291
FDA Medical Device Industry Coalition Quality System Survival: Success Strategies for P&PC and CAPA An Educational Forum 15 April 2016

FDA Medical Device Industry Coalition
Quality System Survival:
Success Strategies for
P&PC and CAPA An Educational Forum
15 April 2016

FDA Medical Device Industry Coalition
Introduction
Disclaimers, housekeeping, logistics,
laws, regulations, and guidance
documents

Disclaimer
The information provided in this forum does
not take the place of the laws and
regulations enforced by FDA
Any reference to a commercial product,
process, service, or company is not an
endorsement or recommendation by the
U.S. government, HHS, FDA or any of its
components

Disclaimer
FDA is not responsible for the contents of
any outside information referenced in this
forum
This forum does not convey any waiver of
responsibility to the firm, nor impart any
immunity to the firm for violations that may
occur, even if you implement our
recommendations as per 21 CFR 10.85(k)
No Smoking

Cell Phones, Pagers and PDA’s
Please make sure your
cell phones, pagers,
and other devices are
set on silent mode
Please step outside if
you must answer or
make a call

Logistics
Restrooms
Breaks
Lunch
Questions
Evaluation form
Certificate of
Attendance

FDA Medical Device Industry Coalition
Quality System Overview
John C. Criscione, MD, PhD
Associate Professor
Department of Biomedical Engineering
Texas A&M University, College Station
Big Picture
High Quality Product!

High Quality Definition of Quality
What is Quality?

High Quality Definition of Quality
Quality is “Realizing Expectations”
A Quality Product or Service will Perform as
Expected
A Low Quality Item Fails to Meet
Expectations

High Quality Definition of Quality
Quality is “Realizing Expectations”
A Quality Product or Service will Perform as
Expected

Expectations for Food, Drugs,
Cosmetics and Devices

Quality System
A Systematic Approach to Achieving Quality
Without Quality Assurance there is no
Quality System…

Is a Quality System Necessary
Yes!
Low Risk of
Injury or Death
High Risk of
Injury or Death
Few Procedures
Few People Maybe Yes
Many Procedures
Many People Yes Absolutely

Is a Quality System Necessary
Yes!
Low Risk of
Injury or Death
High Risk of
Injury or Death
Few Procedures
Few People Maybe Yes
Many Procedures
Many People Yes Absolutely
Bottom Line: If a product or service has a
high impact then quality must be assured…

Is a Quality System Necessary
Yes!
Low Risk of
Injury or Death
High Risk of
Injury or Death
Few Procedures
Few People Maybe Yes
Many Procedures
Many People Yes Absolutely

High Quality Attitude
The Most Critical Component to Attaining
Quality Products and Services
Basis of TQM—Total Quality Management

High Quality Attitude
Quality Attitude Attenuates Down the
Leadership Ladder
Executive Officers Production Staff

High Quality Attitude
In a Company that Values Quality, a Quality
Attitude will Lead to Promotion…
Executive Officers Production Staff

High Quality Attitude
If a Company Does Not Value Quality, There
are Worse Things than a Warning Letter…
Executive Officers Production Staff

High Quality Attitude
Having a High Quality Attitude is Difficult
AND Necessary
Can You Say “Thank You” when a Co-Worker
Inspects Your Work and Reports the Flaws?
It is Human Nature to be Defensive when
Criticized, and yet “to Err is Human”…

Sec. 820.20 Management responsibility.
(a) Quality policy. Management with executive responsibility shall establish its policy and objectives for, and commitment to,
quality. Management with executive responsibility shall ensure that the quality policy is understood, implemented, and
maintained at all levels of the organization.
(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices
are designed and produced in accordance with the requirements of this part.
(1) Responsibility and authority. Each manufacturer shall establish the appropriate responsibility, authority, and interrelation
of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority
necessary to perform these tasks.
(2) Resources. Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for
management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of
this part.
(3) Management representative. Management with executive responsibility shall appoint, and document such appointment of,
a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility
for:
(i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this
part; and
(ii) Reporting on the performance of the quality system to management with executive responsibility for review.
(c) Management review. Management with executive responsibility shall review the suitability and effectiveness of the quality
system at defined intervals and with sufficient frequency according to established procedures to ensure that the quality
system satisfies the requirements of this part and the manufacturer's established quality policy and objectives. The dates and
results of quality system reviews shall be documented.
(d) Quality planning. Each manufacturer shall establish a quality plan which defines the quality practices, resources, and
activities relevant to devices that are designed and manufactured. The manufacturer shall establish how the requirements for
quality will be met.
(e) Quality system procedures. Each manufacturer shall establish quality system procedures and instructions. An outline of the
structure of the documentation used in the quality system shall be established where appropriate.

Sec. 820.25 Personnel.
(a) General. Each manufacturer shall have sufficient personnel with the
necessary education, background, training, and experience to assure that all
activities required by this part are correctly performed.
(b) Training. Each manufacturer shall establish procedures for identifying
training needs and ensure that all personnel are trained to adequately
perform their assigned responsibilities. Training shall be documented.
(1) As part of their training, personnel shall be made aware of device defects
which may occur from the improper performance of their specific jobs.
(2) Personnel who perform verification and validation activities shall be made
aware of defects and errors that may be encountered as part of their job
functions.

Sec. 820.50 Purchasing controls.
Each manufacturer shall establish and maintain procedures to ensure that all purchased or
otherwise received product and services conform to specified requirements.
(a) Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and
maintain the requirements, including quality requirements, that must be met by suppliers,
contractors, and consultants. Each manufacturer shall:
(1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability
to meet specified requirements, including quality requirements. The evaluation shall be
documented.
(2) Define the type and extent of control to be exercised over the product, services, suppliers,
contractors, and consultants, based on the evaluation results.
(3) Establish and maintain records of acceptable suppliers, contractors, and consultants.
(b) Purchasing data. Each manufacturer shall establish and maintain data that clearly describe or
reference the specified requirements, including quality requirements, for purchased or otherwise
received product and services. Purchasing documents shall include, where possible, an agreement
that the suppliers, contractors, and consultants agree to notify the manufacturer of changes in the
product or service so that manufacturers may determine whether the changes may affect the
quality of a finished device. Purchasing data shall be approved in accordance with 820.40.

Sec. 820.70 Production and process controls. (Slide 1 of 2)
(a) General. Each manufacturer shall develop, conduct, control, and monitor production processes to ensure that a
device conforms to its specifications. Where deviations from device specifications could occur as a result of the
manufacturing process, the manufacturer shall establish and maintain process control procedures that describe any
process controls necessary to ensure conformance to specifications. Where process controls are needed they shall
include:
(1) Documented instructions, standard operating procedures (SOP's), and methods that define and control the manner
of production;
(2) Monitoring and control of process parameters and component and device characteristics during production;
(3) Compliance with specified reference standards or codes;
(4) The approval of processes and process equipment; and
(5) Criteria for workmanship which shall be expressed in documented standards or by means of identified and
approved representative samples.
(b) Production and process changes. Each manufacturer shall establish and maintain procedures for changes to a
specification, method, process, or procedure. Such changes shall be verified or where appropriate validated
according to 820.75, before implementation and these activities shall be documented. Changes shall be approved in
accordance with 820.40.
(c) Environmental control. Where environmental conditions could reasonably be expected to have an adverse effect
on product quality, the manufacturer shall establish and maintain procedures to adequately control these
environmental conditions. Environmental control system(s) shall be periodically inspected to verify that the system,
including necessary equipment, is adequate and functioning properly. These activities shall be documented and
reviewed.
(d) Personnel. Each manufacturer shall establish and maintain requirements for the health, cleanliness, personal
practices, and clothing of personnel if contact between such personnel and product or environment could reasonably
be expected to have an adverse effect on product quality. The manufacturer shall ensure that maintenance and other
personnel who are required to work temporarily under special environmental conditions are appropriately trained or
supervised by a trained individual.

Sec. 820.70 Production and process controls. (Slide 2 of 2)
(e) Contamination control. Each manufacturer shall establish and maintain procedures to prevent contamination of
equipment or product by substances that could reasonably be expected to have an adverse effect on product quality.
(f) Buildings. Buildings shall be of suitable design and contain sufficient space to perform necessary operations,
prevent mixups, and assure orderly handling.
(g) Equipment. Each manufacturer shall ensure that all equipment used in the manufacturing process meets specified
requirements and is appropriately designed, constructed, placed, and installed to facilitate maintenance,
adjustment, cleaning, and use.
(1) Maintenance schedule. Each manufacturer shall establish and maintain schedules for the adjustment, cleaning,
and other maintenance of equipment to ensure that manufacturing specifications are met. Maintenance activities,
including the date and individual(s) performing the maintenance activities, shall be documented.
(2) Inspection. Each manufacturer shall conduct periodic inspections in accordance with established procedures to
ensure adherence to applicable equipment maintenance schedules. The inspections, including the date and
individual(s) conducting the inspections, shall be documented.
(3) Adjustment. Each manufacturer shall ensure that any inherent limitations or allowable tolerances are visibly
posted on or near equipment requiring periodic adjustments or are readily available to personnel performing these
adjustments.
(h) Manufacturing material. Where a manufacturing material could reasonably be expected to have an adverse effect
on product quality, the manufacturer shall establish and maintain procedures for the use and removal of such
manufacturing material to ensure that it is removed or limited to an amount that does not adversely affect the
device's quality. The removal or reduction of such manufacturing material shall be documented.
(i) Automated processes. When computers or automated data processing systems are used as part of production or
the quality system, the manufacturer shall validate computer software for its intended use according to an
established protocol. All software changes shall be validated before approval and issuance. These validation activities
and results shall be documented.

Sec. 820.72 Inspection, measuring, and test equipment.
(a) Control of inspection, measuring, and test equipment. Each manufacturer shall ensure that all
inspection, measuring, and test equipment, including mechanical, automated, or electronic
inspection and test equipment, is suitable for its intended purposes and is capable of producing
valid results. Each manufacturer shall establish and maintain procedures to ensure that equipment
is routinely calibrated, inspected, checked, and maintained. The procedures shall include
provisions for handling, preservation, and storage of equipment, so that its accuracy and fitness for
use are maintained. These activities shall be documented.
(b) Calibration. Calibration procedures shall include specific directions and limits for accuracy and
precision. When accuracy and precision limits are not met, there shall be provisions for remedial
action to reestablish the limits and to evaluate whether there was any adverse effect on the
device's quality. These activities shall be documented.
(1) Calibration standards. Calibration standards used for inspection, measuring, and test equipment
shall be traceable to national or international standards. If national or international standards are
not practical or available, the manufacturer shall use an independent reproducible standard. If no
applicable standard exists, the manufacturer shall establish and maintain an in-house standard.
(2) Calibration records. The equipment identification, calibration dates, the individual performing
each calibration, and the next calibration date shall be documented. These records shall be
displayed on or near each piece of equipment or shall be readily available to the personnel using
such equipment and to the individuals responsible for calibrating the equipment.

