PRIMARY AMYLOIDOSIS JAMES BROWN, M.D., F.F.R. Department of Radiology, Royal Lancaster Infirmary, Lancaster IT has long been recognised that amyloidosis may occur in the absence of any apparent predisposing cause. Once considered of great rarity, this primary form of the disease has been shown to be far commoner than was at one time supposed and is now frequently diagnosed during the lifetime of the patient. The demonstration of amyloid in biopsy material is the most conclusive method of establishing the diagnosis and the value of rectal biopsy has recently been reported (Fentem et al 1962). Radiographic examination may also offer help in diagnosis and it is the purpose of this paper to discuss the radiological features encoun- tered in primary amyloidosis and to illustrate them by reference to six new cases in which the diagnosis was suspected neither clinically nor radiologically. In one case the disease was apparently localised to one site but in the other five cases there was widespread systemic involvement. The existence of atypical amyloidosis was first recorded by Wilks (1856) who separated lardaceous disease into five groups, in two of which he failed to demonstrate any associated illness. The major credit for its recognition, however, belongs to Wild who in 1886 described a case of diffuse amyloidosis in a woman of fifty-six years and stressed its independence of the usual chronic illnesses normally associated with amyloid disease. During the following four decades there were only sporadic reports and it was not until 1939 that a major contribution to the subject was made by Koletsky and Stecher in their review of twenty-four cases, to be followed by papers by Eisen (1946), Higgins and Higgins (1950) and Mathews (1954). The review of 154 published cases by Rukavina etal(1956)isparticu!arlyvaluable. Their investiga- tions of a large family pedigree provided evidence that the disease may occur in a familial form being transmitted as a Mendelian dominant characteristic. With its tendency to involve a wide variety of tissues, it is not surprising that the clinical features of primary amyloidosis are not only varied but capable of simulating many other diseases. The localised form usually presents less difficulty for the lesions are most frequently encountered in the upper and lower respiratory tracts and are readily available for biopsy study. In this type of case clinical study usually reveals a single tumour deposit which may remain unchanged for some years and be unassociated with any sign of con- stitutional disturbance. The natural history of the disseminated form, however, is generally characterised by a progressive afebrile illness associated with particular signs and symptoms most frequently affecting the cardiovascular and alimentary systems. In the case of the former, intractible cardiac failure may result and this is the most common clinical manifestation. Indeed, primary amyloidosis should always be considered when confronted with cardiac failure of uncertain cause. When the alimentary system is involved, dysphagia, vomiting, haematemesis, abdominal pain or rarely a sprue-like syndrome may be present; macroglossia is a not infrequent finding and has occurred in about a third of reported cases. Pulmonary, hepatic, splenic and renal involvement have all been described frequently and neurological manifestations have also been recorded. Other tissues less frequently affected include skeletal muscle, lymph nodes and bone. CASE REPORTS Case 1.--Mrs H. C., aged seventy years at death, was first seen at the Royal Lancaster Infirmary in June 1956 when she complained of anorexia, biliousness and a heavy feeling in the abdomen for about one year. On physical examination she was pale but well nourished; the tongue was normal and no enlarged lymph nodes were palpable; the cardio- vascular system was normal; abdominal examination revealed considerable splenomegaly. The haemoglobin was 71 per cent (10"5 gr./100 ml.), R.B.C. 4"24 m./cu, mm., and W.B.C. 6,000/cu. mm., the differential count being normal; urinalysis was normal. Radiological examination of the chest and abdomen was not remarkable apart from demon- strating a large splenic shadow. It was considered likely that the patient was suffering from a myeloproliferative syndrome or reticulosis and in view of the severe distress occasioned by the splenomegaly it was decided to remove the spleen. The spleen, which was much enlarged and found to weigh 854 gr., contained within its substance a large spherical tumour measuring 13 cm. in maximum diameter (Fig. 1). The tumour showed a variegated pattern being for the most part of a dull red hue but having areas of paler gelatinous material. The surrounding splenic tissue was firmer and more homogeneous than usual. Microscopic examination showed the tumour to be composed of massive deposits of amyloid while in the rest of the spleen there was also exten- sive amyloid infiltration with a quite striking peri-follicular distribution. It stained well with both Congo red and methyl violet. Apart from a mild degree of anaemia and varicose ulceration of her legs the patient remained well for the ensuing five years. During 1961 barium studies of the 358
Transcript
P R I M A R Y A M Y L O I D O S I S
JAMES BROWN, M.D., F.F.R.
Department of Radiology, Royal Lancaster Infirmary, Lancaster
IT has long been recognised that amyloidosis may occur in the absence of any apparent predisposing cause. Once considered o f great rarity, this pr imary form of the disease has been shown to be far commoner than was at one time supposed and is now frequently diagnosed during the lifetime of the patient. The demonstra t ion of amyloid in biopsy material is the most conclusive method of establishing the diagnosis and the value of rectal biopsy has recently been reported (Fentem et al 1962). Radiographic examination may also offer help in diagnosis and it is the purpose of this paper to discuss the radiological features encoun- tered in pr imary amyloidosis and to illustrate them by reference to six new cases in which the diagnosis was suspected neither clinically nor radiologically. In one case the disease was apparently localised to one site but in the other five cases there was widespread systemic involvement.
The existence o f atypical amyloidosis was first recorded by Wilks (1856) who separated lardaceous disease into five groups, in two o f which he failed to demonstrate any associated illness. The major credit for its recognition, however, belongs to Wild who in 1886 described a case o f diffuse amyloidosis in a woman of fifty-six years and stressed its independence o f the usual chronic illnesses normally associated with amyloid disease. During the following four decades there were only sporadic reports and it was not until 1939 that a major contr ibution to the subject was made by Koletsky and Stecher in their review of twenty-four cases, to be followed by papers by Eisen (1946), Higgins and Higgins (1950) and Mathews (1954). The review of 154 published cases by Rukavina etal(1956)isparticu!arlyvaluable. Their investiga- tions o f a large family pedigree provided evidence that the disease may occur in a familial form being transmitted as a Mendelian dominant characteristic.
