Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038www.elsevier.com/locate/yjmcc
Original article
Ranolazine decreases diastolic calcium accumulation caused byATX-II or ischemia in rat hearts
Heather Fraser a,⁎, Luiz Belardinelli a, Lianguo Wang b, Peter E. Light b,Jeffrey J. McVeigh a, Alexander S. Clanachan b
a CV Therapeutics Inc., 3172 Porter Drive, Palo Alto, CA 94304, USAb Department of Pharmacology, Cardiovascular Research Group, University of Alberta, Edmonton, Alberta, Canada
Received 27 June 2006; received in revised form 11 August 2006; accepted 18 August 2006Available online 5 October 2006
Abstract
Cardiac pathologies are associatedwith increased late INa that contributes to the dysregulation of ion homeostasis and causes electrical and contractiledysfunction. This studywas designed to test the hypothesis that an increased late sodium channel current (INa) leads to Ca
2+ overload and left ventricular(LV) dysfunction, and thereby inhibition of late INa (e.g., by ranolazine) improves Ca2+ homeostasis and reduces LV dysfunction. IntracellularCa2+ ([Ca2+]i) and LV function were measured simultaneously in rat isolated perfused hearts. Augmentation of late INa with sea anemone toxin-II(ATX-II, 12 nM) increased diastolic [Ca2+]i (d[Ca
2+]i), and impaired LV mechanical function, but had no effect on [Ca2+]i transient amplitude.Although ranolazine (4 and 9 μM), an inhibitor of late INa, had no direct effects on d[Ca2+]i or LV function, it significantly reduced the deleteriouseffects of ATX-II. Global ischemia increased d[Ca2+]i and inhibited Ca2+ transient amplitude. During reperfusion, Ca2+ transient amplituderecovered fully, but d[Ca2+]i remained elevated and LV function was depressed, indicative of Ca2+ overload. Ranolazine (9 μM) reduced d[Ca2+]iaccumulation during ischemia as well as reperfusion and improved recovery of LV function. These results show that augmentation of late INa withATX-II or by ischemia is associated with diastolic Ca2+ overload and LV dysfunction. The beneficial effects of ranolazine in reducing Ca2+
Voltage-gated sodium channels (VGSCs) play a fundamentalrole in the propagation of action potentials in the myocardium.VGSC activation is triggered by membrane depolarization andresults in a rapid influx of Na+ leading to further depolarization,Ca2+ entry and the initiation of excitation-contraction coupling.Under normal conditions, once activated, VGSCs rapidlyinactivate (within a few ms); hence, influx of Na+ is transient[1]. The resulting increase in the intracellular concentration ofNa+ ([Na+]i) is small and Na+ homeostasis can be restored byactivity of the Na+-K+-ATPase [1].
The VGSC inactivation process is slowed and/or is incompleteunder certain conditions, causing a sustained/persistent influx of
Na+, herein referred to as late INa [2–4]. Variant 3 of the long QTsyndrome (LQT3) is caused by mutations in the cardiac VGSCgene SCN5A [3] that destabilize the INa inactivation process andcause an increase in late INa. Likewise, phosphorylation of cardiacVGSCs by stress-activated kinases is also associated withincreased late INa [5]. An increase in late INa may increase actionpotential duration (APD), APD dispersion and induce early afterdepolarizations (EADs) that may lead to the development of life-threatening polymorphic ventricular tachycardias such as torsadede pointes [6]. Moreover, the increase in [Na+]i and resultantextrusion of the excess Na+ ions via reverse-mode Na+-Ca2+
exchange (NCX) may cause simultaneous Ca2+ entry and maycause Ca2+ overload and subsequent mechanical dysfunction[4,7–10].
VGSCs may contribute to hypoxia-induced Na+-dependentCa2+ loading after cells become unexcitable [8], and augmen-tation of late INa is associated with pathological conditions such
1032 H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
as heart failure [11,12] and hypoxia [13,14], and with exposureof the myocardium to reactive oxygen species [15,16], ischemicmetabolites [17,18] or glycolytic intermediates [19].
Despite accumulating evidence for a beneficial effect ofinhibition of late INa and Na+-dependent Ca2+ overload in thetreatment of myocardial ischemia [20], evidence for theinvolvement of late INa in intracellular Ca2+ accumulation andLV mechanical dysfunction at the whole heart level islacking. Therefore, an aim of the present study was toinvestigate the role of late INa on Ca2+ accumulation and LVfunction in intact heart.
