CHAPTER FOUR Recent Advances in 11 B Solid-State Nuclear Magnetic Resonance Spectroscopy of Crystalline Solids Ying-Tung Angel Wong, David L. Bryce University of Ottawa, Ottawa, ON, Canada Contents 1. Introduction 214 2. Theoretical Background 215 2.1 Nuclear Magnetic Shielding Interaction 216 2.2 Nuclear Electric Quadrupolar Interaction 217 2.3 Indirect Spin–Spin (J) Coupling Interaction 218 3. Experimental Methods 218 3.1 Measurement of CS and EFG Tensor Parameters 218 3.2 Measurement of Indirect Spin–Spin (J) Coupling Constants 220 4. Survey of Available Data 222 4.1 EFG and CS Tensor Parameters 222 4.2 Indirect Spin–Spin (J) Coupling Constants 264 5. Concluding Remarks 272 References 273 Abstract We review the progress made in 11 B solid-state nuclear magnetic resonance (SSNMR) spectroscopy of crystalline materials over the past 20 years, with a focus on the appli- cations of 11 B NMR observables in providing electronic and structural information. A brief description of some of the common SSNMR methods for measuring 11 B chemical shift (CS) tensor parameters, electric field gradient (EFG) tensor parameters, and indirect spin–spin (J ) coupling constants is first provided. Recent 11 B SSNMR studies on crys- talline boron systems, such as diboron compounds, boronic esters and acids, borates, and boron-containing Lewis acid/base adducts, are then summarized, and the corresponding experimentally obtained 11 B NMR parameters are presented. In general, data from studies that only report isotropic CSs are not tabulated. Our survey highlights the ability of 11 B SSNMR spectroscopy to provide an abundance of diverse chemical information, ranging from the coordination environment of the boron, to ligand iden- tity, bond strengths, bond orders and bond angles, and the potential of this technique to characterize inorganic and organic crystalline solids. Owing to the sensitivity of 11 B Annual Reports on NMR Spectroscopy, Volume 93 # 2018 Elsevier Ltd ISSN 0066-4103 All rights reserved. https://doi.org/10.1016/bs.arnmr.2017.08.005 213
Transcript
CHAPTER FOUR
Recent Advances in 11B Solid-StateNuclear Magnetic ResonanceSpectroscopy of Crystalline SolidsYing-Tung Angel Wong, David L. BryceUniversity of Ottawa, Ottawa, ON, Canada
Contents
1. Introduction 2142. Theoretical Background 215
2.1 Nuclear Magnetic Shielding Interaction 2162.2 Nuclear Electric Quadrupolar Interaction 2172.3 Indirect Spin–Spin (J) Coupling Interaction 218
3. Experimental Methods 2183.1 Measurement of CS and EFG Tensor Parameters 2183.2 Measurement of Indirect Spin–Spin (J) Coupling Constants 220
4. Survey of Available Data 2224.1 EFG and CS Tensor Parameters 2224.2 Indirect Spin–Spin (J) Coupling Constants 264
5. Concluding Remarks 272References 273
Abstract
We review the progress made in 11B solid-state nuclear magnetic resonance (SSNMR)spectroscopy of crystalline materials over the past 20 years, with a focus on the appli-cations of 11B NMR observables in providing electronic and structural information.A brief description of some of the common SSNMRmethods for measuring 11B chemicalshift (CS) tensor parameters, electric field gradient (EFG) tensor parameters, and indirectspin–spin (J ) coupling constants is first provided. Recent 11B SSNMR studies on crys-talline boron systems, such as diboron compounds, boronic esters and acids, borates,and boron-containing Lewis acid/base adducts, are then summarized, and thecorresponding experimentally obtained 11B NMR parameters are presented. In general,data from studies that only report isotropic CSs are not tabulated. Our survey highlightsthe ability of 11B SSNMR spectroscopy to provide an abundance of diverse chemicalinformation, ranging from the coordination environment of the boron, to ligand iden-tity, bond strengths, bond orders and bond angles, and the potential of this techniqueto characterize inorganic and organic crystalline solids. Owing to the sensitivity of 11B
Annual Reports on NMR Spectroscopy, Volume 93 # 2018 Elsevier LtdISSN 0066-4103 All rights reserved.https://doi.org/10.1016/bs.arnmr.2017.08.005
SSNMR spectroscopy to chemical structures and the suitability of the 11B nuclide forhigh-resolution techniques such as MQMAS and DOR, we anticipate that 11B SSNMRspectroscopy will continue to evolve as an indispensable tool for solid-state character-ization of boron-containing systems and for the advancement of various fields, such asNMR crystallography, the synthesis of novel boron reagents, and the development ofboron-based hydrogen storage materials.
Keywords: 11B solid-state NMR spectroscopy, Boron-11, Electric field gradient, Chemicalshift anisotropy, Chemical shifts, J coupling constants
1. INTRODUCTION
Boron-containing compounds comprise an important class of reagents
for synthetic chemistry [1] and boron is also a fundamental building block for
various types of materials, such as glasses, ceramics, and minerals. Conse-
quently, NMR studies on boron nuclei are of significant importance as they
can provide reactivity and structural information. There are twoNMRactive
boron isotopes, 10B and 11B, and their NMR properties are given in Table 1.
Both 10B and 11B are quadrupolar nuclei (I>1/2), with 10B possessing a spin
of 3 and 11B possessing a spin of 3/2, and their corresponding nuclear electric
quadrupole moments,Q, are both relatively modest (Q(10B)¼8.459 fm2 and
Q(11B)¼4.059 fm2) [2]. Nevertheless, it is more advantageous to perform
NMR studies on 11B instead of 10B since the former exhibits a higher natural
abundance (NA) and magnetogyric ratio (γ), a smallerQ, as well as a straight-
forwardly observable central transition (CT) (i.e., mI¼1/2$ mI¼�1/2).
The latest reviews on the application of 11B solid-state nuclear magnetic
resonance (SSNMR) in the investigation of crystalline solids have mostly
been featured as brief subsections in surveys which describe the spectro-
scopic characterization of a specific class of compounds (e.g., pharmaceuti-
cals, boronate and benzoxaborolate ligands, and frustrated Lewis pairs (FLPs)
and/or materials (e.g., zeolites) [3–9]. To the best of our knowledge, the
most recent review which broadly discusses the application of 11B SSNMR
Table 1 NMR Properties of the Boron IsotopesI NA (%)a γ (107 rad s21 T21)a Q (fm2)a Ξ (%)a,b
10B 3 19.9 2.8746786 8.459 10.743658
11B 3/2 80.1 8.5847044 4.059 32.083974
aFrom Ref. [2].bThe ratio between the resonance frequency of the reference (BF3�Et2O) and that of the protons of TMSin CDCl3 (at infinite dilution).
214 Ying-Tung Angel Wong and David L. Bryces
to the study of various crystalline compounds was published more than
10 years ago by MacKenzie and Smith [10], and in this work only 11B elec-
tric field gradient (EFG) tensor parameters (the quadrupolar coupling con-
stant (CQ) and the asymmetry parameter (η)) and isotropic chemical shift
(CS) (δiso) measurements were surveyed. Owing to growing interest in
boron chemistry, a considerable number of 11B SSNMR studies have been
conducted on crystalline samples since the survey by MacKenzie and Smith,
and therefore an updated perspective on this field is warranted. Here, we
review the 11B SSNMR studies of crystalline samples reported in the past
20 years, with a specific focus on studies where tensorial information
has been reported rather than only isotropic values. 11B NMR studies on
solutions, as well as amorphous solids (e.g., glasses) are not considered. Inter-
ested readers can refer to the monograph by N€oth andWrackmeyer [11] for
extensive discussions on 11B solution NMR and various reviews [10,12–14]for detailed surveys on the expansive field of 11B NMR of glasses. We focus
on the advances made in measuring the EFG tensor parameters, the CS ten-
sor parameters (δiso, the span (Ω) and the skew (κ)) and the indirect spin–spin(J) coupling constants in crystalline solids, and how these interactions can be
employed to obtain electronic and structural information. A brief theoretical
background on the nuclear magnetic shielding, the nuclear electric
quadrupolar, and the indirect spin–spin (J) coupling interactions is first pro-
vided in Section 2, followed by a brief discussion in Section 3 on experimen-
tal SSNMR methods that are commonly employed to measure the EFG
tensor parameters, the CS tensor parameters, and the J coupling constants
of 11B. In Section 4, we tabulate and discuss the available 11B SSNMR data
for the review period. This section is broken down into two subsections
which provide (1) the EFG and CS tensors parameters and (2) J coupling
constants that were reported over the past 2 decades, respectively. Conclud-
ing remarks are then presented in Section 5.
2. THEORETICAL BACKGROUND
When a quadrupolar nucleus in a diamagnetic molecule is placed in an
external static magnetic field, the corresponding nuclear spin Hamiltonian,bH , can be expressed as a sum of various interactions:
bH ¼ bHZ + bH σ + bHQ + bHJ + bHD (1)
where bHZ is the Zeeman Hamiltonian originating from the interaction
between the nucleus and the external static magnetic field; bH σ describes
215Recent Advances in 11B SSNMR Spectroscopy
the nuclear magnetic shielding interaction which arises from the sur-
rounding electrons; bHQ is the quadrupolar coupling Hamiltonian which
accounts for the interaction betweenQ and the EFG generated by the sur-
rounding electrons and nuclei; bHJ describes the indirect spin–spin (J) cou-
pling interactions, and bHD corresponds to the direct through-space
dipolar coupling interactions. In this section, a brief background on the
magnetic shielding interaction, the electric quadrupolar interaction and
the J coupling interaction is given, together with important definitions
and equations.
2.1 Nuclear Magnetic Shielding InteractionThe nuclear magnetic shielding (σ) interaction describes the magnetic
shielding of the nucleus due to the local magnetic field generated by the sur-
rounding electrons. As proposed by Ramsey [15], this interaction can be
expressed as a sum of diamagnetic (σd) and paramagnetic (σp) contributions.σd depends on the ground electronic state of the systemwhile σp results frommixing of the ground electronic state and the excited states. In general, the σtensor is anisotropic and antisymmetric, and three tensor components
(i.e., the principal components) are required to describe the appearance
of the corresponding NMR spectra. Following the Herzfeld–Berger con-vention [16], the three components are σ11, σ22, and σ33, and can be
reexpressed in terms of the isotropic value (σiso), Ω, and κ, which are
defined as follows:
σiso ¼ σ11 + σ22 + σ333
(2)
Ω¼ σ33�σ11 (3)
κ¼ 3 σiso�σ22ð ÞΩ
(4)
where σ33�σ22�σ11. Ω describes the breadth of the powder pattern and
can be any positive value. κ gives the asymmetry and the corresponding
value varies from �1 to +1, where a κ of 1 or �1 signifies an axially sym-
metric CS tensor.
Experimentally, the CS (δ) is measured with respect to a reference com-
pound. The relationship between the tensor components of δ and σ is given
by:
δii ¼ σii,reference�σii1�σii,reference
�106ppm (5)
216 Ying-Tung Angel Wong and David L. Bryces
where ii is a particular element of the tensor, and δ11�δ22�δ33. Further-more, Ω and κ can also be expressed in terms of δ:
Ω¼ δ11�δ33 (6)
κ¼ 3 δ22�δisoð ÞΩ
(7)
The IUPAC-recommended CS reference for 11B is 15% by volume of
BF3�Et2O in CDCl3 [2,17]. However, in SSNMR experiments,
NaBH4(s) is often employed instead as a secondary reference, where the
corresponding 11B CT resonance is found at �42.06 ppm relative to
BF3�Et2O.
2.2 Nuclear Electric Quadrupolar InteractionA quadrupolar nucleus (I>1/2) will have a nonspherical nuclear charge dis-
tribution and therefore an electric quadrupole moment, Q. The nuclear
electric quadrupolar interaction arises from the coupling between Q and
the EFG at the site of the nucleus. The EFG is a symmetric, traceless
second-rank tensor quantity, and the magnitude of the interaction can be
described by CQ and η, which are defined as follows:
CQ¼ eQV 33
h(8)
η¼V11�V22
V33
(9)
where e is the charge of an electron, h is Planck’s constant, and V11,V22, and
V33 are the principal components of the EFG tensor such that jV33 j� jV22 j� jV11 j andV33+V22+V11¼0 (i.e., traceless). η can take on any valuebetween 0 and 1.
Since the nuclear electric quadrupolar interaction is dependent on the
EFG tensor, it is therefore strongly influenced by the electronic symmetry
at the site of the nucleus. For instance, nuclei in a cubic environment (e.g.,
tetrahedral symmetry) will have aCQ of zero, while nuclei in an axially sym-
metric environment will have a η of zero. Consequently, in favourable cases,structural insights can be easily obtained by assessing the values of CQ and η,and nuclei in different structural environments can be differentiated via the
nuclear electric quadrupolar interaction.
217Recent Advances in 11B SSNMR Spectroscopy
2.3 Indirect Spin–Spin (J) Coupling InteractionThe J coupling interaction arises from the coupling between two nuclei as
mediated by the intervening electrons. The second-rank tensor which
describes this interaction (J) is anisotropic and antisymmetric. The isotropic
(Jiso) component is the most commonly measured, while the anisotropic (ΔJ)component is only available under favourable circumstances since it is often
small and experimentally indistinguishable from the dipolar coupling inter-
action. Furthermore, for quadrupolar nuclei in the solid state, Jiso is still
rarely reported as the corresponding spectra are often dominated by
quadrupolar interactions. Nonetheless, as described in the next sections, var-
ious techniques can be employed for extracting the homonuclear and het-
eronuclear J coupling constants, and the coupling constants can provide
valuable insights into electronic structure as this interaction arises from
the orbital overlap of two atoms.
3. EXPERIMENTAL METHODS
3.1 Measurement of CS and EFG Tensor Parameters3.1.1 Single-Crystal MethodThough experimentally tedious, single-crystal SSNMR spectroscopy is
extremely powerful for obtaining EFG and CS tensor information since it
can provide the magnitude of the tensors as well as the orientation of the
tensors with respect to the crystal frame and relative to each other. Further-
more, this technique allows for accurate measurement of small 11B chemical
shift anisotropy (CSA) parameters for systems with large first-order 11B
quadrupolar coupling if a sum frequency analysis is performed on the
observed satellite transitions (STs) [18]. Using 11B single-crystal SSNMR,
the EFG and/or CS tensors have been measured for various boron systems,
ranging from borates to metal borides [18–22]. A detailed description of the
experimental aspects and data analysis pertaining to single-crystal SSNMR
spectroscopy can be found in Refs. [23–25]. Briefly, a single crystal is rotatedat an axis that is either perpendicular or at a general angle from B0, and a
series of NMR spectra are recorded as a function of the rotation angle
[23–25]. The resonances of the spectral peaks depend on the orientation
of the crystal with respect to B0, and either the CT and/or the STs are mon-
itored. Depending on the angle of the rotational axis with respect to B0, the
required number of rotation axes (e.g., rotation about three orthogonal axes
vs rotation about two axes) and the range of rotation angles (e.g., 0–180 vs
0–360 degrees) will differ. Plots corresponding to the position of the
218 Ying-Tung Angel Wong and David L. Bryces
resonance(s) are constructed with respect to the crystal rotation angle about a
given axis and the resulting plots are fitted. The fitting parameters can be
mathematically related to the matrix elements of the EFG and CS tensors
in the reference frame of the sample holder. Transformation of the tensor
from the sample holder frame to the principal axis system frame then pro-
vides the principal components and tensor orientations.
3.1.2 Powdered SamplesIn powdered samples, a distribution of crystallite orientations with respect to
B0 is present, resulting in SSNMR spectra that consist of powder patterns.
The line shape of the powder pattern is governed by the corresponding CS
and EFG tensors; therefore, one general method to obtain the CS and EFG
tensor parameters is to fit the CT resonance acquired under bothmagic angle
spinning (MAS) and static conditions at multiple fields. The CT is preferred
since it is generally less broadened and therefore more easily detected as
compared to the STs, and the CT can be optimally observed by the use
of a ‘solid π/2’ pulse, in which the π/2 pulse width obtained from solution
and/or from samples where the symmetry around the 11B nucleus is cubic
(e.g., in NaBH4) is scaled by 1/(I+1/2). The MAS spectrum is first simu-
lated in order to obtain the values for δiso, CQ, and η, and these values are
then employed in the static spectrum simulation in order to extract the
values for Ω, κ, and the Euler angles (α, β, and γ) between the EFG and
CS tensors. The use of multiple fields can improve the accuracy of the
CS tensor parameters as an increase in B0 would decrease the influence of
second-order quadrupolar coupling while increasing the influence of the
CS tensor on the SSNMR spectra. Moreover, SSNMR probes often con-
tain boron nitride and the corresponding 11B signal may encumber spectral
fitting. In this case, spin echo experiments (i.e., π/2-τ1-π-τ2-acquire) can beperformed in order to suppress the background signal.
The CS and EFG tensor parameters cannot always be directly extracted
from the CT line shape acquired using simple one-pulse and/or Hahn echo
experiments. For instance, for systems with small CQ(11B) values, the
corresponding CT would be sharp and featureless. In these situations, the
EFG tensor parameters can be acquired by simulating the spinning sideband
manifold obtained using satellite transition spectroscopy (SATRAS) [26,27].
