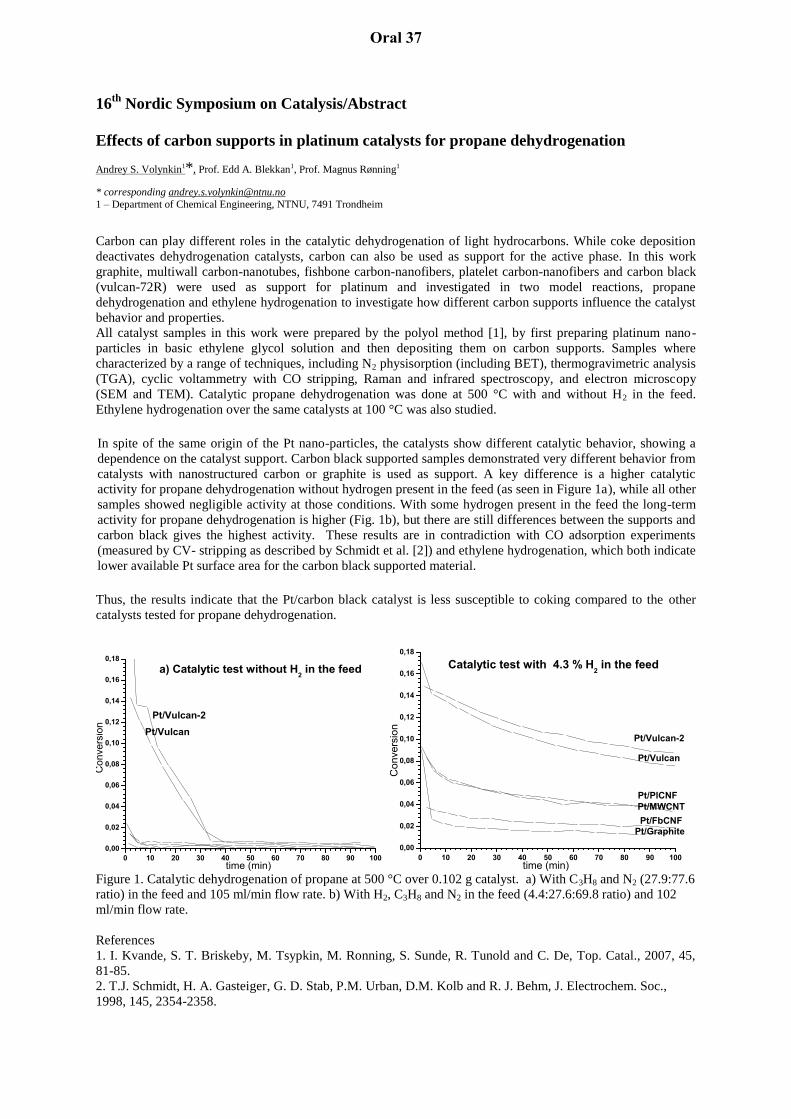
Pt-based Diesel Oxidation Catalysts: Oxidation state of Platinum and the origin of CO oscillations in real Pt/Al 2 O 3 catalysts Andreas Gänzler 1 , Alexey Boubnov 1 , Maria Casapu 1 , Henning Lichtenberg 1 , Jan-Dierk Grunwaldt 1 * 1 Institute for Chemical Technology and Polymer Chemistry (ITCP) and Institute of Catalysis Research and Technology (IKFT), KIT, Kaiserstr. 12, 76131 Karlsruhe, Germany, [email protected]Diesel engines are more efficient than conventional gasoline engines, however, diesel exhaust gas aftertreatment represents a significantly more challenging task: The abatement system needs to oxidize CO and unburned hydrocarbons, but also to reduce NOx in a net oxidizing atmosphere. Therefore multi-component systems are used with Pt or Pd based diesel oxida- tion catalysts (DOC) as first stage with CO oxidation as major task. However, even though studied for decades, the noble metal catalyzed reaction is not yet fully understood, especially with respect to more applied conditions. In this regard, the occurrence of CO conversion os- cillations in such catalysts is controversially debated (cf. [1-3] and refs. therein). Here, we demonstrate for the first time how a combination of spatially resolved catalytic reactivity studies and structural analysis can provide new insight into Pt-based DOC catalysts during CO-oxidation and its oscillating conditions by extending our previous studies [3]. Our approach is based on a combination of spatially resolved X-ray absorption spectroscopy at the Pt L 3 -edge with IR-thermography and net catalytic performance measurements at the end of the catalyst bed. Results obtained from IR-thermography at one specific temperature are shown in Figure 1. Along with oscillations in temperature (1.5-2 K) also changes in the oxidation state of Pt were observed at the same location in the catalyst bed. Fig. 1: Thermography images during oscillatory CO oxidation: (a) temperature hot-spots visualized by difference imaging, (b) temperature in the middle of the catalyst bed. By this novel approach - correlating processes on a macroscopic level (temperature, catalytic performance) with structural information on the microscopic level (oxidation state, atomic structure) obtained from spatially resolved measurements - we can unambiguously conclude that on Pt-based catalysts the reduced surface is more active compared to oxidized platinum, which is in contrast to some recent studies on Pt clusters on model oxide surfaces. During a short period of high catalytic activity the Pt particles obviously deactivate, which can be di- rectly correlated to a transition from reduced to oxidized Pt. Subsequently the catalyst slowly regenerates (slow increase in catalytic activity), which can be linked to the regeneration of reduced Pt sites. The observed oscillatory behavior originates from a strong tendency of small particles to oxidize, whereas larger platinum particles tend to remain reduced and therefore do not show any or much less pronounced oscillatory behavior. These large particles also ex- hibit higher turnover frequencies for CO conversion, in agreement with earlier studies [4]. [1] R. Imbihl and G. Ertl, Chem. Rev. 95, 697 (1995). [2] R. Jensen, T. Andersen, A. Nierhoff, T. Pedersen, O. Hansen, S. Dahl, I. Chorkendorff, Phys. Chem. Chem. Phys. 15, 2698 (2013). [3] A. Boubnov, A. Gänzler, S. Conrad, M. Casapu, and J.-D. Grunwaldt, Top. Catal. 56, 333 (2013). [4] A. Boubnov, S. Dahl, E. Johnson, A.P. Molina, S.B. Simonsen, F.M. Cano, S. Helveg, L.J. Lemus-Yegres, and J.-D. Grunwaldt, Appl. Catal. B, 126 315 (2012). Oral 1
Transcript
Pt-based Diesel Oxidation Catalysts: Oxidation state of Platinum and the origin of CO
oscillations in real Pt/Al2O3 catalysts
Andreas Gänzler1, Alexey Boubnov
1, Maria Casapu
1, Henning Lichtenberg
1, Jan-Dierk
Grunwaldt1*
1Institute for Chemical Technology and Polymer Chemistry (ITCP) and Institute of Catalysis Research and
Diesel engines are more efficient than conventional gasoline engines, however, diesel exhaust
gas aftertreatment represents a significantly more challenging task: The abatement system
needs to oxidize CO and unburned hydrocarbons, but also to reduce NOx in a net oxidizing
atmosphere. Therefore multi-component systems are used with Pt or Pd based diesel oxida-
tion catalysts (DOC) as first stage with CO oxidation as major task. However, even though
studied for decades, the noble metal catalyzed reaction is not yet fully understood, especially
with respect to more applied conditions. In this regard, the occurrence of CO conversion os-
cillations in such catalysts is controversially debated (cf. [1-3] and refs. therein). Here, we
demonstrate for the first time how a combination of spatially resolved catalytic reactivity
studies and structural analysis can provide new insight into Pt-based DOC catalysts during
CO-oxidation and its oscillating conditions by extending our previous studies [3].
Our approach is based on a combination of spatially resolved X-ray absorption spectroscopy
at the Pt L3-edge with IR-thermography and net catalytic performance measurements at the
end of the catalyst bed. Results obtained from IR-thermography at one specific temperature
are shown in Figure 1. Along with oscillations in temperature (1.5-2 K) also changes in the
oxidation state of Pt were observed at the same location in the catalyst bed.
Fig. 1: Thermography images during oscillatory CO oxidation: (a) temperature hot-spots visualized by difference imaging, (b) temperature in the middle of the catalyst bed.
By this novel approach - correlating processes on a macroscopic level (temperature, catalytic
performance) with structural information on the microscopic level (oxidation state, atomic
structure) obtained from spatially resolved measurements - we can unambiguously conclude
that on Pt-based catalysts the reduced surface is more active compared to oxidized platinum,
which is in contrast to some recent studies on Pt clusters on model oxide surfaces. During a
short period of high catalytic activity the Pt particles obviously deactivate, which can be di-
rectly correlated to a transition from reduced to oxidized Pt. Subsequently the catalyst slowly
regenerates (slow increase in catalytic activity), which can be linked to the regeneration of
reduced Pt sites. The observed oscillatory behavior originates from a strong tendency of small
particles to oxidize, whereas larger platinum particles tend to remain reduced and therefore
do not show any or much less pronounced oscillatory behavior. These large particles also ex-
hibit higher turnover frequencies for CO conversion, in agreement with earlier studies [4].
[1] R. Imbihl and G. Ertl, Chem. Rev. 95, 697 (1995). [2] R. Jensen, T. Andersen, A. Nierhoff, T. Pedersen, O. Hansen, S. Dahl, I. Chorkendorff, Phys. Chem. Chem. Phys. 15, 2698 (2013).
[3] A. Boubnov, A. Gänzler, S. Conrad, M. Casapu, and J.-D. Grunwaldt, Top. Catal. 56, 333 (2013).
[4] A. Boubnov, S. Dahl, E. Johnson, A.P. Molina, S.B. Simonsen, F.M. Cano, S. Helveg, L.J. Lemus-Yegres, and J.-D. Grunwaldt, Appl. Catal. B, 126 315 (2012).
Oral 1
0 10 20 30 40 50 60 70 80
0
20
40
60
80
100
HCOOH
conversion
CO2 selectivity
Blue: Fresh
Red: Aged 10 h
Yie
ld/C
on
vers
ion
/Sele
cti
vit
y, %
Time (h)
CO selectivity
NH3 yield
16th
Nordic Symposium on Catalysis
Ammonium formate decomposition over Au/TiO2: A highly stable catalyst with unique
selectivity against NH3 oxidation
Manasa Sridhar1, Jeroen Anton van Bokhoven
1,2, Oliver Kröcher
1,3*
1 – Paul Scherrer Institut, 5232 Villigen, Switzerland
2 – ETH Zurich, Institute for Chemical and Bioengineering, 8093 Zurich, Switzerland
3 – École Polytechnique Fédérale de Lausanne (EPFL), Institute of Chemical Sciences and Engineering, 1015
Lean NOx reduction over Ag/Al2O3 has been widely studied for several types of reducing agents [1]. However,
there are only a limited number of detailed reports on methanol as reductant for NOx [2]. Methanol is today one
of the most promising renewable fuels for transports both on land and at the sea. In addition, methanol is a small
molecule and thus methanol-SCR provides a model system for investigations of HC-SCR reactions in general.
The objective of the present study is to investigate the influence of hydrogen and silver loading on the activity
and selectivity for lean NOx reduction with methanol over Ag-Al2O3 catalysts. The aim is to contribute to a
fundamental understanding of methanol-SCR reactions over Ag-Al2O3.
Ag-Al2O3 samples (1-4 wt% Ag) were prepared according to a previously described sol-gel method including
freeze-drying [3]. The catalytic performance of coated monolith samples was studied in a continuous flow
reactor where the outlet gas composition was analyzed by gas-phase FTIR spectroscopy [4]. The silver species
were characterized by temperature programmed reduction with hydrogen (H2-TPR) and UV-Vis spectroscopy.
