plexus
which nurtures the epidermis.
The deepest layer of the skin is a far coarser fibrous matrix, the reticular
dermis, which is the main structural element of the skin. Equally importantly,
the microcirculation which subserves the skin is entirely housed in the dermis.
Blood flow through skin can vary by a factor of 100 fold depending on
environmental conditions. The dermis is also penetrated by sensory nerve
endings and an extensive lymphatic network. Moreover, skin appendages such
as sweat gland, sebaceous glands, hair follicles, and arrector pili muscles are
anchored within the dermis.
Page 26
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 12
The main cell inhabitants of the dermis are fibroblasts, mast cells and
macrophages. Fibroblasts synthesize the structural fibers, while mast cells are
thought to synthesize the ground substance. Macrophages work as immune
response. In addition, plasma cells, chromatophores, fat cells, nerve cells and
endings can also be found along with blood vessels, nerves and lymphatics.
1.3.4 Skin appendages
Skin appendages include hair follicles and their associated sebaceous
glands, eccrine glands, apocrine glands, and arrector pili muscles. The hair
follicle unit is composed of the hair, hair follicle, associated sebaceous gland,
and pili muscles. Hair is a compact of keratinized structures, which consist of
three layers, including an outermost cuticle, a cortex of densely packed
keratinized cells, and a medulla of loose flattened cells. Hairs can be found
mostly everywhere on the body except for the soles of the feet, the palms of the
hand, and mucocutaneous junctions. There are100 follicles per square
centimeter, representing one thousandth of the skin’s surface. A hair emerges
from a follicle, which is set within the dermis at a slight angle. The hair follicle
consists of three major components, including internal root sheath, external root
sheath, and dermal papilla (Figure 7). This arrangement results in a solid
implantation of the hair root in the hair follicle. In addition, each follicle is
anchored to the surrounding connective tissue by an individual strand of
arrector pili muscle.
Furthermore, each follicle is associated with one or more flask like
sebaceous glands, which secret an oily secretion, sebum. Then the sebum is
forced upward around the hair shaft and onto the skin surface. Sebum mainly
consists of squalene, cholesterol esters, wax esters, and triglycerides. It has
several biological functions including the regulation of steroidogenesis and
androgen synthesis, and providing antibacterial and water resistance to skin.
Page 27
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 13
Fig.7 Skin appendages: (a) Structure of the skin (b) Structure of the hair follicle
(c) Cross-section of the hair, source from Ref. (Wosicka and Cal, 2010)
Eccrine glands (sweat glands) are distributed over the entire body except the
genitalia and lips. These simple tubular glands open directly on the skin
surfaces and extend to the footings of the dermis. There are between 150 and
600 glands per square centimeter of body surface depending on body site
.However, the estimated number of actual sweating glands is much less than
that value, since many of these glands remains dormant. Thus, these glandular
openings occupy approximately one ten thousandth of the skin surface. Eccrine
sweat is slightly acidic (pH=5) due to traces of lactic acid, which is moderately
bacteriostatic.
1.3.5 Skin penetration routes
When a skin drug delivery system is applied topically, drug-containing
carriers or free drug in the system could interact with either the stratum
corneum or the sebum filled ducts of the pilosebaceous glands.
Thus, two principle absorption routes are involved, including the
transepidermal route, where the drug delivery system interacts with or diffuses
Page 28
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 14
through stratum corneum, and transfollicular route, where they interact with or
diffuse through the follicles.
In the case of the transepidermal route, since the impermeable character of the
corneocytes, the intercellular space of corneocytes provides the only continuous
phase, which is also the predominant penetration pathway (intercellular route or
intercorneocyte pathway) from the skin surface to the viable epidermis.
However, the tortuous zigzag bestowed by staggered corneocytes arrangement
(typically 18–21), corneocyte layers as well as the highly organized crystalline
lamellae structures of the mortar lead to an outstanding barrier property of the
labyrinthine intercellular route.
The transportation of molecules across this layer is primarily passive diffusion,
in accordance with Fick’s law, and no active transport processes have been
identified to date.Thus the permeability of stratum corneum as a penetration
resistor is proportional to the diffusive mobility of drug molecules within it
(diffusion coefficient, Dsc, also proportional to the capacity of the SC to
solubilize the drug molecules relative to vehicle (partition coefficient, Ksc) but
inversely proportional to the thickness of stratum corneum (hsc). Consequently,
at the steady state and sink condition, drug permeation can be described as
following:
where Jsc (μg cm-2
h-1
) is the steady state flux through stratum corneum. C is
the concentration of drug in the topical drug delivery system. When considering
transfollicular route, initially it was not considered to be a significant skin
penetration route, as evidence suggested that they accounted for only
approximately 0.1% of the skin surface area .Recently, it has been demonstrated
that hair follicles may act as a significant penetration pathway and/or potential
Page 29
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 15
reservoirs for topically applied compound. As mentioned before, owing to the
presence of sebum in follicles, the permeation through follicular route can be
described as following:
Where J sebum (μg cm-2
h-1
) is the steady state flux through sebum/hair follicle.
C is the concentration of drug in the topical drug delivery system. Ksebum and
Dsebum are diffusion coefficient through sebum and drug partition coefficient
in sebum/water, respectively.
In short, either or both routes can be important depending on the
physicochemical properties of a drug as well as the condition of the skin, since
the percutaneous absorption is a spontaneous passive diffusion process which
takes the path of least resistance.
1.4. Sustained drug delivery system
Modified release delivery systems may be divided conveniently into the
following categories 25
Delayed release
Sustained release
Site specific targeting
Receptor targeting
Sustained release, sustained action, prolonged action, controlled release,
extended action, timed release, depot and repository dosage forms are terms
used to identify drug delivery systems that are designed to achieve a prolonged
therapeutic effect by continuously releasing medication over an extended period
of time after an administration of single dose.
Page 30
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 16
The term “sustained release” is used to describe a dosage form
formulated to retard the release of a therapeutic agent such that its appearance
into systemic circulation is delayed and/or prolonged and its plasma profile is
sustained in duration. The onset of pharmacological action is often delayed and
duration of its therapeutic effect is often sustained. Controlled release dosage
forms are designed to release drug in vivo according to predictable rates that
can be verified by in vitro measurements. Controlled release technology implies
a quantitative understanding of the physicochemical mechanism of drug
availability to the extent that the dosage form release rate can be specified.26
Various designations such as smart, targeted, intelligent, novel and therapeutic
have been given to sustained release systems.
The sustained release dosage forms continue to lure both the market and
the researchers by virtue of improved patient compliance and reduced
incidences of adverse drug reactions. The field of sustained release technology
is vastly growing and as a consequence has witnessed a remarkable
sophistication. Many new technologies and devices are continuously being tried
for providing a more reliable control and precision over the release of the
actives.27
1.4.1 Rationale of sustained drug delivery systems:
In general, the goal of sustained release dosage form is to maintain
therapeutic blood or tissue level of the drug for extended period of time. This is
generally accomplished by attempting to obtain “zero order” release from the
dosage form. Zero order release constitutes drug release from the dosage form
which is independent of the amount of drug in the delivery system. Sustained
release system generally do not attain this type of release and usually try to
mimic zero order release by providing drug in slow “first order” fashion (i.e.
Page 31
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 17
concentration dependent). Thus sustained release dosage form consists of two
parts:
An immediately available dose to establish the blood level quickly in an amount
sufficient to produce the desired pharmacological response (i.e. Loading dose).
The remaining amount of total dose (maintenance dose) is then gradually
released to maintain constant blood level of the drug. 28
Figure 8: Plasma drug concentration profiles for conventional tablet or capsule
formulation, a sustained release formulation and a zero order controlled release
formulation.
1.4.2. Factors influencing the design and performance of sustained
release products:
To establish criteria for the design of controlled release products a
number of variables must be considered such as:
T
I
M
E
Page 32
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 18
1.4.2.1. Drug properties:
Physicochemical properties of drug including stability, solubility,
partitioning characteristics, charge and protein drug binding play a dominant
role in the design and performance of controlled release systems.
1.4.2.2. Route of drug delivery:
Physiological constraints imposed by particular route, such as, first pass
metabolism, GI motility, blood supply and sequestration of small foreign
particles by the liver and spleen.
1. Target sites.
2. Acute or chronic therapy.
3. The disease.
4. The patient29
.
1.4.3.3. Advantages of sustained drug delivery systems over conventional
dosage forms
Improved patient compliance and convenience due to less frequent drug
administration.
Reduction in fluctuations in steady state levels and therefore better control
of disease condition and reduced intensity of local or systemic side effects.
Increased safety margin of high potency drugs due to better control of
plasma levels.
Maximum utilization of drug enabling reduction in total amount of dose
administered.
Page 33
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 19
Reduction in health care costs through improved therapy, shorter treatment
period, less frequency of dosing, reduction in personal time to dispense,
administer and monitor patients.
Improved bioavailability of some drugs. 30
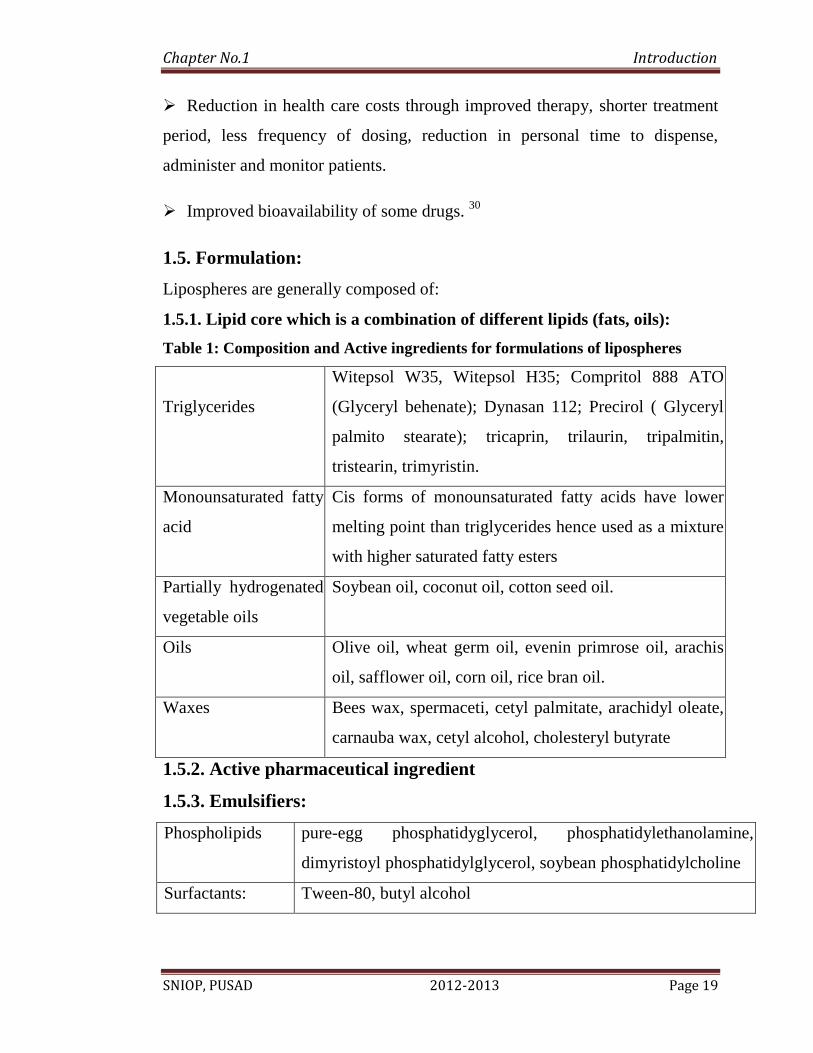
1.5. Formulation:
Lipospheres are generally composed of:
1.5.1. Lipid core which is a combination of different lipids (fats, oils):
Table 1: Composition and Active ingredients for formulations of lipospheres
Triglycerides
Witepsol W35, Witepsol H35; Compritol 888 ATO
(Glyceryl behenate); Dynasan 112; Precirol ( Glyceryl
palmito stearate); tricaprin, trilaurin, tripalmitin,
tristearin, trimyristin.
Monounsaturated fatty
acid
Cis forms of monounsaturated fatty acids have lower
melting point than triglycerides hence used as a mixture
with higher saturated fatty esters
Partially hydrogenated
vegetable oils
Soybean oil, coconut oil, cotton seed oil.
Oils Olive oil, wheat germ oil, evenin primrose oil, arachis
oil, safflower oil, corn oil, rice bran oil.
Waxes Bees wax, spermaceti, cetyl palmitate, arachidyl oleate,
carnauba wax, cetyl alcohol, cholesteryl butyrate
1.5.2. Active pharmaceutical ingredient
1.5.3. Emulsifiers:
Phospholipids pure-egg phosphatidyglycerol, phosphatidylethanolamine,
dimyristoyl phosphatidylglycerol, soybean phosphatidylcholine
Surfactants: Tween-80, butyl alcohol
Page 34
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 20
1.5.4. Stabilizers:
Gelatin, pectin, carrageenan, polyvinyl alcohol, polyoxyethylene sorbitan
trioleate, Pluronic PE 8100, lauryl sarcosine.