The potential role of late INa in ischemia-related cardiacpathologies is supported by studies with ranolazine, acompound recently approved for the chronic treatment ofangina pectoris. Ranolazine reduces the severity of anginapectoris at plasma concentrations that cause no or minimalchanges in cardiac function or systemic hemodynamics[21,22]. It is approximately 38-fold more potent at inhibitinglate INa than inhibiting peak INa in ventricular myocytes fromcanine failing hearts [23]. Consistent with these findings,ranolazine has been shown to suppress EADs and cardiacarrhythmias in guinea pig and rabbit models of long-QTsyndrome [24,25], but it has yet to be demonstrated thatranolazine will limit Ca2+ accumulation in the heart andthereby reduce Ca2+ overload and LV dysfunction when lateINa is enhanced.
The studies reported here used isolated ejecting/workinghearts from rats to test the following hypotheses: (1) the seaanemone toxin ATX-II, known to increase, selectively, late INa,causes Ca2+ accumulation and LV dysfunction; (2) ranolazine,known to inhibit late INa, reduces Ca
2+ accumulation and limitsLV dysfunction caused by ATX-II; and (3) ischemia-inducedCa2+ accumulation and LV dysfunction, which may in part bedue to an increase in late INa, is reduced by ranolazine.
2. Materials and methods
2.1. Heart perfusions
Male Sprague–Dawley rats (300–400 g) were anesthetizedwith pentobarbital (150 mg/kg, i.p.) according to the Universityof Alberta Animal Policy and Welfare Committee and theGuide for the Care and Use of Laboratory Animals (NIHPublication No. 85-23, revised 1996). Each heart was rapidlyremoved, the aorta was cannulated and a non-working(Langendorff) perfusion was promptly (within 30 s) initiatedwith Krebs–Henseleit (KH) solution. After 10 min ofLangendorff perfusion, working mode perfusion was initiatedas described previously [26] and hearts were paced at 5 Hz. Theperfusate (recirculating volume of 100 mL, 37 °C, pH 7.4,gassed with a 95% O2–5% CO2 mixture) consisted of amodified KH solution containing the following (in mM): KCl(4.7), NaCl (118), KH2PO4 (1.2), MgSO4 (1.2), CaCl2 (2.5),NaHCO3 (25), glucose (11), palmitate (1.2) and insulin100 mU/L. Palmitate was pre-bound to bovine serum albumin(3%). Perfusions were performed at a constant workload(preload, 11.5 mm Hg; afterload, 80 mm Hg).
2.2. Measurement of LV mechanical function
Heart rate, systolic and diastolic aortic pressures (mm Hg),cardiac output (mL/min) and aortic flow (mL/min) wereacquired digitally using Chart V5.0 software (AD Instruments,Colorado Springs, CO). LV minute work (L/min/mm Hg),calculated as cardiac output×LV developed pressure served as acontinuous index of LV function. Coronary flow (mL/min) wascalculated as the difference between cardiac output and aorticflow. CVC (mL/min/mm Hg) was calculated as coronary flow/aortic diastolic pressure.
2.3. Measurement of intracellular Ca2+
Hearts were loaded with the fluorescent Ca2+ indicator, indo-1AM (5 μM) and indo-1 fluorescence was measured from theepicardial surface of a∼0.3 cm2 area of the LV-free wall using aspectrofluorometer (Photon Technology International, London,Ontario, Canada) fitted with a bifurcated fiber optic cablecontaining both excitation (354 nm) and emission bundles [27].Signals were acquired at 500 Hz and the ratio of indo-1fluorescence emitted at 405 nm and 485 nm was calculated toprovide an index of intracellular d[Ca2+]i and s[Ca2+]i. Ca
2+
transient amplitude was calculated as s[Ca2+]i minus d[Ca2+]i.
2.4. Activation and inhibition of late INa
To increase late INa, the sea anemone toxin II (ATX-II) wasadded to the recirculating perfusate of aerobic hearts to inhibitselectively Na+ channel inactivation [28,29]. After 10 min ofATX-II exposure, hearts were treated with either vehicle (DMSOfinal concentration ≤0.25%, n=11), 4 μM (n=9) or 9 μMranolazine (n=9). In other experiments, hearts (n=4) weretreated with 9 μM ranolazine to determine the direct effects ofranolazine on [Ca2+]i and LV function. Thereafter, the effects ofATX-II in ranolazine-pretreated hearts (n=4) were measuredand compared to the effects of ATX-II in untreated hearts(n=11).
2.5. Ischemia-reperfusion (IR) protocol
This comprised a 15-min period of baseline aerobic perfusion(hearts paced at 5 Hz), 20 min of global, no-flow ischemia (nopacing), followed by 30 min of aerobic reperfusion (pacing at5 Hz re-attempted within 2 min of reperfusion). One group ofhearts was exposed to vehicle alone (DMSO final concentration≤0.25%, n=9) whereas a second group was pretreated (5 minprior to the onset of ischemia) with ranolazine (9 μM, n=6).
