32
Structural Bioinformatics (C3210) Principles of Structure-Based Design

Structural Bioinformatics(C3210)
Principles of Structure-Based Design

2
Design of Drug Candidates: An Iterative Process The design of new ligands is carried out as a step-by-step
procedure The state-of-the art design process is based in large part, on a
good understanding of the molecular recognition of protein-ligand complexes relying upon analogies to other systems and using advanced computerized molecular design programs

3
Steps in Structure-Based Drug Design The steps used in structure-based drug design for designing new
lead compounds are:• Obtaining 3D structure of protein• Active site identification• Ligand-receptor fit analysis• Design of new leads
Note: A lead (i.e. the "leading" compound) in drug discovery is a chemical compound that has pharmacological or biological activity and whose chemical structure is used as a starting point for chemical modifications in order to improve potency, selectivity, or pharmacokinetic parameters.

4
Small Changes Can Produce Huge Effects When structural changes are made that modify the energy of
interaction of a ligand with its receptor, one can observe a large change in the biological properties
For example the SB203580 molecule has a Ki of 100 nM on the protein kinase p38. However the molecule looses its activity if residue Thr-106 is mutated by Met-106. The same molecule is entirely inactive on the kinase ERK2, however if the residue Glu-105 of the protein is mutated by Thr-105, the Ki of the molecule becomes 13 nM.
Fig.: SB203580 molecule
Note: Ki (inhibition constant) is the concentration of inhibitor which would occupy 50% of the enzyme molecules if no substrate were present

5
Increasing Biological Activity Each small amount that is gained in the binding energy of a
ligand is translated into significant improvements in its biological activity
The following graph illustrates the relative values of Kd as a function of the binding energies, at 300°K

6
Beginning the Design Phase Once the phase of analysis is complete, the design phase can
start One has to identify candidate scaffolds with appropriate
substituents that can ensure enhanced interactions with selected sites of the protein
In the case of the optimization of a known series, the information is used to design new analogs

7
Eight Golden Rules in Receptor-Based Ligand Design
The important considerations for receptor-based ligand design can be summarized into the following eight rules:
1. Coordinate to key anchoring sites2. Exploit hydrophobic interactions3. Exploit hydrogen bonding capabilities4. Exploit electrostatic interactions5. Favor bioactive form & avoid energy strain6. Optimize vdW Contacts and avoid bumps7. Structural water molecules and solvation8. Consider entropic effect
8
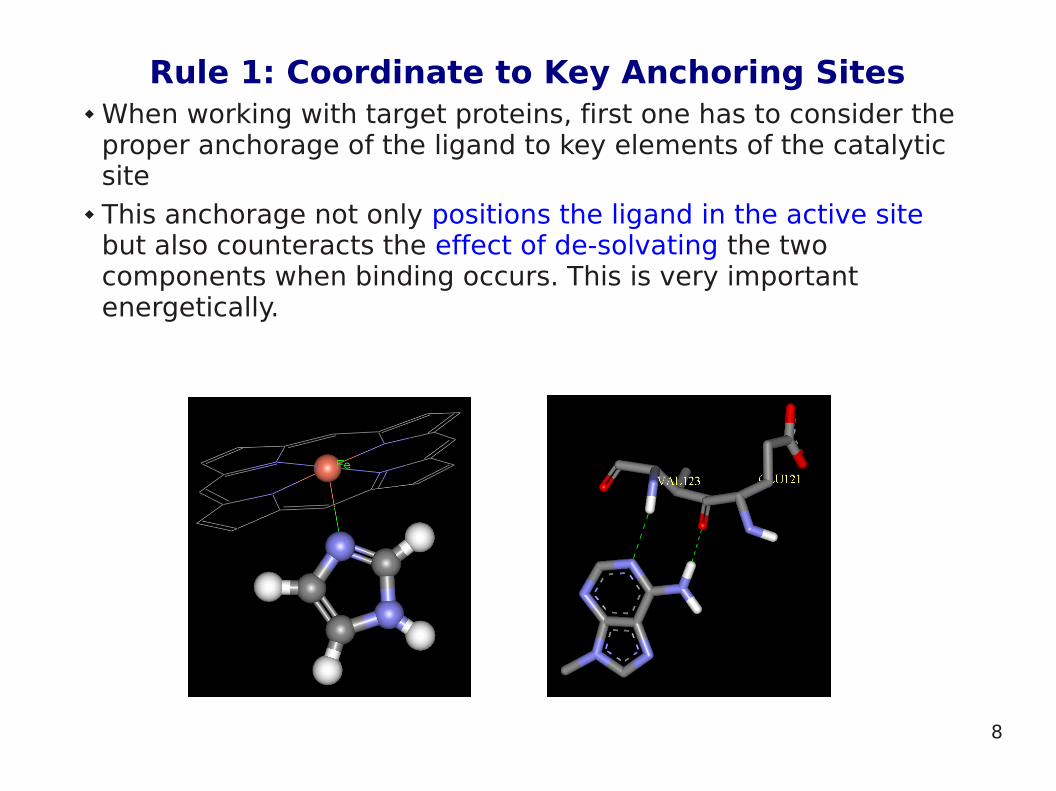
Rule 1: Coordinate to Key Anchoring Sites When working with target proteins, first one has to consider the
proper anchorage of the ligand to key elements of the catalytic site
This anchorage not only positions the ligand in the active site but also counteracts the effect of de-solvating the two components when binding occurs. This is very important energetically.