Furthermore, for systems with multiple 11B sites, peak overlap due to the
presence of second-order quadrupolar coupling can hinder spectral fitting.
Consequently, high-resolution techniques may be utilized to accurately
extract the relevant NMR parameters. A commonly employed method is
the two-dimensional experiment known as multiple-quantum MAS
219Recent Advances in 11B SSNMR Spectroscopy
(MQMAS) NMR spectroscopy [28]. In MQMAS, the second-rank ele-
ments of the first- and second-order quadrupolar interactions are scaled
to zero via MAS, while the fourth-rank elements of the second-order
quadrupolar interactions are removed by allowing the signals to evolve as
multiple quantum coherences. The end result is a high-resolution ‘isotropic’
spectrum in the indirect dimension that is free from second-order quad-
rupolar broadening. The values for CQ, η, and δiso for each site can then
be extracted by simulating the direct dimension spectrum (which is still
under the influence of second-order quadrupolar broadening) at a given
indirect frequency. Furthermore, since the CSA interaction is scaled in
the indirect dimension of the MQMAS spectrum by the multiple-quantum
order, small 11B CSA parameters can then be obtained from MQMAS
experiments as illustrated by Hansen et al. [18].
Another high-resolution technique which can be employed is double
rotation (DOR) spectroscopy [29]. In DOR, the sample is rotated about
two axes simultaneously, thereby removing the anisotropic broadening aris-
ing from CS, dipolar, and quadrupolar interactions. The resulting spectrum
is then influenced by δiso, the second-order quadrupolar-induced shifts
(δQIS; for11B, δQIS¼ 1
40
C2Q
ν20
1 + η2
3
� �), the residual dipolar shifts, and Jiso
[30,31]. The value for δQIS extracted from the DOR spectra can be
employed to facilitate the simulation of the corresponding MAS spectra,
thereby allowing for the accurate extraction of CQ and η. Nonetheless,
for boron nuclei with small quadrupolar coupling interaction, this method
might not be applicable since the corresponding value for δQIS would be
small and difficult to extract.
3.2 Measurement of Indirect Spin–Spin (J) Coupling ConstantsThe measurement of J(11B, X) coupling constants can be difficult since
anisotropic quadrupolar spectral broadening can mask the J coupling effects
on the CT spectra. Nevertheless, several methods allow for accurate mea-
surement of J(11B, X) coupling constants. In the case of heteronuclear J cou-
pling with a spin-1/2 nucleus (i.e., IX¼1/2), the coupling constant can be
obtained from the MAS spectrum of the spin-1/2 nucleus if the coupling
interaction is large enough to give an observable multiplet structure
[6,7,32–36]. On the other hand, if the J coupling is too small to be detectable
from the MAS spectrum, heteronuclear J-resolved spectroscopy can be used
instead. In this 2D experiment, the J coupling multiplets are given in the
indirect dimension without the influence of anisotropic line broadening,
220 Ying-Tung Angel Wong and David L. Bryces
and consequently, the J(11B, X) coupling can be easily measured [6,7,34].
Moreover, in cases where the boron nucleus is located in a highly spherical
electronic environment, it may also be possible to directly extract the J cou-
pling constants from the 11B MAS spectrum since the corresponding
quadrupolar interaction would be minimal.
For the measurement of J(11B, X) coupling constants where X is also
quadrupolar (i.e., heteronuclear J coupling where IX>1/2, or homonuclear
J coupling), the removal of second-order quadrupolar coupling effects can
be crucial. This can be accomplished by using eitherMQMAS or DOR, and
by simulating the corresponding spectrum, J coupling information can be
extracted. These techniques have been shown to be successful for measuring
heteronuclear [30,37–40] and homonuclear 11B J couplings [30,37].
Though powerful, these two methods can be difficult to perform since
the acquisition of MQMAS spectra can be time consuming, while special-
ized probe hardware is required for DOR.
An easily implementable series of techniques which can be used for the
extraction of J(11B, 11B) coupling constants are the 11B double quantum fil-
tered (DQF) J-resolved experiments [41], which can provide higher spectral
resolution than that of MQMAS and DOR [42]. Similar to a regular
J-resolved experiment, the J coupling information is extracted into the indi-
rect dimension via a CT selective echo [31,41,43]. However, in the DQF
J-resolved experiments, a DQF (typically an INADEQUATE block) is also
implemented before the echo in order to suppress the large zero frequency
signal observed in the indirect dimension of the regular J-resolved spectra
[31,41,43]. The resulting 11BDQF J-resolved spectra thus consist of a simple
doublet in the indirect dimension for each pair of boron spins, and the J cou-
pling constants can be accurately measured since the zero frequency signal
which oftenmasks theweak J doublets is now removed. Themagnitude of the
DQF J splitting is governed by the J coupling constant, as well as the crystal-
lographic symmetry and molecular dynamics of the system [31,41–45]. For apair of crystallographically inequivalent 11B nuclei that are not under the influ-
ence of dynamics, the doublet would be split by J (Fig. 1) [41,42,44]. On the
other hand, if the 11B nuclei are crystallographically equivalent or crystallo-
graphically inequivalent but magnetically equivalent on the timescale of the
experiment due to molecular motions, a splitting of 3J would be observed
(Fig. 1) [41,42,44,45]. Therefore, both the crystallographic symmetry and
the presence of dynamics must be taken into consideration when analysing
DQF J-resolved spectra in order to correctly determine the J coupling
constants.
221Recent Advances in 11B SSNMR Spectroscopy
4. SURVEY OF AVAILABLE DATA
4.1 EFG and CS Tensor ParametersOver the course of the review period, 11B SSNMR studies have been per-
formed on a variety of crystalline materials, ranging from important synthetic
reagents to valuable geological samples. In this section, we present the
experimental 11B EFG and CS tensor parameters acquired in the past 2
decades, which are summarized in Tables 2–11. Note that in general, data
from studies that only reported δiso(11B) are not tabulated here, but some
earlier works are included due to their importance. The section is divided
in terms of the classes of compounds, and selected results are discussed in
detail in order to illustrate how 11B EFG and CS tensor parameters can
be used to elucidate the electronic and molecular environment of the
boron atom.
Fig. 1 11B DQF J-resolved SSNMR spectra of (A) bis(catecholato)diboron and (B) [bis(catecholato) diboron]�IMes. Neither of these samples exhibit dynamic disorder. Inbis(catecholato) diboron, the borons are crystallographically equivalent due to the pres-ence of an inversion centre; therefore a splitting of 3J is observed. In [bis(catecholato)diboron]�IMes, the borons are crystallographically inequivalent, and therefore a splittingof J is observed. Adapted from F.A. Perras, D.L. Bryce, Symmetry-amplified J splittings forquadrupolar spin pairs: a solid-state NMR Probe of homoatomic covalent bonds, J. Am.Chem. Soc. 135 (2013) 12596–12599.
222 Ying-Tung Angel Wong and David L. Bryces
Table 2 11B EFG and CS Tensor Parameters for Borane Derivatives, Diboron Compounds, Tetrahedral Boron Anions, and Boron-Containing ZwitterionsMeasured Using 11B SSNMR SpectroscopyCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
Trimesitylborane 77.4�0.5 121�1 1.0 4.75�0.01 0.0 MAS and
static
Bryce et al. [46]
Pt(dbbpy)(C^CC6H4BMes2)2�4CH2Cl2 74�1.5 90�40 1.0�0.5 4.65�0.10 0.00�0.05 MAS Hudson et al. [47]
Continued
Table 2 11B EFG and CS Tensor Parameters for Borane Derivatives, Diboron Compounds, Tetrahedral Boron Anions, and Boron-Containing ZwitterionsMeasured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
Dimesitylborinium cation 91.2�0.2 130�1 0.90�0.10 5.44�0.08 0.02�0.02 MAS and
static
Alain et al. [48]
Bis(catecholato)diboron 30.5�1.0 — — 2.85�0.05 0.85�0.05 MAS Perras and Bryce [30]
Bis(catecholato) diboron�IMes
three-coordinate site
34.3�0.5 — — 3.0�0.1 0.89�0.05 MAS Perras and Bryce [41]
Bis(catecholato) diboron�IMes
four-coordinate site
1�1 — — — — MAS Perras and Bryce [41]
Bis(catecholato) diboron�dipicoline 11.0�0.5 — — 2.2�0.05 0.15�0.10 MAS Perras and Bryce [42]
Bis(pinacolato) diboron 31.5�0.5 — — 2.70�0.05 0.85�0.10 MAS Perras and Bryce [41]
— — — 2.5�0.1 0.9�0.1 MQMAS Wi and Frydman [37]
Pinacolato bis(2-hydroxypropyl)amino
diboron four-coordinate site
7.0�0.5 — — 1.7�0.1 0.7�0.1 MAS Perras and Bryce [42]
Pinacolato bis(2-hydroxypropyl)amino
diboron three-coordinate site
34�1 — — 2.9�0.1 0.90�0.05 MAS Perras and Bryce [42]
Tetrahydroxy diboron 29.5�1.0 — — 3.2�0.1 0.58�0.05 MAS Perras and Bryce [42]
Tetrakis(pyrrolidino) diborane site 1 31.2�0.5 — — 2.7�0.1 1.00�0.05 MQMAS Perras and Bryce [42]
Tetrakis(pyrrolidino) diborane site 2 35.2�0.5 — — 2.9�0.1 0.50�0.08 MQMAS Perras and Bryce [42]
9-BBN 29�1 — — 2.7�0.1 1.00�0.15 MAS Perras and Bryce [42]
aConverted from Haeberlen convention.
Table 3 11B EFG and CS Tensor Parameters for Acids, Esters, and Related Boron Systems Measured Using 11B SSNMR SpectroscopyCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
Boric acid 19.6�0.3 — — 2.85�0.05 0.40�0.10 MAS Weiss and Bryce [49]
— — — 2.5 0 MAS Gervais et al. [50]
2-(tert-Butyldimethylsilyloxy)
naphthalene-6-boronic acid
29.0�1.0 33�11 �0.50�0.20 3.29�0.10 0.40�0.10 MAS and static Weiss and Bryce [49]
2-Chloropyridine-3-boronic acid 27.5�1.0 32�2 0.40�0.40 3.05�0.10 0.10�0.20 MAS and static Weiss and Bryce [49]
2-Acetyl-3-thiopheneboronic acid 26.0�2.0 30�6 0.20�0.20 2.83�0.25 0.10�0.30 MAS and static Weiss and Bryce [49]
2,6-Dibromophenylboronic acid 30.0�1.4 40�10 0.40�0.60 3.10�0.20 0.30�0.20 MAS and static Weiss and Bryce [49]
Phenylboronic acid site 1 30.5�1.0 17.7�1.3 0.80�0.20 3.00�0.10 0.45�0.10 MAS and static Oh et al. [32]
Phenylboronic acid site 2 31.0�1.0 18.7�1.3 1.00�0.20 3.03�0.10 0.40�0.10 MAS and static Oh et al. [32]
4-Methoxyphenylboronic acid 28.6�0.6 23.0�1.2 0.73�0.13 3.02�0.05 0.43�0.05 MAS and static Oh et al. [32]
3,4-Dimethoxyphenylboronic acid 28.7�0.6 27.0�1.2 0.35�0.12 2.97�0.05 0.40�0.05 MAS and static Oh et al. [32]
4-Methylphenylboronic acid site 1 30.2�1.0 17.7�1.4 0.80�0.20 3.00�0.10 0.42�0.10 MAS and static Oh et al. [32]
4-Methylphenylboronic acid site 2 30.7�1.0 18.7�1.4 1.00�0.20 3.03�0.10 0.40�0.10 MAS and static Oh et al. [32]
2-Methylphenylboronic acid 30.4�0.5 28.3�1.0 0.38�0.13 3.05�0.05 0.43�0.05 MAS and static Oh et al. [32]
4-Chlorophenylboronic acid 29.7�0.5 26.0�1.0 0.52�0.10 3.03�0.05 0.40�0.05 MAS and static Oh et al. [32]
2-Chloro-5-(trifluoromethyl)
phenylboronic acid
28.9�0.5 24.0�1.0 0.57�0.10 2.97�0.05 0.30�0.05 MAS and static Oh et al. [32]
4-(Methylthio)phenylboronic acid 28.8�0.5 25.7�1.0 0.40�0.10 2.95�0.05 0.37�0.05 MAS and static Oh et al. [32]
Continued
Table 3 11B EFG and CS Tensor Parameters for Acids, Esters, and Related Boron Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
5-Fluoro-1,3-dihydro-1-hydroxyl-2,1-
benzoxaborole (AN2690)
31.1�0.1 — — 2.8�0.02 0.51�0.03 MAS Sene et al. [51]
(S)-3-(Aminomethyl)-7-(3-
hydroxypropoxy)benzo[c][1,2]
oxaborol-1(3H)-ol hydrochloride
form 1
30.6 — — 2.79 0.41 MAS Vogt et al. [52]
2-(Hydroxymethyl)phenylboronic acid
cyclic monoester (BBzx)
31.0�2.0 19�1 �0.90�0.10 2.80�0.10 0.45�0.10 MAS and static Weiss and Bryce [49]
31.1�0.1a — — 2.9�0.02a 0.48�0.03a MAS Sene et al. [51,53]
LDH-BBzx (benzoxaborole form of
BBzx)
31.8�0.4 — — 2.9�0.2 0.36�0.08 MAS Sene et al. [53]
LDH-BBzx (tetrahedral
benzoxaborolate form of BBzx)
8.3�0.2 — — 1.2�0.1 0.5�0.3 MAS Sene et al. [53]
MgBBZx�10H2O (tetrahedral
benzoxaborolate form of BBzx)
8.5�0.1 — — 1.27�0.04 0.39�0.06 MAS Sene et al. [53]
CaBBzx�3H2O (tetrahedral
benzoxaborolate form of BBzx)
9.3�0.1b — — 1.30�0.10b 0.5�0.3b MAS Sene et al. [53]
CaBBzx�3H2O (planar benzoxaborolate
form of BBzx)
30.7�0.1 — — 2.87�0.03 0.68�0.05 MAS Sene et al. [53]
Ca[Oct-B(OH)3]2 site 1 4.8 — — 1.46 0.28 MAS Berthomieu et al. [54]
Ca[Oct-B(OH)3]2 site 2 4.5 — — 1.47 0.13 MAS Berthomieu et al. [54]
Sr[Bu-B(OH)3]2 site 1 4.60�0.04 — — 1.39�0.04 0.11�0.08 MAS Berthomieu et al. [54]
Sr[Bu-B(OH)3]2 site 2 4.38�0.07 — — 1.35�0.04 0.25�0.07 MAS Berthomieu et al. [54]
Ca(C4H9-B(OH)3)2 site 1 4.78�0.03 — — 1.46�0.02 0.28�0.03 MQMAS Sene et al. [55]
Ca(C4H9-B(OH)3)2 site 2 4.54�0.04 — — 1.47�0.03 0.13�0.02 MQMAS Sene et al. [55]
Ca(PhB(OH)3)2 site 1 3.8�0.3 — — 1.4�0.2 0.4�0.2 MAS Reinholdt et al. [56]
Ca(PhB(OH)3)2 site 2 4.0�0.2 — — 1.3�0.1 0.2�0.1 MAS Reinholdt et al. [56]
10-Hydroxy-10,9-
boroxophenanthrene
27 — — 2.9 0.6 MAS Carnevale et al. [57]
Bis(boroxophenanthryl)ether 25 — — 2.9 0.5 MAS Carnevale et al. [57]
Triphenyl borate 17.9�0.5 <10 0.0�0.3 2.32�0.02 0.0 MAS and static Bryce et al. [46]
Triethanolamine borate 56 — — 1.20 0 MAS Wu and Yamada [58]
4DBF2 26.0�0.2 28.0�0.5 0.0�0.2 2.65�0.1 0.52�0.02 MAS and static Borsacchi et al. [59]
4-Fluorophenylboronic acid
neopentylglycol ester
26.5�1.0 23�2 0.30�0.10 2.79�0.10 0.40�0.15 MAS and static Weiss and Bryce [49]
4-Nitrophenylboronic acid pinacol ester 29.8�1.0 12�2 0.60�0.10 2.83�0.20 0.51�0.10 MAS and static Weiss and Bryce [49]
1H–Indazole-5-boronic acid pinacol
ester
30.3�1.0 14�2 �0.25�0.10 2.76�0.20 0.59�0.10 MAS and static Weiss and Bryce [49]
4-(2-Propynylcarbamoyl)
phenylboronic acid pinacol ester
30.2�1.0 10�2 0.40�0.10 2.66�0.10 0.68�0.10 MAS and static Weiss and Bryce [49]
Continued
Table 3 11B EFG and CS Tensor Parameters for Acids, Esters, and Related Boron Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
2,4,6-Trimethoxyphenylboronic acid
neopentyl glycol ester
27.3�1.0 30�2 0.60�0.10 2.89�0.10 0.37�0.05 MAS and static Weiss and Bryce [49]
Phenylboronic acid catechol cyclic ester 30.8�0.5 26.0�1.0 �0.25�0.10 2.71�0.05 0.78�0.05 MAS and static Oh et al. [32]
4-Methoxyphenylboronic acid catechol
cyclic esterc30.8�0.1 23�1 �0.45�0.05 2.68�0.17 0.80�0.10 MAS and static Oh et al. [32]
3,4-Dimethoxyphenylboronic acid
catechol cyclic ester
31.5�0.5 25.5�1.0 �0.85�0.10 2.68�0.05 0.78�0.05 MAS and static Oh et al. [32]
4-Methylphenylboronic acid catechol
cyclic ester
30.4�0.5 28.0�1.0 �0.15�0.10 2.68�0.05 0.78�0.05 MAS and static Oh et al. [32]
2-Methylphenylbornic acid catechol
cyclic ester
30.1�0.5 28.0�1.0 �0.15�0.10 2.64�0.05 0.80�0.05 MAS and static Oh et al. [32]
4-Fluorophenylboronic acid catechol
cyclic ester
30.0�0.7 27.5�1.0 �0.22�0.10 2.64�0.05 0.74�0.05 MAS and static Oh et al. [32]
4-Chlorophenylboronic acid catechol
cyclic ester
30.3�0.5 27.7�1.0 �0.12�0.10 2.69�0.05 0.76�0.05 MAS and static Oh et al. [32]
aThe two crystallographically inequivalent borons were unresolved by 11B MAS NMR spectroscopy.bAverage value for sites 1, 2, 3, 4, and 6.cData from J.W.E. Weiss, M.Sc. thesis, University of Ottawa, 2011.
