Quantum Monte Carlo The QMC=Chem code Quantum Monte Carlo simulations in chemistry at the petascale level and beyond A. Scemama 1 , M. Caffarel 1 , E. Oseret 2 , W. Jalby 2 1 Laboratoire de Chimie et Physique Quantiques / IRSAMC, Toulouse, France 2 Exascale Computing Research / Intel, CEA, GENCI, UVSQ Versailles, France 28 June 2012 A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Transcript
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo simulations in chemistryat the petascale level and beyond
A. Scemama1, M. Caffarel1, E. Oseret2, W. Jalby2
1Laboratoire de Chimie et Physique Quantiques / IRSAMC,Toulouse, France
2Exascale Computing Research / Intel, CEA, GENCI, UVSQVersailles, France
28 June 2012
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo methods
Solve the Schrödinger equation with random walksState-of-the-art and routine approaches in physics : nuclearphysics, condensed-matter, spin systems, quantum liquids,infrared spectroscopy . . .Still of confidential use for the electronic structure problem ofquantum chemistry (as opposed to post-HF and DFT)Reason : Very high computational cost for small/mediumsystems
But :Very favorable scaling with system size compared to standardmethodsIdeally suited to extreme parallelism
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo methods
Solve the Schrödinger equation with random walksState-of-the-art and routine approaches in physics : nuclearphysics, condensed-matter, spin systems, quantum liquids,infrared spectroscopy . . .Still of confidential use for the electronic structure problem ofquantum chemistry (as opposed to post-HF and DFT)Reason : Very high computational cost for small/mediumsystems
But :Very favorable scaling with system size compared to standardmethodsIdeally suited to extreme parallelism
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo methods
Solve the Schrödinger equation with random walksState-of-the-art and routine approaches in physics : nuclearphysics, condensed-matter, spin systems, quantum liquids,infrared spectroscopy . . .Still of confidential use for the electronic structure problem ofquantum chemistry (as opposed to post-HF and DFT)Reason : Very high computational cost for small/mediumsystems
But :Very favorable scaling with system size compared to standardmethodsIdeally suited to extreme parallelism
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo methods
Solve the Schrödinger equation with random walksState-of-the-art and routine approaches in physics : nuclearphysics, condensed-matter, spin systems, quantum liquids,infrared spectroscopy . . .Still of confidential use for the electronic structure problem ofquantum chemistry (as opposed to post-HF and DFT)Reason : Very high computational cost for small/mediumsystems
But :Very favorable scaling with system size compared to standardmethodsIdeally suited to extreme parallelism
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo methods
Solve the Schrödinger equation with random walksState-of-the-art and routine approaches in physics : nuclearphysics, condensed-matter, spin systems, quantum liquids,infrared spectroscopy . . .Still of confidential use for the electronic structure problem ofquantum chemistry (as opposed to post-HF and DFT)Reason : Very high computational cost for small/mediumsystems
But :Very favorable scaling with system size compared to standardmethodsIdeally suited to extreme parallelism
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Quantum Monte Carlo for molecular systems
Problem : Solve stochastically the Schrödinger equation for Nelectrons in a molecule
r1, . . . , rN : Electron coordinatesΦ : Exact wave functionΨ : Trial wave function
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
QMC Algorithm
Walker : vector of R3N containing the electron positionsDrifted diffusion of walkers with birth/death process togenerate the 3N-density (Ψ× Φ) (needs Ψ, ∇Ψ, ∆Ψ)
Compute HΨ(r1,...,rN)Ψ(r1,...,rN) for all
