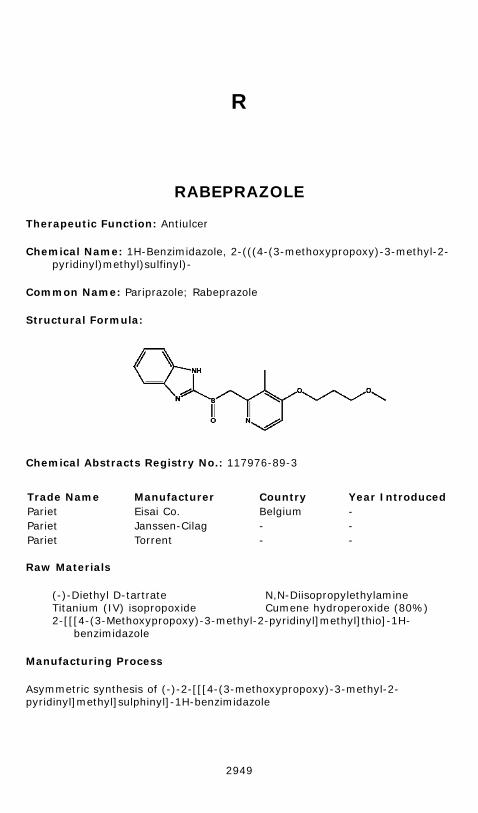
Asymmetric synthesis of (-)-2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulphinyl]-1H-benzimidazole
Trade Name Manufacturer Country Year IntroducedPariet Eisai Co. Belgium -Pariet Janssen-Cilag - -Pariet Torrent - -
2949
2.1 g (6.3 mmol) of 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]methyl]thio]-1H-benzimidazole was dissolved in 50 ml of toluene. Tothe solution was added 40 µl (2.2 mmol) of water, 1.6 ml (9.4 mmol) of (-)-diethyl D-tartrate and 1.1 ml (3.8 mmol) of titanium (IV) isopropoxide. Themixture was stirred for 60 minutes at 50°C and then cooled to roomtemperature. 0.44 ml (2.6 mmol) of N,N-diisopropylethylamine and 1.1 ml(6.0 mmol) of cumene hydroperoxide (80%) were added. After stirring for 2 hat room temperature the mixture consisted of 9% sulphide, 4% sulphone and86% sulphoxide according to achiral HPLC. To the mixture toluene (50 ml)was added and the resultant solution was extracted three times with anaqueous ammonia (12%) solution with a total volume of 150 ml. Thecombined aqueous layers were neutralized by the addition of concentratedacetic acid (30 ml). Thereafter, the workup procedure employed extraction,evaporation and flash chromatography yielding 1.62 g of the title compoundwith a purity of 99.9% (achiral analysis) and with an enantiomeric excess of90% (chiral analysis). After treating the material with acetonitrile there was aprecipitate that could be removed by filtration. Concentrating the filtrateafforded 1.36 g (60%) of the title compound as an oil with an optical purity of91.5%.
Asymmetric synthesis of (+)-2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulphinyl]-1H-benzimidazole
2.1 g (6.3 mmol) of 2-[[[4-(3-methoxypropoxy)-3-methyl-2-pyridinyl]methyl]thio]-1H-benzimidazole was dissolved in 50 ml of toluene. Tothe solution was added 40 µl (2.2 mmol) of water, 1.6 ml (9.4 mmol) of (+)-diethyl L-tartrate and 1.1 ml (3.8 mmol) of titanium (IV) isopropoxide. Themixture was stirred for 60 minutes at 50°C and then cooled to roomtemperature. 0.44 ml (2.6 mmol) of N,N-diisopropylethylamine and 1.1 ml(6.0 mmol) of cumene hydroperoxide (80%) were added to the solution. Afterstirring for 2 h at room temperature the mixture consisted of 9% sulphide,4% sulphone and 85% sulphoxide according to HPLC. To the mixture toluene(50 ml) was added and the resultant solution was extracted three times withan aqueous ammonia (12%) solution with a total volume of 150 ml. Thecombined aqueous layers were neutralized by the addition of concentratedacetic acid (30 ml). Thereafter, the workup procedure employed extraction,evaporation and flash chromatography yielding 1.63 g of the title compoundwith a purity of 99.9% (achiral analysis) and with an enantiomeric excess(e.e.) of 91% (chiral analysis). After treating the material with acetonitrile,there was a precipitate that could be removed by filtration. Concentrating thefiltrate afforded 1.1 g (49%) of the title compound as an oil with an opticalpurity of 96%.
References
Larson et al; US Patent No. 5,948,789; Sep. 7, 1999; Assigned to Astra Aktiebolag, Sodertalje, Sweden
RALOXIFENE HYDROCHLORIDE
Therapeutic Function: Antiestrogen
2950 Raloxifene hydrochloride
Chemical Name: Methanone, (6-hydroxy-2-(4-hydroxyphenyl)benzo(b)thien-3-yl)(4-(2-(1-piperidinyl)ethoxy)phenyl)-, hydrochloride
Common Name: Keoxifen hydrochloride; Radoxifene hydrochloride
Structural Formula:
Chemical Abstracts Registry No.: 82640-04-8; 84449-90-1 (Base)
A 183 g portion of methyl 4-(2-piperidinoethoxy)benzoate was dissolved in600 ml of methanol, and 200 ml of 5 N sodium hydroxide was added. Themixture was stirred at ambient temperature for 48 hours, the solvent wasevaporated, and the residue was dissolved in 1 liter of water. The solution wascooled to below 10°C, and was acidified with cold 6 N hydrochloric acid. Theproduct crystallized, and was collected by filtration and washed with methanolat -40°C. The solids were recrystallized from 3400 ml of methanol to obtain167 g of the expected product, melting point 274°-277°C.
6-Hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophene
A 100 g portion of 3-methoxybenzenethiol and 39.1 g of potassium hydroxidedissolved in 300 ml of water were added to 750 ml of denatured ethanol, andthe flask was put in a cooling bath. A total of 164 g of α-bromo-4-methoxyacetophenone was then added in small portions, and the mixture wasstirred for 10 minutes in the cooling bath after the addition was complete andthen for 3 hours at ambient temperature. The solvent was then evaporated offin vacuum, and 200 ml of water was added. The mixture was extracted withethyl acetate, and the organic layer was washed twice with water, twice withaqueous sodium bicarbonate solution, and twice with aqueous sodium chloridesolution. The organic layer was then dried over magnesium sulfate, filtered
Raloxifene hydrochloride 2951
Trade Name Manufacturer Country Year Introduced
Evista Eli Lilly USA -
and evaporated under vacuum to obtain 202 g of crude α-(3-methoxyphenylthio)-4-methoxyacetophenone, which was recrystallized frommethanol and washed with hexane to obtain 158 g of purified product, m.p.53°C.
A 124 g portion of the above intermediate was added in small portions to 930g of polyphosphoric acid at 85°C. The temperature rose to 95°C during theaddition, and the mixture was stirred at 90°C for 30 minutes after theaddition was complete, and was then stirred an additional 45 minutes while itcooled without external heating. One liter of crushed ice was then added tothe mixture, and an external ice bath was applied to control the temperaturewhile the ice melted and diluted the acid. 500 ml of additional water wasadded, and the light pink precipitate was filtered off and washed, first withwater and then with methanol. The solids were dried under vacuum at 40°Cto obtain 119 g of crude 6-methoxy-2-(4-methoxyphenyl)-benzo[b]thiophene.The crude product was slurried in hot methanol, filtered, and washed with coldmethanol, and the solids were recrystallized from 4 liters of ethyl acetate,filtered, washed with hexane and dried to obtain 68 g of purified intermediateproduct, m.p. 187°-190.5°C.
90 g of pyridine hydrochloride was added to a flask equipped with a distillationhead, condenser and collecting flask, and was heated with stirring until thetemperature in the distillation head was 220°C. The distillation apparatus wasthen removed, the pot was cooled to 210°C, and 30 g of the above prepared6-methoxy-2-(4-methoxyphenyl)-benzo[b]thiophene was added. The mixturewas stirred at 210°C for 30 minutes, and was then poured into 250 ml of ice-water. The precipitate was extracted into 500 ml of ethyl acetate, and theorganic layer was washed with 150 ml of saturated aqueous sodiumbicarbonate and then with 150 ml of saturated aqueous sodium chloride. Theorganic layer was then dried over magnesium sulfate, filtered and evaporatedto dryness under vacuum to obtain 25.5 g of the desired intermediateproduct, m.p. >260°C.
Under a nitrogen blanket, a mixture of 3 g of 4-(2-piperidinoethoxy)benzoicacid hydrochloride, 2 drops of dimethylformamide, 2.5 ml of thionyl chlorideand 40 ml of chlorobenzene was heated at 70°-75°C for about one hour. Theexcess thionyl chloride and 15-20 ml of solvent were then distilled off. Theremaining suspension was cooled to ambient temperature, and to it wereadded 100 ml of dichloromethane, 2.7 g of 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene and 10 g of aluminum chloride. The solution was stirred forabout one hour, 7.5 ml of ethanethiol was added, and the mixture was stirredfor 45 minutes more. Then 40 ml of tetrahydrofuran was added, followed by15 ml of 20% hydrochloric acid, with an exotherm to reflux. Fifty ml of waterand 25 ml of saturated aqueous sodium chloride was added. The mixture wasstirred and allowed to cool to ambient temperature. The precipitate wascollected by filtration and washed successively with 30 ml of water, 40 ml of25% aqueous tetrahydrofuran, and 35 ml of water. The solids were then driedat 40°C under vacuum to obtain 5.05 g of product, which was identified byNMR as 6-hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-piperidinoethoxy)benzoyl]benzo[b]thiophene hydrochloride; melting point 217°C.
2952 Raloxifene hydrochloride
Purification of 6-hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-piperidinoethoxy)benzoyl]benzo[b]thiophene hydrochloride
200 g of crude 6-hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-piperidinoethoxy)benzoyl]benzo[b]thiophene hydrochloride, typical of theproduct of Example 16 above, was added to 4400 ml of methanol and 60 mlof deionized water in a 5-liter flask. The slurry was heated to reflux,whereupon most of the crude product went into solution. The remaining solidwas removed by filtration under vacuum, using a filter aid pad. A distillationhead was then attached to the flask, and solvent was distilled off until thevolume of the remaining solution was about 1800 ml. The heating mantle wasthen turned off, and the solution was cooled very slowly overnight, withconstant stirring. The crystalline product was then collected by vacuumfiltration, and the flask was washed out with filtrate to obtain all of theproduct. The crystals were washed on the filter with two 100 ml portions ofcold (below 0°C) methanol, and the washed product was dried at 60°C undervacuum to obtain 140 g of dried product. The product was slurried in 3000 mlof methanol and 42 ml of water, heated to reflux and cooled very slowly. Theproduct was filtered and dried as above to obtain 121 g of highly purifiedproduct, melting point 259°-260°C.
References
Jones C.D.; US Patent No. 4,418,068; Nov. 29, 1983; Assigned to Eli Lilly and Company, Indianapolis, IN
RAMIPRIL
Therapeutic Function: Antihypertensive
Chemical Name: Cyclopenta(b)pyrrole-2-carboxylic acid, octahydro-1-(2-((1-(ethoxycarbonyl)-3-phenylpropyl)amino)-1-oxopropyl), (2S-(1(R*(R*)),2-α,3a-β,6a-β))-
269 g of methyl 3-chloro-2-acetylamino-propionate and 257 g ofcyclopentenopyrrolidine in 1.5 liters of dimethylformamide were kept at roomtemperature for 24 hours. The mixture was concentrated in vacuo, the residuewas taken up in a little water and the aqueous mixture was adjusted to pH 2with concentrated hydrochloric acid and extracted twice with 4 liter portions ofethyl acetate. On concentration of the organic phase, a light yellow oilremained. Yield: 290 g.
270 g of the acetylamino derivative prepared under (1) were refluxed in 1.5liters of 2 N hydrochloric acid for 45 minutes. The mixture was concentrated invacuo, the residue was taken up in glacial acetic acid, 5 g of Pt/C (10% of Pt)were added and hydrogenation was carried out under 5 bar. After filtration,the mixture was concentrated and the residue was crystallized fromchloroform/diisopropyl ether. Melting point 205°-209°C. Yield: 150 g.
40 g of the carboxylic acid prepared under (2) were added to an ice-coldmixture of 390 g of benzyl alcohol and 65 g of thionyl chloride and themixture was left to stand at room temperature for 24 hours. Afterconcentration in vacuo, 47 g of the benzyl ester were crystallized fromchloroform/isopropanol.Melting point: 175°C (hydrochloride).
2954 Ramipril
Trade Name Manufacturer Country Year Introduced Altace Libbs - - Altace Aventis - - Cardace Hoechst Germany - Cardace Aventis Pasteur India - Corpril Ranbaxy Global Consumer
14 g of the benzyl ester prepared according to (3) were reacted with 6.7 g ofHOBt (oxybenztriazol), 13.8 g of N-(1-S-carbethoxy-3-phenyl-propyl)-S-alanine and 10.2 g of dicyclohexylcarbodiimide in 200 ml ofdimethylformamide. After the mixture had been stirred for 3 hours at roomtemperature, the dicyclohexylurea which had precipitated was filtered off thefiltrate was concentrated, the residue was taken up in 1 liter of ethyl acetateand the mixture was extracted by shaking with 3 x 500 ml of 5% NaHCO3
solution. The organic phase was concentrated and the residue waschromatographed over a column of 1 kg of silica gel using ethylacetate/petroleum ether in the ratio 2:1. The isomer eluted first was theS,S,S-compound, and concentration of a later eluate gave the S,S,R-compound. The products were obtained as an oil. The structure of them wasconfirmed NMR.
8.0 g of the L,L,L-benzyl ester from (4) were dissolved in 100 ml of ethanoland were debenzylated hydrogenolytically under normal pressure, withaddition of 0.5 g of 10% Pd/C. This reaction could also have been carried outunder pressure, together with a shortening of the reaction time. After thecalculated amount of hydrogen had been taken up, the catalyst was filteredoff and the residue was concentrated in vacuo. The product crystallized fromether, in almost quantitative yield. Melting point: 110°-112°C(decomposition). The NMR and mass spectra obtained are in agreement withthe given structure; [α]D = +15.6° (c = 1, methanol).
References
Teez et al.; US Patent No. 4,727,160; Feb. 23, 1988; Assigned to Hoechst Aktiengesellschaft, Frankfurt am Main, Fed. Rep. of Germany
RANITIDINE
Therapeutic Function: Antiulcer, Antiallergic
Chemical Name: N-[2-[[[5-(Dimethylamino)methyl-2-furanyl]methyl]thio]ethyl]-N'-methyl-2-nitro-1,1-ethenediamine
N-methyl-1-(methylthio)-2-nitroetheneamine (230 g) in water (400 ml) wasstirred and heated at 45°C to 50°C. 2-[[[5-(Dimethylamino)methyl-2-furanyl]methyl]thio]ethanamine (321 g) was added dropwise over 4 hours andthe resultant solution stirred for a further 3.5 hours.
