76
quantum scattering calculations on chemical reaction 1 st Day

quantum scattering calculationson chemical reaction
1st Day

schedule
day 1.1.Introduction
day 2.2.three-atom reactions
day 3.3. four-atom reations4. five and six-atom reactions5. conclusion
* This article covers the period 1995-2003Up to 1995, Bowman & Schatz, Ann. Rev. Phys. Chem. 46:169-195

scattering
wave function
-incoming plane wave :
-outgoing spherical wave :
-scattering amplitude :
-S function :

cross section
differential cross section : the probability to observe a scattered particle in a given quantum state per solid angle unit
cross section: integral of the differential cross section on the whole sphere of observation

coordinate system
Jacobi coordinate(ri,Ri)
reaction path coordinate(u,s)
hypersphericalcoordinate(θ,ρ)

coordinate system
Jacobi coordinate (3 different ways)

state-to-state reaction dynamics
state-to-state reaction probability (P)
Pαtα’t’ = |Sαtα’t’|2 (α: reagents or products, t: electronic structure)
A schematic diagram of state-to-state reaction dynamics in the vibrationally adiabatic basis
reaction probabilities for H+HD(v=0,j=0) → D+H2(v ’, j ’)

1.introduction

quantum scattering calculation
To get informations from dynamics of chemical reactions
simplist reationA + BC(v, j) -> AB(v', j') + C
objectivesrigorous PES for chemical reaction
It is possible to calculate kinetic quantities, such as rate constants, from the dynamics calculationsand these can have useful applications in models of reacting systems(such as atmospheres, interstellar clouds, and combustion processes).
scope of this articleapplication of quantum molecular collision theory to bimolecular chemical reactions in the gas phase

time-independent Schrődinger eqn.
variational methodsinvolve expansion of wavefunction in basis function for vibrational-rotational statesof reactants and products
hyperspherical methodJacobi coordinates transformed to set of polar coordinatesgeneral code for differential cross section for atom-diatom rxn. (using hyperspherical method)

time-independent Schrődinger eqn.
4-atom reactionsreduced dimensionally(RD) theoryex.rotating bond approximation(RBA)
3-atom reactions(atom-diatiom)accurate state-to-state reaction probablities for J = 0(J > 0, at least twice as many angular basis functions are required)
approximation for J > 0 (J-shifting approximation): PJ(E)(J > 0) from PJ=0(E)(J = 0)
treat explicitly : s, R, r, and θaveraging over : γ,φ(φ: out-of-plane torsional motion of AB w.r.t. CD)
“not rotationally selectivein CD”

time-dependent Schrődinger eqn.
solve TD eqn. -> reaction dynamics on the potential surfaceThe solutions are time-evolving quantum wave packets
Ψ(t) = exp(-iHt/ħ) • Ψ(t=0) (time-evolution operator exp(-iHt/ħ))
exp(-iHt/ħ) : computed by propagation algorithm
Ψ(t) : represented by grid

time-dependent Schrődinger eqn.
split-operator method :
Chebychev series :
Ψ(t) : represented by grid
exp(-iHt/ħ) : computed by propagation algorithm
Lanczos method (iteration method)
In some case, different methods are used by TD Hamiltonian

time-dependent Schrődinger eqn.
advantage physical picture of dynamicsvector-matrix multiplication method (scale : TD→less than N2, TI→N3)available for six-dimensional four-atom, three-atom with large basis set calculation
disadvantageunable to the dynamics involves long-lived resonance (efficient for TI)artificial absorbing potentials in TD typically reflect small part of wave packet(thus, TI often gives more accurate results than TD)if state-to-state probabilities or cross sections are required,
* efficiency of a TD method depends on the basis set and propagation method

time-dependent Schrődinger eqn.
reactant product decoupling(RPD) approachavoid the coordinate problem by splitting the (exact) Schrodinger eqn.