positionsThe energy is the average ofall computed HΨ(r1,...,rN)
Ψ(r1,...,rN)
Extreme parallelism :Independent populations ofwalkers can be distributedon different CPUs
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
QMC Algorithm
Walker : vector of R3N containing the electron positionsDrifted diffusion of walkers with birth/death process togenerate the 3N-density (Ψ× Φ) (needs Ψ, ∇Ψ, ∆Ψ)
Compute HΨ(r1,...,rN)Ψ(r1,...,rN) for all
positionsThe energy is the average ofall computed HΨ(r1,...,rN)
Ψ(r1,...,rN)
Extreme parallelism :Independent populations ofwalkers can be distributedon different CPUs
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
QMC Algorithm
Walker : vector of R3N containing the electron positionsDrifted diffusion of walkers with birth/death process togenerate the 3N-density (Ψ× Φ) (needs Ψ, ∇Ψ, ∆Ψ)
Compute HΨ(r1,...,rN)Ψ(r1,...,rN) for all
positionsThe energy is the average ofall computed HΨ(r1,...,rN)
Ψ(r1,...,rN)
Extreme parallelism :Independent populations ofwalkers can be distributedon different CPUs
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
QMC Algorithm
Walker : vector of R3N containing the electron positionsDrifted diffusion of walkers with birth/death process togenerate the 3N-density (Ψ× Φ) (needs Ψ, ∇Ψ, ∆Ψ)
Compute HΨ(r1,...,rN)Ψ(r1,...,rN) for all
positionsThe energy is the average ofall computed HΨ(r1,...,rN)
Ψ(r1,...,rN)
Extreme parallelism :Independent populations ofwalkers can be distributedon different CPUs
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
QMC Algorithm
Walker : vector of R3N containing the electron positionsDrifted diffusion of walkers with birth/death process togenerate the 3N-density (Ψ× Φ) (needs Ψ, ∇Ψ, ∆Ψ)
Compute HΨ(r1,...,rN)Ψ(r1,...,rN) for all
positionsThe energy is the average ofall computed HΨ(r1,...,rN)
Ψ(r1,...,rN)
Extreme parallelism :Independent populations ofwalkers can be distributedon different CPUs
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Implementation in QMC=Chem
Block : Nwalk walkers executing Nstep stepsCompute as many blocks as possible, as quickly aspossibleBlock averages have a Gaussian distribution
Nstep
Nproc
Nwalk
CPU time
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Implementation in QMC=Chem
Block : Nwalk walkers executing Nstep stepsCompute as many blocks as possible, as quickly aspossibleBlock averages have a Gaussian distribution
Nstep
Nproc
Nwalk
CPU time
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Implementation in QMC=Chem
Block : Nwalk walkers executing Nstep stepsCompute as many blocks as possible, as quickly aspossibleBlock averages have a Gaussian distribution
Nstep
Nproc
Nwalk
CPU time
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
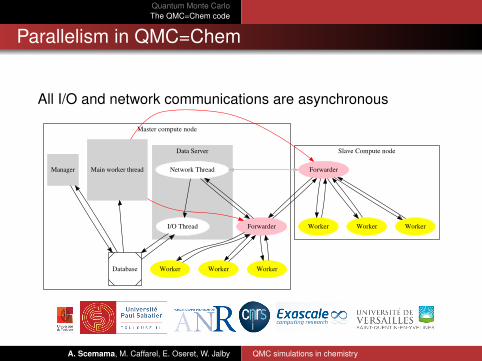
Quantum Monte CarloThe QMC=Chem code
Parallelism in QMC=Chem
All I/O and network communications are asynchronous
Master compute node
Data Server Slave Compute node
Manager
Database
Main worker thread
Forwarder
Forwarder
Worker WorkerWorker
Network Thread
I/O Thread Worker WorkerWorker
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Fault-tolerance
Extreme parallelism −→ possible system failuresBlocks are Gaussian→ losing blocks doesn’t change theaverageSimulation survives to removal of any nodeRestart always possible from data base
Efficiency of the matrix products :Static analysis (MAQAO) : Full-AVX (no scalar operations),inner-most loops perform 16 flops/cycleDecremental analysis (DECAN) : good balance betweenflops and memory operationsUp to 64% of the peak measured on Xeon E5
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Efficiency of the matrix products :Static analysis (MAQAO) : Full-AVX (no scalar operations),inner-most loops perform 16 flops/cycleDecremental analysis (DECAN) : good balance betweenflops and memory operationsUp to 64% of the peak measured on Xeon E5