1.6. Methods of preparation:
1.6.1 Melt dispersion technique 31
In this method, drug is dissolved or dispersed in the melted lipidic phase (figure
9). Aqueous phase is composed of water or suitable buffer which is heated to the
same temperature as lipid phase. The aqueous phase is kept under stirring during
which emulsifier is added. To the aqueous phase containing emulsifier, lipid
phase containing drug is added drop by drop while maintaining the temperature
and stirring speed. After this “hot emulsification phase”, the temperature of the
mixture is rapidly brought down to room temperature or below room
temperature by adding ice cold water or ice under continuous stirring. This cold
resolidification results in the formation of discrete lipospheres which can be
filtered. Several drugs like bupivacaine, glypizide , aceclofenac , retinyl acetate,
progesterone,sodium cromglycate, diclofenac , carbamazepine , C14-diazepam,
proteins like somatostatin , thymocartin , casein , bovine serum albumin ,
R32NS1 malaria antigen , tripalmitin based lipospheres for labon-chip
applications have been prepared by melt dispersion methods. Lipids carrying
antigens exert their adjuvant effect to immunogenicity of antigens and the effect
was found to decrease in the following order for the lipids studied: ethyl
stearate>olive oil>tristearin>tricaprin>corn oil>stearic acid. Also inclusion of
negatively charged lipids like dimyristoyl phosphotidyglycerol in the lipid core
was found to improve the antibody response to encapsulated malaria antigen.
Page 35
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 21
1.6.2 Solvent evaporation method 31
In this method, lipid is dissolved in an organic solvent. Commonly used organic
solvents include ethyl acetate, ethanol, acetone or dichloromethane. This lipid
phase is emulsified into aqueous phase containing emulsifier. Organic solvent is
evaporated by stirring the oil in water emulsion for 6-8 h under ambient
conditions. Discrete lipospheres can be collected by filtration through paper
filter after the water rises to the surface. Examples of the drugs formulated as
lipospheres by this method include paclitaxel, thymocartin, bovine serum
albumin, triptorelin leuprolide .
Fig 9: Schematic representation of the methods of production of LS: melt
dispersion and solvent evaporation
Page 36
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 22
1.6.3 Co-solvent solvent evaporation method 31
In this co-solvent - solvent evaporation method employing chloroform and N-
methyl pyrollidone to create a clear solution, although low yield and large
particle size is obtained, which is altered by variation in the solvent used.
Lipospheres made up of polar and non-polar lipids using synthetic stabilizers
instead of phospholipids which are the deviation from the definition of
liposphere reported by Domb in his patent. Although their work is not related to
protein delivery but they tried it with hydrophilic drug and reported around 50%
entrapment by double emulsification method
1.6.4 Sonication method 32
In this technique, the drug is mixed with lipid in a scintillation vial which is pre-
coated with phospholipids. The vial is heated until the lipid melts, and then
vortexed for 2min to ensure proper mixing of the ingredients. A 10 ml of hot
buffer solution is added into the above mixture and sonicated for 10min with
intermittent cooling until it reaches to the room temperature.
1.6.5 Rotoevaporation method 32
In this technique, lipid solution with drug is prepared in a round bottom flask
containing 100 grams of glass beads (3 mm in diameter) mixed thoroughly till a
clear solution is obtained. Then, the solvent is evaporated by using
rotoevaporizer under reduced pressure at room temperature and a thin film is
formed around the round bottom flask and the glass beads. Raise the temperature
upto 40 °C until complete evaporation of the organic solvent. Known amount of
0.9 % saline is added to the round bottom flask and the contents are mixed for 30
min at room temperature and then the temperature is lowered to 10 °C by
Page 37
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 23
placing in ice bath and mixing is continued for another 30min until lipospheres
are formed.
1.6.6. Microfluidizer method 32
Lipospheres can also be prepared by using a microfluidizer which is equipped
with two separate entry ports. From one entry port, a homogenous melted
solution or suspension of drug and carrier is pumped and from second entry port,
an aqueous buffer is pumped. The liquids are mixed in the instrument at elevated
temperatures where the carrier is melted and rapidly cooled to form the
lipospheres. The temperature of the microfluidizer can also be changed at any
stage of the lipospheres processing to manipulate the particle size and
distribution.
1.6.7 Polymeric lipospheres 32
Polymeric biodegradable lipospheres can also be prepared by solvent or melt
processes. The difference between polymeric lipospheres and the standard
liposphere formulations is the composition of the internal core of the particles.
Standard lipospheres, as those previously described, consist of a solid
hydrophobic fat core that is composed of neutral fats like tristearin, while in the
polymeric lipospheres, biodegradable polymers such as polylactide (PLD) or
PCL substitute the triglycerides. Both types of lipospheres are thought to be
stabilized by one layer of phospholipid molecules embedded in their surface.
1.6.8 Microemulsion 32
In this method, drug is added to the melted lipid. Aqueous phase is prepared by
adding surfactant like Tween 80 into water maintained at same temperature as
lipid phase. This is followed by the addition of co-surfactant like butyl alcohol to
the aqueous phase. The aqueous phase containing surfactant and co-surfactant is
Page 38
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 24
added to lipid phase kept under stirring. Rapid cooling of the above mixture
results on formation of discrete lipid particles. Flurbiprofen lipospheres
prepared by this method. Presence of Tween80 at 2%, butyl alcohol at 2ml and
water at 50ml found to give discrete lipospheres of superior quality.
1.6.9 Multiple Emulsions 32
In this method, drug solution (aqueous phase) is added to melted lipid. The
primary emulsion formed as a result is added into aqueous solution containing
emulsifier kept at the same temperature as primary emulsion. The multiple
emulsions formed as a result is subjected to rapid cooling to form lipospheres.
Morel et al reported a 90 % entrapment efficiency of D-Trp-6- LHRH peptide
from stearic acid-egg lecithin based lipospheres prepared by this technique.
Drugs like thymopentin, cyclosporine and peptides like papain were investigated
for liposphere formulations by this method.
Page 39
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 25
1.7. Factors influencing quality attributes of lipospheres
1.7.1. Factors influencing morphology of lipospheres
Table 2: Factors influencing morphology of lipospheres
S.No. Factors Influence
1.
Drug
loading
Proportion of larger particles formed was high on
increasing the drug amount. At maximum drug: lipid
(1:1)33
insufficient coating of drug by lipid leads to the
formation of aggregates during cooling phase resulting in
irregular, fluffy and fragile particles.
2.
Type of
lipid
Combination of apolar (tristearin, tripalmitin or tribehenin)
with polar lipids (glycery monostearate, glyceryl
monooleate) gave lipospheres satisfactory in terms of size,
shape and recovery.
3.
Type of
impeller
Lipospheres were produced using different impeller types
33 and particle characteristics of formed lipospheres were
studied. Impellers used were of rotor (2-blade, 3-blade)
type, helicoidal rotor (4-blade) type, double truncated cone
rotor. Lipospheres could not be produced using 2-blade
rotor and resulted in the formation of elliptical particles.
Page 40
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 26
1.7.2. Factors influencing entrapment efficiency
Table 3: Factors influencing entrapment efficiency
S.No. Factors Influence
1.
Type of
lipid
Hydrophobicity of lipids promotes entrapment of
drugs. Long chain triglycerides (tristearin and
triarachidin) are generally more hydrophobic than short
chain triglycerides like tricaprin and trilaurin.
Accordingly the free drug contents of formulations
containing the long chain triglycerides were found to
be lower than short chain triglycerides34
. Also long
chain triglycerides were found to increase the
bioavailability of drug as they increase in
gastrointestinal residence time of drug compared to
medium chain and short chain fatty acids35
Lipid
excipients reduce the activity of P-glycoprotein and
MDR (multi drug resistant) associated protein 2 by
down regulating the protein expression and increase in
cell membrane permeability in addition to lymphatic
uptake.
2.
Amount of
Phospholipid
As the phospholipid (coat) amount increases, formation
of alternative systems like liposomes was observed
which will compromise drug entrapment. Experiments
with triglyceride, phospholipid at a 1:0.5 to 1: 0.25
w/w 36
revealed that 70-90% of phospholipid polar
heads were accessible on liposphere surface thus
enhancing the loadability of drug.
Page 41
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 27
3.
Effect of
method of
preparation
Melt dispersion method was found to be superior over
solvent evaporation method in terms of entrapment
efficiency as melt method promotes drug incorporation
core where as solvent evaporation promotes drug
incorporation in coat.
1.7.3. Factors influencing drug release
Table 4: Factors influencing drug release
S.No. Factor Influences
1.
Release
pattern
The release mechanism of drugs namely tetracaine,
etomidate an prednisolone37
entrapped in lipid particles.
Dynasan 112 (glycerol trilaurate), Compritol 888 ATO
(glycerol behenate) were used as lipid carriers and Pluronic
F 68 (Poloxamer 188), Lipoid S 75 (soy lecithin), Lipoid KG
were used as emulsifiers. Tetracaine and etomidate
lipospheres have shown burst release and prednisolone
lipospheres gave prolonged release.
2.
Effect of
particle
size
Smaller particles have larger surface area exposed to
dissolution medium and higher diffusion coefficient. If the
drug resides in the outer shell diffusion distance becomes
shorter resulting in fast (burst) release.
3.
Type of
lipid
Highest T8h value was obtained with stearyl alcohol
lipospheres compared to fatty acids like stearic acid. Stearyl
alcohol possesses hydroxyl groups promoting matrix
hydration by providing a hydrophilic pathway for water
molecules to solubilize the drug and increase in dissolution
rate. Lowest T 8h value was obtained from stearic acid
lipospheres because of interaction of stearic acid with metal
Page 42
Chapter No.1 Introduction
SNIOP, PUSAD 2012-2013 Page 28
ions in medium forming sodium soaps which are crystals
that contain fatty acid and metal carboxylate ion pairs
retarding the release.
4
Effect of
stabilizer
Lipospheres formulated with gelatin as stabilizer released
80% of total drug in 8hrs resulting in sigmoid mode of
release whereas formulations with Poloxamer 40738
resulted
in a biphasic pattern (burst release followed by slow release)
1.8. Applications of liposphers
1.8.1. Parenteral route
Lipospheres have been exploited for the delivery of anesthetics like lidocaine
bupivacaine for the parenteral delivery of antibiotics like ofloxacin, norfloxacin,
chloramphenicol palmitate and oxytetracycline , and antifungal agents, such as
nystatin and amphotericin B for the parenteral delivery of vaccines and
adjuvants.
1.8.2. Transdermal route 39
Properties of lipospheres like film forming ability, occlusive
properties;controlled release from solid lipid matrix resulting in prolonged
release of drug and retarded systemic absorption of drugs; increasing the
stability of drugs which are susceptible to extensive hepatic metabolism, make
them attractive candidates for topical delivery.
1.8.3. Oral delivery 40, 41
Several categories of drugs like antibiotics, anti-inflammatory compounds,
vasodilators, anticancer agents, proteins and peptides are being formulated as
oral lipospheres.
Page 43
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 29
2. Literature Review
Sandipan dasgupta et al (2012)42
Nanostructured Lipid carriers (NLC)
based gel for Topical Delivery of aceclofenac preparation
,characterization ,and in vivo evaluation.stearic acid as the solid lipid and
oleic acid as the liquid lipid ,pluronic F68 as the sur factant and
phospholipon 90G as the co-surfactant were used NLC prepared by melt
–emulsification and high speed homogenization methods.the anti-
inflammatory effect of NLC gel was assesed by rat paw edema
technique and compared to marketed aceclofenac gel.
Esimone et al (2012) 43
Formulation and evaluation of goat fat and shea
butter based lipospheres of benzyl penicillin lipospheres of benzyl
penicillin were formulated using the conventional thin film hydration
technique five different combinations of shea butter, surfactant (span 80)
and goat fat were the key variables employed in the formulations the
presence of goat fat however seems to impact negatively on the in vivo
stability of liposphere.
Abraham J. Domb et al (2012)44
Preparation and characterization of an
oral pro-dispersion liposphere formulation for cyclosporin, a water
insoluble drug with limited bioavailability. Pro-dispersion formulation
consisted of a solid fat, dispersing agents and amphiphilic solvents as the
major components besides cyclosporin A (CsA) were prepared in the
present work. For preparation of this formulation, phospholipid was
dissolved in pharmaceutically acceptable water soluble organic solvent,
thereafter CsA along with other components was added and formulation
optimization was carried out. After formulation preparation, particle size
determination and in vitro release study was carried out. Additionally,
Page 44
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 30
ultracentrifugation, TEM, Cryo-TEM and DSC techniques were used for
in vitro characterization of formulation. The prepared system was also
compared with marketed Neoral® microemulsion formulation.
Vignesh Muruganandham et al (2012)45
Formulation, Development &
Characterization of Ofloxacin Microspheres Ofloxacin is anti bacterial
agent that has a wide range of activity against gram (-ve) and gram (+ve)
microorganisms. Multiple doses of Ofloxacin are required to attain
steady state concentration. The main objective of this study was to
formulate, develop and characterize Ofloxacin microspheres to prolong
the release rate so as to decrease the necessity of multiple dosings
especially in patients with renal impairment. The Ofloxacin
microspheres were prepared using natural polymers by non-ionic
crosslinking technique. Five different formulations were prepared with
respective quantities of the polymer (Chitosan) with copolymer (Gelatin
and sodium alginate) with drug in different drug-polymer ratio of 1:0.5,
1:0.75 and 1:1. The prepared microspheres were evaluated for
percentage drug loading, entrapment efficiency, surface morphology,
and in-vitro release characteristics to identify the effect of addition of
these polymers.Cumulative release data were fitted into kinetic models.