Verification Vs. Validation
User Requirement or
Process Functionality
Assumption
Testable Specification
Test Data
Functionality Results
Verification Validation

Sec. 820.75 Process validation.
(a) Where the results of a process cannot be fully verified by subsequent
inspection and test, the process shall be validated with a high degree of
assurance and approved according to established procedures. The validation
activities and results, including the date and signature of the individual(s)
approving the validation and where appropriate the major equipment
validated, shall be documented.
(b) Each manufacturer shall establish and maintain procedures for monitoring
and control of process parameters for validated processes to ensure that the
specified requirements continue to be met.
(1) Each manufacturer shall ensure that validated processes are performed by
qualified individual(s).
(2) For validated processes, the monitoring and control methods and data, the
date performed, and, where appropriate, the individual(s) performing the
process or the major equipment used shall be documented.
(c) When changes or process deviations occur, the manufacturer shall review
and evaluate the process and perform revalidation where appropriate. These
activities shall be documented.

Sec. 820.80 Receiving, in-process, and finished device acceptance.
(a) General. Each manufacturer shall establish and maintain procedures for acceptance activities. Acceptance activities
include inspections, tests, or other verification activities.
(b) Receiving acceptance activities. Each manufacturer shall establish and maintain procedures for acceptance of incoming
product. Incoming product shall be inspected, tested, or otherwise verified as conforming to specified requirements.
Acceptance or rejection shall be documented.
(c) In-process acceptance activities. Each manufacturer shall establish and maintain acceptance procedures, where
appropriate, to ensure that specified requirements for in-process product are met. Such procedures shall ensure that in-
process product is controlled until the required inspection and tests or other verification activities have been completed, or
necessary approvals are received, and are documented.
(d) Final acceptance activities. Each manufacturer shall establish and maintain procedures for finished device acceptance to
ensure that each production run, lot, or batch of finished devices meets acceptance criteria. Finished devices shall be held in
quarantine or otherwise adequately controlled until released. Finished devices shall not be released for distribution until:
(1) The activities required in the DMR are completed;
(2) the associated data and documentation is reviewed;
(3) the release is authorized by the signature of a designated individual(s); and
(4) the authorization is dated.
(e) Acceptance records. Each manufacturer shall document acceptance activities required by this part. These records shall
include:
(1) The acceptance activities performed;
(2) the dates acceptance activities are performed;
(3) the results;
(4) the signature of the individual(s) conducting the acceptance activities; and
(5) where appropriate the equipment used. These records shall be part of the DHR.

Sec. 820.90 Nonconforming product.
(a) Control of nonconforming product. Each manufacturer shall establish and maintain
procedures to control product that does not conform to specified requirements. The
procedures shall address the identification, documentation, evaluation, segregation, and
disposition of nonconforming product. The evaluation of nonconformance shall include a
determination of the need for an investigation and notification of the persons or
organizations responsible for the nonconformance. The evaluation and any investigation shall
be documented.
(b) Nonconformity review and disposition.
(1) Each manufacturer shall establish and maintain procedures that define the responsibility
for review and the authority for the disposition of nonconforming product. The procedures
shall set forth the review and disposition process. Disposition of nonconforming product shall
be documented. Documentation shall include the justification for use of nonconforming
product and the signature of the individual(s) authorizing the use.
(2) Each manufacturer shall establish and maintain procedures for rework, to include
retesting and reevaluation of the nonconforming product after rework, to ensure that the
product meets its current approved specifications. Rework and reevaluation activities,
including a determination of any adverse effect from the rework upon the product, shall be
documented in the DHR.

Sec. 820.100 Corrective and preventive action.
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and
preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records,
service records, complaints, returned product, and other sources of quality data to identify existing
and potential causes of nonconforming product, or other quality problems. Appropriate statistical
methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality
system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product
and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is
effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent
identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated
to those directly responsible for assuring the quality of such product or the prevention of such
problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and
preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.

Sec. 820.100 Corrective and preventive action.
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and
preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records,
service records, complaints, returned product, and other sources of quality data to identify existing
and potential causes of nonconforming product, or other quality problems. Appropriate statistical
methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality
system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product
and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is
effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent
identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated
to those directly responsible for assuring the quality of such product or the prevention of such
problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and
preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.


FDA Medical Device Industry Coalition
Getting It Wrong:
Warning Letter Learnings
Jeff R. Wooley, Compliance Officer
Dallas District, FDA

Common Challenges
Scope
Systemic Corrective Actions
Risk
Poor Inputs

Scope
CAPA should identify scope of impacted
product
Initial determination and review due to new
information
Should be documented and supported.
Investigators will evaluate this.

Scope
Preamble Comment 158
“These procedures must provide for control and
action to be taken on devices distributed, and
those not yet distributed, that are suspected of
having potential nonconformities.”

Scope
Distributed
Recall?
Safety Notice?
Revised labeling?
All evaluations or decisions should be
documented and supported.
Consider HHE or risk analysis

Scope
In-Process or Awaiting Distribution
Rework?
Additional testing or inspections?
Quarantine?
Cessation of production?
Any rework should be controlled
Decisions documented and supported

Scope
Timeframes of problem
Timeframes for manufacture/distribution
Product families
Multiple products made on same line
Multiple products made with same
components
Documentation is critical

Scope
Failure to establish procedures for corrective and
preventive action, as required by 21 CFR
820.100(a)…Specifically, your firm removed your
Liquid Bone Glue lot number 1111 from the
market due to your CAPA investigation
identifying contamination in the raw material,
used in the manufacture of this finished device.
However, your firm did not take any corrective
action with respect to Liquid Bone Glue lot 2222
that was also manufactured with the same lot of
raw material.

Warning Letter Citations…
“… Your firm’s CAPA investigation did not
review and evaluate additional lots of raw
material in similar drums to ensure all
affected drums/lots were identified…”

Scope
Firm’s CAPA procedure and records do not
specifically address scope
Firm did not address other lots that may
have been impacted
Firm did not consider other component lots
may have been impacted.
As a result, firm’s corrective action and
response to 483 was incomplete and
received WL.

Systemic Corrective Actions
CAPA intended to link data from multiple
subsystems
Intended to correct potential for multiple root
causes across quality system and subsystems
Rare for complete corrective action to be
limited to one process or procedure.

Systemic Corrective Actions
Consider mechanism to review previous
investigations
Distinguish symptoms from root causes
Fishbone
Cause and effect diagrams
5 Why’s

Systemic Corrective Actions
Bone Glue
Contaminated finished product sent to
customers. CAPA confirmed raw material
contaminated.
Purchasing Controls?
Incoming Acceptance?
Finished Product Acceptance?
Nonconforming Material Process?
Investigation (complaint & CAPA)?

Risk
Regulation does not specifically require risk
assesments, etc. as part of CAPA.
However, FDA is clear in preamble you
cannot effectively implement CAPA program
without assessing risk.

Risk
Preamble Comment 159
“…FDA does expect the manufacturer to develop
procedures for assessing the risk, the actions
that need to be taken for different levels of risk,
and how to correct or prevent the problem from
recurring, depending on that risk assessment.”

Risk
Scope and depth of CAPA activities should
be commensurate with risk
Overly exhaustive investigation may unduly
delay correction
Well documented risk assessment and evaluation
may help support limiting investigation.
Consider defining endpoint for investigation

Risk
It may be necessary to take initial
corrections (e.g. containment, stop of
shipment/supply, issuance of advisory
notice) in order to address an immediate
risk or safety issue.
Initial action may be necessary prior to
completion of investigation
Consider risk assessments may need to be
reevaluated as new information comes to light

Risk
Do corrections introduce new risks?
Should be considered as part prior to making
corrections and confirmed during verification.
Consider CAPA findings should also feedback
to design risk analysis

Risk
Firm’s procedure states “As soon as
sufficient information is available to
establish the problem statement and scope,
the CAPA owner initiates the HHE”
At time of inspection firm had documented scope
and problem…but had not conducted HHE.
Resulted in no action by firm
Resulted in multiple avoidable recalls

Data Sources
Good data sources and inputs into CAPA
system are critical
Poor data sources fail to identify issues
Poor data sources fail to provide tools to
facilitate investigation and review
Poor data sources fail to support corrective
actions were effective.

Data Sources
Data should be analyzed against established
criteria.
Consider statistical and non-statistical
Level of analysis across data sources

Data Sources
Failure to establish and maintain procedures
for implementing corrective and preventive
actions for analyzing sources of quality data
…For example your quality procedures do
not contain sufficient instructions for the
analysis and monitoring of quality data using
appropriate statistical methodology to
identify potential sources of nonconforming
product.

Data Sources
“Multiple complaints involving serious injury
are attributed to user error; however, the
complaint information has not been
adequately analyzed to identify potential
quality problems with the device.”

59
Valuable References
http://www.imdrf.org/docs/ghtf/final/sg3/technical-docs/ghtf-sg3-n18-2010-qms-guidance-on-corrective-preventative-action-101104.pdf
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/QualitySystemsRegulations/ucm230127.htm


FDA Medical Device Industry Coalition
Process Planning
Strategies for Effective
Monitoring and Control Cheryl Boyce, Principal,
Boyce Consulting, Inc.

Analyzing Metrics Required by
the CAPA Process 820.100 (a) (1)
Requires statistical analyses of quality metrics
Product, processes, service, returns, quality system
processes, quality records, work operations,
complaints, other……
Section 8 of ISO 13485:2016 requires
“monitoring, measurement, analysis and
improvement of processes” for product and the
quality system
Should your firm have a procedure for quality
system metrics?

Opportunities for Improvements
in this Area Yes, we need a Quality Metrics Procedure
Metrics must be determined that are meaningful
Every company won’t have the same metrics
Important Inclusions to the Procedure:
Metric Category, Specific Metric Name, Owner of
the Metric, Purpose of the Metric, Frequency of
Data Collection, Statistical Analysis Method,
Results and Conclusions.
Emphasis!!! This is a TEAM effort – all
departments and functional areas involved.