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Efficiency of the matrix products :Static analysis (MAQAO) : Full-AVX (no scalar operations),inner-most loops perform 16 flops/cycleDecremental analysis (DECAN) : good balance betweenflops and memory operationsUp to 64% of the peak measured on Xeon E5
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Efficiency of the matrix products :Static analysis (MAQAO) : Full-AVX (no scalar operations),inner-most loops perform 16 flops/cycleDecremental analysis (DECAN) : good balance betweenflops and memory operationsUp to 64% of the peak measured on Xeon E5
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
First step in our scientific project : All-electron calculation of theenergy difference between the β-strand and the α-helixconformations of amyloid peptide Aβ(28-35)
122 atoms, 434 electrons, cc-pVTZ basis set (2960 basisfunctions)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
Scientific results (cc-pVTZ basis set) :Standard DFT (B3LYP) : 10.7 kcal/molDFT with empirical corrections (SSB-D) : 35.8 kcal/molAll-electron MP2 : 39.3 kcal/molCCSD(T) would require at least 100 million CPU hoursQMC in < 2 million CPU hours (1 day) : 39.7 ± 2. kcal/molQMC calculations can be made on these systems −→ study ofthe interaction of Copper ions with β-amyloids
Technological results :Sustained 960 TFlops/s (Mixed SP/DP) on 76 800 cores ofCurie∼ 80% parallel speed-up. (Today, it would be > 95 % : runtermination was optimized)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
Scientific results (cc-pVTZ basis set) :Standard DFT (B3LYP) : 10.7 kcal/molDFT with empirical corrections (SSB-D) : 35.8 kcal/molAll-electron MP2 : 39.3 kcal/molCCSD(T) would require at least 100 million CPU hoursQMC in < 2 million CPU hours (1 day) : 39.7 ± 2. kcal/molQMC calculations can be made on these systems −→ study ofthe interaction of Copper ions with β-amyloids
Technological results :Sustained 960 TFlops/s (Mixed SP/DP) on 76 800 cores ofCurie∼ 80% parallel speed-up. (Today, it would be > 95 % : runtermination was optimized)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
Scientific results (cc-pVTZ basis set) :Standard DFT (B3LYP) : 10.7 kcal/molDFT with empirical corrections (SSB-D) : 35.8 kcal/molAll-electron MP2 : 39.3 kcal/molCCSD(T) would require at least 100 million CPU hoursQMC in < 2 million CPU hours (1 day) : 39.7 ± 2. kcal/molQMC calculations can be made on these systems −→ study ofthe interaction of Copper ions with β-amyloids
Technological results :Sustained 960 TFlops/s (Mixed SP/DP) on 76 800 cores ofCurie∼ 80% parallel speed-up. (Today, it would be > 95 % : runtermination was optimized)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
Scientific results (cc-pVTZ basis set) :Standard DFT (B3LYP) : 10.7 kcal/molDFT with empirical corrections (SSB-D) : 35.8 kcal/molAll-electron MP2 : 39.3 kcal/molCCSD(T) would require at least 100 million CPU hoursQMC in < 2 million CPU hours (1 day) : 39.7 ± 2. kcal/molQMC calculations can be made on these systems −→ study ofthe interaction of Copper ions with β-amyloids
Technological results :Sustained 960 TFlops/s (Mixed SP/DP) on 76 800 cores ofCurie∼ 80% parallel speed-up. (Today, it would be > 95 % : runtermination was optimized)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
Amyloid β peptide simulation on Curie
Scientific results (cc-pVTZ basis set) :Standard DFT (B3LYP) : 10.7 kcal/molDFT with empirical corrections (SSB-D) : 35.8 kcal/molAll-electron MP2 : 39.3 kcal/molCCSD(T) would require at least 100 million CPU hoursQMC in < 2 million CPU hours (1 day) : 39.7 ± 2. kcal/molQMC calculations can be made on these systems −→ study ofthe interaction of Copper ions with β-amyloids
Technological results :Sustained 960 TFlops/s (Mixed SP/DP) on 76 800 cores ofCurie∼ 80% parallel speed-up. (Today, it would be > 95 % : runtermination was optimized)
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry
Quantum Monte CarloThe QMC=Chem code
A. Scemama, M. Caffarel, E. Oseret, W. Jalby QMC simulations in chemistry