Table 4 11B EFG and CS Tensor Parameters for Borates, Borosilicates, Thioborates, and Related Systems Measured Using 11B SSNMR SpectroscopyCompound δiso (ppm) CQ (MHz) η Method References
NaBO2 18.4�0.3 2.41�0.02 0.80�0.05 MAS Stebbins et al. [60]
Na4B2O5 21.8�0.3 2.63�0.02 0.50�0.05 Stebbins et al. [60]
Al4B2O9 three-coordinate site 17.4 2.66 0.10 MAS Fisch et al. [61]
16.3 2.6 0.11 MAS Fischer et al. [62]
Al4B2O9 four-coordinate site �1.0 — — MAS Fisch et al. [61]
�1.8 — — MAS Fischer et al. [62]
xAl2O3:yB2O3 x:y¼9:2, 5:1, 1:2, 1.2:1, 1:3.3 16.7–16.8 2.61–2.62 0.08–0.09 MAS Fisch et al. [61]
B2O3a 14.6�0.1 2.690�0.005 <0.05 MAS Kroeker and Stebbins [63]
Cs2O�9B2O3 three-coordinate site 16.7�0.2 2.50�0.05 0.2b MAS Kroeker and Stebbins [63]
Cs2O�9B2O3 four-coordinate site 0.95 0.200�0.050 >0.5 SATRAS Kroeker and Stebbins [63]
CaO�B2O3c 17.15�0.05 2.53�0.01 0.63�0.02 MAS Kroeker and Stebbins [63]
2MgO�B2O3d 18.7�0.1 2.78�0.02 0.48�0.02 MAS Kroeker and Stebbins [63]
2MgO�B2O3 site 1 18.6�0.1 2.71�0.02 0.50�0.02 MAS Hansen et al. [64]
2MgO�B2O3 site 2 19.0�0.1 2.81�0.02 0.45�0.02 MAS Hansen et al. [64]
3MgO�B2O3 22.5�0.1 2.94�0.02 <0.05 MAS Kroeker and Stebbins [63]
22.6�0.1 2.93�0.01 0.03�0.02 MAS Hansen et al. [64]
Li2O�B2O3 17.08�0.06 2.56�0.01 0.60�0.03 MAS Kroeker and Stebbins [63]
La2O3�B2O3e 20.4�0.1 2.67�0.01 0.05�0.05 MAS Kroeker and Stebbins [63]
Continued
Table 4 11B EFG and CS Tensor Parameters for Borates, Borosilicates, Thioborates, and Related Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
β-BaB2O4 �1.1�0.3 2.50�0.02 0.74�0.02 MAS Maia et al. [65]
High-pressure KB3O5 four-coordinate site 1 0.31�0.02 0.95�0.05 0.55�0.04 MQMAS Neumair et al. [66]
High-pressure KB3O5 four-coordinate site 2 1.75�0.02 1.2�0.1 0.74�0.05 MQMAS Neumair et al. [66]
High-pressure KB3O5 three-coordinate site 16.8�0.2 2.5�0.1 0.17�0.03 MQMAS Neumair et al. [66]
CsB3O5 three-coordinate site 1 17.81�0.20 2.50�0.05 0.27�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
CsB3O5 three-coordinate site 2 19.93�0.20 2.78�0.05 0.23�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
CsB3O5 four-coordinate site 1.20�0.20 0.17�0.05 0.25�0.05 SATRAS Alderman et al. [67]
α–CsB5O8 four-coordinate site 2.89�0.20 0.29�0.05 >0.5 SATRAS Alderman et al. [67]
α–CsB5O8 three-coordinate site 1 18.70�0.20 2.58�0.05 0.23�0.05 MAS and DOR Alderman et al. [67]
α–CsB5O8 three-coordinate site 2 16.08�0.20 2.54�0.05 0.25�0.05 MAS and DOR Alderman et al. [67]
α–CsB5O8 three-coordinate site 3 16.08�0.20 2.54�0.05 0.25�0.05 MAS and DOR Alderman et al. [67]
α–CsB5O8 four-coordinate site 4 18.79�0.20 2.58�0.05 0.23�0.05 MAS and DOR Alderman et al. [67]
K2B4O7 four-coordinate site 1 1.84�0.20 �0.52�0.05 — SATRAS Alderman et al. [67]
K2B4O7 four-coordinate site 2 2.39�0.20 �0.52�0.05 — SATRAS Alderman et al. [67]
K2B4O7 four-coordinate site 3 0.50�0.20 �0.52�0.05 — SATRAS Alderman et al. [67]
K2B4O7 four-coordinate site 4 0.97�0.20 �0.52�0.05 — SATRAS Alderman et al. [67]
K2B4O7 three-coordinate sites 1 and 2 18.91�0.20 2.52�0.05 0.12�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
K2B4O7 three-coordinate site 3 16.20�0.20 2.55�0.05 0.06�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
K2B4O7 three-coordinate site 4 19.90�0.20 2.66�0.05 0.07�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
α–BaB4O7 four-coordinate site 1 1.79�0.20 — — — Alderman et al. [67]
α–BaB4O7 four-coordinate site 2 0.47�0.20 — — — Alderman et al. [67]
α–BaB4O7 four-coordinate site 3 0.47�0.20 — — — Alderman et al. [67]
α–BaB4O7 four-coordinate site 4 0.47�0.20 — — — Alderman et al. [67]
α–BaB4O7 three-coordinate site 1 19.23�0.20 2.75�0.05 0.18�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
α–BaB4O7 three-coordinate site 2 17.23�0.20 2.48�0.05 0.07�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
α–BaB4O7 three-coordinate site 3 19.14�0.20 2.74�0.05 0.18�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
α–BaB4O7 three-coordinate site 4 16.15�0.20 2.46�0.05 0.33�0.05 MAS, DOR, and
MQMAS
Alderman et al. [67]
Li2B4O7f three-coordinate site 18.1�0.2 2.56�0.03 0.21�0.04 MAS Hansen et al. [18,64]
— 2.600�0.007 0.167�0.004 Single crystal Lim and Kim [21]
Li2B4O7 four-coordinate site 2.3�0.1 0.52�0.02 0.51�0.02 MAS Hansen et al. [64]
2.2�0.1 0.52�0.02 0.51�0.02 SATRAS Hansen et al. [18]
— 0.5268�0.0026 0.530�0.011 Single crystal Lim and Kim [21]
Continued
Table 4 11B EFG and CS Tensor Parameters for Borates, Borosilicates, Thioborates, and Related Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
KZnB3O6 four-coordinate site 2.4�0.1 0.8�0.1 0.4�0.1 MQMAS Neumair et al. [66]
KZnB3O6 three-coordinate site 18.1�0.3 2.4�0.1 0.3�0.2 MQMAS Neumair et al. [66]
Ga1�xFexBO3 x¼0, 0.01, 0.02 24.5�0.1
–27.6�0.1
2.84�0.02
–3.16�0.02
0 MAS Seleznyova et al. [68]
BaTi(BO3)2 �2.5�0.2 2.635�0.003 0.00�0.01 MAS Maia et al. [65]
Li2B3O4F3 four-coordinate site 1 �1.9�1 1.2�0.2 0.67 MAS Br€auniger et al. [69]
�2.9�1 1.1�0.2 0.71 MQMAS Br€auniger et al. [69]
Li2B3O4F3 four-coordinate site 2 �0.2�1 1.0�0.2 0.90 MAS Br€auniger et al. [69]
0.2�1 1.0�0.2 0.90 MQMAS Br€auniger et al. [69]
Li2B3O4F3 three-coordinate site 14.9�1 2.5�0.2 0.10 MAS Br€auniger et al. [69]
14.2�1 2.5�0.2 0.10 MQMAS Br€auniger et al. [69]
Li2B6O9F2 three-coordinate sites 1 and 2 13.5�1 2.6�0.2 0.27 MAS Br€auniger et al. [69]
12.9�1 2.4�0.2 0.10 MQMAS Br€auniger et al. [69]
Li2B6O9F2 three-coordinate sites 3 and 4 16.8�1 2.7�0.2 0.00 MAS Br€auniger et al. [69]
17.2�1 2.8�0.2 0.25 MQMAS Br€auniger et al. [69]
Li2B6O9F2 four-coordinate site 1 �2.9�1 0.6�0.2 1.00 MAS Br€auniger et al. [69]
�2.2�1 0.9�0.2 1.00 MQMAS Br€auniger et al. [69]
Li2B6O9F2 four-coordinate site 2 0.8�1 0.7�0.2 1.00 MAS Br€auniger et al. [69]
1.5�1 0.8�0.2 1.00 MQMAS Br€auniger et al. [69]
Na2B4O7�10H2O three-coordinate siteg 18.2�0.1 2.565�0.003 0.105�0.002 Single crystal Hansen et al. [18]
Na2B4O7�10H2O four-coordinate siteh 2.1�0.1 0.497�0.001 0.624�0.003 Single crystal Hansen et al. [18]
NaCa[B5O6(OH)6]�5H2O four-coordinate
site 1, 2, and 3
1.2�0.1i 0.42�0.01i 0.42�0.15i MAS Zhou et al. [70]
NaCa[B5O6(OH)6]�5H2O three-coordinate
site 1
18.0�0.1 2.57�0.01 0.15�0.05 MAS Zhou et al. [70]
NaCa[B5O6(OH)6]�5H2O three-coordinate
site 2
18.9�0.1 2.51�0.01 0.08�0.05 MAS Zhou et al. [70]
Calcium borohydroxyapatite (10CaO�(6 �x)
PO2.5�xBO1.5�t H2O; x¼0.5) site 1
1.4 2.72 0.07 MAS Ternane et al. [71]
Calcium borohydroxyapatite (10CaO�(6 �x)
PO2.5�xBO1.5�t H2O; x¼0.5) site 2
3.8 2.62 0.06 MAS Ternane et al. [71]
1% Doped Ce calcium borohydroxyapatite
(10CaO�(6 �x)PO2.5�xBO1.5�t H2O: 1%
Ce3+; x¼0.5) site 1
1.4 2.72 0.07 MAS Ternane et al. [71]
1% Doped Ce calcium borohydroxyapatite
(10CaO�(6 �x)PO2.5�xBO1.5�t H2O: 1%
Ce3+; x¼0.5) site 2
3.8 2.62 0.06 MAS Ternane et al. [71]
CaB3O4(OH)3�H2O three-coordinate sitej 17.1�0.3 2.543�0.005 0.055�0.004 Single crystal Hansen et al. [18]
CaB3O4(OH)3�H2O four-coordinate site 1k 1.2�0.2 0.440�0.001 0.499�0.003 MAS Hansen et al. [18]
CaB3O4(OH)3�H2O four-coordinate site 2l 1.2�0.2 0.312�0.001 0.809�0.003 Single crystal Hansen et al. [18]
CaB2Si2O8m �0.2�0.1 0.39�0.02 0.43�0.03 SATRAS Hansen et al. [18]
— 0.392�0.002 0.429�0.004 Single crystal Hansen et al. [18]
Continued
Table 4 11B EFG and CS Tensor Parameters for Borates, Borosilicates, Thioborates, and Related Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
BO3 unit in CaSi1/3B2/3O8/3 19.5�0.2 2.62�0.05 0.51�0.01 MAS V�eron et al. [72]
CaBSiO4(OH)n 0.2�0.2 0.17�0.01 0.65�0.02 SATRAS Hansen et al. [18]
— 0.172�0.001 0.647�0.005 Single crystal Hansen et al. [18]
B(OSi)3 group in borosilicate zeolites 9.1–11.0 2.3–2.75 0–0.2 MAS Fild et al. [73], Lezcano-
Gonzalez et al. [74], Wiper et al.
[75], Fild et al. [76], Koller et al.
[77], and Marthala et al. [78]
B(OSi)4 group in borosilicate zeolites �1.9 to
�4.1
<1 0 MAS Mathala et al. [78], Fild et al.
[73], and Tong and Koller [79]
B(OH)3 group in borosilicate zeolites 18–19.2 2.2–2.6 0–0.2 MAS Fild et al. [73], Tong and Koller
[79], Hwang et al. [80], and
Koller et al. [77]
B(OSi)2OH group in borosilicate zeolites 12.1–15.4 2.2–2.6 0–0.1 MAS Tong and Koller [79] and
Lezcano-Gonzalez et al. [74]
B(OSi)(OH)2/B(OSi)2(OH) in borosilicate
zeolites
14.6–15.5 2.3–2.46 0.2–0.29 MAS Hwang et al. [80], Koller et al.
[77], Tong and Koller [79], and
Wiper et al. [75]
BPO4 �3.3 0.030 0 SATRAS Raskar et al. [81]
NH4[ZnBP2O8] �3.3 0.150 0.6 SATRAS Raskar et al. [81]
Rb3[B2P3O11(OH)2] sites 1 and 2 �0.6 0.750 0.3 SATRAS Raskar et al. [81]
K3[BP3O9(OH)3] �3.3 0.150 0.6 SATRAS Raskar et al. [81]
Tricoordinated surface boron unit in
B-doped, (B,N)-codoped and (Ag,B)-
codoped TiO2
19.7–20.6 2.20–2.37 0.16–0.40 MQMAS and
MAS
Feng et al. [82,83]
Tetracoordinated surface boron unit in
B-doped and (B,N)-codoped TiO2
4.0 1.03 — MQMAS Feng et al. [83]
Hydrated tetracoordinated surface boron unit
in B-doped and (B,N)-codoped TiO2
14.2 — — MQMAS Feng et al. [83]
Tetracoordinated interstitial boron unit in
B-doped, (B,N)-codoped and (Ag,B)-
codoped TiO2
2.8–2.9 1.30 — MQMAS and
MAS
Feng et al. [82,83]
Tricoordinated interstitial boron unit in
B-doped, (B,N)-codoped and (Ag,B)-
codoped TiO2
18.2–18.9 2.38–2.45 0.17–0.40 MQMAS and
MAS
Feng et al. [82,83]
BO3/2 network polymer in B-doped and
(B,N)—codoped TiO2
14.8–15.9 2.51–2.68 0.08–0.26 MQMAS and
MAS
Feng et al. [82,83]
Boroxol ring polymer in B-doped, (B,N)-
codoped and (Ag,B)-codoped TiO2
16.4–16.9 2.00–2.42 0.20–0.75 MQMAS and
MAS
Feng et al. [82,83]
Metathioborate unit in Na2S�B2S3 60�1 2.42�0.05 0.43�0.04 MQMAS and
MAS
Hwang et al. [84]
BS4 unit in Na2S�2B2S3 site 1 �1.5 0.8�0.05 — MAS Hwang et al. [84]
BS4 unit in Na2S�2B2S3 site 2 �3 0.8�0.05 — MAS Hwang et al. [84]
BS33� unit in 3Na2S�B2S4 site 1 67�3 — — MQMAS Hwang et al. [84]
Continued
Table 4 11B EFG and CS Tensor Parameters for Borates, Borosilicates, Thioborates, and Related Systems Measured Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
BS33� unit in 3Na2S�B2S4 site 2 71�3 — — MQMAS Hwang et al. [84]
B2S3 site 1 60.4�0.5 2.40�0.05 0.05�0.01 MAS Hwang et al. [85]
B2S3 site 2 63.6�0.5 2.15�0.05 0.13�0.01 MAS Hwang et al. [85]
aCSA parameters were obtained by Kroeker and Stebbins [63] using 11B MAS SSNMR spectroscopy with Ω¼15�2 ppm and κ¼1.bValue was fixed in the simulation.cΩ was found to be 38�5 ppm by Kroeker and Stebbins [63] using 11B MAS SSNMR spectroscopy.dΩ was found to be 33�5 ppm by Kroeker and Stebbins [63] using 11B MAS SSNMR spectroscopy.eCSA parameters were obtained by Kroeker and Stebbins [63] using 11B MAS SSNMR spectroscopy with Ω¼17�3 ppm and κ¼1.fCSA parameters were obtained by Hansen et al. [18] using 11B MQMAS NMR spectroscopy with δσ¼�12�3 ppm and ησ¼0.9�0.1.gCSA parameters were obtained by Hansen et al. [18] using 11B single-crystal SSNMR spectroscopy with Ω¼5.4�0.6 ppm and κ¼0.8�0.2 as converted from Haeberlen convention.hCSA parameters were obtained byHansen et al. [18] using 11B single-crystal SSNMR spectroscopy withΩ¼13.0�0.9 ppm and κ¼�0.2�0.1 as converted fromHaeberlen convention.iAverage value of the three sites.jCSA parameters were obtained by Hansen et al. [18] using 11B single-crystal SSNMR spectroscopy with Ω¼8.6�1 ppm and κ¼0.2�0.1 as converted from Haeberlen convention.kCSA parameters were obtained by Hansen et al. [18] using 11B MAS SSNMR spectroscopy with Ω¼10.6�2.5 ppm and κ¼�0.6�0.1 as converted from Haeberlen convention.lCSA parameters were obtained by Hansen et al. [18] using 11B single-crystal SSNMR spectroscopy with Ω¼8.0�0.7 ppm and κ¼0.2�0.2 as converted from Haeberlen convention.mCSA parameters were obtained by Hansen et al. [18] using 11B single-crystal SSNMR spectroscopy with δσ¼6.0�0.6 ppm and ησ¼0.4�0.1.nδσ was obtained by Hansen et al. [18] using 11B SATRAS (δσ¼�5�3 ppm) and single-crystal SSNMR spectroscopy (δσ¼�4.6�0.4). ssb intensities of the SATRAS spectrum werefound to be insensitive to changes in ησ, while ησ was found to be 0.4�0.2 by 11B single-crystal SSNMR.
