Purification and Properties of an NADPH-dependent Carbonyl Reductase from Human Brain RELATIONSHIP TO PROSTAGLANDIN 9-KETOREDUCTASE AND XENOBIOTIC KETONE REDUCTASE* (Received for publication, September 2, 1980) A nonspecific NADPH-dependent carbonyl reductase fromhumanbrain(formerly designated as aldehyde reductase 1; Ris, M. M., and von Wartburg, J. P. (1973) Eur. J. Biochem 37,69-77) has been purified to homo- geneity. The enzyme reduces a number of biologically and pharmacologically active carbonyl compounds. Quinones, e.g. menadione, ubiquinone, and tocopher- olquinoneare the best substrates, followed by alde- hydes containing an activated carbonyl moiety, e.g. 4- nitrobenzaldehyde or methylglyoxal. The enzyme also reduces ketones, e.g. prostaglandins of the E and A class, the anthracycline antibiotic daunorubicin and 3- ketosteroids. During catalysis the pro 4s hydrogen atom of the nicotinamide ring of NADPH is transferred to the substrate. Flavonoids, e.g. quercetin and rutin, indomethacin, ethacrynic acid, and dicoumarol inhibit the enzyme activity. 4-Hydroxymercuribenzoate and iodoacetate inactivate the enzyme. NADPH and sub- strate do not protect against the loss of activity. Car- bonyl reductase consists of a single polypeptide chain with a molecular weight of 30,000. The native enzyme occurs in three molecular forms with similar substrate specificity and inhibitor sensitivity. The isoelectric points of the three enzyme species are 6.95, 7.85, and 8.5. In the presence of coenzyme the isoelectric points are shifted to 5.2 to 5.9. The comparison of structural and enzymic features of carbonylreductase with other monomeric oxidoreductases suggests a close relation- ship of carbonyl reductase with prostaglandin 9-keto- reductase and xenobiotic ketone reductase. Carbonyl compounds are widely distributed in living matter. They are encountered as metabolic intermediates and as mediators of highly specific biological effects. Generally, car- bonyl compounds, such as the metabolic intermediates of the energy-generating pathways are further oxidized. However, in some cases reduction appears to be favored. Enzyme activities which have been characterized by their ability to reduce particular aldehydes and ketones havebeen reported in var- ious species. A number of these reductases share typical features, in that they require NADPH as coenzyme, display a slightly acid pH optimum, are localized in the cytoplasm of the cell, and are composed of a single peptide chain. Aldehyde reductase (EC 1.1.1.2) and aldose reductase (EC 1.1.1.21) occur in a number of tissues of various species (1, 2). They have been implicated in the metabolism of aldehydes derived * This work was supported by Grant 3.404-0.78 from the Swiss National Science Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article musttherefore be herebymarked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. from biogenic amines (3,4), the reduction of sugar aldehydes (5), and the biosynthesis of L-ascorbate (6). Prostaglandin 9- ketoreductase, another member of the “aldehyde-ketone re- ductase family,” catalyzes the interconversion of the E and F class prostaglandins. This enzyme, too, occurs in a number of tissues and species (7-14). Finally, a group of similar enzymes catalyze the reduction of xenobiotic aldehydes and ketones (15-17). One of them has been purified to homogeneity from rat liver (15) and is identicalwith the classified aldehyde reductase (16). Another activity which has been characterized by its ability to reduce the ketone group of several drugs is referred to as xenobiotic ketone reductase (17). Four enzymes capable of reducing both aldehydes and ketones have also been demonstrated in human brain (18). Two of the activities correspond to aldehyde and aldose reductase, respectively, and a third enzyme has been charac- terized as succinicsemialdehyde reductase (4). The fourth enzyme differs from the other aldehyde reductases by its substrate specificity and inhibitor sensitivity and has not yet been further characterized. In this paper the purification and characterization of this enzyme are described, and evidence is presented that it has properties similar to prostaglandin 9-ketoreductase and xe- nobiotic ketone reductase. A preliminary account of this work has been presented.’ EXPERIMENTAL PROCEDURES Materials Human tissue from both sexes was obtained from legal medical autopsies; causes of death were accidents, homicides, suicides, and cardiovascular diseases. The tissue was frozen 6 to 20 h after death and stored at -20°C. Proteins used for Sephadex column and poly- acrylamide gel calibration were purchased from Boehringer (Mann- heim, Germany) and Sigma (St. Louis, Mo.). Sigma also furnished neuraminidase from Clostridium perfringens, all coenzyme nucleo- tides, dicoumarol, and warfarin (3-[a-acetonylbenzyl]-4-hydroxycou- marin). Menadione was bought from F. Hoffmann-La Roche (Basel, Switzerland), and daunorubicin (daunomycin) was obtained from Serva (Heidelberg, Germany). The compounds listed below were gifts from the following companies: ubiquinones and a-tocopherolquinone (F. Hoffmann-La Roche); PGE,‘, PGE2 and PGAt (Sandoz-Wander, Basel, Switzerland); ethacrynic acid and indomethacin (Merck, Sharp and Dohme, Rahway, N.J.); flavonoids and D-catechin &ma, Nyon, Switzerland); Cibacron 3 G-A (Ciba-Geigy, Basel Switzerland). Menadiol and ubiquinol-1 were synthesized from the corresponding quinones by reduction with sodiumdithionite following the procedure of Mayer and Isler (19). PGFI, and PGFz,, were synthesized from Hoffman, P. L., Wermuth, B., and von Wartburg, J. P. (1979) Third International Symposium on Alcohol and Aldehyde Metabo- lizing Systems, Toronto, Abstr. 81. The abbreviations used are: PGE, prostaglandin E; PGF, prosta- glandin F; PGA, prostaglandin A. 1206
Transcript
Purification and Properties of an NADPH-dependent Carbonyl Reductase from Human Brain RELATIONSHIP TO PROSTAGLANDIN 9-KETOREDUCTASE AND XENOBIOTIC KETONE REDUCTASE*
(Received for publication, September 2, 1980)
A nonspecific NADPH-dependent carbonyl reductase from human brain (formerly designated as aldehyde reductase 1; Ris, M. M., and von Wartburg, J. P. (1973) Eur. J. Biochem 37,69-77) has been purified to homo- geneity. The enzyme reduces a number of biologically and pharmacologically active carbonyl compounds. Quinones, e.g. menadione, ubiquinone, and tocopher- olquinone are the best substrates, followed by alde- hydes containing an activated carbonyl moiety, e.g. 4- nitrobenzaldehyde or methylglyoxal. The enzyme also reduces ketones, e.g. prostaglandins of the E and A class, the anthracycline antibiotic daunorubicin and 3- ketosteroids. During catalysis the pro 4s hydrogen atom of the nicotinamide ring of NADPH is transferred to the substrate. Flavonoids, e.g. quercetin and rutin, indomethacin, ethacrynic acid, and dicoumarol inhibit the enzyme activity. 4-Hydroxymercuribenzoate and iodoacetate inactivate the enzyme. NADPH and sub- strate do not protect against the loss of activity. Car- bonyl reductase consists of a single polypeptide chain with a molecular weight of 30,000. The native enzyme occurs in three molecular forms with similar substrate specificity and inhibitor sensitivity. The isoelectric points of the three enzyme species are 6.95, 7.85, and 8.5. In the presence of coenzyme the isoelectric points are shifted to 5.2 to 5.9. The comparison of structural and enzymic features of carbonyl reductase with other monomeric oxidoreductases suggests a close relation- ship of carbonyl reductase with prostaglandin 9-keto- reductase and xenobiotic ketone reductase.