Figure 1a shows a step-response experiment where H2 is introduced and removed from the feed gas
composition. The results show that H2 is formed during methanol-SCR conditions. In accordance with Johnson
et al. [5], this availability of H2 is suggested to contribute to the high low-temperature activity often observed when using alcohols as reducing agents. Always when H2 is added to the feed in Figure 1a, more oxidized
reaction products are formed. Interestingly, only diminutive amounts of HCHO and CO are observed during
methanol oxidation, compared to during SCR conditions. Consequently their formation must be highly
influenced by the presence of NO, and maybe part of the NOx reduction reactions. Moreover, the addition of H2
results in a temperature increase in the catalyst, likely to a large extent owing to oxidation of H2 to H2O.
However, it is concluded from the methanol-SCR experiments in Figure 1b that the temperature increase alone
cannot cause the higher NOx reduction observed in the presence of H2. Comparison of the H2 consumption
during H2-TPR with the N2 formation during the experiments in Figure 1b, shows that the silver species reduced
by H2 cannot be directly associated with the N2 formation. Furthermore, the UV-Vis analysis indicates a higher
proportion of large metallic particles in the high-loaded samples, which can explain the lower N2 formation at
high temperatures, owing to a more extensive combustion of the reducing agent. In the present study we show
that H2 actually is formed during methanol-SCR conditions. This availability of hydrogen is suggested to result in a similar effect as H2 addition to HC-SCR, when not using an oxygenated reducing agent.
Figure 1. a) Step-response experiment with (3 wt% Ag) Ag-Al2O3 at 260 °C. b) Methanol-SCR during cooling
ramp, with and without H2, over Ag-Al2O3 (1-4% Ag). Inlet gas concentration (when used): 10 % O2, 1,700 ppm
methanol, 500 ppm NO, 1000 ppm H2, Ar bal.
[1] R. Burch, Catal. Rev.-Sci. Eng. 46 (2004) 271.
[2] M. Männikkö, M. Skoglundh, H. Härelind, Top. Catal. 56 (2013) 145.
[3] H. Kannisto, H.H. Ingelsten, M. Skoglundh, J. Mol. Catal. A: Chem. 302 (2009) 86.
[4] M. Männikkö, M. Skoglundh, H.H. Ingelsten, Appl. Catal. B 119-120 (2012) 256.
Oral 3
Combining HC-SCR over Ag/Al2O3 and hydrogen generation over Rh/CeO2-ZrO2 using
bio-fuels: an integrated system approach for real applications
Fredrik Gunnarsson1*, Moa Z. Granlund
2*, Mattias Englund
1, Jazaer Dawody
3, Lars J.
Pettersson2, Hanna Härelind1
1 Competence Centre for Catalysis, Dept. of Chemical and Biological Engineering, Chalmers University of
Technology, SE-412 96 Göteborg, Sweden 2
Chemical Engineering and Technology, Chemical Technology, KTH Royal Institute of Technology, SE-100 44,
Stockholm, Sweden 3
Volvo Group Trucks Technology, Advanced Technology and Research, SE-412 96 Göteborg, Sweden
We report on a high NOx reduction activity over Ag/Al2O3 catalysts, using hydrogen from a reformer with a
Rh/CeO2-ZrO2 catalyst. The focus of the study is to evaluate the performance of hydrocarbon selective catalytic
reduction (HC-SCR) catalyst in real conditions. Initially, the catalytic materials for fuel reformer (Rh/CeO2-ZrO2 [1])
and the HC-SCR (Ag/Al2O3 [2]) were evaluated in separate bench scale reactor setups. The two bench scale reactor
setups were subsequently combined into one reactor setup with the aim to evaluate the reformate’s influence on the
HC-SCR process in a controlled environment. The reducing agent and reforming fuel used in the combined bench
scale reactor setup was commercial biodiesel (NexBTL). The final phase of the study was to evaluate the
performance of the HC-SCR catalyst in real exhaust conditions. A pilot-scale version of the 4 wt. % Ag/Al2O3
catalyst doped with 100 ppm Pt was used together with a single cylinder genset engine (Yanmar, L100). The system
can evaluate the performance in real exhausts together with real reformate from the separate fuel reformer.
The results from the combined bench-scale reactor setup show that a significant improvement in the NOx reduction
can be achieved when using reformate hydrogen (Figure 1). The improvement is highest at low temperatures and
increase with increasing hydrogen addition. The effect decreases as the temperature is increased, with negligible
effect above 350 °C. The largest improvement is seen over the 4 wt. % Ag/Al2O3 catalyst doped with 100 ppm Pt.
The trends from the pilot-scale reactor can be seen to mimic those from the bench-scale reactor. The NOx reduction
data from the real exhaust experiments using reformate hydrogen mimic those when using bottled hydrogen. The
study gives an excellent link between evaluation of the catalytic materials, controlled bench-scale experiments and
applied engine experiments, proving a viable concept for onboard hydrogen production.
[1] M.Z. Granlund, K. Jansson, M. Nilsson, J. Dawody, L.J. Pettersson, Appl. Catal. B, Environ. (2014),
http://dx.doi.org/10.1016/j.apcatb.2014.02.043
[2] F Gunnarsson, H Kannisto, M Skoglundh, H Härelind, Appl. Catal. B: Environ. 152–153 (2014) 218–225
Figure 1. NOx reduction results from bench
scale reactor experiments 4 wt. % with 100
ppm Pt Ag/Al2O3 samples. Hydrogen
concentrations are denoted as 0 ppm (■),
1000 ppm (●), 1500 ppm (▲) and 3250 ppm
(▼). Inlet gas composition: 200 ppm NO, 10
% O2, 5 % H2O, N2,bal and NExBTL as
reducing agent with a C/N molar ratio of 6
and GHSV = 33,200 h-1.
Oral 4
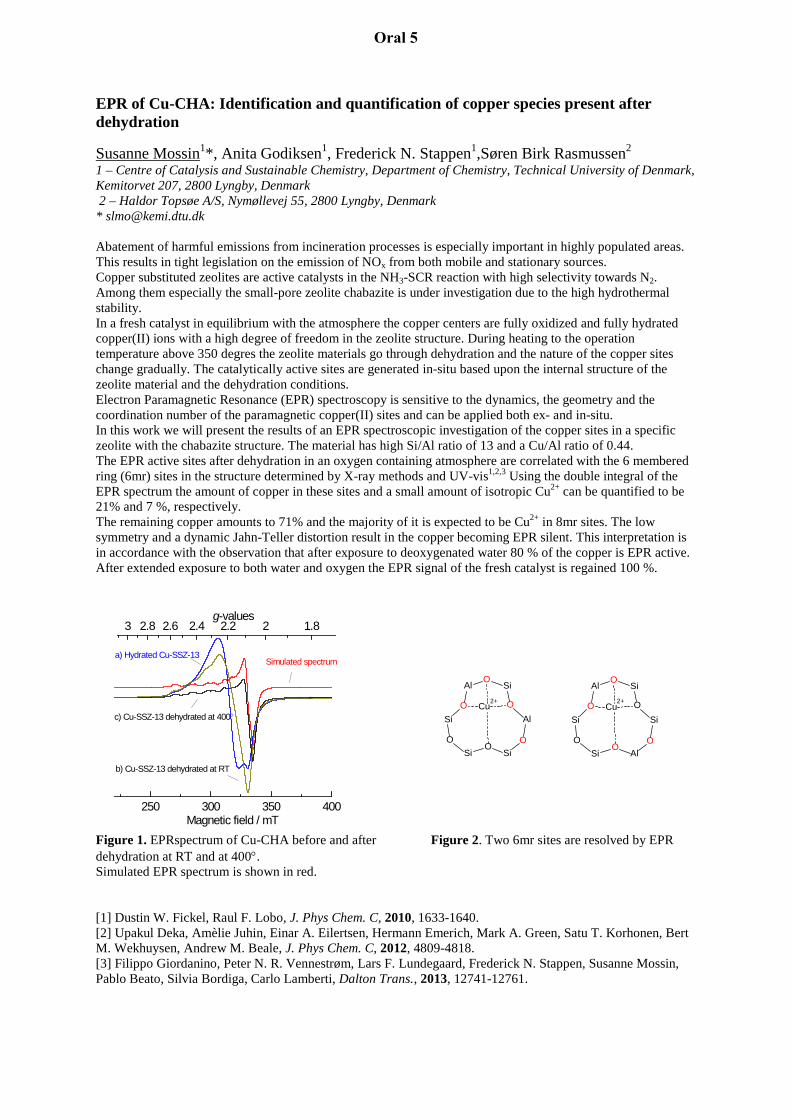
EPR of Cu-CHA: Identification and quantification of copper species present after dehydration
Susanne Mossin1*, Anita Godiksen1, Frederick N. Stappen1,Søren Birk Rasmussen2 1 – Centre of Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800 Lyngby, Denmark 2 – Haldor Topsøe A/S, Nymøllevej 55, 2800 Lyngby, Denmark * [email protected] Abatement of harmful emissions from incineration processes is especially important in highly populated areas. This results in tight legislation on the emission of NOx from both mobile and stationary sources. Copper substituted zeolites are active catalysts in the NH3-SCR reaction with high selectivity towards N2. Among them especially the small-pore zeolite chabazite is under investigation due to the high hydrothermal stability. In a fresh catalyst in equilibrium with the atmosphere the copper centers are fully oxidized and fully hydrated copper(II) ions with a high degree of freedom in the zeolite structure. During heating to the operation temperature above 350 degres the zeolite materials go through dehydration and the nature of the copper sites change gradually. The catalytically active sites are generated in-situ based upon the internal structure of the zeolite material and the dehydration conditions. Electron Paramagnetic Resonance (EPR) spectroscopy is sensitive to the dynamics, the geometry and the coordination number of the paramagnetic copper(II) sites and can be applied both ex- and in-situ. In this work we will present the results of an EPR spectroscopic investigation of the copper sites in a specific zeolite with the chabazite structure. The material has high Si/Al ratio of 13 and a Cu/Al ratio of 0.44. The EPR active sites after dehydration in an oxygen containing atmosphere are correlated with the 6 membered ring (6mr) sites in the structure determined by X-ray methods and UV-vis1,2,3 Using the double integral of the EPR spectrum the amount of copper in these sites and a small amount of isotropic Cu2+ can be quantified to be 21% and 7 %, respectively. The remaining copper amounts to 71% and the majority of it is expected to be Cu2+ in 8mr sites. The low symmetry and a dynamic Jahn-Teller distortion result in the copper becoming EPR silent. This interpretation is in accordance with the observation that after exposure to deoxygenated water 80 % of the copper is EPR active. After extended exposure to both water and oxygen the EPR signal of the fresh catalyst is regained 100 %. Figure 1. EPRspectrum of Cu-CHA before and after Figure 2. Two 6mr sites are resolved by EPR dehydration at RT and at 400°. Simulated EPR spectrum is shown in red. [1] Dustin W. Fickel, Raul F. Lobo, J. Phys Chem. C, 2010, 1633-1640. [2] Upakul Deka, Amèlie Juhin, Einar A. Eilertsen, Hermann Emerich, Mark A. Green, Satu T. Korhonen, Bert M. Wekhuysen, Andrew M. Beale, J. Phys Chem. C, 2012, 4809-4818. [3] Filippo Giordanino, Peter N. R. Vennestrøm, Lars F. Lundegaard, Frederick N. Stappen, Susanne Mossin, Pablo Beato, Silvia Bordiga, Carlo Lamberti, Dalton Trans., 2013, 12741-12761.