9
Rule 2: Exploit Hydrophobic Interactions With hydrophobic pockets, placing a hydrophobic surface of the
ligand in hydrophobic sites of the target protein provides an important driving force in complex formation because it reduces non-polar surface areas exposed to water
Although individually small, the total contribution of hydrophobic forces to drug-receptor interactions is substantial
Empirical data suggests that the free energy contribution due to hydrophobic forces is approximately 2.9 kJ/mol per methylene group and 8.4 kJ/mol for a benzene ring
Unlike hydrogen bonds, the hydrophobic interactions are not directional

10
Rule 3: Exploit Hydrogen Bonding Capabilities Unsatisfied hydrogen bond donors and acceptors are rarely seen
in proteins and protein-ligand complexes because this would be highly energetically unfavorable
A carbonyl oxygen is optimally satisfied when it accepts two different hydrogen bonds with C=O --- H angles close to 120°. However hydrogen bonds to carbonyl oxygen atoms with a C=O --- H angle close to 180° form the basis for β-sheet formation and are quite favorable. The average N-H --- O angle is about 155° (with 90% lying between 140° and 180°).
Almost all protein groups are capable of forming hydrogen bonds like this. Where groups are not explicitly hydrogen bonded, they are probably solvated.

11
Rule 4: Exploit Electrostatic Interactions The optimization of ligand-protein electrostatics can be achieved
by placing a positive charge in close vicinity to an enzyme negative charge

12
Rule 5: Favor Bioactive Form & Avoid Energy Strain
Conformational energy calculations are performed on each design idea in order to determine the internal penalty required for the new ligand to attain its bioactive binding conformation inside the protein. The internal energy that is required for the small molecule to reach its binding conformation is energy lost in binding.
Restricting the conformation space of an inhibitor can be beneficial to binding when the conformation is biased towards the bioactive conformer.