quantum scattering calculationson chemical reaction
2nd Day

2. Three-atom reactions

H+H2 → H2+H
quantum scattering calculation (1975)
Liu-Siegbahn-Truhlar-Horowitz(LSTH) PES (late 1980s)converged integral and differential cross sections
geometric phase(GP) associated with conical intersection(1990)

geometric phase(Berry phase) associated with conical intersectionex. Jahn-Teller effect conical intersection
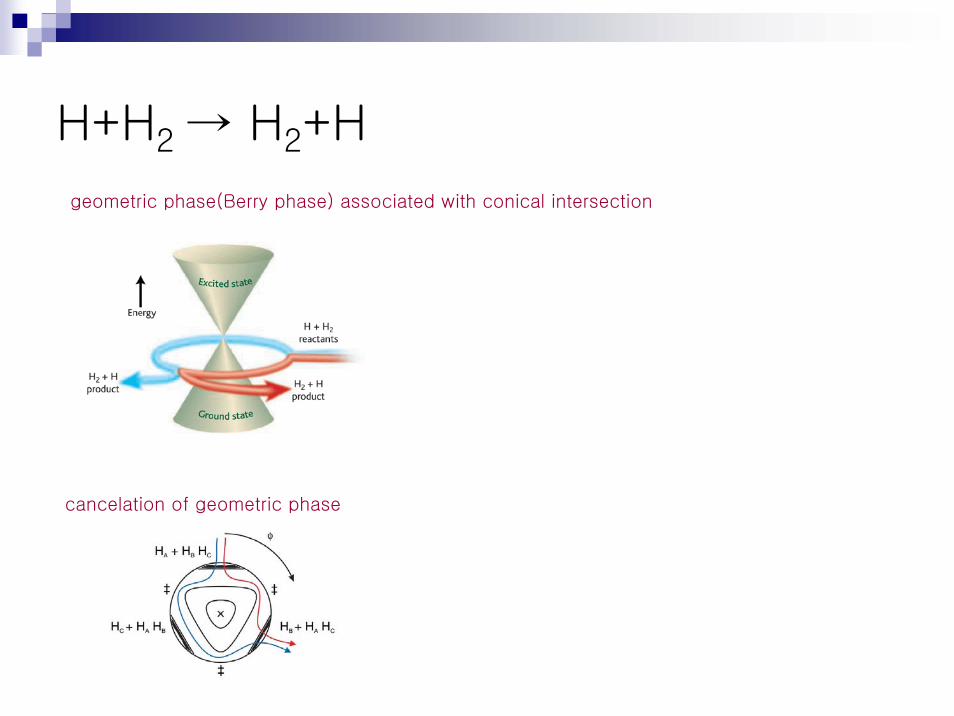
H+H2 → H2+H
geometric phase(Berry phase) associated with conical intersection
cancelation of geometric phase

H+H2 → H2+Hcancelation of geometric phaserepresentative classical trajectories for H+H2(v=1,j=0) reaction

H+D2 / H+HD → HD + D/H
qunatum scattering calculation (since 1995)
BKMP2 PES
methods used in BKMP2 calculationsKohn variational methodhyperspherical coupled channel methodTD wave packet mathod
overall behavior of the cross sectionsusing quantum and quasiclassical trajectory(QCT)backward scattering dominates for low value of j’shifting to sideway scattering at higher j’
forward scatteringtime-delay in forward scattering

H+D2 / H+HD → HD + D/H
quasiclassical trajectory(QCT)“In the quasiclassical trajectory method, molecules are prepared in discrete internal energy states corresponding to the quantum state of the molecule.Once the trajectory is begun, this quantum restriction is relaxed so that the time evolution of the system is governed solely by classical mechanics. Asimilar “quantization” is often employed on the analysis of product moleculeinternal energy state” Truhlar and Muckerman, ‘Reactive scattering cross section’
*exampleO(3P) + HCl → OH + Cl

H+D2 / H+HD → HD + D/H
forward scatteringschematic illustration of the time-delayed reaction mechanism

F+H2 → HF+H
importancebenchmark exothermic reaction
Stark-Werner(SW) surface first accurate ab initio potential energy surfaceStark & Werner, 1996correctly predict a bent TSrealistic barier hightagreement with exp. of Neumark group
QCT/quantum scattering calculation on SW surface
Feschbach resonance
spin-orbit coupling