AlO
Si
OAl
OSi
OSi
O
SiO Cu
AlO
Si
OSi
OAl
OSi
O
SiO Cu
2+ 2+
250 300 350 400
b) Cu-SSZ-13 dehydrated at RT
a) Hydrated Cu-SSZ-13
c) Cu-SSZ-13 dehydrated at 400°
Magnetic field / mT
3 2.8 2.6 2.4 2.2 2 1.8
Simulated spectrum
g-values
Oral 5
16th Nordic Symposium on Catalysis Effective Mn/TiO2 catalyst synthesized by deposition-precipitation method for low-temperature selective catalytic reduction of NO with NH3
1 – DTU Department of Chemistry, Kemitorvet, Building 207, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark 2 – Combustion and Harmful Emission Control Research Centre, Department of Chemical and Biochemical Engineering, Building 229, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark Waste incineration is a proven technology to reduce net greenhouse gas emissions because it avoids methane emissions from landfills. The incineration of waste releases CO2, however, the generated heat can be used to make electricity and for district heating, thus replacing coal or gas. One challenge of waste incineration is the flue gas containing high levels of Hg, Zn, dust etc. due to which the SCR unit is preferably placed at the tail-end position which requires costly reheating of the flue gas. Over the last decade there was a great interest in development of low-temperature SCR catalysts containing transition metal oxide catalysts like V2O5/TiO2, Fe/TiO2, Cu/TiO2, and Mn/TiO2. Among them Mn supported on titania and promoted with Fe is a promising, non-toxic candidate [1]. The most commonly used method of preparation is impregnation. However, high loadings of metals can cause them to be present in crystalline phase which causes unselective oxidation of NH3 instead of reduction of NO. We have developed a deposition-precipitation (DP) based method of preparation for MnFe/TiO2 yielding activities significantly higher than when using the traditional impregnation (IMP) method, see figure 1. The catalysts were thoroughly characterized using NH3-TPD, H2-TPR, XRD, N2-physisorption, XPS and TGA, see table 1. Compared to the IMP method the DP method leads to more surface acid sites, improved redox properties, no crystalline MnOx, higher surface area and more surface adsorbed oxygen. The higher content of surface adsorbed oxygen as evidenced by XPS is increasing the rate of NO to NO2 oxidation and might make the material also interesting for VOC removal.
150 200 250 300
0
20
40
60
80
100
NO
Con
vers
ion
[%]
T [°C]
IMP DP
150 200 250 3000
20
40
60
80
100
NO
Con
vers
ion
[%]
T [°C]
IMP DP
References [1]: Fu, M.; Li, C.; Lu, P.; Qu, L.; Zhang, M.; Zhou, Y.,; Yua, M.; Fange, Y., Catal. Sci. Technol. (2014), 4, 14-25
Table 1. Summary of important characteristics of IMP- and DP-prepared catalysts.
Figure 1. NO conversion over MnFe/Ti prepared by impregnation (IMP) and deposition-precipitation (DP).
Oral 6
16th
Nordic Symposium on Catalysis
Fundamental deactivation and regeneration mechanisms of Fe-BEA as NH3-SCR
catalyst for NOX reduction
Soran Shwan1*, Jonas Jansson
2, Louise Olsson
1 and Magnus Skoglundh
1
1 – Competence Centre for Catalysis, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden.
2 – Volvo Group Trucks Technology, SE-40508 Gothenburg, Sweden.
16th Nordic Symposium on Catalysis/Abstract template Title: CO and CO2 Hydrogenation to Methanol
Felix Studt1* 1 – SUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, CA * [email protected] Methanol is among the top ten petrochemicals produced in the world and has been discussed as a transportation fuel in a methanol-based economy [1]. Industrially, methanol is produced from synthesis gas, a mixture of CO, CO2 and H2, at 230-280 °C and 50-120 bar employing a Cu/ZnO/Al2O3 catalyst [2]. Despite several decades of research, the active site of the industrially employed Cu/ZnO/Al2O3 catalyst was only identified recently [3]. It was found that defects and step sites are responsible for both CO and CO2 hydrogenation due to the increase of binding of reaction intermediates and their hydrogenation barriers when compared to terraces. This work also elucidated the reaction mechanism of the conversion of a mixture of CO2, CO and H2 to form methanol. Combined theoretical and experimental work found that the industrial catalyst comprised of Cu and ZnO allows for the fast conversion of CO2 to methanol while bare copper is superior at hydrogenating CO [4]. Performance catalysts will therefore expose a distribution of different sites that maximize the total conversion based on the feed composition (CO2 relative to CO) and reaction conditions. This analysis of Cu/ZnO/Al2O3 catalysts was extended towards other alloy compositions, theoretically investigating their ability to hydrogenate CO2 at low pressures. NiGa alloys were predicted by theory to exhibit catalytic properties close to or superior of those found for Cu/ZnO/Al2O3 (see Figure 1), a finding that was confirmed experimentally [5]. .
Figure 1. Theoretical activity volcano for CO2 hydrogenation to methanol.[5] The turnover frequency (TOF) is plotted as a function of the oxygen binding energy (∆EO) on stepped (211) surfaces. ∆EO for Ni-Ga intermetallic compounds are depicted in red. References [1] G. A. Olah, Angew. Chem. Int. Ed. 52 (2013) 104. [2] J. B. Hansen, P. E. H. Nielsen, in: Handbook of Heterogeneous Catalysis, eds. G. Ertl, H. Közinger, F. Schüth (Wiley VCH, Weinheim, 2008). [3] M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov, R. Schlögl, Science 336 (2012) 893. [4] F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov, R. Schlögl, manucscript in preparation. [5] F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff, J. K. Nørskov, Nature Chem. in press. DOI: 10.1038/NCHEM.1873
-2 -1 0 1 2-1
0
1
2
∆EO-∆EO (eV)
Pd
Cu
Ni
Cu+ZnNi3Ga
NiGa
Cu
-2 -1 0 1 2
-1
0
1
-2
log
(TO
F/TO
FCu ) Ni5Ga3
Ni3Ga
Ni5Ga3
NiGa
Oral 8
Mixed alcohols synthesis over K-Ni-MoS2 catalysts prepared by conventional co-
precipitation and by microemulsion. Effect of alcohols co-feeding.
Rodrigo Suárez París1*, Magali Boutonnet1, Sven Järås1 1 Department of Chemical Engineering and Technology, KTH - Royal Institute of Technology, Stockholm, 10044,
Water and CO on monoclinic zirconia: Experimental and computational insights
Sonja Kouva1*, Karoliina Honkala2, Juha Lehtonen1, Jaana Kanervo1 1 – Dept. of Biotechnology and Chemical Technology, Aalto University, P.O. Box 16100, FI-00076 Aalto 2 – Dept. of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä * [email protected] Zirconium oxide (zirconia, ZrO2) is an interesting catalyst material for processing of bio-based chemicals and fuels. In this work we have studied the surface of monoclinic zirconia first calcined at 600 °C and then treated with H2 reduction and/or water vapor. CO was introduced to the sample at 100 °C and then the sample was heated in CO up to 550 °C. We have combined in situ DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy) and temperature-programmed surface reaction (TPSR) to connect identities and stabilities of surface species to a dynamic, quantitated reactor response. Density functional theory (DFT) calculations complement the experimental data in terms of surface species configurations and their energetics.
Our results indicate that especially the low-temperature interaction between CO and monoclinic zirconia is affected by the pretreatment selection. On reduced zirconia, adsorbed linear CO (ca. 2190 cm-1) is observed on the surface during initial CO adsorption at 100 °C (shown on the left in Fig. 1) with decreasing intensity over time, the same decrease is observed in terminal OH intensity. Simultaneously, formate intensity (on the right, 1569 cm-1) slowly increases. Based on these observations, it seems that at low temperatures where linear CO is formed on the surface, formate formation can take place via terminal surface OH group and adsorbed linear CO, as previously suggested by Bianchi et al. [1]. Water vapor pretreatment blocks linear CO formation, as expected [2], and the onset of formate formation is delayed until 175-200 °C. This is in agreement with computational results showing that formate formation from gas-phase CO is highly activated, Eact is ca. 150 kJ/mol.
The gas-phase response for reduced ZrO2 during heating in CO is shown in Fig. 2., level below zero equals to uptake on the sample and level above zeros equals to release from the sample. Before heating the sample was stabilized in CO flow for 2 hours.
CO uptake takes place up to 330 °C, in accordance with the DRIFTS experiments showing formates at their highest intensity at 325 °C. Water release above 300 °C is due to dehydration of the sample, a blank run in helium shows a similar water release. Formate decomposition is observed after 330 °C: Formate species decompose reversibly to CO and irreversibly to CO2 and H2 with the contribution of the surface hydroxyl groups. The calculated reaction energies for irreversible formate decomposition are ca. 180-360 kJ/mol and in addition to that the calculations show that the process is activated. Based on the observed high reaction temperatures this seems reasonable. All three methods, DRIFTS, TPSR and DFT, give concurring results on formate formation and decomposition. References [1] D. Bianchi, T. Chafik, M. Khalfallah and S. J. Teichner, Appl. Catal., A 105 (1993) 223. [2] C. Morterra, L. Orio and C. Emanuel, J. Chem. Soc., Faraday Trans. 86 (1990) 3003.
Figure 1. 2300-2000 cm-1 and 1650-1450 cm-1 regions duringCO adsorption at 100 °C on reduced ZrO2. The inset showsthe linear CO (2192 cm-1) intensity during CO adsorption.
Figure 2. CO-TPSR for reduced ZrO2.Level 0 is the feed level, 3·10-7 mol/(g s)for 2% CO and none for the othercomponents.
Oral 10
16th Nordic Symposium on Catalysis In situ monitoring of cobalt supported catalysts for Fischer-Tropsch synthesis under realistic activation, reaction and regeneration conditions
Nikolaos E. Tsakoumis1*, Magnus Rønning1, Erling Rytter1, Anders Holmen1 1 –Department of Chemical Engineering, Norwegian University of Science and Technology (NTNU), NO-7491 Trondheim, Norway. * Corresponding: [email protected] Fischer–Tropsch synthesis (FTS) is a tool for the utilization of synthesis gas derived from different feedstocks, i.e. natural gas, coal, and biomass [1]. In FTS on cobalt supported catalysts, carbon monoxide and hydrogen are converted into linear paraffinic hydrocarbons and water. Although the process counts many decades of existence and different commercial applications, still a lack of understanding exists regarding reaction, deactivation and regeneration mechanisms [2,3]. In order to address these questions a capillary based set-up that can accommodate X-ray based techniques [4,5] and Raman spectroscopy [6] was modified in order to withstand realistic conditions of activation, reaction and regeneration. Several different Co based catalysts were evaluated. Carbon based supports were used in addition to more industrially relevant Al2O3.
Figure 1. Flow diagram of the in situ set-up, 3D representation of Raman spectra, X-ray powder
diffractogramms and normalized X-ray absorption spectra obtained during catalyst activation together with signal from the MS showing H2O production and quantitative determination of the involved phases.
With the proposed experimental configuration the catalyst activation could be followed in detail and all the intermediate phases could be identified for several Co supported catalysts. The reaction was also performed at realistic conditions of high pressure (10-18 bar) and FTS relevant temperatures (210-220oC). Results highlight the importance of the induction period in FTS in which phase transformations were detected coinciding with changes in the reactor environment on the way to the pseudo-steady state. During the first days of reaction the signal from X-ray based bulk techniques and Raman spectroscopy was rather stable and only simulation of temperature runaways up to 400oC showed significant sintering and crystalline carbon formation. Phase gradients along the reactors length at different stages of the experiment were also detected. For the evaluation of the long term exposure of the catalytic material into FTS environment, catalysts were withdrawn from fixed-bed and slurry reactors and characterized in a pseudo-in situ manner. Results showed that one of the main deactivation mechanisms is sintering of Co nanoparticles although other deactivation mechanisms co-exist. Ex situ characterization techniques were applied to support the findings. Regeneration attempts of the deactivated catalyst through reducing and oxidative-reducing environments have been performed. The activity and selectivity of the catalyst was regained after specific treatments. However the structure activity/selectivity relations are still to be explained. [1] M.E. Dry, Catal. Today 71 (2002) 227–241. [2] N.E. Tsakoumis, M. Rønning, Ø. Borg, E. Rytter, A. Holmen, Catal. Today 154 (2010) 162–182. [3] N.E. Tsakoumis, A. Voronov, M. Rønning, W. van Beek, Ø. Borg, E. Rytter, A. Holmen, J. Catal. 291 (2012) 138–148. [4] J.W. Couves, J.M. Thomas, D. Waller, R.H. Jones, A.J. Dent, G.E. Derbyshire, G.N. Greaves, Nature 354 (1991) 465–468. [5] B.S. Clausen, H. Topsøe, Catal. Today 9 (1991) 189. [6] N.E. Tsakoumis, R. Dehghan, R.E. Johnsen, A. Voronov, W. van Beek, J.C. Walmsley, Ø. Borg, E. Rytter, D. Chen, M. Rønning, A.