With its tendency to involve a wide variety o f tissues, it is not surprising that the clinical features o f primary amyloidosis are not only varied but capable o f simulating many other diseases. The localised form usually presents less difficulty for the lesions are most frequently encountered in the upper and lower respiratory tracts and are readily available for biopsy study. In this type of case clinical study usually reveals a single tumour deposit which may remain unchanged for some
years and be unassociated with any sign of con- stitutional disturbance. The natural history of the disseminated form, however, is generally characterised by a progressive afebrile illness associated with particular signs and symptoms most frequently affecting the cardiovascular and alimentary systems. In the case of the former, intractible cardiac failure may result and this is the most c o m m o n clinical manifestation. Indeed, pr imary amyloidosis should always be considered when confronted with cardiac failure of uncertain cause. When the alimentary system is involved, dysphagia, vomiting, haematemesis, abdominal pain or rarely a sprue-like syndrome may be present; macroglossia is a not infrequent finding and has occurred in about a third of reported cases. Pulmonary, hepatic, splenic and renal involvement have all been described frequently and neurological manifestations have also been recorded. Other tissues less frequently affected include skeletal muscle, lymph nodes and bone.
CASE REPORTS
Case 1.--Mrs H. C., aged seventy years at death, was first seen at the Royal Lancaster Infirmary in June 1956 when she complained of anorexia, biliousness and a heavy feeling in the abdomen for about one year. On physical examination she was pale but well nourished; the tongue was normal and no enlarged lymph nodes were palpable; the cardio- vascular system was normal; abdominal examination revealed considerable splenomegaly. The haemoglobin was 71 per cent (10"5 gr./100 ml.), R.B.C. 4"24 m./cu, mm., and W.B.C. 6,000/cu. mm., the differential count being normal; urinalysis was normal. Radiological examination of the chest and abdomen was not remarkable apart from demon- strating a large splenic shadow. It was considered likely that the patient was suffering from a myeloproliferative syndrome or reticulosis and in view of the severe distress occasioned by the splenomegaly it was decided to remove the spleen.
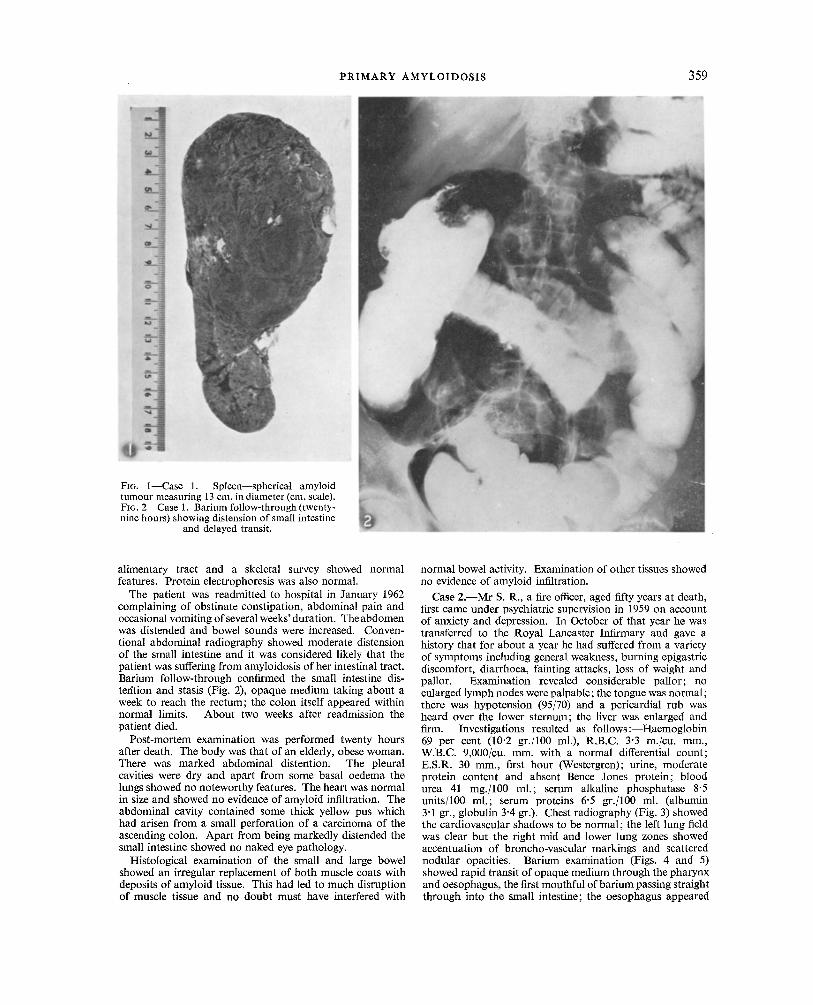
The spleen, which was much enlarged and found to weigh 854 gr., contained within its substance a large spherical tumour measuring 13 cm. in maximum diameter (Fig. 1). The tumour showed a variegated pattern being for the most part of a dull red hue but having areas of paler gelatinous material. The surrounding splenic tissue was firmer and more homogeneous than usual. Microscopic examination showed the tumour to be composed of massive deposits of amyloid while in the rest of the spleen there was also exten- sive amyloid infiltration with a quite striking peri-follicular distribution. It stained well with both Congo red and methyl violet.