The solution was then heated at reflux for 1/2 hour, cooled to 70°C and 4-methylpentan-2-one (2 liters) added. The water was removed by azeotropicdistillation under reduced pressure (260 torrs) and the resultant solutiontreated with charcoal (10 g) at 50°C. The solution was filtered and cooled to10°C. N-[2-[[[5-dimethylamino)methyl-2-furanyl]methyl]thio]ethyl]-N'-methyl-2-nitro-1,1-ethenediamine (380 g) was filtered off and dried, meltingpoint 69°C to 70°C.
References
Merck Index 8019 DFU 4 (9) 663 (1979) PDR p. 919 OCDS Vol. 3 p. 131 (1984)
2956 Ranitidine
Trade Name Manufacturer Country Year Introduced Zantac Glaxo UK 1981 Zantac Glaxo Italy 1981 Zantic Glaxo Switz. 1982 Zantac Glaxo France 1982 Sostril Cascan W. Germany 1982 Zantic Glaxo W. Germany 1982 Zantac Glaxo Netherlands 1982 Zantac Glaxo Sweden 1983 Zantac Glaxo Canada 1983 Zantac Glaxo US 1983 Acidex Syncro Argentina - Ranidil Duncan Italy - Taural Roemmers Argentina - TorioI Vita Spain - Ulcex Guidotti Italy - Vizerul Montpellier Argentina -
DOT 18 (12) 665 (1982) I.N. p. 839 REM p. 798 Price, B.J., Clitherow, J.W. and Bradshaw, J.; US Patent 4,128,658; December
5, 1978; assigned to Allen and Hanburys Ltd.
RAZOXANE
Therapeutic Function: Antitumor
Chemical Name: dl-1,2-Bis(3,5-dioxopiperazin-1-yl)propane
Common Name: -
Structural Formula:
Chemical Abstracts Registry No.: 21416-87-5
Raw Materials
Formamide 1,2-Diaminopropane tetraacetic acid
Manufacturing Process
1,2-Diaminopropane tetraacetic acid (100 g) and formamide (400 ml) areheated together at reduced pressure under nitrogen at 100°C to 110°C for 1hour, and then at 150°C to 155°C for 4 hours. The brown solution isevaporated under reduced pressure at 80°C to 90°C and the residue is takenup in methanol (120 ml) and cooled in the refrigerator overnight. Filtration,followed by washing with methanol and vacuum drying at 65°C gives dl-1,2-bis(3,5-dioxopiperazin-1-yl)propane (62 g, 70%) as a very pale creammicrocrystalline solid, melting point 237°C to 239°C.
References
Merck Index 8026
Razoxane 2957
Trade Name Manufacturer Country Year IntroducedRazoxin I.C.I. UK 1977
DFU 2 (7) 473 (1977) Kleeman and Engel p. 800 DOT 13 (12) 546 (1977) Creighton, A.M.; US Patents 3,941,790; March 2, 1976; and 4,275,063;
June 23, 1981; both assigned to National Research Development Corp.
REBOXETINE MESYLATE
Therapeutic Function: Antidepressant
Chemical Name: ()-(2R*)-2-((αR*)-α-(o-ethoxyphenoxy)benzyl)morpholine, monomethanesulfonate
Common Name: Reboxetine mesilate
Structural Formula:
Chemical Abstracts Registry No.: 98769-82-5; 98769-81-4 (Base)
To 2-[α-(2-ethoxyphenoxy)benzyl]-morpholin-3-one in 60 ml of anhydrousTHF there was added dropwise under stirring a solution of BH3 in THF. Thewhole was heated at reflux for 3 hours and dropwise addition under coldconditions (0-(-5)°C) was then made of 3 ml of methanol and then of 3 ml of23% HCl. The solvent was removed under reduced pressure. The residue was
2958 Reboxetine mesylate
Trade Name Manufacturer Country Year IntroducedEdronax Pharmacia and Upjohn - -Lonol Promeco - -Davedax Bracco S.p.A. - -Norebox Pharmacia and Upjohn - -
diluted with H2O, made alkaline and extracted with chloroform. The organicextracts were washed to neutrality, dried and evaporated to dryness, to obtain2-[α-(2-ethoxyphenoxy)benzyl]-morpholine. Yield >90%; one diastereoisomer,M.P. 170-171°C.
In practice it is usually used as monomethanesulfonate salt.
References
Melloni P. et al.; US Patent No. 4,229,1980; Assigned to Farmitalia Carlo Erba, S.p.A., Milan, Italy
RELAXIN
Therapeutic Function: Ovarian hormone
Chemical Name: Polypeptide of approximately 6,000 molecular weight
Common Name: Releasin
Structural Formula: See Chemical name
Chemical Abstracts Registry No.: 9002-69-1
Raw Materials
Hog ovaries Acetone
Manufacturing Process
500 pounds of frozen hog ovaries (relaxin content: 20,200 G.P.U./lb) areground with 50 pounds of solid carbon dioxide (Dry Ice) in a Fitzpatrick millusing a 1/4 inch screen. The resulting finely divided tissue-carbon dioxidehomogenate at a temperature of -20°C is stirred into a 1.6 N HCl solutionprepared by mixing 15 liters of concentrated (12 N) HCl with 100 liters ofwater. The homogenate is added to the aqueous acid over a period ofapproximately 1 hour so that the temperature of the mixture does not fallbelow -5°C. The resulting slurry is stirred for 6 hours and then allowed tostand overnight.
The following day, a quantity of 200 gallons of acetone is added to thesuspension followed by stirring for 8 hours. The mixture is again allowed tostand overnight. The following day, the clear supernatant liquid is decantedfrom the suspension and the tissue residue is removed by filtration. The filter
Relaxin 2959
Trade Name Manufacturer Country Year IntroducedReleasin Warner Lambert US 1956Cervilaxin National US 1957
cake (tissue residue) is repulped with 35 gallons of a mixture of 0.3 volume12 N HCl, 9.7 volumes water and 30.0 volumes acetone and the resultingsuspension is filtered. The filtrates are combined with the supernatant liquidobtained by decantation to form the acid-acetone extract with a volume of275 gallons. The relaxin content of the extract is 9.4 G.P.U./ml or 19,600G.P.U./lb ovaries extracted, an activity yield of about 97 percent.
References
Merck Index 8031 I.N. p.841 Doczi, J.; US Patent 3,096,246; July 2, 1963; assigned to Warner-Lambert
Pharmaceutical Co.
REMIFENTANIL HYDROCHLORIDE
Therapeutic Function: Analgesic, Anesthetic
Chemical Name: 1-Piperidinepropanoic acid, 4-(methoxycarbonyl)-4-((1-oxopropyl)phenylamino)-, methyl ester, monohydrochloride
Common Name: Remifentanil hydrochloride
Structural Formula:
Chemical Abstracts Registry No.: 132539-07-2; 132875-61-7 (Base)
To a solution of 4-methoxycarbonyl-4-[(1-oxopropyl)phenylamino]-piperidine(200 mg, 0.68 mmol) in acetonitrile (1.1 ml) is added methyl acrylate (124 µl,1.36 mmol) at room temperature. The solution is stirred at 50°C for 2 hours,cooled to room temperature, and concentrated to an oily residue. The residueis chromatographed on silica gel (ethyl acetate) to give 3-[4-methoxycarbonyl-4-[(1-oxopropyl)phenylamino]-1-piperidine]propanoic acid,methyl ester as an oil: 253 mg, 97%. An equimolar amount of oxalic acid isadded to a solution of the free base in ethyl acetate. The precipitated salt isrecrystallized by adding methanol and heating until the solid goes back intosolution. Upon cooling the salt precipitates as a white solid; oxalate salt. It isrecrystallized from methanol and 2-butanone; m.p. 170°-172°C.
The hydrochloride salt may be make by dissolving the free base in toluene,saturating the solution with dry hydrogen chloride and concentrating to asolid. The solid is then recrystallized from appropriate solvent.
References
Feldman et al.; US Patent No. 5,019,583; May 28, 1991; Assigned to Glaxo Inc., Research Triangle Park, N.C.
REPAGLINIDE
Therapeutic Function: Antidiabetic
Chemical Name: Benzoic acid, 2-ethoxy-4-(2-((3-methyl-1-(2-(1-piperidinyl)phenyl)butyl)amino)-2-oxoethyl)-, (S)
3 g (11.9 mmols) of 3-ethoxy-4-ethoxycarbonyl-phenylacetic acid, 3.7 g (14.3mmols) of triphenylphosphine, 3.3 ml (23.8 mmols) of triethylamine and 1.15ml (11.9 mmols) of carbon tetrachloride were added successively to a solutionof 2.9 g (11.9 mmols) of 3-methyl-1-(2-piperidino-phenyl)-1-butylamine in 29of acetonitrile. The mixture was then stirred for 15 hours at roomtemperature, the solvent was removed in vacuo, and the residue was taken upin a mixture of ethyl acetate and water. The organic phase was dried oversodium sulfate, filtered and concentrated by evaporation in vacuo. Theevaporation residue was purified by column chromatography on silaca gel(toluene/acetone = 10/1). Yield: 4.9 g (85% of theory). M.p. 143°-145°C(petroleum/ether). High-melting-point form (B) of 2-ethoxy-4-[N-{1-(2-piperidinophenyl)-3-methyl-1-butyl}-aminocarbonylmethyl]-benzoic acid.
A mixture of 4.7 g (9.7 mmols) of ethyl 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl)-aminocarbonylmethyl]-benzoate and 14.7 ml of 1 Nsodium hydroxide was stirred in 47 ml of ethanol for 2 hours at 60°C, thenneutralized with 14.7 ml of 1 N hydrochloric acid and cooled to 0°C. Themixture was filtered to remove the precipitated colorless crystals, and thecrystals were washed with ice water and with a little ice cold ethanol and thendried at 100°C/1 Torr. Yield: 3.9 g (88% of theory). M.p. 140°-142°C. Uponfurther recrystallization from ethanol/water (2/1) the melting point remainedconstant.
Low-melting-point form (A) of 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl}-aminocarbonylmethyl]-benzoic acid.
1 g of the high-melting-point form (B) of 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl}-aminocarbonyl-methyl]-benzoic acid was dissolvedat room temperature in 5 ml of acetone, and 5 of petroleum ether (m.p. 60°-70°C) were added. Upon trituration, crystallization gradually set in. The samequantity of petroleum ether was added again, and after crystallization hadended, the mixture was filtered. The crystals were washed with petroleumether, and the almost colorless crystals were dried for 2 hours at 60°C/0.1Torr. Yield: 0.7 g. M.p. 95°-98°C (clear beginning at 135°C). The IR-spectrafor this form are identical to the IR-spectra for the form (A), melting point90°-92°C.
2962 Repaglinide
Trade Name Manufacturer Country Year IntroducedEurepa Torrent Pharmaceuticals Ltd. India -NovoNorm Novo Nordisk Denmark -Prandin Medley USA -Repaglinide Novo Nordisk Denmark -
High-melting-point form (B) of 2-ethoxy-4-[N-{1-2-piperidino-phenyl)-3-methyl-1-butyl}-aminocarbonylmethyl]-benzoic acid
1 g of the low-melting-point form (A) of 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl}-amino-carbonylmethyl]-benzoic acid was dissolvedin 10 ml of ethanol/water (2/1) while heating over a steam bath. The solutionwas then cooled to 0°C, whereupon crystallization began. The mixture wasfiltered, and the residue was washed with a little ice-cold ethanol and dried at100°C/1 Torr. Yield: 0.8 g. M.p. 140°-142°C.
Foamy form (C) of 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl}-aminocarbonylmethyl]-benzoic acid
1.5 g of the high-melting-point form (B) of 2-ethoxy-4-[N-{1-(2-piperidino-phenyl)-3-methyl-1-butyl}-amino-carbonylmethyl]-benzoic acid was dissolvedin 5 ml of methanol while heating. The solution was then cooled to 0°C withtrituration. The crystals precipitated thereby were separated by filtration,washed with a little cold methanol, and dried for 2 hours at 60°C/0.1 Torr.Yield of adduct (with 1 times CH3OH): 1.2 g. M.p. 85°-90°C. The adduct wasconverted into the methanol-free foamy form (C) by heating for 24 hours at60°C/5 Torr over phosphorus pentoxide. Melting range: 75°-85°C.
References
Grell et al.; US Patent No. 5,216,167; Jun. 1, 1993; Assigned to Dr. Karl Thomae GmbH, Biberach an der Riss, Fed. Rep. of Germany
Grill et al.; US Patent No. 5,312,924; May 17, 1994; Assigned to Dr. Karl Thomae GmbH, Biberach an der Riss, Fed. Rep. of Germany
REPROTEROL
Therapeutic Function: Bronchodilator
Chemical Name: 7-[3-[[2-(3,5-Dihydroxyphenyl)-2-hydroxyethyl]amino]propyl]-3,7-dihydro-1,3-dimethyl-1H-purine-2,6-dione
Common Name: -
Structural Formula:
Reproterol 2963
Chemical Abstracts Registry No.: 54063-54-6; 13055-82-8 (Hydrochloride salt)
Raw Materials
Theophylline 1-Bromo-3-chloropropane Palladium on carbon 3,5-Dihydroxy-ω-bromoacetophenone Benzylamine Hydrogen
Manufacturing Process
Theophylline is reacted first with 1-bromo-3-chloropropaneto give chloropropyltheophylline, then with benzylamine to give benzylaminopropyltheophylline.That is reacted with 3,5-dihydroxy-ω-bromoacetophenone to give the startingmaterial.
500 g of 7-[3-[2-(3,5-dihydroxyphenyl)-2-oxoethyl-benzylamino]-propyl]-theophylline hydrochloride obtained as above were dissolved in 5 liters ofdimethyl acetamide. There were added 25 g of a 10% palladium-carboncatalyst, the mixture heated to 70°C and hydrogenated with stirring at thistemperature and 2 bar pressure until the speed of hydrogenation perceptiblyslowed (about 2 hours). Subsequently, the mixture was filtered and afteraddition of a further 25 g of the palladium catalyst hydrogenated at 6 bar tothe end (2 to 3 hours). The mixture was filtered, the greatest part of thesolvent distilled off at a water jet vacuum, and the residue treated with 8liters of ethanol. The solution was cooled for 12 hours with flowing water andthe precipitated material filtered off with suction. Then it was boiled for onehour with 2 liters of methanol with stirring and the passing through ofnitrogen, allowed to cool to 25°C and filtered off with suction. After drying ina vacuum at 55°C there were obtained 391 g (= 94.5% of theory) of pure 7-[3-[2-(3,5-dihydroxyphenyl)-2-hydroxyethylamino]propyl]-theophyllinehydrochloride. Melting point 263°C to 265°C.