F+H2 → HF+H
resonance in F+HD reactionexperimenatal data

F+H2 → HF+H
resonance in F+HD reactionexperimenatal data / calculation

F+H2 → HF+H
spin-orbit coupling

Cl+H2
importanceimpotant model for transition state theoryatmospherically important reaction
PES G3 (Allison et al.)Bian-Werner(BW)
Bian-Werner(BW) surface well describes experiment (compare to others)-van der Waals well

insertion-type reations
insertion-type reactions1. O(1D)+H2 → OH(2Π)+H2. N(2D)+H2 → NH+H 3. C(1D)+H2 → CH+H 4. H+O2 → OH+O 5. O(1D)+HCl → OH+Cl / ClO+H6. N(4S)+O2 → NO+O
featuredeep wellsabsence or near absence of reaction barriers

heavy-light-heavy reactions
reactionsO(3P) + HCl → OH + ClCl + HCl → HCl + ClCl + HBr → HCl + BrF + HCl → HF + Cl
featurebarriersvery low skew angles
O-H-Cl angle(a) 10°, (b) 80.4°, (c) 131.4°, (d) 180°
O(3P) + HCl → OH + Cl

metal included reactions
reactionsLi+HF → LiF+HH+LiH → Li+H2 / HLi+H
featurelate, noncollinear TSwell in both the reagent and product channel
PES for Li + HF
Li-F-H angle(a) 180°, (b) 106°, (c) 74°, (d) 45°

ion-molecule reactionsinvolving three atoms
reactionsD++H2 → D+H2
+ / HD+H+ / HD++HHe+H2
+ → HeH++H
featuredeep wellslong-range potetialspossibility of strong nonadiabatic effect associated with charge transfer

reactive collisions at ultracold temp.
aimBose-Einstein condensation between molecules
reactionsNa + Na2 (Soldan et al.) F + H2F + D2
featureultracold temperature (<10-3K)
calculation (Na+Na2)hyperspherical close-coupling methodobtained J=0 cross sections down to 10-9Kaccordance with Wigner threshold laws

quantum scattering calculationson chemical reaction
3rd Day

3. Four-atom reactions

OH+H2 → H2O+H
Potential Energy SurfaceSchatz & Elgersma PESCollins PES Ochoa de Aspuru & Clary PES
calculation (1995)TD wave packet method
reduced dimensionrotating bond approximation(RBA)semi-rigid vibrating rotor target(SVRT)

OH+H2 → H2O+H
rotating bond approximation(RBA)

OH+H2 → H2O+H
rotating bond approximation(RBA)
exact rovibrational Hamiltionian for the isolated ABC molecule
Hamiltonian(J=0,Ω=0)

OH+H2 → H2O+H
rotating bond approximation(RBA)
R-matrix propagator methoddiagonalize the internal Hamiltionian Hl
fix,
Gaussian basis function
expanding V1 in Legendre series nL
Hθ is diagonalized
oprimized basis set,

OH+H2 → H2O+H
rotating bond approximation(RBA)
configuration intereraction(CI)
initial state k is expanded
close-coupling equation

OH+H2 → H2O+H
rotating bond approximation(RBA)
reaction cross sections obtained form S matrixinitial state
initial translational energy
S matrix
hyperspherical basis function