Table 5 11B EFG and CS Tensor Parameters for Boron Heterocycles Obtained Using 11B SSNMR SpectroscopyCompound δiso (ppm) CQ (MHz) η Method References
(4-MeC6H4)3B3O3 24.7 3.0 0.5 MAS Beckett et al. [86]
(3,5-Me2C6H3)3B3O3 25.1 3.0 0.5 MAS Beckett et al. [86]
Boroxine cycle of bortezomib form I site 1 9.5�0.2 1.7�0.3 — MQMAS Brus et al. [87]
Boroxine cycle of bortezomib form I site 2 24.9�0.2 2.9�0.3 — MQMAS Brus et al. [87]
Boroxine cycle of bortezomib form I site 3 28.9�0.2 3.0�0.3 — MQMAS Brus et al. [87]
Boroxine cycle of bortezomib form II site 1 10.7�0.2 1.5�0.3 — MQMAS Brus et al. [87]
Boroxine cycle of bortezomib form II site 2 16.5�0.2 2.1�0.3 — MQMAS Brus et al. [87]
Boroxine cycle of bortezomib form II site 3 27.5�0.2 3.0�0.3 — MQMAS Brus et al. [87]
Hexamethylborazinea,b 36.0�0.4 2.98�0.03 0.01�0.01 MAS Forgeron et al. [36]
Hexaphenylborazine 36 3.12 0.24 MAS T€onshoff et al. [88]
B-Triphenylborazine — 3.09�0.03 0.18�0.04 MAS Forgeron et al. [36]
Trialcynylborazine 24 2.8 0 MAS Gervais et al. [50]
Trichloroborazine 31.0�0.5 2.47�0.03 0.78�0.05 MAS Perras and Bryce [30]
Continued
Table 5 11B EFG and CS Tensor Parameters for Boron Heterocycles Obtained Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
B3S3(SH)3c 50 2.54 0 Static Conrady-Pigorsch et al. [89]
B3S3(C6H5)3d 45 3.03 0.58 Static Conrady-Pigorsch et al. [89]
B2Se3(C6H5)2e 42 2.65 0.7 Static Conrady-Pigorsch et al. [89]
B2Se3I2 �14 0.25 — Static Conrady-Pigorsch et al. [89]
aSample exhibits molecular dynamics; reported parameters were extracted from spectra obtained at 298 K.bCSA parameters were obtained by Forgeron et al. [36] using 11B SSNMR spectroscopy under static conditions with Ω¼55�15 ppm and κ¼1.00�0.01.cCSA parameters were obtained by Conrady-Pigorsch et al. [89] using 11B SSNMR spectroscopy under static conditions with Ω¼88 ppm and κ¼0.43 as convertedfrom Haeberlen convention.dCSA parameters were obtained by Conrady-Pigorsch et al. [89] using 11B SSNMR spectroscopy under static conditions with Ω¼96 ppm and κ¼0.75 as convertedfrom Haeberlen convention.eCSA parameters were obtained by Conrady-Pigorsch et al. [89] using 11B SSNMR spectroscopy under static conditions with Ω¼82 ppm and κ¼0.87 as convertedfrom Haeberlen convention.
Table 6 Isotropic 11B Chemical Shifts and the Corresponding 11B EFG Tensor Parameters for Boron-Containing Lewis Acid/Base AdductsObtained Using 11B SSNMR SpectroscopyCompound δiso (ppm) CQ (MHz) η Method References
(C6F5)3B�PPh3 �7.4�0.5 1.59�0.05 — MAS Wiegand et al. [33]
— 1.63�0.05 0.15�0.1 MQMAS Wiegand et al. [33]
(C6F5)3B�PPh2C^CMe �8.0�0.5 1.54�0.05 0.16�0.1 MAS Wiegand et al. [6]
(C6F5)3B�PPh2C^CPr �7.4�0.5 1.64�0.05 0.24�0.1 MAS Wiegand et al. [6]
H3B�P(PhO)3 3.0�0.1 1.22�0.02 0.10�0.05 MAS Wu et al. [39]
H3B�NH3 �23 1.5 0 MAS Gervais et al. [50]
H3B�NH2Me �18 1.6 0 MAS Gervais et al. [50]
H3B�NHMe2 �15 1.4 0 MAS Gervais et al. [50]
H3B�NMe3 �8 1.6 0 MAS Gervais et al. [50]
— 1.67�0.05 0 MQMAS Wi and Frydman [37]
H3B�NH2tBu �22 1.6 0 MAS Gervais et al. [50]
H3B�N(C5H3)Me2 — 1.8 0.2 MQMAS Wi and Frydman [37]
(4-MeC6H4)3B3O3�cyclohexylamine three-coordinate
site
31.7 2.4 1.0 MAS Beckett et al. [86]
(4-MeC6H4)3B3O3�cyclohexylamine four-coordinate site 3.0 1.6 0.5 MAS Beckett et al. [86]
(4-MeC6H4)3B3O3�isoquinoline three-coordinate site 32.5 2.5 1.0 MAS Beckett et al. [86]
(4-MeC6H4)3B3O3�isoquinoline four-coordinate site 5.8 1.5 0.6 MAS Beckett et al. [86]
(4-MeC6H4)3B3O3�benzylamine three-coordinate site 31.0 2.4 1.0 MAS Beckett et al. [86]
(4-MeC6H4)3B3O3�benzylamine four-coordinate site 1.9 1.5 0.6 MAS Beckett et al. [86]
Continued
Table 6 Isotropic 11B Chemical Shifts and the Corresponding 11B EFG Tensor Parameters for Boron-Containing Lewis Acid/Base AdductsObtained Using 11B SSNMR Spectroscopy—cont’dCompound δiso (ppm) CQ (MHz) η Method References
(3,5-Me2C6H4)3B3O3�4-picoline three-coordinate site 31.3 2.4 1.0 MAS Beckett et al. [86]
(3,5-Me2C6H4)3B3O3�4-picoline four-coordinate site 4.8 1.5 0.6 MAS Beckett et al. [86]
(3,5-Me2C6H4)3B3O3�morpholine three-coordinate site 31.1 2.4 1.0 MAS Beckett et al. [86]
(3,5-Me2C6H4)3B3O3�morpholine four-coordinate site 3.3 1.5 0.4 MAS Beckett et al. [86]
BF3�pyridine 0.10 0.280�0.01 0.25�0.05 SATRAS Ch�enard et al. [90]
BF3�4-picoline �0.10 0.270�0.01 0.10�0.05 SATRAS Ch�enard et al. [90]
BF3�quinoline 0.50 0.310�0.01 0.20�0.05 SATRAS Ch�enard et al. [90]
BF3�isoquinoline 0.40 0.270�0.01 0.10�0.05 SATRAS Ch�enard et al. [90]
1,4-bis-BF3�pyrazine 0.00 0.650�0.01 0.05�0.05 SATRAS Ch�enard et al. [90]
1,4-bis-BF3�quinoxaline 0.50 0.680�0.01 0.05�0.05 SATRAS Ch�enard et al. [90]
4-BF3�2-methylquinoxaline 0.75 0.460�0.01 0.10�0.05 SATRAS Ch�enard et al. [90]
H3B�AsPh3 �35.4 1.9 0 MQMAS Wi et al. [38]
(C6F5)3B�AsMe3 �13.2�0.5 1.30�0.01 0�0.06 MAS Faucher et al. [91]
(C6F5)3B�AsEt3 �12.2�0.5 1.34�0.02 0�0.1 MAS Faucher et al. [91]
(C6F5)3B�AsPh3 �3.1�0.5 2.05�0.02 0�0.05 MAS Faucher et al. [91]
Ph3B�AsMe3 �0.25�0.5 2.0�0.1 0�0.1 MAS Faucher et al. [91]
Ph3B�AsPh3 9.3�0.5 2.8�0.1 0�0.1 MAS Faucher et al. [91]
H3B�AsPh3 �34.6�0.5 1.85�0.05 0�0.1 MAS Faucher et al. [91]
Table 7 11B EFG and CS Tensor Parameters for Boron-Containing Frustrated Lewis Pairs (FLPs) and Corresponding Adducts and OligomersCompound δiso (ppm) CQ (MHz) η Method References
(C6F5)2B-(C6F5)C]C(Ph)-PMes2 0.3�0.5 1.55�0.05 0.19�0.1 MQMAS Wiegand et al. [33]
— 1.54�0.05 — MAS
(C6F5)2B-(C6F5)C]C(Ph)-PPh2 site 1 �7.6�0.5 1.25�0.04 0.18�0.1 MQMAS Wiegand et al. [33]
(C6F5)2B-(C6F5)C]C(Ph)-PPh2 site 2 �5.9�0.5 1.36�0.04 0.15�0.1 MQMAS Wiegand et al. [33]
(C6F5)2B-(C6F5)C]C(n-Pr)-PPh2 �6.6�0.5 1.35�0.04 0.15�0.1 MQMAS Wiegand et al. [33]
— 1.34�0.04 — MAS
(C6F5)2B-(C6F5)C]C(PPh2)-PPh2 �4.7�0.5 1.31�0.04 0.15�0.1 MQMAS Wiegand et al. [33]
— 1.31�0.04 — MAS
(C6F5)2B-(C6F5)C]C(Me)-PPh2 �7.5�0.5 1.27�0.04 0.16�0.1 MQMAS Wiegand et al. [33]
— 1.25�0.04 — MAS
(C6F5)2B-(C6F5)C]C(Tol)-PMes2 0.3�0.5 1.57�0.05 0.17�0.1 MQMAS Wiegand et al. [33]
— 1.55�0.05 — MAS
(C6F5)2B-(C6F5)C]C(p-Tol)-PPh2 �8.4�0.5 1.2�0.1 0.2�0.1 MAS Ekkert et al. [34]
tBuN^C:B(C6F5)2-(C6F5)C]C(p-Tol)-PPh2 �19.5�0.5 1.0�0.1 0.4�0.1 MAS Ekkert et al. [34]
(C6F5)2B-(C6F5)C]C(p-Tol)-PPh2/nBuNC cyclic
product
�14.0�0.5 0.1�0.1 0.5�0.1 SATRAS and
MAS
Ekkert et al. [34]
(C6F5)2B-(Me)C]C(p-Tol)-PPh2 �9.3�0.5 1.4�0.1 0.3�0.1 MAS Ekkert et al. [34]
Continued
Table 7 11B EFG and CS Tensor Parameters for Boron-Containing Frustrated Lewis Pairs (FLPs) and Corresponding Adducts and Oligomers—cont’dCompound δiso (ppm) CQ (MHz) η Method References
tBuN^C:B(C6F5)-(Me)C]C(p-Tol)-PPh2 �18.8�0.5 1.1�0.1 0.6�0.1 MAS Ekkert et al. [34]
(C6F5)2B-((CH2)2PMes2)C]C(Me3Si)-PPh2 �7.5�0.5 1.34�0.04 0.30�0.1 — Wiegand et al. [7]
(C6F5)2B-((CH2)2P(O)Mes2)C]C(Me3Si)-PPh2 �7.4�0.5 1.31�0.04 0.32�0.1 — Wiegand et al. [7]
(C6F5)2B-((CH2)2P(O)Mes2)C]C(Me3Si)-PPh2/
NOH adduct
�6.2 0.71 0.82 — Wiegand et al. [7]
(C6F5)2B-((CH2)2Ph)C]C(Me3Si)-PPh2 �7.1�0.5 1.31�0.04 0.41�0.1 — Wiegand et al. [7]
(C6F5)2P–B(C6F5)2C]CH-Me site 1 60.1�0.5 4.15�0.12 0.15�0.1 MAS Wiegend et al. [6]
(C6F5)2P–B(C6F5)2C]CH-Me site 2 59.0�0.5 4.25�0.13 0.2�0.1 MAS Wiegend et al. [6]
(C6F5)2P–B(C6F5)2C]CH-Pha 57.9 4.2 0.14 MAS Rosorius et al. [92]
(C6F5)2B-(CH2)3-PPh2 �9.1�0.5 1.43�0.04 0.55�0.1 MAS Wiegand et al. [33]
(C6F5)2B-(CH2)2-PMes2 3.3�0.5 1.8�0.05 0.6�0.1 MAS Wiegand et al. [33]
(C6F5)2B-(CH2)2-PMes2/SO2 cyclic adduct
(i.e., [B]-O-(O)S-[P]) epimer 1
1.0�0.5 1.62�0.16 0.46�0.1 MAS Sajid et al. [93]
(C6F5)2B-(CH2)2-PMes2/SO2 cyclic adduct
(i.e., [B]-O-(O)S-[P]) epimer 2
1.0�0.5 1.92�0.19 0.48�0.1 MAS Sajid et al. [93]
1-B(C6F5)2-2-PMes2 cyclohexane 8.6�0.5 2.10�0.06 0.43�0.1 MAS Wiegand et al. [33]
1-B(C6F5)2-2-PMes2 cyclohexane/cyclic SO2 adduct
([B]-O-(O)S-[P] isomer)
0.9�0.5 1.88�0.18 0.50�0.1 MAS Sajid et al. [93]
2-B(C6F5)2-3-PMes2-norbornaneb 75.2�0.5 4.66�0.23 0.22�0.1 MAS Sajid et al. [94]
2-B(C6H5)2-3-PMes2-norbornane/SO2 cyclic adduct
[B]-O-S(O)-[P] epimer 1
2.4�0.5 2.15�0.11 0.47�0.1 MAS Sajid et al. [94]
2-B(C6H5)2-3-PMes2-norbornane/SO2 cyclic adduct
[B]-O-S(O)-[P] epimer 2
�0.2�0.5 1.71�0.09 0.38�0.1 MAS Sajid et al. [94]
2-B(C6H5)2-3-PMes2-norbornane/NO cyclic adduct �6.6�0.5 1.06�0.03 0.72�0.1 MAS Wiegand et al. [95]
2-B(C6H5)2-3-PMes2-norbornane/NOH cyclic adduct �6.6�0.5 0.97�0.03 0.83�0.1 MAS Wiegand et al. [95]
H(C6F5)B-CH2-C(Me)H-P(C6F5)2 cyclotrimer �14.6�0.5 1.69�0.05 0.35�0.1 MAS Erdmann et al. [96]
aCSA parameters were obtained by Rosorius et al. [92] using 11B MAS SSNMR spectroscopy with Δσ¼91.3 ppm and ησ¼0.16.bCSA parameters were obtained by Sajid et al. [94] using 11B MAS SSNMR spectroscopy with Δσ¼122.5�20 ppm and ησ¼0.60�0.1.