References
Merck Index 8035 Kleeman and Engel p. 800 OCDS Vol. 3 p. 231 (1984) DOT 13 (2) 552 (1977) I.N. p. 842 Klingler, K.H. and Bickel, E.; US Patent 4,150,227; April 17, 1979; assigned to
Degussa (Germany)
Trade Name Manufacturer Country Year Introduced Bronchospasmin Homburg W. Germany 1977 Bronchospasmin Farmades Italy 1981 Bronchodil Berlimed UK 1981 Asmaterol Lusofarmaco Italy - Tiffen Tosi Italy -
2964 Reproterol
RESCIMETOL
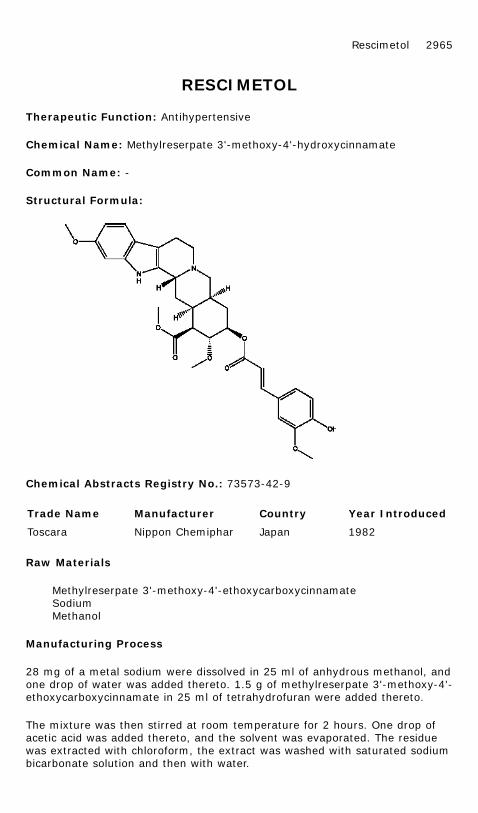
Therapeutic Function: Antihypertensive
Chemical Name: Methylreserpate 3'-methoxy-4'-hydroxycinnamate
28 mg of a metal sodium were dissolved in 25 ml of anhydrous methanol, andone drop of water was added thereto. 1.5 g of methylreserpate 3'-methoxy-4'-ethoxycarboxycinnamate in 25 ml of tetrahydrofuran were added thereto.
The mixture was then stirred at room temperature for 2 hours. One drop ofacetic acid was added thereto, and the solvent was evaporated. The residuewas extracted with chloroform, the extract was washed with saturated sodiumbicarbonate solution and then with water.
Rescimetol 2965
Trade Name Manufacturer Country Year Introduced
Toscara Nippon Chemiphar Japan 1982
The chloroform layer was dried over sodium sulfate, and the solvent wasevaporated, so that there was obtained a brown amorphous matter. This wasrecrystallized from chloroform-hexane, and there was then obtained 1.0 g(78% of yield) of methylreserpate 3'-methoxy-4'-hydroxycinnamate which wascharacterized as pale yellow needles having a melting point of 259°C to260°C.
References
Merck Index 8038 DFU 3 (3) 183 (1978) (As CD-3400) and 5 (12) 635 (1980) DOT 18 (10) 551 (1982) Kametani, T.; US Patent 3,898,215; August 5, 1975; assigned to Nippon
Chemiphar Co., Ltd.
RESCINNAMINE
Therapeutic Function: Antihypertensive
Chemical Name: 11,17α-Dimethoxy-18β-[[1-oxo-3-(3,4,5-trimethoxyphenyl)-2-propenyl]-oxy]-3β,20α-yohimban-16α-carboxylic acid methyl ester
Common Name: 3,4,5-Trimethoxycinnamoyl methyl reserpate
4.0 grams of 3,4,5-trimethoxycinnamic acid, MP 125.5° to 127°C was refluxedfor 35 minutes under anhydrous conditions with 6.0 parts by volume ofredistilled thionyl chloride.
The excess thionyl chloride was removed under vacuum and by distilling fromthe residue two portions of dry benzene. The crystalline residue wascrystallized twice from hexane-ether to yield 3,4,5-trimethoxycinnamoylchloride which was obtained in the form of bright yellow prisms, MP 95° to96°C.
Trade Name Manufacturer Country Year Introduced Moderil Pfizer US 1956 Aldatense Searle France - Anaprel Servier France - Apolon Toyama Japan - Aporecin Kayaku Japan - Aporesin A.L. Norway - Apotension Santen Japan - Apoterin Seiko Japan - Atension Santen Japan - Caniramine Hokuriku Japan - Cartric Sanwa Japan - Cinnaloid Taito Pfizer Japan - Colstamin Kowa Japan - Daisaloid Mohan Japan - Isocalsin Kowa Japan - Paresinan Wakamoto Japan - Rescamin Pharmacia Sweden - Rescimin Torlan Spain - Rescinate Ohta Japan - Rescisan Pharmacia Sweden - Rescitens Fargal Italy - Resiloid Nippon Shoji Japan - Rosex Teikoku Japan - Rozex Teisan Japan - Sciminan Kotani Japan - Seripinin Fuji Zoki Japan - Sinselpin Kobayashi Japan -
Rescinnamine 2967
To a solution of 0.80 part by weight of methyl reserpate in 10 parts by volumeof dry distilled pyridine at 10° to 15°C were added in portions during 20minutes with stirring and external cooling 1.1 parts by weight of 3,4,5-trimethoxycinnamoyl chloride. The reaction was carried out under nitrogen.After standing at room temperature for 65 hours the pyridine was removedunder reduced pressure and at a temperature of 50° to 60°C. A brown solidfroth-like material was obtained which was chromatographed on 30 parts byweight of alumina (activity II-III). The fractions eluted with benzene-acetonemixtures, on crystallization from benzene yielded 3,4,5-trimethoxycinnamateof methyl reserpate in the form of needles, which on recrystallization frommethanol melted at 232° to 234°C as described in US Patent 2,854,454.
The 3,4,5-trimethoxycinnamic ester of methyl reserpate is also present inRauwolfia plants and obtainable in purified form therefrom by extraction asdescribed in US Patents 2,974,144 and 2,876,228.
References
Merck Index 8039 Kleeman and Engel p. 801 PDR p. 1422 OCDS Vol. 1 p. 319 (1977) I.N. p. 843 REM p. 909 Ulshafer, P.R.; US Patent 2,854,454; September 30, 1958 Ordway, H.W. and Guercio, P.A.; US Patent 2,876,228; March 3, 1959;
assigned to Chas. Pfizer and Co., Inc. Klohs, M.W., Draper, M.D. and Keller, F.; US Patent 2,974,144; March 7, 1961;
assigned to Riker Laboratories, Inc.
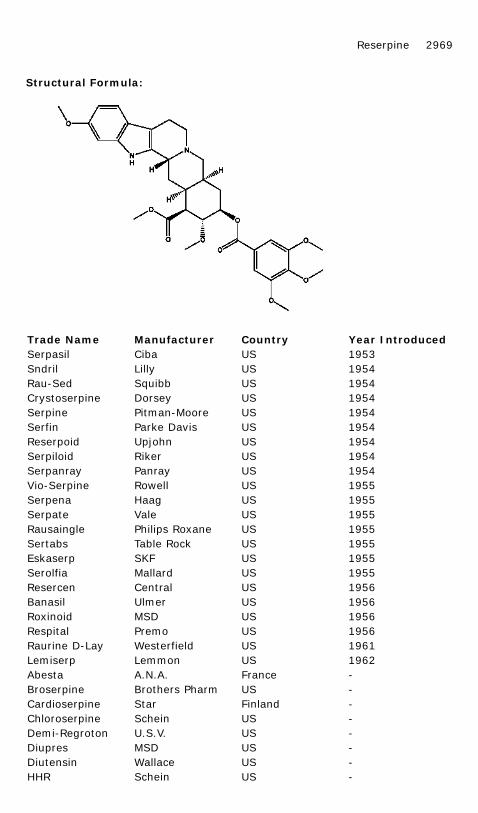
RESERPINE
Therapeutic Function: Antihypertensive
Chemical Name: 11,17α-Dimethoxy-18β-[(3,4,5-trimethoxybenzoyl)oxy]-3β,20α-yohimban-16β-carboxylic acid methyl ester
Common Name: 3,4,5-Trimethoxybenzoyl methyl reserpate
Chemical Abstracts Registry No.: 50-55-5
Raw Materials
Rauwolfia plant bark Methanol
2968 Reserpine
Structural Formula:
Trade Name Manufacturer Country Year Introduced Serpasil Ciba US 1953 Sndril Lilly US 1954 Rau-Sed Squibb US 1954 Crystoserpine Dorsey US 1954 Serpine Pitman-Moore US 1954 Serfin Parke Davis US 1954 Reserpoid Upjohn US 1954 Serpiloid Riker US 1954 Serpanray Panray US 1954 Vio-Serpine Rowell US 1955 Serpena Haag US 1955 Serpate Vale US 1955 Rausaingle Philips Roxane US 1955 Sertabs Table Rock US 1955 Eskaserp SKF US 1955 Serolfia Mallard US 1955 Resercen Central US 1956 Banasil Ulmer US 1956 Roxinoid MSD US 1956 Respital Premo US 1956 Raurine D-Lay Westerfield US 1961 Lemiserp Lemmon US 1962 Abesta A.N.A. France - Broserpine Brothers Pharm US - Cardioserpine Star Finland - Chloroserpine Schein US - Demi-Regroton U.S.V. US - Diupres MSD US - Diutensin Wallace US - HHR Schein US -
Reserpine 2969
Trade Name Manufacturer Country Year Introduced Hydro-Fluserpine Schein US - Hydromox Lederle US - Hydropres MSD US - Hydroserpine Schein US - Key-Serpine Key US - Lemiserp Lemmon US - Metatensin Merrell Dow US - Naquival Schering US - Neo-Serp Neo Canada - Raulen Paul Maney Canada - Rausan Wassermann Spain - Rausedan Arzneimittelwerk
Dresden E. Germany -
Rauvilid Pharmacia Sweden - Rauwita Lifasa Spain - Regroton U.S.V. US - Renese-R Pfipharmecs US - Resedril Estedi Spain - Rese-Lar Perga Spain - Reser-Ar Luar US - Resercrine Casgrain and
Charbonneau Canada -
Reserfia Medic Canada - Reserpur A.F.I. Norway - Resine Kirk US - Resomine Bonjean Belgium - Rivasin Giulini W. Germany - Slutensin Bristol US - Ser-Ap-Es Ciba US - Serolfia Ascher US - Serpalan Lannett US - Serpax Verdun Canada - Serpedin Pharmacia Sweden - Serpena Haag US - Serpentil Pliva Yugoslavia - Serpipur Kwizda Austria - Serpivite Vitarine US - Serpoid Canfield US - Serpone Hartz Canada - Serpresan Maipe Spain - Sertina Fellows-Testagar US - SK-Reserpine SKF US - Unipres Reid-Rowell US - Vio-Serpine Rowell US - V-Serp Vangard US -
2970 Reserpine
Manufacturing Process
7,000 parts by weight of powdered bark from the root of Rauwolfia serpentinaBenth, are percolated with about 35,000 parts by volume of methanol. Afterevaporating the methanol extract, 1,050 parts by weight are obtained of adark colored powder which is treated several times with water for removal ofsoluble constituents. The insoluble residue remaining from this operation issubsequently masticated five times, in each case with 1,500 parts by volumeof 10% aqueous acetic acid, the solution being best separated from thesmeary residue by centrifuging. The brown acetic acid solution, which forfurther working up can be concentrated at low temperature to a small volumeor be diluted with half the volume of water, possesses a pH of about 3.9. Thissolution is extracted by shaking with 3,500 to 4,000 parts by volume ofchloroform divided into 3 to 4 portions. These chloroform extracts are washedonce with potassium carbonate solution and twice with water, dried withsodium sulfate and evaporated to dryness under reduced pressure. Theresidue, amounting to 70 to 80 parts by weight, forms a green-brown coloredpowder. For further purification, this residue is dissolved in benzene andchromatographed over 1,000 to 1,200 parts by weight of neutral aluminumoxide (activity H-III according to Brockmann). On elution with benzene thereare first obtained small quantities of a yellow oil and 0.9 part by weight of aninactive crystallizate of melting point 238°C to 239°C, after which thesubstance of sedative activity follows. As soon as the major quantity of theactive substance has been eluted, further elution is carried out with a mixtureof 2 parts by volume of benzene and 1 part by volume of acetone. In thismanner the residue of the sedative substance is obtained and after that afurther inactive crystallizate of melting point 141°C to 143°C. The eluatefractions containing the sedative substance are evaporated to dryness. Byrecrystallization of the residue from hot acetone or a mixture of chloroformand ether.6.5 to 7 parts by weight of reserpine are obtained in the form ofalmost colorless crystals of melting point 262°C to 263°C (withdecomposition).
References
Merck Index 8042 Kleeman and Engel p. 802 PDR pp. 710, 812, 993, 1011, 1168, 1185, 1231, 1409, 1449, 1606, 1634,
1723, 1820, 1876, 1999 I.N. p. 843 REM p. 908 Schwyzer, R. and Mueller, J.; US Patent 2,833,771; May 6, 1958; assigned to
Ciba Pharmaceutical Products, Inc.