Apart from a mild degree of anaemia and varicose ulceration of her legs the patient remained well for the ensuing five years. During 1961 barium studies of the
358
PRIMA RY AMYLOIDOSIS 359
alimentary tract and a skeletal survey showed normal features. Protein electrophoresis was also normal.
The patient was readmitted to hospital in January 1962 complaining of obstinate constipation, abdominal pain and occasional vomiting of several weeks' duration. Theabdomen was distended and bowel sounds were increased. Conven- tional abdominal radiography showed moderate distension of the small intestine and it was considered likely that the patient was suffering from amyloidosis of her intestinal tract. Barium follow-through confirmed the small intestine dis- tefftion and stasis (Fig. 2), opaque medium taking about a week to reach the rectum; the colon itself appeared within normal limits. About two weeks after readmission the patient died.
Post-mortem examination was performed twenty hours after death. The body was that of an elderly, obese woman. There was marked abdominal distention. The pleural cavities were dry and apart from some basal oedema the lungs showed no noteworthy features. The heart was normal in size and showed no evidence of amyloid infiltration. The abdominal cavity contained some thick yellow pus which had arisen from a small perforation of a carcinoma of the ascending colon. Apart from being markedly distended the small intestine showed no naked eye pathology.
Histological examination of the small and large bowel showed an irregular replacement of bo th muscle coats with deposits of amyloid tissue. This had led to much disruption of muscle tissue and no doubt must have interfered with
normal bowel activity. Examination of other tissues showed no evidence of amyloid infiltration.
Case 2 . - -Mr S. R., a fire officer, aged fifty years at death, first came under psychiatric supervision in 1959 on account of anxiety and depression. In October of that year he was transferred to the Royal Lancaster Infirmary and gave a history that for about a year he had suffered from a variety of symptoms including general weakness, burning epigastric discomfort, diarrhoea, fainting attacks, loss of weight and pallor. Examination revealed considerable pallor; no enlarged lymph nodes were palpable; the tongue was normal; there was hypotension (95/70) and a pericardial rub was heard over the lower sternum; the liver was enlarged and firm. Investigations resulted as follows :--Haemoglobin 69 per cent (10'2 gr./100 ml.), R.B.C. 3"3 m./cu, mm., W.B.C. 9,000/cu. mm. with a normal differential count; E.S.R. 30 mm., first hour (Westergren); urine, moderate protein content and absent Bence Jones protein; blood urea 41 mg./100 ml.; serum alkaline phosphatase 8"5 units/100 ml.; serum proteins 6"5 gr./100 ml. (albumin 3"1 gr., globulin 3"4 gr.). Chest radiography (Fig. 3) showed the cardiovascular shadows to be normal; the left lung field was clear but the right mid and lower lung zones showed accentuation of broncho-vascular markings and scattered nodular opacities. Barium examination (Figs. 4 and 5) showed rapid transit of opaque medium through the pharynx and oesophagus, the first mouthful of barium passing straight through into the small intestine; the oesophagus appeared
360 C L I N I C A L R A D I O L O G Y
rigid; there was free gastro-oesophageal incompetence but no evidence of hiatus hernia or oesophagitis; the stomach showed atrophy of mucosa and was virtually aperistaltic particularly in its distal third. Follow-through examination revealed nothing remarkable. The combination of gastro- oesophageal incompetence, mucosal atrophy and aperistalsis suggested a diffuse infiltrative condition seen in scirrhous cancer. Barium enema examination showed normal features of the large bowel. At laparotomy, the liver was grossly enlarged, waxy in appearance and was found to dent on pressure. The condition was thought to be amyloidosis and a biopsy was taken. The stomach was not grossly abnormal.
Microscopic examination of the liver showed a remarkable
and widespread distribution of amyloid. Instead of the more frequently encountered lobular deposition, the amyloid was everywhere deposited within columns of liver ceils in a regular and even pattern (Fig. 6). The material could be stained both by methyl violet and Congo red.
The patient's condition slowly deteriorated and he died at home in June 1960. Unfortunately a post-mortem examination was not obtained.
Case 3 . - -Mr W. B., a boilerman, aged sixty-three years at death, was admitted to the Queen Victoria Hospital, Morecambe in June 1960 complaining of loss of weight, diarrhoea and vomiting of about five weeks' duration. He had been previously healthy. Clinical examination revealed a thin patient who showed marked muscle twitching and
PRIMARY A M Y LO ID O S IS 361
hyperpnoea; the heart was slightly enlarged to the left and the blood pressure was 140/70; there was border-line venous jugular engorgement; the liver was enlarged and tender but no ankle oedema was present; the spleen was not palpable. The haemoglobin was 61 per cent (9"I gr./100 ml.) and the R.B.C. 3'6 m./cu, ram.; the blood urea was 263 rag./100 ml. and the urine contained a moderate amount of protein. Radiological examination of the chest (Fig. 7) showed a normal cardiac shadow; there was old pleural thickening at the left lung base; the right mediastinal shadow showed a smooth crescentic widening at the level of the ascending aorta, not apparently vascular. Abdominal radiography revealed splenic enlargement; the renal shadows appeared normal. Barium meal and enema examinations showed no abnormality of the intestinal tract.
The patient failed to respond to supportive therapy, gradually became comatose and died in uraemia three weeks following admission.
Post-mortem examination was performed eighteen hours after death. The left thoracic cavity was obliterated by old dense adhesions but the right pleura was normal. The lungs showed much oedema and there was extensive pneumonia affecting the left lower lobe. The principal finding in the thoracic organs was gross enlargement of the mediastinal lymph nodes. These measured up to 4 cm. in diameter.