Table 8 11B EFG and CS Tensor Parameters for Metal Borohydrides Obtained Using 11B SSNMR SpectroscopyCompound δiso (ppm) CQ (MHz) η Method References
α-Y(BH4)3a �17.3 0.0774 0.97 MAS Ravnsbæk et al. [97]
Y(BH4)3�7NH3a �36.6�0.2b–37.1�0.2b 0.084�0.010c 0.40�10c SATRAS Jepsen et al. [98]
Y(BH4)3�6NH3a �37.4�0.2 — — MAS Jepsen et al. [98]
Y(BH4)3�5NH3a �28.6�0.2 — — MAS Jepsen et al. [98]
Y(BH4)3�4NH3a �27.5�0.2 0.402�0.020 0.23�0.05 SATRAS Jepsen et al. [98]
NaY(BH4)4 site 1 �23.9�0.2 0.635�0.004 0.07�0.03 SATRAS Roedern et al. [99]
NaY(BH4)4 site 2 �26.9�0.2 0.445�0.004 0.17�0.03 SATRAS Roedern et al. [99]
LiBH4a �41.2 0.090 0.94 MAS Ravnsbæk et al. [97]
o-LiBH4a,d �41.0�0.5 0.099�0.003 0.91�0.2 MAS Arnbjerg et al. [100]
LiZn2(BD4)5 site 1a �42.3 0.945 0.09 SATRAS Ravnsbæk et al. [101]
LiZn2(BD4)5 site 2a �43.1 0.748 0.32 SATRAS Ravnsbæk et al. [101]
LiZn2(BD4)5 site 3a �46.0 0.553 0.19 SATRAS Ravnsbæk et al. [101]
LiZn2(BD4)5 site 4a �43.3 0.879 0.06 SATRAS Ravnsbæk et al. [101]
aData obtained at ambient and/or room temperature.bSites 1 and 2.cAverage values for different 11B sites.dCSA parameters were obtained by Arnbjerg et al. [100] using 11BMAS SSNMR spectroscopy withΩ¼59�6 ppm and κ¼�0.07�0.15 as converted fromHaeberlenconvention.
Table 9 11B EFG and CS Tensor Parameters for Metal Borides Obtained Using 11B SSNMRSpectroscopy
Compoundδiso(ppm) CQ (MHz) η Method References
YB4 site 4e — 1.03�0.06a 0.00
+0.02/�0.00
Static J€ager et al. [102]
YB4 site 4h — 1.40�0.06a 0.02�0.02 Static J€ager et al. [102]
YB4 site 8j — 1.03�0.08a 0.46�0.08 Static J€ager et al. [102]
YB6 — 1.20�0.03a 0 Static J€ager et al. [102]
YB12 — 1.06�0.08b 0.93 Static J€ager et al. [103]
— 1.08a 0.94 Single
crystal
Fojud et al. [19]
ZrB12 — 1.08�0.08b 0.94 Static J€ager et al. [103]
LuB12 — 1.12�0.08b 0.98 Static J€ager et al. [103]
ReB2c 5 0.550 0 Static Koumoulis et al.
[104]
7.6 — 0 MAS Koumoulis et al.
[104]
— 0.552�0.006a 0 Single
crystal
Zogał et al. [22]
OsB2 — 0.570�0.020a 0.8–0.9 Static Suh et al. [105]
RuB2 — 0.570�0.020a 0.8–0.9 Static Suh et al. [105]
MgB2 — 1.67�0.02a 0 Static Baek et al. [106]
AlB2 — 1.08�0.04a 0 Static Burkhardt et al.
[107]
— 1.08�0.02a 0 Static Baek et al. [106]
LaB6 — 1.03a 0 Single
crystal
Herzig et al. [20]
LaB4 site 4e 42 0.69 0.00 SATRAS Schmitt et al.
[108]
LaB4 site 4h 47 1.1 0.05 SATRAS Schmitt et al.
[108]
LaB4 site 8j 18 0.8 0.5 SATRAS Schmitt et al.
[108]
CaB4�xCx
site 8j
11 0.8 0.3 SATRAS Schmitt et al.
[108]
aConverted from νQ using CQ¼2νQ.bCalculated from the experimentally observed value for jV33 j.cCSA parameters were obtained by Koumoulis et al. [104] using 11B MAS SSNMR spectroscopy withΩ¼150 ppm and κ¼�1.
4.1.1 Borane Derivatives, Diboron Compounds, Tetrahedral BoronAnions, and Boron-Containing Zwitterions
Data for this section are reported in Table 2. One of the earliest experimental
characterizations of 11B CSA parameters was performed on trimesitylborane
by Bryce et al. [46]. The boron CS and EFG tensor parameters were
extracted from the MAS and static spectra of a powdered sample at multiple
magnetic fields. CSA at the boron site was clearly present as reflected by the
large Ω(11B) value (121 ppm, Fig. 2), which covered the range of the
δiso(11B) values known at the time of the study for tricoordinate boron com-
pounds. On the other hand, the Ω(11B) of the analogous borate ester com-
pound, triphenyl borate, was found to be significantly smaller (Ω of
�10 ppm, Fig. 2). The authors rationalized the large difference in anisotropic
boron shielding between the two compounds via Ramsey’s theory of nuclear
magnetic shielding. For trimesitylborane, there is a significant paramagnetic
contribution to σ11 and σ22 due to the mixing of the vacant boron pz orbital
with the occupied orbitals, as indicated by the favourable overlap of the pzorbital with occupied orbitals by virtual rotation in the xy plane. Conversely,
the lone pairs on the oxygen of the borate ester can donate electron density
into the empty boron pz orbital, therefore the corresponding boron pz orbital
Table 10 11B EFG and CS Tensor Parameters for Boron Carbides, Nitrides, and RelatedSystems Measured Using 11B SSNMR Spectroscopy
Compoundδiso(ppm) CQ (MHz) η Method References
LiBC 49 2.80�0.05 0.1�0.1 MAS Langer et al.
[109]
B4C sites 6h1and 6h2
�5.0 0.84 0 MAS and
MQMAS
Kanehashi and
Saito [110]
SiB4 sites 6h1and 6h2
— 1.24 0 MQMAS Kanehashi and
Saito [110]
h-BN — 3.2 0 MQMAS Kanehashi and
Saito [110]
SrBa8[BN2]6 26.6�1 3.30�0.05 0.2�0.03 MAS Seidel et al.
[111]
EuBa8[BN2]6 26.6�1a 3.10�0.05a 0.03�0.03a MAS Seidel et al.
[111]
24.3�1b 3.10�0.05b 0.03�0.03b
aExtracted from spectrum recorded at B0¼4.7 T.bExtracted from spectrum recorded at B0¼11.7 T.
246 Ying-Tung Angel Wong and David L. Bryces
Table 11 11B EFG and CS Tensor Parameters for Boron Metallocene Complexes Measured Using 11B SSNMR SpectroscopyCompound δiso (ppm) Ω (ppm) κ CQ (MHz) η Method References
[Cp∗2B][AlCl4]a �41.3 �0.1 73.0 �0.3 0.98 �0.02 1.14 �0.01 0.10 �0.04 MAS and Static Schurko et al. [112]
Cp∗2BMe 81.9 �0.1 146.1 �0.3 0.75 �0.04 4.52 �0.02 0.11 �0.01 MAS and Static Schurko et al. [112]
aSample exhibits molecular dynamics.
is unavailable for virtual rotation. Consequently, trimesitylborane exhibits a
larger Ω(11B) compared to triphenyl borate.
Since the study by Bryce et al. [46], 11B CSA parameters have been mea-
sured for several compounds. For instance, Alain et al. [48] characterized the
CS and EFG tensors for the dimesitylborinium cation, and similar to trime-
sitylborane, a largeΩ(11B) was observed and attributed to paramagnetic con-
tributions. Experimental values were compared to those obtained from
ab initio and DFT calculations on an isolated dimesitylborinium cation
(i.e., using RHF and B3LYP methods), as well as the full unit cell crystal
structure of the sample (i.e., using GIPAW calculations). The authors note
that while the calculated CQ(11B) value is influenced by the presence of the
counteranion, this effect is less significant for the 11B CS tensors. Moreover,
a comparison between the dimesitylborinium cation and other systems with
boron directly bonded to carbon ligands (i.e., trimesitylborane [46] and
Cp∗2B+½ AlCl4�½ [112]) showed that the orientation and magnitude of
the CS tensors are sensitive to the identity of the ligand. Analogous conclu-
sions were also obtained for EFG tensors. These results were supported by
theoretical studies, where the 11B magnetic shielding and EFG tensors were
found to be sensitive to the C–B+–C angle (Fig. 3), as well as the identity of
the ligand attached to the boron centre. The ability of EFG tensor param-
eters to reflect the boron bonding environment was also illustrated for
diboron compounds [42]. It was found that by coordinating an additional
ligand to one of the boron centres (e.g., by adding a ligand to one of the
three-coordinate boron centres in bis(catecholato) diboron, resulting in
Fig. 2 11B static SSNMR spectra of trimesitylborane (left) and triphenyl borate (right)acquired at B0¼17.63 T. The corresponding simulated spectra obtained using param-eters shown in Tables 2 and 3 (middle trace), and by assuming a span of 0 ppm are alsoprovided (bottom trace). From D.L. Bryce, R.E. Wasylishen, M. Gee, Characterization oftricoordinate boron chemical shift tensors: definitive high-field solid-state NMR evidencefor anisotropic boron shielding, J. Phys. Chem. A 105 (2001) 3633–3640.
248 Ying-Tung Angel Wong and David L. Bryces
the formation of [bis(catecholato) diboron]�IMes which consists of three-
and four-coordinate boron sites), the CQ and η values for the remaining
three-coordinate boron site were measured to increase significantly even
though the immediate bonding interactions at this site were unaltered.
This was rationalized based on the polarization of the BdB bond. By adding
an extra ligand, a large positive charge is induced on the resulting
Fig. 3 Theoretical dependence of the principal components of the 11B EFG (V11, V22, andV33) and magnetic shielding (σ11, σ22, and σ33) tensors on the C–B+–C angle (θ) in thediphenylborinium cation calculated by Alain et al. [48] using the B3LYP level of theorywith the 6–311++G(d,p) basis set. The CBCC dihedral angle was fixed at 90 degrees, andthe fitted trend lines are given as follows: V33¼ (�1.53�10�5)(θ2)+ (5.50�10�3)(θ)+1.43�10�1, R2¼1.00; V22¼ (�3.43�10�6)(θ2)+ (1.22�10�3)(θ) �4.28�10�1, R2¼1.00; V11¼ (1.87�10�5)(θ2)� (6.72�10�3)(θ)+2.85�10�1, R2¼1.00; σ33¼ (�5.22�10�3)(θ2)+ (1.82�100)(θ) �6.18�101, R2¼0.991; σ22¼ (�6.07�10�3)(θ2)+ (2.21�100)(θ) �2.28�102, R2¼0.999; and σ11¼ (�1.01�10�2)(θ2)+ (3.67�100)(θ) �3.62�102,R2¼0.999. From A.E. Alain, Y. Shoji, T. Fukushima, D.L. Bryce, 11B solid-state NMR interac-tion tensors of linear two-coordinate boron: the dimesitylborinium cation, Inorg. Chem.54 (2015) 11889–11896.
249Recent Advances in 11B SSNMR Spectroscopy
four-coordinate boron centre, thereby increasing the magnitude of jV22 j forthe three-coordinate boron (which points along the BdB bond), resulting
in a larger value of η (Eq. 9).
4.1.2 Acids, Esters, and Related SystemsAcids and esters which contain boron have been the focus of several 11B
SSNMR investigations, and the measured tensors have been shown to
reflect the immediate bonding environments of the borons in these com-
pounds (see data in Table 3). A systematic study by Weiss and Bryce [49]
on a series of boronic acid and boronic esters with aromatic substituents
showed that the experimental CQ(11B) values are, in general, larger for
the acids. An analogous trend was also observed for the spans. Furthermore,
a comparison between theΩ values for boronic acids and esters to those of a
borane and a borate ester reveals that an increase in the number of oxygens
bonded to the boron drastically decreases the value of Ω (Fig. 4) since the
140
120
100
80
W (p
pm
)
60
Borate
Boronicester
Boronicacid
Borane
40
20
0
Fig. 4 Graphical representation of the experimentally measured Ω for the boronicesters and acids investigated by Weiss and Bryce [49], and the borane and borate ester(labelled as borate) studied by Bryce et al. [46], illustrating the decrease inΩ as the num-ber of oxygen atoms bonded to the boron centre increases. The data points for theboronic acids and esters indicate the average values for different compounds andthe vertical bars show the range of Ω, while the data points for the borate esterand the borane indicate single experimental measurements and the vertical bars showthe experimental measurement errors. From J.W.E. Weiss, D.L. Bryce, A solid-state 11B NMRand computational study of boron electric field gradient and chemical shift tensors inboronic acids and boronic esters, J. Phys. Chem. A 114 (2010) 5119–5131.
250 Ying-Tung Angel Wong and David L. Bryces
presence of the lone pair on the oxygen decreases the paramagnetic contri-
bution to the total σ. Using phenylboronic acid as a model system, the
authors performed computational investigations on how the dihedral angle
(ΦCCBO), the presence of hydrogen bonding interactions, and the identity of
the substituents on the aromatic ring would influence the value of Ω(11B).
They found that the calculatedΩ correlates positively with ΦCCBO, and the
presence of hydrogen bonding interactions and electron-withdrawing sub-
stituents would also result in a larger Ω (Fig. 5). However, the presence of
aromatic rings with electron-donating groups has a less pronounced effect.
Therefore, it was concluded that the magnitude of the Ω is dependent on
multiple factors, and this 11B NMR parameter is the most diagnostic for
the boron bonding environment in boronic acids and esters. The depen-
dence of the Ω on ΦCCBO has also been observed for arylboronic acid
45.0
40.0
35.0
30.0
25.0
20.0
15.00 10 20 30 40 50
Dihedral (degree)
60 70
OH
BOH
80 90
W (p
pm
)
Fig. 5 Correlation between the theoretical Ω(11B) and the ΦCCBO, as defined by theatoms highlighted in red, for monomeric and dimeric forms of phenylboronic acid.Results obtained from Gaussian calculations (B3LYP) for a monomer and a dimer arerepresented by diamonds and squares, respectively, while results obtained from ADF(GGA-revPBE) calculations for a monomer and a dimer are represented by trianglesand circles, respectively. From J.W.E. Weiss, D.L. Bryce, A solid-state 11B NMR and compu-tational study of boron electric field gradient and chemical shift tensors in boronic acidsand boronic esters, J. Phys. Chem. A 114 (2010) 5119–5131.
251Recent Advances in 11B SSNMR Spectroscopy
and the corresponding catechol cyclic esters, and the behaviour in which the
Ω varies as a function of theΦCCBO was found to be dependent on the func-
tional group substituted on the phenyl ring, as well as the position of the
substitution [32].
Besides boronic acids and boronic esters, benzoxaboroles, benzoxa-
borolates (the anion of benzoxaboroles), and boronates (the anion of boronic
acids) have also been characterized using 11B SSNMR spectroscopy [51–56],and a detailed discussion on these compounds has been provided in Ref. [9].
The quadrupolar nature of 11B makes it an ideal probe for the characteriza-
tion of these compounds since it provides different NMR observables for11B atoms that are in a tetrahedral environment and those that are in a planar
environment. For example, tetrahedral benzoxaborolates and boronates,
in which the 11B atoms are in a tetrahedral environment, have smaller
CQ(11B) values compared to boronic acids and benzoxaboroles, in which
the 11B atoms are in a planar environment (CQ1.1–1.5 MHz vs CQ2.8–3.3 MHz) [9]. The 11B NMR parameters for a planar benzoxaborolate
also differ from that of the corresponding tetrahedral benzoxaborolate [53].
For instance, in CaBBzx�3H2O, the BBzx benzoxaborolate anions were
found to exist in planar and tetrahedral configurations, and the δiso(11B)
and the CQ(11B) values were measured to be significantly different bet-
ween the two. The planar anion gave δiso and CQ values that are closer
to those of the neutral analogue (δiso¼30.7 ppm and CQ¼2.87 MHz for
the planar benzoxaborolate, δiso¼31.1 ppm and CQ¼2.90 MHz for the
benzoxaborole), while the δiso and the CQ of the tetrahedral anion were
observed to be significantly smaller (δiso¼9.3 ppm and CQ¼1.30 MHz,
see Fig. 6) [53].
4.1.3 Borates, Borosilicates, Thioborates, and Related Systems11B SSNMR is a powerful tool for characterizing different boron species in
borates since the NMR observables for BO3 units can differ significantly
from those of BO4� units. Data for this section are reported in Table 4.
For BO3, the δiso(11B) values are typically in the range of c.12–19 ppmwhile
the boron nuclei in BO4� are more shielded, resulting in δiso(
11B) values
ranging from c.2 to �4 ppm [10]. Besides the boron coordination number,11B CS tensor parameters can also be correlated to other structural features of
the borates. For instance, insights into the number of nonbridging oxygens
in tricoordinate borates can be obtained as illustrated by Kroeker and
Stebbins [63]. First, it was observed that the δiso(11B) value generally
increases as the number of nonbridging oxygen increases. For compounds
252 Ying-Tung Angel Wong and David L. Bryces
with the same number of nonbridging oxygens, borons which reside in a
ring structure are more deshielded as compared to those in a chain. There-
fore, within the collection of the compounds studied, δiso(11B) was found to
have the general trend of δiso(T0)�δiso(T
1)>δiso(T2(ring))>δiso(T
2)�δiso(T
3(ring))>δiso(T3), where the superscript n in Tn denotes the number
of bridging oxygens. However, no trend was observed between δiso and
average bond length or B–O–B and O–B–O angles. By accounting for sec-
ond neighbour effects using bond valence sums, in which the cation–oxygenbond strengths surrounding a given tricoordinate boron are added, the
authors explained that the trend between δiso(11B) and the number of
B
CaBBzx·3H2OA MgBBzx·10H2O MgBBzx·7H2O
BCHMg/CaO
CaBBzx·3H2O
O
B
OHOH
MgBBzx·7H2O
B
O
O
B
OH
O
Planarbenzoxaborolate
Benzoxaborole(acid from)
40 30 20 10
11B chemical shift (ppm)
BBzx
Tetrahedralbenzoxaborolate
0 −10
−
−
Fig. 6 Coordination modes of the benzoxaborolates in CaBBzx �3H2O, MgBBzx �10H2O,andMgBBzx �7H2O (A) and the 11B SSNMR spectra for CaBBzx �3H2O, MgBBzx �7H2O, andBBzx recorded under MAS (B). From S. Sene, S. B�egu, C. Gervais, G. Renaudin, A. Mesbah,M.E. Smith, P.H. Mutin, A. van der Lee, J.-M. Nedelec, C. Bonhomme, D. Laurencin, Interca-lation of benzoxaborolate anions in layered double hydroxides: toward hybrid formulationsfor benzoxaborole drugs, Chem. Mater. 27 (2015) 1242–1254.