OH+H2 → H2O+H
rotating bond approximation(RBA)
real calculation
convergence w.r.t. number of basis functions
calculated cross sections σ(0,0→n,m)
OH+H2 → H2O+H
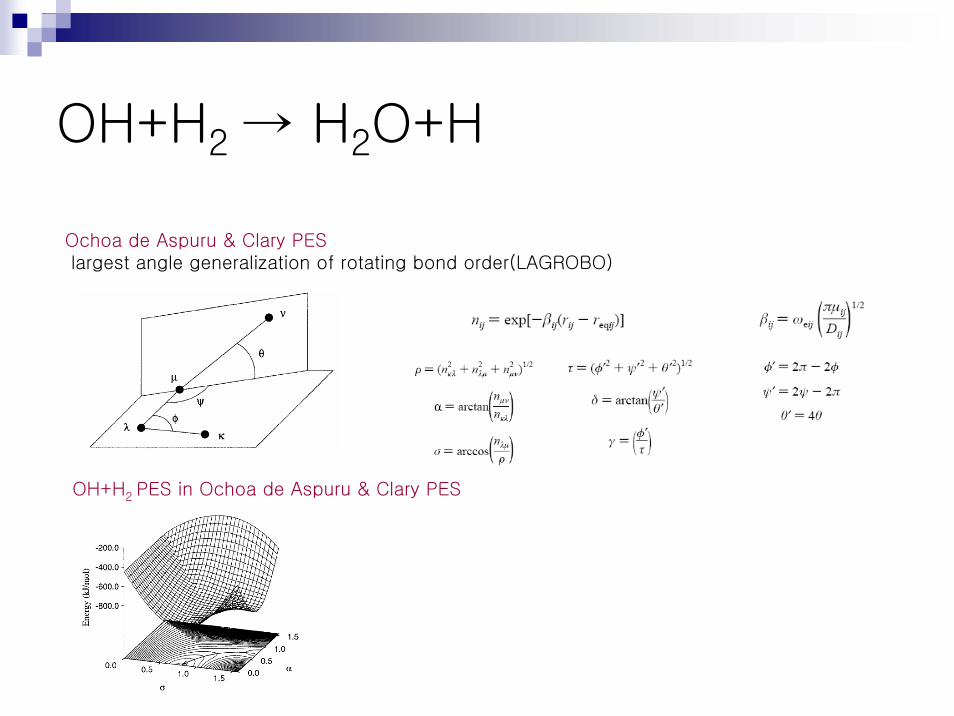
Ochoa de Aspuru & Clary PESlargest angle generalization of rotating bond order(LAGROBO)
OH+H2 PES in Ochoa de Aspuru & Clary PES

OH+D2 → HOD+D
mode specific behavor
energy level diagram HOD product are labeled (m,n)
m : quantum number for bending moden : OD local stretching mode
D atom product time-of-flight spectrasolid line : simulation based on best-fit
translation energy and angulardistribution

OH+D2 → HOD+D
mode specific behavor
comparision of three different theoretical predictionenergy level diagram HOD product are labeled (m,n)
m : quantum number for bending moden : OD local stretching mode

H2+CN → HCN+H
calculationfirst quantum-dynamical calculation (Clary et al.)temperature dependent J-shifting procedure(Zhang)wave packet calculation(Zhu)SVRT model(Ma et al.)
reduced dimensionalityL2 eigenstate method(Skokov & Bowman)RBA calculationextended RBA(Takayanagi & Schatz)-include CN stretching motion (CN bond have some effect)

OH+CO → CO2+H
importancemain reaction for producing CO2 in flame and in the Earth’s atmosphere
calculationfirst 6D wave packet calculation-initially state-selected reaction probabilities-similar to 5D result
resonancelong-lived “HOCO” intermediates.complex potential method was developed to characterize resonances

OH+HCl → Cl+H2O
importanceimportant source of Cl in the Earth’s atmosphere
calculation3D theory-Born-Oppenheimer type separation(for light and heavy nuclear motion)-comparison to RBA-extended to planar treatment of the Cl+H2Ovibrationally adiabatic model

other four-atom reaction
calculationTI method
..

4. five and six atom reactions
featuremore than four atoms are very hard to carry outapproximations are normally needed

H+CH4→ H2+CH3
reducing dimensionalitySVRT, RBA, modified-RBA, RLU
calculationTD reduced dimensionality method-consider rotational motion of CH4
-agree with the rate constants at higher temperature

O(3P)+CH4→ CH3+OH
importancekey reaction of CH4 in flame
reducing dimensionalityRBA calculation-large mode-selective effect-very low vibrational excitation of CH3
-cross sections about twice as large (Fig.7)RLU calculationSVRT calculation

Cl+CH4→ CH3+HCl
importancemajor source of the HCl in the atmosphere
reducing dimensionalityRLU calculationwave packet calculation

Cl-+CH3Cl→ CH3Cl+Cl-
Cl-+CH3Br→ ClCH3+Br-
importanceimportant prototype reaction in physical organic chemistry
PESdeep “ion-dipole” wells in reaction and product region
reducing dimensionalityRBA calculationnew TI quantum scattering theory

C(3P)+C2H2→ C3H+H

other polyatomic reactions

conclusion
in this paperbimolecular chemical reactionspast seven yearsover 40 different reactions
time-dependent methodwave packet method
free radicals.
potential energy surface.