Carbonyl compounds are widely distributed in living matter. They are encountered as metabolic intermediates and as mediators of highly specific biological effects. Generally, car- bonyl compounds, such as the metabolic intermediates of the energy-generating pathways are further oxidized. However, in some cases reduction appears to be favored. Enzyme activities which have been characterized by their ability to reduce particular aldehydes and ketones have been reported in var- ious species. A number of these reductases share typical features, in that they require NADPH as coenzyme, display a slightly acid pH optimum, are localized in the cytoplasm of the cell, and are composed of a single peptide chain. Aldehyde reductase (EC 1.1.1.2) and aldose reductase (EC 1.1.1.21) occur in a number of tissues of various species (1, 2). They have been implicated in the metabolism of aldehydes derived
* This work was supported by Grant 3.404-0.78 from the Swiss National Science Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
from biogenic amines (3 ,4) , the reduction of sugar aldehydes (5), and the biosynthesis of L-ascorbate (6). Prostaglandin 9- ketoreductase, another member of the “aldehyde-ketone re- ductase family,” catalyzes the interconversion of the E and F class prostaglandins. This enzyme, too, occurs in a number of tissues and species (7-14). Finally, a group of similar enzymes catalyze the reduction of xenobiotic aldehydes and ketones (15-17). One of them has been purified to homogeneity from rat liver (15) and is identical with the classified aldehyde reductase (16). Another activity which has been characterized by its ability to reduce the ketone group of several drugs is referred to as xenobiotic ketone reductase (17).
Four enzymes capable of reducing both aldehydes and ketones have also been demonstrated in human brain (18). Two of the activities correspond to aldehyde and aldose reductase, respectively, and a third enzyme has been charac- terized as succinic semialdehyde reductase (4). The fourth enzyme differs from the other aldehyde reductases by its substrate specificity and inhibitor sensitivity and has not yet been further characterized.
In this paper the purification and characterization of this enzyme are described, and evidence is presented that it has properties similar to prostaglandin 9-ketoreductase and xe- nobiotic ketone reductase. A preliminary account of this work has been presented.’
EXPERIMENTAL PROCEDURES
Materials
Human tissue from both sexes was obtained from legal medical autopsies; causes of death were accidents, homicides, suicides, and cardiovascular diseases. The tissue was frozen 6 to 20 h after death and stored at -20°C. Proteins used for Sephadex column and poly- acrylamide gel calibration were purchased from Boehringer (Mann- heim, Germany) and Sigma (St. Louis, Mo.). Sigma also furnished neuraminidase from Clostridium perfringens, all coenzyme nucleo- tides, dicoumarol, and warfarin (3-[a-acetonylbenzyl]-4-hydroxycou- marin). Menadione was bought from F. Hoffmann-La Roche (Basel, Switzerland), and daunorubicin (daunomycin) was obtained from Serva (Heidelberg, Germany). The compounds listed below were gifts from the following companies: ubiquinones and a-tocopherolquinone (F. Hoffmann-La Roche); PGE,‘, PGE2 and PGAt (Sandoz-Wander, Basel, Switzerland); ethacrynic acid and indomethacin (Merck, Sharp and Dohme, Rahway, N.J.); flavonoids and D-catechin &ma, Nyon, Switzerland); Cibacron 3 G-A (Ciba-Geigy, Basel Switzerland).
Menadiol and ubiquinol-1 were synthesized from the corresponding quinones by reduction with sodium dithionite following the procedure of Mayer and Isler (19). PGFI, and PGFz,, were synthesized from
Hoffman, P. L., Wermuth, B., and von Wartburg, J. P. (1979) Third International Symposium on Alcohol and Aldehyde Metabo- lizing Systems, Toronto, Abstr. 81.
The abbreviations used are: PGE, prostaglandin E; PGF, prosta- glandin F; PGA, prostaglandin A.
1206
Human NADPH-dependent Carbonyl Reductase 1207
PGEl and PGE,, respectively, with sodium borohydride according to Bishai and Coceani (12). Biogenic aldehydes were prepared as de- scribed by Tabakoff et al. (20) by incubating the amines with mono- amine oxidase-containing preparations of lysed mitochondria from calf liver. All other chemicals (of the highest grade commercially available) were obtained from Fluka (Buchs, Switzerland) and Merck (Darmstadt, Germany). Substrates and inhibitors which were not sufficiently soluble in water were dissolved in methanol/water to a final methanol concentration not exceeding 2%. This concentration had no effect on the catalytic activity of carbonyl reductase.
Cellulose resins were obtained from Whatman (Maidstone, Great Britain). Sephadex G-100 and DEAE-Sepharose were purchased from Pharmacia (Uppsala, Sweden). 8-(6-Aminohexyl)-amino-2"phos- phoadenosine diphosphoribose-Sepharose was a gift from Dr. C. Y. Lee (National Institute of Environmental Health Sciences, Research Triangle Park, N.C.).
Methods
Enzyme Assay Reaction mixtures consisted of 60 mM sodium phosphate, pH 6.5,
enzyme, 0.08 mM NADPH, and substrates at various concentrations as indicated in tables and figures in a total volume of 1.0 ml. Reactions were initiated by the addition of substrate, and the decrease in absorbance at 340 nm was monitored with an SP6-300 or SP 1800 recording spectrophotometer (Pye Unicam, Cambridge, England) a t 25'C. Blanks without substrate and blanks without enzyme were routinely included. Inhibitors were added to the reaction mixtures after the change in absorbance had been monitored for an initial period, so that the activity in the presence of inhibitor could be directly compared. One enzyme unit is defined as the change in absorbance at 340 nm corresponding to the oxidation of 1 pnol of NADPH.
Protein Concentration
This was estimated from the absorbance at 280 and 260 nm (21).
Isoelectric Focusing
Isoelectric focusing was carried out in a Uniphor column electro- focusing system (LKB Producter AB, Bromma, Sweden) with pH gradients of pH 3 to 10. Solutions of 1% Ampholine were used, and as a support a density gradient from 5 to 50% sucrose was prepared. A 5-ml enzyme sample in 10 mM sodium phosphate buffer, pH 7.0, containing 20% (w/v) sucrose, was applied in the middle of the column. Electrofocusing was performed for 2 days a t a maximum electrical output of 3 watts. In addition, isoelectric focusing of purified carbonyl reductase was performed on thin layer polyacrylamide gels (Ampholine PAG plates, LKB Producter AB) as described by the manufacturer. Enzyme bands were visualized by staining with Coo- massie brilliant blue.
Polyacrylamide Gel Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed on 10 and 12.5% gels prepared in 0.375 M Tris/CI, pH 8.9, containing 0.1% sodium dodecyl sulfate as described by Laemmli (22). Protein bands were stained with Coomassie brilliant blue.
Polyacrylamide gel electrophoresis in the absence of sodium do- decyl sulfate was accomplished with 7.5% acrylamide gels using the same buffer system. Enzyme activity was revealed by immersing the gels in 0.1 M sodium phosphate buffer, pH 7.0, containing 0.1 mM NADPH, 0.2 mM menadione, and nitroblue tetrazolium (0.5 mg/ml). In this system menadiol, which is produced by the enzymic reaction, reduces nitroblue tetrazolium to formazan, and enzyme activity is detected by the appearance of a purple hand.
Stereochemistry of Hydrogen Transfer Tritium-labeled (4S)-[4-"H]NADPH and (4R)-[4-"H]NADPH were
prepared enzymically from ~- [ I -~H]ghcose (New England Nuclear, Dreieichen, Germany) and NADP+ as described by Wermuth et al. (23). In order to determine which of the two hydrogen atoms of NADPH was transferred during catalysis, carbonyl reductase was incubated with 0.5 mM (4s)- or (4R)-[4-3H]NADPH and 1 mM car- bonyl substrate in 20 mM sodium phosphate, pH 7.0. When no further decrease in absorbance at 340 nm was observed, the incubation mixture was diluted with H20 to a conductivity of 5 ms cm" and applied to a column packed with DEAE-Sepharose, equilibrated
against 10 mM sodium phosphate, pH 6.0. NADP' was eluted with a gradient 0 to 400 mM NaCl in 10 mM phosphate, pH 6.0, and radio- activity was determined by scintillation counting.