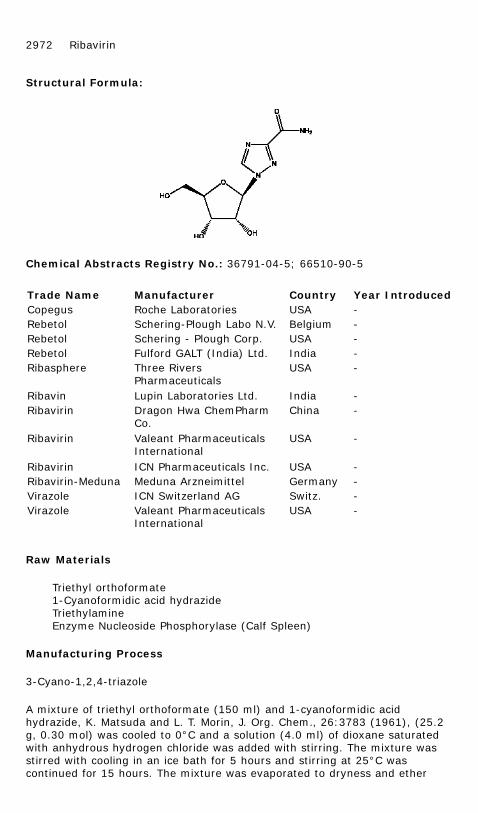
RIBAVIRIN
Therapeutic Function: Antiviral
Chemical Name: 1,2,4-Triazole-3-carboxamide, 1-β-D-ribofuranosyl-
Common Name: Ribavirin; Tribavirin
Ribavirin 2971
Structural Formula:
Chemical Abstracts Registry No.: 36791-04-5; 66510-90-5
A mixture of triethyl orthoformate (150 ml) and 1-cyanoformidic acidhydrazide, K. Matsuda and L. T. Morin, J. Org. Chem., 26:3783 (1961), (25.2g, 0.30 mol) was cooled to 0°C and a solution (4.0 ml) of dioxane saturatedwith anhydrous hydrogen chloride was added with stirring. The mixture wasstirred with cooling in an ice bath for 5 hours and stirring at 25°C wascontinued for 15 hours. The mixture was evaporated to dryness and ether
Trade Name Manufacturer Country Year Introduced Copegus Roche Laboratories USA - Rebetol Schering-Plough Labo N.V. Belgium - Rebetol Schering - Plough Corp. USA - Rebetol Fulford GALT (India) Ltd. India - Ribasphere Three Rivers
Pharmaceuticals USA -
Ribavin Lupin Laboratories Ltd. India - Ribavirin Dragon Hwa ChemPharm
Co. China -
Ribavirin Valeant Pharmaceuticals International
USA -
Ribavirin ICN Pharmaceuticals Inc. USA - Ribavirin-Meduna Meduna Arzneimittel Germany - Virazole ICN Switzerland AG Switz. - Virazole Valeant Pharmaceuticals
International USA -
2972 Ribavirin
(500 ml) was added to the residue. The solution was filtered, washed withwater, and the organic layer was dried over magnesium sulfate. The solutionwas filtered and the ether was removed. Crystallization of the product fromethyl acetate-benzene provided 16.0 g (56.8%) of 3-cyano-1,2,4-triazole witha melting point of 185°-187°C. All properties of the compound were identicalwith those of a sample prepared by the method of Cipens and Grinsteins,Latvijas PSR Zinatnu Adad. Vestis., Kim Ser., 1965 (2), 204-208. Chem Abst.,63, 13243 (1965).
1,2,4-Triazole-3-thiocarboxamide
A mixture of 3-cyano-1,2,4-triazole (4.7 g, 0.050 mol), ethanol (50 ml) andtriethylamine (8.0 ml) was stirred at room temperature while hydrogen sulfidegas was bubbled into the mixture for 4 hours. The solvent was removed andwater was added to the residue to provide 2.7 g of product. Recrystallizationfrom water afforded pure 1,2,4-triazole-3-thiocarboxamide with a meltingpoint of >350°C.
1-β-D-Ribofuransyl-1,2,4-triazole-3-carboxamide
Synthesis of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide from 1,2,4-triazole-3-carboxamide via Purified Calf Spleen Nucleoside Phosphorylase.
The samples were incubated at 25°C for 5 minutes and then frozen in dryice/isopropanol to stop the reaction. Aliquots of the thawed samples were thenspotted on silica gel together with standard solutions of 1,2,4-triazole-3-carboxamide and 1-β-D-ribofuranosyl-1,2,4-triazole- 3-carboxamide andseparated in isopropanol:NH4OH:H2O (7:1:2). Areas of the chromatogramscoinciding with 1,2,4-triazole-3-carboxamide were removed and counted todetermine the percent of conversion of 1,2,4 -triazole-3-carboxamide to 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide.
1,2,4-Triazole-3-carboxamide may be also converted to 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide by reaction with the enzyme NucleosidePhosphorylase at a pH within the range of about 7 to 8, at an enzymeconcentration about 0.15 mg/ml, and a temperature approximately 25° toabout 35°C. Satisfactory results have been obtained when the triazole base ispresent in a concentration greater than 5 x 10-5M and ribose-1-phosphate ispresent at a concentration greater than 2 x 105M. Generally are required forthe reactionabout 0.5 to about 1 hour. The source of the enzyme may beanimal, tissue, or bacteria. The principal bacterial sources are E. coli andyeast, Brevebacterium, while a variety of animal sources exist, including beefspleen, rat liver, calf liver, calf thymus, beef liver, monkey brain, horse liver,calf spleen, human erythrocytes, fish skin, and fish muscle.
References
Witkowski et al.; US Patent No. 3,976,545; Aug. 24, 1976; Assigned to ICN Pharmaceuticals, Inc., Irvine, Calif.
Fujishima et al.; US Patent No. 4,614,719; Sep., 30, 1986; Assigned to Yamasa Shoyu Kabushiki Kaisha, Chiba, Japan
Ribavirin 2973
RIBOFLAVIN
Therapeutic Function: Enzyme cofactor vitamin
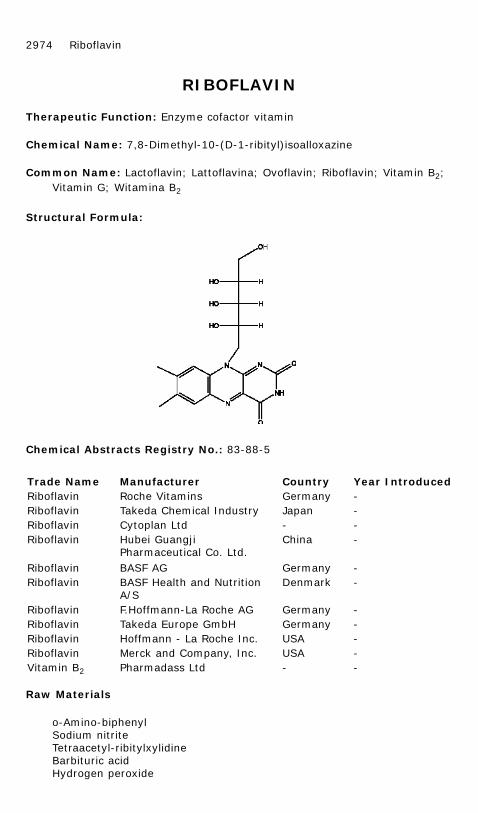
Chemical Name: 7,8-Dimethyl-10-(D-1-ribityl)isoalloxazine
Trade Name Manufacturer Country Year Introduced Riboflavin Roche Vitamins Germany - Riboflavin Takeda Chemical Industry Japan - Riboflavin Cytoplan Ltd - - Riboflavin Hubei Guangji
Pharmaceutical Co. Ltd. China -
Riboflavin BASF AG Germany - Riboflavin BASF Health and Nutrition
A/S Denmark -
Riboflavin F.Hoffmann-La Roche AG Germany - Riboflavin Takeda Europe GmbH Germany - Riboflavin Hoffmann - La Roche Inc. USA - Riboflavin Merck and Company, Inc. USA - Vitamin B2 Pharmadass Ltd - -
2974 Riboflavin
Manufacturing Process
A solution of o-biphenyl diazonium sulfate is prepared as follows: o-amino-biphenyl 21.1 g (0.125 mole), is dissolved by warming in a mixture of 100 mlof glacial acid and 136 ml of water. To this solution is added a mixture: 11.2ml of concentrated sulfuric acid and 25 ml of water. The temperature is thenlowered to 0° to 5°C and 8.65 g (0.125 mole) of sodium nitrite is added insmall portions to the: stirred solution over a one hour period. The solution isstirred for an additional two hours at 0° to 5°C after the addition of the nitriteis complete.
A solution of ribitylxylidine is prepared as follows: a stirred suspension of 42.3g (0.10 mole) of tetraacetylribitylxylidine and 85 ml of water is heated to 95°to 100°C. To this material is added, over a five to 10 minute period, exactly0.40 mole of aqueous 25 to 30% sodium hydroxide solution, the temperaturebeing held at 95° to 100°C. Shortly after the addition of the caustic solution,the mixture becomes homogeneous, the temperature rises suddenly, and arapid evolution of steam occurs. The light, straw-colored solution is thenstirred for a one hour at 95° to 100°C to insure completeness of reaction.With the resulting ribitylxylidine solution at a temperature of 90°C, 166 ml of57.5 percent (v./v.) aqueous acetic acid is added, and the solution is thencooled to 0° to 5°C.
The solution of o-biphenyl diazonium sulfate is now added to the ribitylxylidinesolution at such a rate as to maintain the temperature of the resulting mixturebelow 8°C. The mixture is then stirred at 0° to 5°C for 24 hours. After thestirring period is complete the crude product is isolated by filtration; the wetproduct is slurried in 500 ml of water, refiltered, washed with 200 ml of water,and air-dried at 50° to 60°C. The dried material is pulverized to give about 44g of 2-(o-biphenylazo)-4,5-dimethyl-1-ribitylamino-beiizene which is obtainedas a granular, orange product; m.p. 134°-139°C, dec.
To a mixture of 225 ml of ethyl acetate and 40.5 ml of glacial acetic acid isadded 43.6 g (0.10 mole) of 2-(o-biphenylazo)-4,5-dimethyl-l-ribitylamino-benzene, prepared as described above, and 16.9 g (0.132 mole) of barbituricacid. The resulting mixture is refluxed with stirring for about 90 hours, at theend of which period an 0.5 ml test sample of the reaction solution, whenmixed with 15 ml of 26% aqueous hydrochloric acid, gives a light straw colorshowing completion of the reaction. If the reaction is incomplete, the reactionmixture is refluxed for additional five hours periods until a test for completionof the reaction is obtained. The reaction mixture is cooled to 10°C for onehour and filtered. The crude product is washed successively with 50 ml of coldethyl acetate and 50 ml of cold water, then air-dried at 70°C to give about 37g of crude riboflavin, which is obtained as a brownish-yellow powder; yieldapproximately 98.5% of theory.
Crude riboflavin, prepared as described above starting with 43.6 g of 2-(o-biphehylazo)-4,5-dimethyl-1-ribityl amino-benzene, is washed with 50 ml ofcold ethyl acetate, slurried with 180 ml of methanol at 65°C for thirtyminutes. The methanol slurry is cooled to 10°C for thirty minutes, filtered,and the filtered material washed with 40 ml of cold methanol. The methanolwashed riboflavin is then slurried with 180 ml of water at 80°C for thirtyminutes, the slurry is cooled to 70°C, filtered, and the filtered material iswashed with 40 ml of hot (70°C) water. The hot water-washed riboflavin is
Riboflavin 2975
dissolved in a mixture of 70 ml of hydrochloric acid and 23 ml of water bywarming to 45°C. The aqueous hydrochloric acid solution is treated with a 1.5ml of 30% hydrogen peroxide solution to oxidize impurities and filteredthrough a filter precoated with diatomaceous silica (Super-Cel). The oxidized,filtered solution is poured, with vigorous agitation, into 900 ml of water at 95°to 100°C. The resulting riboflavin slurry is cooled slowly to 10°C, filtered, andthe filtered material is washed with 2 x 50 ml of water and 4 x 50 ml portionsof methanol. The washed material is air-dried, at 70°C, to give about 30 g ofriboflavin; yield about 80% of theory based on the 42.3 g oftetraacetylribitylxylidine used as starting material.
References
Howe Ch.A. et al.; US Patent No. 2,807,611; Sept. 24, 1957; Assignted to Merck and Co., Inc., Rahway, N.J., a corporation of New Jersey
RIBOSTAMICIN
Therapeutic Function: Antibiotic
Chemical Name: O-2,6-Diamino-2,6-dideoxy-α-D-glucopyranosyl-(1-->4)-O-[β-D-ribofuranosyl-(1-->5)]-2-deoxy-D-streptamine
Streptomyces thermoflavus SF-733 strain was inoculated to 15 liters of aliquid medium (pH 7.0) containing glucose 2.5%, soybean meal 3.5%, solublevegetable protein 1.0% and NaCl 0.25% and shake-cultured in a jar-fermenter at 28°C for 3 days. 10 liters of culture filtrate (potency, 200meg/ml) obtained by filtering culture broth at pH 4.0 was adjusted to pH 7.0and applied to a column filled with 1 liter of Amberlite IRC 50 (NH4
+type,Rohm and Haas) to adsorb active ingredient on ion-exchange resin. Afterwashing with water the column was eluted with 0.5 N ammonia water. Activefractions were concentrated in vacuo and freeze-dried. 5.9 g of crude powderthus obtained was dissolved in 10 ml of water, applied to a column filled with400 ml of Dowex 1 x 2 (OH-type, Dow Chemicals) and developedchromatographically with water to give 250 ml of active fraction which wasconcentrated in vacuo, whereby 2.1 g of light yellow powder of SF-733substance was obtained. 2.0 g of this powder was dissolved in 3 ml of water,applied to a column filled with 100 ml of Amberlite CG 50 (NH4
+type) washedwith water and eluted with 0.2 N ammonia water. 400 ml of active fractionwas collected, concentrated in vacuo and freeze-dried to give 600 mg of whitepowder of free base of SF-733 substance. This powder was dissolved in about5 ml of water and concentrated to syrup and added with about 50 ml ofethanol. The mother liquor together with white precipitate thus formed wasconcentrated in vacuo to dryness. 650 mg of ethanol-solvate-like whitepowder was dissolved in 65 ml of methanol. The solution became cloudyimmediately after dissolution and crystals were gradually separated. Aftertightly sealed and left alone at 30°C overnight crystals were collected bymeans of glass filter and washed with about 1 ml of methanol. The crystalswere held on calcium chloride as a drying agent at room temperature in vacuoand then dried on phosphorus pentoxide as a drying agent at 60°C for 19hours in vacuo to give 440 mg of free base crystals of SF-733 substance.Yield: 73%.