2.6. Sources of drugs
Ranolazine 1-piperazineacetamide, N-(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-, (±)-, Lot # E4-NE-002, from CV Therapeutics Inc. was dissolved in DMSO.Concentrations of ranolazine in perfusate samples were analyzedby CV Therapeutics using LC/MS/MS. ATX-II (Lot # AT-05)was purchased from Alomone Labs (Jerusalem, Israel) and was
Fig. 1. Effects of dobutamine and verapamil on intracellular Ca2+ and LVfunction. (A) d[Ca2+]i, (B) Ca
2+ transient amplitude and (C) LV function weremeasured during aerobic perfusion (0 to 15 min), and then in the presence ofeither dobutamine (Dob, •, 10 nM) or verapamil (Ver, ○, 100 nM). Valuesshown are the mean±SE (n=4 per group).
1033H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
initially dissolved in dH2O. Indo-1AM (Lot # 305) was obtainedfrom Tef Labs (Austin, TX) and was dissolved in DMSO on theday of the experiment. Insulin was purchased from NovoNordisk Canada Inc. (Mississauga, Ontario, Canada) and fattyacid-free bovine serum albumin (Fraction V) was purchasedfrom Equitech-Bio, Inc. (Kerrville, TX). All other constituentsof the KH solution were reagent grade and purchased fromFisher Scientific, Ltd. (Ottawa, Ontario, Canada).
2.7. Statistical analysis
All values are expressed as mean±SE for the indicatednumber of independent observations (n). The significance of thedifference among groups was determined by using eitherANOVA (ranolazine 0, 4 and 9 μM groups) followed by aBonferroni multiple comparison test or a Students t-test(ranolazine 0 and 9 μM groups in pretreatment protocol and IRprotocol). Differences were judged to be significant whenp<0.05.
3. Results
3.1. Effects of dobutamine and verapamil on intracellular Ca2+
and LV function
Simultaneous measurements of LV mechanical function andintracellular Ca2+ in working/ejecting rat hearts have not beenreported previously. In the first series of experiments, hearts wereexposed to drugs known to affect Ca2+ channel function. Diastolic(d)[Ca2+]i, Ca
2+ transient amplitude and LV function were stableduring baseline aerobic perfusion (Fig. 1). Dobutamine (10 nM)and verapamil (100 nM) elicited the expected increases anddecreases, respectively, in LV function and Ca2+ transientamplitude while having no marked effect on d[Ca2+]i.
3.2. Effects of ATX-II on intracellular Ca2+ and LV function
ATX-II (12 nM) caused a rapid increase in d[Ca2+]i thatreached a plateau after ∼15 min, but Ca2+ transient amplitudewas minimally affected. These effects were associated with aninitial and transient (∼3 min) increase in LV function that wasfollowed by a gradual decline over the remaining 30-min periodof ATX-II treatment by 86±8% (n=11) of the baseline level(Fig. 2). The gradual decline in LV function was not due to aneffect on coronary perfusion and O2 availability because coronaryflow (Fig. 2) and CVC (data not shown) were not significantlyaltered until LV function had declined by more than 50%.
3.3. Reversal by ranolazine of the effects of ATX-II on [Ca2+]iand LV function
Ranolazine (9 μM) added after 10 min of ATX-II exposurenot only prevented any further increases in d[Ca2+]i (p<0.001)but actually reduced d[Ca2+]i relative to ATX-II alone (Fig. 3).Although ranolazine had no effect on Ca2+ transient amplitudeat 9 μM, it also prevented deterioration in LV function; after30 min of ATX-II exposure, LV function was depressed by
only 32±11% (n=9) of the baseline level (p<0.001)compared with 86±8% (n=11) in untreated hearts. Similarly,a lower concentration of ranolazine (4 μM, n=9), added after10 min of ATX-II exposure, also prevented further increasesin d[Ca2+]i (p<0.05) and reduced values relative to ATX-IIalone. Ranolazine (4 μM) also attenuated the deterioration inLV function, which was 86±8% without ranolazine (n=11)and 50±11% with ranolazine (n=9, p<0.05). In contrast tohearts treated with ATX-II alone, hearts treated with ATX-IIplus ranolazine (4 and 9 μM) had stable coronary flow andCVC (data not shown).
3.4. Attenuation by ranolazine of the effects of ATX-II on[Ca2+]i and LV function
Ranolazine alone (9 μM) had no effect on d[Ca2+]i, Ca2+
transient amplitude, or LV function (n=4). As shown in Figs. 2
Fig. 3. Reversal by ranolazine of the ATX-II-induced effects on intracellularCa2+ and LV function. (A) d[Ca2+]i, (B) Ca
2+ transient amplitude and (C) LVfunction were measured during aerobic perfusion (0 to 15 min), and then inthe presence of ATX-II (12 nM, •, n=11). Ranolazine (4 μM, ▲, n=9 or9 μM, ○, n=9) was added 10 min following ATX-II. *Significant differences(p<0.05, ANOVA and Bonferroni post-test) between ranolazine-treated andATX-II alone values at the end of perfusion. Values are means±SE.