The Scanning Electron Microscope analysis revealed a smooth and
spherical surface morphology with mean particle size of the
microspheres ranging from 7 to 14 μm. Drug loading was found to
increase with the increase in the concentration of encapsulating polymer,
chitosan, sodium alginate and gelatin concentration. Drug release obeyed
the first order kinetics45
.
Sanming Li et al (2012)46
Nanostructured lipid carriers (NLC)-based gel
was developed as potential topical system for flurbiprofen (FP) topical
Page 45
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 31
delivery. The characterizations of the prepared semisolid formulation for
topical application on skin were assessed by means of particle size
distribution, zeta potential analysis, X-ray analysis, in vitro percutaneous
penetration, rheological study, skin irritation test, in vivo
pharmacodynamic evaluation and in vivo pharmacokinetic study. The
NLC remained within the colloidal range and it was uniformly dispersed
after suitably gelled by carbopol preparation. It was indicated in vitro
permeation studies through rat skin that FP-NLC-gel had a more
pronounced permeation profile compared with that of FP loaded
mcommon gel. Pseudoplastic flows with thixotropy were obtained for all
NLC-gels after storage at three different temperatures. No oedema and
erythema were observed after administration of FPNLC- gel on the
rabbit skin, and the ovalbumin induced rat paw edema could be inhibited
by the gel.
Satheesh Babu et al (2011)47
Manufacturing techniques of lipospheres
Lipid microspheres, often called lipospheres (LS), have been proposed as
new type of lipid-based encapsulation system for drug delivery of
bioactive compounds especially lipophilic compounds. LS consist of
solid microparticles with a mean diameter usually the size range between
0.2 to 500μm, composed of a solid hydrophobic fat matrix, where the
bioactive compound(s) is dissolved or dispersed. The lipospheres have
several advantages over other colloidal delivery systems (including nano
& micro emulsions, nanaoparticles, hydrogels and liposomes).
Loganathan Veerappan et al (2010)48
formulation development and
evaluation of flurbiprofen liposphere microencapsulation is a rapidly
expanding technology in the production of controlled release dosage
Page 46
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 32
forms. Most of the non-steroidal antiinflammatory drugs (Nsaid) have
been widely used for the treatment of acute and chronic arthritic
conditions. flurbiprofen appears to be more active as an anti-
inflammatory agent than other nsaid products and is usually well
tolerated. flurbiprofen is frequently prescribed for the treatment of
rheumatoid arthritis, osteoarthritis and ankylosing spondylitis. By
formulating sustained release dosage form of this drug leads to
minimization of damage to the gastro intestinal mucosa. Development of
formulation was made with different formulation variables and suitable
formulation was selected for further evaluations.
Kamal Dua et al (2010)49
Aceclofenac is a new generation non-steroidal
anti-inflammatory drug showing effective anti-inflammatory and
analgesic properties. It is available in the form of tablets of 100 mg.
Importance of aceclofenac as a NSAID has inspired development of
topical dosage forms. This mode of administration may help avoid
typical side effects associated with oral administration of NSAIDs,
which have led to its withdrawal. Furthermore, aceclofenac topical
dosage forms can be used as a supplement to oral therapy for better
treatment of conditions such as arthritis. Ointments, creams, and gels
containing 1 % (m/m) aceclofenac have been prepared. They were tested
for physical appearance, pH, spreadability, extrudability, drug content
uniformity, in vitro diffusion and in vitro permeation. Gels prepared
using Carbopol 940 (AF2, AF3) and macrogol bases (AF7) were
selected after the analysis of the results. They were evaluated for acute
skin irritancy, anti-inflammatory and analgesic effects using the
carrageenan-induced thermal hyperalgesia and paw edema method.
Page 47
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 33
Maha Nasr et al (2008)50
liposphere as a carrier for topical Delivery of
Aceclofenac preparation characterization and in vivo. The aim of
exploiting the favorable properties of this carrier system and developing
a sustained release formula to overcome the side effects resulting from
aceclofenac oral administration. Lipospheres were prepared using
different lipid cores and phospholipid coats adopting melt and solvent
techniques. liposphere systems were found to possess superior anti-
inflammatory activity compared to the marketed product in both lotion
and paste consistencies. Liposphere systems proved to be a promising
topical system for the delivery of aceclofenac.
Jia-You Fang et al (2007)51
acoustically active lipospheres (AALs)
were prepared using perfluorocarbons and coconut oil as the cores of
inner phase. These AALs were stabilized using coconut oil and
phospholipid coatings. A lipophilic antioxidant, resveratrol, was the
model drug loaded into the AALs. AALs with various percentages of
perfluorocarbons and oil were prepared to examine their
physicochemical and drug release properties. Co-emulsifiers such as Brij
98 and Pluronic F68 (PF68) were also incorporated into AALs for
evaluation. AALs with high resveratrol encapsulation rates (_90%) were
prepared, with a mean droplet size of 250–350 nm. The AALs produced
with perfluorohexane as the core material had larger particle sizes than
those with perfluoropentane. Resveratrol in these systems exhibited
retarded drug release in both the presence and absence of plasma in
vitro; the formulations with high oil and perfluorocarbon percentages
showed the lowest drug release rates.
Hagalavadi Nanjappa Shivakumar et al (2007)52
Design and statistical
optimization of glipizide loaded A 32 factorial design was employed to
Page 48
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 34
produce glipizide lipospheres by the emulsification phase separation
technique using paraffin wax and stearic acid as retardants. The effect of
critical formulation variables, namely levels of paraffin wax (X1) and
proportion of stearic acid in the wax (X2) on geometric mean diameter
(dg), percent encapsulation efficiency (% EE), release at the end of 12 h
(rel12) and time taken for 50% of drug release (t50), were evaluated
using the F-test. Mathematical models containing only the significant
terms were generated for each response parameter using the multiple
linear regression analysis (MLRA) and analysis of variance (ANOVA).
Both formulation variables studied exerted a significant influence (p <
0.05) on the response parameters. Numerical optimization using the
desirability approach was employed to develop an optimized formulation
by setting constraints on the dependent and independent variables. The
drug release from lipospheres followed first-order kinetics and was
characterized by the Higuchi diffusion model. The optimized liposphere
formulation developed was found to produce sustained anti-diabetic
activity following oral administration in rats.
Vandana B. Patravale et al (2007)53
to develop solid lipid nanoparticles
(SLN) of tretinoin (TRE) with the help of facile and simple
emulsification-solvent diffusion (ESD) technique and to evaluate the
viability of an SLN based gel in improving topical delivery of TRE. The
feasibility of fabricating SLN of TRE by the ESD method was
successfully demonstrated in this investigation. The developed SLN
were characterized for particle size, polydispersity index, entrapment
efficiency of TRE and morphology. Studies were carried out to evaluate
the ability of SLN in improving the photostability of TRE as compared
to TRE in methanol. Encapsulation of TRE in SLN resulted in a
significant improvement in its photostability in comparison to
Page 49
Chapter No.2 Literature Review
SNIOP, PUSAD 2012-2013 Page 35
methanolic TRE solution and also prevented its isomerization.
Furthermore, the skin irritation studies carried out on rabbits showed that
SLN based TRE gel is significantly less irritating to skin as compared to
marketed TRE cream and clearly indicated its potential in improving the
skin tolerability of TRE. In vitro permeation studies through rat skin
indicated that an SLN based TRE gel has permeation profile comparable
to that of the marketed TRE cream.
Page 50
ChapterNo.4 Plan of Work
SNIOP, PUSAD 2012-2013 Page 37
4. Plan of Work
Present proposed research work was planned as follows-
A) Literature survey.
B) Selection of drug, lipid core material and coat material.
C) Procurement of drug, core material and coat material.
D) Preliminary study of drug, core material and coat material.
E) Formulation of Liposphere.
F) Evaluation of Liposphere.
1. Photo microscopic analysis.
2. Scanning Electron microscopy.
3. Particle size analysis.
4. Differential scanning Calorimetry.
5. Fourier transforms infrared spectroscopy (FTIR).
6. In vitro dissolution test.
H) Formulation of lipospheres based gel.
I) Evaluation of Lipospheres based gel.
1. pH.
2. Drug content.
3. Viscosity.
4. Spreadability.
5. In vitro permeation of liposphere based gel.
6. Skin-irritation testing (Draize patch test).
7. In vivo Anti-inflammatory Study of Liposphere.
8. Stability study.
Page 51
Chapter No. 3 Aim and Objective
SNIOP, PUSAD 2012-2013 Page 36
33.. Aim and Objectives
Aim
Preparation and evaluation of liposphere based topical drug delivery system
containing a NSAID drug.
Objectives
1. To study formulation and evaluation of liposphere as a carrier for
topical delivery of NSAID drug.
2. To optimize the formulation using suitable experimental technique,
regarding particle size, stability, release property, surface morphology,
hydrophobicity, drug entrapment efficiency etc.
3. To study in vitro release of NSAID from liposphere
4. In vivo Anti-inflammatory study of liposphere.
Page 52
Chapter No.6 List of Material and Equipment
SNIOP, PUSAD 2012-2013 Page 45
6. List of material and equipment
Table 5: List of Materials Used
Sr. No. Instrument Used (Model No.) Make
1. FTIR Spectrophotometer Aligent Cary 630 ATR
2. UV–1700 Spectrophotometer,
double beam
Shimadzu, 1700, Japan
3. Electronic balance Citizen scale (CY 104),
Germany
4. Brookfield viscometer (Dial type) Middleboro, MA-02346, USA
5. Differential Scanning
Calorimetry (DSC 60)
Mettler Toledo, Zaventem
(U.S.)
6. Mechanical Stirrer Remi lab stirrer, Mumbai
7. Magnetic Stirrer Remi lab stirrer, Mumbai
8. Intel play Qx3 microscope
(200X magnification)
Edmund Scientific (U.S.)
9. Dissolution test apparatus Electro lab , Mumbai
10. Scanning Electron Microscope
(JSM 6380A)
JOEL, Japan
Page 53
Chapter No.6 List of Material and Equipment
SNIOP, PUSAD 2012-2013 Page 46
11 Centrifuge machine Remi, Mumbai
12. Digital pH Meter Chemi line (CL-110)
13. Plethysmometer (UGO-Basile, 7140,Comerio,
Italy
14. Stability chamber Skylab,Mumbai
List of chemicals and reagents used
Aceclofenac was procured as a gift sample from Concept
pharmaceutical, india. Soy phosphatidylcholine-35% was kindly gifted by
perfect Biotech industries Pvt Ltd, Nagpur. LIPOVA-E120 (Egg
phosphatidylcholine) gifted by VAV Life sciences Pvt Ltd. Mumbai. All other
chemicals were procured from Research lab fine Chem industries, Mumbai.
They are-
Table 6: List of chemical used
Chemicals Grade
Absolute Ethanol AR grade
Methanol LR grade
Potassium chloride LR grade
Sodium chloride AR grade
Page 54
Chapter No.6 List of Material and Equipment
SNIOP, PUSAD 2012-2013 Page 47
AR grade-Analytical reagent, LR grade-Laboratory reagent
Potassium dihydrogen phosphate AR grade
Acetone LR grade
disodium hydrogen phosphate AR grade
Acetic anhydride AR grade
Disodium hydrogen phosphate AR grade
Carbopol 940P AR grade
Triethanolamine (TEA) AR grade
Page 55
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 38
5. Drug Profile
5.1. Drug: Aceclofenac 54,55,56,57
5.1.1. Structure:
Chemical Name [(2, 6-dichlorophenyl)amino]
phenylacetoxyacetic acid.
Molecular formula C16H13Cl2NO4
Molecular weight 354.18
Melting point 149-153 0C
Description A white or almost white, crystalline
powder. Insoluble in water, soluble in
ethanol and acetone
Mode of action Highly selective β2 agonist
Dose 100 mg
Plasma half-life 2 to 3 h
Plasma protein binding 40-50%
Category Anti inflammatory
Analgesic
Page 56
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 39
Adverse reactions
Gastrointestinal System – Duodenal ulcer, gastrointestinal perforation
Urinary System – Interstitial nephritis
Central and Peripheral Nervous System – Optic neuritis
Psychiatric – Hallucination, Drowsiness, Confusion
Skin and Appendages – Epidermal necrolysis, Erythema multiforme,
dermatitis
Respiratory – Aggravated asthma
Haematological – Aplastic anaemia
Contraindications
1. Active peptic ulceration
2. Recurrent indigestion (relative contraindication)
3. Care should be taken in patients on anticoagulants
4. Care should be taken in patients with hypertension or heart failure
5. Pregnancy and lactation
6. History of sensitivity to aspirin or other NSAISD drugs.
Drug Interactions
1. Anticoagulants
2. Alcohol and smoking
3. Lithium
4. Diuretics
5. Antihypertensive drugs
6. Diflunisal
7. Anti-platelet agents and selective serotonin reuptake inhibitors
Page 57
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 40
5.2. Lipid profile
5.2.1. Lecithin58
5.2.1.1. Structure:
CH2
CH
OCH
2
P
O
OCH2CH
2N(CH
3)3
O-CO-R1
O-CO-R2
O
R1 and R
2 are fatty acids
+
_
Non proprietary names Lecithin
Synonyms Soya lecithin, soyabean lecithin,
phosphatidylcholine.