When Would these Metrics be
Used? During Working Monthly CAPA Meetings with Responsible
Departments.
Remember to have a procedure for these meetings requiring who shall
attend, what is required, and that analyses, conclusions, assigned tasks
& responsibilities shall be documented – opportunity for improvement.
What if there are no assigned tasks?
Inputs to Management Reviews
The primary foundation for a successful quality system is
management’s understanding of the status of the Quality System to
ensure….?
The quality system is appropriate for the specific medical devices
designed or manufactured, and that it meets the requirements of 820,
that the Quality Policy and Objectives are met, that there are
adequate resources. 820.5, 820.20

Why are Metrics Related to
Production Processes Important? To reveal the on-going process capability to
meet product conformance to
specifications;
To reveal costs of quality such as scrap and
rework; and,
To identify opportunities for investigations,
corrective actions and improvements.

So…What would be some Metrics Related to
Production Processes? Possibilities:
Normalized scrap data via a run chart on-going over 12
months.
Normalized rework data via a run chart on-going over
12 months.
Unscheduled Equipment Maintenance events per work
station, Pareto & Run Chart per month traveling 12
months.
# of NCRs (normalized) from manufacturing and
assembly areas, Pareto and Run Chart per month
traveling 12 months.
(Note: Your data must be customized to your needs.)

Production - Production Process Owner
Purpose of Metric Frequency Of
Reporting Statistical
Analysis Method Responsible
Department
Number of rejects sorted
by product failure codes
for each product#.
Measures the
effectiveness of the
manufacturing
operation
Monthly Pareto Chart &
Run Chart - per
month for the
current and
previous 12
months
Manufacturing
# of Scrap and rework
dispositions by product #
and work station/process
type
Measures the
effectiveness of the
manufacturing
operation
Monthly Pareto Chart
and Run Chart -
per month for
the current and
previous 12
months
Manufacturing
Unscheduled preventive
maintenance occurrences
by workstation/process
Measures the
effectiveness of the
PM program to
prevent unplanned
process shut-downs
Monthly Pareto and Run
Chart – per
month for the
current and
previous 12
months
Manufacturing
Revealing Metrics to Monitor Production Areas –
How might these be included in an SOP?
(Your data must be customized to your needs.)

Resource for Statistical
Production Monitoring
Utilization of Statistical Method for
Production Monitoring Technical Report No.
59, PDA
www.pda.org/bookstore

A Monthly CAPA meeting or
Management Review Meeting
Observed an increase in the number of
NCRs on the run chart for the heat
sealing packaging process over the last 3
months. (Were alert and action levels
established?)
The data was normalized.

Initiate Investigation and Remediation
Develop a Team
Develop an Investigation Plan including:
Defining the problem
Understanding the problem and its scope
Brainstorming its possible causes and analyzing causes and
effects and devising a solution to the problem.
Document all findings and results
Investigation reveals that the equipment qualification had been
over-looked as part of the validation.
There was no Manufacturing and Product Quality Plan as part of the Design
Transfer Phase which required the firm to plan including, but not limited to: all
equipment qualifications, test method validations, process validations,
equipment calibrations.
Is this actually the root cause?

Why Validate the Process &
Include Equipment Qualification?
Why would it require validation?
Each output quality characteristic cannot be
fully verified via inspection and testing
Why does that matter?
Instill quality into the process and product, cannot
inspect it in!!

Why Validate this Process..
Could result in inadequate packaging: Excerpt from a Hazards and
Harms Analysis for sterile product.
ID Hazard(s) Initiating Event Hazardous Situation Harm Severity
H6-
001A Bacteria and/or
viruses One or more components
are not sterile. Non-
sterile component
introduced or implanted
Patient acquires
infection Patient
hospitalized and
treated with
antibiotics (May
require revision)
3
H6-001B Bacteria and/or
viruses One or more components
are not sterile. Non-
sterile component
introduced or implanted
Patient acquires
infection
Patient Death 5
3 Severe
Injury requiring medical or surgical intervention to preclude
permanent impairment of a body function or permanent damage to a
body structure.
5 Catastrophic Patient Death

Why Validate the Process?
Costs of Quality
Rework, Scrap, Unscheduled Maintenance
The elements required to successfully validate
a process, such as Multi-Vari Analysis and DOE
techniques, reveal the key variables and
interaction of variables to instill consistency.
These become the variables you will monitor
along with the process parameters, which helps
with your production and process controls!.

Why Validate the Process?
A formal PFMEA- is part of a company’s quality system,
but will not achieve maximum benefits if used without
a compliant quality system such as a documented
design and development process, operating procedures,
training process and procedures, product
specifications, packaging specifications, etc.(Robin E.
McDermott, Raymond J. Mikulak, Michael R.
Beauregard, 1996)
An integral element of the process documentation
should be the design FMEA (DFMEA). It is extremely
difficult (if not impossible) to do a thorough PFMEA
without completion of a DFMEA (D.H. Stamatis, 1995)

Validate the Process
Process validation should be performed
along with your product design and
development.
Part of Design Transfer
Validation ensures a robust process and is
defined in 21 CFR 820.3.
establishing by objective evidence that a process
consistently produces a result or product
meeting its predetermined specifications.

Concept / Feasibility (Preliminary
Prototypes)
Design and Development
Design Verification
Pilot Production (= verification prototypes)
Production (=Actual Prod.
Units)
Distribution
Design Changes
Design Validation
Process Validatio
n
Design Control
Design
Transfer

Validation Resources
GHTF (now IMDRF)/SG3/N99-10:2004
(Edition 2) Quality Management Systems-
Process Validation Guidance;
Guidance for Industry Process Validation:
General Principles and Practices, U.S FDA,
CDER, CBER, CVM
CDRH not included in this, but is still a good
resource.

Investigation Results…
Based on the Investigation, a team shall
create and implement a corrective action to
fill the design transfer gap.
We won’t cover this today, but this is an
important point as previously discussed.
However we will discuss how to validate the process to
instill consistency to predetermined criteria utilizing
Multivari Analysis along with developing proper process
monitoring methods.

Develop the Proper Team
QA, Engineering, Manufacturing, Regulatory,
Purchasing….
Develop Validation Protocols per the
requirements of your Validation
Procedure(s) and your Protocol Development
and Maintenance Procedure
As part of your investigation, ensure that the
Validation Procedures are adequate.
Do you gain much benefit from monitoring a
process that is not in control?

Validation Elements, IQ
IQ (Installation Qualification) The primary purpose of the installation qualification is to ensure
that the actual physical equipment, raw materials, ancillary
equipment, and environmental requirements all meet the
engineering specifications.
Ensure that all of the equipment is properly installed according
to the company’s specs and that it operates according to
specifications.
Electrical and Air pressure requirements are met.
Ensure the environmental controls are in place and not
affected.
Should product and packaging specifications already be
established and verified?
Supplier Qualifications & Contracts for packaging?

Now is the Time To Ensure: Package Seal Strength has been specified
All equipment is calibrated and on schedule
All preventive maintenance is established
Test methods are validated
Personnel qualified
Test Method Validation Resources:
Concepts for R&R Studies, Larry Barrentine, ASQ
ASTM F1469
Want to ensure the repeatability and reproducibility of your test methods
so you know you are obtaining consistent results within each operator and
between operators within a certain confidence level during the OQ and
production process monitoring.
Your workmanship standards, including approved representative samples,
e.g., visual standard for seal integrity.
Software functional testing has been performed.

Validation Elements, OQ
The PRIMARY purpose of the operational
qualification is:
to determine the process variation;
to reduce the variability of the process within
some acceptable limits;
and to optimize the process so it is consistently
capable.

What is Process Variation..
Variation is the amount of change of a
quality characteristic. For example, the
amount of change in the package
tensile strength sampled at different
times over a specified time period.
First we are interested in the type of
variation the process has.
This helps to narrow the possible causes of
process variation to simplify the DOE
process and optimize your process
specifications.

What is Process Variation..
Using Multi-Vari Analysis one can understand the major
type of variation:
Time to time
Piece to piece, and
Within piece
Each type is associated with certain factors that cause
that variation, which can then be used in your DOE.
Performed via the selection and measurement of a
stratified sample of the product which usually consists
of several groups of consecutive pieces selected over a
period of several hours, shifts, or days.

Summary of Multi-Vari Analysis for
Heat Sealing Process
Capture worst-case or widest variation
conditions.
8 Cavities per die shot.
Took 2 sets of 8 samples after “steady state
obtained”, samples taken within 5 – 10 minutes
(Documented Machine time, temp., & pressure.)
Determined which 3 cavities had the widest
variation in tensile strength.

Summary of Multi-Vari Analysis for
Heat Sealing Process
Used those 3 cavities for rest of analysis
Took 3 samples from one die shot and then 3 samples immediately
from the next die shot. (Using the pre-identified “widest variation”
cavities.)
Waited 5 minutes and obtained another set of 6 samples.
Recorded the time and machine settings with each set of 3
samples.
Tested each sample on the side seal and the end seals.
Tested 3 samples immediately from each set of 6 and held the
other 3 for testing 24 hours later
Performed this 3 times during the shift, and one sampling was right
after lunch
Yielded 3 pairs of 6 samples where half of the sample were tested
right away and the other half were tested in 24 hours

Summary of Multi-Vari Analysis for Heat
Sealing Process
The side and end seals were treated as two different
data sets.
Determined the greatest variation was between the
outer and inner seals,
and between the Cavity that was located on the
outer-most location of the sealing machine vs. the
cavity that had both side seals in the inner locations
of the sealing machine.
Data showed the whole packaging machine was
weaker towards the middle – almost like how an old
mattress sags in the middle

Why Would that Be….
Performed some investigative work
We worked with the maintenance personnel
and the maintenance manual and discovered
that there were Teflon gaskets used to
support each cavity that had not been
replaced.
These were replaced.

What next?
Updated the PM Procedure and Log for the Heat Sealer.
Ensured Maintenance Personnel understood the importance of this
change through documented training session.
Did we find the root cause of variation in this heat sealing process?
Using the time, temp. and pressure specifications that had already
been established by the company, we randomly collected 50
samples each day for one work week. (company had 1 shift) using
the trained personnel and current operating procedures for a
Process Capability Study.
We ensured samples were collected right after lunch as well as
randomly throughout the day.

Process Capability
Sample size was based on a 95% Confidence level with
an Alpha Risk of .05 which required a Minimum Sample
Cpk of 1.598.
Cpk incorporates the centering of the process and its
dispersion.
Process was computer controlled and operators ensured
time, temp. and pressure were monitored to ensure the
process parameters were within the tolerances already
established.
However, no adjustments to the machine were made.
Tensile testing was performed. Calculated Cpks for
each day. (Had ensured test method was validated.)