13
Rule 6: Optimize VDW Contacts and Avoid Bumps
Attractive van der Waals interactions occur over a short distance range and attraction decreases as 1/r6. As a result, optimization of attractive van der Waals interactions occurs as the shape of the protein binding site and the shape of the ligand match well.
Calculations of steric fit are difficult because of possible flexing motions of the protein backbone and especially the residue side-chains
14
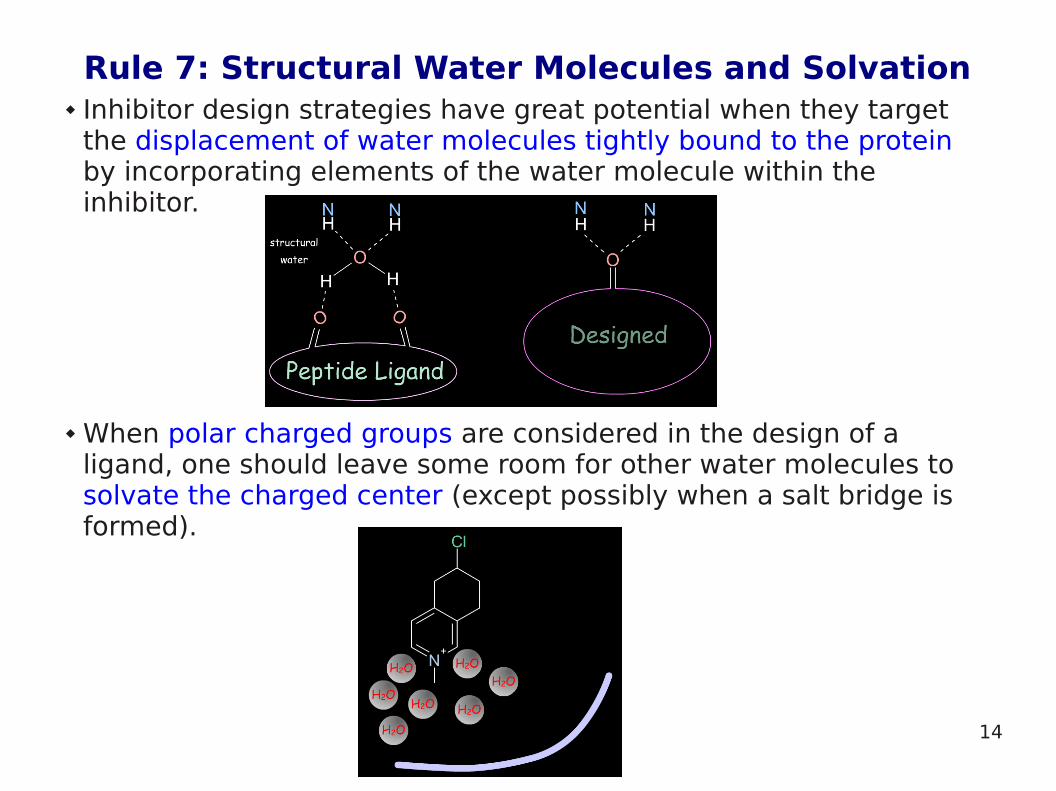
Rule 7: Structural Water Molecules and Solvation Inhibitor design strategies have great potential when they target
the displacement of water molecules tightly bound to the protein by incorporating elements of the water molecule within the inhibitor.
When polar charged groups are considered in the design of a ligand, one should leave some room for other water molecules to solvate the charged center (except possibly when a salt bridge is formed).

15
Rule 8: Consider Entropic Effect A flexible molecule has a better chance of finding an optimal fit
into a receptor, but this is achieved at the cost of large conformational entropy
Sufficient conformational rigidity is essential to ensure that the loss of entropy upon ligand binding is acceptable
A rigid molecule has little conformational entropy but is unlikely to fit optimally into the receptor
An analysis of the contributions of various functional groups to protein-ligand binding demonstrates that each freely rotating bond in a ligand reduces binding free energy by about 2.9 kJ/mol
Making a flexible molecule more rigid will lead to enhanced activity if the right conformation is maintained

16
Example of Ligand Rigidification Making a ligand more rigid will lead to enhanced activity if the
right conformation is maintained For example freezing of two rotational degrees of freedom in
flexible antiulcer agents in a rigid analog resulted in a 150-fold enhancement in activity

