Page 1
ORIGINAL PAPER
Quantum Chemical Determination of Stable Intermediateson CO2 Adsorption Onto Metal(Salen) Complexes
Maria C. Curet-Arana • Paul Meza •
Radames Irizarry • Rafael Soler
Published online: 21 April 2012
� Springer Science+Business Media, LLC 2012
Abstract Coupling reactions of CO2 and epoxides to
produce either cyclic carbonates or polycarbonates are
environmentally friendly reactions that allow the use of an
inexpensive and renewable feedstock while achieving
carbon efficiency. In this study, density functional theory
calculations were used to understand the role the
metal(salen) catalyst on CO2 adsorption. We have per-
formed a systematic analysis of the plausible interactions
of CO2 with metal(salen) catalysts and ethylene oxide/
metal(salen) complexes. Adsorption reactions were ana-
lyzed on six metal(salen) complexes: Co, Cr, Mn, Fe, Zn,
and Al, using the unrestricted OPBE functional. Geometry
optimizations were carried out beginning with a variety of
different conformations and frequency calculations were
used to verify that structures lie in an energy minimum.
Our results demonstrate that CO2 does not bind to the metal
atom of the bare metal(salen). The adsorption of CO2 onto
metal(salen) complexes is an endothermic reaction and the
lowest energy adsorbed complex involves the interaction of
CO2 with the adsorbed opened-epoxide.
Keywords CO2 adsorption � Coupling reaction �Quantum mechanical calculations � Metal(salen)
complexes � Al � Fe � Mn � Co � Cr � Zn
1 Introduction
Since petroleum resources are predicted to be depleted in
the near future, there is a growing effort to develop new
chemical processes using biorenewable resources, such as
CO2. A chemical process that uses CO2 for the production
of valuable chemicals would benefit from using an inex-
pensive, nontoxic, abundant, and renewable feedstock.
However, the thermodynamic stability of CO2 has ham-
pered its use as a reagent for chemical synthesis.
Inoue and coworkers demonstrated in 1969 that it was
possible to catalyze the reaction of CO2 with propylene
oxide in the presence of a zinc based catalyst [1]. The
coupling of CO2 with epoxides could yield cyclic carbon-
ates or polycarbonates via two competing reactions.
OO O
O
CO2catalyst+
1
O
O O
OCO2
n
catalyst+
2
The products from Reactions 1 and 2 are widely used in
industry. Cyclic carbonates are used as precursors in fine
chemical and pharmaceutical synthesis due to their cyclic
chiral nature. Cyclic carbonates are also used as aprotic
solvents, as electrolytes in high-energy density batteries,
and as raw materials for the synthesis of polymers [2].
Polycarbonate, on the other hand, is one of the most widely
used plastics, with an annual worldwide production of 2
million tons, and this number increases between 5 and
Electronic supplementary material The online version of thisarticle (doi:10.1007/s11244-012-9802-6) contains supplementarymaterial, which is available to authorized users.
M. C. Curet-Arana (&) � P. Meza � R. Irizarry � R. Soler
Department of Chemical Engineering, University of Puerto Rico,
Mayaguez, PR 00680, USA
e-mail: [email protected]
123
Top Catal (2012) 55:260–266
DOI 10.1007/s11244-012-9802-6
Page 2
10 % every year [3]. The production of polycarbonates
involves the use of toxic reactants, such as phosgene, and
the formation of side products, such as hydrogen chloride.
Therefore, using an alternate route to produce polycar-
bonates based on CO2 and epoxides would greatly decrease
the environmental concerns associated with this process.
Numerous catalysts have been tested for Reactions 1
and 2, but metal(salen) complexes exhibit some advantages
over other catalytic materials for these systems [4–8]. A
schematic diagram of a metal(salen) complex is shown in
Fig. 1. Metal(Salen) complexes are cheap and easy to
synthesize, they can be easily modified, tailored, and
immobilized on a solid support, and they have excellent
thermal and chemical stability. Nevertheless, while Reac-
tions 1 and 2 are exothermic (DHrxn for Reaction 1 is
-56.9 kJ/mol), typical experimental conditions for
metal(salen) catalyzed reactions involve high pressures
(20–150 atm) and temperatures (80–130 �C) [9–12].