Chemical name and CAs
registry no
Lecithin [8002-43-5]
Empirical formula CH2OCOR’- - - CHOCOR’’- - - CH2OPOO-
OCH2CH2N (CH3)3
Functional category Emollient; Emulsifying agent; Solubilizing
agent
Description They may vary in color from brown to light
yellow, depending upon whether they are
bleached or not or on the degree of purity.
Page 58
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 41
when they are exposed to air, rapid oxidation
occurs, also resulting in a dark yellow or
brown color
Solubility Insoluble but swells up in water and in Nacl
solution forming a colloidal suspension,
soluble in about 12 parts cold, absolute
alcohol; soluble in chloroform, ether,
petroleum ether, in mineral oil and sparingly
soluble in benzene
Incompatibility Incompatible with esterase owing to
hydrolysis
Regulatory status Gras listed included in the FDA inactive
ingredient guide. included in nonparenteral
and parenteral medicine licensed in UK
Safety It is highly biocompatible and oral doses of
up to 80 g daily have been given
therapeutically in the treatment of Tardive
dyskinesia.
Page 59
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 42
5.2.2. Stearyl alcohol
5.2.2.1. Structure
Chemical Name octadecyl alcohol or 1-octadecanol)
Molecular formula CH3(CH2)16CH2OH
Molecular weight 270.49
Melting point 55-58°C
Description Freely soluble in chloroform and in
ether; soluble in ethanol (95 per cent);
practically insoluble in water
5.2.3. Bees wax
Structure
CAS No 8012-89-3
Chemical Name Bees wax
Molecular formula C15H31COOC30H61
Page 60
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 43
Molecular weight 415
Melting point 62 to 64 °C
Description white,solid Insoluble in water, soluble
in alcohol
Category Cosmetic Ingredients & Chemicals
5.2.4. Carnauba wax
A coumpound is a pure substance and Carnuba is a mixture. Carnauba wax
contains mainly esters of fatty acids (80-85 %), fatty alcohols (10-15 %), acids
(3-6 %) and hydrocarbons (1-3 %). Specific for carnauba wax is the content of
esterified fatty diols (about 20%), hydroxylated fatty acids (about 6 %) and
cinnamic acid (about 10 %). Cinnamic acid, an antioxidant, may be
hydroxylated or methoxylated. The major components of carnauba wax are
aliphatic and aromatic esters of long-chain alcohols and acids, with smaller
amounts of free fatty acids and alcohols, and resins. Another site claim common
components as C30 alcohols and C26 acids.
CAS No 8015-86-9
Synonyms Carnuba;carnauba, brazil wax, carnubawax, carnaba
wax, wax, carnaubawachs, carnauba wax yellow,
carnauba wax flakes.
Molecular formula -------
Molecular weight ----
Page 61
Chapter No.5 Drug Profile
SNIOP, PUSAD 2012-2013 Page 44
Melting point 81-86 °C
Description Soluble on warming in chloroform, in ethyl acetate and
in xylene; practically insoluble in water and in ethanol
Category Cosmetic Ingredients & Chemicals
Page 62
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 48
7. Experimental work
7.1. Preformulation study
Identification test
7.1.1. Aceclofenac
Organoleptic properties
Aceclofenac was tested for organoleptic properties such as
appearance, color, odor, taste, etc.
7.1.2. Melting point
The melting point of the aceclofenac was determined by capillary
method.
7.1.3. Solubility determination59
The approximate solubility of Pharmacopeial substance is indicated
by descriptive terms in accompanying table.
7.1.4. Ultra-violet scanning
The scanning of Aceclofenac was performed in 7.4 pH phosphate
buffer saline and λmax was found to be 276 nm which was complies with the
λmax reported in BP.
7.1.5. Fourier transforms infrared (FTIR) spectroscopy
Procedure: Small quantity of aceclofenac 1 mg was placed on diamond
ATR crystal then it was scanned between 4000-500 cm-1
range.
7.1.6. Lipids
7.1.6.1. Soyalecithin
Saponification value60
About 35 g Potassium hydroxide was weighed, dissolved in 20 ml of
water; sufficient ethanol was added (96 %) to produce 1000 ml and allowed
to stand overnight. Clear liquid was poured off. About 2 gm of soya lecithin
Page 63
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 49
was accurately weighed and taken into a 200 ml conical flask, 25 ml of
ethanolic solution of KOH was added, boiled under a reflux condenser for 1
h with rotating the contents frequently. When the solution was still hot, the
excess of alkali was titrated with 0.5 M Hcl using 1 ml of phenolphthalein
solution as indicator.
Blank determination was carried out excluding the substance being
examined. The saponification value was calculated from expression
Saponification value = 28.5 V/W
Where, V- difference in ml between titration
W- Weight in g of substance taken.
7.2. Preparation of standard calibration curve of aceclofenac
7.2.1. Preparation of phosphate buffer saline pH 7.4
About 2.38 g of disodium hydrogen phosphate (Na2Po4), 0.19 g of
potassium dihydrogen phosphate (KH2PO4) and 8.0 g of sodium chloride
(Nacl) was taken in volumetric flask and volume was made with water to
produce 1000 ml. This solution had a pH of about 7.461
.
7.2.2. Determination of λmax for aceclofenac
Stock solutions of Aceclofenac was prepared by dissolving in 100 ml
of phosphate buffer saline solution pH (7.4), solutions were further diluted
and analyzed spectrophotometrically between 220 to 370 nm to determine
λmax.
7.2.3. Preparation of standard calibration curve of aceclofenac
The calibration curve was plotted within the concentration range of 2-10
µg/ml. Appropriate dilutions were prepared and absorbance was measured
for each solution at 276 nm since maximum absorbance was observed at this
wavelength. Graph was plotted for absorbance vs. concentration.
Page 64
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 50
y = 0.0109x R² = 0.994.
Correlation co-efficient value indicated the linear correlation between
concentration and absorbance.62
Where
Y is absorbance
X is concentration
R2 is coefficient of regression.
7.3. Formulation of liposphere by melt dispersion technique
Lipospheres (LS) were prepared by melt dispersion method. In this
method different lipid materials (Carnauba wax, Bees wax and stearyl
alcohol) along with the drug aceclofenac were used to form core material in
the ratio 2:1 and 3:1, egg phosphatidylcholine and soy phosphatidylcholine
were used as the coat material so as to give the core to coat ratio (cr/ct) of
2:1 and 3:1. The lipid core material was melted at 75 ºC using water bath
and then added with the required amount of aceclofenac by dispersing it in
the molten lipid. Separately 1000 mg of phospholipid was added in 100 ml
of phosphate buffer saline, which was preheated at 75 ºC and the given
mixture was homogenized using mechanical stirrer until a uniform
dispersion was obtained. To this dispersion at 75 ºC the previously prepared
drug was added to molten lipid at 75 ºC and dispersed with the help of
mechanical stirrer at 4000 rpm speed. The homogenized milky solution was
then rapidly cooled down to 10-20 ºC with continued stirring for another 5
min for formation of uniform dispersion of lipospehers63
.
Page 65
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 51
Table 7: Formulation codes of lipospheres (F1-F12)
Egg pc –Egg phosphatidylcholine, soy pc-soya phosphatidylcholine
Cr-core material, Ct-coat material
7.4. Evaluations of Lipospehers
7.4.1. Separation of Unentrapped Aceclofenac from the Prepared
Lipospheres
Aceclofenac lipospheres were separated from free unentrapped
aceclofenac by centrifugation at 20,000 rpm for 30 min. The pellets formed
were washed with 10 ml phosphate buffered saline and recentrifuged again
for 30 min64-66
. The lipospheres were decanted and kept in the refrigerator
for further investigations.
7.4.2. Determination of entrapment efficiency
The entrapped drug concentration was determined by lysis of the
lipospheres with absolute alcohol65-66
. Accurately weighed amount of loaded
lipospheres (50 mg) was dissolved in 10 ml absolute alcohol and covered
well with aluminum foil to prevent evaporation. The solution was sonicated
for 15 min to obtain a clear solution. An aliquot of 1 ml of this solution was
added to 9 ml of absolute alcohol. The solution was sonicated for another
15 min. The concentration of aceclofenac in absolute alcohol was
Code Lipid core
Material
Lipid
coat
material
Quantity
of lipid
core (mg)
Quantity
of lipid
coat(mg)
Drug
(mg)
Ratio
(Cr/Ct)
F1 Stearyl alcohol Egg pc 2000 1000 500 2:1
F2 Carnauba wax Egg pc 2000 1000 500 2:1
F3 Bees wax Egg pc 2000 1000 500 2:1
F4 Stearyl alcohol Soya pc 2000 1000 500 2:1
F5 Carnauba wax Soya pc 2000 1000 500 2:1
F6 Bees wax Soya pc 2000 1000 500 2:1
F7 Stearyl alcohol Egg pc 3000 1000 1000 3:1
F8 Carnauba wax Egg pc 3000 1000 1000 3:1
F9 Bees wax Egg pc 3000 1000 1000 3:1
F10 Stearyl alcohol Soya pc 3000 1000 1000 3:1
F11 Carnauba wax Soya pc 3000 1000 1000 3:1
F12 Bees wax Soya pc 3000 1000 1000 3:1
Page 66
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 52
determined spectrophotometrically at wavelength 276 nm after appropriate
dilution. Each sample was analyzed in triplicate. The entrapment efficiency
was calculated through the following relationship:
Entrapment efficiency percentage = Entrapped drug/ Total drug×100
7.4.3. Photo microscopic analysis
A drop of lipospheres preparation was placed on a slide for morphological
examination under optical stereo microscope and photographed at a
magnification of ×200 by means of a fitted camera.
7.4.4. Particle size analysis
The size of the prepared Lipospheres was measured by the optical
microscopy method using a calibrated stage micrometer. It is carried out by
using a compound microscope at 10 x lances. Dried lipospheres were first
re-dispersed in distilled water and placed in a glass slide and the number of
division of calibrated eye piece was counted by a micrometer using a stage
micrometer 67
.
7.4.5. Fourier transforms infrared (FTIR) spectroscopy
FTIR spectra of pure Aceclofenac and the optimized liposphere formulation
were recorded with a FTIR spectrophotometer.
FTIR Spectroscopy: There is always a possibility of drug Lipid interaction
in any formulation due to their intimate contact. The technique employed in
the present study for this purpose is IR spectroscopy.IR spectroscopy is one
of the most powerful analytical techniques, which offers the possibility of
chemical identification.
Page 67
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 53
7.4.6. Differential scanning calorimetry (DSC)
Samples of aceclofenac, and drug loaded lipospheres of the selected
formulation were submitted to DSC analysis using differential scanning
calorimeter calibrated with indium. The analysis was carried out on 1 mg
samples sealed in standard aluminum pans. Thermograms were obtained at a
scanning rate of 10 °C/min using dry nitrogen flow of (25 ml/min). Each
sample was scanned between zero and 400 °C.
7.4.7. Scanning electron microscopy (SEM)
The detailed surface characteristics of the selected aceclofenac
lipospheres formulation were observed using a scanning electron
microscope. The lipospheres sample was attached to the specimen holder
using a double coated adhesive tape and gold coated (~20 nm thickness)
under vacuum using a sputter coater for 5–10 min at 40 mA and then
investigated at 30 kV 68
.
7.4.8. In vitro release of aceclofenac from lipospheres
Prepared Lipospheres equivalent to 100 mg of aceclofenac lipospheres
were accurately weighed and filled into gelatin capsules or tight in the
Muslin cloth. Degassed 7.4 Phosphate buffer saline (PBS) medium (900 ml)
was placed into the dissolution tester jars and the temperature was
maintained at 37 ±0.5 °C. A USP II (paddle) dissolution apparatus
(Electrolab) at 100 rpm was used. Samples of 5 ml were drawn at time
points of 30, 60, 120, 180, 240, 300, 360, 420 and 480 min and an equal
amount of fresh dissolution medium was replaced each time. After suitable
dilution, the samples withdrawn were analyzed spectrophotometrically at a
wavelength 276 nm 69
. Results were the mean of three runs. The amounts of
drug present in the samples were calculated with the help of appropriate
calibration curve constructed from reference standard. Percentage drug
release was calculated.
Page 68
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 54
7.5. Formulations of lipospheres based gel
Lipospheres based gel was prepared according to the formula (Table 9).
The suitable lipospheres formulation for the topical delivery of aceclofenac
was selected based on the evaluation of characteristics like: particle size,
entrapment efficiency, and in vitro release. It was found that the formulation
F7 and F8 were more suitable among the other formulations. Different
gelling agents such as: carbopol 940 P, xanthan gum, chitosan, and
hydroxypropyl methyl cellulose (HPMC) were used for the conversion of
the lipospheres dispersion into the gel formulation. Based on the
compatibility with the lipospheres dispersion and the ease of spreadability,
carbopol was selected as the gelling agent. Appropriate quantity of carbopol
940P was soaked in water for a period of 2 h. Carbopol was then neutralized
with triethanolamine (TEA) with stirring. Then specified amount of
liposphere, glycerin and permeation enhancer (Mineral oil) was mixed.