Preparation and Isolation of Reaction Products
Enzymically reduced metabolites of substrates were prepared by the reaction of parent carbonyl compounds with purified carbonyl reductase in the presence of NADPH. The reaction mixtures (2 to 4 ml) consisted of 0.1 M sodium phosphate, pH 7.0, 1 to 2 mM (48-14- 'HJNADPH, enzyme solution, and 1 to 5 mM carbonyl compound (menadione, ubiquinone-1, PGE,, and 4-nitrobenzaldehyde). The re- action mixtures were incubated at 25°C until no further decrease in absorbance at 340 nm was observed (1 to 4 h). The reaction products of menadione, ubiquinone-I, and 4-nitrobenzaldehyde were extracted with twice the volume CHC13. The reaction mixture containing .pros- taglandin was acidified with 1 M citric acid, and the products were extracted with ethyl acetate. The organic phases were separated and concentrated by evaporating the solvent under nitrogen. Controls of each substrate were similarly prepared without enzyme. Aliquots (10 to 20 pl) of the organic extracts and of reference compounds were applied to silica thin layer plates containing fluorescence indicator (E. Merck AG, Darmstadt, Germany). Menadione and ubiquinone-1 and their metabolites were chromatographed in a solvent of benzene/ methanol (85:15). Prostaglandins were chromatographed in a solvent of benzene/dioxane/acetic acid (50:502), and 4-nitrobenzaldehyde and its metabolites were separated in chloroform. The carbonyl compounds and metabolites were observed as quenching spots on the fluorescent background. In addition, prostaglandins were visualized by spraying with a 10% solution of phosphomolybdic acid in ethanol and gentle heating. Spots were scraped into counting vials, and radioactivity was determined by scintillation counting.
Purification of Carbonyl Reductase
To avoid bacterial growth all buffers contained 1 mM NaN9 (which has no effect on the catalytic activity of carbonyl reductase), and the whole purification was carried out a t 4'C. Elution of ultraviolet light absorbing material was monitored with a Uvicord I1 (LKB Producter AB).
Step 1: Extraction-Approximately 500 g of human tissue (brain, liver) were homogenized with an equal amount (v/w) of 0.1 M sodium phosphate buffer, pH 7.4, in a Waring Blendor for 2 to 3 min. The homogenate was centrifuged for 2 h a t 20,000 X g. The precipitate was re-extracted with the same volume of buffer and recentrifuged. The precipitate was discarded. The pooled supernatants were dialyzed for 24 h against three times 10 liters of 5 mM sodium phosphate buffer, pH 7.4, and centrifuged for 1 h a t 20,000 X g.
Step 2: DEAE-Cellulose Chromatography-The dialyzed extract was applied to a column (2.4 X 50 cm) packed with DEAE-cellulose, equilibrated against 5 mM sodium phosphate buffer, pH 7.4. Carbonyl reductase did not adsorb to the ion exchanger under these conditions and was recovered in the eluate.
Step 3: CM-Cellulose Chromatography-The eluate was dialyzed against three times 10 liters of 5 mM sodium phosphate buffer, pH 6.5, and precipitated protein was removed by centrifugation for 1 h at 20,000 X g. The supernatant was applied to a column (2.4 X 50 cm) packed with CM-cellulose equilibrated against 5 mM sodium phos- phate, pH 6.5. The column was percolated with starting buffer to remove nonadsorbed inactive proteins. The adsorbed enzyme was eluted with a linear gradient of 5 to 100 mM sodium phosphate buffer, pH 6.5. One asymmetric peak of reductase activity was obtained, and all active fractions were pooled.
Step 4: Gel Filtration-The pool from the CM-cellulose containing carbonyl reductase activity (maximum 200 m l ) was applied to a Sephadex G-100 column (7 X 200 cm). Elution was performed with 10 mM Tris/phosphate buffer, pH 7.6. Fractions with enzyme activity were pooled.
Step 5: Affinity Chromatography on 8-(6-Amznohexyl)-amLno-2'- phosphoadenosine Diphosphoribose-Sepharose-The pool from Step 4 was applied on a column (1 X 50 cm) packed with 8-(6- aminohexyl)-amino-2'-phosphoadenosine diphosphoribose-Sepha- rose equilibrated against 10 mM Tris/phosphate buffer, pH 7.6. The column was washed with 50 mM Tris/phosphate buffer, pH 7.6, until the eluate showed no absorbance at 280 nm. Carbonyl reductase was eluted by a pulse of 10 PM NADPH in 20 mM Tris/phosphate, pH 7.6. Fractions containing carbonyl reductase were pooled and dialyzed against 10 mM Tris/phosphate, pH 8.6.
Step 6: DEAE-Sepharose Chromatography-The dialysate was
1208 Human NADPH-dependent Carbonyl Reductase
applied to a column (1 X 50 cm) packed with DEAE-Sepharose, equilibrated against dialysis buffer. The adsorbed enzyme was eluted by a linear gradient of 0 to 100 m~ NaCl in 10 mM Tris/phosphate, pH 8.6. Fractions containing carbonyl reductase activity were pooled and stored at 4OC.