Many research groups have postulated reaction mecha-
nisms for Reactions 1 and 2 based on kinetic experiments
[11, 13–21]. While most of the debate in the literature
centers on the step involving the opening of the epoxide
ring, most of the postulated reaction mechanisms assume
the same intermediate for CO2 adsorption, shown as the
product in Fig. 2. The coupling reaction of epoxides with
CO2 catalyzed by metal(salen) complexes involve the
interaction of both reactants with the catalyst. While the
adsorption of epoxides on metal(salen) complexes has been
extensively studied computationally [22–24], the interac-
tion of CO2 with metal(salen)/epoxide complexes is not
fully understood. To fill this void, we used density func-
tional theory (DFT) calculations to investigate proposed
intermediates for the adsorption of CO2 onto six
metal(salen) catalysts: Mn, Fe, Co, Cr, Zn, and Al. The
main focus of the calculations was the stability and
reactivity of the proposed CO2/salen intermediates. Dif-
ferent spin multiplicities were examined, as well as the
effect of a surrounding solvent. We also calculated the
geometry and energetics of the bare salen catalysts in order
to calculate reaction energies.
2 Methodology
Spin-unrestricted DFT calculations were performed to
obtain optimized geometries for metal(salen) complexes
and stable intermediates for the adsorption of CO2. We
have performed a systematic study on the adsorption of
CO2 on six different metal(salen) complexes: chromium,
manganese, iron, cobalt, zinc and aluminum. The specific
metal(salen) complexes analyzed are shown in Fig. 1. Two
models of the salen catalyst (labeled m1 and m2 in Fig. 1)
were used in the calculations. Model-1 consists of 14 heavy
atoms, while model-2 consists of 22. These models are a
mimic of the metal(salen) complexes that have been
studied in previous experiments for the coupling of CO2
and epoxides[13, 14]. Unchlorinated Zn(salen) was used to
analyze the adsorption in these complexes.
The OPTX-based exchange functional [25, 26] and the
Perdew–Becke –Ernzerhof (PBE) correlation functional
[27, 28] were used to analyze this system. The selection for
both functionals was based on the work published by
Ghosh and coworkers, where they assessed the perfor-
mance of different functionals for the electronic structure
of transition metal salens [29]. Among the functionals that
they analyzed, they report that O-PBE exchange–correla-
tion functional accurately captures the electronic structure
of the transition metals in the salens. The effective core
potential LANL2DZ was used as the basis set [30–33].
This is a double-f quality Dunning basis set that replaces
core electrons with a potential field to save computational
cost. This basis set has been shown by many research
groups to yield accurate energies of reaction with great
computational efficiency on these systems, and additional
test calculations using a larger basis set on similar systems
provide similar results [23, 34, 35].
Geometries and energies were obtained by performing
full geometry optimization with no symmetry constraints.
Optimizations were performed on the three lowest multi-
plicities for each structure. The optimization of stable
model 1 (m1) model 2 (m2)
Fig. 1 Metal(salen) complexes used in the DFT calculations.
M = metal atom, such as Co, Cr, Mn, Fe, Zn, Al
...
X XXX
Fig. 2 Elementary steps
involving coupling reaction of
ethylene oxide and CO2
catalyzed by a metal(salen)
complex
Top Catal (2012) 55:260–266 261
123
Page 3
species was verified with frequency calculations to ensure
that the structures lied in a minimum energy configuration.
Standard statistical mechanics was used to calculate the
translational, vibrational, rotational, and electronic contri-
butions to the partition function. The rigid-rotor, harmonic-
oscillator approximation was used to obtain the vibrational
frequencies. Wavefunction stability was confirmed using
procedure described by Seeger and Pople, and Bauernsch-
mitt and Ahlrichs [36, 37]. The \S2[ values in all of the
calculations were verified to ensure that the results do not
have spin contamination. Natural bond orbital population
analysis was performed for all intermediates to extract
electronic structure information [38, 39]. A polarizable
continuum model (PCM) was used to study solvent effects
on the energies of the stable intermediates [40]. For these
calculations, dichloromethane was used as the solvent,
which has a dielectric constant of 8.93. All calculations were
carried out with the Gaussian 09 program package [41].