Solvent blend was transferred to carbopol container and agitated for
additional 20 min. The dispersion was then allowed to hydrate and swell for
60 min, finally adjusted the pH with 98 % TEA until the desired pH value
was approximately reached (6.8-7). During pH adjustment, the mixture was
stirred gently with a spatula until homogeneous gel was formed. All the
samples were allowed to equilibrate for at least 24 hours at room
temperature prior to performing rheological measurements70-76
.
Page 69
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 55
Table 8: Formulation of gel optimized Batch
Ingredients
(1.5%w/w)
Formulations Gels 50g
F7 F8 LF7 LF8 Blank Lipospheres
gel
Carbopol 940P 0.5 0.5 0.5 0.5 0.5
Lipospheres 0.75 0.75 0.75 0.75 --
Glycerin 5 5 5 5 5
Triethanolamine
(TEA)
q.s q.s. q.s. q.s. q.s.
Permeation
enhancer
--- --- 0.5 0.5 ---
Distilled water 43.7
5
43.75 43.18 43.18 44.5
7.6. Characterization of lipospheres based topical gel
7.6.1. pH
pH of gel was determined using digital pH meter. About 1 g of gel was
stirred in distilled water till a uniform suspension effected. The volume was
made up to 40 ml and pH of the solution was measured 77
.
7.6.2. Drug content78
Take 1g gel in a 100 ml of volumetric flask and dissolve with little amount
of methanol and mixture was shaken till solution was affected. The volume
was made up to 100 ml with methanol. The solution was filtered through
Whatman filter paper (No. 41). Further dilute 5 ml to 50 ml with methanol.
The absorbance of the solution was measured at 276 nm against reagent
blank.
7.6.3. Viscosity79
Viscosity of the gel was determined by using Brookfield viscometer (Dial
type). As the system is non-Newtonian spindle no. 4 is used. Viscosity is
measured for the fixed time 2 min for 100 rpm.
Page 70
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 56
7.6.4. Spreadability
80-82
The spreadability of the gel was determined using the following
technique: 0.5 g gel was placed within a circle of 1 cm diameter premarked
on a glass plate over which a second glass plate was placed. A weight of 500
g was allowed to rest on the upper glass plate for 5 min. The increase in the
diameter due to spreading of the gels was noted.
It was calculated using formula,
S = M. L / T
Where, S = spreadability
M = weight tied to upper slide
L = length of glass slide
T = time taken
Shorter time interval, to cover distance of 6.5 cm, indicates better
spreadability.
7.6.5. In vitro permeation of liposphere based gel83, 84
This study was carried out for the optimized batch selected, based on all
above evaluation parameters. One of the batches among them was
formulation prepared without permeation enhancer F8 and other was
formulation prepared with permeation enhancer LF8 was studied through
cellophane membrane using a fabricated dissolution testing apparatus. The
dissolution medium used was artificial tear fluid freshly prepared (pH 7.4).
Cellophane membrane, previously soaked overnight in the dissolution
medium, was tied to one end of a specifically designed glass cylinder (open
at both ends and of 5 cm diameter). A 5 ml volume of the formulation was
accurately pipetted into this assembly. The cylinder was suspended in 50 ml
of dissolution medium maintained at 37 ºC so that the membrane just
touched the receptor medium surface. The dissolution medium was stirred at
50 rpm using magnetic stirrer. Aliquots, each of 1 ml volume, were
Page 71
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 57
withdrawn at regular intervals and replaced by an equal volume of the
receptor medium. The aliquots were suitablely diluted with the receptor
medium and analyzed by UV-Vis spectrophotometer at 276 nm.
7.6.6. Skin irritation testing (Draize patch test)85
The lipospheres gel in comparison to marketed aceclofenac gel was
evaluated by carrying out the Draize patch test on rabbits. Animal care and
handling throughout the experimental procedure were performed in
accordance to the CPCSEA guidelines. The experimental protocol was
approved by the institutional animal ethical committee. White rabbits
weighing 2.5–3 kg and were acclimatized before the beginning of the study.
Animals were divided into four groups as follows:
Group 1: No application (Control).
Group 2: Marketed formulation (Audigel 1.5% w/w gel)
Group 3: Lipospheres based gel containing LF7 (1.5% w/w)
Group 4: Lipospheres based gel containing LF8 (1.5%, w/w)
The back of the rabbits were clipped free of hair, 72 h prior to the
application of the formulations. Formulations, 0.5 g, were applied on the
hair free skin of rabbits by uniform spreading. Four rabbits will be use for
the skin irritancy study. All the test formulation will apply on single rabbit,
so total four surface areas will create on the skin of rabbit (3 cm×3 cm)
using hair depletion. To avoid biological variation, the study will perform on
four rabbits. The respective test sample will apply on specified area for
seven day and simultaneous observation for skin irritation such as redness,
edema and skin rash. The results will interpret in the form of grading scale:
A-no reaction; B-slight, patchy erythema; C-moderate but patchy erythema;
D-moderate erythema, and E-severe erythema with or without edema.
Page 72
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 58
7.6.7. In vivo anti-inflammatory study of lipospheres47
The anti-inflammatory activity of the selected lipospheres formulation
(LF7 and LF8) applied and compared to the marketed product using the rat
paw edema test. The protocol of the present work was approved by the
experimental protocol was approved by the institutional animal ethical
committee. Male Wistar rats (130–150 g) were randomly divided into four
groups of 3 rats each. Group I control application, group II for marketed
formulation (AUDIGEL), group III for lipospheres based gel (LF7), group
IV for lipospheres based gel (LF8) formula the volume of paw edema
(milliliter) was measured in each animal using a plethysmometer to a
precision of two decimal places. The rats were marked on the left hind paw
just beyond the tibiotarsal junction, so that every time the paw was dipped in
the electrolyte fluid column up to a fixed mark to ensure constant paw
volume. The tested preparations were applied to the left hind paws of rats
using an amount equivalent to 1 mg of aceclofenac. After 1 h of topical
application, initial paw volume of the rats was measured by dipping the rat
paw into the electrolyte column just before carrageenan injection and the
increase in volume due to fluid displacement was noted from a digital
display, followed by the injection of 0.1 ml of 1% (w/v) carrageenan
solution in saline in the subplantar region of left hind paw of the rats.
Measurement of paw volume was done after 1, 2, 3, 4, 5, 6, 7 and 8 h. The
edema rate and inhibition rate of each group was calculated as follows: The
edema rate and percentage inhibition of each group were calculated as
follows:
.
The % inhibition of edema was calculated by formula:
% inhibition= 1-(a-x/b-y)*100
Page 73
Chapter No.7 Experimental Work
SNIOP, PUSAD 2012-2013 Page 59
Where,
A= paw thickness of test animal after treatment
X= initial paw thickness of test animal
B=paw thickness of control animal after treatment
Y= initial paw thickness of control animal.
7.8. Stability studies86
Stability study was performed as per ICH guideline. The purpose of stability
testing is to provide evidence on how the quality of a drug substance or drug
product varies with time under the influence of a variety of environmental
factors such as temperature, humidity and light. Therefore, stability studies
provide data to justify the storage condition and shelf-life of the drug product.
For drug substance, such studies establish the retest date in addition to the
storage condition of raw material. Initial formulation are packed in the
aluminum lacquered tube and kept in the stability chamber, at different
stability condition.
All the selected formulations were subjected to a stability testing for
three months as per ICH norms at a temperature of 40 ºc ± 2 ºc /75 % ± 5
% RH. All selected formulations (LF7 and LF8) were analyzed for the
change in appearance, pH and drug content by procedure stated earlier.
Page 74
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 60
8. Results and Discussion
8.1 Identification test
8.1.1. Aceclofenac
Physical characters of aceclofenac were found as
Table 9: Result of physical characters of aceclofenac drug
S.no. Characters Inference
1 Nature Amorphous powder
2 Color white
3 Odor Odorless
4 Taste Slightly Bitter
5
Solubility-
In methanol
In water
In ethanol
Soluble
Practically insoluble
Freely soluble
8.1.2. Melting point
The melting point of the aceclofenac was determined by capillary
method and found to 149-153 0C which compiled with melting point
reported in BP.
8.1.3. Ultra-violet scanning
The scanning of aceclofenac was performed in 7.4 pH phosphate
buffer saline and λmax was found to be 276 nm which was compiles with the
λmax reported in BP.
Page 75
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 61
Fig.10: UV Spectrum of aceclofenac
8.1.4. Fourier Transforms Infrared (FTIR) spectroscopy-Scanning was
performed between 4000-500 cm-1
range.
Fig.11: FTIR of aceclofenac
Page 76
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 62
Table 10:- Interpretation of aceclofenac FTIR
Sr. No. Functional Group Wave Number (cm-1
)
1 Amino group, OH, Aliphatic
and Aromatic CH 3600-2300
2 Carboxylic acid salt 1580
3 Aromatic ring 1580, 1515
4 Aromatic ether 1250, 1015
5 Isopropyl group 1180
6 Aliphatic ether, sec.alcohol 1100
7 1,4-disubstituted benzene 820
8.1.5. Lipids
8.1.5.1. Soyalecithin
Saponification value60
Saponification value = 28.5 V/W
Where, V- Difference in ml between titration
W- Weight in g of substance taken.
Result: Saponification value was found to be 190-195 which complies with
Merck Index76
.
Page 77
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 63
8.2. Preparation of calibration curve of aceclofenac
Table 11: UV calibration curve reading of aceclofenac in phosphate buffer
saline pH 7.4
Sr. No. Concentration
(µg/ml)
Absorbance
1 0 0
2 2 0.0259
3 4 0.0452
4 6 0.0616
5 8 0.0896
6 10 0.1072
Result: The prepared dilutions obey’s Beers Lambert’s law in the entire
concentration range selected and the coefficient of correlation was found to
be 0.994.
Fig.12: UV Calibration curve of aceclofenac
From the scanning of drug in phosphate buffer pH 7.4, it was
concluded that the drug had λmax of 276 nm. From the standard calibration
curve of aceclofenac in pH 7.4 buffer (Figure), it was concluded that drug
y = 0.0109x R² = 0.9946
0
0.02
0.04
0.06
0.08
0.1
0.12
0 2 4 6 8 10 12
Ab
sorb
ance
Concentration μg/ml
Calibration curve
Page 78
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 64
obeys Beer-Lamberts law in concentration range of 0-10 µg/ml. The linear
equation in pH7.4 Phosphate buffer was obtained as:
y = 0.0109x R² = 0.994.
Correlation co-efficient value indicated the linear correlation between
concentration and absorbance.
8.3. Evaluations of lipospehers
Table 12: Results of entrapment efficiency
DP=dispersed particle, AP= aggregated particle, PVA –Polyvinyl alcohol
Fig.13: Result of entrapment efficiency of liposphere (F1-F12)
96.3 70.42 76.35
101.65
32.86
88.84 78.07
96.92
55.48
120.2
50.2 49.38
1 2 3 4 5 6 7 8 9 10 11 12
Entrapment Efficiency
Code Lipid core
Material
Entrapment
±SD (n=3)
Stabilizer
(q.s.)
Appearance
F1 Stearyl alcohol 96.35±0.941 Pectin DP
F2 Carnauba wax 70.42.±1.38 Pectin DP
F3 Bees wax 76.35±2.92 Pectin AP
F4 Stearyl alcohol 101.65±0.99 ------ DP
F5 Carnauba wax 32.86±2.52 ----- DP
F6 Bees wax 88.84±1.19 Gelatin DP
F7 Stearyl alcohol 78.07±0.97 PVA DP
F8 Carnauba wax 96.92±2.51 PVA DP
F9 Bees wax 55.48±0.59 PVA AP
F10 Stearyl alcohol 120.2±2.40 PVA DP
F11 Carnauba wax 50.29±1.73 PVA DP
F12 Bees wax 49.38±1.81 Gelatin AP
Page 79
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 65
8.3.1. Photomicroscopic analysis
The photomicrographs of aceclofenac lipospehers of formula F8 was
represented by Fig. 14 & 15. It reveals the uniform spherical shape of
lipospehers showing the solid lipid core and the phospholipid coat.
Fig.14 Fig. 15
Fig. Photomicrograph of optimized aceclofenac lipospehers prepared by melt
method at a magnification of 60 X (Fig.14) and 200X (Fig.15)
8.3.2. Particle size analysis
From Table 13 it was evident that the combination of carnauba wax (F8)
are stearyl alcohol (F7) with the highest amount the smallest particles. Use
of soy lecithin and egg phosphatidylcholine resulted in the smallest particle
size but the lipospehers were found to be aggregated with soya lecithin when
observed under microscope. Carnauba wax with egg phosphatidylcholine
gives the smallest particle size was decreased. As the core/coat ratio
increased, the particle size of lipospehers. This decrease in particle size was
probably due to the availability of higher amounts of carrier material and
emulsifier for the formation of discrete spherical particles.