RESULTS
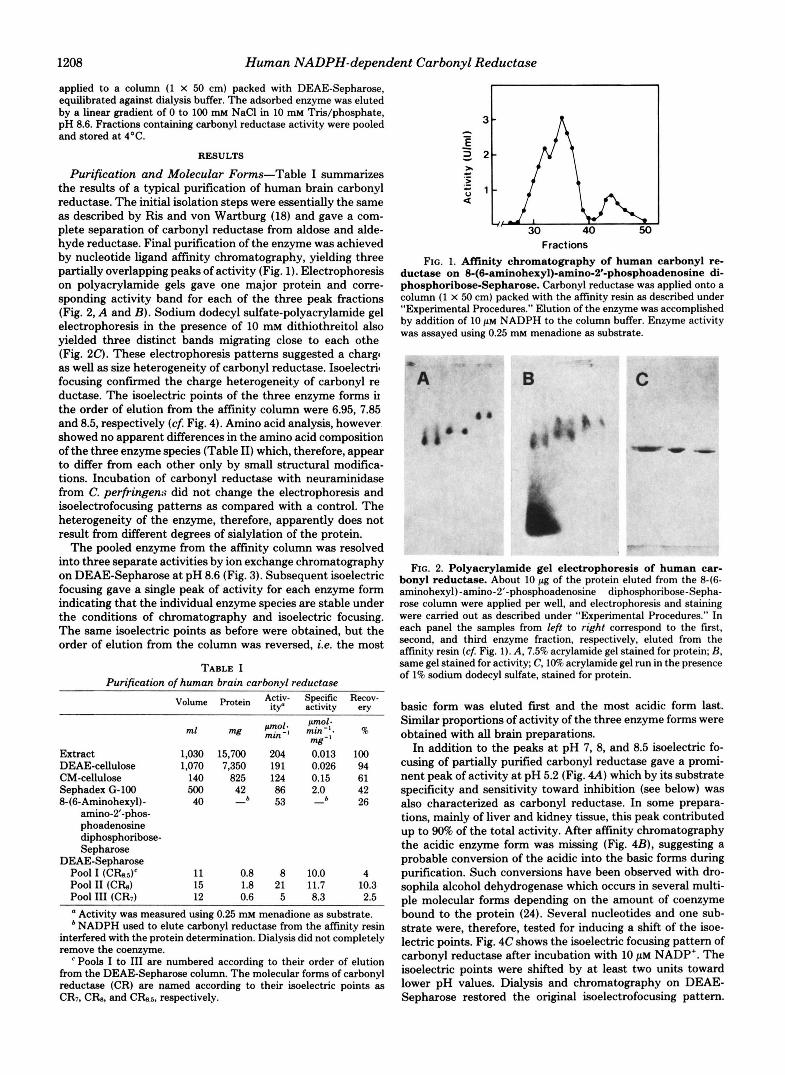
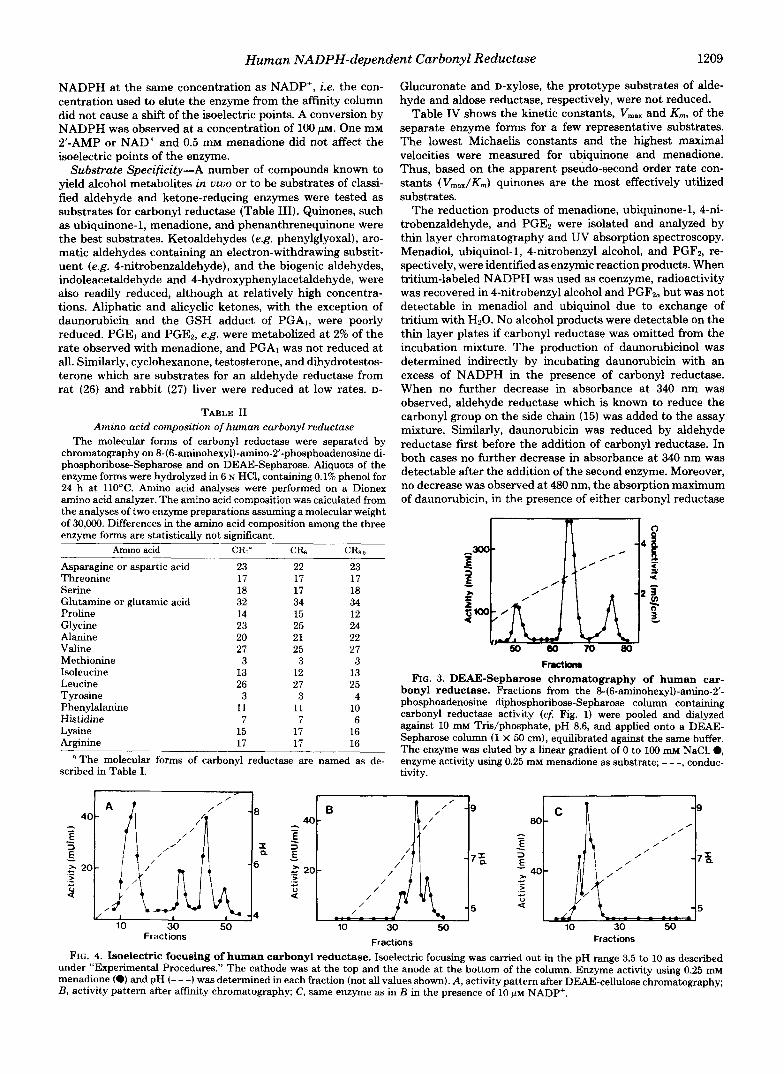
Purification and Molecular Forms-Table I summarizes the results of a typical purification of human brain carbonyl reductase. The initial isolation steps were essentially the same as described by Ris and von Wartburg (18) and gave a com- plete separation of carbonyl reductase from aldose and alde- hyde reductase. Final purification of the enzyme was achieved by nucleotide ligand aftinity chromatography, yielding three partially overlapping peaks of activity (Fig. 1). Electrophoresis on polyacrylamide gels gave one major protein and corre- sponding activity band for each of the three peak fractions (Fig. 2, A and B) . Sodium dodecyl sulfate-polyacrylamide gel electrophoresis in the presence of 10 mM dithiothreitol also yielded three distinct bands migrating close to each othe (Fig. 20. These electrophoresis patterns suggested a chargc as well as size heterogeneity of carbonyl reductase. Isoelectril focusing confirmed the charge heterogeneity of carbonyl re ductase. The isoelectric points of the three enzyme forms ir the order of elution from the affinity column were 6.95, 7.85 and 8.5, respectively (cf. Fig. 4). Amino acid analysis, however showed no apparent differences in the amino acid composition of the three enzyme species (Table 11) which, therefore, appear to differ from each other only by small structural modifica- tions. Incubation of carbonyl reductase with neuraminidase from C. perfringen:r did not change the electrophoresis and isoelectrofocusing patterns as compared with a control. The heterogeneity of the enzyme, therefore, apparently does not result from different degrees of sialylation of the protein.
The pooled enzyme from the affinity column was resolved into three separate activities by ion exchange chromatography on DEAE-Sepharose at pH 8.6 (Fig. 3). Subsequent isoelectric focusing gave a single peak of activity for each enzyme form indicating that the individual enzyme species are stable under the conditions of chromatography and isoelectric focusing. The same isoelectric points as before were obtained, but the order of elution from the column was reversed, i.e. the most
TABLE I Purification of human brain carbonyl reductase
DEAE-Sepharose Po01 I (CRs.s)‘ 11 0.8 8 10.0 4 Pool I1 ( C h ) 15 1.8 21 11.7 10.3 Po01 111 (CR7) 12 0.6 5 8.3 2.5
Activity was measured using 0.25 mM menadione as substrate. NADPH used to elute carbonyl reductase from the affinity resin
interfered with the protein determination. Dialysis did not completely remove the coenzyme.
Pools I to 111 are numbered according to their order of elution from the DEAE-Sepharose column. The molecular forms of carbonyl reductase (CR) are named according to their isoelectric points as CR7, C b . and Cb.5, respectively.
Fractions FIG. 1. Affinity chromatography of human carbonyl re-
ductase on 8-(6-aminohexyl)-amino-2’-phosphoadenosine di- phosphoribose-Sepharose. Carbonyl reductase was applied onto a column (1 X 50 cm) packed with the affinity resin as described under “Experimental Procedures.” Elution of the enzyme was accomplished by addition of 10 p~ NADPH to the column buffer. Enzyme activity was assayed using 0.25 mM menadione as substrate.
FIG. 2. Polyacrylamide gel electrophoresis of human car- bonyl reductase. About 10 pg of the protein eluted from the 846- aminohexyl)-amino-2’-phosphoadenosine diphosphoribose-Sepha- rose column were applied per well, and electrophoresis and staining were carried out as described under “Experimental Procedures.” In each panel the samples from left to right correspond to the first, second, and thud enzyme fraction, respectively, eluted from the aftinity resin (cf Fig. 1). A, 7.5% acrylamide gel stained for protein; B, same gel stained for activity; C, 10% acrylamide gel run in the presence of 1% sodium dodecyl sulfate, stained for protein.
basic form was eluted first and the most acidic form last. Similar proportions of activity of the three enzyme forms were obtained with all brain preparations.
In addition to the peaks at pH 7, 8, and 8.5 isoelectric fo- cusing of partially purified carbonyl reductase gave a promi- nent peak of activity at pH 5.2 (Fig. 4A) which by its substrate specificity and sensitivity toward inhibition (see below) was also characterized as carbonyl reductase. In some prepara- tions, mainly of liver and kidney tissue, this peak contributed up to 90% of the total activity. After affinity chromatography the acidic enzyme form was missing (Fig. 4B), suggesting a probable conversion of the acidic into the basic forms during purification. Such conversions have been observed with dro- sophila alcohol dehydrogenase which occurs in several multi- ple molecular forms depending on the amount of coenzyme bound to the protein (24). Several nucleotides and one sub- strate were, therefore, tested for inducing a shift of the isoe- lectric points. Fig. 4C shows the isoelectric focusing pattern of carbonyl reductase after incubation with 10 p~ NADP’. The isoelectric points were shifted by at least two units toward lower pH values. Dialysis and chromatography on DEAE- Sepharose restored the original isoelectrofocusing pattern.