3 Results
Table 1 describes the systems that are discussed in this
section. This section first presents an analysis of CO2
adsorption onto the bare metal(salen) catalysts. Next,
results for CO2 adsorption onto epoxide/metal(salen)
complexes are presented followed by the interaction of
CO2 with the opened epoxide ring adsorbed onto the cat-
alysts. All systems were analyzed for six metal(salen)
complexes: Cr, Mn, Fe, Co, Zn, and Al. Geometry opti-
mizations were performed beginning with several config-
urations using model 1 (m1). Adsorbed stable
intermediates found with m1 were then used as initial
configurations in optimizations with model 2 (m2).
3.1 CO2 Interaction with Bare Metal(salen) Complexes
Several geometry optimizations were performed in order to
elucidate the possible interactions between CO2 and the
bare metal(salen) complexes. Geometry optimizations
performed on the interaction of CO2 with m1 yielded four
stable adsorbed complexes, with no imaginary frequencies,
and they are labeled as 1-m1, 2-m1, 3-m1 and 4-m1 in
Table 2. Complex 4-m1, in which the CO2 interacts
through the oxygen atom with the metal atom of m1 was
only obtained for Aluminum-m1. As it can be observed
from this table, in complexes 1-m1 and 2-m1, the carbon
and the oxygen atoms of CO2 interacts with the aromatic
ring of the salen complex. The optimized geometries
obtained for 1-m1 and 2-m1 do not vary significantly
among the different metals. In 1-m1, the CO2 interacts
through the nitrogen and the carbon atoms of the salen ring.
For the six metal atoms considered in this model, the dis-
tance between the CO2 and the interacting atoms in 1-m1 is
1.55 ± 0.01 A, and the CO2 is bent with an angle of
126.7 ± 0.2�. In complex 2-m1, the interaction of CO2
with the salen model is through two of the carbon atoms of
the aromatic ring, and the distance between the CO2 and
the interacting atoms of the salen model, m1, is
1.61 ± 0.05 A. The angle of CO2 in this intermediate is
130.5 ± 0.2�. Similar results were obtained when model 2
was used in the calculations. Optimized geometries for the
cobalt intermediate in 1-m2 and 2-m2 are shown in
Table 3. The geometries of these intermediates are very
similar to the results obtained in 1-m1 and 2-m1. The
distance between CO2 and the interacting atoms of the
salen model-2 is 1.55 ± 0.02 A in 1-m2, and
1.64 ± 0.05 A. As it can be observed in the figures of
Table 3, the salen ring in m2 is bent downwards upon
adsorption of CO2.
Table 3 illustrates the only configurations that resulted
in adsorbed CO2 with the bigger salen model, m2. As
shown in this table, when expanding the salen model into
m2, CO2 remained adsorbed only in complexes in which
CO2 interacts with the aromatic ring of the salen complex.
Configurations in which CO2 interacts with the metal atom
of the salen complex, such as in 3-m1 and 4-m1, did not
yield an adsorbed complex using m2.
Table 1 Systems considered
for the adsorption of CO2CO2 / bare metal(salen) CO2 / adsorbed epoxide onto
metal(salen)CO2 / opened-epoxidering adsorbed onto metal(salen)
Cl
Cl
ClCl
Cl
Cl
+
ClCl
Cl
ClClCl
262 Top Catal (2012) 55:260–266
123
Page 4
It is interesting to notice that the spin ground state of the
salen complex is conserved upon adsorption of CO2 to
form either 1-m1 and 2-m1. This is illustrated in Table 4,
where the relative energies of the spin states are tabulated
for the six metal complexes studied. A natural bond orbital
population analysis in 1-m2 and 2-m2 reveals that the
natural charge of the carbon atom of CO2 decreases from
0.947 (on the isolated CO2 molecule) to 0.779 and 0.801 on
1-m2 and 2-m2, respectively (See Supplementary Infor-
mation). Even when the CO2 does not bind to the metal
atom in 1-m2 and 2-m2, the charges of the metal atoms
change upon adsorption, and they slightly increase on both
1-m2 and 2-m2.