The glands were discrete and of a pale pink colour with a waxy consistency. The inguinal, cervical and axillary nodes seemed normal. The heart showed some enlargement due to left ventricular hypertrophy and weighed 450 gr. The abdominal cavity was normal and there was no enlargement of the mesenteric and para-aortic glands. The alimentary tract, liver and pancreas appeared normal but the spleen was much enlarged, weighing 350 gr. Its cut surface pre- sented an appearance of multiple, grey-pink nodules scattered haphazardly throughout the splenic parenchyma; these nodules were small, none measuring more than a few millimetres in diameter. The kidneys were grossly abnormal and although normal in size (125 gr. and 135 gr.) showed marked distortion of their finer structure. The cortico- medullary junction was no longer recognisable, the whole parenchyma being replaced by fluffy grey tissue. The surfaces were finely granular although the capsules stripped easily. The brain and endocrine glands appeared normal except for minimal irregular thickening of the adrenal glands.
Microscopic examination showed widespread deposits of amyloid in the mediastinal nodes, spleen and adrenal glands; the pituitary, thyroid, pancreas, liver, lungs and brain were not involved. The kidneys showed massive glomerular deposits (Fig. 8) which had enlarged these structures some-
362 CLINICAL RADIOLOGY
times to twice their normal size. There was an associated fibrosis of the interstitial tissue with atrophy and dilatation of tubules. The renal arterioles were diffusely thickened with amyloid which was everywhere stained by Congo red but did not stain metachromatically with crystal violet. In the spleen the deposits were of focal nodular type (Fig. 9) and here again the splenic arterioles were involved. The deposits in the adrenal glands were more extensive than was apparent on naked eye inspection and favoured the zona reticularis and medullary areas.
Case 4.--Mrs P. T., aged forty years at death, was first seen in the Out-Patient Department of the Chesterfield Royal Hospital in September 1960. Fifteen days later she was admitted under the care of Dr R. T. Gaunt. She gave a history of epigastric pain, anorexia and loss of weight (4 st.) during the preceding six months, but thought that her gradually failing health could be dated from the birth of her eighth and last child, two years previously. She thought she was becoming darker in colour but this was difficult to evaluate as she was of Indian origin. Her only other complaint was of an indolent ulcer of the tongue which had been present for two years. Clinical examination revealed a weak and emaciated woman with scanty pubic and axillary hair and a chronic indurated ulcer on the upper surface of the tongue; apart from the finding of hypotension the respiratory and cardiovascular systems were normal (B.P. 90/60); there was no buccal pigmentation; the epigastrium was tender and there were increased bowel sounds and visible peristalsis; there was no splenic, hepatic or lymph node enlargement; the central nervous system was normal. The haemoglobin was 45 per cent (6"7 gr./100 ml.), R.B.C, 2"5 m./cu, mm., colour index 0"9, and E.S.R. 35 mm., first hour (Westergren); faecal examination showed occult blood; the urine contained a trace of protein. Radiological examina-
tion (Dr Ralph) showed no abnormality of the oesophagus, stomach or duodenum; after forty-five minutes some pooling of barium occurred in the upper jejunum which seemed to have lost its normal motility and mucosal pattern (Figs. 10 and 11). Chest radiography and barium enema examinations were normal.
The patient was treated with a high protein diet, vitamins, iron by mouth and cortisone and made some temporary improvement. Following discharge on her own wish she was readmitted shortly afterwards suffering from broncho- pneumonia and died.
Autopsy was carried out four hours after death. There was inflammatory consolidation of both lower lobes. Significant naked eye changes were confined to the alimentary tract. The oesophagus and stomach were normal. The greater part of the small intestine appeared normal but there was an extensive length of jejunum measuring approximately 4 ft. which showed multiple small areas of mucosal ulcera- tion; over about 2 ft. the ulceration had coalesced and the mucosa was completely denuded and the gut appeared locally thickened. The mesentery was studded with mod- erately large fleshy glands, the largest being some 2 cm. in diameter.
Microscopical sections of jejunum showed the presence of extensive amyloidosis associated with ulceration of the mucosa. Sections of stomach were normal. A minor degree of interstitial amyloid was demonstrated in the spleen, and in the kidney some of the glomeruli showed hyalinisation due to amyloid deposits; in addition, extensive deposits of amyloid were evident between the tubules in the medulla. The liver, spleen, thyroid, adrenals and femoral marrow were microscopically normal.
Case 5.--Mrs A. N., aged seventy-three years, was referred to the Ophthalmic Department, Royal Lancaster Infirmary
FIG. 10 Fm. 11 Fins. 10 and I 1--Case 4. Barium meal showing pooling of barium in upper jejunum with loss of motility and mucosal pattern.
PRIMARY AMYLOIDOSIS 363
in December 1960 complaining that for six months her right eye had become prominent. Five years previously a rodent ulcer on the right cheek had required treatment by x-rays. On clinical examination the patient looked well and the only abnormal findings were a right-sided proptosis and hyper- tension (200/90). Radiological examination of the frontal sinuses (Fig. 12) showed evidence of an expanding lesion on the right side thought to be a mucocele, which was later explored and drained into the nasal cavity. The surgeon noted that the lesion was loculated and not typical of mucocele. A biopsy was obtained (Fig. 13) and microscopic examination of the fragments showed it to be composed almost entirely of amyloid tissue. Several arterioles showed marked amyloid infiltration of their walls. The material stained well with Congo red but poorly with methyl violet. The patient was later readmitted for further clinical assess- ment. Urine and blood examinations were normal; the E.S.R. was 44 mm., first hour (Westergren); the intravenous Evans blue test was negative. Chest radiography showed no lesion of the heart, lungs or great vessels; barium studies of the intestinal tract revealed normal features and a skeletal survey showed no osseous changes. The patient has been seen at intervals and remains well.