Human NADPH-dependent Carbonyl Reductase 1209
NADPH at the same concentration as NADP’, ie. the con- centration used to elute the enzyme from the affinity column did not cause a shift of the isoelectric points. A conversion by NADPH was observed at a concentration of 100 p ~ . One mM 2”AMP or NAD’ and 0.5 mM menadione did not affect the isoelectric points of the enzyme.
Substrate Specificity-A number of compounds known to yield alcohol metabolites in vivo or to be substrates of classi- fied aldehyde and ketone-reducing enzymes were tested as substrates for carbonyl reductase (Table 111). Quinones, such as ubiquinone-1, menadione, and phenanthrenequinone were the best substrates. Ketoaldehydes (e.g. phenylglyoxal), aro- matic aldehydes containing an electron-withdrawing substit- uent (e.g. 4-nitrobenzaldehyde), and the biogenic aldehydes, indoleacetaldehyde and 4-hydroxyphenylacetaldehyde, were also readily reduced, although at relatively high concentra- tions. Aliphatic and alicyclic ketones, with the exception of daunorubicin and the GSH adduct of PGA1, were poorly reduced. PGEl and PGE2, e.g. were metabolized at 2% of the rate observed with menadione, and PGA, was not reduced at all. Similarly, cyclohexanone, testosterone, and dihydrotestos- terone which are substrates for an aldehyde reductase from rat (26) and rabbit (27) liver were reduced at low rates. D-
TABLE I1 Amino acid composition of human carbonyl reductase
The molecular forms of carbonyl reductase were separated by chromatography on 8-(6-aminohexyl)-amino-2”phosphoadenosine di- phosphoribose-Sepharose and on DEAE-Sepharose. Aliquots of the enzyme forms were hydrolyzed in 6 N HCI, containing 0.1% phenol for 24 h at 110°C. Amino acid analyses were performed on a Dionex amino acid analyzer. The amino acid composition was calculated from the analyses of two enzyme preparations assuming a molecular weight of 30,000. Differences in the amino acid composition among the three enzyme forms are statistically not significant.
scribed in Table I. The molecular forms of carbonyl reductase are named as de-
Glucuronate and D-xylose, the prototype substrates of alde- hyde and aldose reductase, respectively, were not reduced.
Table IV shows the kinetic constants, V,,, and Km, of the separate enzyme forms for a few representative substrates. The lowest Michaelii constants and the highest maximal velocities were measured for ubiquinone and menadione. Thus, based on the apparent pseudo-second order rate con- stants ( Vmax/K,,,) quinones are the most effectively utilized substrates.
The reduction products of menadione, ubiquinone-1, 4-ni- trobenzaldehyde, and PGE2 were isolated and analyzed by thin layer chromatography and UV absorption spectroscopy. Menadiol, ubiquinol-1, 4-nitrobenzyl alcohol, and PGF2, re- spectively, were identified as enzymic reaction products. When tritium-labeled NADPH was used as coenzyme, radioactivity was recovered in 4-nitrobenzyl alcohol and PGF2, but was not detectable in menadiol and ubiquinol due to exchange of tritium with H20. No alcohol products were detectable on the thin layer plates if carbonyl reductase was omitted from the incubation mixture. The production of daunorubicinol was determined indirectly by incubating daunorubicin with an excess of NADPH in the presence of carbonyl reductase. When no further decrease in absorbance at 340 nm was observed, aldehyde reductase which is known to reduce the carbonyl group on the side chain (15) was added to the assay mixture. Similarly, daunorubicin was reduced by aldehyde reductase first before the addition of carbonyl reductase. In both cases no further decrease in absorbance at 340 nm was detectable after the addition of the second enzyme. Moreover, no decrease was observed at 480 nm, the absorption maximum of daunorubicin, in the presence of either carbonyl reductase
Frutkmm FIG. 3. DEAE-Sepharose chromatography of human car-
bonyl reductase. Fractions from the 8-(6-aminohexyl)-amino-2’- phosphoadenosine diphosphoribose-Sepharose column containing carbonyl reductase activity (cf . Fig. 1) were pooled and dialyzed against 10 m Tris/phosphate, pH 8.6, and applied onto a DEAE- Sepharose column (1 X 50 cm), equilibrated against the same buffer. The enzyme was eluted by a linear gradient of 0 to 100 mM NaC1.0, enzyme activity using 0.25 mM menadione as substrate; - - -, conduc- tivity.
, B - 9 /’
40 - - - 3 E
/
E - /
- /(, - 7 % g 20-
4 > .- +
- 5
10 30 50 Fractions Fractions Fractions
FIG. 4. Isoelectric focusing of human carbonyl reductase. Isoelectric focusing was carried out in the pH range 3.5 to 10 as described under “Experimental Procedures.” The cathode was at the top and the anode at the bottom of the column. Enzyme activity using 0.25 mM menadione (0) and pH (- - -) was determined in each fraction (not all values shown). A, activity pattern after DEAE-cellulose chromatography; B, activity pattern after affinity chromatography; C, same enzyme as in B in the presence of 10 p~ NADP+.
1210 Human NADPH-dependent Carbonyl Reductase
TABLE I11 Substrate specificity of human carbonyl reductase
Enzyme activity was measured as described under "Experimental Procedures." The pooled enzyme fractions after the affinity chroma- tography step were used as enzyme source. The activity with 4- nitrobenzaldehyde was set as 100% allowing the direct comparison of the results with those from previous work on human brain aldehyde reductases (4, 18).
Determined in the presence of 0.1% Triton X-100. PGAl and 10 IIIM GSH were incubated in the assay mixture for 10
Determined at 410 nm, using an extinction coefficient of 10' M"
Determined at 600 run, using an extinction coefficient of 20. lo3
min before carbonyl reductase was added.
cm".
M" cm".
or aldehyde reductase, indicating that neither enzyme reduces the ring carbonyl groups.
Carbonyl reductase required NADPH as coenzyme. No activity was measurable with menadione or 4-nitrobenzalde- hyde when NADPH was replaced by the same concentration of NADH. The reverse reaction, i.e. the dehydrogenation of the alcohol or quinol to the corresponding carbonyl group was tested in the presence of 0.5 mM NADP* with menadiol and 4-nitrobenzyl alcohol as substrates. No NADPH was produced at pH 7 or 9. Similarly, testosterone, 5a- and 5P-androstan- 17@-01-3-one, which are substrates for guinea pig liver alde- hyde reductase (28) were not oxidized. However, NADPH production was observed in the presence of 1 mM PGAI, a substrate of 15-hydroxyprostaglandin dehydrogenase (13). At pH 7 and 9 the activity was 12 and 25% respectively, of the one obtained with 4-nitrobenzaldehyde under standard con- ditions. Two micromolar quercetin inhibited the dehydroge- nase activity by about 40%. Unlike PGA,, PGEI was not oxidized.
pH Optimum-Carbonyl reductase exhibited a relatively broad pH optimum between pH 5 and 7. In sodium phosphate buffer menadione, 4-nitrobenzaldehyde, and daunorubicin were best reduced at pH 6. Oxidation of PGAl occurred twice as fast a t pH 9 than at 7.