17
The Four Design Methods There are four basic methods in receptor-based drug design:
• Analog design• Database searching• Computerized de novo design• Manual design
18
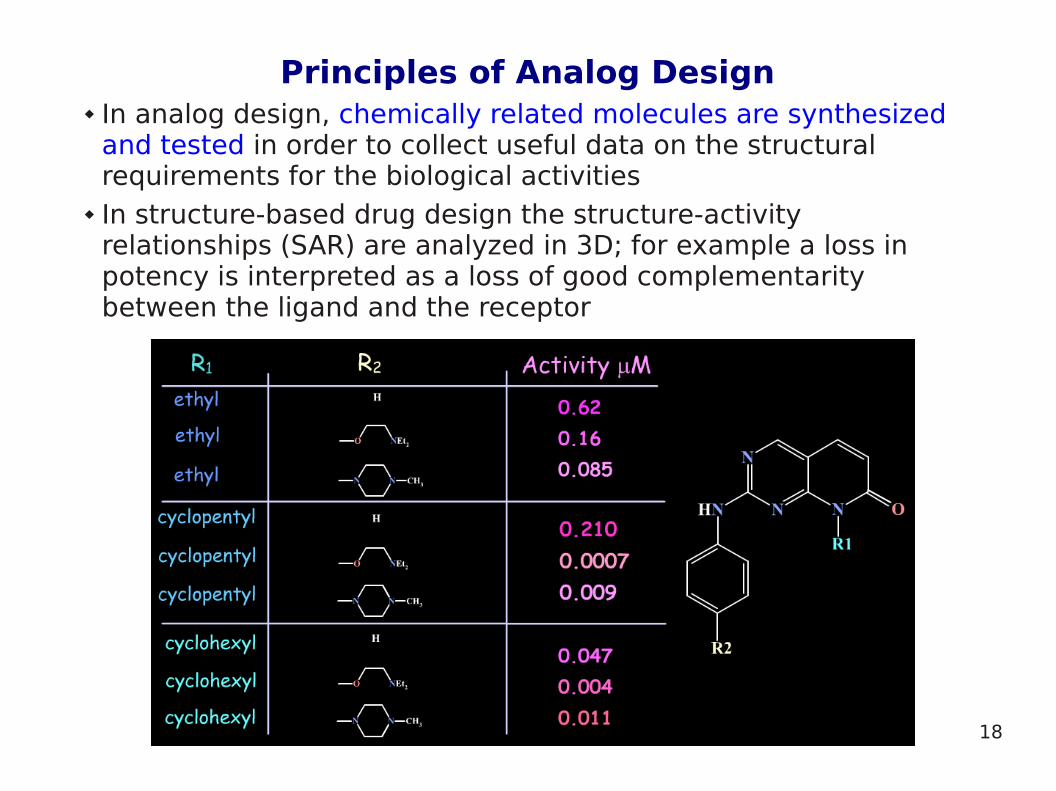
Principles of Analog Design In analog design, chemically related molecules are synthesized
and tested in order to collect useful data on the structural requirements for the biological activities
In structure-based drug design the structure-activity relationships (SAR) are analyzed in 3D; for example a loss in potency is interpreted as a loss of good complementarity between the ligand and the receptor

19
Example of Analog Design The analysis of the crystal structure of the complex between
trimethoprim and dihydrofolate reductase (DHFR) reveals the existence of possible additional binding sites. In particular, a basic arginine residue could be considered for increasing the binding.
The analysis indicated that a carboxyl function introduced in the chemical structure of trimethoprim could form an ionic linkage with the arginine residue. The carboxyl analog proved to be significantly more potent than the reference trimethoprim and its X-ray structure confirmed the validity of this finding.

20
Database Searching It is sometimes a challenge to design de novo leads on the basis
of protein structure alone. The database approach provides an easy way to arrive at leads. It consists of a computerized search of a database of synthesized or naturally occurring compounds and testing them in silico (e.g. using docking methods).
The result of this treatment is a list of compounds ranked by a score based on the overall match with the receptor

21
Advantages and Problems of Database Searching An advantage of database searching method is that it generally
does not require special synthetic efforts (the molecule is available) but on the other hand it is highly dependent on the content of the database
The problem of database searching is that due to the conformational complexity of the current molecules, only a few of their conformations are actually stored in the databank. Unless complicated conformational generators are incorporated in the calculations, database searching cannot be exhaustive and therefore many molecules may be overlooked in the process.
Throughout the world there are thousands of chemical suppliers offering more than 3 million of organic molecules. The advantage of using commercially available molecules in database searching is that they offer a great diversity and can be purchased and tested rapidly.
The main purpose of a 3D-database search is not to find compounds with ultrapotent activities. A realistic goal of a 3D-database search should be the discovery of possibly active molecules, which can subsequently be used as the basis for further synthetic modifications.