253Recent Advances in 11B SSNMR Spectroscopy
nonbridging oxygen results from the long-range atomic structures rather
than the degree of network connectivity of the boron atoms (Fig. 7). More-
over, a full shielding tensor analysis revealed that Ω changes considerably
between compounds bearing one or two nonbridging oxygens and com-
pounds with zero or three nonbridging oxygens. κ was also found to vary
between these borates. Therefore, the authors concluded that the small
changes observed in δiso were a result of significant changes in some or all
of the CS principal tensor components, and in order to understand the rela-
tion between magnetic shielding and structure, a full tensor analysis would
be the most informative. On the other hand, Alderman et al. [67] showed
that correlations between NMR parameters and the local geometry of the
boron can be obtained if high-resolution NMR techniques were employed.
In this study, they investigated systems where correlations between δiso andstructural parameters are difficult to obtain by MAS due to peak overlap as a
result of the small CS range (<4 ppm) and the quadrupolar nature of boron.
The samples consisted of borates with trigonal and tetrahedral boron sites,
24
22
20
18d iso
(pp
m)
16
145.8 6.0 6.2
Sum of cation–oxygenbond strengths (valence units)
LaB
2MgBNaB
CaBCs9B
B2O3
LiB
KB
2NaB
3MgB
6.4
Fig. 7 Relationship between δiso(11B) and the sum of cation–oxygen bond strengths
for binary borates (LaB¼ La2O3 �B2O3; 2MgB¼2MgO �B2O3; 3MgB¼3MgO�B2O3;NaB¼ Na2O�B2O3; 2NaB¼2Na2O�B2O3; KB¼ K2O�B2O3; CaB¼ CaO �B2O3; LiB¼ Li2O�B2O3;Cs9B¼ Cs2O�9B2O3). Unless indicated, uncertainties in δiso(
11B) are within the bounds ofthe mark. The data point for KB is from Ref. [113]. Reprinted from S. Kroeker, J.F. Stebbins,Three-coordinated boron-11 chemical shifts in borates, Inorg. Chem. 40 (2001) 6239–6246.
254 Ying-Tung Angel Wong and David L. Bryces
where the trigonal borate is in a T3(ring) and/or T3 structure and non-
bridging oxygens are absent. In order to overcome peak overlap, MQMAS
and DOR were employed together with isotopic dilution (see Fig. 8 for
example). This allowed for highly accurate measurements of CSs and EFG
tensor parameters, and excellent agreement between experiment and theo-
retical values was obtained. As such, peak assignments to specific sites were
accomplished, thereby enabling the correlation between δiso(11B) and (1)
the average B–O–B bond angle for both tricoordinate and tetracoordinate
boron sites and (2) the T3(ring) unit trigonal planar angular deviation.
δiso(11B) can also be employed for identifying boron species with dif-
ferent second nearest neighbour environments. For instance, it was found
that δiso(11B) can be used to discriminate between borophosphates with
Fig. 8 11B 3QMAS spectra of (A) CsB3O5, (B) K2B4O7, and (C) α-BaB4O7 recorded at11.75 T. Blue spectra in (D) to (F) are the corresponding 3QMAS slices at the indirect fre-quencies of the resolved tricoordinate boron resonance, and the associated fits aregiven in red. MAS spectra (black) are also provided for comparison. From O.L.G.Alderman, D. Iuga, A.P. Howes, K.J. Pike, D. Holland, R. Dupree, Spectral assignmentsand NMR parameter–structure relationships in borates using high-resolution 11B NMRand density functional theory, Phys. Chem. Chem. Phys. 15 (2013) 8208–8221.
255Recent Advances in 11B SSNMR Spectroscopy
different numbers of B–O–P linkages [81]. In the case of borosilicate
zeolites, it has been observed that boron species with different numbers
of B–O–Si linkages can form by dehydration and/or hydration
[73,77,79,80] and that they can be differentiated based on their δiso(11B)
values [73,77]. For example, in B-beta zeolites, B(OSi)3 which forms from
the dehydration of B(OSi)4 can be converted to boron species with differ-
ent numbers of B–O–Si bonds (i.e., B(OSi)x(OH)3�x, x¼0–3) via hydro-lysis [77,79]. These intermediate species have been identified in various11B SSNMR experiments and it was found that the CSs can be used to
distinguish between borons with different degrees of anchoring in the zeo-
lite framework [73,77].11B EFG tensor parameters can also provide valuable information on the
coordination state of borates. For instance, in borosilicate zeolites, frame-
work borons can be trigonally (B[3]) and/or tetrahedrally coordinated
(B[4]). These two species can be easily differentiated by their corresponding
CQ(11B) values since the resonance for B[3] can be expected to be dominated
by second-order quadrupolar broadening, while a sharp line can be antici-
pated for B[4]. In a study performed by Fild et al. [73], which investigated the
boron coordination in a series of borosilicate zeolites using 11B SSNMR, it
was found that for zeolites with larger countercations (e.g., Li+, Na+, NH4+,
and H+(H2O)n clusters), the boron is tetrahedrally coordinated as suggested
by the sharp 11B resonance (see Fig. 9 for example). On the other hand,
for zeolites with H+ as the charge compensator, a quadrupole pattern
(CQ¼2.3–2.6 MHz, η¼0.1–0.2) was observed in the 11B SSNMR spectra,
indicating that the borons are trigonally coordinated (see Fig. 9 for example).
These results are in agreement with the notion that in the dehydrated proton
form of the zeolites, the proton interacts with the framework oxygens more
strongly as compared to larger cations, which consist of weak covalent inter-
actions with the framework oxygens. Therefore, the proton can break the
BdO bond, resulting in B[3]. The coordination state of the boron can also
be altered by the addition of probe molecules and their effects on
borozeolites have been investigated using 11B SSNMR [74,75,78]. Marthala
et al. [78] performed a systematic study on how the adsorption of probe mol-
ecules with proton affinities of 812–930 kJ/mol influences the coordination
of borons in H-[B]ZSM-5. The authors observed that for molecules with
proton affinities �854 kJ/mol, the coordination of the boron changes from
trigonal to tetrahedral, as identified by the decrease of CQ(11B) from 2.7 to
�0.85 MHz.
256 Ying-Tung Angel Wong and David L. Bryces
Thioborate (B2S3), the sulfur analogue of boron trioxide, has also been
studied using 11B SSNMR.Hwang et al. [85] illustrated that by using a com-
bination of MQMAS and MAS, structural characterization of crystalline
thioborate (c-B2S3) can be accomplished. Owing to the high resolution
of MQMAS spectra, the authors were able to identify various boron species
(i.e., B2O3, BS2O, BSO2, and BS4) that were present in the sample in
addition to the two trigonally coordinated boron sites (one in a four-
membered ring and one in a six-membered borosulfol ring) of B2S3.
A comparison to the corresponding spectra of glassy B2S3 (v-B2S3) showed
that the two tricoordinate boron sites of c-B2S3 are in similar environments
to those found in v-B2S3 as the corresponding NMR parameters are iden-
tical (δiso¼60.4 ppm, CQ¼2.40 MHz, η¼0.05; and δiso¼60.6 ppm,
CQ¼2.41 MHz, η¼0.02 for boron site 1 of c-B2S3 and v-B2S3, respec-
tively; δiso¼63.6 ppm, CQ¼2.15 MHz, η¼0.13; and δiso¼64.1 ppm,
B[4]A D
B E
C F
B[4]
B[3]
0
(ppm)
20 0
(ppm)
20
0
(ppm)
20 0
(ppm)
20
0
(ppm)
20 0
(ppm)
20
−H2O
+H2O
−H2O
+H2O
−H2O
+H2O
[Na, H2O]
[Na, H, H2O]
[H, H2O]
[Na]
[H]
[Na, H]
Fig. 9 11B MAS SSNMR spectra of B-Beta zeolite measured at 11.75 T. Spectra on the leftare of the hydrated samples, which were ion-exchanged and calcined in air; spectra onthe right were recorded after dehydration of the samples under vacuum at elevatedtemperatures. From C. Fild, D.F. Shantz, R.F. Lobo, H. Koller, Cation-induced transformationof boron-coordination in zeolites, Phys. Chem. Chem. Phys. 2 (2000) 3091–3098.
257Recent Advances in 11B SSNMR Spectroscopy
CQ¼2.20 MHz, η¼0.16 for boron site 2 of c-B2S3 and v-B2S3, respec-
tively). Investigation of the structural changes of polycrystalline xNa2S
+(1 �x)B2S3 as a function of x has also been conducted using 11B MAS
and MQMAS experiments [84]. As expected, at x¼0, the boron atoms
reside in four-membered and/or six-membered rings. However, at
x¼0.33, the boron environment was observed to change from trigonal
to tetrahedral as indicated by the small CQ (0.8 MHz). Based on the 11B
MAS spectrum, the structure was concluded to consist of B4S6 units that
are analogous to adamantane. A further increase of x to 0.5 resulted in a
δiso(11B) value of approximately 60 ppm, which is within experimental error
of the δiso value for the six-membered borosulfol ring in B2S3. Therefore,
the boron atoms were concluded to be in metathioboroxyl rings, where they
are once again trigonally coordinated. Lastly, at x greater than 0.5, the pres-
ence of BS33� was detected and the compound was concluded to consist of
orthothioborate units.
4.1.4 Boron HeterocyclesCompared to the other classes of boron compounds, there aremuch fewer 11B
SSNMR data for cyclic boron systems (Table 5). Nonetheless, investigations
have been performed on boron–chalcogen rings (i.e., X¼ O, S, and Se)
[86,87,89], as well as boron–pnictogen rings (i.e., X¼ N) [30,36,50,88]
(Table 5). Beckett et al. [86] studied the triarylboroxines (4-MeC6H4)3B3O3
and (3,5-Me2C6H3)3B3O3 and their corresponding amine adducts. The
δiso(11B) values for the boroxines are approximately 25 ppm (Table 5),
and the formation of amine adducts resulted in a change in these values.
For the three-coordinated boron sites, the δiso(11B) was measured to be
approximately 31–32 ppm, while for the four-coordinated boron sites,
the δiso(11B) were found to be approximately 3–6 ppm (Table 6). Boron–
chalcogen rings with heavier chalcogens (i.e., S and Se) have also been inves-
tigated by Conrady-Pigorsch et al. [89] using 11B SSNMR spectroscopy. By
applying the Townes–Dailey theory to the EFG tensor parameters,
they were able to derive the electron population in the boron pz orbitals rel-
ative to the σ orbitals, therefore acquiring information on the degree of
π-bonding present in the systems. The authors found that the presence of
the phenyl group in B3S3(C6H5)3 does not contribute at all to the π-bondorder, while systems with SedSe bonds were observed to increase the elec-
tron population of the pz orbitals in the boron atoms. In the case of cyclic
boron–pnictogens, different borazine derivatives have been characterized
using 11B SSNMR [30,36,50,88]. Forgeron et al. [36] measured the CS
258 Ying-Tung Angel Wong and David L. Bryces
and EFG tensor parameters of hexamethylborazine at room temperature.
The experimental η was found to be approximately zero, which deviated
from the calculated values (0.13–0.19) and the previously reported values
obtained at 77 K (0.133) [114,115]. They explained that the difference in
η is due to the rapid motion in the borazine ring at room temperature,
resulting in an averaging of the EFG tensor components. Evidence for
motion was also provided by variable temperature experiments, in which
the CQ(11B) was observed to increase by 2% at 110 K as compared to the
respective value at room temperature due to a decrease in the rate of motion.
4.1.5 Lewis Acid/Base Adducts11B EFG tensor parameters have been extracted for a plethora of Lewis acid/
base adducts, including BdP [6,33,39], BdN [37,50,86,90], and BdAs
motifs [38,91] (Table 6). As compared to their Lewis acid analogues, the
complexation with a Lewis base can promote a decrease in the value of
CQ(11B) [7,33,91]. For instance, the CQ(
11B) of B(C6F5)3 is measured to
be 4.26 MHz [33] and upon the formation of a Lewis acid/base adduct
with phosphorus and/or arsenic compounds, the corresponding CQ(11B)
has been observed to range from ca. 1.3 to 2 MHz [7,33,91]. For boron–ammonia systems, the δiso(
11B) was found to increase as the number of methyl
groups bonded to nitrogen increases (i.e., from H3B�NH3 to H3B�NMe3,
Table 6) [50]. Ch�enard et al. [90] performed a series of SATRAS experiments
in order to extract the EFG tensor parameters for pyridine– and pyrazine–BF3complexes (Fig. 10) and illustrated thatCQ(
11B) is highly sensitive to the pres-
ence of steric crowding, as well as the chemical structures of the Lewis acid/
base adducts. For instance, BF3�pyridine, which consists of a distorted tetra-
hedral geometry at the boron site, has a CQ(11B) of 0.280 MHz, whereas the
quinoline analogue (BF3�quinoline) was found to have a slightly larger
CQ(11B) (0.310 MHz). This increase in CQ(
11B) was due to peri interac-
tion effects (a steric interaction between the C8dH bond and the BF3group) from the addition of the benzo group, thereby altering the local boron
geometry. A similar observation was also made in 1,4-bis-BF3�pyrazine and1,4-bis-BF3�quinoxaline (CQ¼0.650 and 0.680 MHz, respectively). The
CQ(11B) was also observed to increase when one of the carbons is substi-
tuted with a nitrogen (i.e., BF3�quinoline vs 4-BF3�2-methylquinoxaline,
CQ¼0.310 and 0.460 MHz, respectively) and when an additional N–BF3group is present (e.g., BF3�pyridine vs 1,4-bis-BF3�pyrazine, CQ¼0.280 vs
0.650 MHz, respectively). Furthermore, the authors found that the 11B
quadrupolar coupling constants reflect the electrochemical features of the
259Recent Advances in 11B SSNMR Spectroscopy
adducts. In general, an increase in CQ(11B) indicates a higher reduction
potential since the distortion in the tetrahedral coordination of the boron
due to the presence of peri interaction leads to an ease of reduction.
4.1.6 FLPs and Corresponding Adducts and OligomersExtensive 11B SSNMR analyses have been performed on FLPs by Erker,
Eckert, Grimme, and coworkers [6,7,33,34,92–96] (Table 7). The topic
has been reviewed in detail [6,7]; therefore, only a brief summary will be
given here. FLPs are systems with a Lewis acid and a Lewis base that are
unable to form the classical Lewis acid/base adducts due to steric hindrance.
These compounds have attracted much attention in the past few years due to
their ability to bind with and/or activate small molecules (e.g., H2, CO2,
alkenes, etc.). FLPs can be differentiated from the classical adducts based
on EFG tensor orientations [6,7]. For the classical adducts,V33 overlaps with
the Lewis acid/base bond (i.e., the BdP bond vector for boron–phosphorusadducts), whereas a�20 degree angle is found between the BdV33 and the
BdP vectors for intramolecular vicinal P/B FLPs [6,7,33]. The orientation
of V33 can also provide insights into the reactivity of FLPs since it has been
proposed that the observed H2 activation by these systems is due to the elec-
tric field generated by the corresponding Lewis acid/base [116]. The direc-
tions of the V33 components then suggest that FLPs have a more easily
Fig. 10 Structures of the pyridine– and pyrazine–BF3 complexes studied by Ch�enardet al. and the corresponding experimental CQ(
11B) values. Data taken from E. Ch�enard,A. Sutrisno, L. Zhu, R.S. Assary, J.A. Kowalski, J.L. Barton, J.A. Bertke, D.L. Gray, F.R.Brushett, L.A. Curtiss, J.S. Moore, Synthesis of pyridine– and pyrazine–BF3 complexes andtheir characterization in solution and solid state, J. Phys. Chem. C 120 (2016) 8461–8471.
260 Ying-Tung Angel Wong and David L. Bryces
accessible reactive pocket as compared to the classical adducts since the EFG
points away from the BdP bond [6,7]. Furthermore, the strength of the
interaction between the Lewis acid and the Lewis base in FLPs can be cor-
related to 11B NMR parameters [33]. For instance, as the distance between
the boron and phosphorus increases,CQ increases and η decreases due to lessdistortion to the trigonal geometry of the boron site (Fig. 11). A similar effect
has also been observed in the case of δiso, where an increase in BdP distance
results in an increase in δiso(11B) (Fig. 12). FLP adducts, oligomers, and
aggregates have also been characterized using 11B SSNMR spectroscopy.