Inhibition-Table V lists a number of compounds which were tested as inhibitors of carbonyl reductase. The best
inhibitors were flavonoids and structurally related com- pounds, which are also good inhibitors of aldose reductase (29). A similar inhibition was obtained with all three enzyme forms, but the extent of inhibition depended on the substrate used. With quinones and aldehydes a 50% inhibition was obtained at a flavonoid concentration of 0.8 to 1 p ~ , whereas with ketone substrates, e.g. daunorubicin and PGE,, 3 to 5 times higher concentrations were needed to equal this inhi- bition. Dicoumarol and warfarin which are potent inhibitors of NAD(P)H dehydrogenase (EC 1.6.99.2, menadione reduc- tase, DT-diaphorase) (30, 31) inhibited carbonyl reductase by 50% at a concentration of 0.1 to 0.2 mM, using either menadi- one or 4-nitrobenzaldehyde as substrates. Indomethacin and ethacrynic acid inhibit prostaglandin 9-ketoreductase from various sources (10, 14, 32). Similarly, these drugs inhibited
TABLE IV Michaelis constants and relative maximal velocities of the
molecular forms of human brain carbonyl reductase The molecular forms were separated by chromatography on
DEAE-Sepharose. The nomenclature as described in Table I is used. K,,, and V,,, values were estimated from Lineweaver-Burk double reciprocal plots (25). For K , values of substrates the concentration of NADPH was held constant at 0.08 mM; for the determination of K , for NADPH 4-nitrobenzaldehyde was held constant at 0.5 mM.
Substrate
' Relative velocity Michaelis constant Concen-
tration CRns CR, CR, CRni CRx CR: V l M % V l M
Menadione
50 48 52 2 1.6 1.8 w 4-Nitrohenzalde- 58 6 0 5 5 5 4 m 5 Phenylglyoxal 87 84 85 0.016 0.017 0.015 m Ubiquinone-1
100 100" 100 0.06 0.045 0.057 w
Daunorubicin 0.25 0.13 19 16 (25)b
0.01 0.005 0.004 NADPH
17 hyde
PGEl 1.3 1.1 (1.7) 1.0 0.45 1
The absolute specific activity of the three enzyme forms under standard assay conditions (0.25 mM menadione) was between 7 and 13 units/mg depending on the enzyme preparation (cf. Table I). No apparent differences were detectable among the three enzyme species.
Numbers in parentheses are maximal relative velocities.
TABLE V Inhibition of human carbonyl reductase
Inhibition studies were carried out as described under "Experimen- tal Procedures" using 0.5 mM 4-nitrobenzaldehyde as substrate. The pooled fractions after the affinity chromatography step were used as enzyme source.
Similar inhihition was obtained with the flavonoids quercetin,
The enzyme was incubated with the inhibitor for 10 min before quercitrin, luteolin, kaempferol, and naringenin.
the reaction was started (cf. text).
Human NADPH-dependent Carbonyl Reductase 1211
the prostaglandin E reduction by carbonyl reductase. Essen- tially the same inhibition was observed if menadione or 4- nitrobenzaldehyde were the substrates. Chelating agents, such as 1,lO-phenanthroline and a,a-dipyridyl were inhibitory only at millimolar concentrations, and EDTA showed no effect at all. On the contrary, EDTA appeared to stabilize the enzyme during storage. No effect on the enzyme activity was observed in the presence of barbiturates and hydantoins which are inhibitors of aldehyde reductase.
Inactivation of human brain carbonyl reductase by 4-hy- droxymercuribenzoate has previously been reported (18). In this study, 10 p~ 4-hydroxymercuribenzoate inactivated car- bonyl reductase by more than 90% within 10 min. The enzyme was not protected against the inactivation by the coenzyme, NADPH, and/or the substrate, 4-nitrobenzaldehyde. One mil- limolar iodoacetic acid inactivated the enzyme by 50% after an incubation time of 10 min.
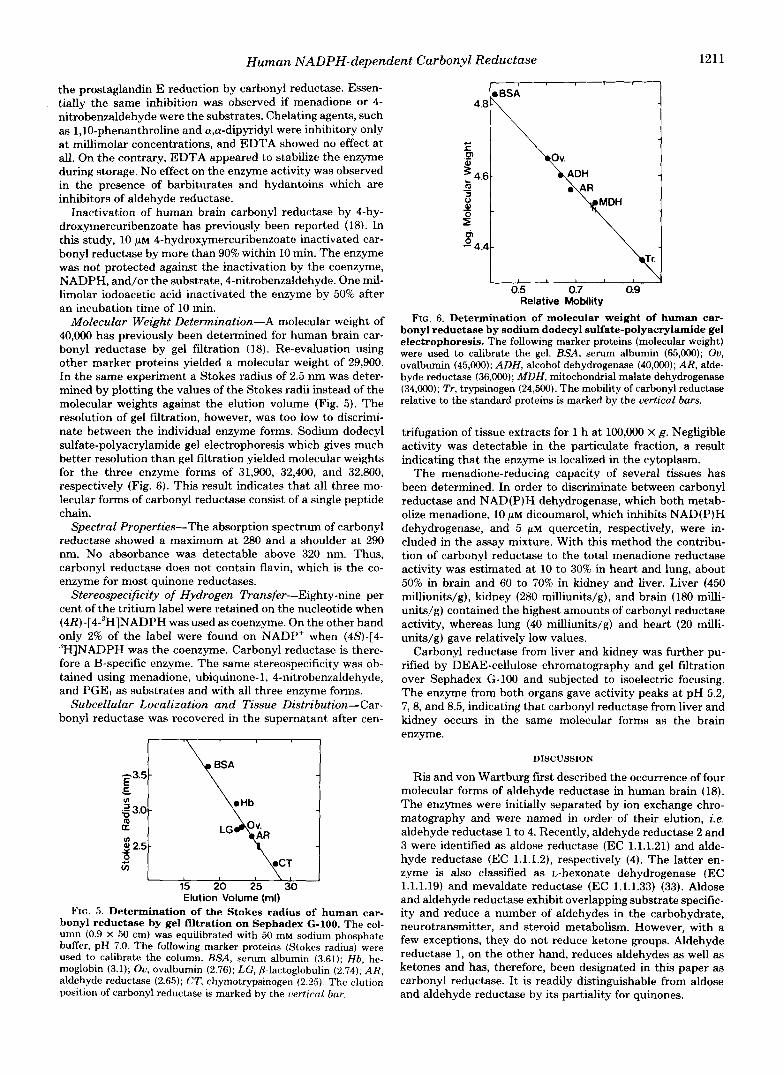
Molecular Weight Determination-A molecular weight of 40,000 has previously been determined for human brain car- bonyl reductase by gel filtration (18). Re-evaluation using other marker proteins yielded a molecular weight of 29,900. In the same experiment a Stokes radius of 2.5 nm was deter- mined by plotting the values of the Stokes radii instead of the molecular weights against the elution volume (Fig. 5). The resolution of gel fitration, however, was too low to discrimi- nate between the individual enzyme forms. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis which gives much better resolution than gel filtration yielded molecular weights for the three enzyme forms of 31,900, 32,400, and 32,800, respectively (Fig. 6). This result indicates that all three mo- lecular forms of carbonyl reductase consist of a single peptide chain.
Spectral Properties-The absorption spectrum of carbonyl reductase showed a maximum a t 280 and a shoulder at 290 nm. No absorbance was detectable above 320 nm. Thus, carbonyl reductase does not contain flavin, which is the co- enzyme for most quinone reductases.
Stereospecificity of Hydrogen Transfer-Eighty-nine per cent of the tritium label were retained on the nucleotide when (4R)-[4-'H]NADPH was used as coenzyme. On the other hand only 2% of the label were found on NADP' when (4S)-[4- "]NADPH was the coenzyme. Carbonyl reductase is there- fore a B-specific enzyme. The same stereospecificity was ob- tained using menadione, ubiquinone-1, 4-nitrobenzaldehyde, and PGEl as substrates and with all three enzyme forms.