Process Capability Results & Process
Monitoring
All Cpks were between 3.58 - 4.33,
Have Removed the Root Cause Variable.
So maintain the current time, temp, pressure
specifications. Monitoring them throughout the
production runs.
Software provides process parameter print out for
each run.
Ensured visual seal inspection workmanship
standard was available, and all personnel qualified.

Monitoring Methods, cont’d. Visual seal inspections are performed and documented evenly
spaced throughout each run.
Visual inspection monitoring includes:
Proper Seal Width
Lack of Wrinkles, fold-overs, cracks
Continuity (uninterrupted, no channels)
Cleanliness, i.e., lack of debris, any fluids
Pinholes
Tears
Oversealed areas, such as a transparent or translucent seal
areas
Any problem found, either with process specifications or
seal/package specifications, NCR documented, evaluation and
investigation performed commensurate to the risk.

Visual Standard for Seal Integrity
ASTM F1886 testing provides a qualitative (accept / reject) visual
inspection method for evaluating the appearance characteristics of
unopened, intact seals in order to determine the presence of
defects that may affect the integrity of the package. ASTM F1886
covers the determination of channels in the package seal down to
a width of 75 μm (0.003 in.) with a 60 – 100 % probability.
See ASTM.com for additional package testing methods.

Monitoring
What data might we collect from this
process for our monthly CAPA meetings
and/or Management Reviews?
# of NCRs (normalized) from manufacturing
and assembly areas, Pareto and Run Chart per
month traveling 12 months.
May want to consider the failure codes for each
type of seal problem.

DOE, Design of Experiments
If multi-vari analysis hadn’t been so powerful in
this case, then would have followed with DOEs
to determine the process operating limits to
optimize the process.
Would multi-vari analysis be a good
investigation tool?
Note: Most of the time, the interaction of time
and temperature will be the critical variables
to control for heat sealing.
DOEs would be analyzed with ANOVA
techniques.

PFMEA
Can conduct PFMEA more easily, since we
now more thoroughly understand the
potential cause(s) of failures.
The cause of potential process failures
should be the “root causes”
The likelihood of the failure occurrence(s)
will be lower with a Cpk equal to or greater
than 1.33.
Remember, if there is a high severity, must
still investigate the problem, regardless.

News to Consider……. http://www.packagingdigest.com/testing/inspecting-heat-seals-
form-fill-seal-line-151209
The Induction Integrity Verification System (I2VS) from DIR Technologies
(which stands for Dynamic Infra-Red Technologies) uses non-contact
infrared imaging for inspecting heat seals.
DIR Technologies has introduced the DIR Eye for monitoring and inspecting
the heat-sealing process on form-fill-seal machines, including those
commonly used for medical device packaging.
The technology can be used to detect and analyze the heat signature
generated from the sealing of Tyvek/poly seals of form-fill-seal packages.
“The DIR Eye monitors the uniformity of the heat seal—in each individual
package and in contrast to other packages in the same batch,” he says.
“The width of the sealing area is monitored. Inspection checks that the
seal is correctly aligned, continuous, and uninterrupted. The DIR Eye can
‘see’ whether the product or any debris (for example, errant pieces of the
Tyvek) has entered the sealing area.

News, Cont’d.
100% of the packages are inspected at the speed of the packaging
line.
We see what cannot be seen by the naked eye or a regular camera.
For example, clear cooling fluid that can drip from the sealing
press on to the sealing area of the package and interfere with the
ability for the package to be sealed. The Tyvek, for example, can
be too thick, making it impossible for the sterilizing gas to
penetrate the material. In some cases overheating can damage
the Tyvek in such a way that particles can enter the package and
contaminate it. None of these issues can be seen with a standard
visual test.”
Note: This is food for thought.

Bonus Material!
Extra reading from Cheryl Boyce
Read at your leisure – not discussed
here Improving Process Quality Through Variation
Research
MultiVari Analysis for Heat Sealing Process








MultiVari Analysis for Heat
Sealing





FDA Medical Device Industry Coalition
Break
10:30 – 10:45 am

FDA Medical Device Industry Coalition
Efficient Validation Strategies
and VMPs
Anne Holland Wilson

Agenda
History and Recent Trends
Why Validate
21 CFR 820.75 Process Validation
Standards and Guidance
Definitions and Terminology
Processes Requiring Validation
Validation Planning
Validation Sequence
Process Monitoring, Control and Revalidation

FDA Medical Device Industry Coalition
History and Recent Trends

What is a Process? ANSI/ISO/ASQ 9000
A process is defined as a set of interrelating or interacting
activities which transforms inputs to outputs

Process Validation History
In practice…
De facto validations in American Industry since 1950’s; Bell
Telephone, Aerospace Industry, Chemical and Paper Industry
For Medical Products…
1st documented Process Validation Studies were for sterilization
(steam, then EtO) in the pharmaceutical industry during the
1960’s.

2015 Trends
Process Validation, 21 CFR 820.75, moves to
5th Place in Warning Letters
Production and Process Controls, Subpart G,
moves to 2nd Place in Warning Letter
Deficiencies
From GMP News- Medical Device Warning Letter Statistics

FDA Medical Device Industry Coalition
Why Validate?

Why Validate?
Economic Reasons Customer Satisfaction: Non-conforming product
can lead to lost customers.
Product Liability: Product Specifications must be maintained.
Reduced Production Costs: PV leads to reduced inspections, testing, scrap, and rework. Shifts costs from production to prevention.

Why Validate? 1976 GMP, converted to law in 1979, was substantive (i.e.,
violation was a criminal act). Message was validate.
1987 Guideline on General Practices of Process Validation. “This guideline…states principles and practices of general applicability that are not legal requirements but are acceptable to the FDA.” Message was how to validate.
1999 (Edition 1)/ 2004 (Edition 2) Global Harmonization Task Force- Quality Management Systems- Process Validation Guidance. Message was to define process validation principles and methods such that the resulting product or service can be practically guaranteed.

FDA Medical Device Industry Coalition
21 CFR 820.75 Process
Validation

cGMP Requirements 21 CFR 820.75
Applies to processes where the results cannot
be fully verified by subsequent inspection and
test, the process shall be validated with a high
degree of assurance and approved according to
established procedures
Activities, Results & Equipment validated shall
be documented
Date and signature of individual approving the
validation shall be documented

cGMP Requirements 21 CFR 820.75 continued
Each manufacturer shall ensure that:
Written procedures are in place for monitoring and
control of process parameters for validated processes
Process validation is performed by qualified
individuals
Monitoring and control methods and data, the date
performed and the people associated are documented
When changes or process deviations occur, the
manufacturer shall review and evaluate the process
and perform revalidation where appropriate. These
activities shall be documented.

Automated Processes 820.70
Are computers or automated data processing
systems used as part of production or the
quality system?

Automated Processes 820.70
If yes… then the manufacturer shall validate the computer software for its intended use according to an established protocol.
All software changes shall be validated.

What May Happen if You Don’t
Validate or Do It Poorly?
REGULATORY IMPACT
483 Observation
Warning Letter
Worse

Warning Letter (June 12, 2015)
Your firm failed to adequately validate the
equipment:
Installation Qualification (IQ) not approved
No Operational Qualification (OQ) conducted
Performance Qualification (PQ) consisted of only (b)(4)
lot of production which is inadequate to demonstrate
the reproducibility of the production line and only
(b)(4) samples from the production lot. Further the PQ
documentation for the line verification was incomplete.

Warning Letter (August 12, 2015)
Failure to validate a process whose results cannot be
fully verified by subsequent inspection and test as
required by 21 CFR 820 .75(a). For example:
Your firm did not validate the complete range of process
parameters used for (b)(4) of the duodenoscope bending section
assembly…
Your firm did not document the statistical rationale for the sample
size used in the validation
…you did not segregate or determine the worst case materials
during EO/ECH residual testing as part of the sterilization
validation… to determine proper aeration time.

483 Common Themes
Missing SOP for Process Validation
Missing Statistical Rationale
Lack of Consecutive Runs
Not Following Protocol
Acceptance Criteria Not Met
They didn’t do what they said they would….

FDA Medical Device Industry Coalition
Standards and Guidance

Standards
Provide guidance to manufacturers to promote industry
consistency.
FDA: US Food and Drug Administration
ASTM: American Society for Testing and Materials
ISO: International Organization for Standards
AAMI: Association for Advancement of Medical Instrumentation
NOTE: If standards are not followed during process validation,
the rationale must be provided and approved prior to
validation.

Worldwide Validation Guidance
Title: Quality Management Systems – Process Validation Guidance
Authoring Group: SG3
Endorsed by: The Global Harmonization Task Force
(International Medical Device Regulators Forum 2011)
Date: Edition 2 - January 2004

FDA Medical Device Industry Coalition
Definitions and Terminology

Validation
FDA-Establishing documented evidence which
provides a high degree of assurance that a
specific process will consistently produce a
product meeting its pre-determined
specifications and quality characteristics.
GHTF- Establishing by objective evidence that a
process consistently produces a result or
product meeting its predetermined
requirements.

Worst Case Conditions
FDA: Worst Case- A set of conditions encompassing
upper and lower processing limits and circumstances,
including those within standard operating procedures,
which pose the greatest chance of process or product
failure when compared to ideal conditions.

Qualification
QUALIFICATION
OPERATING
CONDITIONS
FAULT
SEEDING
WORST CASE
CONDITIONS
Definition: To establish confidence that a process, process equipment, and ancillary equipment are capable of consistently operating within established limits and tolerances.
More narrowly focused than validation

FDA vs GHTF Terminology
Concept Relevant Questions FDA's 1987 Guidance GHTF 2004 Guidance
Is the equipment installed
correctly?
Does the equipment perform
as expected?
Are the process factors that
influence resulting product
quality understood?
Has "worst case" testing been
performed to establish
process control limits?
Is there consistent process
output under normal operating
conditions?
When operating under
controlled conditions, does the
process deliver product that
meets its specifications?
Equipment Capability
Process Characterization
Process Adequacy
Equipment Installation
Qualification
Installation Qualification
(IQ)
Preliminary
Considerations
Operational
Qualification (OQ)
Process Performance
Qualification / Product
Performance
Qualification
Performance
Qualification (PQ)
Comparison of Process Validation Terminology

FDA Medical Device Industry Coalition
Processes Requiring Validation

Which Processes Require
Validation? Processes whose outputs can NOT be fully verified by subsequent
inspection or test.
Processes whose routine end product testing may have
insufficient sensitivity to verify the safety and efficacy of
finished devices.
Processes for which validation might be more cost-effective than
verification.
Processes for which validation is performed due to the potential
impact on the product. Consider incorporating risk analysis into
decision-making.
Supplier processes for components and/or devices

Processes Requiring Validation
Process
Test methods Cleanrooms Air systems
Water systems Cleaning, sanitation, degreasing Calibration
Aseptic processing Unique filtration processes Filling operations
Plastic bonding Plastic injection
molding/extrusion
Wave/hand
soldering
Utilities Dipping plastic and rubber Mixing
Lyophilization Sterile packaging operations Sterilization
Formulation methods Software-controlled processes Shelf Life

Validation Decision Tree

Which Processes Do Not Require
Validation? Processes that can be fully verified by subsequent
inspection and/or testing.
Even though validation may not be required, the company may still decide to validate the process.
Examples:
Output of a machining process which contains a precision-bored hole. The hole can be fully verified by inspection.
Production-line test procedure for quality characteristic can be conducted 100% in an economical fashion.