22
De-Novo Design The purpose of automated construction approaches is to
discover a new chemical framework that fits to the active site of the target receptor or enzyme
Some methods are based on an existing moiety and additional fragments are appended by a step-by-step build up procedure
Other methods consist of assembling novel molecules from pieces that are positioned optimally in favorable regions of the active site

23
Manual Design It is a major challenge to design de novo leads on the basis of a
protein structure alone The manual design is greatly facilitated by starting from an
existing "solution" which consists of the visualization of a complex
This procedure is currently undertaken either by the medicinal chemist or the molecular modeler

24
Example of Successful Structure-Based Design The use of the crystallographic structure of the HIV-1 protease in
drug design represents one of the more impressive success stories in the structure-based drug design field. Structure-based design studies has resulted in the identification of distinct classes of inhibitors and several successful drug candidates have emerged from these studies and are used in the control of AIDS.
The HIV-1 protease plays a crucial part in the life cycle of the HIV virus. Inhibitor drugs block the action of the protease and the virus perishes because it is unable to mature into its infectious form.
The HIV-1 protease is a small dimer enzyme comprising two identical folded 99 amino-acid chains A and B

25
HIV-1 Protease Structure The assembly of the two chains of the HIV-1 protease forms a
hydrophobic cavity at the center of the catalytic site of the enzyme
An interesting feature of the structure is the "flap region" in each chain that arches over the active site. Relative to the free enzyme, the complex exhibits backbone movements as the flap regions close jaw-like over the substrate.
26
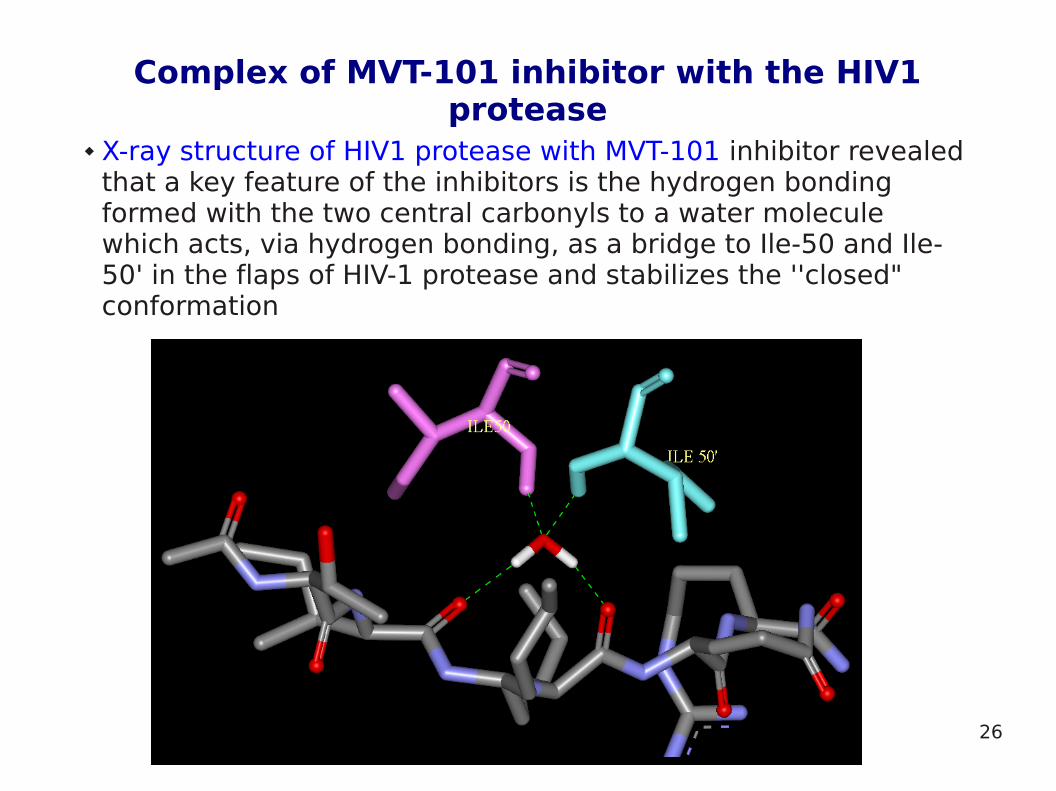
Complex of MVT-101 inhibitor with the HIV1 protease
X-ray structure of HIV1 protease with MVT-101 inhibitor revealed that a key feature of the inhibitors is the hydrogen bonding formed with the two central carbonyls to a water molecule which acts, via hydrogen bonding, as a bridge to Ile-50 and Ile-50' in the flaps of HIV-1 protease and stabilizes the ''closed" conformation