Fig. 2. Effect of ATX-II (12 nM) on intracellular Ca2+ and LV function. (A)d[Ca2+]i, (B) Ca2+ transient amplitude and (C) LV function (•) and coronaryflow (○) were measured during aerobic perfusion (0 to 15 min), and then for afurther 30 min in the presence of ATX-II (12 nM). Values are means±SE (n=11).
1034 H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
and 3, exposure to ATX-II alone for 30 min increased d[Ca2+]iand reduced LV function from baseline by 86±8% (n=11). Incomparison, in hearts pretreated with ranolazine (9 μM,10 min prior to addition of ATX-II), the ATX-II-inducedincrease in d[Ca2+]i was significantly (p<0.001) attenuated(Fig. 4). Moreover, LV dysfunction caused by ATX-II was alsoattenuated by pre-treatment with ranolazine. Specifically, after30 min of exposure to ATX-II, LV mechanical function wasdepressed by only 24±5% in hearts pretreated with ranolazine(p<0.001, n=4), whereas in hearts not treated with ranolazine,LV function was depressed by 86±8%.
3.5. Effect of global ischemia and reperfusion on [Ca2+]i andLV function
During aerobic conditions (15 min), indices of both d[Ca2+]i,Ca2+ transient amplitude and LV function were stable (Fig. 5).There was a steady increase in d[Ca2+]i throughout the periodof global ischemia by 66±8% relative to values measured
during aerobic baseline. Ischemia gradually abolished Ca2+
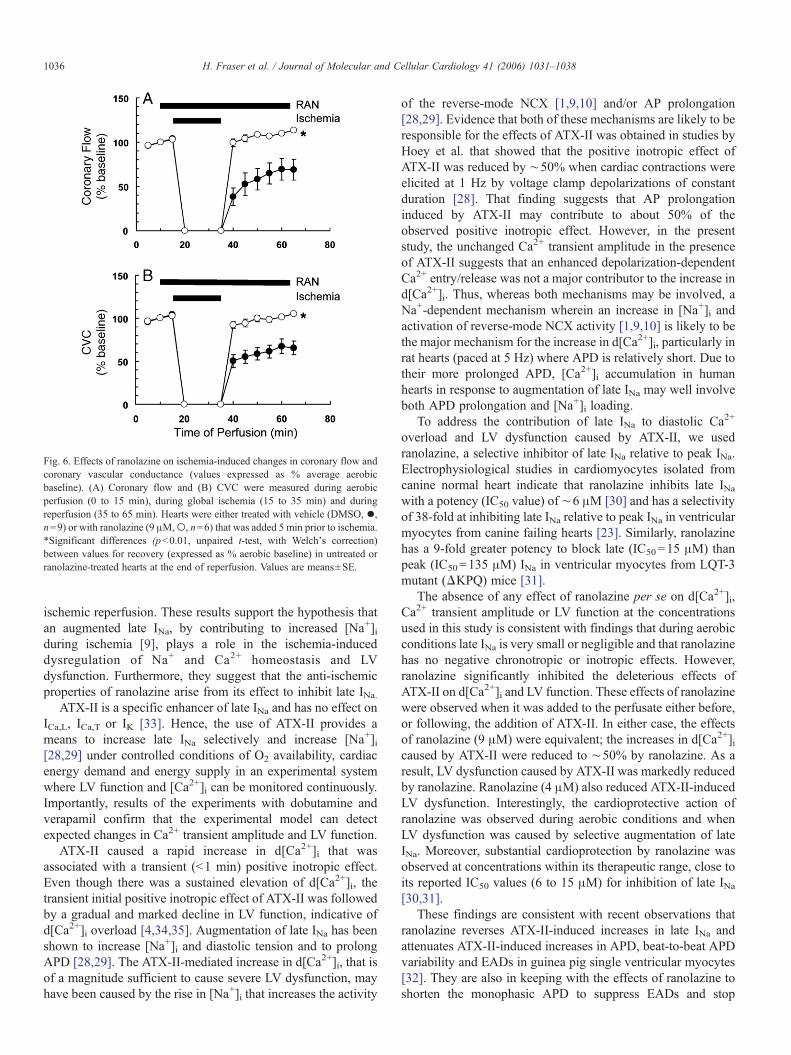
transients within 10 min and LV function ceased. Duringreperfusion, d[Ca2+]i remained elevated by 17±2% (n=9)relative to aerobic values, but Ca2+ transient amplitudequickly recovered to aerobic baseline values. LV functionrecovered poorly during reperfusion and after 30 minreperfusion was still depressed by 77±6% (n=9) of aerobicvalues (Fig. 5). At the end of reperfusion, coronary flow andCVC were reduced by 31±12% and 34±8%, respectively,relative to aerobic values (Fig. 6).