Subcellular Localization a n d Tissue Distribution-Car- bony1 reductase was recovered in the supernatant after cen-
-3 .4 E y
iz 0 eCT
15 20 25 30 Elution Volume (mi)
FIG. 5. Determination of the Stokes radius of human car- bonyl reductase by gel filtration on Sephadex G-100. The col- umn (0.9 X 50 cm) was equilibrated with 50 m M sodium phosphate buffer, pH 7.0. The following marker proteins (Stokes radius) were used to calibrate the column. BSA, serum albumin (3.61); Hb, he- moglobin (3.1); Ou, ovalbumin (2.76); LG, P-lactoglobulin (2.74); AR, aldehyde reductase (2.65); CT, chymotrypsinogen (2.25). The elution position of carbonyl reductase is marked by the vertical bur.
\ 1
24.4i Q
0.5 0.7 0.9 Relative Mobility
FIG. 6. Determination of molecular weight of human car- bonyl reductase by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The following marker proteins (molecular weight) were used to calibrate the gel. BSA, serum albumin (65,000); Ou, ovalbumin (45,000); ADH. alcohol dehydrogenase (40,000); AR, alde- hyde reductase (36,000); MDH, mitochondrial malate dehydrogenase (34,000); Tr, trypsinogen (24,500). The mobility of carbonyl reductase relative to the standard proteins is marked by the vertical bars.
trifugation of tissue extracts for 1 h a t 100,000 x g. Negligible activity was detectable in the particulate fraction, a result indicating that the enzyme is localized in the cytoplasm.
The menadione-reducing capacity of several tissues has been determined. In order to discriminate between carbonyl reductase and NAD(P)H dehydrogenase, which both metab- olize menadione, 10 p~ dicoumarol, which inhibits NAD(P)H dehydrogenase, and 5 p~ quercetin, respectively, were in- cluded in the assay mixture. With this method the contribu- tion of carbonyl reductase to the total menadione reductase activity was estimated at 10 to 30% in heart and lung, about 50% in brain and 60 to 70% in kidney and liver. Liver (450 milliunits/g), kidney (280 milliunits/g), and brain (180 milli- units/g) contained the highest amounts of carbonyl reductase activity, whereas lung (40 milliunits/g) and heart (20 milli- units/g) gave relatively low values.
Carbonyl reductase from liver and kidney was further pu- rified by DEAE-cellulose chromatography and gel filtration over Sephadex G-100 and subjected to isoelectric focusing. The enzyme from both organs gave activity peaks at pH 5.2, 7,8, and 8.5, indicating that carbonyl reductase from liver and kidney occurs in the same molecular forms as the brain enzyme.
DISCUSSION
Ris and von Wartburg fiist described the occurrence of four molecular forms of aldehyde reductase in human brain (18). The enzymes were initially separated by ion exchange chro- matography and were named in order of their elution, i.e. aldehyde reductase 1 to 4. Recently, aldehyde reductase 2 and 3 were identified as aldose reductase (EC 1.1.1.21) and alde- hyde reductase (EC 1.1.1.2), respectively (4). The latter en- zyme is also classified as L-hexonate dehydrogenase (EC 1.1.1.19) and mevaldate reductase (EC 1.1.1.33) (33). Aldose and aldehyde reductase exhibit overlapping substrate specific- ity and reduce a number of aldehydes in the carbohydrate, neurotransmitter, and steroid metabolism. However, with a few exceptions, they do not reduce ketone groups. Aldehyde reductase 1, on the other hand, reduces aldehydes as well as ketones and has, therefore, been designated in this paper as carbonyl reductase. I t is readily distinguishable from aldose and aldehyde reductase by its partiality for quinones.
1212 Human NADPH-dependent Carbonyl Reductase
Quinones play important roles as oxidation-reduction cata- lysts in many biological processes. Ubiquinones (coenzyme Q ) are a constitutive part of the respiratory chain, menaquinones (vitamin K) act as coenzyme in the carboxylation of glutamic acid to y-carboxyglutamic acid in prothrombin and other proteins, and tocopherolquinones (vitamin E) are thought to protect the lipids of biological membranes against oxidation. However, in vivo these quinones are embedded in the hydro- phobic environment of membranes and little is known on their availability as substrates for soluble oxidoreductases. Recent evidence indicates that the cytoplasmic NAD(P)H dehydro- genase (EC 1.6.99.2, quinone reductase, DT-diaphorase) re- duces vitamin K in rat liver (34). Other cytosolic quinone reductases, e.g. carbonyl reductase, may, therefore, also take part in the metabolism of biologically active quinones. En- zymes which oxidize NADPH in the presence of quinones, oxidation-reduction dyes, and ferricyanide have been isolated from several species including man (35, 36). They differ, however, from carbonyl reductase .in that they contain flavin and use FMN or FAD for full activity. In this respect carbonyl reductase is unusual among the quinone reductases which as a rule use flavin as coenzyme (30, 31). An NADPH-linked quinone reductase (NADPH dehydrogenase) which does not contain flavin has been detected in human red blood cells (37). However, this enzyme has not been purified and has only sparsely been characterized with respect to substrate specific- ity and sensitivity to inhibitors, allowing no further compari- son with carbonyl reductase.
In addition to quinones, carbonyl reductase reduces several biologically and pharmacologically important compounds. Prostaglandins, e.g. modulate a number of hormonal, neuro- hormonal, or other stimuli and have a variety of pharmaco- logical effects (38). The reactions catalyzed by carbonyl re- ductase, i.e. the reduction of the oxo group at position 9 and the oxidation of the hydroxyl group at position 15 are thought to be the fmst steps in the inactivation of the prostaglandins of the E and F class. Enzymes with prostaglandin 9-ketore- ductase and 15-hydroxyprostaglandin dehydrogenase activity have been isolated and characterized from mammalian (8, 9, 11, 13, 14) and nonmammalian (10, 12) tissues. In man, a prostaglandin 9-ketoreductase has been isolated from red blood cells (9) and from placenta (13, 14), from which it was obtained in a pure state (14). This enzyme has many of the properties reported for carbonyl reductase. These properties include a molecular weight of about 30,000, requirement for NADPH as coenzyme localization in the cytoplasm of the cell, specificity for various prostaglandins, and inhibition by indo- methacin and ethacrynic acid. Moreover, chromatography on “blue Sepharose” and hydroxylapatite resolved the enzyme activity into three separate fractions (14). Thus, prostaglandin 9-ketoreductase from placenta and carbonyl reductase from brain exhibit essentially the same structural and catalytic properties, suggesting that they are identical. The significance of prostaglandin reduction by carbonyl reductase, however, is unclear. The K , values for prostaglandins are rather high, and the turnover numbers are low. An exception is the GSH adduct of PGA,, which was readily reduced by carbonyl reductase at low concentrations. This adduct is also the best substrate for prostaglandin 9-ketoreductase from chicken heart (39) and rabbit kidney (40). To date no convincing evidence for the natural occurrence of PGA in mammalian systems has been produced. Nevertheless, the preference of carbonyl reductase for the GSH adduct of PGA above all the other prostaglandins suggests that a GSH conjugate which resembles the one of PGA may be a natural substrate for this enzyme.