References
Merck Index 8106 Kleeman and Engel p. 807 DOT 9 (3) 112 (1973) I.N. p. 848 Shomura, T., Ezaki, N., Tsuruoka, T., Niwa, T., Akita, E. and Niida, T.; US
Patent 3,661.892; May 9, 1972; assigned to Meiji Seika Kaisha, Ltd. (Japan)
RIFAMPIN
Therapeutic Function: Antitubercular
Rifampin 2977
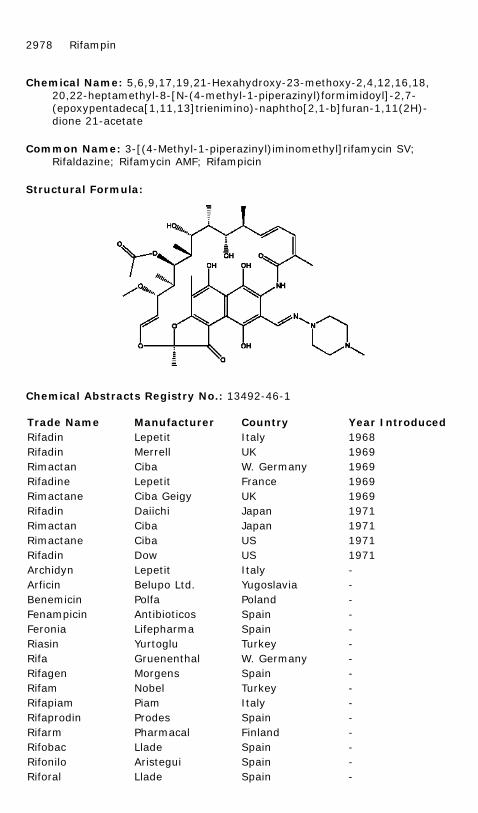
Chemical Name: 5,6,9,17,19,21-Hexahydroxy-23-methoxy-2,4,12,16,18,20,22-heptamethyl-8-[N-(4-methyl-1-piperazinyl)formimidoyl]-2,7-(epoxypentadeca[1,11,13]trienimino)-naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate
Common Name: 3-[(4-Methyl-1-piperazinyl)iminomethyl]rifamycin SV; Rifaldazine; Rifamycin AMF; Rifampicin
Structural Formula:
Chemical Abstracts Registry No.: 13492-46-1
Trade Name Manufacturer Country Year Introduced Rifadin Lepetit Italy 1968 Rifadin Merrell UK 1969 Rimactan Ciba W. Germany 1969 Rifadine Lepetit France 1969 Rimactane Ciba Geigy UK 1969 Rifadin Daiichi Japan 1971 Rimactan Ciba Japan 1971 Rimactane Ciba US 1971 Rifadin Dow US 1971 Archidyn Lepetit Italy - Arficin Belupo Ltd. Yugoslavia - Benemicin Polfa Poland - Fenampicin Antibioticos Spain - Feronia Lifepharma Spain - Riasin Yurtoglu Turkey - Rifa Gruenenthal W. Germany - Rifagen Morgens Spain - Rifam Nobel Turkey - Rifapiam Piam Italy - Rifaprodin Prodes Spain - Rifarm Pharmacal Finland - Rifobac Llade Spain - Rifonilo Aristegui Spain - Riforal Llade Spain -
2978 Rifampin
Raw Materials
3-Formylrifamycin SV 1-Amino-4-methylpiperazine
Manufacturing Process
3-Formylrifamycin SV is treated with 1-amino-4-methylpiperazine intetrahydrofuran to give rifampin.
References
Merck Index 8113 Kleeman and Engel p. 808 PDR pp. 810, 1236 DOT 5 (1) 24 (1969) I.N. p. 848 REM p. 1233 Maggi, N. and Sensi, P.; US Patent 3,342,810; September 19, 1967; assigned
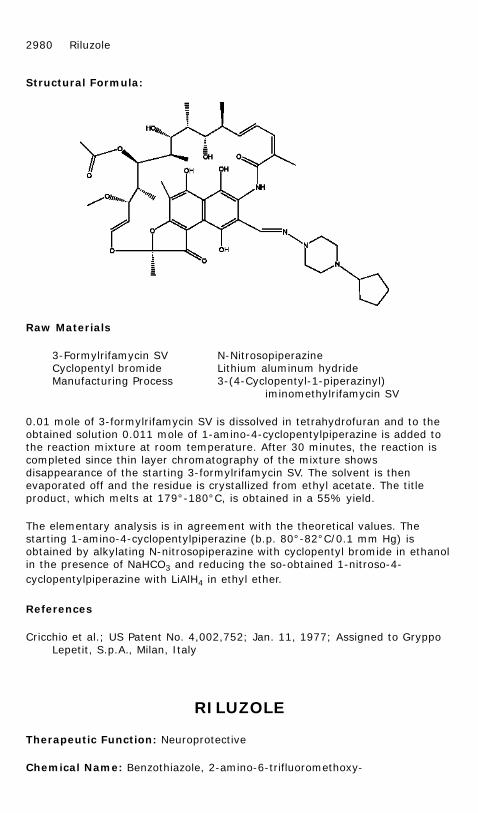
0.01 mole of 3-formylrifamycin SV is dissolved in tetrahydrofuran and to theobtained solution 0.011 mole of 1-amino-4-cyclopentylpiperazine is added tothe reaction mixture at room temperature. After 30 minutes, the reaction iscompleted since thin layer chromatography of the mixture showsdisappearance of the starting 3-formylrifamycin SV. The solvent is thenevaporated off and the residue is crystallized from ethyl acetate. The titleproduct, which melts at 179°-180°C, is obtained in a 55% yield.
The elementary analysis is in agreement with the theoretical values. Thestarting 1-amino-4-cyclopentylpiperazine (b.p. 80°-82°C/0.1 mm Hg) isobtained by alkylating N-nitrosopiperazine with cyclopentyl bromide in ethanolin the presence of NaHCO3 and reducing the so-obtained 1-nitroso-4-cyclopentylpiperazine with LiAlH4 in ethyl ether.
References
Cricchio et al.; US Patent No. 4,002,752; Jan. 11, 1977; Assigned to Gryppo Lepetit, S.p.A., Milan, Italy
RILUZOLE
Therapeutic Function: Neuroprotective
Chemical Name: Benzothiazole, 2-amino-6-trifluoromethoxy-
2980 Riluzole
Common Name: Riluzole; Rilutek
Structural Formula:
Chemical Abstracts Registry No.: 1744-22-5
Raw Materials
Sodium nitrite 4-Amino-3-nitrobenzotrifluoride Potassium thiocyanate Copper thiocyanate Tin
Manufacturing Process
To a stirred solution of 74.6 g (0.5 mol) of benzylisothiocyanate in 500 mLtoluene was added dropwise 46.6 g (0.5 mol) of aniline. The solution wasrefluxed 18 hours. After cooling, the reaction mixture was concentrated underreduced pressure. The residue was recrystallized from an appropriate solventto yield N-phenyl-N'-benzylthiourea. Isothiocyanates which are notcommercially available may be prepared from aliphatic or aryl primary aminesby the following methods: Kurita and Iwakura, Org. Synth. 59, 195; Jochims,Chem. Ber. 101, 1746 (1968); or Castro, Pena, Santos, and Vega, J. Org.Chem. 49, 863 (1984).
To a stirred solution of 20.6 g (0.10 mol) 4-amino-3-nitrobenzotrifluoride in30 mL conc. H2SO4 and 30 mL H2O at 0°C was added dropwise 37.5 mL 20%sodium nitrite. The mixture was stirred for 90 min at 0-5°C. Potassiumthiocyanate (10 g in 20 mL H2O) was added dropwise and stirred 15 min. Thereaction was poured into a vigorously stirred slurry of 18 g (0.148 mol)copper thiocyanate in 60 mL H2O. Gas evolution began and the mixturefoamed. The reaction was stirred two hours at 3°C and then heated to 70°Cfor 20 min. The reaction was cooled to 25°C and stirred an additional 18hours. The solution was filtered and the water was extracted with toluene (3 x100 mL). The toluene layer was dried (Na2SO4), filtered, and concentratedunder reduced pressure to yield 3-nitro-4-thiocyanatebenzotrifluoride as apurple oil. The product was purified by silica gel chromatography. The columnwas eluted initially with hexane followed by hexane/CH2Cl2 (7:3) to yield anoil which was crystallized from heptane to yield a yellow 3-nitro-4-thiocyanatebenzotrifluoride, m.p. 72-73°C.
To a vigorously stirred solution of 4.0 g (0.16 mol) 3-nitro-4-thiocyanatebenzotrifluoride in 50 mL conc. HCl was added 16.0 g (0.135 mol)granulated tin over one hour. The reaction changed from an orange to very
Riluzole 2981
Trade Name Manufacturer Country Year IntroducedRilutek RPR - -PK-26124 EMD Biosciences, Inc. - -
pale yellow to white. The reaction was stirred at 25°C for 20 hours. Thereaction solution was diluted with H2O (250 mL) and conc. NH4OH was addeddropwise. The product precipitated along with the tin salts. The solid wasfiltered and boiled in CHCl3 (3 x 200 mL). The aqueous layer was extractedwith CHCl3. All the CHCl3 washings were combined, dried (MgSO4), filtered,and concentrated under reduced pressure to yield a dark brown solid. Thecrude 2-amino-5-trifluoromethylbenzothiazole hydrochloride was dissolved inhot Et2O and filtered. To the filtrate was added a solution of freshly preparedEt2O/HCl. The product precipitate was filtered and washed with Et2O to yield awhite solid of 2-amino-5-trifluoromethylbenzothiazole hydrochloride, m.p.255.-257°C.
References
Merck Index, Monograph number: 8389, Twelfth edition, 1996, Editor: S. Budavari; Merck and Co., Inc.
Johnson G., Pavia M. R.; US Patent No. 4,826,860; May 2, 1989; Assigned to Warner-Lambert Company (Morris Plaines, NJ)
RIMANTADINE HYDROCHLORIDE
Therapeutic Function: Antiviral
Chemical Name: 1-Adamantanemethylamine, α-methyl-, hydrochloride
Common Name: Remantadine hydrochloride; Rimantadine hydrochloride
Structural Formula:
Chemical Abstracts Registry No.: 1501-84-4; 13392-28-4 (Base)
Raw Materials
1-Adamantylmethylketone Acetamide Formic acid
2982 Rimantadine hydrochloride
Trade Name Manufacturer Country Year IntroducedFlumadine Forest Pharmaceuticals, Inc. - -Rimantadine hydrochloride
Tai Yuan Pharmaceutical Factory
China -
Manufacturing Process
A mixture of 3 g of 1-adamantylmethylketone, 4 g of acetamide and 1.35 mlof 86% formic acid was boiled for 3.5 hours. Then the mixture was cooled,deluted with water and extracted with ether. The ether solution was dried overalkali, filtered and saturated with dry hydrogen chloride. The resultingprecipitate was filtered off and boiled with 30 ml of concentrated hydrochloricacid for 5 hours. The reaction mixture was cooled, and the resultingprecipitate was filtered off. After recrystalliszation from a mixture of absoluteethanol and ether 2.32 g of the desired product was obtained (64% tostarting ketone). MP 345°-347°C.
Rimantadine may be also prepared by refluxing of the 1-adamantylmethylketone and ammonium formate in diethylene glycol or byhydrogenization of 1-adamantylmethyl ketoxime in the presence of a platinumon carbon catalyst at a room temperature and pressure.
References
Polis Y. et al.; US Patent No. 3,852,352; Dec. 3, 1974 Liu J.; US Patent No. 4,551,552; Nov. 5, 1985; Assigned to E.I. Du Pont de
Nemours and Company, Wilmington, Del.
RIMITEROL
Therapeutic Function: Bronchodilator
Chemical Name: 4-(Hydroxy-2-piperidinylmethyl)-1,2-benzenediol
Common Name: Erythro-3,4-dihydroxyphenyl-2-piperidinylcarbinol
Structural Formula:
Chemical Abstracts Registry No.: 32953-89-2; 31842-61-2 (Hydrogen bromide)
To a stirred suspension of 5.0 grams (0.21 gram atom) of magnesiumturnings in 15 ml of tetrahydrofuran under nitrogen is added 43.4 grams (0.2mol) of 4-bromoveratrole to maintain constant reflux. An additional 40 ml ofsolvent is added and the Grignard reagent thus prepared is heated on a steambath for one hour. This solution is then added dropwise to a solution of 20.8grams (0.2 mol) of 2-cyanopyridine in 300 ml of ether. The mixture is stirredovernight at room temperature, decomposed by addition of 250 ml of 10%hydrochloric acid and the separated aqueous layer is made alkaline with 40%sodium hydroxide solution. This mixture is extracted with methylene chlorideand the dried extract concentrated. The residue is distilled and the fraction at190° to 235°C/12 mm is crystallized to give 3,4-dimethoxyphenyl-2-pyridylketone, MP 93° to 94°C.
A solution of 0.5 gram of the above ketone in 15 ml of 48% hydrobromic acidis refluxed for 1.5 hours and then concentrated in vacuo. The residue isdissolved in ethanol, toluene is added, the solution concentrated and theresidue stripped with toluene to yield 3,4-dihydroxyphenyl-2-pyridyl ketonehydrobromide, MP 246° to 247°C (decomposition).
A mixture of 0.5 gram of platinum oxide and a solution of 2.0 grams (0.0067mol) of 3,4-dihydroxyphenyl-2-pyridyl ketone hydrobromide in 20 ml of waterand 80 ml of ethanol is hydrogenated on the Parr apparatus using an initialhydrogen pressure of 50 psi at room temperature. The reaction mixture isfiltered, the filtrate concentrated in vacuo and the residue triturated withacetone to give erythro-3,4-dihydroxyphenyl-2-piperidinylcarbinolhydrobromide, MP 210° to 211°C (decomposition).
Treatment of the above hydrobromide with aqueous sodium bicarbonatefollowed by extraction with ethyl acetate yields the free base of the carbinolMP 203° to 204°C which may be reacted with other acids to give other acidaddition salts.
References
Merck Index 8117 Kleeman and Engel p. 809 OCDS Vol. 2 p. 278 (1980) DOT 10 (11) 272 (1974) I.N. p. 849 Kaiser, C. and Ross, S.T.; US Patent 3,705,169; December 5, 1972; assigned
to Smith Kline and French Laboratories
2984 Rimiterol
RISEDRONATE SODIUM
Therapeutic Function: Bone calcium regulator
Chemical Name: Phosphonic acid, (1-hydroxy-2-(3-pyridinyl)ethylidene)bis-, monosodium salt
Common Name: Risedronate sodium
Structural Formula:
Chemical Abstracts Registry No.: 115436-72-1; 105462-24-6 (Base)
A 3-neck round-bottom flask fitted with a reflux condenser and a magnetic stirbar is charged with 6.94 grams (0.04 mole) 3-pyridine acetic acid 9.84 grams(0.14 mole) phosphorus acid, and 150 ml of chlorobenzene. This reactionmixture is heated on a boiling water bath, and 16.5 grams (0.12 mole)phosphorus trichloride is added dropwise with stirring. This reaction mixture isheated for 2 1/2 hours during which time a viscous yellow oil forms. Thereaction mixture is then cooled in an ice bath and the chlorobenzene solutionis decanted off from the solidified product. The reaction flask containing thissolidified product is charged with 150 ml of water and heated in a boilingwater bath for several hours. The hot solution is then filtered through Celite545 300 ml of methanol is added to the warm filtrate solution, and aprecipitate develops. After cooling in ice for 1 hour, the precipitate is filteredoff and then washed with methanol/water (1/1 volume/volume), methanol,and ether, and air dried. The product may be recrystallized from hot water.Yield is approximately 5.9 grams (52%). The sample is characterized by P-31and C-13 NMR. It may be converted into the sodium salt with equivalent ofNaOH.