27
Complex of MVT-101 inhibitor with the HIV1 protease
The inhibitor have also a very extended hydrogen bond network with other amino acids in the catalytic site. For example MVT-101 inhibitor forms hydrogen bonds with Ile-50, Ile-50', Gly-48, Gly-48', Asp-29, Asp-29', Gly-27 and Gly-27'
28
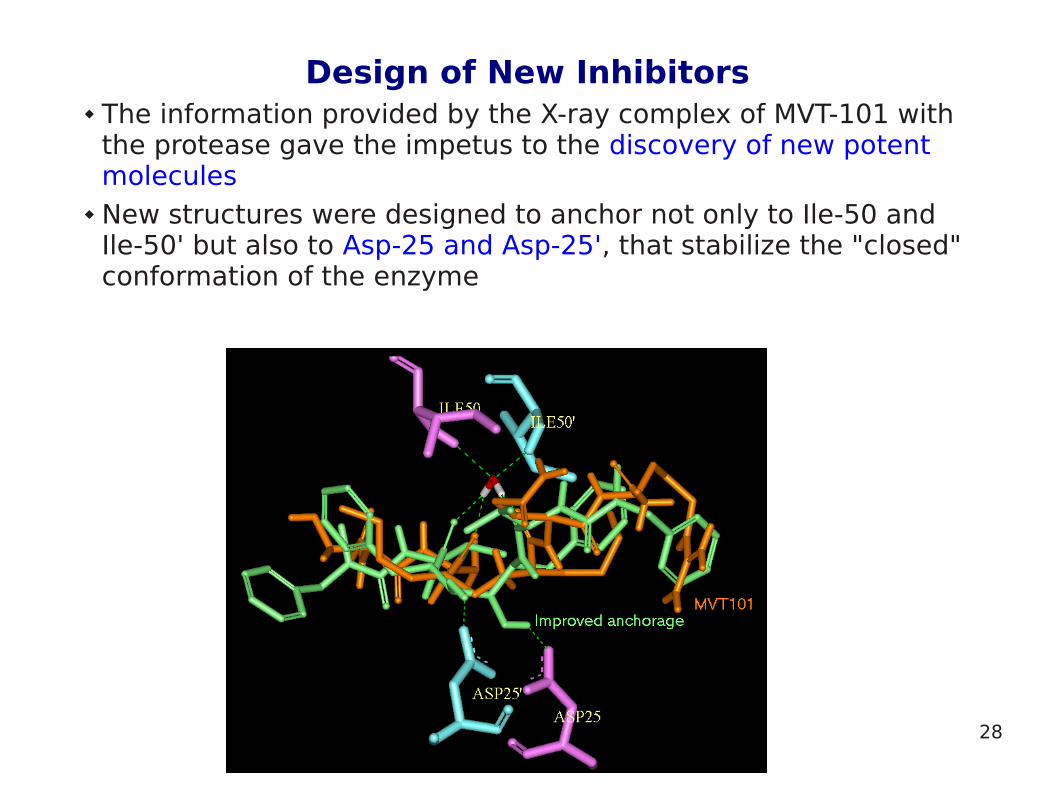
Design of New Inhibitors The information provided by the X-ray complex of MVT-101 with
the protease gave the impetus to the discovery of new potent molecules
New structures were designed to anchor not only to Ile-50 and Ile-50' but also to Asp-25 and Asp-25', that stabilize the "closed" conformation of the enzyme