Page 80
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 66
Table 13: Mean particle size of different batchs (F1-F12)
8.4. Drug lipid compatibility
8.4.1. Fourier transform infrared (FTIR) spectroscopy
Drug-lipid compatibility in optimized lipospehers (F6, F7, and F8) was
evaluated by FTIR and DSC analysis. FTIR spectra are shown in Fig. 16, 17,
18, 19. Interactions between the ingredients used in topical formulations, such
as Aceclofenac, carnauba wax, bees wax, stearyl alcohol, Carbopol were studied
using FTIR spectroscopy. Aceclofenac was identified by the presence of
characteristic bands. The FTIR spectral analysis of aceclofenac alone (Fig. 9)
showed that principle peaks were observed at wave numbers 3316 (N-H
stretching vibration for amine), 2935 (aromatic C-H stretching), 1769 (carbonyl
C=O stretching). The same peaks were observed at 3325, 2954, 1733, 1418,
1234, and 719 in F6 at 3302, 2955, 1471, 1299, and, 718 in F7 at 3320, 2954,
1733, 1467, 1257, 1260 and 719 cm–1 in F8. All the principle characteristic
bands of the drug were also observed in the respective formulations. The
Formulation code Mean particle size (μm)
F1 28.2±0.31
F2 14.2±.61
F3 56.8±.0.32
F4 42.9±0.43
F5 22.3±0.12
F6 85.2±0.32
F7 14.2±0.12
F8 14.2±1.22
F9 42.9±0.65
F10 42.9±0.46
F11 85.2±0.98
F12 96.2±0.12
Page 81
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 67
findings were suggestive of the absence of any major incompatibility between
aceclofenac and the components employed in the preparation of dermatological
bases.
Fig. 16: FTIR of pure drug
Fig. 17: FTIR of formulation (F6)
Fig.18: FTIR of formulation (F7)
Page 82
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 68
Fig. 19: FTIR of formulation F8
8.4.2. Differential scanning calorimetry (DSC)
Figure 20, 21 and 22 showed the results of the DSC analysis of
aceclofenac, and the physical mixture of drug loaded liposphere formulation.
From the DSC thermograms, it was observed that melting point depression
occurred The DSC thermogram of aceclofenac showed an exothermic peak
at 160.44 °C, which is the reported melting point of the aceclofenac. Drug-
loaded liposphere showed a large endothermic peak at 89.07 °C (F6) and
76.22 (F8). It was observed from the DSC thermogram that the exothermic
peak of aceclofenac at about 160.4 °C no longer exists in the DSC, traces of
the drug-loaded liposphere. Taking into consideration the drug-crystal-free
particle surface, it is apparent that aceclofenac is amorphously dispersed
within the liposphere, which is preferable for a controlled release system.
Furthermore, the inclusion of drug molecules in the lipid is normally
accompanied by a depression in the lipid’s melting point.
Page 83
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 69
0.00 1.00 2.00 3.00 4.00
Time [min]
-40.00
-30.00
-20.00
-10.00
0.00
mW
DSC
100.00
150.00
200.00
C
Temp
154.10 x100COnset
170.49 x100CEndset
151.40 x100CStart
172.85 x100CEnd
160.44 x100CPeak
-754.14 x100mJ
-106.82 x100J/g
Heat
-38.56 x100mWHeight
File Name: Pure Drug.tadDetector: DSC60Acquisition Date 13/03/01Acquisition Time 14:49:51Sample Name: Pure DrugSample Weight: 7.060[mg]Annotation:
Thermal Analysis Result
Fig.20: DSC of pure aceclofenac drug
0.00 2.00 4.00 6.00 8.00
Time [min]
-15.00
-10.00
-5.00
0.00
mW
DSC
100.00
200.00
C
Temp
76.22 x100COnset
97.91 x100CEndset
68.71 x100CStart
101.32 x100CEnd
89.32 x100CPeak
-414.84 x100mJ
-56.75 x100J/g
Heat
-13.58 x100mWHeight
File Name: F8.tadDetector: DSC60Acquisition Date 13/03/01Acquisition Time 15:28:40Sample Name: F8Sample Weight: 7.310[mg]Annotation:
Thermal Analysis Result
Fig.21: DSC of Drug loaded liposphere (F8)
Page 84
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 70
0.00 2.00 4.00 6.00 8.00
Time [min]
-10.00
-5.00
0.00
mW
DSC
100.00
200.00
C
Temp
75.80 x100COnset
98.04 x100CEndset
71.47 x100CStart
102.07 x100CEnd
89.07 x100CPeak
-316.77 x100mJ
-41.03 x100J/g
Heat
-9.92 x100mWHeight
File Name: F6 NEW.tadDetector: DSC60Acquisition Date 13/03/01Acquisition Time 15:43:15Sample Name: F6 NEWSample Weight: 7.720[mg]Annotation:
Thermal Analysis Result
Fig.22: DSC of Drug loaded liposphere (F6)
8.4.3. Scanning electron microscopy
Figure 23, 24 illustrates the SEM of optimized F8 batch of prepared
aceclofenac lipospehers. The particles are spherical in shape with irregular
surface as usually obtained when egg phosphatidylcholine is used as the
coat.
Fig.23: Scanning photomicrographs of aceclofenac lipospehers with carnauba
wax: under high magnification (1000X)
Page 85
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 71
Fig.24: Scanning photomicrographs of aceclofenac lipospehers with
Carnauba wax: under low magnification (350X)
The SEM photomicrographs in figure (23 and 24) showed that the
lipid lipospheres of batch have a spherical morphology. In SEM
photomicrographs, the lipid lipospheres were observed at different
magnification value i.e. 350 X, 1000 X which showed surface texture of
lipospehers. Surface texture of the lipospheres is rough because of presence
of crystals of drug on the surface of lipid lipospheres.The lipospheres were
found to be spherical, rounder, free flowing and of the monolithic matrix
type. The SEM photomicrographs of drug-loaded lipospheres showed that
the lipospheres were almost spherical in shape with rough and nonporous
surface and it also indicated that the drug was dispersed at amorphous state
in the lipid matrices.
Page 86
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 72
8.5.4. In vitro release of aceclofenac from lipospehers (F1-F4)
Table 14: Percentage drug release from various formulations of Liposphers
S.No. Cumulative percent drug release (%)
Time
(min)
F1 F2 F3 F4
1. 0 0 0 0 0
2. 30 4.087 14.86 23.23 38.53
3. 60 8.974 21.31 40.54 46.85
4. 120 18.72 38.01 45.59 50.58
5. 180 20.01 52.23 49.22 57.95
6. 240 24.96 66.36 57.53 61.92
7. 300 26.43 80.24 62.41 65.80
8. 360 37.48 94.20 67.39 72
9. 420 47.27 -- 77.46 75.11
10. 480 59.88 -- 79.23 79.48
11. 540 68.13 -- 82.00 82.16
12. 600 75.31 -- 92.52 83.81
(Mean, n=3)
Fig.25: in vitro drug release of formulation batches F1, F2, F3, F4
0
10
20
30
40
50
60
70
80
90
100
0 100 200 300 400 500 600 700
Cu
mu
lati
ve %
dru
g R
ele
ase
Time (min)
F1
F2
F3
F4
Page 87
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 73
8.5.5. In vitro release of aceclofenac from lipospehers (F5-F8)
Table 15: Percentage drug release from various formulations of liposphers
S.No. Cumulative percent drug release (%)
Time
(min)
F5 F6 F7 F8
1. 0 0 0 0 0
2. 30 29.96 25.57 16.94 18.58
3. 60 33.82 27.60 32.76 29.96
4. 120 38.75 31.02 38.02 38.35
5. 180 42.78 32.76 47.27 52.14
6. 240 43.94 37.69 55.34 66.36
7. 300 51.33 48.96 66.36 75.28
8. 360 67.47 49.96 75.36 83.72
9. 420 69.23 56.28 83.81 91.68
10. 480 72.33 58.97 91.68 92.52
11. 540 80.57 69.05 -- --
12. 600 82.88 81.11 -- --
(Mean, n=3)
Fig.26: in vitro drug release of formulation batches F5, F6, F7, F8
0
10
20
30
40
50
60
70
80
90
100
0 100 200 300 400 500 600 700
Cu
mu
lati
ve %
dru
g R
ele
ase
Time (min)
F5
F6
F7
F8
Page 88
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 74
8.5.6. In vitro release of aceclofenac from lipospehers (F9-F12)
Table 16: Percentage drug release from various formulations of liposphers
S.No. Cumulative percent drug release (%)
Time
(min)
F9 F10 F11 F12
1. 0 0 0 0 0
2. 30 8.52 12.72 13.47 17.16
3. 60 16.92 17.1 16.92 25.41
4. 120 18.88 27.93 25.33 31.10
5. 180 27.29 33.81 42.22 36.94
6. 240 37.73 43.91 43.94 40.54
7. 300 46.76 50.63 51.33 42.23
8. 360 55.00 59.13 67.47 50.64
9. 420 66.04 67.38 69.23 59.04
10. 480 75.28 68.22 72.33 62.32
11. 540 80.07 79.23 83.01 73.63
12. 600 83.64 82.60 83.90 83.89
(Mean, n=3)
Fig. 27: in vitro drug release of formulation batches F9, F10, F11, and F13
8.6. Drug release kinetics
The cumulative percent drug release curve of the drug loaded lipospehers. it
is obvious that with all lipospheres prepared using egg phosphatidylcholine,
the percentages of drug released after 8 h (T8hr) were overall significantly
0
10
20
30
40
50
60
70
80
90
0 100 200 300 400 500 600 700
F9
F10
F11
F12
Page 89
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 75
lower than with those prepared with soybean phosphatidylcholine under the
same conditions. In addition, soybean phosphatidylcholine membranes are
known to be more fluid than egg phosphatidylcholine membranes resulting
in faster drug release. The highest T 8h value was obtained with carnauba
wax, formulation F8 (92.52 %) and F7 (91.68 %) formulation, with stearyl
alcohol. The lowest T 8h value was obtained with Bees wax, formulation F6
(58.97 %). The optimized batchs F8 and F7.
Drug release kinetics for formulations F1-F12 was shown in table (18)
shows First Order, Higuchi's and Peppas Korsmeyer's plot respectively.
Table 17: drug release kinetics for the various formulations of lipospheres
Formulation
Code
R2
n Zero
order
equation
First
order
equation
Higuchi’s
equation
Korsmeyer
Peppas
equation
F1 0.9852 0.9321 0.9557 0.9866 0.9364
F2 0.9929 0.8748 0.9571 0.9970 0.7696
F3 0.7947 0.9682 0.9389 0.9787 0.4105
F4 0.4777 0.9515 0.8751 0.9892 0.2745
F5 0.8383 0.9695 0.9591 0.9464 0.3677
F6 0.8855 0.9303 0.9425 0.9287 0.3847
F7 0.9396 0.9712 0.9891 0.9881 0.5719
F8 0.9440 0.9578 0.9925 0.9960 0.6054
F9 0.9934 0.9641 0.9855 0.9874 0.7842
F10 0.9743 0.9790 0.9935 0.9964 0.6625
F11 0.9673 0.9783 0.9917 0.9908 0.6732
F12 0.9364 0.9329 0.9592 0.9771 0.4947
Page 90
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 76
Result: The correlation coefficient (R2) values showed that formulations
follow Korsmeyer peppas model for drug release.
8.6.1. Zero order, First order, Higuchi’s equation,and Korsmeyer
Peppas equation plot of drug release of Lipospheres F7, F8 (a, b)
Optimized batch
a) Fig.28: F7
b) Fig.29: F8
0
20
40
60
80
100
120
0 100 200 300 400 500 600
% D
rug
Re
lea
sed
Time
Release Profile
Zero
1st
Matrix
Peppas
Hix.Crow.
0
20
40
60
80
100
120
140
0 100 200 300 400 500 600
% D
rug
Re
lea
sed
Time
Release Profile
Zero
1st
Matrix
Peppas
Hix.Crow.
Page 91
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 77
The drug release kinetics showed in (Table 17), where majority of the
batches governed by peppas model. All the batches showed release up to 8 h
and above 40-90% of drug released with each formulation. Formulation F8
(92.52%) showed maximum release while other formulation showed less
amount of drug release in 8 h. Formulation of F7, F8 and F11 has highest
correlation coefficient (R2=0.9970, 0.9960, 0.9908) respectively and follows
drug release by peppas model. The drug release from lipospheres depends on
many factors including the composition of lipospheres, the type of drug
encapsulated and nature of the cell. Once released, drug that normally
crosses the membrane of a cell will enter the cell. The drug is released from
lipospheres by one of the possible mechanism i.e., Endocytosis, fusion and
adsorption. Orally administered lipospheres release the drug by endocytosis
as gut epithelial cells take up intact lipospheres by absorptive endocytosis.
8.7. Formulations of lipospheres based gel
Lipospheres based gel was prepared according to the formula (Table 8).
8.8. Characterization of lipospheres based topical gel
Table 18: Result of pH, Viscosity, Drug content and Spreadability
S.
No.
Formulation pH Viscosity
(Cps)
Drug
content
(%)
Time(sec) Spredability
(g.cm/sec)
1 F7 6.5 29600 83.33 20 47.27
2 F8 6.9 30200 96.51 20 37.14
3 LF7 6.9 29200 100.317 20 40
4 LF8 7.0 31000 115.73 20 34.66
Page 92
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 78
Fig.30: Lipospheres based gel of optimized batch
All the Lipospheres based topical gel formulations were having good feel
and showed no clogging and lumps which indicate good texture of system.
pH of lipospshere based topical gel was around the neutral pH and in the
range of 6.5-7.0.
All the formulations showed no significant skin irritation on intact skin (Fig
32).Thus, indicating skin acceptability of these formulations for topical
application.
Viscosity is an important parameter for characterizing the gels as it affects
the spreadibility, and release of the drug. Viscosity of formulations was
ranging between 29000-32000 cps.