Holmen, Catal. Today 205 (2013) 86–93.
Oral 11
16th
Nordic Symposium on Catalysis
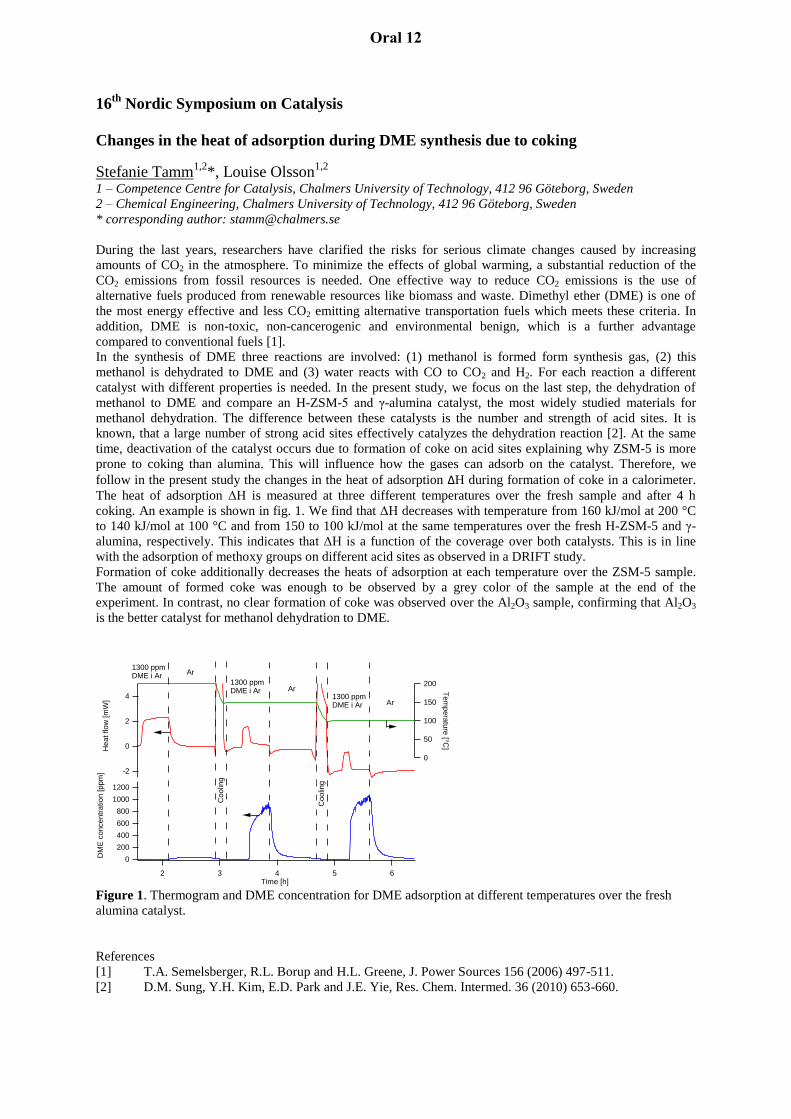
Changes in the heat of adsorption during DME synthesis due to coking
Stefanie Tamm1,2
*, Louise Olsson1,2
1 – Competence Centre for Catalysis, Chalmers University of Technology, 412 96 Göteborg, Sweden
2 – Chemical Engineering, Chalmers University of Technology, 412 96 Göteborg, Sweden
CO2 uptake in supported ionic liquid phase materials
Helene Kolding1, Anders Riisager1, Rasmus Fehrmann1* 1 – Centre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, 2800 Kongens Lyngby, Denmark * [email protected] Carbon dioxide (CO2) has been named a culprit in the struggle against man-made climate change [1]. Abatement and conversion of CO2 is most easily accomplished where the highest concentrations are found: Power plant flue gases. However, flue gas contains many compounds incompatible with most catalyst systems: corrosive and reactive species such as SO2, NOx as well as a lot of water. In order to rule out the sensitivity of most catalysts to the impurities found in flue gases, separation and concentration of CO2 may well be a necessity for a catalyst to work properly. Transformation of CO2 therefore demands a pure source of CO2. One way to accomplish this could be to use an end-tail system with reversible ab- and desorption, where desorption is carried out separately to obtain a pure stream. A number of amino acid-based ionic liquids and their supported ionic liquid phase (SILP) analogs have been tested for reversible CO2 uptake, including tetrahexylammonium prolinate, [N6666][Pro].[2] Most materials showed a stoichiometric uptake of CO2 at ambient temperature and pressure. Reversible absorption/desorption and rapid sorption dynamics were shown to occur for the SILP materials without degradation with desorption at 80°C under a flow of Ar. Even after several cycles, the SILP materials retain >90% of their absorption stoichiometry. A flow study showed a significant silent time when a fixed-bed of [N6666][Pro] SILP was inserted into a dry gas stream containing 9 % CO2.[2] This system is envisioned to be used for CO2 uptake in an end-tail coal-fired power plant [1] S. Solomon, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, M. Tignor, and H. L. Miller, Climate Change 2007, Cambridge University Press, 2007. [2] H. Kolding, A. Riisager, and R. Fehrmann, Science China Chemistry 55 (2012) 1648
Oral 13
16th Nordic Symposium on Catalysis/Abstract template High throughput catalyst discovery for the direct synthesis of propylene carbonates from propylene glycol and CO2
Richard H. Heyn1* 1 – SINTEF Materials and Chemistry, P. O. Box 124 Blindern, 0314 Oslo * [email protected] Utilization of CO2 is of current interest from several perspectives. For large scale applications, the reduction of CO2 to fuels is under consideration as a recyclable energy storage vector and, depending on the overall CO2 balance of the reduction technology, a means of mitigating atmospheric CO2 emissions. Small scale applications focus on the use of CO2 in the synthesis of value-added chemicals and polymers. The effect on atmospheric CO2 emissions in these applications is significantly smaller, due to the overall volumes of these chemicals and polymers. The driving force here is sustainability, as CO2 is a renewable, essentially non-toxic, and inherently cheap C1 feedstock for the chemical industry. New reaction pathways involving CO2 rather than other C1 feedstocks can in principle be cleaner, with fewer byproducts and a smaller environmental footprint. Regardless of the application, radical improvements in both catalysis and process design are required to improve implementation of CO2 utilization technologies. CO2 is a very stable molecule kinetically and thermodynamically, so these improvements are necessary to drag CO2 out of these wells and into the light of chemical productivity. The EU project CyclicCO2 [1] is addressing these issues by a combination of catalyst and process design research for the production of cyclic carbonates from the renewables glycerol and CO2. Cyclic carbonates are envisioned for a number of applications, such as solvents, battery electrolytes, and additives to personal care products and detergents. Our activities and this presentation will discuss the discovery via high throughput technologies and understanding of improved catalysts for reaction 1. We have used this reaction as a surrogate for the carboxylation of glycerol due to experimental and analytical challenges with the use of glycerol. The catalyst screening is performed in the presence of MeCN, which acts as a chemical water trap (producing acetamide and eventually acetoxypropanols), in order to push the equilibrium further toward products. A total of 125 different catalyst combinations have been screened (Figure 1). Selected hits have been studied in a bench scale Parr reactor. Selected combinations show improved catalytic activities as compared to the standard catalyst Zn(OAc)2 + p-chlorobenzenesulfonic acid (Figure 2).
Figure 1. Normalized yields of propylene carbonate from Figure 2. Time vs. yield plot for the synthesis of high throughput experiments. propylene carbonate with Zn(OAc)2 + acid. Details of these experiments and the modelling of catalytic intermediates for this reaction will be presented. Reference [1] This research project has received funding from the European Union Seventh Framework Programme (FP7/2007 – 2013) under grant agreement n° 309497
C OO +
HO OH
O O
O
+ H2O (1)
Oral 14
Selective adsorption in Zirconium based metal-organic frameworks
Sigurd Øien1*, Knut Hylland1, Unni Olsbye1, Richard Blom2, Mats Tilset1, Karl Petter Lillerud1 1 – Department of Chenistry, University of Oslo, P.O. box 1033 Blindern, 0315 OSLO, Norway 2 – SINTEF Materialer og kjemi, P.O.Box 124 Blindern, NO-0314 Oslo, Norway * corresponding [email protected] Zr-based metal-organic frameworks (MOFs) are crystalline, porous materials that recently have gained a lot of attention for their properties as catalysts. The catalytic property comes from the possibility to incorporate active seats as part of the framework itself, and has been demonstrated for many reactions1. Furthermore, this can be used to tune the adsorption properties of the material by designing sterical and chemical composition of the pores with functional groups.
Figure 1: The structure of UiO-67-Me.
Depending on the functional groups on the organic constituent, the BET surface area of Zr biphenyl framework UiO-67 (see figure 1) ranges from 2000 – 3000 m2/g, making it a promising adsorbent. We have shown in initial experiments that by functionalizing UiO-67 with methyl groups, the BET surface area decreases slightly, and the adsorption properties of methane, CO2 and water is altered (see figures 2 and 3).
Figure 2. High pressure adsorption curve of CO2. Figure 3. High pressure adsorption curves of methane. Our initial data suggests that UiO-67-Me has a higher enthalpy of adsorption and a higher storage capacity for methane, than regular UiO-67. For CO2, we observe the opposite. This selectivity is an exciting result, but is also likely improvable by utilizing other functional groups. References [1] M. Kim, S. M. Cohen, CrystEngComm, 2012,14, 4096-4104.
Oral 15
The reactivity of radicals and coke molecules trapped within the HZSM-5 cavities in the Methanol and Ethanol-to-Hydrocarbons reaction. S. Hamieh1, L. Pinard1, K. Ben Tayeb2*, H. Vezin2 1 – IC2MP UMR CNRS 7285, University of Poitiers, 86000 Poitiers, France. 2 – LASIR UMR CNRS 8516, University of Lille 1, 59650 Villeneuve d’Ascq, France. * [email protected]
Ethanol and methanol are converted using HZSM-5 (Si/Al=40) zeolite into identical hydrocarbons and surprisingly with same selectivity. This is only possible owing to the fact that the two reactants have a common reaction intermediate (CRI). The objective of the study is to identify the CRI which will allow to propose a mechanism of the initial C-C bond formation from C1 unit that is an open question addressed many times and since long times.
The HZSM-5 zeolite converts, at 623K and under 3.0 MPa, methanol and ethanol into aromatics, olefins and paraffins molecules during a long time in spite of a high coke content [1, 2]. The mechanism of transformation of alcohols into hydrocarbons is radical and occurs through a common intermediate species; the carbene. :CH2 oligomerizes into olefins following a rake mechanism in which the growth probability factor α, determined by the Flory’s equation, is 0.53. Then olefins are either transformed into aromatics or are isomerized through benzyl carbocations blocked inside pores zeolite (Coke).