3.6. Attenuation by ranolazine of the effects of ischemia andreperfusion on [Ca2+]i and LV function
Ranolazine (9 μM), added 5 min prior to the onset of globalischemia, had no measurable effect on either d[Ca2+]i, Ca
2+
1035H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
transient amplitude, LV function, coronary flow or CVC (Figs.5 and 6). Ranolazine (9 μM) did, however, significantly reduceCa2+ accumulation during ischemia by reducing the rate ofincrease in d[Ca2+]i during ischemia by 50% (p<0.05); valuesat end of ischemia were reduced by 47% (p<0.01). Ranolazinehad no effect on Ca2+ transient amplitude during reperfusion,but significantly reduced d[Ca2+]i (p<0.01) so that after30 min, d[Ca2+]i was increased by only 2.4±2.1%, n=6values (Fig. 6). In hearts treated with ranolazine, LVmechanical function during reperfusion was only depressedby 30±6% (n=6) of aerobic values compared with 77±6%(n=9) in untreated hearts (p<0.001). Ranolazine alsoprevented the decreases in coronary flow (p<0.01) and CVC(p<0.01) measured during reperfusion (Fig. 6); in the presence
Fig. 5. Effects of ranolazine on ischemia-induced changes in intracellular Ca2+
and LV function. (A) d[Ca2+]i, (B) Ca2+ transient amplitude and (C) LV function
were measured during aerobic perfusion (0 to 15 min), during global, no-flowischemia (15 to 35 min), as well as during reperfusion (35 to 65 min). Heartswere either treated with vehicle (DMSO, •, n=9) or with ranolazine (9 μM,○,n=6) that was added 5 min prior to the onset of ischemia. *Significantdifferences (p<0.05, unpaired t-test) between values in untreated or ranolazine-treated hearts at the end of ischemia or at the end of reperfusion. Values aremeans±SE.
Fig. 4. Attenuation by ranolazine of the effects of ATX-II on intracellular Ca2+
and LV function. (A) d[Ca2+]i, (B) Ca2+ transient amplitude and (C) LV function
were measured during aerobic perfusion (0 to 15 min), in the absence (○, n=11,same data as in Fig. 4) or presence (•, n=4) of ranolazine (9 μM). ATX-II(12 nM), administered to each group at 15 min, was present throughout theremainder of perfusion. *Significant difference (p<0.05, Students t-test)between values at end of perfusion. Values are means±SE.
of ranolazine, coronary flow and CVC recovered to 113±2%and 105±2% of aerobic values, respectively.
4. Discussion
Selective augmentation of late INa with ATX-II increasedd[Ca2+]i and impaired LV function of rat isolated working/ejecting hearts but had no effect on Ca2+ transient amplitude.Ranolazine, at concentrations known to inhibit late INa[23,30–32], had no effect on either d[Ca2+]i, Ca
2+ transientamplitude, LV function or coronary flow, but it significantlyreduced diastolic Ca2+ accumulation and LV dysfunctioncaused by ATX-II. In addition, ranolazine reduced Ca2+
accumulation during ischemia and improved recovery of Ca2+
homeostasis, LV function and coronary flow during post-
Fig. 6. Effects of ranolazine on ischemia-induced changes in coronary flow andcoronary vascular conductance (values expressed as % average aerobicbaseline). (A) Coronary flow and (B) CVC were measured during aerobicperfusion (0 to 15 min), during global ischemia (15 to 35 min) and duringreperfusion (35 to 65 min). Hearts were either treated with vehicle (DMSO, •,n=9) or with ranolazine (9 μM,○, n=6) that was added 5 min prior to ischemia.*Significant differences (p<0.01, unpaired t-test, with Welch's correction)between values for recovery (expressed as % aerobic baseline) in untreated orranolazine-treated hearts at the end of reperfusion. Values are means±SE.
1036 H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
ischemic reperfusion. These results support the hypothesis thatan augmented late INa, by contributing to increased [Na+]iduring ischemia [9], plays a role in the ischemia-induceddysregulation of Na+ and Ca2+ homeostasis and LVdysfunction. Furthermore, they suggest that the anti-ischemicproperties of ranolazine arise from its effect to inhibit late INa.
ATX-II is a specific enhancer of late INa and has no effect onICa,L, ICa,T or IK [33]. Hence, the use of ATX-II provides ameans to increase late INa selectively and increase [Na+]i[28,29] under controlled conditions of O2 availability, cardiacenergy demand and energy supply in an experimental systemwhere LV function and [Ca2+]i can be monitored continuously.Importantly, results of the experiments with dobutamine andverapamil confirm that the experimental model can detectexpected changes in Ca2+ transient amplitude and LV function.