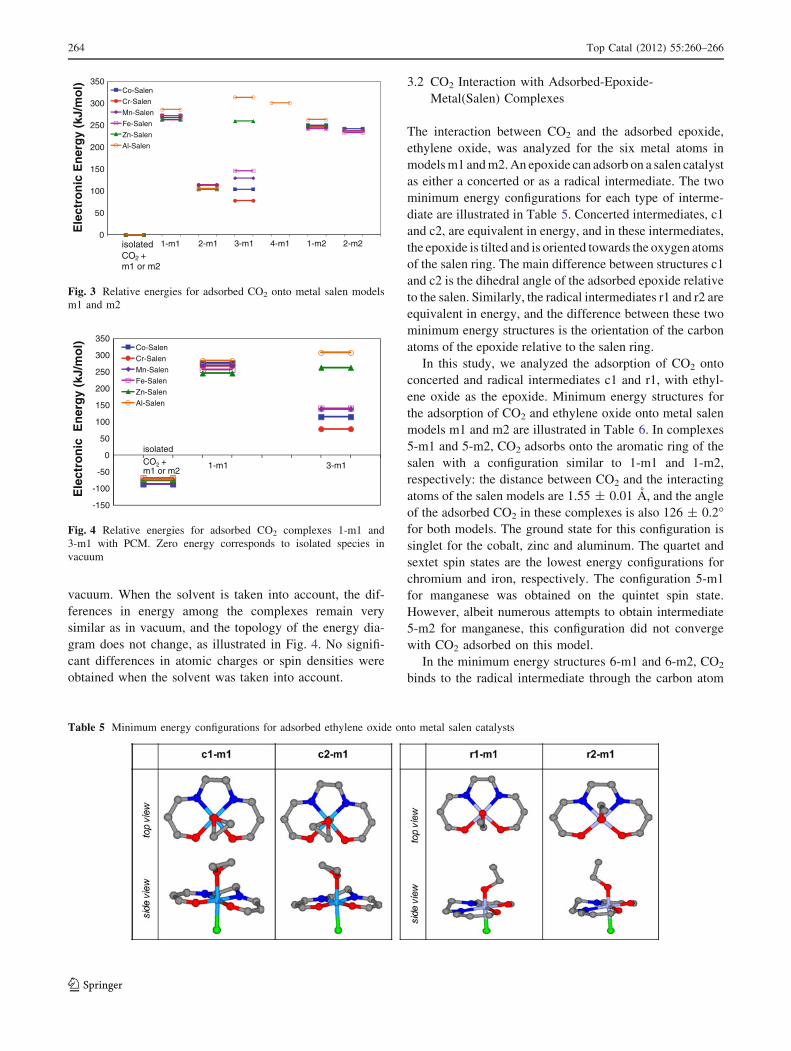
Energies for the adsorbed complexes illustrated in
Tables 2 and 3 relative to the isolated CO2 and salen
models, m1 or m2, are illustrated in Fig. 3 for the six
metals considered in this study. Two interesting features
can be observed in Fig 3: (1) energies of reaction for the
adsorption of CO2 in all the configurations presented in
Tables 2 and 3 are positive, and (2) for the complexes in
which CO2 interacts with the aromatic ring of the salen
models, the energies of reaction are independent of the
metal. It is interesting also to notice the different reaction
energies obtained when expanding the salen model for
complex 2-m1.
A PCM was used to study solvent effects on the energies
of stable complexes: metal-m1, 1-m1 and 3-m1. For these
calculations, dichloromethane was used as the solvent.