The role of coke is paradoxical, it can be a poison of zeolite acid sites or an active site of the isomerization reaction. Finally, the mechanism proposed in Fig. 1 is an alternative to the hydrocarbon pool mechanism, it combines two types of active sites: radicals and coke. It represents a combination of three mechanisms: I- “rake” type mechanism involving carbene species, II- olefins isomerization by active coke and III- transformation of olefins into aromatics.
Figure 1: Mechanism of methanol and ethanol conversion into hydrocarbons on HZSM-5 zeolite (olig. =
oligomerization, cyl. = cyclization and HT = hydrogen transfer, A = aromatics)
References [1] L. Pinard, S. Hamieh, C. Canaff, F. Ferreira Madeira, I. Batonneau-Gener, S. Maury, O. Delpoux, K. Ben Tayeb, Y. Pouilloux, H. Vezin, Journal of Catalysis. 299 (2013) 284-297. [2] L. Pinard, K. Ben Tayeb, S. Hamieh, H. Vezin, C. Canaff, S. Maury, O. Delpoux, Y. Pouilloux, Catalysis Today 218-219 (2013) 57-64.
C2
C=2
C3•• C4
•• C7+••:CH2
CH3OH (C2H5OH)
:CH2:CH2
α=0.53 αα α
Gas phase
ββ
:CH2 :CH2
C3= n-C4
=
C5•• C6
••
β β
n-C5= n-C6
=
Adsorbed phase
Adsorbed phase
HC+
CH3
CH3
CH2+
C+
CH3
CH3
i-C4+ A
olig.
cyl.
HT
olig.
cyl.
HT
A
Gas phase
β=0.53
I
III
II
A(CH3)… A(CH3)2
A(CH3)A(CH3)2
Oral 16
A Reactivity Descriptor in Solid Acid Catalysis: Predicting Turnover Frequencies of Alkene Methylation in Zeotype Materials
Chuan-Ming Wang1,2, Rasmus Y. Brogaard2,3, Bert M. Weckhuysen4, Jens K. Nørskov2,3, Felix Studt2,* 1 – Shanghai Research Institute of Petrochemical Technology SINOPEC, Shanghai 201208, China 2 – SUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USA 3 – Department of Chemical Engineering, Stanford University, CA 94305, USA 4 – Inorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Faculty of Science, Utrecht University, 3584 CG Utrecht, The Netherlands * [email protected] Recent work has reported discovery of metal surface catalysts by employing a descriptor-based approach, establishing a correlation between few well-defined properties of a material and its catalytic activity [1]. This theoretical work aims for a similar approach in solid acid catalysis, focusing on the reaction between alkenes and methanol catalyzed by Brønsted acidic zeotype catalysts. We modify the acidity by isomorphic substitution of metal ions in the zeolite and zeotype frameworks. Experimentally, the ammonia heat of adsorption is often used as a measure of the strength of acid sites [2]. Using periodic DFT calculations, we show that this measure can be used to establish linear scaling relations for the energy of intermediates and transition states of different chemical composition. Importantly, the scaling relations are independent of the chemical composition of the framework, lining up both zeolite and aluminophosphate materials of the same framework topology. These results show that the heat of ammonia adsorption can quantify the reactivity of the Brønsted acid sites. Interestingly, the scaling lines exhibit the same slope for transition states involving alkenes of different size and substitution in the same framework topology. This illustrates that the alkenes are equally sensitive to acid site reactivity, despite an inherent difference in the absolute rates of their methylation in a given material. The scaling lines allow us to use micro-kinetic modeling to predict a quantitative relation between ammonia heat of adsorption and the rate of alkene methylation from first principles [3]. We propose this as a step towards descriptor-based design of solid acid catalysts.
References [1] F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen, J. K. Nørskov, Science 320 (2008) 1320. [2] E. Derouane, J, Vedrine, R. R. Pinto, P. Borges, L. Costa, M. Lemos, F. Lemos, F. R. Ribeiro, Catal. Rev. - Sci. Eng. 55 (2013) 454. [3] C.-M. Wang, R. Y. Brogaard, B. M. Weckhuysen, J. K. Nørskov, F. Studt (2014), submitted.
-150 -125 -100 -75 -50-75
-50
-25
0
25
50
75
100
Tran
sitio
n)state)en
ergy
)/)kJmol
21
∆HNH3)/)kJmol21
2125
250
2100
50
275
100
2150 275
225 concerted
stepwise))step)1
0
25
75
stepwise))step)2
y=1.09x+97.9
y=0.80x+117.9
y=0.84x+147.1R2=0.98
R2=0.95
R2=0.98
AlPO234
CHA
250
Figure 2. Predicted rate of propene methylation as a function of calculated heat of ammonia adsorption in zeotype materials of the CHA framework.
-200 -175 -150 -125 -100 -75 -50-4
-3
-2
-1
0
1
2
3
4
log$TO
F$/$site
-1 s-1
∆HNH3$/$kJmol-1
-125
-3
-100
2
-4
4
-200 -75
-2
0
1
3
Co-AlPO-34
-175 -150 -50
-1
Zn-AlPO-34
Si-AlPO-34Al-MFI
Al-CHANi-AlPO-34
Mg-AlPO-34
Figure 1. Scaling relations of transition states involved in the stepwise and concerted mechanisms of propene methylation in zeotype materials of the CHA framework topology.
Oral 17
Observation of Coke formation during the Methanol-to-Olefin Process using in situ
UV/Vis and Confocal Fluorescence Microspectroscopy
Emily C. Corker,1 Elena Borodina,
2 Javier Ruiz-Martínez,
2,* Rasmus Fehrmann
1 and Bert M.
Weckhuysen2,*
1 – Centre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of
Denmark, DK-2800, Kgs. Lyngby, Denmark
2 – Group of Inorganic Chemistry and Catalysis, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht,
Dynamics of catalytic Pd nano-particles studied by time resolved high energy XRD
Davide Ferri1*, Mark A. Newton2, Marco di Michiel2, Oliver Kröcher1 1 – Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland 2 – ESRF, F-38043 Grenoble, France *Corresponding: [email protected] Automotive catalysts work typically under severe conditions with respect to temperature and reaction environment. Moreover, due to the dynamics of engine operation the atmosphere to which the catalysts are exposed also changes periodically. Obtaining information about relevant structural changes occurring under such operation conditions at lab scale is challenging in terms of time and spectral resolution of the selected spectroscopic method. When time resolution is met, identification of relevant dynamic spectral features may be hindered by their typically weaker response compared to static signals not relevant to the process under study. In this contribution, we use the concentration modulation excitation approach (ME) [1] to extract structural information of small Pd nano-particles under fast transients from synchrotron X-ray diffraction (XRD). ME consists in the alternate and repeated switch between reaction conditions followed by phase sensitive analysis enabling separation of signals of possibly relevant components from those of the bulk response [2]. The advantage of using the ME approach is emphasized in the case of 2 wt% Pd/CeZrO2 (Pd/CZ) that is typically silent in the XRD with respect to the Pd-PdO phase. The time-resolved high energy XRD data collected during CO-O2 experiments designed to simulate oxygen storage capacity (OSC) experiments for three-way catalysis applications exhibit the reflections of cubic CZ but hardly display any tangible change. Also, given the fine dispersion of PdO induced by CZ, no information is available about the state of Pd. The phase-resolved XRD data exhibit only the very subtle changes associated with the signals responding to the modulation experiment. All signals of the CZ support shift repeatedly to lower and back to higher Q values in response to the CO and the O2 pulses, respectively. This behaviour qualitatively describes the reduction and the re-oxidation of CZ associated with its OSC. Importantly, the data also exhibit additional broad features (e.g. at ca 2.8 Å-1) that correspond to reflections of Pd nano-particles of 2 nm. The sole intensity change is associated with the relative increase of long range order attributed to PdO reduction. Therefore, the modulation data can capture the dynamic reduction and re-oxidation of PdO and CZ. Reduction of both PdO and CZ occurs much slower than re-oxidation. Comparison with the same experiment on 2 wt% Pd/Al2O3 confirms that this is the effect of the OSC of CZ. Hence, it is demonstrated that subtle structural changes of the XRD patterns can be captured and their temporal response precisely assessed. This provides access to the detailed structural-dynamic behaviour of the system.
Figure 1. (a) Colour map representation and (b) time-resolved hard-XRD patterns of 2 wt% Pd/CZ during a full CO/O2 modulation experiment at 573 K (T= 50 s). (c) Corresponding phase-resolved data. (d-f) High energy-XRD data for an identical experiment performed on 2 wt% Pd/Al2O3. (○) Pd and (�) PdO. [1] D. Baurecht, U.P. Fringeli, Rev. Sci. Instrum. 72 (2001) 3782. [2] D. Ferri, S.K. Matam, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff, M.A. Newton, PCCP 12 (2010) 5634; D. Ferri, M.A. Newton, M. Di Michiel, S. Yoon, G.L. Chiarello, V. Marchionni, S.K. Matam, M. Aguirre, A. Weidenkaff, F. Wen, J. Gieshoff, PCCP 115 (2013) 1231; D. Ferri, M.A. Newton, M. Nachtegaal, Top. Catal. 54 (2011) 1070.
Oral 20
16th Nordic Symposium on Catalysis Catalytic application of metal nanoparticles confined in porous materials
Jacob Abildstrøm1, Agata Gallas-Hulin1, Jerrik Mielby1, Søren Kegnæs1* 1 – Department of Chemistry, Technical University of Denmark, Kemitorvet 207, Kgs. Lyngby 2800 * Corresponding authors [email protected], In spite of the great technological, environmental and economic interests, general methods for the stabilization of metal nanoparticles against sintering are missing. Although for some specific systems it has been achieved by optimizing the interaction of nanoparticles with a support material or by encapsulation of the metal particles [1-3]. However, these known catalytic systems are in general rather expensive and difficult to synthesize and they cannot be produced in industrial scale. Therefore, the development of novel sintering stable heterogeneous nanoparticle catalysts, which find use in the chemical industry, is of great importance. Recently, we have developed several different catalytic systems where metal nanoparticles are confined in different porous materials. As an example, in figure 1, is shown gold nanoparticles trapped inside a silicalite-1 zeolite. The aim with encapsulation metal nanoparticles in a porous matrix, like a zeolite, is first of all to prevent the metal nanoparticle from sintering during a high temperature catalytic reaction. Furthermore, the porous matrix can also contribute active to the catalytic reaction. We have for instance shown that the encapsulated metal nanoparticles only are accessible through the pores which give highly size-selective reactions. Here, we present the progress, which we have made, on the synthesis of metal nanoparticles confined in different porous materials like zeolites, metal oxides and polymers. We have tested the materials as catalysts in different selective reactions. Furthermore, we have characterized the materials with various techniques including SEM, in situ TEM, STEM, TEM tomography, XPS, XRF, and XRD among others
Figure 1. STEM image of gold nanoparticles encapsulated in silicalite-1.
References [1] P. M. Arnal, M. Comotti, F. Schüth, Angew. Chem., 118 (2006), 8404. [2] Y. Dai, B. Lim, Y. Yang, C. M. Cobley, W. Li, E. C. Cho, B. Grayson, P. T. Fanson, C. T. Campbell, Y. Sun, and Y. Xia, Angew. Chem. Int. Ed. 49 (2010) 1. [3] A. B. Laursen, K. T. Højholt, L. F. Lundegaard, S. B. Simonsen, S. Helveg, F. Schüth, M. Paul, J.-D. Grunwaldt, S. Kegnæs, C. H. Christensen, K. Egeblad, Angew. Chem. Int. Ed. 49 (2010) 3504.