ATX-II caused a rapid increase in d[Ca2+]i that wasassociated with a transient (<1 min) positive inotropic effect.Even though there was a sustained elevation of d[Ca2+]i, thetransient initial positive inotropic effect of ATX-II was followedby a gradual and marked decline in LV function, indicative ofd[Ca2+]i overload [4,34,35]. Augmentation of late INa has beenshown to increase [Na+]i and diastolic tension and to prolongAPD [28,29]. The ATX-II-mediated increase in d[Ca2+]i, that isof a magnitude sufficient to cause severe LV dysfunction, mayhave been caused by the rise in [Na+]i that increases the activity
of the reverse-mode NCX [1,9,10] and/or AP prolongation[28,29]. Evidence that both of these mechanisms are likely to beresponsible for the effects of ATX-II was obtained in studies byHoey et al. that showed that the positive inotropic effect ofATX-II was reduced by ∼50% when cardiac contractions wereelicited at 1 Hz by voltage clamp depolarizations of constantduration [28]. That finding suggests that AP prolongationinduced by ATX-II may contribute to about 50% of theobserved positive inotropic effect. However, in the presentstudy, the unchanged Ca2+ transient amplitude in the presenceof ATX-II suggests that an enhanced depolarization-dependentCa2+ entry/release was not a major contributor to the increase ind[Ca2+]i. Thus, whereas both mechanisms may be involved, aNa+-dependent mechanism wherein an increase in [Na+]i andactivation of reverse-mode NCX activity [1,9,10] is likely to bethe major mechanism for the increase in d[Ca2+]i, particularly inrat hearts (paced at 5 Hz) where APD is relatively short. Due totheir more prolonged APD, [Ca2+]i accumulation in humanhearts in response to augmentation of late INa may well involveboth APD prolongation and [Na+]i loading.
To address the contribution of late INa to diastolic Ca2+
overload and LV dysfunction caused by ATX-II, we usedranolazine, a selective inhibitor of late INa relative to peak INa.Electrophysiological studies in cardiomyocytes isolated fromcanine normal heart indicate that ranolazine inhibits late INawith a potency (IC50 value) of ∼6 μM [30] and has a selectivityof 38-fold at inhibiting late INa relative to peak INa in ventricularmyocytes from canine failing hearts [23]. Similarly, ranolazinehas a 9-fold greater potency to block late (IC50=15 μM) thanpeak (IC50=135 μM) INa in ventricular myocytes from LQT-3mutant (ΔKPQ) mice [31].
The absence of any effect of ranolazine per se on d[Ca2+]i,Ca2+ transient amplitude or LV function at the concentrationsused in this study is consistent with findings that during aerobicconditions late INa is very small or negligible and that ranolazinehas no negative chronotropic or inotropic effects. However,ranolazine significantly inhibited the deleterious effects ofATX-II on d[Ca2+]i and LV function. These effects of ranolazinewere observed when it was added to the perfusate either before,or following, the addition of ATX-II. In either case, the effectsof ranolazine (9 μM) were equivalent; the increases in d[Ca2+]icaused by ATX-II were reduced to ∼50% by ranolazine. As aresult, LV dysfunction caused by ATX-II was markedly reducedby ranolazine. Ranolazine (4 μM) also reduced ATX-II-inducedLV dysfunction. Interestingly, the cardioprotective action ofranolazine was observed during aerobic conditions and whenLV dysfunction was caused by selective augmentation of lateINa. Moreover, substantial cardioprotection by ranolazine wasobserved at concentrations within its therapeutic range, close toits reported IC50 values (6 to 15 μM) for inhibition of late INa[30,31].
These findings are consistent with recent observations thatranolazine reverses ATX-II-induced increases in late INa andattenuates ATX-II-induced increases in APD, beat-to-beat APDvariability and EADs in guinea pig single ventricular myocytes[32]. They are also in keeping with the effects of ranolazine toshorten the monophasic APD to suppress EADs and stop
1037H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
ventricular tachycardia caused by ATX-II in guinea pig andrabbit isolated perfused hearts [24,25]. By inhibition of late INa,ranolazine reduces [Ca2+]i, likely by shortening AP durationand/or reducing [Na+]i accumulation and [Ca2+]i overload.
IR increases [Na+]i and [Ca2+]i and impairs LV function[9,10]. Although the mechanism(s) underlying the rise in [Na+]iduring ischemia remain unresolved, it appears that increases inlate INa as well as Na+-H+ exchanger (NHE) activity likelycontribute to Na+ influx [7–10]. Thus, because an increased lateINa contributes to the rise in [Na+]i and subsequent Ca2+
overload and LV dysfunction in IR, we examined the effects ofranolazine on IR-induced changes in d[Ca2+]i and LV function.