Daunorubicin is used in cancer chemotherapy, and its me-
tabolism has been thoroughly investigated. A major step in the degradation of this drug is the conversion to the respective alcohol metabolite, daunorubicinol. Enzymes effecting this reduction have been isolated from human, rabbit, and rat tissues (15-17, 41). In man two daunorubicin reductases can be distinguished (17). They differ from each other with respect to the molecular weight and the pH optimum of daunorubicin reduction. One enzyme, which has a molecular weight of 38,700 and a pH optimum at 8.5, is identical with the aldehyde reductase (17,33). The other reductase has a molecular weight of 34,500 and a pH optimum at 6. This enzyme is referred to as pH 6 daunorubicin reductase or xenobiotic ketone reduc- tase. Similar to carbonyl reductase it is localized in the cyto- plasm of the cell, requires NADPH as coenzyme, is inhibited by quercetin, and is resolved into several peaks of activity by isoelectric focusing. Though it has only partially been purified and only anthracycline antibiotics were used as substrates, the available data indicate a close relationship or even identity of xenobiotic ketone reductase and carbonyl reductase.
Ketoaldehydes represent another class of aldehyde reduc- tase substrates of biological and pharmacological interest. These compounds, similar to daunorubicin, interfere with cell growth and have been used as antitumor agents (42). Whether carbonyl reductase may decrease the potency of these drugs by inactivating them is not known and it may be worth further investigation. Another aspect in the role of ketoaldehydes, e.g. glyoxal and methylglyoxal, in living matter is also related to the regulation of cell growth. According to Szent-Gyorgyi et al., a universal mechanism exists in living systems which regulates growth and is governed by the concentration of certain ketoaldehydes (43). Carbonyl reductase could thus interfere with the regulatory system by reducing such modu- lator aldehydes.
Carbonyl reductase also reduces steroid hormones and might be involved in the catabolism of these compounds. Aldehyde reductases exhibiting 3-ketosteroid reductase activ- ity have been described from rat (26), guinea pig (28), and rabbit liver (27). Although some similarities between carbonyl reductase and one of the rabbit enzymes (Fa in Ref. 27) are evident, further characterization is needed to establish the relationship between the two enzymes.
Acknowledgments- I thank Professor J. P. von Wartburg for his continuous interest in this work, Miss B. Affolter and Miss U. Kiotzsche for their skillful technical assistance, and Dr. B. Dewald for providing the prostaglandins used in this study. I also thank Dr. P. Lavanchy of the Eidgenossische Versuchsanstalt fur Milchwirt- schaft, Bern, for carrying out the amino acid analyses.
REFERENCES 1. Bosron, W. F., and Prairie, R. L. (1973) Arch. Biochem. Biophys.
2. Clements, R. S., Weaver, J . P., and Winegrad, A. I. (1969) Bio- chem. Biophys. Res. Commun. 37,347-353
3. Tabakoff, B. (1977) in Structure and Function of Monoamine Enzymes (Usdin, E., Weiner, N., and Youdim, M. B. H., eds) pp. 629-649, Marcel Dekker, Inc., New York
4. Hoffman, P. L., Wermuth, B., and von Wartburg, J. P. (1980) J. Neurochem. 35,354-366
5. Gabbay, K. H., and Khoshita, J. H. (1975) Methods Enzymol.
6. Burns, J. J. (1967) in Metabolic Pathways (Greenberg, D. M., ed) 3rd Ed, Vol. 1, pp. 394-411, Academic Press, New York
7. Lee, S.-C., and Levine, L. (1974) J . Biol. Chem. 249, 1369-1375 8. Lee, S.-C., Pong, S.-S., Katzen, D., Wu, K.-Y., and Levine, L.
9. Kaplan, L., Lee, S.-C., and Levine, L. (1975) Arch. Biochem.
154, 166-172
41, 159-165
(1975) Biochemistry 14, 142-145
Biophys. 167,287-293 10. Hassid, A., and Levine, L. (1977) Prostaglandins 13, 503-516 11. Stone, K. J., and Hart, M. (1975) Prostaglandins 10,273-288 12. Bishai, I., and Coceani, F. (1976) J. Neurochem. 26, 1167-1174
Human NADPH-dependent Carbonyl Reductase 1213
13. Westbrook, C., Lin, Y . M., and Jarabak, J . (1977) Biochem.
14. Lin, Y. M., and Jarabak, J . (1978) Biochem. Biophys. Res. Com-
15. Felsted, R. L., Gee, M., and Bachur, N. R. (1974) J. Biol. Chem.
16. Felsted, R. L., Richter, D. R., and Bachur, N. R. (1977) Biochem.
17. Ahmed, N. K., Felsted, R. L., and Bachur, N. R. (1978) Biochem.
18. Ris, M. M., and von Wartburg, J . P. (1973) Eur. J. Biochem. 37,
19. Mayer, H., and Isler, 0. (1971) Methods Enzymol. 18,527 20. Tabakoff, B., Anderson, R., and Alivisatos, S. G . A. (1973) Mol.
21. Layne, E. (1957) Methods Enzymol. 3,447-454 22. Laemmli, U. K. (1970) Nature 227,680-685 23. Wermuth, B., Munch, J. D. B., and von Wartburg, J. P. (1979)
24. Jacobson, K. B., Murphy, J. B., Knopp, J. A,, and Ortiz, J . R.
25. Lineweaver, H., and Burk, D. (1934) J. Am. Chem. SOC. 56,658-
26. Pietruszko, R., and Chen, F.-F. (1976) Biochem. Pharmacol. 25,
27. Sawada, H., Hara, A., Nakayama, T., and Kato, F. (1980) J.
28. Sawada, H., Hara, A,, Hayashibara, M., and Nakayama, T. (1979)
29. Varma, S. D., Mizuno, A., and Kinoshita, J. H. (1977) Science
Biophys. Res. Commun. 76,943-949
mun. 81, 1227-1234
249,3672-3679
Pharrnacol. 26, 1117-1124
Pharmacol. 27,2713-2719
69-77
Pharmacol. 9,428-437
Experientia 35, 1288-1289
(1972) Arch. Biochem. Biophys. 149,22-35
666
2721-2725
Biochem. 87, 1153-1165
J. Biochem. 86,883-892
195, 205-206 30. Emster, L. (1967) Methods Enzymol. 10, 309-317 31. Martius, C. (1963) in The Enzymes (Boyer, P. D., ed) 2nd Ed.,
Vol. 7, pp. 517-532, Academic Press, New York 32. Stone, K. J., and Hart, M. (1976) Prostaglandins 12, 197-207 33. von Wartburg, J . P., and Wermuth, B. (1980) in Enzymatic Basis
of Detoxication (Jakoby, W. B., ed) Vol. 1, pp. 249-260, Aca- demic Press, New York
34. Wallin, R., Gebhardt, O., and Prydz, H. (1978) Biochem. J. 169,
35. Levine, W., Giuditta, A., Englard, S., and Strecker, H. J. (1960) J. Neurochem. 6,28-36
36. Koli, A. K., Yearby, C., Scott, W., and Donaldson, K. 0. (1969) J, Biol. Chem. 244,621-629
37. Scott, E. M., Duncan, I. W., and Ekstrand, V. (1965) J. Biol. Chem. 240,481-485
38. Samuelsson, B., Goldyne, M., Granstrom, E., Hamberg, M., Ham- marstrom, s., and Malmsten, C. (1978) Annu. Rev. Biochem. 47,997-1092
39. Cagen, L. M., and Pisano, J . J . (1979) Biochim. Biophys. Acta 573, 547-551
40. Toft, B. S., and Hansen, H. S. (1979) Biochim. Biophys. Acta 574, 33-38
41. Felsted, R. L., Richter, D. R., Jones, D. M., and Bachur, N. R. (1980) Biochem. Pharmacol. 29, 1503-1516
42. Schauenstein, E., Esterbauer, H., and Zollner, H. (1977) Alde- hydes in Biological Systems, pp. 126-129, Pion Limited, Lon- don
43. Szent-Gyorgyi, A., Egyud, L., and McLaughlin, J. (1967) Science 155, 539-541