quantum scattering calculations on chemical reactions
1.Introduction
2.three-atom reactions
2-1. H+H2,
2-2. F+H2,
2-3. Cl+H2,
2-4. O+H2, N+H2, C+H2, H+O2, O+HCl, N+O2
2-5. O+HCl, Cl+HCl, Cl+HBr, F+HCl
2-6. Li+HF, Na+HF, Mg+HF, Li+H2
2-7. ion-molecule reactions involving three atoms
2-8. reactive collisions at ultracold temperatures
3. four-atom reations
3-1. OH+H2
3-2. H2+CN
3-3. OH+CO
3-4. OH+HCl
3-5. other four-atom reactions
4. five and six-atom reactions
4-1. H+CH4
4-2. O+CH4
4-3. Cl+CH4
4-4. Cl-+CH3Cl, Br-+CH3Cl
4-5. C+C2H2
4-6. other polyatomic reactions
5. conclusion

0. preliminary
scattering
* wave function
-incoming plane wave :
-outgoing spherical wave :
-scattering amplitude :
-S function :
Cross section
* differential cross section
: the probability to observe a scattered particle in a given quantum state
per solid angle unit
* cross section
: integral of the differential cross section on the whole sphere of observation

Jacobi coordinate(ri,Ri)
reaction path coordinate(u,s)
hypersphericalcoordinate(θ,ρ)
Jacobi coordinate (3 different ways)
state-to-state reaction probability (P)
Pαtα’t’ = |Sαtα’t’|2 (α: reagents or products, t: electronic structure)
A schematic diagram of state-to-state reaction dynamics in the vibrationally adiabatic basis
Coordinate system
state-to-state reaction dynamics

1. introduction
* This article is for the period 1995-2003
Up to 1995, Bowman & Schatz, Ann. Rev. Phys. Chem. 46:169-195
quantum scattering calculation
* To get informations from dynamics of chemical reactions
* simplist reation
A + BC(v, j) -> AB(v', j') + C
* objectives
-rigorous PES for chemical reaction
-It is possible to calculate kinetic quantities, such as rate constants,
from the dynamics calculations and these can have useful applications
in models of reacting systems
(such as atmospheres, interstellar clouds, and combustion processes).
*scope of this article
-application of quantum molecular collision theory to bimolecular chemical
reactions in the gas phase
time-independent Schrődinger eqn. (TI)
* variational method
-involve expansion of wavefunction in basis function for vibrational-rotational
states of reactants and products
* hyperspherical method
: Jacobi coordinates transformed to set of polar coordinates
-general code for differential cross section for atom-diatom rxn.
(using hyperspherical method)
* 4-atom reactions
-reduced dimensionally(RD) theory
rotating bond approximation(RBA)
treat explicitly : s, R, r, and θaveraging over : γ,φ(φ: out-of-plane torsional motion of AB w.r.t. CD)
“not rotationally selectivein CD”

* 3-atom reactions(atom-diatiom)
-accurate state-to-state reaction probablities for J = 0
(J > 0, at least twice as many angular basis functions are required)
approximation for J > 0 (J-shifting approximation)
: PJ(E)(J > 0) from PJ=0(E)(J = 0)
time-dependent Schrődinger eqn. (TD)
* solve TD eqn. -> reaction dynamics on the potential surface
-solutions are time-evolving quantum wave packets
Ψ(t) = exp(-iHt/ħ) * Ψ(t=0) (time-evolution operator exp(-iHt/ħ))
exp(-iHt/ħ) : computed by propagation algorithm
1.split-operator method :
2.Chebychev series :
3.Lanczos method (iteration method)
4.In some case, different methods are used by TD Hamiltonian
Ψ : represented by grid
* advantage
-physical picture of dynamics
-vector-matrix multiplication method (scale : TD→less than N2, TI→N3)
available for six-dimensional four-atom, three-atom with large basis set
calculation
* disadvantage
-unable to the dynamics involves long-lived resonance (efficient for TI)
-artificial absorbing potentials in TD typically reflect small part of wave packet
(thus, TI often gives more accurate results than TD)
-if state-to-state probabilities or cross sections are required,