Oral 21
Investigation of Active Sites for the Oxygen Reduction Reaction on Nitrogen-doped
Carbon Nanomaterials
Marthe E. M. Buan1*, Navaneethan Muthuswamy
1, Ida Hjorth
1, De Chen
1 and Magnus
Rønning1
1 – Department of Chemical Engineering, Norwegian University of Science and Technology (NTNU),
Near-Ambient Pressure XPS study of CO and H2 Oxidation over Pd model surfaces
Anne Borg1*, Vasco R. Fernandes1, Mari. H. Farstad1, Jan Knudsen2, Johan Gustafson2, Edvin Lundgren2, Hilde J. Venvik3 1 – Department of Physics, Norwegian University of Science and Technology, NO-7491 Trondheim, NORWAY 2 – Division of Synchrotron Radiation Research, Lund University, Box 117, SE-221 00 Lund, SWEDEN 3 – Department of Chemical Engineering, Norwegian University of Science and Technology, NO-7491 Trondheim, NORWAY * corresponding author: [email protected] Palladium is a versatile oxidation catalyst, among others finding applications in CO removal from car exhaust [1] and total oxidation of hydrocarbons [2]. Furthermore, Pd and its alloys have high solubility, permeability and selectivity for hydrogen, making them suitable as hydrogen separation membrane materials [3]. Pd/Ag is a commonly selected alloy [4]. In the present work, we have investigated single crystal surfaces of Pd(100) and Pd75Ag25(100) as model systems for addressing oxide formation during CO and H2 oxidation reactions at Pd surfaces as well as the influence of Ag as alloying element on these reactions under near ambient, oxygen rich conditions. The experimental tools applied have been near ambient X-ray photoelectron spectroscopy (NAPXPS) in combination with quadropole mass spectrometry (QMS). The experiments were performed at beamline I511-1 of the MAX IV Laboratory [5]. An example of recorded data during CO oxidation over Pd(100) is presented in Fig. 1. The activation energy for CO oxidation over this surface was determined to be 1.0 eV, while a value of 1.1 eV was obtained for Pd75Ag25(100). CO inhibition of the reaction is observed at low temperatures for both surfaces upon CO oxidation only as well as in the case of simultaneous oxidation of CO and H2. In the latter case, the CO oxidation reaction is determining the overall reaction behaviour. While high activity for H2 oxidation over Pd(100) is demonstrated already at room temperature, low activity for this reaction is observed below a critical temperature over Pd75Ag25(100). Ag as alloying element thus significantly affects the oxidation reactions. While the characteristic surface oxide [6] is the highly active surface in the reactions for Pd(100), chemisorbed oxygen plays this role for Pd75Ag25(100). In summary, the presence of silver in the outermost surface layer significantly affects the surface chemistry and thereby the reaction mechanism.
Figure 1. CO oxidation over the Pd(100) surface at 0.7 mbar total pressure and O2:CO ratio 10:1. Left: The O 1s core level region recorded as a function of sample temperature along with the corresponding QMS data for O2, CO and CO2. Right: Decomposition of the O 1s core level spectra at two different stages (marked with dashed lines in the left part) during the CO oxidation experiment, low activity (lower panel) and high activity (upper panel) towards CO2 production. References [1] R. Heck et al., Catalytic Air Pollution Control: Commercial Technology, 3.ed, Van Nostand Reinhold, 2009. [2] P. Henry, Palladium Catalyzed Oxidation of Hydrocarbons, Springer 1980. [3] See eg. N. Itoh andR. Govind, Ind. Eng. Chem. Res. 28 (1989) 1554. [4] A. K. M. Fazle Kibria et al., Int. J. Hydrogen Energy 25 (2000) 853. [5] J. Schnadt et al., J. Synchrotron Rad. 19 (2012) 701. [6] M. Todorova et al., Surf. Sci. 541 (2003) 101.
Gas phase O2
Pd 3p
Surface oxide
CO
Gas phase CO2
Oral 24
16th
Nordic Symposium on Catalysis/Abstract template
Inhibition and Deactivation of Ni-MoS2 for Hydrodeoxygenation by Bio-oil Impurities
Peter M. Mortensen1, Diego Gardini
2, Hudson W. P. de Carvalho
3 Christian D. Damsgaard
2,
Jan-Dierk Grunwaldt3, Peter A. Jensen
1, Jakob B. Wagner
2, Anker D. Jensen
1*
1 – Department of Chemical and Biochemical Engineering, Technical University of Denmark, Søltofts Plads
229, DK-2800, Denmark
2 – Center for Electron Nanoscopy, Technical University of Denmark, Fysikvej 307, DK-2800, Denmark
3 – Institute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT),
A prospective route to biofuels usable in the current infrastructure is the combination of flash pyrolysis and hydrodeoxygenation (HDO). However, little work has been devoted to evaluating long term stability or
resistance of catalysts toward feed impurities during HDO [1]. Traditional hydrotreating catalysts, as Co-MoS2
and Ni-MoS2, have been among the most tested catalysts [1]. This group of catalysts is already industrially
established catalysts in hydrodesulfurization (HDS) [1]. In the current study the stability of Ni-MoS2/ZrO2 has
been evaluated in a simulated bio-oil system.
The long term stability of the Ni-MoS2/ZrO2 catalyst is highly dependent on the presence of sulfur as evidenced
by two experiments in which sulfur was added from different sources and different concentrations. Figure 1
shows that 0.3 vol% 1-octanethiol in the feed was found insufficient to maintain stability of the catalyst, as the
degree of deoxygenation (DOD) decreased from 74% to 44% over 109 h of operation. Much better stability was
obtained when adding 2 vol% DMDS to the feed, here the DOD only dropped from 90% to 82% over 96 h of
operation.
Figure 1: Stability of Ni-MoS2/ZrO2 during HDO of
phenol and 1-octanol in two cases with different types
and concentrations of feed sulfur. T = 280 °C, P = 100
bar, WHSV = 4.0 h-1.
Figure 2: DOD over a Ni-MoS2/ZrO2 catalyst during
HDO of phenol in 1-octanol exposed to chlorine or
potassium. 1-chlorooctane was added to the feed in a
concentration of 0.05 wt% Cl. KNO3 was
impregnated on fresh catalyst in stoichiometric
amounts. T = 280 °C, P = 100 bar, WHSV = 4.0 h-1.
Analysis of the spent catalyst samples by XRD, TEM, and elemental analysis revealed that the catalyst co-fed
with 0.3 vol% 1-octanethiol was partly oxidized to MoO3.
Adding chlorine or alkali metal to the catalyst was found to cause a decrease in the DOD, as shown in Figure 2. Potassium caused a severe persistent deactivation, decreasing the DOD to ca. 5%. DFT calculations by
Andersen et al. [2] showed that potassium saturates the vacancy sites along the edges of the MoS2 slabs, which
probably explains the severe deactivation. Thus, both Ni and MoS2 based catalysts are severely deactivated by
potassium, indicating that this should be removed from the bio-oil before long term operation can be achieved.
Addition of chlorine to the feed was found to inhibit the reaction, and only decreased the DOD from 91% in the
un-poisoned case to 74% in the chlorine poisoned case. Removing the chlorine from the feed increased the
activity toward the level of the un-poisoned case again, showing a non-persistent nature of the chlorine
deactivation mechanism.
This work highlights the potential deactivation mechanisms relevant during hydrodeoxygenation of bio-oil.
[1] P.M. Mortensen, J.-D. Grunwaldt, P.A. Jensen, K.G. Knudsen, A.D. Jensen, Appl. Catal. A, 407 (1-2) (2011) 1
[2] A. Andersen et al., J. Phys. Chem. C, 115 (2011) 9025
Comparative study of Pt and Ni-Mn supported catalysts in dry reforming of methane Tatyana Kuznetsova*, Tamara. Kriger, Evgenii Paukshtis, Vladimir Rogov, Dmitrii Arendarskii, Arcadii Ishchenko, Vladislav Sadykov Boreskov Institute of Catalysis, Pr. Lavrentieva, 5, 630090, Novosibirsk, Russia * corresponding [email protected] For Pt supported on γ-Al2O3 or Ce-Zr-(La)-O and NiyMnMezOx (20%)/γ-Al2O3 composites (Ni/Mn ratio =0.2÷1.0, Me=Cu, Mg, Mo, La, Zr or Ce, z=0.2), catalytic properties in dry reforming of CH4 (DR) in concentrated feeds were studied. NiMnOx-based composites are characterized by XRD and HRTEM. The specific features of reaction mechanism for different types of catalysts are elucidated by pulse microcalorimetry and kinetic transients and discussed with a due regard for their real structure and surface properties. Steady-state catalytic activity strongly depends upon composition of active components and supports (Fig. 1). For supported Pt catalysts (loading 1.4 wt.%), the highest activity is observed for CeZrLax (x=0.2-0.3) fluorite–like supports. At pulsing more diluted reaction mixture (7% CH4 + 7% CO2), the difference in activity of oxygen -pretreated Pt catalysts is not so dramatic. For both catalysts, conversions of CH4 and especially CO2 in pulses of one reagent (CH4 or CO2) are significantly lower than those observed in mixed pulses. This indicates the important role of conjugated activation of both reagents for the oxidative transformation of CH4 into CO and H2. Up to 1-3 oxygen monolayers (mobile oxygen of doped ceria-zirconia with the heat of O2 adsorption < 620-640 kJ/mol) are removed from oxidized Pt/CeZrLa catalyst until reaching the quasi steady-state, thus creating sites for efficient CO2 activation on reduced surface. Co-adsorption and activation of CH4 (on Ptδ+ centers) and CO2 (on anion vacancies sites) along with O transfer from activated CO2 to CHx fragments effectively proceed at Pt/CeZrLaxO interface without participation of strongly bound surface/bulk oxygen. The most optimal structure of such interface is realized for Pt/CeZrLax (x=0.2-0.3) catalyst due to partial incorporation of Pt into surface positions in vicinity of domain boundaries of fluorite (forming complex cluster defects) which helps to stabilize cationic Pt species. A weak interaction of Pt with alumina provides only a low concentration of active Ptδ+ sites at Pt/support interface and, hence, a low activity. Oxidized NiMnOx/alumina composites are slowly (~ 1h) activated in the reaction mixture at 900oC due to segregation of Ni nanoparticles from the mixed oxide precursor. Composite with Ni/Mn ratio =0.2 (Fig. 1) is less active as compared to Pt/CeZrLa catalyst. It confirms well-known data on a higher activity of Pt species in comparison with Ni in the activation of C-H bonds in CH4. Substitution of Mn by different cations decreases activity. In spent catalysts, Ni0, NiO, MnOx and NiMnOx clusters stabilized on the surface of Al-Mn and Al-Ni spinels are observed. Surface species Ni/NiO+MnOx could be considered as active centers for DR. The total scheme of DR mechanism on composites is close to that for Pt catalysts. CH4 activation proceeds on Ni0/Niδ+ clusters stabilized on NiMnOx surface domains which provide anion vacancies for CO2 activation and O transfer. Thus, for various catalysts, DR efficiently proceeds at metal/reducible support interface with a similar mechanism of conjugated activation of both reagents. NiMnO-based composites supported on alumina can be suggested as active and inexpensive catalysts for DR. A somewhat lower efficiency of Ni centers as compared to Pt in activation of C-H bonds in CH4 would be compensated by a higher Ni loading.
Figure 1. CH4 conversion for Pt and NiMnOx supported catalysts in DR at 700 and 800oC. 31.5% CH4 + 46.0% CO2 + 22.5% He, 2.3 l/h.
Figure 2. Possible models of active clusters for Pt/CeZrLaO (1), Pt/Al2O3 (2) and NiMnOx/Al2O3 (3) catalysts in DR.
Support by FP7 OCMOL and BIOGO Projects is gratefully acknowledged.