Case 6.--Mrs M. H., aged forty-five years at death, was referred to Stepping Hill Hospital, Stockport in June 1961 under the care of Dr E. R. Smith. For several months she had been losing weight (1 st.) and appetite and later noticed increasing dyspnoea and slight cough. Her husband had suffered from pulmonary tuberculosis and the patient attended for periodic surveillance during the years 1956 to
1958 during which time chest radiography was normal. Physical examination indicated some weight loss; there was no dyspnoea at rest; the fingers were clubbed and several soft lymph nodes were palpable in the left axilla; chest expansion was good but fine crepitations were audible over the right lung; the patient was normotensive (130/85) and the heart was clinically normal; abdominal examination was not remarkable and the central nervous system was normal. The haemoglobin was 81 per cent, W.B.C. 9,000/cu. mm. with a normal differential count; serum proteins were 6.4 gr./ 100 ml. (albumin 2"9 gr., globulin 3-5 gr.); the E.S.R. was 50 mm., first hour (Westergren); the Mantoux test was positive to 1/1,000 dilution. Chest radiography (Fig. 14) revealed coarse, partly confluent mottling throughout both lung fields maximal towards the lung bases with some enlargement of both hilar shadows. Sputum examination was persistently negative for tubercle bacilli and malignant cells. Liver biopsy showed no suggestion of sarcoid.
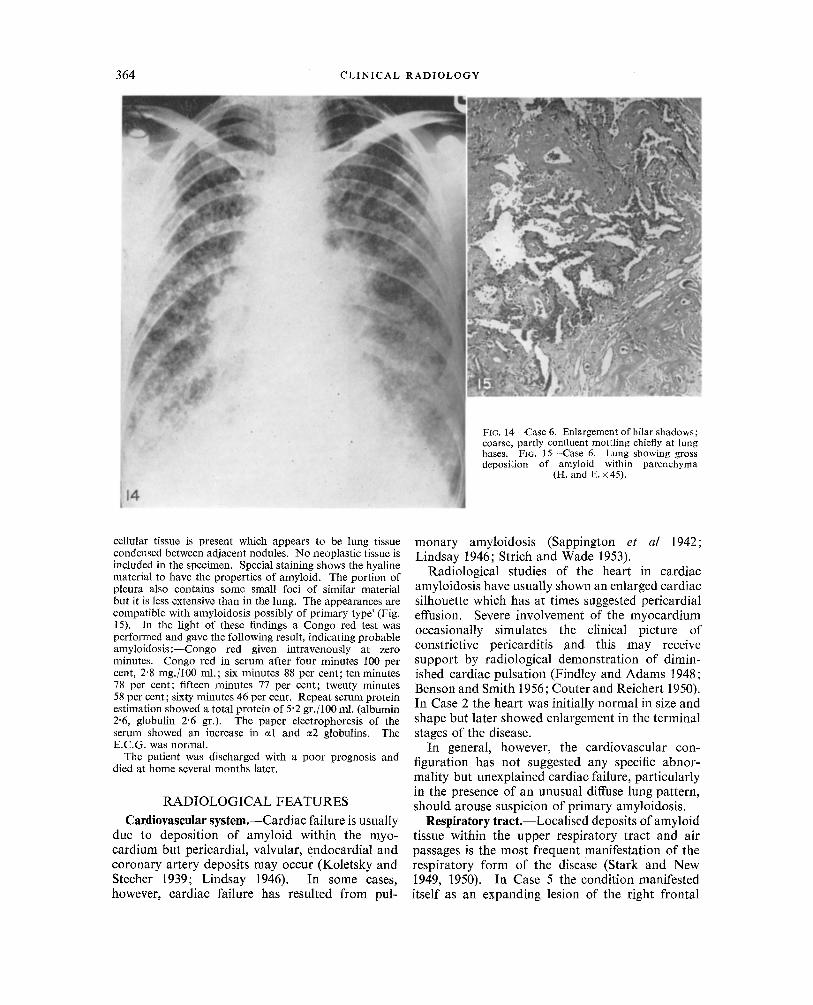
The patient was placed on anti-tuberculous therapy which did not influence the low grade pyrexia but some gain in weight and general improvement was observed. It was felt that lung biopsy was justifiable to establish the diagnosis and this was undertaken by Mr Gordon D. Jack on 8th August 1961. The right lung was exposed through a small 7th interspace incision. It felt extremely hard, was nodular and pinkish grey in colour. The parietal pleura was also nodular. The histological report (Dr Kathleen V. Lodge) was : - - 'The alveolar pattern of the lung is lost and replaced by nodular loci of amorphous hyaline material. There is only slight inflammatory cell infiltration. In some parts more
364 C L I N I C A L R A D I O L O G Y
cellular tissue is present which appears to be lung tissue condensed between adjacent nodules. No neoplastic tissue is included in the specimen. Special staining shows the hyaline material to have the properties of amyloid. The portion of pleura also contains some small loci of similar material but it is less extensive than in the lung. The appearances are compatible with amyloidosis possibly of primary type' (Fig. 15). In the light of these findings a Congo red test was performed and gave the following result, indicating probable amyl0idosis:--Congo red given intravenously at zero minutes. Congo red in serum after four minutes 100 per cent, 2'8 rag./100 ml.; six minutes 88 per cent; ten minutes 78 per cent; fifteen minutes 77 per cent; twenty minutes 58 per cent; sixty minutes 46 per cent. Repeat serum protein estimation showed a total protein of 5"2 gr. / 100 ml. (albumin 2"6, globulin 2"6 gr.). The paper electrophoresis of the serum showed an increase in ~1 and ~2 globulins. The E.C.G. was normal.
The patient was discharged with a poor prognosis and died at home several months later.