Single point calculations were performed with PCM using
OPBE/LANL2DZ on optimized structures obtained in
Table 2 Minimum energy
configurations for adsorption of
CO2 onto a bare salen catalysts
with model 1
Table 3 Minimum energy configurations for adsorption of CO2 onto
a bare salen catalysts with model 2
Table 4 Relative energy between spin states for adsorbed CO2
intermediates onto bare salen catalysts with model m1
Esinglet Etriplet EquintetMn-m1 141 80 01-Mn-m1 - - 02-Mn-m1 - 83 03-Mn-m1 - - 0Co-m1 16 0 371-Co-m1 19 0 -2-Co-m1 25 0 -3-Co-m1 0 - -Zn-m1 0 243 4931-Zn-m1 0 - -2-Zn-m1 0 - -3-Zn-m1 - 0 -Al-m1 0 238 4971-Al-m1 0 - -2-Al-m1 0 243 -3-Al-m1 - 0 -4-Al-m1 - 0 246
Edoublet Equartet EsextetCr-m1 91 0 1551-Cr-m1 - 0 -2-Cr-m1 - 0 -3-Cr-m1 - 0 -Fe-m1 57 18 01-Fe-m1 - 30 02-Fe-m1 70 31 03-Fe-m1 0 - -
Top Catal (2012) 55:260–266 263
123
Page 5
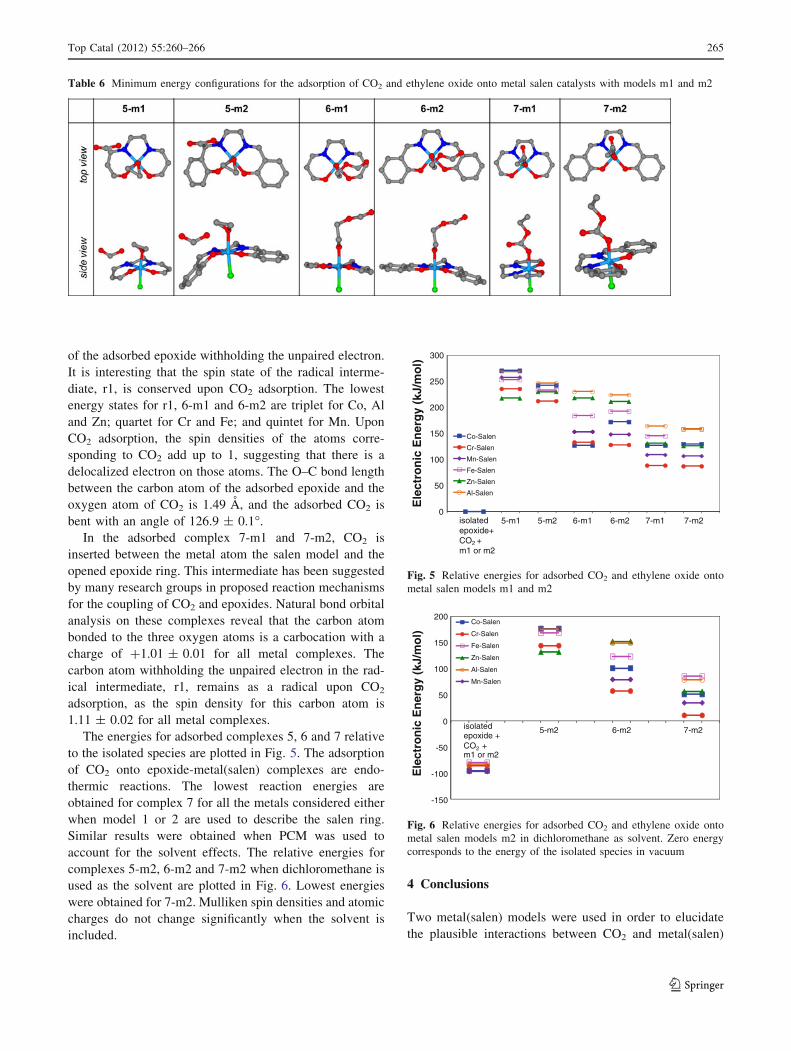
vacuum. When the solvent is taken into account, the dif-
ferences in energy among the complexes remain very
similar as in vacuum, and the topology of the energy dia-
gram does not change, as illustrated in Fig. 4. No signifi-
cant differences in atomic charges or spin densities were
obtained when the solvent was taken into account.