Risedronate sodium 2985
Trade Name Manufacturer Country Year Introduced
Actonel Aventis - -
Actonel Procter and Gamble USA -
References
Benedict et al.; US Patent No. 5,583,122; Dec. 10, 1996; Assigned to The Procter and Gamble Company, Cincinnati, Ohio
RISPERIDONE
Therapeutic Function: Antipsychotic, Neuroleptic
Chemical Name: 4H-Pyrido(1,2-a)pyrimidin-4-one, 6,7,8,9-tetrahydro-3-(2-(4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl)ethyl)-2-methyl-
To a stirred mixture of 65 parts of 1,3-difluorobenzene, 130 parts ofaluminium chloride and 195 parts of dichloromethane was added dropwise asolution of 95 parts of 1-acetyl-4-piperidine-carbonyl chloride in 65 parts ofdichloromethane while cooling. Upon completion, stirring was continued for 3
Trade Name Manufacturer Country Year Introduced
Rasin Pfizer - -
Respidon Torrent Pharmaceuticals Ltd. India -
Risperdal Janssen-Ortho Inc. - -
Risperidone Janssen-Cilag - -
Rispolept Janssen-Cilag SpA - -
2986 Risperidone
hours at room temperature. The reaction mixture was poured into a mixtureof crushed ice and hydrochloric acid. The product 1-acetyl-4-(2,4-difluorobenzoyl)piperidine as a residue. A mixture of 48 parts of 1-acetyl-4-(2,4-difluorobenzoyl)-piperidine and 180 parts of a hydrochloric acid solution 6N was stirred and refluxed for 5 hours. The reaction mixture was evaporatedand the residue was stirred in 2-propanol. The product was filtered off anddried, yielding 39 parts (83%) of (2,4-di-fluorophenyl)(4-piperidinyl)methanone hydrochloride. A mixture of 12 parts of above product,12 parts of hydroxylamine hydrochloride and 120 parts of ethanol was stirredat room temperature and 10.5 parts of N,N-diethylethanamine were added.The whole was stirred and 25 refluxed for 3 hours. After cooling, theprecipitated product was filtered off and dried, yielding 11 parts (100%) of(2,4-di-fluorophenyl)(4-piperidinyl)methanone oxime.
A mixture of 11 parts of (2,4-difluorophenyl)(4-piperidinyl)-methanone oxime,25 parts of potassium hydroxide and 25 parts of 30 water was stirred andrefluxed for 2 hours. The reaction mixture was cooled and extracted withmethylbenzene. The extract was dried, filtered and evaporated. The residuewas crystallized from petroleum ether, yielding 6;8 parts of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole.
A mixture of 5.3 parts of 3-(2-chloroethyl)-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one monohydrochloride, 4.4 parts of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole, 8 parts of sodium carbonate, 0.1 parts ofpotassium iodide and 90 parts of N,N- dimethylformamide was stirredovernight at 85°-90°C. After cooling the reaction mixture was poured intowater. The product was filtered off and crystallized from a mixture of N,N-dimethylformamide and 2-propanol. The product was filtered off and dried,yielding 3.8 parts (46%) of 3-[2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl] ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]-pyrimidin- 4-one: melting point 170°C.
References
Kennis et al.; European Patent Office No. 0 196 132 A2, 13.03.86
RITODRINE
Therapeutic Function: Muscle relaxant (obstetric)
Chemical Name: erythro-p-Hydroxy-α-[1-[(p-hydroxyphenethyl)amino]ethyl]benzyl alcohol
Common Name: N-(p-Hydroxyphenylethyl)-4-hydroxynorephedrine
Chemical Abstracts Registry No.: 26652-09-5; 23239-51-2 (Hydrochloride salt)
A solution of 44 grams of 2-bromo-4'-benzyloxypropiophenone and 44 gramsof 2-(4-methoxyphenyl)ethylamine in 270 ml of ethanol was refluxed for 3hours. Then the ethanol was distilled off in vacuo and the concentrate mixedwith ether. The resulting crystallizate was sucked off after which the filtratewas mixed with an excess of 2 N hydrochloric acid. As a result of this thehydrochloride of 4'-benzyloxy-2-[2-(4-methoxyphenyl)ethylamino]-propiophenone slowly crystallized. This substance was also sucked off, washedwith water and alcohol, and dried in vacuo. After recrystallization from dilutealcohol the yield was 25.5 grams of a product with a melting point of 217° to218°C.
12 grams of the product thus obtained were dissolved in a mixture of 300 mlof ethanol and 90 ml of water. After 42 ml of 1% palladium chloride solutionand 3.9 grams of Norit had been added to this solution it was hydrogenated atroom temperature and at a pressure of 1.1 atmospheres until approximately760 ml of hydrogen had been taken up. Then the catalyst was removed byfiltration and the solvent of the filtered solution was evaporated entirely invacuo.
The resulting residue, which consisted of the hydrochloride of 4'-hydroxy-2-[2-(4-methoxyphenyl)ethylamino]propiophenone, was mixed with 30 ml of a48% hydrobromic acid solution and the mixture was boiled until nomethylbromide developed any more, which was the case after approximately
Trade Name Manufacturer Country Year Introduced Pre-Par Duphar Italy 1975 Yutopar Duphar UK 1976 Pre-Par Duphar France 1976 Pre-Par Duphar/Thomae W. Germany 1976 Yutopar Merrell Dow US 1980 Yutopar Astra US 1980 Miolene Lusofarmaco Japan - Utopar Ferrosan Denmark -
2988 Ritodrine
45 minutes. Then the reaction mixture was stored in the refrigerator, afterwhich the hydrobromide of 4'-hydroxy-2-[2-(4-hydroxyphenyl)ethylamino]propiophenone crystallized. It was sucked off andconverted into the hydrochloride by again dissolving the resulting substance inwater, discoloring the solution with a little Norit and then adding an equalvolume of concentrated hydrochloric acid. As a result of this the hydrochloridecrystallized. The yield was 9.6 grams of a product with a melting point of 136°to 138°C. After this product had been recrystallized once again it was reducedto the amino alcohol.
For this purpose a solution of 3.2 grams of the hydrochloride in 160 ml ofdistilled water was provided with 0.5 gram of Norit and 8 ml of 1% palladiumchloride solution and the mixture was hydrogenated at room temperature andat a pressure of 1.1 atmospheres until no hydrogen was taken up any more.The catalyst was then removed by filtration, after which the filtrate wasconcentrated in vacuo. To the concentrated solution of the reduced productwas then added an excess of dilute ammonia, as a result of which the base ofthe 1-(4-hydroxyphenyl)-2-[2-(4-hydroxyphenyl)ethlamino] propanolprecipitated as a tough mass. After the mixture had been stored in therefrigerator for some time, the product was sucked off, washed with water anddried in vacuo. This base was a resinous mass with a melting point ofapproximately 88° to 90°C. Yield was 2.3 grams.
References
Merck Index 8121 Kleeman and Engei p. 810 PDR p. 609 OCDS Vol. 2 p. 39 (1980) DOT 10 (1) 23 (1974) I.N. p. 850 Claassen, V., Van Dijk, J. and Moed, H.D.; US Patent 3,410,944; November
12, 1968; assigned to North American Philips Company, Inc.
RITONAVIR
Therapeutic Function: Antiviral
Chemical Name: 2,4,7,12-Tetraazatridecan-13-oic acid, 10-hydroxy-2-methyl-5-(1-methylethyl)-1-(2-(1-methylethyl)-4-thiazolyl)-3,6-dioxo-8,11-bis(phenylmethyl)-, 5-thiazolylmethyl ester, (5S-(5R*,8R*,10R*,11R*))-
Common Name: Ritonavir
Chemical Abstracts Registry No.: 155213-67-5
Ritonavir 2989
Trade Name Manufacturer Country Year Introduced Norvir Abbott Laboratories USA - Ritomune Cipla Limited India - Ritonavir Abbott Laboratories USA -
The synthesis of (2S,3S,5S)-5-(N-(N-((N-Methyl-N-((2-isopropyl-4-thiazolyl)methyl)amino)carbonyl)valinyl)amino)-2-(N-((5-thiazolyl)methoxycarbonyl)amino)-1,6-diphenyl- 3-hydroxyhexane consists of 20 steps.
1. N-((Benzyl)oxy)carbonyl)-L-phenylalaninal. It was prepared from 50 g(0.175 mol) of N-(((benzyl)oxy)-carbonyl-L-phenylalaninol in 200 ml ofdichloromethane under N2 atmosphere at -60°C by treating with 131 ml of a 2M solution of oxalyl chloride in dichloromethane, a solution of 24.5 ml ofanhydrous dimethyl sulfoxide in 870 ml of anhydrous dichloromethane forabout 2 hours. The resulting solution was stirred at -60°C for 1 h, thentreated over a period of 15 min with 97 ml of triethylamine in order that theinternal temperature remained below -50°C. After addition the solution wasstirred at -60°C for 15 min, then, with the cooling bath in place, was treatedrapidly with a solution of 163 g of citric acid in 550 ml of water. The resultingslurry was stirred vigorously for 10 min, allowed to warm, diluted to 1 literwith water, and separated. The organic layer was washed with 700 ml ofwater followed by a mixture of 550 ml of water and 150 ml of saturatedaqueous NaHCO3, dried over MgSO4, and concentrated in vacuo at 20°C togive the crude desired compound as a light yellow solid.
2990 Ritonavir
2. (2S,3R,4R,5S)-2,5-Bis-(N-(((benzyl)oxy)carbonyl)amino)-3,4-dihydroxy-1,6-diphenylhexane and (2S,3S,4S,5S)-2,5-bis-(N-(((benzyl)oxy)carbonyl)amino)-3,4-dihydroxy-1,6-diphenylhexane.
A suspension of 78.5 g of VCl3 (tetrahydrofuran)3and 16 g of zinc dust in 400ml of dry dichloromethane was stirred under N2 atmosphere for 1 h at 25°C.A solution of 0.175 mol of N-(((benzyl)oxy)carbonyl)-L-phenylalaninal in 200ml of dichloromethane was then added and the resulting mixture was stirredat ambient temperature under N2 for 16 h. The resulting mixture was addedto 500 ml of 1 M aqueous HCl, diluted with 500 ml of hot chloroform, andshaked vigorously for 2 min. The layers were separated, and the organic layerwas washed with 1 M aqueous HCl and separated. Filtration of the organicphase provided the crude desired product as a solid residue. The residue wasslurried in 1.25 liters of acetone, treated with 5 ml of concentrated H2SO4,and stirred for 16 h at ambient temperature. The resulting mixture wasfiltered, and the residue (residue A) was washed with 50 ml of acetone. Thecombined filtrate was concentrated to a volume of 250 ml, diluted with 1000ml of dichloromethane, washed three times with water and once withsaturated brine, dried over MgSO4 and concentrated to give a viscous oil. Theoil was taken up in 1000 ml of 1 M HCl in methanol (prepared from 71 ml ofacetyl chloride and 1000 ml of methanol) and stirred at ambient temperaturefor 2 h. The resulting precipitate was filtered, washed with methanol, and air-dried on the filter to provide 26.7 g of the desired compound as a white solid.The filtrate was concentrated and filtered to give a second crop (8.3 g) of(2S,3R,4R,5S)-2,5-bis-(N-(((benzyl)oxy)carbonyl)amino)-3,4-dihydroxy-1,6-diphenylhexane.
Residue A (above, 2.65 g) was suspended in 75 ml of tetrahydrofuran (THF)and 75 ml of 1 M aqueous HCl and refluxed for 24 h. After concentration ofthe resulting solution in vacuo, the residue was taken up in 10% methanol inchloroform, washed two times with water, dried over Na2SO4 and concentratedin vacuo to provide (2S,3S,4S,5S)-2,5-bis-(N-(((benzyl)oxy)carbonyl)amino)-3,4-dihydroxy-1,6-diphenylhexane as a white solid
A suspension of 25 g (44 mmol) of the compound prepared in the step 2 in500 ml of 2:1 dichloromethane/hexane was treated with 23 g of a-acetoxyisobutyryl bromide. The resulting mixture was stirred at ambienttemperature until the reaction clarified, washed twice with 200 ml portions ofsaturated NaHCO3, dried over MgSO4, and concentrated in vacuo to give 30.8g of the crude desired compound. A portion was purified by silica gelchromatography using 9:1 dichloromethane:ethyl acetate to provide the puredesired compound as a white solid.
A solution of 35.56 g (52.8 mmol) of the above compound (step 3) in 375 mlof dioxane was treated with 255 ml of 1 N aqueous sodium hydroxide andstirred at ambient temperature for 16 h, during which the desired compoundprecipitated. The resulting mixture was filtered, and the residue was washed
Ritonavir 2991
with water and dried to provide 22.23 g (76%) of the desired compound as awhite solid.
A mixture of 39.2 g (71.2 mmol) of (2S,3R,4R,5S)-2,5-bis-(N-(((benzyl)oxy)carbonyl)amino)-3,4-epoxy-1,6-diphenylhexane in 600 ml ofTHF was treated under N2 with 13 g (0.36 mol) of sodium borohydride. Theresulting mixture was treated dropwise with 27.7 ml (0.36 mol) oftrifluoroacetic acid. After being stirred for 3.5 h at ambient temperature, theresulting mixture was quenched with 1 N aqueous HCl, diluted with water, andstirred for 16 h. The resulting mixture was filtered, washed with water, anddried to provide 22.85 g (58%) of the hydroxide octahydrate in 400 ml of 1,4-dioxane and 400 ml desired compound as a white solid.
A suspension of 32 g of the crude resultant compound of step 5 and 55.5 g(176 mmol) of barium hydroxide octahydrate in 400 ml of water was refluxedfor 4 h. The resulting mixture was filtered, and the residue was rinsed withdioxane. The combined filtrates were concentrated to a volume ofapproximately 200 ml and extracted with 4 x 400 ml portions of chloroform.The combined organic layers were dried over Na2SO4, filtered, andconcentrated in vacuo. The residue was purified by silica gel chromatographyusing first 2% isopropylamine in chloroform and then 2% isopropylamine/2%methanol in chloroform to provide 10.1 g (81%) of the pure desiredcompound as a white solid.
A solution of 11.28 g (40 mmol) of (2S,3S,5S)-2,5-diamino-1,6-diphenyl-3-hydroxyhexane and 4.88 g (40 mmol) of phenylboric acid in 1 liter of toluenewas refluxed and the water azeotropically removed with the aid of a DeanStark trap until the distillate was clear. The solvent was then removed invacuo to provide the crude desired compound which was used immediatelywithout further purification.
8. Thioformamide.
To a cooled (0°C) solution of formamide (30.5 mL, 0.76 mol) in 1 L of diethylether was added 89 g (0.19 mol) of phosphorous pentasulfide in smallportions with stirring. The reaction mixture was allowed to warm to ambienttemperature, stirred for 2 h, filtered, and concentrated in vacuo to affordthioformamide as a yellow offensive smelling oil which was used withoutpurification.
9. Ethyl 2-chloro-2-formylacetate.
To 0.5 mol of potassium t-butoxide (500 mL of a 1 M solution in THF) and 500mL of dry THF cooled to 0°C was added dropwise a solution of ethylchloroacetate (0.5 mol, 53.5 mL) and ethyl formate (0.5 mol, 40.4 mL), in
2992 Ritonavir
200 mL of THF over 3 hours. After completion of addition, the reactionmixture was stirred for 1 hour and allowed to stand overnight. The resultingsolid was diluted with diethyl ether and cooled in an ice bath. Then, the pHwas lowered to approximately 3 using 6 N HCl. The organic phase wasseparated, and the aqueous layer was washed 3 times with diethyl ether. Thecombined ethereal portions were dried over Na2SO4 and concentrated invacuo. The crude desired compound was stored at -30°C and used withoutfurther purification.