RADIOLOG1CAL FEATURES
Cardiovascular system.--Cardiac failure is usually due to deposition of amyloid within the myo- cardium but pericardial, valvular, endocardial and coronary artery deposits may occur (Koletsky and Stecher 1939; Lindsay 1946). In some cases, however, cardiac failure has resulted from pul-
monary amyloidosis (Sappington et al 1942; Lindsay 1946; Strich and Wade 1953).
Radiological studies of the heart in cardiac amyloidosis have usually shown an enlarged cardiac silhouette which has at times suggested pericardial effusion. Severe involvement of the myocardium occasionally simulates the clinical picture of constrictive pericarditis and this may receive support by radiological demonstration of dimin- ished cardiac pulsation (Findley and Adams 1948; Benson and Smith 1956; Couter and Reichert 1950). In Case 2 the heart was initially normal in size and shape but later showed enlargement in the terminal stages of the disease.
In general, however, the cardiovascular con- figuration has not suggested any specific abnor- mality but unexplained cardiac failure, particularly in the presence of an unusual diffuse lung pattern, should arouse suspicion of primary amyloidosis.
Respiratory tract.--Localised deposits of amyloid tissue within the upper respiratory tract and air passages is the most frequent manifestation of the respiratory form of the disease (Stark and New 1949, 1950). In Case 5 the condition manifested itself as an expanding lesion of the right frontal
P R I M A R Y A M Y L O I D O S I S 365
sinus which on clinical and radiological grounds was regarded as a mucocele (Fig. 12).
Involvement of the lower respiratory tract is less common and has been reviewed by Prowse (1958). In this event chest radiography may show tumour- like masses within the lung and there-may be concomitant pulmonary collapse due to bronchial occlusion. Whitwell (1953) described wedge-shaped and rounded pulmonary lesions, frequently multiple and bilateral, and Schmidt et al (1953) reported three cases in which tumour-like shadows strongly suggested carcinoma; in two of these cases dys- trophic calcification was present. The most common manifestation of lower respiratory amyloidosis, however, is a diffuse stenosing infiltration of the trachea and bronchi giving a characteristic broncho- scopic appearance (Prowse 1958). The broncho- graphic features of tracheo-bronchial amyloidosis have recently been described by Kamberg et al (1962) whose two cases showed characteristic indentations on the walls of the main air passages.
Amyloid infiltration of the pulmonary alveoli and vessels may be extreme. In the case described by Sappington et al (1942) the lungs were said to resemble a ~ne rubber sponge and the radiographs were reported as showing indefinite haziness of the lung fields. At other times pulmonary involvement may lead to pleural effusion or a stippled or nodular appearance or result in diffuse accentuation of the broncho-vascular pattern (Ferris 1936; Dirske 1946; Eisen 1946; Wang and Robbins 1956). Enlargement of hilar shadows may be due to lymph node involvement (Dirske 1946) or, more usually, occasioned by extensive infiltration of the great vessels (Reimann et al 1935). In Case 2, there was initially barely discernible stippling of the right lower lung zone (Fig. 3) which was later obscured by pulmonary congestion, while in Case 6 the hilar shadows were seen to be enlarged and the lung fields were affected by a coarse, partly con- fluent mottling, maximal towards the lung bases (Fig. 14). An unusual case of pleural amyloidosis was described by Lundin et al (1961) in which rounded opacities were present at both lung apices.
It is doubtful if a confident radiological inter- pretation of these diffuse lung changes is possible and a wide variety of conditions enter into the differential diagnosis. Similarly, solitary pulmonary masses may defeat all attempts at diagnosis without recourse to biopsy.
Alimentary tract.--Amyloidosis of the gastro- intestinal tract may range from minor deposition within the walls of blood vessels t O gross involve- ment when segments of intestine may become greatly thickened and rigid.
Radiological studies of the oesophagus have been rare and in those cases examined abnormal reten- tion of barium in the vallecula and pyriform sinuses and narrowing of the lower gullet, suspected as malignant, have been described (Korelitz and Spindell 1956; Heitzman et al 1962). Oesophageal varices, secondary to extensive hepatic amyloidosis, were recorded by Pocock and Dickens (1953). In Case 2 the oesophagus appeared dilated and aperistaltic and there was free gastro-oesophageal reflux (Figs. 4 and 5).
On the other hand, reports of the radiological findings in gastric amyloidosis have been much more frequent and Lubarsch (1929) was the first worker to contribute in this field. In one of his cases of mild amyloid infiltration of the gastric wall he could detect no definite radiological abnormality but in his other case he demonstrated pyloric narrowing which suggested malignant disease. In subsequent reports the most frequent finding has been pyloric stenosis or spasm (Gottron 1932; Clausen 1935; Golden 1945; de Wolf and Clarke 1950). Mucosal atrophy was noted by Gottron (1932) and Iverson and Morrison (1948) whilst the converse, mucosal hypertrophy, was described by Shipps and Brannan (1952) and Wang and Robbins (1956). Gastric ulceration, in all cases involving the lesser curvature, has been described by Lindsay and Knorp (1945), Orloff and Felder (1946), Iverson and Morrison (1948) and Cooley (1953). The only other findings of import- ance include antral lesions, and Schnider and Burka (1955) reported appearances resembling annular carcinoma of the antrum. Gastric stasis and aperistalsis were marked features in the cases recorded by Gottron (1932), de Wolf and Clarke (1950), Cooley (1953) and Intriere and Brown (1956). In Case 2 oesophageal rigidity was accom- panied by gastric mucosal atrophy and aperistalsis, barium being seen to pass rapidly through the Gesophagus and stomach into the intestine (Figs. 4 and 5).