3.2 CO2 Interaction with Adsorbed-Epoxide-
Metal(Salen) Complexes
The interaction between CO2 and the adsorbed epoxide,
ethylene oxide, was analyzed for the six metal atoms in
models m1 and m2. An epoxide can adsorb on a salen catalyst
as either a concerted or as a radical intermediate. The two
minimum energy configurations for each type of interme-
diate are illustrated in Table 5. Concerted intermediates, c1
and c2, are equivalent in energy, and in these intermediates,
the epoxide is tilted and is oriented towards the oxygen atoms
of the salen ring. The main difference between structures c1
and c2 is the dihedral angle of the adsorbed epoxide relative
to the salen. Similarly, the radical intermediates r1 and r2 are
equivalent in energy, and the difference between these two
minimum energy structures is the orientation of the carbon
atoms of the epoxide relative to the salen ring.
In this study, we analyzed the adsorption of CO2 onto
concerted and radical intermediates c1 and r1, with ethyl-
ene oxide as the epoxide. Minimum energy structures for
the adsorption of CO2 and ethylene oxide onto metal salen
models m1 and m2 are illustrated in Table 6. In complexes
5-m1 and 5-m2, CO2 adsorbs onto the aromatic ring of the
salen with a configuration similar to 1-m1 and 1-m2,
respectively: the distance between CO2 and the interacting
atoms of the salen models are 1.55 ± 0.01 A, and the angle
of the adsorbed CO2 in these complexes is also 126 ± 0.2�for both models. The ground state for this configuration is
singlet for the cobalt, zinc and aluminum. The quartet and
sextet spin states are the lowest energy configurations for
chromium and iron, respectively. The configuration 5-m1
for manganese was obtained on the quintet spin state.
However, albeit numerous attempts to obtain intermediate
5-m2 for manganese, this configuration did not converge
with CO2 adsorbed on this model.
In the minimum energy structures 6-m1 and 6-m2, CO2
binds to the radical intermediate through the carbon atom
0
50
100
150
200
250
300
350E
lect
ron
ic E
ner
gy
(kJ/
mo
l) Co-Salen
Cr-Salen
Mn-Salen
Fe-Salen
Zn-Salen
Al-Salen
isolated CO2 +m1 or m2
1-m1 2-m1 3-m1 4-m1 1-m2 2-m2
Fig. 3 Relative energies for adsorbed CO2 onto metal salen models
m1 and m2
-150
-100
-50
0
50
100
150
200
250
300
350
Ele
ctro
nic
En
erg
y (k
J/m
ol) Co-Salen
Cr-Salen
Mn-Salen
Fe-Salen
Zn-Salen
Al-Salen
isolated
CO2 +m1 or m2
3-m11-m1
Fig. 4 Relative energies for adsorbed CO2 complexes 1-m1 and
3-m1 with PCM. Zero energy corresponds to isolated species in
vacuum
Table 5 Minimum energy configurations for adsorbed ethylene oxide onto metal salen catalysts
264 Top Catal (2012) 55:260–266
123
Page 6
of the adsorbed epoxide withholding the unpaired electron.
It is interesting that the spin state of the radical interme-
diate, r1, is conserved upon CO2 adsorption. The lowest
energy states for r1, 6-m1 and 6-m2 are triplet for Co, Al
and Zn; quartet for Cr and Fe; and quintet for Mn. Upon
CO2 adsorption, the spin densities of the atoms corre-
sponding to CO2 add up to 1, suggesting that there is a
delocalized electron on those atoms. The O–C bond length
between the carbon atom of the adsorbed epoxide and the
oxygen atom of CO2 is 1.49 A, and the adsorbed CO2 is
bent with an angle of 126.9 ± 0.1�.
In the adsorbed complex 7-m1 and 7-m2, CO2 is
inserted between the metal atom the salen model and the
opened epoxide ring. This intermediate has been suggested
by many research groups in proposed reaction mechanisms
for the coupling of CO2 and epoxides. Natural bond orbital
analysis on these complexes reveal that the carbon atom
bonded to the three oxygen atoms is a carbocation with a
charge of ?1.01 ± 0.01 for all metal complexes. The
carbon atom withholding the unpaired electron in the rad-
ical intermediate, r1, remains as a radical upon CO2
adsorption, as the spin density for this carbon atom is
1.11 ± 0.02 for all metal complexes.