FDA Medical Device Industry Coalition
Validation Planning

Validation Timing PROSPECTIVE VALIDATION (The Preferred Approach)
FDA: Prospective Validation-Validation conducted
prior to the distribution of either a new product,
or product made under a revised manufacturing
process, where the revisions may affect the
product’s characteristics.
Prospective validation involves proving that the
process does what it is supposed to do by
performing an experimental plan, otherwise
known as a validation protocol, before the
process is actually implemented.

Validation Planning
Form
validation
team
Select
methods &
tools for
validation
Document
rational for
not
validating
Establish
“Master
Validation
Plan”
Identify &
describe
processes
Plan
validation
approach
Apply GHTF
decision
approach to each
process
Establishing the Process Validation Program
AAMI Quality System Compendium, 2004

Validation Planning

Process Validation Master Plan
Top level document that defines the
approach to validation.
Roadmap for accomplishing related process
validations
Answers how much validation is enough?
Why?

Validation Master Plan- Example
Operation/
Process
Description SOP/WI Validation
Required?
Validation Category
& Reference
Comments
N/A Order Issuance WI-8.2-1 YES
Category B
PR-2016-XXX
ERP System is validated to assure
that when an order is received a
production schedule is created to
specific part numbers, lot numbers
and quantities to be produced.
A control plan and check sheets for
each lot number are generated. It is
reviewed, signed and dated prior to
issuance to production .
10 Machine WI-7.5-9
Yes A
Verifiable process controlled
through training and procedures.
Acceptability is based on
dimensional inspection.
IQ, OQ and PQ

VMP Example Cont’d Operation/
Process
Description SOP/WI Validation
Required?
Validation Category
& Reference
Comments
40 Wash
WI-7.5-XX
WI-7.5-XX
WI-7.5-XX
Yes A
PR-XXXX
IQ, OQ and PQ to demonstrate
cleanliness per ASTM F 2459-05
and ISO 10993-5.
45 DI Water
System
Service Agreement
with Supplier;
WI-7.5-XX
Yes A
PR-XXXX
IQ, OQ and PQ to demonstrate
process consistently meets cleaning
process and AAMI TIR34:2007
requirements.
Ongoing monitoring consists of
water sample collection performed
by trained personnel and analyzed
by an outside laboratory at defined
intervals.
70 Final Inspection SOP-8.2-X No N/A
Verifiable manual process controlled
through training and procedures.

Validation Process
Generally consists of (3) steps:
Installation Qualification
Operational Qualification
Performance Qualification
Elements of each qualification will vary-depending on
the process. Use standard templates for efficiency and
to minimize the possibility of omissions.
Note: Usually process validation coexists with, and
supplements in-process and end product testing.

Process Validation Flowchart
Individual Process Validation Steps
Create
Validation
Protocols
Determine
On-going
Process
Controls
Monitor &
Control
Process
Perform Process Validation
IQ OQ PQ

Process Validation Overview
Generate Installation
Qualification (IQ/OQ)
Protocol
Conduct IQ and OQ on
both the Killion and Wellex
Are Acceptance
Criteria Achieved?
Generate Performance Qualification
(OQ) Protocol
Determine Root Cause and
Develop Action Plan to Continue
Analyze Operational and QC Data
From Each Run
Are Acceptance
Criteria Achieved?
YES
NO
NO
Conduct the PQ (3 Runs at
Nominal Conditions) for Largest &
Smallest Diameters
Are Acceptance
Criteria Achieved?EndYES
NO
Conduct
Feasibility
Analysis
YES
Is Process
Capable?
YES
NO
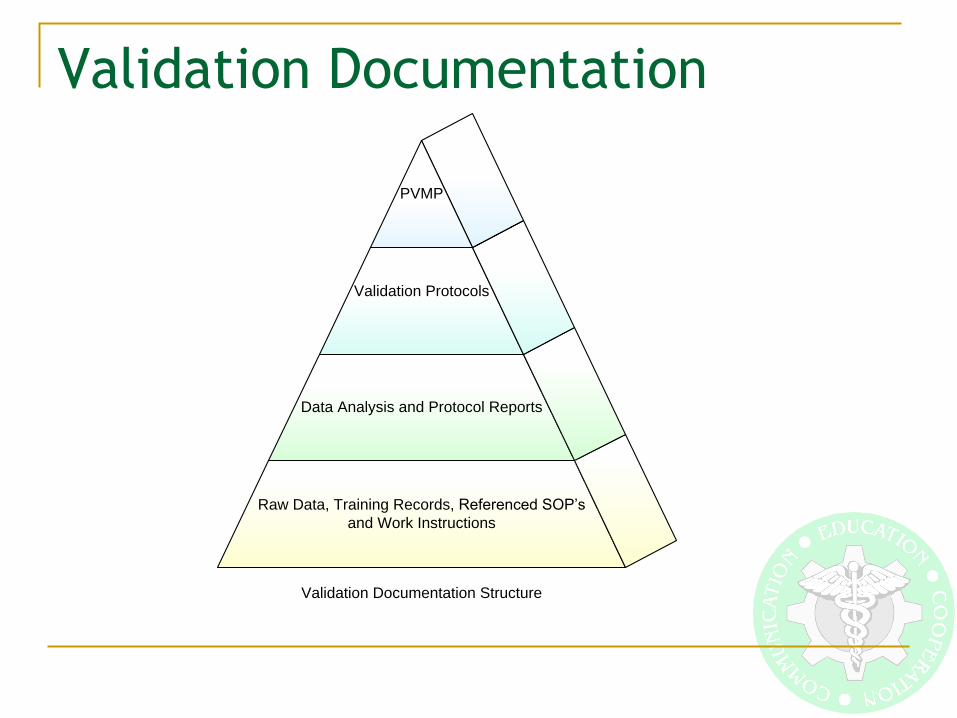
Validation Documentation
Raw Data, Training Records, Referenced SOP’s
and Work Instructions
Data Analysis and Protocol Reports
Validation Protocols
PVMP
Validation Documentation Structure

What Should Be in the Protocol?
Purpose
Scope
Sample Size with statistical rationale for
selection
Reference Documents
Method
Acceptance Criteria

What This Means:
Process and product specification should be
determined before starting validation.
Validations must be in writing.
Confidence level should be based on
criticality.
For product performance qualification, at
least three (3) sequential groups of product
are evaluated.

Agree on Analysis Techniques Up
Front How good is your evaluation?
What test for distribution assumptions is appropriate?
Will you have access to the required number of samples?
What assumptions are being made about the process or the data?
Agree on process criticality and determine appropriate α value.
Will samples/material be “representative” of production?

Tools Used to Identify Key
Process Variables Based on Risk Fishbone Diagram
Process Flow Chart
Hazard Analysis
Fault Tree Analysis
Failure Modes, Effects and Critical Analysis
Screening Designs of Fractional Factorial
Experiments

FDA Medical Device Industry Coalition
Validation Sequence
IQ/OQ/PQ

Installation Qualification (IQ)
Definitions FDA
Establishing confidence that process equipment and ancillary
systems are capable of consistently operating within
established limits and tolerances.
GHTF
Establishing by objective evidence that all key aspects of the
process equipment and ancillary system installation adhere to
the manufacturer’s approved specification and that the
recommendation of the supplier of the equipment are suitably
considered

Items Generally Included in an IQ
Utility requirements
Safety features
Environmental conditions required
Equipment identity, serial numbers, location,
model numbers
Calibration
Preventative Maintenance

IQ- How to Perform
Verify equipment configuration and schematics exists
Verify any custom fixtures meet print(s)
Verify ancillary systems (air , water) are connected and operate as specified
Measure the equipment/systems to determine if they meet design specification tolerances.
Establish preventative maintenance procedures, and repair lists.
Develop and implement calibration methods and procedures, if required.
Document all results

Operational Qualification (OQ) Demonstrate it works over anticipated range and worst case conditions
In this phase, process parameters should be challenged to assure they will result in a product that meets requirements under all anticipated conditions of manufacturing. Also, action level(s) should be developed to monitor routine production.
OQ Considerations include: Should simulate actual production conditions and worst cases
Process control limits (time, temperature, pressure, setup conditions, etc.)
Software parameters
Raw material specs
Process operating procedures
Material handling requirements
Process change control
Training
Short term stability and capability of process (think process performance)
Potential failure modes, action levels (think FMEA, risk management)

Items Generally Included in an OQ
Process capability study
Design of Experiments
Gage R&R
Test method validation
Verification and testing of
circuit breakers and fuses
for proper operation.
Exercise of all moving
parts of the equipment to
assure that they perform
properly.
Exercise of any sensors or
control elements to make
certain they perform
properly.
Confirmation of the safety
shielding or other safety
devices to assure that
they perform properly and
that they prevent the
danger they are designed
to eliminate.
Verification and testing of
electrical connections for
possibility of power surge
or loss of power.