* reactant product decoupling(RPD) approach
-avoid the coordinate problem by splitting the (exact) Schrodinger eqn.
* efficiency of a TD method depends on the basis set and propagation method
-various propagation methods
1. TI wave packet
-Lippmann-Schwinger eqn.
2. damped Chebyshev
-Chebyshev Operator : transform time/energy to order/angle formulation
-numerical advantage
3. real wave packet
4. filter diagonalization
5. symplectic integrator
6. L2 methods
* reagent to product coordinate transformation
-applying it at just one time-step
often difficult, owing to spreading of the wave packet

2.three-atom reactions
H+H2 → H2+H
quantum scattering calculation (1975)
Liu-Siegbahn-Truhlar-Horowitz(LSTH) PES (late 1980s)
-converged integral and differential cross sections
geometric phase(GP) associated with conical intersection(1990)
ex. Jahn-Teller effect conical intersection
-cancelation of geometric phase
-representative classical trajectories for H+H2(v=1,j=0) reaction

-cancelation of geometric phase
H+D2 / H+HD → HD + D/H
qunatum scattering calculation (since 1995)
BKMP2 PES
methods used in BKMP2 calculations
-Kohn variational method
-hyperspherical coupled channel method
-TD wave packet mathod
overall behavior of the cross sections
-using quantum and quasiclassical trajectory(QCT)
-quasiclassical trajectory(QCT)
'In the quasiclassical trajectory method, molecules are prepared in discrete
internal energy states corresponding to the quantum state of the molecule.
Once the trajectory is begun, this quantum restriction is relaxed so that the
time evolution of the system is governed solely by classical mechanics. A
similar "quantization" is often employed on the analysis of product molecule
internal energy state' Truhlar and Muckerman, 'Reactive scattering cross section'
-backward scattering dominates for low value of j'
-shifting to sideway scattering at higher j'
forward scattering
-time-delay in forward scattering

F+H2 → HF+H
importance
-benchmark exothermic reaction
Stark-Werner(SW) surface
-first accurate ab initio potential energy surface
-Stark & Werner, 1996
-correctly predict a bent TS
-realistic barier hight
-agreement with exp. of Neumark group
-QCT/quantum scattering calculation on SW surface
Feschbach resonance
-resonance in F+HD reaction
* experimenatal data
* experimenatal data / calculation
spin-orbit coupling

Cl+H2
importance
-impotant model for transition state theory
-atmospherically important reaction
PES
-G3 (Allison et al.)
-Bian-Werner(BW)
Bian-Werner(BW) surface
-well describes experiment (compare to others)
*van der Waals well
insertion-type reations
insertion-type reactions
1. O(1D)+H2 → OH(2Π)+H
2. N(2D)+H2 → NH+H
3. C(1D)+H2 → CH+H
4. H+O2 → OH+O
5. O(1D)+HCl → OH+Cl / ClO+H
6. N(4S)+O2 → NO+O
feature
-deep wells
-absence or near absence of reaction barriers

Li-F-H angle(a) 180°, (b) 106°, (c) 74°, (d) 45°
heavy-light-heavy reactions
reactions
1. O(3P) + HCl → OH + Cl
2. Cl + HCl → HCl + Cl
3. Cl + HBr → HCl + Br
4. F + HCl → HF + Cl
feature
-barriers
-very low skew angles
metal included reactions
reactions
Li+HF → LiF+H
H+LiH → Li+H2 / HLi+H
PES for Li + HF
ion-molecule reactions involving three atoms
reactions
1. D++H2 → D+H2+ / HD+H+ / HD++H
2. He+H2+ → HeH++H
feature
-deep wells
-long-range potetials
-possibility of strong nonadiabatic effect associated with charge transfer
O-H-Cl angle(a) 10°, (b) 80.4°, (c) 131.4°, (d) 180°
O(3P) + HCl → OH + Cl

reactive collisions at ultracold temp.
aim
Bose-Einstein condensation between molecules
reactions
1. Na + Na2
2. F + H2
3. F + D2
feature
ultracold temperature (<10-3K)
calculation (Na+Na2)
hyperspherical close-coupling method
obtained J=0 cross sections down to 10-9K
accordance with Wigner threshold lows