The energies for adsorbed complexes 5, 6 and 7 relative
to the isolated species are plotted in Fig. 5. The adsorption
of CO2 onto epoxide-metal(salen) complexes are endo-
thermic reactions. The lowest reaction energies are
obtained for complex 7 for all the metals considered either
when model 1 or 2 are used to describe the salen ring.
Similar results were obtained when PCM was used to
account for the solvent effects. The relative energies for
complexes 5-m2, 6-m2 and 7-m2 when dichloromethane is
used as the solvent are plotted in Fig. 6. Lowest energies
were obtained for 7-m2. Mulliken spin densities and atomic
charges do not change significantly when the solvent is
included.
4 Conclusions
Two metal(salen) models were used in order to elucidate
the plausible interactions between CO2 and metal(salen)
Table 6 Minimum energy configurations for the adsorption of CO2 and ethylene oxide onto metal salen catalysts with models m1 and m2
0
50
100
150
200
250
300
Ele
ctro
nic
En
erg
y (k
J/m
ol)
Co-Salen
Cr-Salen
Mn-Salen
Fe-Salen
Zn-Salen
Al-Salen
isolated epoxide+CO2 +m1 or m2
5-m1 6-m1 7-m15-m2 6-m2 7-m2
Fig. 5 Relative energies for adsorbed CO2 and ethylene oxide onto
metal salen models m1 and m2
-150
-100
-50
0
50
100
150
200
Ele
ctro
nic
En
erg
y (k
J/m
ol)
Co-Salen
Cr-Salen
Fe-Salen
Zn-Salen
Al-Salen
Mn-Salen
isolated epoxide +CO2 +m1 or m2
5-m2 6-m2 7-m2
Fig. 6 Relative energies for adsorbed CO2 and ethylene oxide onto
metal salen models m2 in dichloromethane as solvent. Zero energy
corresponds to the energy of the isolated species in vacuum
Top Catal (2012) 55:260–266 265
123
Page 7
complexes with six different metals. Our calculations with
the bigger metal(salen) model demonstrate that CO2 does
not bind to the metal atom of the bare metal(salen). The
only possible interactions between CO2 and the bare
meta(salen) catalysts are through the aromatic rings of the
salen. However, the energy of creating such adsorbed
complexes are in the order of 200 kJ/mol even when eth-
ylene oxide is adsorbed onto the metal atom of the
metal(salen) catalyst. Moreover, energies of reaction do not
change significantly when a PCM is used to take into
account the solvent effect on the energies. Interactions of
CO2 with a radical intermediate formed with an adsorbed-
opened-epoxide onto the metal(salen) complex can yield in
two possible stable configurations: adsorption through the
radical atom or ‘‘insertion’’ between the metal and the
oxygen atom of the epoxide. The latter complex, which is a
carbo-cation-radical intermediate, is the lowest energy
configuration, but it is still above 85 kJ/mol relative to the
isolated species.
Acknowledgments Support from the National Science Foundation
(DMR-0934115) and the Puerto Rico Institute for Functional Mate-
rials under the National Science Foundation Award No. EPS-1002410
is gratefully acknowledged. PM, RS and RI have been partially
supported by NSF (HDR-0833112). The authors thank IFN for pro-
viding computational resources on the High Performance Computing
Facility. MCCA thanks Prof. Randall Snurr for helpful discussions.