Oral 32
Supported nickel based catalysts, Ni/Mg(Al)O, for natural gas conversion, prepared via
delamination and restacking of MgAl- and NiAl- nanosheets
J. Karthikeyan, H. Song, U. Olsbye, H. Fjellvåg, A. Olafsen Sjåstad*
Department of Chemistry and Centre for Materials Science and Nanotechnology, University of Oslo, P. O. Box
Due to cost and availability of nickel metal relative to noble metals such as rhodium, nickel based catalysts are normally preferred for converting light alkanes to synthesis gas (H2 + CO). Råberg et al. [1] and Olafsen et al.
[2] have in the past shown that nanometer sized nickel particles supported on an Mg(Al)O mixed oxide give
very active and stable reforming catalysts. The superior performance of this type of catalysts is believed to be
linked to an optimal combination of support basisity and Ni particle size, e.g. [1,2]. Layered double hydroxides
or hydrotalcite like materials are used as precursors when synthesizing the Ni/Mg(Al)O catalysts; Figure 1.
Various LDH synthesis approaches have been explored for optimizing the nickel dispersion on the Ni/Mg(Al)O
catalysts; e.g. incorporation of nickel in the brucite-like layers as a solid solution [1,2] as well as incorporating
anionic nickel complexes to the LDH interlayer gallery [3,4] followed by calcination- and reduction steps.
It is well know that nitrate based LDHs can be delaminated in formamide into positively charged brucite-like
sheets with a thickness of less than one nanometer, without disturbing the atomic arrangement and the chemical
composition of the brucite-like layers [5]. The obtained stable suspension from the delamination process can in a
next step be utilized by stacking chemically distinct layers (e.g. layers of MgAl and NiAl stabilized in different suspensions) in an ordered or disordered manner, to form a nanocompositite [6]; see Figure 2.
In this work we have explored the potential of controlling nickel particle size and particle size distribution of
Ni/Mg(Al)O catalysts through a novel delamination – reconstruction – calcination/activation route. Good metal
dispersion is achieved through restacking of MgAl and NiAl nanosheets in a way that the NiAl layers are highly
separated in the reconstructed nanocomposite (Figure 2). We present procedures for preparation of the catalytic
materials, detailed materials characterization as well as catalytic testing results for a reference Ni/Mg(Al)O
catalyst and the new Ni/(Mg(Al)O catalyst produced from delaminated LDHs. Test reaction used is dry
reforming of propane.
Figure 1 a) Schematic drawing of the structural
arrangement of LDHs. b) TEM images showing
morphology of as-syn 3MgAl-NO3 and 3NiAl-
NO3.
Figure 2 Schematics showing principle for
delamination of two different LDHs and
subsequent restacking into a nanocomposite.
References
[1] L. Råberg, M. Jensen, U. Olsbye, C. Daniel, S. Haag, C. Mirodatos, A.O. Sjåstad, J. Catal. 249 (2007) 250.
[2] A. Olafsen, Å. Slagtern, I.M. Dahl, U. Olsbye, Y. Schuurman, C. Mirodatos, J. Catal. 229 (2005) 163.
[3] C. Gerardin, D. Kostadinova, N. Sanson, B. Coq, D. Tichit, Chem. Mater. 17 (2005) 6473.
[4] C. Gerardin, D. Kostadinova, N. Sanson, B. Coq, D. Tichit, Chem. Mater. 20 (2008) 2086.
[5] Q. Wu, A. Olafsen, Ø.B. Vistad, J. Roots, P. Norby, J. Mater. Chem. 15 (2005) 4695. [6] R.E. Johnsen, Q. Wu, A.O. Sjåstad, Ø.B. Vistad, F. Krumeich, P. Norby, J. Phys. Chem. C 112 (2008)
16733.
Oral 33
16th Nordic Symposium on Catalysis Studies on perovskite-type oxide LaCoO3±δ as methanol steam reforming catalyst: Effect of Pd and Zn substitution on the CO2 selectivity
J. Kuc1, M. Neumann2, M. Armbrüster2, S. Yoon1, A. Weidenkaff1,3, Santhosh K. Matam1* 1 – Empa, Swiss Federal Laboratories for Materials Science and Technology, CH-8600 Dübendorf, Switzerland 2 – Max-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Str. 40, DE-01187 Dresden, Germany 3 – Institute for Materials Science, University of Stuttgart, Heisenbergstr. 3, DE-70569 Stuttgart, Germany * corresponding: [email protected] Steam reforming of methanol (SRM) got a significant attention as a promising process for on-board H2 generation for fuel cell (FC) applications [1]. Methanol is easier and safer to handle than the compressed H2 gas cylinder. The main drawback of SRM is the formation of CO as a by-product, which can poison Pt electrodes of a FC. Thus, extensive studies are dedicated to develop materials (ranging from classical metal oxides to intermetallic compounds) that can selectively perform SMR reaction as shown in eq. 1 [2]. CH3OH + H2O → 3H2 + CO2 ∆H° = 49 kJmol-1 (1) The present study investigates the novel multifunctional perovskite-type oxides as potential SRM catalysts to produce selectively H2 and CO2. To this end, a series of LaCo1-xBxB’yO3-1/2x (B = Pd2+ and B’ = Zn2+; x = y = 0.025, 0.075 and 0.0127: denoted as LCPZO-1, -2 and -3, respectively), together with the reference materials (LCO, LaCoO3-δ; LCPO, LaCo0.873Pd0.127O3-δ; LCZO, LaCo0.89Zn0.11O3-δ), were prepared by the amorphous citrate sol-gel method and characterized by various physico-chemical techniques. SRM activity of the catalysts was determined in a plug flow reactor equipped with mass spectrometer (MS) and gas chromatograph (GC). Prior to the experiments, catalysts were reductively pretreated at 590°C for 2 h. XRD results reveal that un-substituted LCO contains a single phase rhombohedral perovskite crystal structure with the 𝑅3�𝑐 space group. The structure is retained after partial substitution of Co by Pd and/or Zn ions. In situ XRD and TG-DTA during the reduction and H2-TPR data indicate that the onset reduction temperature of Co ions decreases significantly with Pd substitution, which is attributed to the hydrogen spillover effect [3].
Figure 1. Temperature dependent CH3OH conversion (left) and CO2 selectivity (right) of LCO ( ), LCPO ( ), LCZO ( ), LCPZO-0 ( ), LCPZO-1 ( ), LCPZO-2 ( ) and LCPZO-3 ( ). To evaluate the CO2 selectivity of the catalysts and to eliminate the effect of reverse water gas shift reaction (RWGS) on the selectivity, comparable CH3OH conversion at a given temperature over the catalysts is obtained by changing the catalyst mass. As evident from Figure 1, catalysts LCPZO-1, LCPZO-2 and LCPZO-3 with Pd/Zn show better CO2 selectivity (in the whole temperature range) than the reference materials LCO, LCPO and LCZO. At comparable CH3OH conversion of around 8% at 325°C over the catalysts (except LCPO that shows higher conversion due to Pd), the CO2 selectivity increases from 12 to 80% for reference and Pd/Zn substituted catalysts, respectively. The CO2 selectivity patterns in the high temperature range resemble the CO2 equilibrium curve. The improved selectivity of the catalysts can be attributed to ZnPd-like species [2]. References [1] J. Agrell, M. Boutonnet, and J.L.G. Fierro, Applied Catalysis A: General 253 (2003) 213. [2] M. Armbrüster, M. Behrens, K. Föttinger, S.K. Matam, et al., Catal. Rev. Sci. Eng. 55 (2013) 289. [3] Y.J. Huang, J. Xue, and J.A. Schwarz, Journal of Catalysis 111(1988) 59.
Oral 34
SUPPORTED IONIC LIQUID PHASE (SILP) CATALYZED GAS PHASE ETHYLENE
METHOXY CARBONYLATION
Santosh G. Khokarale*, Eduardo J. Garcia-Suarez, Anders Riisager, Rasmus Fehrmann
Centre for Catalysis and Sustainable Chemistry, Department of Chemistry,
Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark
Supported ionic liquid phase (SILP) is an attractive way to heterogenized homogeneous catalyst. The SILP
technology consists basically in an ionic liquid (IL) film immobilize on a porous solid material (e.g. silica) and a
homogeneous catalyst dissolved in a supported IL layer (Figure 1). In practice, SILP is the perfect combination
of the benefits of both homogeneous catalysts allowing high activity and selectivity and heterogeneous catalysts
due to the large interfacial reaction areas and good product separation. Indeed, SILP catalysis has potential for
efficient catalyst recycling and it makes possible the application of homogeneous catalysis in fixed-bed reactor
technology. Furthermore, the resulting ionic liquid catalyst film is only a few nanometers thick allowing the
complete utilization of both ionic liquid and catalyst since the mass transport resistance from the gas into the
liquid phase is minimized compared with the biphasic systems where ILs are employed as reaction media [1]. In
addition, the very low vapor pressure of ionic liquids makes these catalytic systems optimum to be used in
continuous gas-phase processes minimizing the catalyst deactivation [2]. Therefore, SILP catalysis has attracted
much attention in the last years to the scientific community due to their attractive and successful performance in
many gas-phase catalytic processes with high industrial relevance such as, carbonylation, hydroformylation,
hydroamination, hydrogenation, Suzuki coupling etc., with excellent results in terms of catalytic activity and
catalyst stability [3-6].
Figure.1 SILP catalysis technology
In this work, we reported for the first time the application of the SILP catalysis technology to the continuous
gas-phase methoxycarbonylation of ethylene for methylpropanoate (MP) production (scheme 1) where MP is an
essential monomer in the industrial production of the highly world demanded methylmethacrylate (MMA).
Preliminary screening of the influence of different reactions parameters such as, ionic liquid loading, nature of
the support, metal loading, temperature and GHSV (h-1
), in both catalyst activity and selectivity is performed in
this study.
Scheme 1. Ethylene methoxycarbonylation
[1] A. Riisager, B. Jørgensen, P. Wasserscheid and R. Fehrmann, Chem. Commun. (2006)994.
[2] A. Riisager, R. Fehrmann, M. Haumann, and P.Wasserscheid, Angew. Chem. Int. Ed. 44(2005)815.
[3] R. Fehrmann, A. Riisager and M. Haumann (Eds.), Supported Ionic Liquids: Fundamentals and
Applications, Wiley-VHC, Weinheim, 2014
[4] M. Haumann and A. Riisager, Chem. Rev. 108(2008)1474.
[5] S. Breitenlechner, M. Gleck, T.E. Muller and A. Suppan, J. Mol.Catal. A: Chem. 214(2004)175.
[6] H. Hagiwara, Y. Sugawara, K. Isobe, T.Suzuki, Org. Lett. 6(2004)2325.
Reactant
SILP
Particle
Porous support
Product Metal complex
Ionic Liquid Film
Oral 35
Sonogashira coupling reaction over supported gold nanoparticles
Sheetal Sisodiya1, Joachim Schnadt2, Ola F. Wendt1* 1 - Centre for Analysis and Synthesis, Department of Chemistry, Lund University Box 124, SE-221 00, Lund, Sweden. 2 - Division of Synchrotron Radiation Research, Department of Physics, Lund University Box 118, SE-221 00 Lund, Sweden. * [email protected] The Sonogashira coupling reaction, comprising the carbon-carbon (C-C) bond formation between a terminal alkyne and an aryl or vinyl halide (Scheme 1), represents one of the most important classes of reaction for synthetic organic chemistry and pharmaceuticals. Supported Pd, Ru and/or Pt-based nanoparticles have been extensively studied as catalysts [1,2]. However, their performance is challenged by practical issues, such as, metal leaching, formation of undesired byproducts, and use of cocatalysts. Lately, gold nanoparticles supported on metal oxides have been demonstrated as very selective and efficient catalysts for the title reaction [3]. Still, important aspects, namely: (i) support effect on the dispersion and nature of gold species, (ii) method of deposition of gold, (iii) optimization of reaction conditions, and (iv) reaction mechanism have not yet been addressed in full details. Our work on the Sonogashira coupling of phenylacetylene (PA) with iodobenzene (IB) (Scheme 1) aims to shed light on these crucial points.