10. Ethyl thiazole-5-carboxylate.
250 mL of dry acetone, 7.5 g (0.123 mol) of thioformamide, and 18.54 g(0.123 mol) of ethyl 2-chloro-2-formylacetate were refluxed for 2 hours. Thesolvent was removed in vacuo, and the residue was purified bychromatography (SiO2, 6 cm o.d. column, 100% CHCl3, Rf = 0.25) to provide11.6 g (60%) of the desired compound as a light yellow oil.
11. 5-(Hydroxymethyl)thiazole.
To a precooled (ice bath) lithium aluminum hydride (76 mmol) in 250 mL ofTHF was added ethyl thiazole-5-carboxylate (11.82 g, 75.68 mmol) in 100 mLof THF dropwise over 1.5 hours to avoid excess foaming. The reaction wasstirred for an additional hour, and treated cautiously with 2.9 mL of water, 2.9mL of 15% NaOH, and 8.7 mL of water. The solid salts were filtered, and thefiltrate set aside. The crude salts were refluxed in 100 mL of ethyl acetate for30 min. The resulting mixture was filtered, and the two filtrates werecombined, dried over Na2SO4 and concentrated in vacuo. The product waspurified by silica gel chromatography eluting sequentially with 0%-2%-4%methanol in chloroform, to provide the desired compound, Rf = 0.3 (4%methanol in chloroform), which solidified upon standing in 75% yield.
A solution of 3.11 g (27 mmol) of 5-(hydroxymethyl)thiazole and excess N-methyl morpholine in 100 ml of methylene chloride was cooled to 0°C andtreated with 8.2 g (41 mmol) of 4-nitrophenyl chloroformate. After beingstirred for 1 h, the reaction mixture was diluted with CHCl3, washed with 1 NHCl, saturated aqueous NaHCO3, and saturated brine, dried over NaSO4 andconcentrated in vacuo. The residue was purified by silica gel chromatography(SiO2, 1-2% MeOH/CHCl3, Rf = 0.5 in 4% MeOH/CHCl3) to yield 5.9 g (78%)of the desired compound as a yellow solid.
A solution of 500 mg (1.76 mmol) of (2S,3S,5S)-2,5-diamino-1,6-diphenyl-3-hydroxyhexane and 480 mg (1.71 mmol) of ((5-thiazolyl)methyl)-(4-nitrophenyl)carbonate in 20 ml of THF was stirred at ambient temperature for4 hours. After removal of the solvent in vacuo, the residue was purified bysilica gel chromatography using first 2% then 5% methanol in chloroform toprovide a mixture of the two desired compounds. Silica gel chromatography of
Ritonavir 2993
the mixture using a gradient of 0-1-2% methanol in 93:2 isopropylamine:chloroform provided 110 mg (16%) of (2S,3S,5S)-5-amino-2-(N-((5-thiazolyl)-methoxycarbonyl)amino)-1,6-diphenyl-3-hydroxyhexane (Rsubfsub0.48, 96:2:2 chloroform:methanol:isopropylamine) and 185 mg (28%) of(2S,3S,5S)-2-amino-5-(N-((5-thiazolyl)methoxycarbonyl)amino)-1,6-diphenyl-3-hydroxyhexane (Rf 0.44, 96:2:2 chloroform:methanol:isopropylamine).
A solution of 40 mmol of crude (4S,6S,1'S)-6-(1-amino-2-phenylethyl)-4-benzyl-2-phenyl-3-aza-2-bora-1-oxa cyclohexane in 700 ml of anhydrous THFwas cooled to -40°C and treated dropwise for 1 h with a solution of 7.83 g(27.9 mmol) of ((5-thiazolyl)methyl)-(4-nitrophenyl)carbonate in 300 ml ofdry THF. The resulting solution was allowed to warm to 0°C for 3 h, then toambient temperature for 16 h. The solvent was removed in vacuo, and theresidue was taken up in 700 ml of ethyl acetate, washed with 3 x 150 ml of 1N NaOH and one 150 ml of brine. The organic phase was dried over Na2SO4
and concentrated in vacuo. Purification of the residue by silica gelchromatography using methanol/chloroform mixtures provided the desiredcompound mixed with its regioisomer. A second chromatography using 1-3%isopropylamine in chloroform provided 5.21 g of the desired compound whichsolidified upon standing.
14. 2-Methylpropane-thioamide.
A suspension of 100 g (1.15 mol) of isobutyramide in 4 L of diethyl ether wasstirred vigorously and treated in portions with 51 g (0.115 mol) of P4S 10.The resulting mixture was stirred at ambient temperature for 2 h, filtered, andconcentrated in vacuo to provide 94.2 g (80%) of the crude desiredcompound.
A mixture of 94.0 g (0.91 mol) of 2-methylpropane-thioamide, 115.7 g (0.91mol) of 1,3-dichloroacetone, and 109.7 g (0.91 mol) of MgSO4 in 1.6 liters ofacetone was refluxed for 3.5 h. The resulting mixture was allowed to cool,filtered, and the solvent was removed in vacuo to provide the crude desiredcompound as a yellow oil.
A solution of 40 g of 4-(chloromethyl)-2-isopropylthiazole hydrochloride in 100ml of water was added dropwise with stirring to 400 ml of 40% aqueousmethylamine. The resulting solution was stirred for 1 h, then concentrated invacuo. The residue was taken up in chloroform, dried over Na2SO4, andconcentrated in vacuo. Purification of the residue by silica gel chromatographyusing 10% methanol in chloroform provided 21.35 g (55%) of the desiredcompound.
A solution of 66.1 g (0.328 mol) of 4-nitrophenyl chloroformate in 1.2 liters of
2994 Ritonavir
CH2Cl2 was cooled to 0°C and treated with L-valine methyl esterhydrochloride. The resulting mixture was treated slowly, with stirring, with68.9 ml (0.626 mol) of 4-methylmorpholine. The resulting solution wasallowed to slowly warm to ambient temperature and was stirred overnight.After washing with 3 portions of 10% NaHCO3, the solution was dried overNa2SO4 and concentrated in vacuo. The residue was purified by silica gelchromatography by eluting with chloroform to provide the desired compound.
A solution of 15.7 g (92 mmol) of 2-isopropyl-4-(((N-methyl)amino)-methyl)thiazole in 200 ml of THF was combined with a solution of 20.5 g (69mmol) of N-(((4-nitrophenyl)oxy)carbonyl)-L-valine methyl ester. The resultingsolution was treated with 1.6 g of 4-dimethylaminopyridine and 12.9 ml (92mmol) of triethylamine, heated at reflux for 2 h, allowed to cool, andconcentrated in vacuo. The residue was taken up in CH2Cl2, washedextensively with 5% aqueous K2CO3, dried over Na2SO4, and concentrated invacuo. The resulting product mixture was purified by silica gelchromatography using chloroform as an eluent to provide 16.3 g (54%) of thedesired compound.
A solution of 1.42 g (4.3 mmol) of the resultant compound of step 18 in 17 mlof dioxane was treated with 17.3 ml of 0.50 M aqueous LiOH. The resultingsolution was stirred at ambient temperature for 30 min, treated with 8.7 ml of1 M HCl, and concentrated in vacuo. The residue was taken up indichloromethane, washed with water, dried over Na2SO4, and concentrated invacuo to provide 1.1 g (81%) of the desired compound.
A solution of 70 mg (0.223 mmol) of N-((N-methyl-N-((2-isopropyl-4-thiazolyl)methyl)amino)carbonyl)-L-valine, 79 mg (0.186 mmol) of(2S,3S,5S)-5-amino-2-(N-((5-thiazolyl)methoxycarbonyl)amino)-1,6-diphenyl-3-hydroxyhexane, 30 mg (0.223 mmol) of 1-hydroxybenzotriazole hydrate,and 51 mg (0.266 mmol) of N-ethyl-N'-dimethylaminopropyl carbodiimide in 2ml of THF was stirred at ambient temperature for 16 h. The resulting solutionwas concentrated in vacuo, and the residue was purified by silica gelchromatography using 97:3 CH2Cl2/CH3OH to provide 100 mg (74%) of thedesired compound (Rf 0.4, 95:5 CH2Cl2:CH3OH) as a solid, melting point 61°-63°C.
The structure of the described compounds was confirmed by NMR and massspectrum analysis.
2,4,7,12-Tetraazatridecan-13-oic acid, 10-hydroxy-2-methyl-5-(1-methylethyl)-1-(2-(1-methylethyl)-4-thiazolyl)-3,6-dioxo-8,11-bis(phenylmethyl)-, 5-thiazolylmethyl ester, (5S-(5R*,8R*,10R*,11R*))- and
Ritonavir 2995
(2S,3S,5S)-5-(N-(N-((N-methyl-N-((2-isopropyl-4-thiazolyl)methyl)amino)carbonyl)valinyl)amino)-2-(N-((5-thiazolyl)methoxycarbonyl)amino)-1,6-diphenyl-3-hydroxyhexane have thesame structural formula.
References
Al-Razzak et al.; US Patent No. 5,484,801; Jan. 16, 1996; Assigned to Abbott Laboratories, Abbott Park, III
RIZATRIPTAN BENZOATE
Therapeutic Function: Serotoninergic
Chemical Name: 1H-Indole-3-ethanamine, N,N-dimethyl-5-(1H-1,2,4-triazol-1-ylmethyl)-, monobenzoate
Common Name: Rizatriptan benzoate
Structural Formula:
Chemical Abstracts Registry No.: 145202-66-0
Raw Materials
1,2,4-Triazole sodium salt Palladium on carbon Tin(II) chloride dihydrate 4-Hydrazine Formaldehyde Sodium cyanoborohydride Benzoic acid
2996 Rizatriptan benzoate
Trade Name Manufacturer Country Year Introduced
Maxalt Merck Sharp and Dohme UK -
Maxalt-MLT Merck Sharp and Dohme UK -
Manufacturing Process
1. 1-(4-Nitrophenyl)methyl-1,2,4-triazole
4-Nitrobenzylbromide (21.6 g, 0.1 mol) was added to a rapidly stirredsuspension of 1,2,4-triazole sodium salt (9.1 g, 0.1 mol) in anhydrous DMF(100 ml) and the mixture stirred at room temperature for 16 h. Ethyl acetate(400 ml) was added followed by water (250 ml) and the layers separated. Theorganic phase was washed with water (3 x 250 ml), dried (MgSO4) andevaporated. The residue was chromatographed on silica gel eluting with ethylacetate to give the title-product (10.6 g, 52%); m.p. 98°-100°C.
A solution of 1-(4-nitrophenyl)methyl-1,2,4-triazole (10.0 g, 49 mmol) inethanol (50 ml), ethyl acetate (50 ml), 5 N HCl (10 ml) and water (10 ml)was hydrogenated over 10% Pd/C (1.0 g) at 40 p.s.i., in a Parr apparatus,until an uptake of 188 p.s.i., had been observed (approx 10 mins). Thecatalyst was removed by filtration and the solvent removed under vacuum.The residue was azeotroped with ethanol (2 times) to give the titleaminehydrochloride (10.6 g, 100%).
3. 1-(4-Hydrazinophenyl)methyl-1,2,4-triazole
A solution of sodium nitrite (3.28 g, 48 mmol) in water (20 ml) was added toa solution of the preceding amine hydrochloride (10.0 g, 48 mmol), inconcentrated HCl (40 ml), at such a rate that the temperature did not exceed-10°C. After addition was complete the solution was stirred at 0°C for 0.25 hand then added portionwise to a rapidly stirred solution of SnCl2 x 2H2O (40g) in concentrated HCl (40 ml). The solution was warmed to roomtemperature and basified with 20% aqueous NaOH solution. The solution wasextracted with ethyl acetate (3 x 250 ml) and the combined extracts dried(MgSO4) and filtered. The solution was evaporated to dryness to give thedesired hydrazine (5.0 g, 56%) m.p. 109°-112°C.
4-Chlorobutanal dimethylacetal (3.22 g, 21.1 mmol) was added to a stirredsolution of the preceding hydrazine (5.0 g, 26.4 mmol) in ethanol/water (5:1,180 ml) and 5 N HCl (4.5 ml) and the solution refluxed for 4 h. The solventswere removed under vacuum and the residue chromatographed on silica gel,eluting with CH2Cl2/EtOH/NH3 (30:8:1) to give the desired tryptamine (2.4 g,38%).
A solution of formaldehyde (37% w/w solution, 0.19 g), in methanol (10 ml),was added to a mixture of the preceding tryptamine (0.36 g, 1.5 mmol),NaCNBH3 (0.225 g, 3.6 mmol) and glacial acetic acid (0.45 g), in methanol(10 ml). The mixture was stirred at room temperature for 2 h before addingsaturated K2CO3 (50 ml) and evaporating the methanol. The residue was
Rizatriptan benzoate 2997
extracted with ethyl acetate (3 x 100 ml) and the combined extracts washedwith brine (100 ml), dried (K2CO3), and evaporated. The crude product waschromatographed on silica gel eluting with CH2Cl2/EtOH/NH3 (20:8:1) to givethe free base of the title-compound (0.21 g, 52%).The benzoate salt of N,N-dimethyl-2-[5-(1,2,4-triazol-1-yl-methyl)-1H-indol-3-yl]ethylamine wasprepared by addition of a solution of benzoic acid in diethyl ether to a solutionof the free base in ethanol/diethyl ether (1:4). The precipitated salt wasrecrystallised from ethanol, mp 178°-180°C.
References
Baker et al.; US Patent No. 5,298,520; Mar. 29, 1994; Assigned to Merck Sharp and Dohme Limlted, Hertfordshire, England
ROCIVERINE
Therapeutic Function: Spasmolytic
Chemical Name: 1-(Diethylamino)-2-propyl-cis-2-hydroxy-2-cyclohexyicyclohexane-1-carboxylate
5.6 g of 2-phenyl-2-hydroxy-cyclohexane-carboxylic acid were dissolved in 75cc of glacial acetic acid and reduced in the presence of 0.1 g of platinum oxideunder hydrogen pressure of 22 kg/cm2 at a temperature of 70°C to 80°C.
Hydrogen absorption being completed, the solution was filtered andevaporated to one-fifth of its volume and cooled in a refrigerator. Theprecipitate was filtered, washed with water, and then crystallized from ligroin,thus yielding 4 g of 2-cyclohexyl-2-hydroxy-cyclohexane-carboxylic acid,melting point (Kofler) 122°C to 124°C. This material was esterified with 1-bromo-2-propanol by means of 85% H2SO4 yielding 1-bromoisopropyl-2-cyclohexyl-2-hydroxycyclohexanecarboxylate. Finally this compound wastreated with diethylamine and triethylamine at 120°C to give rociverine.