Measurement/Gage Qualification REPEATABILITY
Variation in measurements obtained with one gage when used several
times by one operator while measuring a characteristic on one part.
REPRODUCIBILITY Variation in the average of the measurements made by different
operators using the same gage when measuring a characteristic on one
part.
EXAMPLES GM
Chrysler
Ford
Barrentine

Sources of Variation
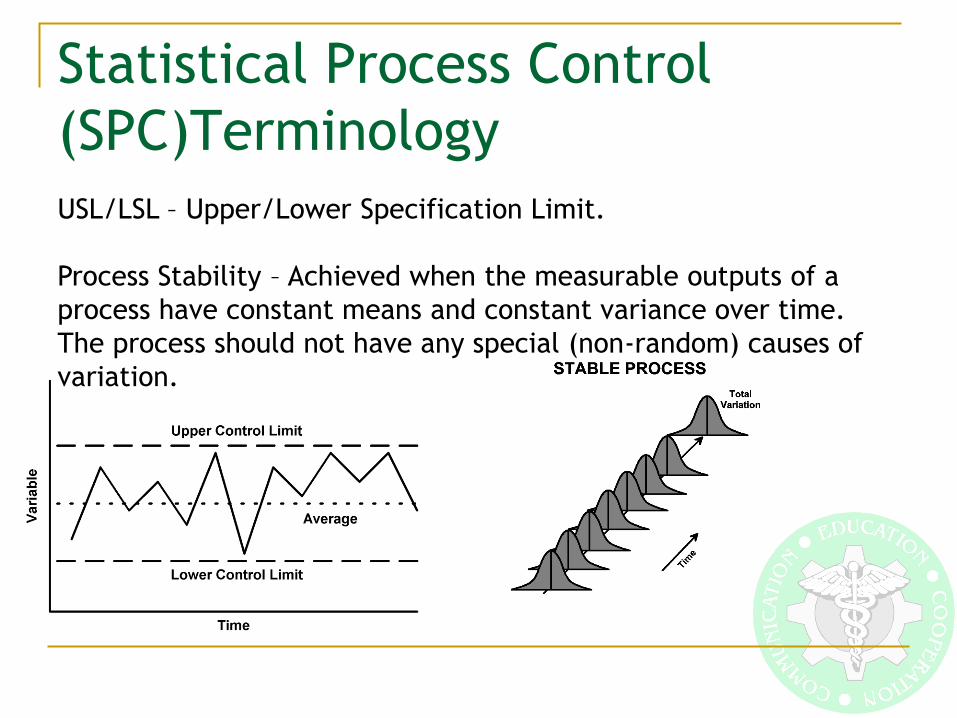
Statistical Process Control
(SPC)Terminology
USL/LSL – Upper/Lower Specification Limit.
Process Stability – Achieved when the measurable outputs of a
process have constant means and constant variance over time.
The process should not have any special (non-random) causes of
variation.

If Stable Then Assess Capability Process Capability – The ability to produce products/services that
meet specifications defined by the customer’s needs, or a
measure of the inherent uniformity of the process and the ability
to direct the process to a defined target.
An unstable process is unpredictable. Process stability must be
ensured before analyzing process capability.

Process Capability Performed
During OQ PROCESS CAPABILITY- The statistical measure of the
common cause variation of a process under controlled conditions (short term).
PROCESS CAPABILITY STUDY – A process capability study is a desirable element for validations involving measurable process outputs. It is a statistical method to determine the capability of a process is in a state of control. These studies can provide the basis for establishing evidence that a process is validated.

Process Capability Studies
Assumptions Statistical Stability
Normal Distribution
Specifications Based on Customer Requirements
Accept index/ration as “true” number
Many types of Indices (always look at >1) Cp and Pp-Process Variation Only
CPU, CPL, Cpk, Ppk-Process Variation & Centering
CR and PR-Process Variation Ratio Only
Goal: To align process with customer requirements Continually reduce variation and minimize loss

Graphical Interpretation of
Process Capability

Interpretation of Process
Capability Higher process capability index results in a more capable
process.
The following table lists expected defects for various
process capability indices:
Process Capability Index
(Cpk)
Estimated Defects per
Million
0.67 45,500
1.00 2,700
1.33 63
1.67 1

Performance Qualification (PQ) In this phase, the key objective is to demonstrate the qualified
process will consistently produce acceptable product under normal operating conditions.
PQ Considerations include: Actual product and process parameters, procedures established in OQ
Acceptability of product
Raw materials variation
Operator and shift variation
Assurance of process capability established in OQ
Process repeatability and long-term process stability
Challenges to process simulating conditions during actual production, including range of operating conditions established during OQ
Development of attributes for continuous monitoring and maintenance of process

FINAL REPORT At the conclusion of all validation activities,
prepare a final report.
Summarize and reference all protocols and
results.
Derive conclusions regarding the validation
status of the process.
Validation team should review and approve, as
well as appropriate management. Update the
VMP.

FDA Medical Device Industry Coalition
Process Monitoring, Control and
Revalidation

Validation Results Are not
Meaningful If… Measurement Systems are not appropriate
for the Job
Repeatable and reproducible
Process is not capable and stable
Maintenance is not established and deployed
Monitoring is not established to verify
process performs as validated
Changes are not revalidated as planned

Ongoing Controls
Attributes for monitoring process should be developed during
OQ/PQ.
Use SPC for routine monitoring of process output(s). Use tools
like:
Control charts, histograms, check sheets, Pareto charts, scatter diagrams, etc.
Sampling plans
Control plans
Also look at other Quality Management System trends, consider:
Nonconforming material trends
Customer feedback/complaint trends
Material Testing/Verification
When negative trends occur, investigate the cause and consider
corrective/preventive action to correct. If necessary, also
consider revalidation.

Revalidation Per the FDA, A system which requires revalidation with
a change to: Packaging
Formulation
Equipment
Sterilization
Process
Water System
which could impact product effectiveness or product
characteristics AND with a change to product
characteristics

Revalidation continued
Revalidate when…
Any significant change in product specifications,
process parameters, equipment type, function or
location, control system, raw materials,
manufacturing materials, major repairs to
process equipment, etc.
Scheduled, planned or otherwise anticipated
recurring validations to demonstrate continuing
compliance of validated process or operation to
meet its intended specifications (e.g.,
sterilization).

FDA Medical Device Industry Coalition
Take Away Message
Validation is the tool to meet high
product quality consistently…it’s your
reputation


FDA Medical Device Industry Coalition
Personnel Strategies:
Effective Training and
Selection Mona Elkhatib, TMAC

Session Topics
The training process stages
Determining needs
Design & Plan
Provide training
Evaluate training effectiveness
Share audit experience

Quote
“Recently I was asked if I was
going to fire an employee
who made a mistake that cost
the company $600,000.
No, I replied, I just spent
$600,000 training him!
Why would I want somebody
to hire his experience?
Thomas J. Watson
Chairman & CEO IBM

Does Training Need to be
Effective?
$1,229
$61.8 Billion

Why Train?
Ensures that employees can do current jobs
Improve employee performance
Bridge the gap between existing and
required:
Skills
Knowledge
Attitudes
Regulatory Requirement

Training Process ISO 10015
1. Define
Training
Needs
2. Design
& Plan
Training
3. Provide
Training
4. Evaluate
Training Monitor

Training Needs Analysis
Task Analysis:
Assessing New Employees’
Training Needs
Performance Analysis:
Assessing Current
Employees’ Training Needs
Detailed study of a job to
identify specific skills
required.

Task Analysis Record
Task list
When & how often performed
Quantity & quality of performance
Conditions under which performed
Skills or knowledge required
Where best learned

Training Needs Analysis
Task Analysis:
Assessing New Employees’
Training Needs
Performance Analysis:
Assessing Current
Employees’ Training Needs
Verify if there is a
performance deficiency
and determine if it can be
corrected thru training or
other means.

Identifying Training Needs
Organizational or technological change
Data collected from past or current training
Performance evaluations
High turnover or seasonal temporary
employees
Internal or external certification needed to
perform specific tasks

Identifying Training Needs
Request from employees as opportunity for
personal development
Results of process reviews and corrective
actions, as a result of customer complaints
Observations
Tests
Regulations

Assessment
Review existing competence
Define the competence gaps
Identify solutions to close gaps
Define the specification for training needs
Training objectives
Expected outcomes of the training

Design & Plan Stage
Address the competence gaps identified
Include a criteria for evaluating the training
outcomes

Define Constraints
These might include:
Regulatory & policy requirements
Financial considerations
Timing and scheduling requirements
Availability and motivation of personnel to
be trained
Availability of in-house resources to provide
training

Training Methods Selection
The appropriate form of training will depend
on:
Resources
Constraints
Objectives

Training Methods Types
On-the-Job Training (OJT)
Courses and workshops (on or off site)
Audiovisual training
Simulated training
Self-training
Distance & on-line learning

Selection Criteria
Date & location
Facilities
Cost
Training objectives
Target group of trainees
Duration of training
Forms of assessment, evaluation &
certification

The Plan
Training objectives aligned with
organization’s objectives & goals
Trainees (target group)
Training method
Duration & resource requirements
Criteria and methods developed for
evaluation of training outcomes

Training Material
Make training material meaningful
Make skills transfer to job easy
Motivate the learner
Explain why the training is important and how it
will benefit the learner

Training Provider
Internal/External?
Do they have the necessary expertise and
competency to deliver the training as
specified by the plan?
“If the employee has not learned, then the
trainer has not taught!”

Provide Training Stage
Train the employees who need to learn the
skill
Structure the training to minimize fatigue

Evaluate Training Stage
Organizations should have evidence that the
training is effective and employee
performance improves as a result of the
training

Kirkpatrick Evaluation Model
Level 1
Level 2
Reaction
Learning
Level 3 Behavior
Level 4 Results

Reaction: Did They Like it?
Observe trainees
Get feedback in writing following session
Use uniform forms to tabulate results
Anonymous- honest reaction

Learning: Did They Learn?
Pre-and-post knowledge and/or skills testing
Use objective measurements to assess what
trainees learnt
Where practical, use a control group to
compare to trainees

Behavior: Are They Using Skill?
These must be met for behavior change:
Desire to change
Knowledge of what to do and how to do it
The right job climate
Help in applying what was learned during
training
Rewards for changing behavior

Behavior: Are They Using Skill?
Systematic appraisal before & after
Performance made by one or more:
Trainee, supervisor, peers, subordinates (if any)
Post training appraisal >3 months after
training
Control group used, where practical

Results: Did Performance Improve?

Monitoring
Ensure that the training process is
implemented
Ensure that the training process is effective

Recap
1. Define
Training
Needs
2. Design
& Plan
Training
3. Provide
Training
4. Evaluate
Training Monitor

Sharing Audit Experience
Adequacy of defining needs
Records of training
Evaluating training effectiveness
Planning for evaluation
Timing
Use of “Training” or “Re-Training” in the
CAPA process


FDA Medical Device Industry Coalition
Lunch
12:00 – 1:00

FDA Medical Device Industry Coalition
Managing Facility Expansions,
Equipment Modifications, &
Continued Growth
Failing to Plan is Planning to Fail
Presented by:
www.an-answer.com

Foundational Stuff
Interactive process…volunteer or be
victimized!
The need for an inquisitive nature
Why…
What…
How…
Who?
CFR 820 Part 21 & standards like ISO 13485
are intended to make organizations think in
terms of closed loop processes!