Easy spreadability is one of the important characteristics of any topical
preparation as far as patient compliance is concerned. Liposphere based gel
is considered to be good if it takes minimum time to spread on the surface.
Among the various gels studied LF8 aceclofenac liposphere gel was find to
show better spreadability. The values of spreadability indicated that the gel
is easily spreadable by small amount of shear. Drug content uniformity of all
formulations were observed and F7, F8 without permeation enhancer and
LF7,LF8 with permeation enhancer batch showed the 83.33 %, 96.51 %,
100.317 % and 115.73 % drug content respectively. Finally selected
formulations were subjected to a stability testing for three months and drug
content of batch F7, F8, LF7 and LF8 was found to be 83.33 %, 96.51 %,
100.317 % and 115.73 % respectively. Depending upon different evaluation
Page 93
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 79
parameters made on all formulations, batch LF8 was declared as an
optimized batch.
8.8.1. In vitro permeation of liposphere based gel
The release profile of a drug predicts how a delivery system might function
and gives valuable insight into its in vivo behavior. The optimized (F8 and
LF8) were subjected to in vitro release studies. These in vitro release studies
were carried out using simulated tear fluid (STF) of pH 7.4 as the dissolution
medium. The drug release data obtained for formulations shows the
cumulative percent drug released. It was found that cumulative percent drug
release was 82.42 %, 91.68 % for F8 and LF8 formulation respectively.
Table 19: Result of cumulative percent (%) drug release of optimize batch
(F8, LF8)
S.NO. Cumulative percent (%) drug release
Time (hr) Batch F8 Batch LF8
1 0 0 0
2 1 25.33 24.84
3 2 33.74 35.03
4 3 43.85 47.32
5 4 55.69 58.49
6 5 64.12 66.02
7 6 71.12 75.40
8 7 79.32 79.23
9 8 82.42 91.68
Page 94
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 80
Fig.31: in vitro release profile of optimized formulation F8 and LF8
8.8.2. Skin irritation testing (Draize patch test)
The results of the skin irritation study revealed that following 72 h
application of LF7, LF8 and marketed preparation (AUDIGEL) and blank
gel, there was no reaction found on the skin. Therefore, it can be assured that
the gel formulation can be used for topical application.
Fig.32: The skin irritation results of lipospheres based topical gel to skin of
four rabbits (number A,B,C,D) after administration of 72 h (1) No application
(2) Marketed aceclofenac gel (AUDIGEL) (3) LS based topical gel (LF7) (4)
LS based topical gel (LF8)
0
10
20
30
40
50
60
70
80
90
100
0 2 4 6 8 10
cum
ula
tive
Pe
rce
nt
dru
g re
leas
e
time (hr)
F8
LF8
Page 95
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 81
8.8.3. In vivo anti-inflammatory study of lipospheres
Table 20: Result of Percentage inhibitions for anti-inflammatory activity
Edema degree (ml) at different movement (h)
Group
0
1
2
3
4
Control
0.7±00.12 1.28±0.48 1.32±0.043 1.33±.09 1.46±0.266
Marketed
0.77±0.22
0.9±0.40 0.91±0.41 0.86±0.4 0.84±0.46
LF7 0.89±0.24 0.98±0.36 1.08±0.37 1.12±0.3 1.06±0.40
LF8 0.65±0.077 0.82±0.075 0.84±0.077 1.02±0.19 0.72±0.36
Edema degree (ml) at different movement (h)
Group
5
6
7
8
% Inhibition after
8 hr
Control 0.7±00.12
1.28±0.48 1.32±0.043 1.33±.09 ----
Marketed 0.77±0.22
0.9±0.40 0.91±0.41 0.86±0.4 65%
LF7 (III) 0.89±0.24 0.98±0.36 1.08±0.37 1.12±0.3 81%
LF8 (IV) 0.65±0.077 0.82±0.075 0.84±0.077 1.02±0.19 96.78%
Data were expressed as mean±S.D and statistically assessed by one way
analysis of variance (ANOVA). Values for edema rate percentage for
lipospheres were compared to the saline control and the differences were
determined statistically using Dunnett’s t test. P<0.05 was considered
significant.
Page 96
Chapter No.8 Results and Discussion
SNIOP, PUSAD 2012-2013 Page 82
The anti-inflammatory activity of the optimized formulation was evaluated
by the carrageenan-induced hind paw inflammation method on wistar rats.
The percentage inhibition value of LF7 and LF8 was compared to marketed
gel. Both of the formulations LF7 and LF8 not only decreased the
inflammation by a larger magnitude, but also sustained the effect for a
prolonged period. After 8 h, the percent edema for LF7 and LF8 was found
to be 81 % and 96.78 %, respectively (as shown in Table 20). While in case
of marketed gel, percentage edema inhibition were found to be 65 %. Hence
the lipospheres based gel formulation of aceclofenac remained superior to
the marketed product in its ability to suppress edema and sustained the anti-
inflammatory activity.
8.9. Stability studies
All the selected formulations were subjected to a stability testing for
three months as per ICH norms at a temperature of 40 ºC ± 2 ºC / 75% ±
5% R H. All selected formulations (LF7 and LF8) were analyzed for the
change in appearance, pH and drug content by procedure stated earlier.
(Table: 21 stability studies)
Table 21: Result of stability study of optimized batch
S No. Month Appearance pH Drug content (%)
Batches Months Appearance pH Drug
content
(%)
1 LF7 Initial white 6.9 100.317
1 white 6.8 100.2
2 white 6.9 99.5
3 white 7.0 99.30
2 LF8 Initial white 7.0 115.73
1 white 6.9 105.21
2 white 6.8 106.2
3 white 7.0 102.1
Page 97
Chapter No.10 Future scope
SNIOP, PUSAD 2012-2013 Page 86
10. Future scope
we can Increase drug loading efficiency
We can improve the bioavailability of poorly water soluble drug by lipid
drug delivery system.
Increasing physical and chemical storage stability
Minimizing overall costs
Page 98
Chapter No.9 Summary and Conclusion
SNIOP, PUSAD 2012-2013 Page 83
9. Summary
The lipospheres system is a newly introduced lipid-based carrier system
developed for parenteral and topical drug delivery of bioactive compounds.
Lipospheres consist of water-dispersible solid microparticles of particle size
between 0.2–500 μm in diameter and composed of a solid hydrophobic fat core
stabilized by one monolayer of phospholipid molecules embedded in their
surface which is a potential group of penetration enhancers. Both egg and
soybean phosphatidylcholine contain unsaturated fatty acids which may be
responsible for the penetration enhancement. The packing nature of unsaturated
fatty acids disrupts the stratum corneum lipid structure and enhances the
percutaneous penetration of drugs, also they strongly raise the fluidity of the
stratum corneum. In addition, lecithin has a high affinity for epidermal tissue
and even seems to improve skin hydration. Being biodegradable and composed
of natural body constituents, topically administered phospholipids can be
generally considered as safe. Lipospheres like solid lipid nanoparticles are one
of the carriers of choice for topically applied drugs because their lipid
components have an approved status or excipients used in commercially
available topical cosmetic or pharmaceutical preparations. The small size of the
lipid particles ensures close contact to the stratum corneum and can increase the
amount of the drug penetrating into the mucosa or skin. Due to their solid lipid
matrix, controlled release from these carriers is possible which is important to
supply the drug over a prolonged period of time and to reduce systemic
absorption, increased drug stability can be achieved and finally lipospheres
possess a film forming ability leading to occlusive properties.
The purpose of this study was to prepare lipospheres containing
aceclofenac intended for topical skin delivery with the aim of exploiting the
favorable properties of this carrier system and developing a sustained release
formula to overcome the side effects resulting from aceclofenac oral
Page 99
Chapter No.9 Summary and Conclusion
SNIOP, PUSAD 2012-2013 Page 84
administration. Lipospheres were prepared using different lipid cores (carnauba
wax, bees wax, steryl alcohol) and phospholipids coats (egg phosphatidylchoine
and soya phosphatidylcholine) by melt dispersion technique. Characterization
of the prepared lipospheres formulation carried out through photomicroscopy,
scanning electron microscopy (SEM), particle size analysis, diffential scanning
caorimetry (DSC), and in vitro drug release and stability study. It was
uniformly dispersed after suitably gelled by Carbopol 940 preparation. The
characterization of the prepared lipospheres based topical gel rheological study,
pH, Spreadability, drug content, skin irritation test. No oedema and erythema
were observed after administration of lipospheres based aceclofenac gel on
rabbit skin, the anti-inflammatory effect of liposphere systems was assessed by
the rat paw edema technique and compared to the marketed product. Results
revealed that liposphere systems were able to entrap aceclofenac at very high
levels (101.65 %). The particle size of liposphere systems was well suited for
topical drug delivery. DSC revealed the molecular dispersion of aceclofenac
when incorporated in lipospheres. Lipospheres were very stable after 3 months
storage at 2–8 °C. Liposphere topical gel was found to possess superior anti-
inflammatory activity compared to the marketed product.
Page 100
Chapter No.9 Summary and Conclusion
SNIOP, PUSAD 2012-2013 Page 85
Conclusion
This present work indicates that the Lipospheres based aceclofenac gel could be
successfully prepared by the melt dispersion technique. The melt dispersion
method produced smaller particles. This study also indicates that the amount of
lipids and lecithin significantly affects the particle size as well as entrapment
efficiency. It can be concluded that the optimized lipospheres gel exhibit faster
onset and prolonged action as compared to the marketed product. Further, in
vivo anti-inflammatory and skin irritation studies are necessary to assess the
improvement of therapeutic efficacy of the LS gel compared to the marketed
product. Findings of this investigation suggest that lipospheres can be
considered a promising delivery system for topical aceclofenac delivery.
Lipospheres were able to entrap the drug at very high levels and sustained its
release over a prolonged time. Lipospheres possessed a suitable size for topical
route and being based on non irritating and non toxic lipids, lipospheres seemed
to be well suited for use on damaged or inflamed skin. Furthermore, lipospheres
possessed a very high stability as well as superior anti-inflammatory activity
compared to the marketed product.
Page 101
Chapter 12 Errata
SNIOP, PUSAD 2012-2013 Page 98
ERRATA
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
Page 102
Chapter 12 Errata
SNIOP, PUSAD 2012-2013 Page 99
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
__________________________________________________________
_________________________________________________________
Page 103
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 87
11. References
1. Domb, A.J. et al., Degradable polymers for site-specific drug delivery,
Polymer Adv. Technol, (1992):3, 279.
2. Barke, S.A. and Khossravi, D., Drug delivery strategies for the new
millennium, Drug Del. Technol, (2001):6, 75.
3. Mizushima, Y., Lipid microspheres (lipid emulsions) as a drug carrier: an
overview, Adv. Drug Del. Rev, (1996):20, 113.
4. Ravi Kumar, M.N.V., Nano and microparticles as controlled drug delivery
devices, J. Pharm. Pharmaceut. Sci, (2000):3, 234.
5. Gombotz, W. et al., Prolonged release of GM-CSF, United States Patent,
(1995):5, 253.
6. Wake, M.C. et al., Effects of biodegradable polymer particles on rat marrow-
derived stromal osteoblasts in vitro, Biomaterials, (1998):19, 1255.
7. Cortesi, R. et al., Preparation of liposomes by reverse-phase evaporation
using alternative organic solvents, J. Microencapsul,( 1999):16, 251.
8. Vyas, S.P., Singh, R. and Dimitrijevic, D., Development and characterization
of nifedipine lipospheres, Pharmazie, (1997):52, 403.
9. Jenning, V., Thünemann, A.F. and Gohla, S.H., Characterization of a novel
solid lipid nanoparticle carrier system based on binary mixtures of liquid and
solid lipids, Int. J. Pharm,(2000):199, 167.
10. Schwarz, C. and Mehnert, W., Solid lipid nanoparticles (SLN) for
controlled drug delivery. II. Drug incorporation and physicochemical
characterization, J. Microencapsul (1999):16, 205.
Page 104
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 88
11. Müller, R.H., Mäder, K. and Gohla, S., Solid lipid nanoparticles (SLN) for
controlled drug delivery: a review of the state of the art, Eur. J. Pharm.
Biopharm,( 2000):50, 161.
12. Takenaga, M., Application of lipid microspheres for the treatment of cancer,
Adv. Drug Del. Rev, (1996):20, 209.
13. Yang, S.C., et al., Body distribution of camptothecin solid lipid
nanoparticles after oral administration, Pharm. Res, (1999): 16, 751.
14. Inoue, K. et al, ex vivo anti-platelet effects of isocarbacyclin methyl ester
incorporated in lipid microspheres in rabbits, Arzneim-Forsch/Drug. Res,
(1995):45, 980.
15. Müller, R.H. et al., Solid lipid nanoparticles (SLN) as potential carrier for
human use: interaction with human granulocytes, J. Control. Rel, (1997):47,
261.
16. Schwarz, C. et al., Solid lipid nanoparticles (SLN) for controlled drug
delivery: production, characterization and sterilization, J. Control. Rel, (1994):
30, 83.
17. Bodmeier, R., Chen, H. and Bhagwatwar, H., Polymer and wax
microspheres prepared by emulsification techniques, Bull. Tech. Gattefossé,
(1990):83.