There have been few radiological studies of the small intestine in primary amyloidosis. Three cases were carefully investigated by Golden (1954) and the features he described include segments of narrowing and dilatation, coarsening of mucosal folds, mucosal ulceration and a very slow transit time of barium which in one instance remained in the small intestine at forty-eight hours. Thicken- ing of mucosal folds and slow transit time were also referred to by Korelitz and Spindell (1956). A sub-serosal haemorrhage gave rise to a filling defect on the second and third portions of the duodenum in a case reported in the records of the Massachusetts
366 CLINICAL
General Hospital (1949). In Case 4 there was pool- ing of barium in the upper jejunum which showed loss of motility and mucosal ulceration (Figs. 10 and 11). In Case 1 there was gross distention and atony of the small intestine and while at autopsy there was evidence of peritonitis it was considered that amyloid infiltration of the muscle coats con- tributed to the severe state of dysfunction (Fig. 2).
Radiological descriptions of the colon in primary amyloidosis are rare (Heeren 1941; Andrade 1952; Wang and Robbins 1956). These authors referred to prominent mucosal folds and features which simulated those of ulcerative colitis.
Differential diagnosis.--In the event of diffuse oesophageal infiltration the resulting disorder of motility may suggest a neuro-muscular abnormality or scleroderma, while in the case of localised disease the condition may simulate neoplasm. The less severe forms of gastric amyloidosis are unlikely to declare themselves to the radiologist. In the case of antral deformity, which has been most com- monly reported, it is not considered that a differen- tiation from malignant disease can readily be made on radiological evidence alone. Indeed, in a case described by Bannick et al (1933) carcinoma of the stomach complicated gastric amyloidosis. Cooley (1953) believes that antral lesions more readily come to light as they are both more likely to cause symptoms and more easily demonstrated. The radiological descriptions of gastric ulceration do not suggest specific appearances. The assessment of mucosal hypertrophy is difficult but the occur- rence of mucosal atrophy seems substantiated and when accompanied by loss of motility suggests a diffuse infiltrative condition. In disease of the small intestine, muscular dysfunction giving rise to dilatation and stasis has also been noted in sclero- derma; when segments of thickening or ulceration are seen, regional enteritis and tuberculosis will be considered in the differential diagnosis. That definite recognisable changes occur in the colon does not as yet appear substantiated.
Bones, joints and soft tissues.~Amyloid infiltra- tion of bones and joints has rarely been recorded. Von Bonsdorff (1933) was the first to demonstrate the radiographic appearance of bone erosion due to amyloid deposits in the peri-articular tissues of the shoulder and elbow joints, and in the following year Gerber (1934) recorded both localised and diffuse infiltration of the bone marrow. In the classical description by Koletsky and Stecher (1939) a fracture of the femoral neck resulted from local soft tissue invasion, a similar case having recently been recorded by Gardner (1961). This author also described a coarse trabecular pattern in the
R A D I O L O G Y
phalanges which was said to mimic the changes found in sarcoidosis. Involvement of the vertebrae with resulting wedging and collapse has been reported by Gerber (1934) and Lindsay and Knorp (1945) and subluxation of the shoulder joint, which was infiltrated by amyloid tissue, was recorded by Golden (1954). Lengh (1937) noted radio-opaque amyloid deposits around the wrists and knees.
These rare manifestations of amyloidosis should be borne in mind when c 9nfronted with unusual bone erosion associated with soft tissue masses around joints.
Urinary tract.--Whilst amyloid infiltration of the kidney is a comparatively common feature of amyloidosis, its radiological assessment is of little value except in the exclusion of associated disease. Localised amyloidosis of the right ureter resulting in hydronephrosis was described by Higbee and Millett (1956) and a circumscribed amyloid tumour of the urinary bladder, which was the subject of radiological investigation and found to measure about the size of a pigeon's egg, was reported by Hartz and Santander (1956).
Liver, spleen, lymph nodes.--Parenchymatous and vascular involvement is common in both liver and spleen and these organs may be considerably enlarged. Local or diffuse enlargement of lymph nodes is unusual (Rukavina et a! 1956), cervical lymph node involvement being the most common. The enlargement may reach up to 5 cm. in diameter and may occur independently of simultaneous disease in the drainage area. Radiographic examina- tion may play some part in confirming hepatic and splenic enlargement and in demonstrating medi- astinal lymphadenopathy as in Case 3 (Fig. 7).
Endocrine glands.--Blood vessel and parenchy- matous deposits of amyloid are not uncommon in the endocrine glands but resulting functional disturbance is rare. A case of amyloid goitre was described by Walker (1942). Amyloid deposits in suprarenal glands have been implicated in a case of Addison's disease (O'Donnell 1950) and diabetes mellitus has been attributed to the condition in a report by Pocock and Dickens (1953). In this latter case abdominal radiography showed extensive pancreatic calcification; the pancreas was normal in size but was grossly calcified and most of the pancreatic tissue was found to be replaced by amyloid.
SUMMARY
A review of the radiological features of primary amyloidosis is given. This study has been illustrated by the clinical and radiological findings in six new cases of the disease. In one patient the condition
PRIMARY AMYLOIDOSIS 367
was a p p a r e n t l y local i sed , whi ls t t he o t h e r five were examples o f sys temic amylo idos i s . A l t h o u g h the d iagnos is is usua l ly es tab l i shed by c l in ico- p a t h o l o g i c a l i nves t i ga t i on it is s h o w n tha t r ad io - log ica l e x a m i n a t i o n m a y offer ass is tance in the e luc ida t ion o f the cond i t i on .