References
1. Inoue S, Koinuma H, Tsuruta T (1969) J. Polym Sci Part B 7:287
2. Sakakura T, Choi J-C, Yasuda H (2007) Chem Rev 107:2365
3. Shaikh A-AG, Sivaram S (1996) Chem Rev 96:951
4. Paddock RL, Nguyen ST (2005) Macromolecules 38:6251
5. Paddock RL, Hiyama Y, McKay JM, Nguyen ST (2004) Tetra-
hedron Lett 45:2023
6. Jutz F, Grunwaldt J-D, Baiker A (2008) J Mol Catal A: Chem
279:94
7. Jing H, Edulji SK, Gibbs JM, Stern CL, Zhou H, Nguyen ST
(2004) Inorg Chem 43:4315
8. Tu M, Davis RJ (2001) J Catal 199:85
9. Lu X-B, Liang B, Zhang Y-J, Tian Y-Z, Wang Y-M, Bai C-X,
Wang H, Zhang R (2004) J Am Chem Soc 126:3732
10. Paddock RL, Nguyen ST (2004) Chem Commun 1622
11. Darensbourg DJ, Rodgers JL, Mackiewicz RM, Phelps AL (2004)
Catal Today 98:485
12. Lu X-B, Feng X-J, He R (2002) Appl Catal A 234:25
13. Darensbourg DJ (2007) Chem Rev 107:2388
14. Coates GW, Moore DR (2004) Angew Chem Int Ed 43:6618
15. Jacobsen EN (2000) Acc Chem Res 33:421
16. Kruper WJ, Dellar DD (1995) J Org Chem 60:725
17. Hansen KB, Leighton JL, Jacobsen EN (1996) J Am Chem Soc
118:10924
18. Cohen CT, Chu T, Coates GW (2005) J Am Chem Soc 127:10869
19. Moore DR, Cheng M, Lobkovsky EB, Coates GW (2003) J Am
Chem Soc 125:11911
20. Paddock RL, Nguyen ST (2001) J Am Chem Soc 123:11498
21. Darensbourg DJ, Moncada AI, Choi W, Reibenspies JH (2008) J
Am Chem Soc 130:6523
22. Oxford GAEC-A, Maria C, Majumder, Debarshi, Gurney, Rich-
ard W, Merlau, Melissa L, Nguyen, SonBinh T, Snurr, Randall Q,
Broadbelt, Linda J (2009) J Catal 266:145
23. Curet-Arana MC, Emberger GA, Broadbelt LJ, Snurr RQ (2008)
J Mol Catal A: Chem 285:120
24. Curet-Arana MC, Snurr RQ,Broadbelt LJ (2008) In: Oyama ST
(ed) Mechanisms in homogeneous and heterogeneous epoxida-
tion catalysis. Elsevier, Amsterdam, p 471
25. Handy NC, Cohen AJ (2001) Mol Phys 99:403
26. Hoe WM, Cohen AJ, Handy NC (2001) Chem Phys Lett 341:319
27. Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
28. Perdew JP, Burke K, Ernzerhof M (1997) Phys Rev Lett 78:1396
29. Conradie J, Ghosh A (2007) J Chem Theory Comput 3:689
30. Dunning TH, Hay PJ (1976) In: Schaeffer III HF (ed) Modern
theoretical chemistry, vol. 3, Plenum press, New York, p 1
31. Hay PJ, Wadt WR (1985) J Chem Phys 82:270
32. Wadt WR, Hay PJ (1985) J Chem Phys 82:284
33. Hay PJ, Wadt WR (1985) J Chem Phys 82:299
34. Sears JS, Sherrill CD (2008) J Phys Chem A 112:3466
35. Takatani T, Sears JS, Sherrill CD (2009) J Phys Chem A 113:
9231
36. Seeger R, Pople JA (1977) J Chem Phys 66:3045
37. Bauernschmitt R, Ahlrichs R (1996) J Chem Phys 104:9047
38. Foster JP, Weinhold F (1980) J Am Chem Soc 102:7211
39. Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899
40. Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
41. Gaussian 09, Revision A.1, Frisch, MJ, Trucks GW, Schlegel HB,
Scuseria GE, Robb MA, Cheeseman, JR, Scalmani, G, Barone, V,
Mennucci, B, Petersson, GA, Nakatsuji, H, Caricato, M, Li X,
Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL,
Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M,
Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery
Jr. JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E,
Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghav-
achari K, Rendell A, Burant JC, Iyengar SS, Tomasi, J, Cossi, M,
Rega, N, Millam, NJ, Klene, M, Knox JE, Cross, JB, Bakken V,
Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O,
Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL,
Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannen-
berg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz
JV, Cioslowski J, Fox DJ Gaussian, Inc., Wallingford, 2009
266 Top Catal (2012) 55:260–266
123