3. Four-atom reactions
OH+H2 → H2O+H
Potential Energy Surface
-Schatz & Elgersma PES
-Collins PES
-Ochoa de Aspuru & Clary PES
calculation (1995)
-TD wave packet method
reduced dimension
-rotating bond approximation(RBA)
-semi-rigid vibrating rotor target(SVRT)
*rotating bond approximation(RBA)
exact rovibrational Hamiltionian for the isolated ABC molecule
Hamiltonian(J=0,Ω=0)

R-matrix propagator methoddiagonalize the internal Hamiltionian Hl
fix,
Gaussian basis function
expanding V1 in Legendre series nL
Hθ is diagonalized
oprimized basis set,
configuration intereraction(CI)
initial state k is expanded
close-coupling equation
reaction cross sections obtained form S matrixinitial state
initial translational energy
S matrix
hyperspherical basis function

real calculation
convergence w.r.t. number of basis functions
calculated cross sections σ(0,0→n,m)
Ochoa de Aspuru & Clary PESlargest angle generalization of rotating bond order(LAGROBO)
OH+H2 PES in Ochoa de Aspuru & Clary PES

OH+D2 → HOD+D
mode specific behavor
energy level diagram HOD product are labeled (m,n)
m : quantum number for bending moden : OD local stretching mode
D atom product time-of-flight spectrasolid line : simulation based on best-fit
translation energy and angulardistribution
comparision of three different theoretical predictionenergy level diagram HOD product are labeled (m,n)
m : quantum number for bending moden : OD local stretching mode
H2+CN → HCN+H
calculation
-first quantum-dynamical calculation (Clary et al.)
-temperature dependent J-shifting procedure(Zhang)
-wave packet calculation(Zhu)
-SVRT model(Ma et al.)
reduced dimensionality
-L2 eigenstate method(Skokov & Bowman)
-RBA calculation
-extended RBA(Takayanagi & Schatz)
include CN stretching motion (CN bond have some effect)

OH+CO → CO2+H
importance
-main reaction for producing CO2 in flame and in the Earth's atmosphere
calculation
-first 6D wave packet calculation
initially state-selected reaction probabilities
similar to 5D result
resonance
-long-lived "HOCO" intermediates.
-complex potential method was developed to characterize resonances
OH+HCl → Cl+H2O
importance
-important source of Cl in the Earth‘s atmosphere
calculation
-Born-Oppenheimer type separation(for light and heavy nuclear motion)
4. five and six atom reactions
feature
-more than four atoms are very hard to carry out
-approximations are normally needed
H+CH4→ H2+CH3
reducing dimensionality
-SVRT, RBA, modified-RBA, RLU
calculation
-TD reduced dimensionality method
consider rotational motion of CH4
agree with the rate constants at higher temperature

O(3P)+CH4→ CH3+OH
importance
-key reaction of CH4 in flame
reducing dimensionality
-RBA calculation
large mode-selective effect
very low vibrational excitation of CH3
-RLU calculation
-SVRT calculation
Cl+CH4→ CH3+HCl
importance
-major source of the HCl in the atmosphere
reducing dimensionality
-RLU calculation
-wave packet calculation
Cl-+CH3Cl→ CH3Cl+Cl- / Cl-+CH3Br→ ClCH3+Br-
importance
-important prototype reaction in physical organic chemistry
PES
-deep ‘ion-dipole’ wells in reaction and product region
reducing dimensionality
-RBA calculation
-time-independent calculation
conclusion
in this paper
-bimolecular chemical reactions
-past seven years
-over 40 different reactions
time-dependent method
-wave packet method as a major technique
free radicals
-open shell calculation
-nonadiabatic reaction calculation
potential energy surface
-functional representation of such surface for polyatomic molecules
remains a major problem


![Quantum Mechanics of Collision Processes [Scattering?]ymambrini.com/My_World/History_files/Born_1.pdf · Quantum Mechanics of Collision Processes∗ [Scattering?] Max Born 21 July](https://static.documents.pub/doc/80x56/5ea0d54b02859f26e34a3cda/quantum-mechanics-of-collision-processes-scattering-quantum-mechanics-of-collision.jpg)