Aeknowledgements.--I am indebted to my colleagues at the Royal Lancaster Infirmary for referring Cases 1, 2, 3 and 5 and for their interest in this paper. Permission to publish Case 4 was kindly given by Dr R. T. Gaunt of the Chesterfield Royal Hospital and I wish to thank Dr L. L. Ralph for his radiological reports. I also wish to thank Dr E. R. Smith, Mr Gordon D. Jack and Dr Kathleen V. Lodge of Stepping Hill Hospital, Stockport for freely making available the clinical findings on Case 6. My grateful thanks are also due to Dr A. G. Rickards of the Royal Lancaster Infirmary for pathological reports, to Mr E. Forster for photographic work and to Mrs I. Fitzpatrick for clerical assistance.
REFERENCES
ANDRADE, C. (1952). Brain. 75, 408. BANNICK, F. G., BERKMAN, J. M. & BEAVER, D. C. (1933).
Arch. intern. Med. 51, 978. BENSON, R. & SMITH, J. F. (1956). Brit. Heart J. 18, 529. Case Records of the Mass. Gen. Hosp. New Engl. J. Med.
(1949). 240, 572. CLAUSEN, A. (1935). Fortsch. Ri~ntgenstr. 51, 528. COOLEY, R. N. (1953). Amer. J. Roentgenol. 70, 428. COUTER, W. T. & REICHERT, R. E., Jun. (1950). Circulation,
2, 441. DE WOLf, H. & CLARKE, B. E. (1950). Amer. J. din. Path.
20, 165. DIRSKE, P. R, (1946). Amer. J. Roentgenol. 56, 577. EISEN, H. N. (1946). Amer. J. Med. 1, 144. FENTEM, P. H., TURNBERG, L. A. & WORMSLEY, K. G. (1962).
Brit. reed. or. 1, 364. FERVdS, H. W. (1936). Amer. J. Path. 12, 701. FINDLEY, J. W., Jun. & ADAMS, W. (1948). Arch. intern.
' ' 7
Med. 81, 342. GARDNER, H. (1961). Brit. J. Radiol. 34, 778. GERBER, I. E. (1934). Arch. Path. 17, 620. GOLDEN, A. (1945). Arch. intern. Med. 75, 413. GOLDEN, R. (1954). Amer. or. Roentgenol. 72, 401. GOTTRON, H. (1932). Arch. Derm. Syph. (Berl.), 166, 584. HARTZ, P. H. & SANTANDER, E. (1956). J. Urol. 75, 687. I-I~E~N, J. G. (1941). Ri~ntgenpraxis. 13, 176.
HEITZMAN, E. J., HEITZMAN, G. C. & ELLIOTT, C. F. (1962). Arch. intern. Med. 109, 595.
HIGBEE, D. R. & MILLETT, W. D. (1956). d. Uro[. 75, 424. HIGGINS, W. H. & HIGGINS, W. H., Jun. (1950). Amer. J.
med. Sci. 220, 610. INTRIERE, A. D. & BROWN, C. H. (1956). Gastroenterology,
30, 833. IVERSON, L. & MORRISON, A. B. (1948). Arch. Path. 45, 1. KAMBERG, S., LOITMAN, B. S. & SUMNER, H. (1962). New
EngL J. Med. 266, 587. KOLETSKY, S. & STECHER, R. M. (1939). Arch. Path. 27, 267. KORELITZ, B. I. & SPINDELL, L. N. (1956). J. M t Sinai Hosp.
23, 683. LENGH, F. (1937). Zbl. allg. Path. path. Anat. 69, 1. LINDSA¥, S. (1946). Amer. HeartJ. 32, 419. LINDSAY, S. & KNORP, W. F. (1945). Arch. Path. (Chicago),
39, 315. LOBARSCH, O. (1929). Virchows Arch. path. Anat. 271, 867. LUNDIN, P., SIMONSSON, B. & W1NBERG, T. (1961). Acta
radiol. (Stockh.), 55, 139. MATHEWS, W. H. (1954). Amer. J. reed. Sci. 228, 317. O'DONNELL, W. M. (1950). Arch. intern. Med. 86, 266. ORLOEF, J. & FELDER, L. (1946). Amer. J. reed. Sci. 212, 275. POCOCK, D. S. & DICKENS, J. (1953). New Engl. d. Med.
248, 359. PROWSE, C. BARRINGTON (1958). Thorax, 13, 308. REIMANN, H. A., KOUCKY, R. F. & EKLUND, C. M. (1935).
Amer. J. Path. 11, 977. RUKAVINA, J. G., BLOCK, W. D., JACKSON, C. E., FALLS,
H. F., CAREY, J. H. & CURTIS, A. C. (1956). Medicine (Balt.), 35, 239.
SAPPINGTON, S. W., DAVIS, J. H. & HORNEFF, J. A. (1942). J. Lab. clin. Med. 27, 882.
SCHM1DT, H. W., McDONALD, J. R. & CLAGETT, O. T. (1953). Ann. Otol (St Louis), 62, 880.
SCHNIDER, B. I. & BURKA, P. (1955). Gastroenterology, 28, 424.
SHIPVS, F. C. & BRANNAN, D. D. (1952). Amer. J. Roentgenol. 68, 204.
STARK, D. B. & NEW, G. B. (1949). Ann. Otol. (St Louis), 58, 117.
STARK, D. B. & NEW, G. B. (1950). Med. Clin. N. Amer. 34, 1145.
STRICH, S. J. & WADE, G. (1953). Lancet, 2, 70. VON BONSDORFF, B. (1933). Finska Li~'k Siillsk. Handl. 75,
447. WALKER, G. A. (1942). Surg. Gynec. Obstet. 75, 374. WANG, C. C. & ROBmNS, L. L. (1956). Radiology, 66, 489. WHITWELL, F. (1953). Thorax, 8, 309. WILD, C. (1886). Beitr. path. Anat. 1, 175. WILKS, S. (1856). Guy's Hosp. Rep. (Series 3), 2, 103.