As expected, ischemia caused cessation of LV function, andafter∼10 min Ca2+ transients were abolished. Average d[Ca2+]iincreased to values higher than those measured for s[Ca2+]iduring aerobic perfusion. During post-ischemic reperfusion,Ca2+ transients quickly became re-established, but d[Ca2+]iremained significantly elevated relative to aerobic values. Thisis consistent with reports of ischemia-induced Ca2+ overloadcontributing to LV dysfunction [4,9,10,34,35] and illustrates theimportance of diastolic Ca2+ overload. Whereas ranolazine hadno direct effect on either d[Ca2+]i, Ca
2+ transient amplitude, LVfunction, coronary flow or CVC during aerobic conditions, itsignificantly reduced the increase in intracellular Ca2+ duringischemia and it enhanced recovery of post-ischemic LVfunction, coronary flow and CVC. That these effects occurredat concentrations less than those required to inhibit NHE andNCX activities [30,36] or L-type Ca2+ current [37] suggests thatthe anti-ischemic properties of ranolazine are likely to arisefrom its effect to inhibit, selectively, late INa. In addition, ourdemonstration of the absence of any direct ranolazine-inducedvasodilator responses during aerobic conditions suggests thatimproved CVC observed during reperfusion is related toreduced Ca2+ overload and improved LV relaxation leading toreduced vascular compression during diastole.
Previous reports have suggested that the anti-ischemicproperties of ranolazine may be due to alterations of fatty acidoxidation [38,39]. It is unlikely that such a mechanism isresponsible for the cardioprotective effects of ranolazinereported here. First, ranolazine exerted a marked reduction indiastolic Ca2+ overload during the actual period of ischemia,when fatty acid oxidation is expected to be inhibited due to theabsence of O2 availability. Secondly, the extent of fatty acidoxidation inhibition achieved by a high concentration ofranolazine (100 μM) was only 12% [40] and is thereforeunlikely to account for the cardioprotective effects oftherapeutic concentrations observed in this and other studies.
In summary, our results show that selective augmentation oflate INa by ATX-II can lead to d[Ca2+]i overload and LVdysfunction in the whole heart, effects that are attenuated bytherapeutic concentrations of ranolazine. Because ranolazineinhibits late INa selectively [23,30–32], the results are also inkeeping with the hypothesis that increased late INa contributes toNa+ and Ca2+ overload observed during IR. Lastly, inhibition oflate INa may be a useful pharmacological target to attenuate thedysregulation of Na+ and Ca2+ homeostasis, and the electricaland mechanical disturbances associated with pathological
conditions where late INa is increased, such as hypoxia[13,14], heart failure [11,12,23] and oxidative stress [15,16].
Acknowledgment
This research was supported by grants to ASC from CVTherapeutics, Inc.
References
[1] Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiacmyocytes. Cardiovasc Res 2003;57:897–912.
[2] Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricularmyocytes. J Physiol 1992;453:219–31.
[3] Clancy CE, TateyamaM, Kass RS. Insights into the molecular mechanismsof bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest2002;110:1251–62.
[4] Noble D, Noble PJ. Late sodium current in the pathophysiology ofcardiovascular disease: consequences of sodium–calcium overload. Heart2006;92(Suppl 4):iv1–5.
[5] Light PE, Wallace CH, Dyck JR. Constitutively active adenosinemonophosphate-activated protein kinase regulates voltage-gated sodiumchannels in ventricular myocytes. Circulation 2003;107:1962–5.
[6] Shimizu W, Antzelevitch C. Sodium channel block with mexiletine iseffective in reducing dispersion of repolarization and preventing torsadedes pointes in LQT2 and LQT3 models of the long-QT syndrome.Circulation 1997;96:2038–47.
[7] Tani M, Neely JR. Deleterious effects of digitalis on reperfusion-inducedarrhythmias and myocardial injury in ischemic rat hearts: possibleinvolvements of myocardial Na+ and Ca2+ imbalance. Basic Res Cardiol1991;86:340–54.
[8] Haigney MC, Miyata H, Lakatta EG, Stern MD, Silverman HS.Dependence of hypoxic cellular calcium loading on Na(+)-Ca2+ ex-change. Circ Res 1992;71:547–57.
[9] Imahashi K, Kusuoka H, Hashimoto K, Yoshioka J, Yamaguchi H,Nishimura T. Intracellular sodium accumulation during ischemia as thesubstrate for reperfusion injury. Circ Res 1999;84:1401–6.
[10] Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, MurphyE. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protectionagainst ischemia/reperfusion injury. Circ Res 2005;97:916–21.
[11] Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gatingof the late Na+ channel in normal and failing human myocardium. J MolCell Cardiol 2002;34:1477–89.
[12] Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al.Increased late sodium current in myocytes from a canine heart failuremodel and from failing human heart. J Mol Cell Cardiol 2005;38:475–83.
[13] Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium currentin rat ventricular myocytes. J Physiol 1996;497:337–47.
[14] Fearon IM, Brown ST. Acute and chronic hypoxic regulation ofrecombinant hNa(v)1.5 alpha subunits. Biochem Biophys Res Commun2004;324:1289–95.