Scheme 1. Sonogashira coupling of phenylacetylene (PA) and iodobenzene (IB) to diphenylacetylene (DPA).
Table 1. Characterization of supported Au catalysts.
a Au content by XRF for impregnated samples. b Surface area (SBET) of the carriers in parenthesis.
Different metal oxide (WO3, TiO2, CeO2, V2O5, and Al2O3) supported gold nanoparticles were synthesized by dry impregnation and deposition-precipitation methods. The catalysts were characterized by various techniques, including powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), electron microscopy, X-ray fluorescence (XRF), and N2 sorption. The synthesized catalysts were tested in Sonogashira coupling of PA and IB using a liquid-phase glass batch reactor set-up, equipped with heater with temperature controller and magnetic stirrer. The reactions were carried out at different reaction temperatures (120-200°C), catalyst amounts, solvents, Au loading, and reaction time. The gold content in the as-prepared catalysts by dry impregnation is slightly less than the nominal value of 4 wt.% (Table 1). The samples prepared by deposition-precipitation (DP) revealed lower gold content (~1-2 wt.%) than those of impregnated, even though the nominal loading was same. The latter could be related to the loss of some gold ions during the washing step in the DP method. XRD patterns (not shown for brevity) of the impregnated samples showed sharper reflections of gold compared to those in the DP samples, likely indicating smaller and better dispersed Au nanoparticles for the latter method. The SBET of the carriers remained similar after deposition of Au, which suggests that the gold is deposited on the surface without altering the pore-structure of the carriers. XPS (not shown) spectra of the fresh Au/WO3 unraveled the formation of Au0. Furthermore, electron microscopy analysis is undertaken. The reaction data demonstrated the superiority of DP method over impregnation for Au/TiO2, Au/CeO2, and Au/V2O5 in term of conversion of IB and selectivity to DPA. However, Au/WO3 prepared by dry impregnation and DP exhibited similar conversion (XIB = 78%) and selectivity (SDPA = 62%). Au/WO3 prepared by dry impregnation was further tested to optimize the reaction conditions. The recyclability tests over Au/WO3 showed a little drop of IB conversion (by ~2%) and DPA selectivity (by ~6%) for the 4th cycle, which suggests the robustness of this catalyst. Also, mechanistic insights gained by XPS studies will be presented. References [1] G.W. Kabalka, L. Wang, and R.M. Pagni, Tetrahedron 57 (2001) 8017. [2] S. Gao, N. Zhao, M. Shu, S. Che, Appl. Catal. A: General 388 (2010) 196. [3] A. Corma, H. Garcia, Chem. Soc. Rev. 37 (2008) 2096.
Oral 36
16th
Nordic Symposium on Catalysis/Abstract
Effects of carbon supports in platinum catalysts for propane dehydrogenation
Andrey S. Volynkin1*, Prof. Edd A. Blekkan1, Prof. Magnus Rønning1
Concerted proton-coupled electron transfer (PCET) plays a critical role for substrate activation in biological
redox catalysis. However, the application of PCET mechanisms to organic synthesis remains largely
unexplored. [1][2] Herein, the first highly enantioselective catalytic protocol for the reductive coupling of
ketones and hydrazones is reported. These reactions proceed through neutral ketyl radical intermediates
generated via a concerted PCET event. Ketyl-formation is mediated by a chiral phosphoric acid and the
photoredox catalyst Ir(ppy)2(dtbbpy)PF6. Interestingly, these neutral ketyl radicals appear to remain H-bonded to
the chiral conjugate base of the Brønsted acid during the course of a subsequent C−C bond-forming step,
furnishing syn 1,2-amino alcohol derivatives with excellent levels of diastereo- and enantioselectivity. This
work illustrates the feasibility and advantages of concerted PCET activation in asymmetric radical chemistry.
References
[1] Tarantino, K. T.; Liu, P.; Knowles, R. R. J. Am. Chem. Soc. 2013, 135, 10022-10025
[2] Rono, L. J.; Yayla, H. G.; Wang, D. Y.; Armstrong, M. F.; Knowles, R. R. J. Am. Chem. Soc. 2013, 135,
17735-17738
Oral 39
16th Nordic Symposium on Catalysis Theory-Assisted Development of Z-Selective Olefin Metathesis Catalysts
Giovanni Occhipinti, Vitali Koudriavtsev, Karl W. Törnroos and Vidar R. Jensen* Department of Chemistry, University of Bergen, Allégaten 4, N-5007 Bergen, Norway * [email protected] Catalysis represents an important application area for molecular-level computational studies. The most important role of these studies has traditionally been to provide valuable insight into the mechanisms with which existing, successful catalysts mediate chemical processes. However, little by little, theory is taking on a more active and leading role in the development of new and better catalysts, exemplified by the advent of fully automated in silico drug-design-style methods for catalyst development [1]. Here, we present hands-on examples of interplay between theory and experiment in development of highly Z-selective catalysts for olefin metathesis. Achieving such catalysts has been a major goal in olefin metathesis for years [2], with reasonably selective catalysts being obtained only in recent years [3,4]. We have developed and used a comprehensive computational approach to develop Z-selective olefin metathesis catalysts. In addition to being based on detailed insight into the mechanism of metathesis as mediated by the ruthenium-based catalysts, the approach also takes into account catalytic activity and catalyst stability. Predictions using this strategy are consistent with experimental observations and have led to the realization of new catalysts with up to 96% Z-selectivity [3,5,6]. Equally important is the fact that the computational focus on catalyst stability has paid off: The most recent of the new catalysts, depicted in Figure 1, can be used in air, with unpurified and non-degassed substrates and solvents, and in the presence of acids [5]. These are traits that are unprecedented among previously reported Z-selective olefin metathesis catalysts. They are also very promising with respect to practical applications of Z-selective olefin metathesis.
Figure 1. New, highly Z-selective and robust olefin metathesis catalyst developed with help from theory References
[1] Y. Chu, W. Heyndrickx, G. Occhipinti, V. R. Jensen, B. K. Alsberg J. Am. Chem. Soc. 2012, 134, 8885–8895. M. Foscato, G. Occhipinti, V. Venkatraman, B. K. Alsberg, V. R. Jensen J. Chem. Inf. Model. 2014, in press. [2] A. Fürstner Science 2013, 341, 1357-1364. [3] G. Occhipinti, F. R. Hansen, K. W. Törnroos, V. R. Jensen J. Am. Chem. Soc. 2013, 135, 3331. [4] B. K. Keitz, K. Endo, M. B. Herbert, R. H. Grubbs J. Am. Chem. Soc. 2011, 133, 9686. M. M Flook, A. J. Jiang, R. R. Schrock, P. Müller, A. H. Hoveyda J. Am. Chem. Soc. 2009, 131, 7962. [5] G. Occhipinti, V. Koudriavtsev, K. W. Törnroos and V. R. Jensen Dalton Trans. 2014, in press. [6] V. R. Jensen, G. Occhipinti, F. Hansen, Novel Olefin Metathesis Catalysts. Int. Patent Appl. WO 2012032131, 2012
Oral 40
16th
Nordic Symposium on Catalysis
Reactivity of Cyclometalated Gold(III) Complexes Toward Alkenes
Eirin Langseth1*
, Eline Aa. Tråseth1, Ainara Nova
2, Richard H. Heyn
3 and Mats Tilset
2
1 – Department of Chemistry, University of Oslo, P.O. Box 1033 Blindern, 0315 Oslo, Norway
2 – Centre for Theoretical and Computational Chemistry (CTCC)
Toluene oxidation mechanism over ZrO2-based gasification gas clean-up catalysts
Tiia Viinikainen*, Sonja Kouva, Juha Lehtonen, Jaana Kanervo Aalto University, School of Chemical Technology, P.O. Box 16100, 00076 Aalto, Finland * [email protected] Biomass gasification is an environmentally attractive way to produce energy and synthesis gas that can be converted into liquid biofuels and chemicals [1]. Before utilizing the product it has to be cleaned from impurities, namely from tar and ammonia [2]. Catalytic hot gas cleaning is a preferable option. Lately ZrO2-based catalysts have shown to preferentially oxidize tar molecules within the gas mixture [3-4]. To enable process design ensuring high-quality gas, detailed knowledge of tar decomposition mechanism needs to be established. Therefore, temperature-programmed oxidation of toluene (a tar model compound) was carried out and the reaction mechanism was postulated. Extensive kinetic data was measured for pure ZrO2, Y2O3-ZrO2 and SiO2-ZrO2 by applying three oxygen/toluene feed ratios of 9.2, 16.7 and 28.6 (≈ 1, 2 and 3.5 x theoretical total oxidation ratio, respectively) and three heating rates (6.7, 10 and 15 °C/min) in the temperature range of 200-600 °C. The temperature-programmed reactor was modelled as a dynamic pseudohomogeneous plug flow reactor and the kinetic parameters were estimated by non-linear regression between the measured and simulated molar flows of the reaction components. Four products were detected in all toluene oxidation experiments over ZrO2-based catalysts: CO2, H2O, CO and H2 (typical gas flow out of the reactor shown for ZrO2 in Fig. 1). The effect of oxygen to toluene feed ratio to CO2/CO formation ratio is shown in Fig. 2. It can be seen that with higher O2/TOL ratio, the CO2/CO ratio increases rapidly at higher temperatures implying potential loss of valuable gasification gas components. The data analysis showed that toluene starts to convert at lower temperatures than oxygen, indicating that surface species are involved. Therefore, the formation of benzyl species [C7H7] from toluene (reaction 1) is suggested to be the first step in toluene oxidation, in accordance with our previous spectroscopic findings [5]. Furthermore, the product formation curves over all three catalysts showed two shapes; CO2 and H2O curves are similar, as are those for CO and H2, suggesting that products are formed in pairs. Including the measured product ratios, a preliminary reaction mechanism is proposed. In this mechanism, benzyl species are first decomposed into 5 CH-units, 1 CH2-unit and 1 C-unit on the surface (reaction 2), products are formed in pairs (reactions 3 and 4) and reactions 5-8 will be used to obtain the measured product ratios. Further data analysis for reaction mechanism hypotheses and the obtained modelling results will be shown in the presentation. C7H8 (g) → [C7H7] + [H] (1) [C7H7] → 5 [CH] + [CH2] + [C] (2) [CH] + 1.25 O2 (g) → CO2 (g) + 0.5 H2O (g) (3) [CH2] + 0.5 O2 (g) → CO (g) + H2 (g) (4)
Figure 1. Typical gas flow out from the reactor Figure 2. The effect of feed ratio to CO2/CO ratio over ZrO2 (O2/TOL: 9.2, heating rate: 10 °C/min). over ZrO2 (heating rate: 10 °C/min). [1] P. Gallezot, A. Kiennemann, in: Ertl, G., Knözinger, H., Schüth, F. and Weitkamp, J. (Eds.), Handbook of Heterogeneous Catalysis, 2nd ed., Wiley-VCH, Weinheim, 2008. [2] D. Sutton, B. Kelleher, J.R.H. Ross, Fuel Process. Technol. 3 (2001) 155. [3] S.J. Juutilainen, P.A. Simell, A.O.I. Krause, Appl. Catal. B 62 (2006) 86. [4] T. Viinikainen, H. Rönkkönen, H. Bradshaw, H. Stephenson, S. Airaksinen, M. Reinikainen, P. Simell, O. Krause, Appl. Catal. A. 362 (2009) 169. [5] T. Viinikainen, E.I. Kauppi, S.T. Korhonen, L. Lefferts, J.M. Kanervo, J. Lehtonen, Appl. Catal. B 142-143 (2013) 769.