In the case of FLP adducts, it was found that 11B NMR parameters can
be employed to differentiate between isomers [34,93,94].
Fig. 11 Correlation between the experimentally determined (■) and the DFT-calculated(«) CQ(
11B) values of various P/B FLPs and the corresponding boron–phosphorus dis-tance illustrating that an increase in distance results in an increase in the CQ valuesdue to less distortion to the trigonal geometry of the boron site (1¼ (C6F5)2B-(C6F5)C]C(Ph)-PMes2; 3¼ (C6F5)2B-(C6F5)C]C(n-Pr)-PPh2; 4¼ (C6F5)2B-(C6F5)C]C(PPh2)-PPh2;5¼ (C6F5)2B-(C6F5)C]C(Tol)-PMes2; 6¼ (C6F5)2B-(C6F5)C]C(Me)-PPh2; 7¼ (C6F5)2B-(CH2)3-PPh2; 8¼1-B(C6F5)2-2-PMes2 cyclohexane). Experimental values were obtainedfrom 11B MAS experiments, while DFT-calculated values were derived using B97-D/def2-TZVP (modified). Error bars indicate an experimental uncertainty of �3%. Linearregression for the experimental values (R2¼0.94) is given by the solid line, while linearregression for the calculated values (R2¼0.91) is given by the dashed line. From T.Wiegand, H. Eckert, O. Ekkert, R. Fr€ohlich, G. Kehr, G. Erker, S. Grimme, New insights into frus-trated Lewis pairs: structural investigations of intramolecular phosphane–borane adducts byusing modern solid-state NMR techniques and DFT calculations, J. Am. Chem. Soc. 134 (2012)4236–4249.
261Recent Advances in 11B SSNMR Spectroscopy
4.1.7 Metal Borohydrides and Metal BoridesIn the past 20 years, there have been significant advances in the charac-
terization of metal borohydrides and metal borides using 11B SSNMR
(Tables 8 and 9). 11B EFG tensor parameters have been measured for differ-
ent types of metal borides, such as MB2 (M¼ Re, Os, Ru, Mg, and Al),
YB4, MB6 (M¼ Y and La), and MB12 (M¼ Y, Zr, and Lu) (Table 9)
[19,20,22,102–108]. Furthermore, various classes of metal borohydrides
have also been analysed using 11B SSNMR, ranging from monometallic
borohydrides to bimetallic borohydrides and ammine metal borohydrides
[97–101] (Table 8). These compounds have attracted much interest lately
due to their potential applications as hydrogen storage materials. 11B
SSNMR can provide valuable information on these systems since it can
Fig. 12 Correlation between the experimentally determined (■) and the DFT-calculated(«) δiso(
11B) values of various P/B FLPs and the corresponding boron–phosphorus dis-tance illustrating that an increase in distance results in an increase in deshielding of theboron. (1¼ (C6F5)2B-(C6F5)C]C(Ph)-PMes2; 3¼ (C6F5)2B-(C6F5)C]C(n-Pr)-PPh2; 4¼(C6F5)2B-(C6F5)C]C(PPh2)-PPh2; 5¼ (C6F5)2B-(C6F5)C]C(Tol)-PMes2; 6¼ (C6F5)2B-(C6F5)C]C(Me)-PPh2; 7¼ (C6F5)2B-(CH2)3-PPh2; 8¼1-B(C6F5)2–2-PMes2 cyclohexane). Theo-retical δiso(
11B) values were calculated as a function of B ���P distance using amodel com-pound (shown in inset) at the BP-86/def-TZVP level of theory. Solid line gives the linearregression for the experimental values (R2¼0.85), while the dashed line gives the linearregression for the DFT-calculated values (R2¼0.99). From T. Wiegand, H. Eckert, O.Ekkert, R. Fr€ohlich, G. Kehr, G. Erker, S. Grimme, New insights into frustrated Lewis pairs:structural investigations of intramolecular phosphane–borane adducts by using modernsolid-state NMR techniques and DFT calculations, J. Am. Chem. Soc. 134 (2012) 4236–4249.
262 Ying-Tung Angel Wong and David L. Bryces
identify noncrystalline decomposition products and phase transitions
[97,100]. For instance, the transition from o-LiBH4 to h-LiBH4 was inves-
tigated using variable temperature 11B MAS SSNMR, and it was observed
that the correspondingCQ(11B) value decreases as the system approaches the
phase transition temperature [100]. Furthermore, 11B SSNMR spectroscopy
can be employed to probe the interaction between the borohydride and the
metal [98]. In the case of Y(BH4)3�nNH3, where n¼4–7, it was found that
for n¼4 and 5, the BH4� anions act as bridging or terminal ligands, while for
n¼6 and 7, the BH4� anions act as counterions. This coordination flexibil-
ity is reflected in the δiso(11B) values, where the increase in covalent inter-
action between Y and BH4� results in an increase in CS for n¼4 and 5
(�27.5 and �28.6 ppm, respectively) as compared to n¼6 and 7 (�37.4
and �36.6 ppm, respectively).
4.1.8 Additional SystemsIn addition to the systems discussed above, other systems such as boron car-
bides [109,110], boron nitrides (Table 10) [110,111], and metallocene com-
plexes [112] (Table 11) have also been analysed using 11B SSNMR. Schurko
et al. [112] recorded the 11B MAS and static spectra of [Cp∗2B][AlCl4],which has a boron that is coordinated to two Cp* rings in a η1 and a η5 fash-ion, and Cp∗2BMe, which has a tricoordinate boron bonded to two η1-Cp*rings and a methyl group. As a result of their different structures, drastically
different 11B NMR parameters were obtained. For example, the boron
nucleus in [Cp∗2B]+ is more shielded (δiso(11B)¼�41.3 ppm) as compared
to Cp∗2BMe (δiso(11B)¼81.9 ppm) due to the presence of a η5-Cp* ring.
The Ω(11B) value for Cp∗2BMe was also observed to be notably larger
(Ω¼146.1 and 73.0 ppm for Cp∗2BMe and [Cp∗2B]+, respectively, seeFig. 13) due to a greater contribution of σpara to σiso in comparison to
[Cp∗2B]+. Furthermore, CQ(11B) was found to be substantially greater
for Cp∗2BMe (4.52 vs 1.14 MHz), reflecting the molecular symmetry of
these systems. Cp∗2BMe has a molecular symmetry of C1, hence its
CQ(11B) can be expected to be large. On the other hand, it has been illus-
trated that for a centrosymmetric polyhedron with V vertices, if V/2 similar
point charges are located on the vertices such that none of them are related
by reflection through the centre, the EFG tensor will be null at the centre
point. Using this, the authors explained that the boron in [Cp∗2B]+ can be
thought of as being in the middle of an icosahedron, with the neighbouring
carbons occupying 6 of the 12 vertices. Although this depiction is not
completely accurate (i.e., the carbons are not exactly located on the vertices
263Recent Advances in 11B SSNMR Spectroscopy
and consist of different charges, and the effects of the second coordination
sphere are neglected), it can still help rationalize the smaller CQ(11B)
observed for [Cp∗2B]+ as compared to Cp∗2BMe. These differences in11B NMR parameters between [Cp∗2B][AlCl4] and Cp∗2BMe clearly show
how the two species can be distinguished using 11B SSNMR.
4.2 Indirect Spin–Spin (J) Coupling ConstantsHomonuclear and heteronuclear J coupling constants have been measured
for various boron systems including borane derivatives [30,40], diboron
compounds [30,37,41,42,44,45], boronic acids and esters [32], and Lewis
acid–base systems and related compounds [6,7,33,34,37–39,90,91]. As theJ coupling interaction arises from the orbital overlap of two atoms, the
corresponding coupling constants can provide valuable information into
the nature of chemical bonding. Selected studies on diboron compounds
and FLPs are discussed in Sections 4.2.1 and 4.2.2, respectively, while the11B J coupling constants for other boron-containing compounds are sum-
marized in Table 12.
4.2.1 Diboron CompoundsDiboron derivatives are an important and versatile class of compounds
with applications in synthetic and materials chemistry. For instance, these
Fig. 13 Experimental and simulated 11B{H} static NMR spectra (with and without theeffects of boron CSA) of (A) [Cp∗2B][AlCl4] and (B) Cp∗2BMe recorded at 8.46 T. FromRef. [112].
264 Ying-Tung Angel Wong and David L. Bryces
Table 12 J(11B, X) Coupling Constants for Crystalline Boron Compounds Measured via SSNMR SpectroscopyCompound YJ(11B, X) X Y Method References
compounds are often employed for reduction, borylation, as well as poly-
merization reactions [1,117,118]. The uses of diboron derivatives depend
on their electronic properties; therefore, insight into their electronic struc-
tures would be highly beneficial for the rational, function-specific designs of
these systems. As illustrated by Bryce and coworkers [42,44,45], one method
of probing the electronic structure of diboron compounds is by the measure-
ment of J(11B, 11B) coupling constants. Using 11B DQF J-resolved experi-
ments, they successfully extracted the J(11B, 11B) coupling constants of
diboron systems with single and/or multiple BdB bonds, such as diborane
derivatives, diborenes, diboracumulene, and diborynes [41,42,44,45]. It was
found that the coupling constants strongly correlate to the hybridization
state of the boron orbital which participates in the BdB bond [42,45].
As illustrated in Fig. 14, an increase in p-character of the boron atomic
orbitals results in a decrease in the magnitude of the J(11B, 11B) coupling
constants, and this can be rationalized by a decrease in the contribution
of the Fermi-contact (FC) mechanism to the J coupling interaction. Since
the J coupling constants reflect the hybridization of the boron orbitals,
Fig. 14 Correlation between the experimentally obtained J(11B, 11B) coupling constantsusing 11B DQF J-resolved SSNMR spectroscopy and (A) the degree of p orbital hybrid-ization, m, of the boron orbitals responsible for the BdB bonds (J¼�63.5 m+215 Hz,R2¼0.97) and (B) the BdB σ-bonding NBO energies (J¼�300ENBO+12.7 Hz, R2¼0.99).The black squares correspond to electron-precise diborane dianions with 2c–2e B(sp3)–B(sp3) bonds studied in Ref. [45], the red squares correspond to the diboranes investi-gated in Ref. [42], and the blue squares correspond to the diborene, diboracumulene,and diboryne compounds studied in Ref. [44]. The lines of best fit were derivedusing diborane data (black and red squares) only. Adapted from Y.T.A. Wong,J. Landmann, M. Finze, D.L. Bryce, Dynamic disorder and electronic structures of electron-precise dianionic diboranes: insights from solid-state multinuclear magnetic resonancespectroscopy, J. Am. Chem. Soc. 139 (2017) 8200–8211.
269Recent Advances in 11B SSNMR Spectroscopy
information on the BdB bond order can then be acquired. For instance, it
was suggested that the NHC-stabilized diboryne does not comprise a BdB
triple bond since the corresponding force constant is lower than what is
expected for a triple bond [119]. However, an investigation into the
J(11B, 11B) coupling constants shows that the coupling constant increases
from the diborenes (75–85 Hz) to diboracumulene (164 Hz) to the diboryne
(187 Hz), which indicates that the bond order of this diboryne is greater than
that of diborenes and diboracumulene [44]. This conclusion was also
supported by an assessment of the corresponding reduced J coupling con-
stants, which was in quantitative agreement with those of the carbon ana-
logues (alkanes, alkenes, and alkynes) [44]. Therefore, insights into the
BdB bond order were obtained via the J(11B, 11B) coupling constants,
and the results suggest that this diboryne does indeed consist of a BdB triple
bond. Not only can the J coupling constant be related to the bond order, it
can also be correlated to the strength of the BdB bond and the electron
withdrawing capacity of the ligands (Figs 14 and 15) [42,45]. Since an
increase in bond order can be expected to result in an increase in BdB bond
strength, the BdB bond energy is then strongly correlated to the magnitude
of the J(11B, 11B) coupling constant. Furthermore, by systematically varying
the ligands on a planar diboron model system (while fixing bond lengths), it
200A B
LB B
L
LL
150
J(11
B,11
B)
(Hz)
180
N–FP–CIAs–Br
160
140
120
100
80
−0.5 −0.4 −0.3sBJB NBO energy (a.u.)
J(11
B,11
B)
(Hz)
100
50
0
Ligand (L)
H CH3 NH2 OH F
Fig. 15 Theoretical dependence of the J(11B, 11B) coupling constant on (A) the ligandidentity and (B) the BdB σ-bonding NBO energy calculated using various heavy ligands.The relations were obtained using DFT calculations and by changing the ligands on aplanar diboron model system with a BdB bond length of 1.74Å. Adapted from F.A.Perras, D.L. Bryce, Boron–boron J coupling constants are unique probes of electronic struc-ture: a solid-state NMR and molecular orbital study, Chem. Sci. 5 (2014) 2428–2437.
270 Ying-Tung Angel Wong and David L. Bryces
was shown through DFT calculations that the presence of more electron
withdrawing ligands would result in a larger J(11B, 11B) coupling constant
[42] (Fig. 15). This is in agreement with the observed correlation between
the J coupling constant and the p-orbital contribution to the BdB bond
since according to Bent’s rule, an electron withdrawing group would
decrease the p-character of the boron orbitals which form the BdB bond,
which would in turn increase the magnitude of the J coupling constant
through the FC mechanism.
4.2.2 Frustrated Lewis PairsBesides the quadrupolar and CS interactions, J coupling interactions have
also been employed to investigate FLPs. Since detailed discussions have been
provided in various reviews [6,7], only a brief discussion will be given here.
Eckert and coworkers [33] conducted 31P CPMAS NMR experiments in
order to probe the covalent BdP interactions in P/B FLPs. They were able
to obtain the first fully resolved J multiplets of a 11B, 31P spin system, and
successfully extract the J(11B, 31P) coupling constants for various FLPs.
The corresponding coupling constants were taken as indication of the pres-
ence of a weak but nonnegligible covalent bond between the Lewis acid and
the Lewis base centres. Furthermore, by systematically varying the BdP dis-
tance on a model Lewis acid/base adduct (B(C6F5)3�PPh3), Wiegand et al.
[7] demonstrated through DFT calculations that the J(11B, 31P) coupling
constants can assess the covalent interaction between the boron and the
phosphorus centres as an increase in BdP distance was found to result in
an exponential decrease in the J(11B, 31P) values. A similar comparison
was also made via experimental data; however, no distinct trend was
observed since the J(11B, 31P) coupling constants were also sensitive to
the identity of the ligands and/or bridging units bonded to the Lewis centres.
The mode of ligand binding on the Lewis centre can also be distinguished
based on the coupling constants [34]. The J(11B, 31P) coupling constants of a
free P/B FLP, the corresponding P/B FLP-isocyanide Lewis adduct and the
cyclic product that forms from the cooperative binding of the isocyanide to
the FLP have been measured using 11B MAS, 31P MAS, and J-resolved
SSNMR experiments. The coupling constants were found to reflect the
mode of binding of the isocyanide, in which the Lewis adduct gave a sig-
nificantly smaller J coupling constant (20–24 Hz) as compared to the
corresponding free FLP (52 Hz) and the cyclic product (43 Hz). A similar
trend was observed via DFT calculations.
271Recent Advances in 11B SSNMR Spectroscopy
5. CONCLUDING REMARKS
Boron can be found in a variety of crystalline materials, ranging from
synthetic reagents to geological samples. As shown in this review, 11B
SSNMR spectroscopy is an indispensable tool for the characterization of
these systems, and significant progress has been reported over the last 2
decades in associating 11B NMR observables with electronic and structural
features. As in the past, a majority of the 11B SSNMR studies have focused
on measuring EFG tensors due to their sensitivity to the charge distribution
surrounding the boron centre. These parameters can provide a variety of
structural information, such as bond angles, bond distances, and coordina-
tion states, and considerable advances have been made in understanding
the relationship between the CQ(11B) and the structures and properties
of boron-containing systems. In comparison, CS tensors have been much
less explored; nevertheless, an increasing amount of attention has been
placed on measuring 11BΩ and κ values in the past few years. This is attrib-
uted to the relatively small magnitude of the CS tensor span for boron,
which can often only be precisely measured using the very high magnetic
field instruments that have become increasingly prominent over the past 2
decades. The magnitudes of these observables have been shown to be
influenced by various factors, such as ligand identity, dihedral angles,
and hydrogen bonding interactions. Furthermore, due to the develop-
ments in high-resolution SSNMR techniques, 11B J coupling constants,
which can be difficult to extract in the solid state due to quadrupolar
broadening, have been accurately measured for several systems, and it
was illustrated that insights into bond strengths and bond order can be
acquired via the J coupling interaction.