The Blended Triumvirate (also known as competence, definition, and control)
The Kaleidoscope Effect!
• Think in terms of a
kaleidoscope,
considering all
and applying each
as needed.
• It may be
different for each
process or activity,
so consider the
kaleidoscope
each time!
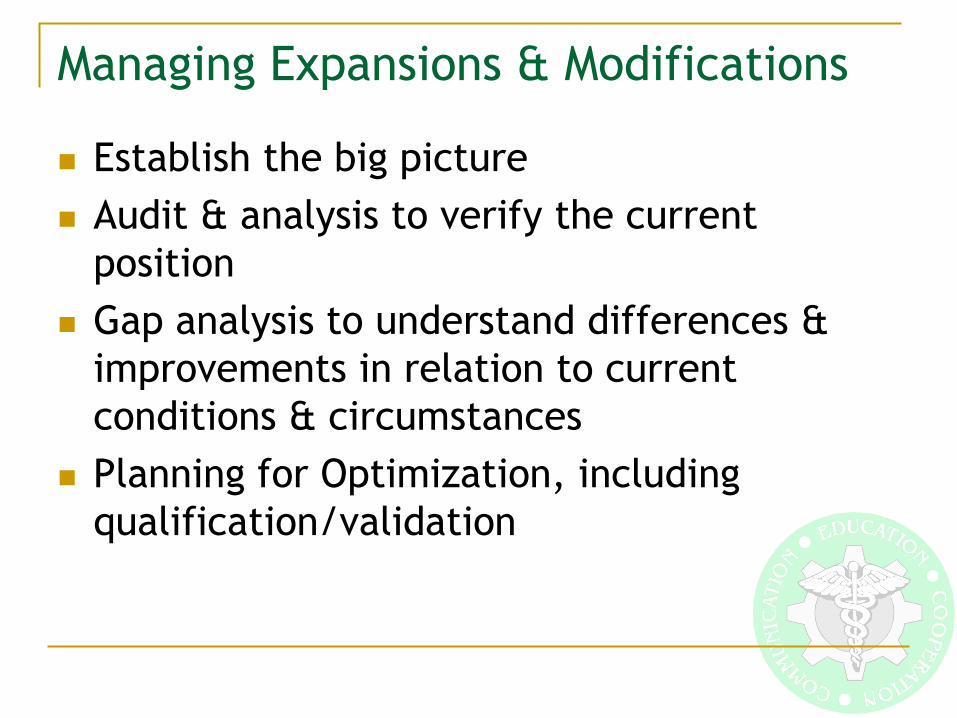
Managing Expansions & Modifications
Establish the big picture
Audit & analysis to verify the current
position
Gap analysis to understand differences &
improvements in relation to current
conditions & circumstances
Planning for Optimization, including
qualification/validation

The Big Picture What are we trying to accomplish?
What do we hope to gain from it?
How soon do we need to have it in place?
If we had to rely on existing methods and
controls, would we be able to meet regulations
and other requirements for the products and
processes involved?
Establish a preliminary plan for the
expansion/modification, including a risk analysis
Not resources…but resourcefulness!

Audits & Analysis of What’s Already There
Think in terms of performance-based auditing
How well do existing processes help us optimize?
How well do existing processes comply with
regulations and other requirements?
Analyze data related to process performance to
understand:
What the evidence says
What the evidence does not say
If what we observed is to be true, this must also be
true
If what we observed is to be true, this cannot be true

Gap Analysis Seeking first to understand…then to
undestand what needs to be updated or
adjusted
Start with an accurate picture (point of
reference) of what is already in place
Using the planned expansion/modification,
compare it with existing and note the gaps
Identify the risks associated with the transition,
including items impacted by the changes being
made
Report on the gaps

Planning For Optimization Establish a plan, considering:
Facility layout
Utility needs, including controls for sustainability
Equipment needs, including qualification/validation
Process controls, to reduce or eliminate risks
People, including required competence
Process instructions
Think in terms of optimization for
organizational benefit or they will struggle to
live it over time.

Coping With Continued Growth Plans and closed-loop process are only effective if they
help with brain cell management
Management’s review of the management systems is to
assess decisions already made, with actions taken intended
to make the organization more suitable (fits who the
organization is), adequate (covers everything the
organization has to live up to) , and effective (driving
behavior)
Changes to the organization, including those related to
facilities, equipment, and products need to be facilitated
by the management systems. Failing to do so affects the
organization’s ability to fulfill its purpose and can have
dire consequences!


FDA Medical Device Industry Coalition
Rework Strategies:
Effective Planning and
Execution Al Alonso
Alonso Quality System Consulting, LLC

NONCONFORMING PRODUCT
Nonconforming (NC) in-process or finished
product is expensive to discard/scrap
If the product can be reworked, what is
required from a product safety and regulatory
perspective?

PURPOSE
My purpose this afternoon is to provide a
path to successfully planning and executing
the rework of NC product into a product
that meets specification and is in regulatory
compliance.

21 CFR 820.90 NONCONFORMING
PRODUCT (a) Control of nonconforming (NC) product.
Each mfr. shall establish and maintain
procedures to control product that does not
conform to specified reqs. The procedures
shall address the identification,
documentation, evaluation, segregation,
and disposition of NC product. The
evaluation of NC shall include a notification
of the persons or organizations responsible
for the NC. The evaluation and any
investigation shall be documented.

21 CFR 820.90 NONCONFORMING
PRODUCT 820.90 (a) Control of NC Product
SOP for NC product
ID, document, evaluate, segregate,
and disposition NC
Evaluation of NC includes determining
need for investigation
And need for notification of the
persons/organization responsible for
the NC

21 CFR 820.90 NONCONFORMING
PRODUCT (b) Nonconformity Review and Disposition. (1)
Each mfr. shall establish and maintain
procedures that define the responsibility for
review and the authority for the disposition of
NC product. The procedures shall set forth the
review and disposition process. Disposition of
NC product shall be documented.
Documentation shall include the justification
for use of NC product and the signature of the
individual(s) authorizing the use.

21 CFR 820.90 NONCONFORMING
PRODUCT 820.90(b)(1) Nonconformity Review and
Disposition
SOP for review of and authority over the
disposition of NC product
Document disposition
Signature of individual(s) authorizing the use

21 CFR 820.90 NONCONFORMING
PRODUCT (2) Each mfr. shall establish and maintain
procedures for rework to include retesting and
reevaluation of the NC product after rework,
to ensure that the product meets its current
approved specifications. Rework and
reevaluation activities, including a
determination of any adverse effect from the
rework upon the product, shall be
documented in the DHR.

21 CFR 820.90 NONCONFORMING
PRODUCT 820.90(b)(2)
SOP for rework including testing and re-
evaluation
Ensure reworked product meets current
approved specs
Document rework and re-evaluation in DHR
Document any adverse effects from the
rework in DHR

DEFINITIONS
REWORK: Means action taken on a NC
product so that it will fulfill the specified
Device Master Record requirements before it
is released for distribution.

DEFINITIONS
REFURBISHING: The processing or
reprocessing to specified requirements of a
medical device which has been previously
released.
Note: Refurbishing applies also to
repackaging and/or re-sterilization of
medical devices, for example when a
container that maintains sterility has been
opened or damaged.

DEFINITIONS
REMANUFACTURER: Any person who
processes, conditions, renovates,
repackages, restores, or does any other act
to a finished device that significantly
changes the finished device’s performance
or safety specifications, or intended use.
Examples are endoscopes and cytoscopes;
also single use devices such as catheters

DEFINITIONS
REPROCESS: Validated processes used to
render a medical device, which has been
previously used or contaminated, fit for a
subsequent single use. These processes are
designed to remove soil and contaminants
by cleaning and to inactivate
microorganisms by disinfection or
sterilization. Examples are surgical
instruments

SOP REQS. FOR REVIEW &
DISPOSITION OF NC PRODUCT Defines the responsibility for review and
authority for disposition of NC product
The disposition shall be documented
Justification for the use of the NC product
Signature of the individual(s) authorizing the
use

SOP REQS. FOR THE REWORK OF
NC PRODUCT Process to define rework, retesting and re-
evaluation of the NC product to ensure that
it meets specs
Document in the DHR rework and re-
evaluation activities
Document in the DHR any determination of
any adverse effects of the rework

ROOT CAUSE ANALYSIS: KEY TO
ELIMINATING REWORK 1. Describe the problem
2. Gather data
3. Identify potential causes
4. Identify which you will remove or change
5. Identify solutions
6. Implement change
7. Monitor to ensure effectiveness
8. The use of your CAPA system is
appropriate

ROOT CAUSE ANALYSIS:
TECHNIQUES The Five Whys: Identify the problem and
continue to dig deeper for the root cause
Failure Modes and Effect Analysis: Aimed
to find various modes within a system. It
involves reviewing as many components as
possible to identify failure modes, and their
cause and effect. It is inductive reasoning
(forward logic) single point of failure
analysis.

ROOT CAUSE ANALYSIS:
TECHNIQUES Pareto Analysis: 20% of the work creates
80% of the results. Collect data and
determine which are the most prevalent
Fault Tree Analysis: At the Top of the Tree
is the problem and all potential causes tree
down from it. It is a Top Down deductive
failure analysis in which the undesired state
is analyzed logically.