18. zur Mühlen, A., Schwarz, C. and Mehnert, W., Solid lipid nanoparticles
(SLN) for controlled drug delivery: drug release and release mechanism, Eur. J.
Pharm. Biopharm, (1998):45, 149.
Page 105
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 89
19. Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, et al.
Doubly amphiphilic poly (2-oxazoline) s as high-capacity delivery systems for
hydrophobic drugs. Biomaterials, (2010):31(18), 4972-4979.
20 Jang JH, Shea LD. Intramuscular delivery of DNA releasing microspheres:
microsphere properties and transgene expression. J Control Release.
(2006):112(1), 120-128.
21. Dey N, Majumdar S, Rao M. Multiparticulate drug delivery systems for
controlled release. Trop. j. pharm. Res, (2008):7(3), 1067-1075.
22. Wichert B, Rohdewald P. A new method for the preparation of drug
containing polylactic acid microparticles without using organic solvents. J
Control Release,(1990):14(3), 269-283.
23. Fricker G, Kromp T, Wendel A, Blume A, Zirkel J, Rebmann H, et al.
Phospholipids and Lipid-Based Formulations in Oral Drug Delivery. Pharm Res
(2010):1-18.
24. MuÈller RH, MaÈder K, Gohla S. Solid lipid nanoparticles (SLN) for
controlled drug delivery-a review of the state of the art. Eur J Pharm Biopharm
(2000):50(1), 161-177.
25. Bangham AD. Techniques in life Science, lipid membrane biochemistry.
Elsevier Scientific Publications. Ireland, (1982):25.
26. Lachman L, Lieberman HA, Kanig JL. In, The Theory and Practice of
Industrial Pharmacy. Varghese Publishing House. Bombay, (1987):417.
27.Vyas SP, Khar RK. Targeted and controlled Drug Delivery, Novel Carrier
system, CBS Publishers and Distributors, (2002): 263-65.
Page 106
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 90
28. Datta S, Karki R. Liposomes encapsulated Rifampicin and Isoniazid. Indian
J Pharm Sci (2000): 62(5), 384.
29. Ramana MV, Chaudhari AD. An approach to minimize pseudo
membranous colitis caused by Clindamycin through liposomal formulation.
Indian J Pharm Sci (2007), 69(3), 390.
30.Qui L, Jing N, Jin Y. Preparation and invitro evaluation of liposomal
chloroquinediphophate loaded by a transmembrane pH-gradient method. Int J
Pharma,(2008), 56-63.
31. Claudio Nastruzzi, editor. Lipospheres in Drug Targets and Delivery
Approaches, Methods, and Applications. New York: Washington, 2005.
32. Domb, Abraham J, Maniar, Manoj, Lipospheres for Controlled Delivery of
Substances. European Patent: EP0502119.
33. Mu L, Feng S. Fabrication, characterization and in vitro release of paclitaxel
(Taxol®) loaded poly (lactic-co-glycolic acid) microspheres prepared by spray
drying technique with lipid/ cholesterol emulsifiers. J Control Release, (2001),
76(3), 239-254.
34.Toongsuwan S, Li LC, Erickson BK, Chang HC.Formulation and
characterization of bupivacaine lipospheres. Int J Pharm.,(2004):280(1-2), 57-
65.
35. Chakraborty S, Shukla D, Mishra B, Singh S. Lipid-An emerging platform
for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm.,
(2009):73(1), 1-15.
36. Wong HL, Rauth AM, Bendayan R, Manias JL, Ramaswamy M, Liu Z, et
al. A new polymer–lipid hybrid nanoparticle system increases cytotoxicity of
Page 107
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 91
doxorubicin against multidrug-resistant human breast cancer cells. Pharm Res.,
(2006):23(7), 1574-1585.
37. zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for
controlled drug delivery-drug release and release mechanism. Eur J Pharm
Biopharm, (1998): 45(2), 149-155.
38. Cortesi R, Esposito E, Luca G, Nastruzzi C. Production of lipospheres as
carriers for bioactive compounds. Biomaterials, (2002): 23(11), 2283-2294.
39. Bhatia A, Singh B, Rani V, Katare O. Formulation, characterization, and
evaluation of benzocaine phospholipid-tagged lipospheres for topical
application. J Biomed Nanotechnol, (2007):3(1), 81-89.
40. Sznitowska M, Janicki S, Gajewska M, Kulik M. Investigation of diazepam
lipospheres based on Witepsol and lecithin intended for oral or rectal delivery.
Acta Pol Pharm–Drug Res, (2000):57, 61–64.
41. Bekerman T, Golenser J, Domb A. Cyclosporin nanoparticulate lipospheres
for oral administration. J Pharm Sci ,004;93(5):1264-1270.
42. Sandipan dasgupta et al, nanostructured lipid carriers (NLC) based gel
for the topical delivery of aceclofenac: preparation, characterization, and in
vivo evaluation, scientia pharamaceutica 2012, 7-8.
43. Esimone et al, formulation and evaluation of goat fat and shea butter
based lipospheres of benzyl penicillin, international journal of
pharmaceutical sciences and research, (2012):4, 1022-1027.
44. Abraham J. Domb et al, cyclosporin pro-dispersion liposphere
formulation, journal of controlled release, (2012):160,406-407.
Page 108
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 92
45. Vignesh muruganandam, formulation development & characterization
of ofloxacin microspheres,indo global journal of pharmaceutical sciences,(
2012):2(2),130-141.
46. Sanming Li et al,Nanostructured lipid (NLC) based topical gel of
flurbiprofen :Design characterization and in Vivo evaluation, international
Journal of pharmaceutics, (2012):439, 349-357.
47. Sateesh Babu N et al, formulation design and in vitro evaluation of
azithromycin loaded lipospheres using melt dispersion technique, journal of
pharmacy research, (2011):4(11), 4069-4071.
48. Loganathan Veerappan et al, formulation development and evaluation
of flurbiprofen Lipospheres,International Journal for the advancement of
science & Arts, (2010): Vol.1 (1).
49. Kamal dua et al,aceclofenac topical dosage forms:In vitro and In vivo
characterization ,Acta pharm, (2010):60, 467-478.
50. Maha Nasr et al, Lipospheres as carriers for topical delivery of
aceclofenac:preparation characterization and In vivoevaluation AAPs
pharmscitech, (2008):19 (1), 154-162.
51. Jia –you fang et al,a study of the formulation design acoustically active
lipospheres as carriers for drug delivery,European Journal of pharmaceutics
and Biopharmaceutics, (2007):67, 67-75.
52. Hagalavadi Nanjappa shivkumar et al, design and statistical
optimization of glipizide loaded lipospheres using response surface
methology, Actapharm, (2007):57, 269-285.
Page 109
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 93
53. vandana B. Pataravale et al, solid lipid nanoparticles (SLN) of tretinoin:
Potential in topical delivery, international journal of pharmaceutics,
(2007):345, 163-171.
54. http://www.tzzy.com/lh6.htm.
55. http://www.chemyq.com/En/xz/xz9/83172xpppl.htm.
56. http://www.medindia.net/doctors/drug_information/aceclofenac.htm.
57.Indian Pharmacopoeia, Ministry of Health and family welfare, govt. of
India, Published by the Drug Controller of India, New Delhi, (2007): vol. II, 63-
64.
58. Dufour P, Vuillemard JC, Laloy E. Characterization of enzyme
immobilization in liposomes prepared from proliposomes. J
Microencapsulation, (1996): 13(2), 185-94.
59. Indian pharmacopoeia, Government of India, Ministry of Health & Family
Welfare, The controller of Publications, New Delhi, (2007): Vol-II, 143.
60.Indian Pharmacopoeia, Ministry of Health and family welfare, govt. of
India, Published by the Drug Controller of India, New Delhi, (2007): Vol-II,
143.
61. Perrett S, Golding M, Williams PW.A simple method for the preparation of
liposome for pharmaceutical applications: Characterization of the liposomes. J
of Pharmacol,(1991): 43, 154-61.
62. Al-Angry AA, Bayomi MA, Khidr SH. Characterization stability and in
vivo targeting of liposomal formulations containing Cyclosporine.Int. J of
Pharmaceutics, (1995): 114, 221-225.
Page 110
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 94
63. A. J. Domb, L. Bergelson, and S. Amselem. Lipospheres for controlled
delivery of substances Microencapsulation, methods and industrial applications,
Marcel Dekker, New York, NY, (1996), 377–410.
64. D. B. Masters and A. J. Domb. Liposphere local anesthetic timedrelease
for perineural site application. Pharm. Res, (1998): 15, 1038–1045.
65. V. Iannuccelli, N. Sala, R. Tursilli, G. Coppi, and S. Scalia. Influence of
liposphere preparation on butyl-methoxydibenzoyl methane photostability.
Eur. J. Pharm. Biopharm, (2006): 63,140–145.
66. R. Tursilli, A. Casolari, V. Iannuccelli, and S. Scalia. Enhancement of
melatonin photostability by encapsulation in lipospheres. J. Pharm. Biomed.
Anal. (2006): 40:910–914.
67. F. Coi, D. Cun, A. Tao, M.Yang , Y.K.Shi and Y.Guan. Preparation
and characterization of melittin- loaded poly (DL-lactic acid) or poly (DL-
lactic –co- glycolic acid )microspheres made by double emulsion method,
Journal of Controlled release, (2005):107 (2), 310-319.
68. I. El Gibaly, and S. K. Abdel-Ghaffar. Effect of hexacosanol on the
characteristics of novel sustained release allopurinol solid lipospheres
(SLS): factorial design application and product evaluation. Int. J. Pharm,
(2005):294, 33–51.
69. J. J. Sheng, P. J. Sirois, J. B. Dressman and G. L. Amidon, Particle
diffusional layer thickness in a USP dissolution apparatus II: a combined
function of particle size and paddle speed, J. Pharm. Sci, (2008): 4815–
4829.
Page 111
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 95
70 Arul B, Sathyamurthy D. Formulation and evaluation of ketorolac
tromethanmine gels.TheEastern Pharmacist (1998):135-6.
71 Chowdary KPR, Kumar PA. Formulation and evaluation of topical drug
delivery systems of ciprofloxacin. Ind J Pharm Sci,(1996): 58 (2),47-50.
72 Sanghavi NM, Mahalaxmi D. Determination in vitro release of
clobetasol propionate from topical bases.Indian Drugs, (1993): 30 (8),364-
70.
73 Mura P, Faucci MT, Bramanti G, Corti P. Evaluation of transcutol as a
clonazepam transdermal penetration enhancer from hydrophilic gel
formulations. Eur J Pharm Sci (2002): 9, 365-372.
74 Arellano A, Santoyo S, Martin C, Ygartua P. Influence of propylene
glycol and isopropyl myristate on the in vitro percutaneous penetration of
diclofenac sodium from carbopol gels. Eur J Pharm Sci, (1998): 7, 129-35.
75 Islam MT, Rodriguez-Hornedo N, Ciotti S, Ackermann C. Rheological
characterization of topical carbomer gels neutralized to different pH. Pharm
Res (2004): 21, 1192-1199.
76. Mohamed MI. Optimization of chlorphenesin emulgel formulation. The
AAPS Journal, (2004):6 (3), 1-7.
77. Tas C, Ozkan Y, Savaser S, Baykara T. In vitro release studies of
chlorpheniramine maleate from gels prepared by different cellulose
derivatives,IL Farmaco, (2003): 58, 605-11.
Page 112
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 96
78. Gendy AME, Jun HW, Kassem AA. In vitro release studies of
flurbiprofen from different topical formulations, Marcel Dekker, New
York, Drug Dev Ind Pharm, (2002):48:823-31.
79. Aejaz A, Azmail , Sanaullah S, Mohsin A.A., Formulation and invitro
evauluation of aceclofenac solid dispersion incorporated gels,International
Journal of Applied Pharmaceutics,(2010): 2(1), 8-12.
80. Nagariya K., Jadon P.S, Naruka P.S., Chauhan C.S., Formulation
development and characterization of aceclofenac gelusing poloxamer 407,
J. Chem. Pharm. Res, (2010): 2(4), 357-363.
81. Mutimer M.N., Riffikin C., Hill J.A., Marry E., Cry N.G., Gliekman
G.J., Am Pharm, Assoc. Sci,(1956):45, 212.
82. Thorat S.P., Rane S.I., Formulation and in vitro evaluation of lecithin
(soya and egg) based aceclofenac organogels, Journal of Pharmacy
Research, (2010): 3(6), 1438-1441.
83. Sirish Vodithala; Sadhna Khatry; Nalini Shastri; M Sadananda.
International Journal of Current Pharmaceutical Research, (2010): 2(3), 33.
84. Basavaraj K. Nanjwade; Manjappa AS; Murthy RSR; Yuvaraj D Pol.
Asian Journal of Pharmaceutical Sciences, (2009):4(3), 189.
85. Draize, J., Woodard, G., Calvery, H., Methods for the study of irritation
and toxicity of substances topically applied to skin and mucous membranes.
J. Pharmacol. Exp. Ther, (1944): 82, 377–390.
86. www.ICH/stability.
Page 113
Chapter No.11 References
SNIOP, PUSAD 2012-2013 Page 97
87. Al-Meshal MA, Khidr SH, Bayomi MA. Oral administration of liposomes
containing Cyclosporine: a pharmacokinetic study. Int. J of Pharmaceutics
(1998): 168, 163-168.