References
Merck Index 8125 DFU 4 (4) 276 (1979) I.N. p. 852 Turbanti, L; US Patents 3,700,675; and 3,700,775; both dated October 24,
1972
ROLITETRACYCLINE
Therapeutic Function: Antibacterial
Chemical Name: 4-(Dimethylamino)-1,4,4a,5,5a,6,11,12a-octahydro-3,6,10,12,12a-pentahydroxy-6-methyl-1,11-dioxo-N-(1-pyrrolidinyl-methyl)-2-naphthacenecarboxamide
1 g (0.00225 mol) of anhydrous tetracycline base, 0.101 g (0.0038 mol) ofparaformaldehyde and 0.302 g (0.0025 mol) pyrrolidine hydrochloride arerefluxed in 25 ml absolute ethanol. After two hours an additional 0.101 gparaformaldehyde is added and refluxing is continued for two more hours. Thesolution is then cooled and two drops of concentrated hydrochloric acid areadded. The product, N'-(1-pyrrolidyl-methyl)-tetracycline hydrochloride, formsand is isolated as a crystalline, antibacterially active solid differing in specificrotation from tetracycline hydrochloride. The product is converted to the freebase by solution in water followed by the addition of one equivalent of sodiumhydroxide. Thus for isolation, the alcoholic solution of N'-(1-pyrrolidyl-methyl)-tetracycline hydrochloride is diluted with 5.0 ml ether to precipitate theproduct, which is collected by filtration and dried in vacuo over P2O5. Theproduct is a crystalline solid melting at about 158°C to 165°C withdecomposition.
References
Merck Index 8127 Kleeman and Engel p. 810 OCDS Vol. 1 p. 216 (1977) I.N. p. 853 Cheney, L.C., Risser, W.C. and Gottstein, W.J.; US Patent 3,104,240;
September 17, 1963; assigned to Bristol-Myers Co.
Trade Name Manufacturer Country Year Introduced Syntetrin Bristol US 1959 Velacycline Squibb US 1960 Transcycline Hoechst France 1961 Anergomycil C.N.N. Italy - Bristacin Bristol Banyu Japan - Farmaciclina Selvi Italy - Hostacyclin-PRM Hoechst Japan - Kinteto Fujita Japan - Quadraciclina Squibb Italy - Reverin Hoechst Italy - Solvocillin Fabr. Antibiot. Rumania - Tetrafarmed Neopharmed Italy - Tetraldina Italsuisse Italy - Tetraverin Polfa Poland -
Chemical Name: 2H-Indol-2-one, 1,3-dihydro-4-(2-(dipropylamino)ethyl)-, monohydrochloride
Common Name: Ropinirole hydrochloride
Structural Formula:
Chemical Abstracts Registry No.: 91374-20-8; 91374-21-9 (Base)
Raw Materials
4-(2-Di-n-propylaminoethyl)-7-hydroxy-2(3H)-indolone hydrobromide 5-Chloro-1-phenyl-1H-tetrazole Phenyl tetrazole ether Palladium on carbon
Manufacturing Process
A mixture of 3.44 g (9.63 mmoles) of 4-(2-di-n-propylaminoethyl)-7-hydroxy-2(3H)-indolone hydrobromide (U. S. Pat. No. 4,314,944), 22 cc ofdimethylformamide, 1.79 g (9.91 mmoles) of 5-chloro-1-phenyl-1H-tetrazole,220 cc of acetone, 10 cc of water and 2.90 g (21 mmoles) of anhydrouspotassium carbonate was refluxed for about 3 hours at which time thin layerchromatographic analysis (silica gel GF, 75-23-2 ethyl acetate-methanol-conc.ammonium hydroxide) indicated that the reaction was complete.
After cooling the reaction mixture in an ice-bath, the inorganic salts wereremoved by filtration and washed with acetone. The combined filtrates wereconcentrated in vacuo. The residual syrup was diluted with saturated brineand extracted with three portions of diethyl ether. The gathered extracts weredried over anhydrous magnesium sulfate, clarified with charcoal and treatedwith ethereal hydrogen chloride until precipitation was complete. The solidwas slurried in diethyl ether and decanted several times, filtered and air-driedto give 3.8 g (86%) of tan 4-(2-di-n-propylaminoethyl)-7-(1-phenyl-1H-tetrazol-5-yloxy)-2(3H)-indolone hydrochloride. Recrystallization from 200 ccof hot acetonitrile gave 2.6 g (59%) of microcrystalline product, m.p. 245°C.
Ropinirole hydrochloride 3001
Trade Name Manufacturer Country Year Introduced
Requip GlaxoSmithKline USA -
Ropinirole Hydrochloride GlaxoSmithKline USA -
Evaporation of the mother liquor and recrystallization of the residue from 25cc of hot acetonitrile gave an additional 400 mg of product, m.p. 244°-245°C.
A mixture of 2.64 g (5.78 mmoles) of the phenyl tetrazole ether, 200 cc ofglacial acetic acid and 1.49 g of 10% palladium-on-carbon was hydrogenatedin a Parr apparatus at 50 p.s.i. for 20 hours at 50°C. The warm reactionmixture was filtered through glass fiber filterpaper and the catalyst washedthoroughly with hot glacial acetic acid. The filtrate was concentrated in vacuo,the pale yellow waxy residue distributed in water and ethyl acetate. Afteracidification of the aqueous phase with 3 N hydrochloric acid, the organicphase was separated and extracted once with 1 N hydrochloric acid. Thecombined aqueous phases were adjusted to pH 8.5 with aqueous 10% sodiumhydroxide and extracted with a mixture of ethyl acetate and diethyl ether. Thecombined organic extract was back-washed once with saturated brine, driedover anhydrous magnesium sulfate, clarified with charcoal, treated withethereal hydrogen chloride and evaporated to dryness in vacuo to give 1.64 g(96%) of pale yellow crystalline solid; 4-(2-di-n-propylaminoethyl)-2(3H)-indolone hydrochloride. Recrystallization from 260 cc of hot acetonitrile whichwas concentrated to about 50 cc gave 1.26 g (74%) of pale yellowmicrocrystalline powder, melting point 240°-242°C. The hydrochloride salt(500 mg) is shaken in the presence of ether/5% sodium carbonate solution.The ether layer is separated, dried and evaporated to give the free base whichis used to prepare other salt forms such as the methanesulfonate,ethanedisulfonate, sulfate or sulfamate by reacting aliquots of the base inether with an excess of each acid.
References
Gallagher, Jr.; US Patent No. 4,452,808; Jun. 5, 1984; Assigned to Smithkline Beckman Corporation, Philadelphia, Pa.
ROPIVACAINE HYDROCHLORIDEMONOHYDRATE
Therapeutic Function: Local anesthetic
Chemical Name: 2-Piperidinecarboxamide, N-(2,6-dimethylphenyl)-1-propyl-, monohydrochloride, (S)-, monohydrate
Common Name: Ropivacaine hydrochloride
Structural Formula:
3002 Ropivacaine hydrochloride monohydrate
Chemical Abstracts Registry No.: 132112-35-7; 84057-95-4 (Base)
The process for preparation of ropivacaine includes three steps.
Step 1, resolution
Pipecoloxylidide hydrochloride (1.0 kg), acetone (3.75 L), and water (0.85 L)were charged. NaOH (aq) was added to pH>11. The phases, thus formed,were separated and the organic phase was diluted with water (1.4 L). L-(-)-Dibenzoyltartaric acid (0.67 kg), dissolved in acetone (3.75 L), was added.The solution was seeded. The crystal slurry was cooled to 2°C. The crystalswere collected by centrifugation and were washed with acetone followed byisobutyl methyl ketone. The product was not dried. The moist crystallineproduct was extracted with isobutyl methyl ketone (3.60 L) and diluted NaOH(2.60 L) at pH>11. The phases were separated. The organic phase waswashed with water (0.6 L) and was used directly in the next step. Yield(calculated on the dry basis) about 0.39 kg of (S)-pipecoloxylidide(about.90%).
Step 2, alkylation and salt precipitation
K2CO3 (0.32 kg), NaI (catalytical amount), and 1-bromopropane (0.28 kg)and about 5% of water, were added to the organic phase from the previousstep. The mixture was heated to reflux to complete the reaction. The excessof bromopropane was removed by distillation. The reaction mixture wasextracted with water (1.70 L). Acetone (1.70 L) was added to the organicphase followed by HCl (aq) to pH about 2. The solution was seeded. Thecrystal slurry was cooled to 9°C. The crystals were collected by centrifugationand were washed with acetone. The product was used directly in the next stepand was not dried. Yield (calculated on the dry basis): 0.47 kg of ropivacainehydrochloride (about 0.90%).
As an alternative, the following procedure was followed:
K2CO3 (0.32 kg), NaI (catalytical amount), 1-bromopropane (0.28 kg) andwater (1.70 L) were added to the organic phase from the previous step. Themixture was heated to reflux to complete the reaction. The excess ofbromopropane was removed by distillation. The reaction mixture wasseparated. Acetone (1.70 L) was added to the organic phase followed by HCl(aq) to pH about 2. The solution was seeded. The crystal slurry was cooled to
Ropivacaine hydrochloride monohydrate 3003
Trade Name Manufacturer Country Year Introduced
Naropin AstraZeneca - -
Ropivacaine hydrochloride AstraZeneca Japan -
9°C. The crystals were collected by centrifugation and were washed withacetone. The product was used directly in the next step and was not dried.Yield (calc. on the dry basis): 0.47 kg of ropivacaine hydrochloride (about0.90%).
Step 3, recrystallisation
Ropivacaine hydrochloride, from the previous step, was slurried in acetone(1.0 L) at reflux temperature. Water (0.60 L) was added. The resultingmixture was filtered and acetone (7.6 L) was added at >40°C. The solutionwas seeded. The slurry of crystals was cooled to 3°C. The crystals werecollected by centrifugation and were washed with acetone. The crystals weredried at 30°-40°C in vacuum. Yield: about 0.42 kg of ropivacainehydrochloride monohydrate (about 80%).
The stucture of the end product was confirmed by NMR analysis.
References
Jaksch P.; US Patent No. 5,959,112; Sep. 28, 1999; Assigned to Astra Aktiebolag, Sodertalje, Sweden
To 100 g of 2-(N-methyl-N-(2-pyridyl)amino)ethanol in 500 ml DMF wasadded 100 g of 4-fluorobenzaldehyde. The reaction mixture was stirred for 10min at room temperature and 80 g of potassium tertiary butoxide was addedto the reaction mixture. The reaction was monitored by TLC. After completionof the reaction, the reaction mixture was cooled to 5-10°C and under the coldconditions, 1.5 L of water was added and stirred for 15 min. The mixture wasextracted with ethyl acetate. The combined organic layer was washed with 3times 1 L water. The organic layer was dried over anhydrous sodium sulfateand concentrated under reduced pressure to give 148 g (88%) of 4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy)benzaldehyde.
4-[2-(N-Methyl-N-(2-pyridyl)amino)ethoxy]benzaldehyde was converted intohighly crystalline 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione by Knoevenegal condensation with 2,4-thiazolidinedione. 800 g of 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione and 280 g maleic acid were dissolved in 1.3 L ofacetone in 5 L three necked round bottom flask. The reaction mixture washeated to 50-55°C and the solution was filtered and slowly cooled to obtain986 g of 5-[4-[2-(N-methyl-N-(2-pyridyl)amino)ethoxy]benzyl]thiazolidine-2,4-dione maleate, (yield 95%; M.P. 120-122°C).
References
Sharad Kumar Vyas; US Patent No. 6,515,132; Feb. 4, 2003; Assigned to Torrent Pharmactuticals, Ltd., Ahmedabad (IN)
To a stirred suspension containing 5.1 g of 57% sodium hydride dispersed inmineral oil and 150 ml of dimethylformamide was added in portions 32.6 g ofethyl 1,4-dihydro-4-oxo-7-(4-pyridyl)-3-quinolinecarboxylate [tautomeric withethyl 4-hydroxy-7-(4-pyridyl)-3-quinolinecarboxylate] followed by the additionof 18.7 g of ethyl iodide. The resulting reaction mixture was heated on asteam bath for three hours with stirring and then concentrated in vacuo toremove the solvent. The semisolid residue was shaken well with a mixture ofchloroform and water, and a small quantity of amorphous brown solid wasfiltered off. The layers were separated and the chloroform layer wasevaporated in vacuo to remove it.
To the oily residue containing ethyl 1-ethyl-1,4-dihydro-4-oxo-7-(4-pyridyl)-3-quinolinecarboxyiate was added excess 10% aqueous sodium hydroxidesolution and ethanol, and the solution was heated on a steam bath for forty-five minutes to hydrolyze the ethyl ester to the corresponding carboxylic acid.The alkaline solution was diluted to a volume of about 500 ml with water,decolorizing charcoal was added and the mixture filtered. The filtrate wasneutralized with acetic acid whereupon the carboxylic acid separated as asolid. The solid was collected and dried in a rotary evaporator. The solid wasboiled with ethanol, the solution chilled and the resulting solid collected. Thesolid was recrystallized from dimethylformamide (about 150 ml) usingdecolorizing charcoal. The filtrate was chilled, diluted with about one-halfvolume of ethanol and the separated crystalline product was collected,recrystallized again from dimethylformamide and dried in vacuo to yield 4.3 g1-ethyl-1,4-dihydro-4-oxo-7-(4-pyridyl)-3-quinolinecarboxylic acid, melting
3006 Rosoxacin
Trade Name Manufacturer Country Year Introduced Eradacin Sterling Winthrop UK 1981 Eracine Winthrop France 1981 Winuron Winthrop W. Germany 1981 Eradacil Winthrop Canada 1983 Winoxacin Winthrop Switz. 1983 Roxadyl Winthrop - -
point 272°C to 273°C raised by further recrystallization to 290°C.
4-(3-nitrophenyl)pyridine is reduced with iron in acetic acid to give 4-(3-aminophenyl)pyridine. That in turn is reacted with ethoxymethylene malonicacid diethyl ester and then thermally rearranged to give the starting material
References
Merck Index 8136 DFU 5 (4) 199 (1980) Kleeman and Engel p. 811 OCDS Vol. 3 p. 185 (1984) DOT 18 (3) 147 (1982) I.N. p. 855 Carabateas, P.M.; US Patent 3,922,278; November 25, 1975; assigned to
Sterling Drug, Inc. Lesher, G.Y. and Carabateas, P.M.; US Patents 3,753,993; August 21, 1973
and 3,907,808; September 23, 1975; both assigned to Sterling Drug, Inc.
Lorenz, R.R. and Thielking, W.H.; US Patent 4,107,167; August 15, 1978; assigned to Sterling Drug, Inc.