ROOT CAUSE ANALYSIS:
TECHNIQUES FISHBONE = ISHIKAWA = CAUSE-and-
EFFECT DIAGRAMS: At the head of the fish
is the problem with the spine having 4 to 8
potential source categories attached to it.
Manpower, Methods, Machines, & Material
Measurements & Environment

REWORK PROCESS
CONSIDERATIONS A high rate of reworks is a strong indication
that the manufacturing process is not in a
state of control
If a non-validated process is used, then it
must be validated before the rework
Rework can effect other components
adversely. Ensure that is not the case.
Evaluate the product to ensure that it meets
the current approved specs

REWORK PROCESS
CONSIDERATIONS Ensure the SOP was followed
The CAPA system is a good tool for
addressing this process
Ensure that all the documentation was
completed and done correctly
Address any unforeseen anomalies with the
appropriate correction

REWORK FOLLOW-UP
BE PROACTIVE
Make management aware of the rework rate
via your monthly Quality report,
Management Review, etc.
As appropriate inform field personnel and
Customer Service
Inform QA and the Complaint Handling area
to alert for potential problems of Reworked
product

REWORK of NC PRODUCT
SUMMARY Understand what Rework means
Know the regulations, 21 CFR 820.90
Write SOPs to comply with the regulations
Follow your procedures
Perform robust and in-depth Root Cause
Analysis
Document
Follow-up for effectiveness


FDA Medical Device Industry Coalition
Effective CAPA: Determining
Appropriate Measures,
Intervals and Evidences
Raja Chatterjee
Director of Quality Assurance

Objectives
Recognize and address the common pitfalls
for the CAPA system.
Look at basic system requirements
Additional considerations

Who likes CAPA?
The process is too burdensome and does
not add value.
It is about blame not about improvement.
I do not know what is expected of me as a
CAPA owner.
Owning a CAPA is like checking into the
roach motel.
CAPA are not initiated using risk based
methods.
I would rather measure success than
failure.
CAPA
Management Leads
Auditors

Basic Steps Analyze quality
metrics to identify
impermissible issues
Stop the bleeding
(containment)
Investigate causes for
impermissible issues
Identify actions to correct
Verify and validate actions
Check effectiveness
• Assessment of issue (what happened)
• Risk Assessment (what is the impact)
• Determine need for CAPA (Corrective Action, Preventive
Action, Correction or No CAPA needed)
• Scope /Problem statement (Is/Is not)
• Containment
• Identification of immediate cause and systemic root-cause (for
Corrective Actions and Preventive Actions)
• Identify actions to address immediate cause and systemic root
cause
• Verify/Validate action (will it solve the problem statement? Will
it break something else?)
• Implement Actions
• Effectiveness check (did it solve the problem statement?)

Basics: Culture to nurture
improvement Avoid discussing fault in CAPA
Its about risk to health and safety of patients
Its about controlling waste and costs
Its about the benefit to the system to avoid
deviations which slow production
CAPA should discuss system improvement
and efficiency
Involve everyone in looking for areas of
weakness or for improvement

Basics: Locating Nonconformity Standard sources: where errors have already
been found Complaints, Nonconforming Product, Audit Findings (internal,
external, and supplier)
Hidden sources: Where potential errors hide Service records
Product returns (non-complaint)
Management Review
Internal Data sources (inspection, preventive maintenance ,etc.)
Employee observation

Filtering
Issues Non-tolerable
AND
Actionable Issues

Product related issues
Complaints and Product Non-Conformances
Evaluate based on risk to patient/user
Impact based filters - Severity versus Occurrence
scales
Can be done for every complaint / NCMR

Product related issues
Complaints and Product Non-Conformances
Evaluate based on risk to patient/user
Control based filters – control chart based
Create “buckets” based on product line as well
as defect types – ‘as reported’ and ‘as analyzed’
Periodic – at minimum quarterly

Quality System Issues
More difficult to monitor because the rules are
not as precise.
Need to build in significance factors (usually base
don potential impact on product) and
occurrence factors (multiple findings and
repeated findings indicate higher risk).
Tier risk using a defined scale like low, moderate
and high as an example.
Low Moderate High
Issue relates to
labeling but does
not affect
identity or usage
Issue affects
identity/usage of
a product but is
isolated.
Issue affects
identity/usage of
a product but is
not isolated.

Defined CAPA Process

Stop the bleeding
Containment the problem does
not need to wait for completion
of root-cause analysis.
Revisit assessment often
Consider all sources product:
customer, in field locations, in-
stock, in-transit, supplier
inventory, demo, and in WIP
Don’t forget to assess risk
related to product that has
already been used

Investigate: Root-cause analysis Develop a clear (and short) problem
statement so a solution can be found.
Defining scope is critical
What product/systems are affected?
What is/is not the problem?
Structured problem solving
Macro tools to identify all problem sources
(Ishikawa, SIPOC or Affinity diagrams)
Micro tools to drill down to specifics
(HACCP, Fault Tree, 5 Why etc.)
A good root cause analysis will
define the plan

Plan your actions
After root cause is determined, reassess
scope and containment of the problem.
It is not uncommon to need both a short-
term and long-term plan
Define responsibilities and time-frames
carefully – the plan should be useful and
have milestones
Plan out how to verify/validate changes
Plan how to check effectiveness of actions

Implementation Should go through every step of the plan
Provide evidence of completion of each step
of the plan
Evidence should include things like Engineering
Change Orders, Completed Rework routers, new
forms or procedures, training records etc.
Make sure that the loop on any open
containment action is closed.
If validations are required, make sure they
are complete and adverse effects on
product will not occur.

Effectiveness check
Should ensure that there is evidence that
the problem has been resolved
Should consider the same set of
circumstances that manifested the
problem in the first place (real life usage)
Examples – complaint follow-ups for a
quarter, incoming inspection results for a
quarter, follow-up audits
Consider that the sample size/ duration
should be sufficient to address the original
occurrence rate

Review and Disclosure

Common pitfalls
Timeliness
Not covering the entire scope of the CAPA
Inadequate verification/validation
Late or no containment
Too many CAPAs for small issues
Unclear accountability for activities
Non-value added steps

Further Considerations
CAPA review board – a group of managers
that can take risk assessment decisions and
allocate resources
CAPA metrics should be a part of
departmental objectives (also for non-QA
groups)
The gateway threshold should be revised
periodically – consider both risk
acceptability and practical criteria such as
organizational bandwidth


FDA Medical Device Industry Coalition
Break
2:50 – 3:05 pm

FDA Medical Device Industry Coalition
Verification of Corrections
Lauren Skokan Priest
Compliance Officer
Denver, CO

CAPA - Review
Scope of CAPA
Immediately contain affected product
Hold distribution and consider options to correct
Determine timeframe of problem to ensure all
affected product is considered
Consider affected raw materials or packaging
shared with similar products
Consider other systems affected

CAPA – Review
Consider risk to the user and patient
Preamble requires consideration of risk
Corrective actions should be commensurate with
risk of the problem
Potential new problems introduced by the
correction must also be considered
Ensure data sources are comprehensive to
identify problems

CAPA Review
Effective CAPA: Determining Appropriate
Measures, Intervals and Evidence
Pointers on how firms can decide on the level of
documentation and appropriate measurement
tools to determine CAPA effectiveness
Approaches to assess risk, scope and measure
effectiveness
Keep documented evidence of every step!

How are corrections verified?
Verification – 820.3(aa)
“means confirmation by examination and
provision of objective evidence that device
specifications conform with user needs and
intended use(s).”
This definition identifies how firms must perform
verification actions
Preamble comment 43 addresses that the
definition was adopted from the ISO 8402:1994
definition

How are corrections verified?
FDA can verify a firm’s corrections during
inspections or during subsequent review by
Compliance or CDRH
During Inspections
Investigator will follow up on observations from
previous inspections
Verify by looking through actions that are
documented
If it isn’t documented, it didn’t happen

How are corrections verified?
During Inspections - Example
Previous inspection found the firm did not have
an adequate system in place for Purchasing
Controls
Next inspection will review corrections
Investigator can review new procedures, and
evaluate evidence of implementation (supplier
qualifications, supplier evaluations, SCARS, etc.)

How are corrections verified?
During Inspections – Use cross cutting approach to
verify records across multiple systems affected by
deficiency
Review receiving acceptance records to verify
raw materials are meeting defined specifications
Can review complaint investigations and CAPAs
for raw material and supplier issues
Review evidence of training to updated
procedures or recurring training

How are corrections verified?
After Inspections – Corrections verified by
Compliance or CDRH
After receiving a 483 during an inspection, a firm
has the option to send in written response(s) to
their local district office describing corrections
Firm may receive correspondence from district
compliance or CDRH describing deficiencies with
those responses which may include: general
letter requesting clarifications, Regulatory
Meeting Request, UTL, WL, etc.

How are corrections verified?
After Inspections – Corrections verified by
Compliance or Center
Firm will then work with the applicable
Compliance Officer in written correspondence to
provide documentation demonstrating
corrections
Firm should supply actual procedures and a
sample of records to demonstrate the new
procedure has been corrected and implemented
Deficiencies will also be assessed during a follow
up inspection

How are corrections verified?
After Inspections – Corrections verified by
Compliance - Example
Firm was cited for
Inadequate CAPA procedure– 820.100
Lack of complaint investigations – 820.198
Inadequate NCR procedures – 820.90
Incomplete Risk Analysis – 820.30

Case Study: What is missing?
After Inspections – Corrections verified by
Compliance - Example
Firm was cited for CAPA, complaints, NCR & RA
Firm sent several responses to the 483 in for
review by compliance to describe their
corrections but ultimately a warning letter was
issued
Some reasons the responses were found
deficient…

What could not be verified?
Response deficiencies Warning Letter
Not all complaints or NCRs were addressed from
the 483 and the firm did not include any
reasoning for why some were not reviewed in the
response
Firm did not conduct root cause investigations
and provide the results for relevant complaints
Firm referred to different CAPAs as justification
for not needing a new investigation, but those
CAPAs were missing necessary testing and data

What could not be verified?
Response deficiencies Warning Letter
Response did not address supplier issues related
to contaminated or otherwise unacceptable raw
materials
Response referred to new CAPAs to address some
concerns but did not include copies of these
CAPA records
Training records for the newly implemented
procedures were not included in response

What could not be verified?
Response deficiencies Warning Letter
NCR procedure was cited as being inadequate,
then firm supplied updated NCRs with
incomplete investigations to demonstrate
corrections
Firm referred to other NCR records to address
CAPAs, but did not include copies of these
records

What could not be verified?
Response deficiencies Warning Letter
Risk analysis did not include review of risks for
products regularly intended to be used with their
device
Response discussed that procedural changes will
occur but did not provide any description or
training to the new procedures
Firm did not commit to reassessing complaints
once changes had been made to assess
effectiveness of correction

Things to remember
FDA can only verify using the records
provided
Provide documentation including procedures
and a sample of records that demonstrate
implementation of the correction
Don’t forget training records!
Opening a CAPA isn’t sufficient if further
documented activities are not provided

Things to remember
Consider re-evaluating complaint rates
at a reasonable timeframe after
correction to assess adequacy
If actions are planned, provide
commitment dates
Perform your own review cross cutting
over various systems to determine how
effective correction was and provide
that information to FDA to show your
systemic correction


FDA Medical Device Industry Coalition
Panel Discussion: Making it
All Work
3:45 – 4:40

FDA Medical Device Industry Coalition
Inputs for 2018 “Big Event”
Future Needs and Desires
3:45 – 4:40

Wrap-Up
Complete and turn-in
Evaluation Form
Pick up thumb drive
Pick up Certificate of
Attendance
Have a safe trip home
Enjoy your weekend!
See you in 2018!

