[15] Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxidein rat ventricular myocytes. J Physiol 1997;500:631–42.
[16] Ma JH, Luo AT, Zhang PH. Effect of hydrogen peroxide on persistentsodium current in guinea pig ventricular myocytes. Acta Pharmacol Sin2005;26:828–34.
[17] Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current atresting potentials in single cardiac myocytes induced by the ischemicmetabolite lysophosphatidylcholine. Circ Res 1992;71:1231–41.
[18] Wu J, Corr PB. Palmitoylcarnitine increases [Na+]i and initiates transientinward current in adult ventricular myocytes. Am J Physiol 1995;268:H2405–17.
[19] Kohlhardt M, Fichtner H, Frobe U. Metabolites of the glycolytic pathwaymodulate the activity of single cardiac Na+ channels. FASEB J 1989;3:1963–7.
1038 H. Fraser et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 1031–1038
[20] Conti CR. Inhibition of sodium-dependent calcium overload to treatmyocardial ischemia. Clin Cardiol 2006;29:141–3.
[21] Chaitman BR, Skettino SL, Parker JO, Hanley P, Meluzin J, Kuch J, et al.Anti-ischemic effects and long-term survival during ranolazine mono-therapy in patients with chronic severe angina. J Am Coll Cardiol2004;43:1375–82.
[22] Chaitman BR, Pepine CJ, Parker JO, Skopal J, Chumakova G, Kuch J, et al.Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercisetolerance and angina frequency in patients with severe chronic angina: arandomized controlled trial. JAMA 2004;291: 309–316.
[23] Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazineimproves abnormal repolarization and contraction in left ventricularmyocytes of dogs with heart failure by inhibiting late sodium current.J Cardiac Electrophysiol 2006;17:S1–9.
[24] Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L.Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther 2004;310:599–605.
[25] Wu L, Shryock JC, Song Y, Belardinelli L. An Increase in late sodiumcurrent potentiates the proarrhythmic activities of low-risk QT-prolong-ing drugs in female rabbit hearts. J Pharmacol Exp Ther 2006;316:718–726.
[26] Neely JR, Liebermeister H, Morgan HE. Effect of pressure developmenton membrane transport of glucose in isolated rat heart. Am J Physiol1967;212:815–22.
[27] Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA,Figueredo V, et al. Preconditioning limits mitochondrial Ca(2+) duringischemia in rat hearts: role of K(ATP) channels. Am J Physiol: Heart CircPhysiol 2001;280:H2321–8.
[28] Hoey A, Harrison SM, Boyett MR, Ravens U. Effects of the Anemoniasulcata toxin (ATX II) on intracellular sodium and contractility in rat andguinea-pig myocardium. Pharmacol Toxicol 1994;75:356–65.
[29] Isenberg G, Ravens U. The effects of the Anemonia sulcata toxin (ATX II)on membrane currents of isolated mammalian myocytes. J Physiol1984;357:127–49.
[30] Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di DiegoJM, Fish JM, et al. Electrophysiological effects of ranolazine, a novelantianginal agent with antiarrhythmic properties. Circulation 2004;110:904–910.
[31] Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine blockof LQT-3 mutant sodium channels: evidence for site of action. Br JPharmacol 2006;148:16–24.
[32] Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine ofthe pro-arrhythmic effects of increasing late INa in guinea pig ventricularmyocytes. J Cardiovasc Pharmacol 2004;44:192–9.
[33] Mantegazza M, Franceschetti S, Avanzini G. Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firingproperties of rat neocortical pyramidal neurones. J Physiol 1998;507:105–116.
[34] Bolli R, Marban E. Molecular and cellular mechanisms of myocardialstunning. Physiol Rev 1999;79:609–34.
[35] Ver Donck L, Borgers M, Verdonck F. Inhibition of sodium and calciumoverload pathology in the myocardium: a new cytoprotective principle.Cardiovasc Res 1993;27:349–57.
[36] Belardinelli L, Shryock JC, Fraser H. The mechanism of ranolazine actionto reduce ischemia-induced diastolic dysfunction. Eur Heart J 2006;8:A10–3.
[37] Allen TJ, Chapman RA. Effects of ranolazine on L-type calcium channelcurrents in guinea-pig single ventricular myocytes. Br J Pharmacol 1996;118:249–54.
[38] McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazinestimulates glucose oxidation in normoxic, ischemic, and reperfusedischemic rat hearts. Circulation 1996;93:135–42.
[40] MacInnes A, Fairman DA, Binding P, Rhodes J, Wyatt MJ, Phelan A, et al.The antianginal agent trimetazidine does not exert its functional benefit viainhibition of mitochondrial long-chain 3-ketoacyl coenzyme A thiolase.Circ Res 2003;93:26–32.