29
Optimization to Fit the hydrophobic pockets Additional efforts were also made to better fill the hydrophobic
pockets (green = MVT-101, red = designed)

30
Example of Optimized Structure A-77003 A typical example of a compound that bears these changes is A-
77003, which is about 5000 times more potent than MVT-101 Analysis of the complex shows that A-77003 anchors well on the
two aspartic acids Asp-25 and Asp-25' A-77003 also binds to Ile-50 and Ile-50' through a water
molecule
31
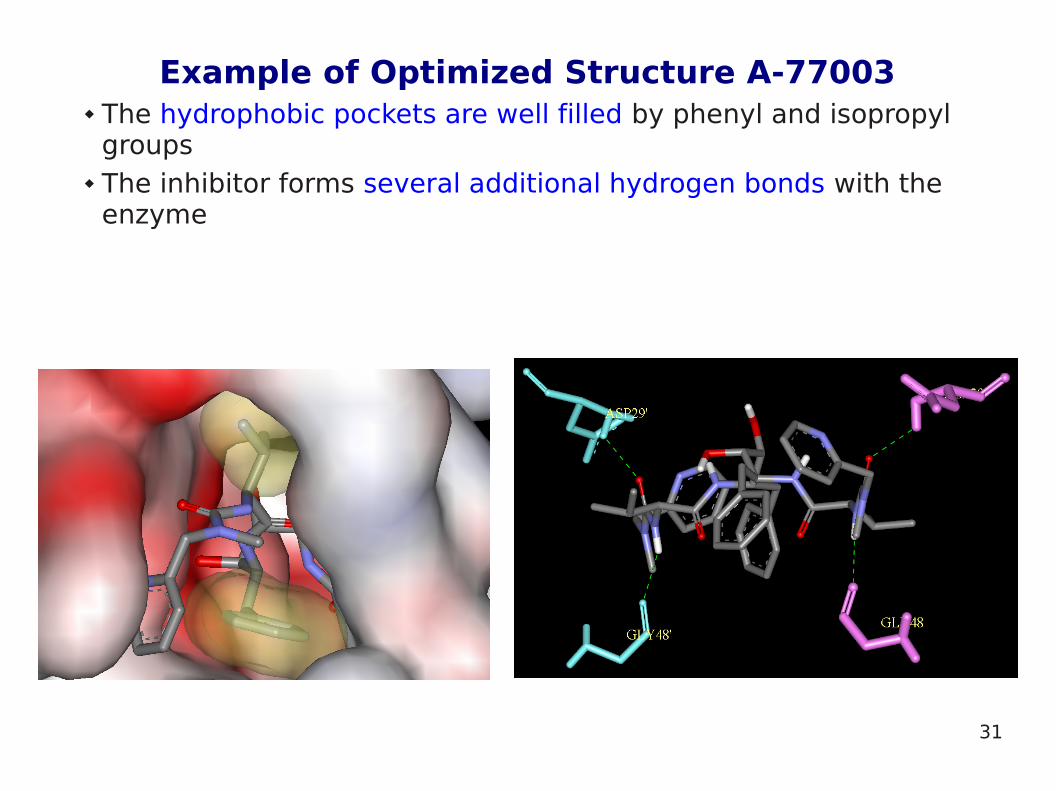
Example of Optimized Structure A-77003 The hydrophobic pockets are well filled by phenyl and isopropyl
groups The inhibitor forms several additional hydrogen bonds with the
enzyme

32
Conclusion The molecular recognition of protein-ligand complexes is highly
complex and the utilization of an iterative design process has greatly advanced the use of protein structure for the design of drug candidates
The state-of-the-art design process relies, in a large part, on a qualitative understanding of the molecular recognition of protein-ligand complexes based upon analogies to other systems and the use of several methods in parallel

