In consideration of the recent developments in the characterization of
crystalline solids using 11B NMR spectroscopy described in this review,
the use of EFG tensor parameters can be anticipated to remain as the focus
of the field in the coming years. Nonetheless, we anticipate a significant
increase in the usage of 11B CSA and J coupling parameters in the study crys-
talline boron systems since these parameters are more experimentally acces-
sible now as compared to the past. Furthermore, the information retrieved
from these NMRobservables (i.e., 11BΩ, κ, and J coupling constants) can becomplementary to those acquired from the nuclear electric quadrupolar
interaction. As such, the continuation of 11B CSA and J coupling investiga-
tions is essential as these parameters will undoubtedly expand our chemical
272 Ying-Tung Angel Wong and David L. Bryces
knowledge of boron-containing compounds, which in turn can facilitate the
advances of various scientific fields due to the wide range of chemical appli-
cation of these compounds.
REFERENCES[1] E. Fernandez, A. Whiting, Synthesis and Application of Organoboron Compounds,
Springer International Publishing, Switzerland, 2015.[2] R.K. Harris, E.D. Becker, S.M. Cabral de Menezes, R. Goodfellow, P. Granger,
NMR nomenclature. Nuclear spin properties and conventions for chemical shifts(IUPAC recommendations 2001), Pure Appl. Chem. 73 (2001) 1795–1818.
[3] L. Mafra, J.A. Vidal-Moya, T. Blasco, Chapter four—structural characterization ofzeolites by advanced solid state NMR spectroscopic methods, Annu. Rep. NMRSpectrosc. 77 (2012) 259–351.
[4] C. Martineau, J. Senker, F. Taulelle, Chapter one—NMR crystallography, Annu.Rep. NMR Spectrosc. 82 (2014) 1–57.
[5] G.A.Monti, A.K. Chattah, Y.G. Linck, Chapter four—solid-state nuclear magnetic res-onance in pharmaceutical compounds,Annu.Rep.NMRSpectrosc. 83 (2014)221–269.
[6] T. Wiegand, H. Eckert, S. Grimme, Solid-state NMR as a spectroscopic tool for char-acterizing phosphane–borane frustrated Lewis pairs, in: G. Erker, D.W. Stephan (Eds.),Frustrated Lewis Pairs I: Uncovering and Understanding, Springer-Verlag, Berlin,Heidelberg, 2013, pp. 291–345.
[7] T. Wiegand, M. Siedow, H. Eckert, G. Kehr, G. Erker, Structural characterization offrustrated Lewis pairs and their reaction products using modern solid-state NMR spec-troscopy techniques, Isr. J. Chem. 55 (2015) 150–178.
[8] T. Br€auniger, M. Jansen, Solid-state NMR spectroscopy of quadrupolar nuclei in inor-ganic chemistry, Z. Anorg. Allg. Chem. 639 (2013) 857–879.
[9] S. Sene, M.A. Pizzoccaro, J. Vezzani, M. Reinholdt, P. Gaveau, D. Berthomieu,S. B�egu, C. Gervais, C. Bonhomme, G. Renaudin, A. Mesbah, A. van der Lee,M.E. Smith, D. Laurencin, Coordination networks based on boronate and ben-zoxaborolate ligands, Crystals 6 (2016) 48.
[10] K.J.D. MacKenzie, M.E. Smith, Multinuclear Solid-State Nuclear Magnetic Reso-nance of Inorganic Materials, Elsevier Science, UK, 2002.
[11] H. N€oth, B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of BoronCompounds, Springer-Verlag, Berlin, Heidelberg, 1978.
[12] P.J. Bray, NMR and NQR studies of boron in vitreous and crystalline borates, Inorg.Chim. Acta 289 (1999) 158–173.
[13] H. Eckert, Structural characterization of noncrystalline solids and glasses using solidstate NMR, Prog. Nucl. Magn. Reson. Spectrosc. 24 (1992) 159–293.
[14] M. Ed�en, NMR studies of oxide-based glasses, Annu. Rep. Prog. Chem., Sect. C:Phys. Chem. 108 (2012) 177–221.
[15] N.F. Ramsey, Magnetic shielding of nuclei in molecules, Phys. Rev. 78 (1950)699–703.
[16] J. Herzfeld, A.E. Berger, Sideband intensities in NMR spectra of samples spinning atthe magic angle, J. Chem. Phys. 73 (1980) 6021–6030.
[17] R.K. Harris, E.D. Becker, S.M. Cabral de Menezes, P. Granger, R.E. Hoffman,K.W. Zilm, Further conventions for NMR shielding and chemical shifts (IUPAC rec-ommendations 2008), Pure Appl. Chem. 80 (2008) 59–84.
[18] M.R. Hansen, T. Vosegaard, H.J. Jakobsen, J. Skibsted, 11B chemical shift anisotropiesin borates from 11B MAS, MQMAS, and single-crystal NMR spectroscopy, J. Phys.Chem. A 108 (2004) 586–594.
[19] Z. Fojud, P.Herzig, O.J. Zogał, A. Pietraszko, A. Dukhnenko, S. Jurga,N. Shitsevalova,Electric-field-gradient tensor and boron site-resolved 11B NMR in single-crystallineYB12, Phys. Rev. B: Condens. Matter Mater. Phys. 75 (2007). 184102.
[20] P. Herzig, Z. Fojud, O.J. Zogał, A. Pietraszko, A. Dukhnenko, S. Jurga,N. Shitsevalova, Electric-field-gradient tensor and charge densities in LaB6:
11Bnuclear-magnetic-resonance single-crystal investigations and first-principles calcula-tions, J. Appl. Phys. 103 (2008). 083534.
[21] A.R. Lim, I.G. Kim, Nuclear quadrupole coupling parameters and structural nature ofthe nonlinear optical material Li2B4O7 by NMR, Solid State Nucl. Magn. Reson.66–67 (2015) 40–44.
[22] O.J. Zogał, Z. Fojud, P. Herzig, A. Pietraszko, A.B. Lyashchenko, S. Jurga,V.N. Paderno, Crystal structure, electric field gradient, and electronic charge densitiesin ReB2: a single crystal X-ray, 11B nuclear magnetic resonance, and first-principlesstudy, J. Appl. Phys. 106 (2009). 033514.
[23] M.A. Kennedy, P.D. Ellis, The single-crystal NMR experiment: an experimentalist’sguide: part II: data analysis and assignment of symmetry-related tensors, ConceptsMagn. Reson. A 1 (1989) 109–129.
[24] M.A. Kennedy, P.D. Ellis, The single-crystal NMR experiment: an experimentalist’sguide: part I: experimental aspects and symmetry considerations, Concepts Magn.Reson. A 1 (1989) 35–47.
[25] T. Vosegaard, E. Hald, P. Daugaard, H.J. Jakobsen, A two-axis goniometer for sen-sitivity enhancement in single-crystal nuclear magnetic resonance spectroscopy, Rev.Sci. Instrum. 70 (1999) 1771–1779.
[26] A. Samoson, Satellite transition high-resolution NMR of quadrupolar nuclei in pow-ders, Chem. Phys. Lett. 119 (1985) 29–32.
[27] J. Skibsted, N.C. Nielsen, H. Bildsøe, H.J. Jakobsen, Satellite transitions in MASNMR spectra of quadrupolar nuclei, J. Magn. Reson. 95 (1991) 88–117.
[28] L. Frydman, J.S. Harwood, Isotropic spectra of half-integer quadrupolar spins frombidimensional magic-angle spinning NMR, J. Am. Chem. Soc. 117 (1995) 5367–5368.
[29] A. Samoson, E. Lippmaa, A. Pines, High resolution solid-state N.M.R. Averaging ofsecond-order effects by means of a double-rotor, Mol. Phys. 65 (1988) 1013–1018.
[30] F.A. Perras, D.L. Bryce, Measuring dipolar and J coupling between quadrupolar nucleiusing double-rotation NMR, J. Chem. Phys. 138 (2013). 174202.
[31] F.A. Perras, D.L. Bryce, Theoretical study of homonuclear J coupling betweenquadrupolar spins: single-crystal, DOR, and J-resolved NMR, J. Magn. Reson.242 (2014) 23–32.
[32] S.-W. Oh, J.W.E. Weiss, P.A. Kerneghan, I. Korobkov, K.E. Maly, D.L. Bryce, Solid-state 11B and 13C NMR, IR, and X-ray crystallographic characterization of selectedarylboronicacids andtheir catechol cyclicesters,Magn.Reson.Chem.50 (2012)388–401.
[33] T. Wiegand, H. Eckert, O. Ekkert, R. Fr€ohlich, G. Kehr, G. Erker, S. Grimme, Newinsights into frustrated Lewis pairs: structural investigations of intramolecularphosphane–borane adducts by using modern solid-state NMR techniques and DFTcalculations, J. Am. Chem. Soc. 134 (2012) 4236–4249.
[34] O. Ekkert, G.G. Miera, T. Wiegand, H. Eckert, B. Schirmer, J.L. Petersen,C.G. Daniliuc, R. Fr€ohlich, S. Grimme, G. Kehr, G. Erker, Remarkable coordinationbehavior of alkyl isocyanides toward unsaturated vicinal frustrated P/B Lewis pairs,Chem. Sci. 4 (2013) 2657–2664.
[35] B. Wrackmeyer, U. Klaus, W. Milius, E. Klaus, T. Schaller, Stannacyclohexanes andspiro-tin compounds with a stannole or a stannolene group, J. Organomet. Chem.517 (1996) 235–242.
[36] M.A.M. Forgeron, D.L. Bryce, R.E. Wasylishen, R. R€osler, A solid-statemultinuclear magnetic resonance investigation of hexamethylborazine, J. Phys. Chem.A 107 (2003) 726–735.
[37] S. Wi, L. Frydman, Residual dipolar couplings between quadrupolar nuclei in highresolution solid state NMR: description and observations in the high-field limit,J. Chem. Phys. 112 (2000) 3248–3261.
[38] S. Wi, V. Frydman, L. Frydman, Residual dipolar couplings between quadrupolarnuclei in solid state nuclear magnetic resonance at arbitrary fields, J. Chem. Phys.114 (2001) 8511–8519.
[39] G. Wu, S. Kroeker, R.E. Wasylishen, R.G. Griffin, Indirect spin–spin coupling inmultiple-quantum magic-angle-spinning NMR spectra of quadrupolar nuclei,J. Magn. Reson. 124 (1997) 237–239.
[40] F.A. Perras, D.L. Bryce, Residual dipolar coupling between quadrupolar nuclei undermagic-angle spinning and double-rotation conditions, J. Magn. Reson. 213 (2011)82–89.
[41] F.A. Perras, D.L. Bryce, Symmetry-amplified J splittings for quadrupolar spin pairs: asolid-state NMRProbe of homoatomic covalent bonds, J. Am. Chem. Soc. 135 (2013)12596–12599.
[42] F.A. Perras, D.L. Bryce, Boron–boron J coupling constants are unique probes of elec-tronic structure: a solid-state NMR and molecular orbital study, Chem. Sci. 5 (2014)2428–2437.
[43] F.A. Perras, D.L. Bryce, Revisiting the concept of equivalence in solid-state NMR,eMagRes 4 (2015) 561–574.
[44] F.A. Perras, W.C. Ewing, T. Dellermann, J. B€ohnke, S. Ullrich, T. Sch€afer,H. Braunschweig, D.L. Bryce, Spying on the boron–boron triple bond using spin–spincoupling measured from 11B solid-state NMR spectroscopy, Chem. Sci. 6 (2015)3378–3382.
[45] Y.T.A. Wong, J. Landmann, M. Finze, D.L. Bryce, Dynamic disorder and electronicstructures of electron-precise dianionic diboranes: insights from solid-state multinuclearmagnetic resonance spectroscopy, J. Am. Chem. Soc. 139 (2017) 8200–8211.
[46] D.L. Bryce, R.E.Wasylishen, M. Gee, Characterization of tricoordinate boron chem-ical shift tensors: definitive high-field solid-state NMR evidence for anisotropic boronshielding, J. Phys. Chem. A 105 (2001) 3633–3640.
[47] Z.M. Hudson, C. Sun, K.J. Harris, B.E.G. Lucier, R.W. Schurko, S. Wang, Probingthe structural origins of vapochromism of a triarylboron-functionalized platinum(II)acetylide by optical and multinuclear solid-state NMR spectroscopy, Inorg. Chem.50 (2011) 3447–3457.
[48] A.E. Alain, Y. Shoji, T. Fukushima, D.L. Bryce, 11B solid-state NMR interaction ten-sors of linear two-coordinate boron: the dimesitylborinium cation, Inorg. Chem.54 (2015) 11889–11896.
[49] J.W.E. Weiss, D.L. Bryce, A solid-state 11B NMR and computational study of boronelectric field gradient and chemical shift tensors in boronic acids and boronic esters,J. Phys. Chem. A 114 (2010) 5119–5131.
[50] C. Gervais, F. Babonneau, J. Maquet, C. Bonhomme, D. Massiot, E. Framery,M. Vaultier, 15N cross-polarization using the inversion–recovery cross-polarizationtechnique and 11B magic angle spinning NMR studies of reference compounds con-taining BdN bonds, Magn. Reson. Chem. 36 (1998) 407–414.
[51] S. Sene, D. Berthomieu, B. Donnadieu, S. Richeter, J. Vezzani, D. Granier, S. B�egu,H. Mutin, C. Gervais, D. Laurencin, A combined experimental–computational studyof benzoxaborole crystal structures, CrstEngComm 16 (2014) 4999–5011.
[52] F.G. Vogt, G.R. Williams, R.C.B. Copley, Solid-state NMR analysis of a boron-containing pharmaceutical hydrochloride salt, J. Pharm. Sci. 102 (2013) 3705–3716.
[53] S. Sene, S. B�egu, C. Gervais, G. Renaudin, A. Mesbah, M.E. Smith, P.H. Mutin,A. van der Lee, J.-M. Nedelec, C. Bonhomme, D. Laurencin, Intercalation of ben-zoxaborolate anions in layered double hydroxides: toward hybrid formulations forbenzoxaborole drugs, Chem. Mater. 27 (2015) 1242–1254.
[54] D. Berthomieu, C. Gervais, G. Renaudin, M. Reinholdt, S. Sene, M.E. Smith,C. Bonhomme, D. Laurencin, Coordination polymers based on alkylboronate ligands:synthesis, characterization, and computational modelling, Eur. J. Inorg. Chem.2015 (2015) 1182–1191.
[55] S. Sene, M. Reinholdt, G. Renaudin, D. Berthomieu, C.M. Zicovich-Wilson,C. Gervais, P. Gaveau, C. Bonhomme, Y. Filinchuk, M.E. Smith, J.-M. Nedelec,S. B�egu, P.H. Mutin, D. Laurencin, Boronate ligands in materials: determining theirlocal environment by using a combination of IR/solid-state NMR spectroscopies andDFT calculations, Chem. A Eur. J. 19 (2013) 880–891.
[56] M. Reinholdt, J. Croissant, L. Di Carlo, D. Granier, P. Gaveau, S. B�egu,J.-M. Devoisselle, P.H. Mutin, M.E. Smith, C. Bonhomme, C. Gervais, A. van derLee, D. Laurencin, Synthesis and characterization of crystalline structures based onphenylboronate ligands bound to alkaline earth cations, Inorg. Chem. 50 (2011)7802–7810.
[57] D. Carnevale, V. del Amo, D. Philp, S.E. Ashbrook, Detecting solid-state reactivity in10-hydroxy-10,9-boroxophenanthrene using NMR spectroscopy, Tetrahedron66 (2010) 6238–6250.
[58] G. Wu, K. Yamada, Residual dipolar couplings in MAS and MQMAS NMR spectraof quadrupolar nuclei, Chem. Phys. Lett. 313 (1999) 519–524.
[59] S. Borsacchi, L. Calucci, M. Geppi, Orientational order of liquid crystals by 11B NMRspectroscopy, Chem. Phys. Lett. 508 (2011) 63–66.
[60] J.F. Stebbins, P. Zhao, S. Kroeker, Non-bridging oxygens in borate glasses: character-ization by 11B and 17O MAS and 3QMAS NMR, Solid State Nucl. Magn. Reson.16 (2000) 9–19.
[61] M. Fisch, T. Armbruster, D. Rentsch, E. Libowitzky, T. Pettke, Crystal-chemistry ofmullite-type aluminoborates Al18B4O33 and Al5BO9: a stoichiometry puzzle, J. SolidState Chem. 184 (2011) 70–80.
[62] R.X. Fischer, V. Kahlenberg, D. Voll, K.J.D. MacKenzie, M.E. Smith, B. Schnetger,H.-J. Brumsack, H. Schneider, Crystal structure of synthetic Al4B2O9: a member ofthe mullite family closely related to boralsilite, Am. Mineral. 93 (2008) 918–927.
[63] S. Kroeker, J.F. Stebbins, Three-coordinated boron-11 chemical shifts in borates,Inorg. Chem. 40 (2001) 6239–6246.
[64] M.R. Hansen, G.K.H. Madsen, H.J. Jakobsen, J. Skibsted, Refinement of boratestructures from 11B MAS NMR spectroscopy and density functional theory calcula-tions of 11B electric field gradients, J. Phys. Chem. A 109 (2005) 1989–1997.
[65] L.J.Q. Maia, V.R. Mastelaro, J.F. Schneider, P. Parent, C. Laffon, Structural studies inthe BaO–B2O3–TiO2 system by XAS and 11B-NMR, J. Solid State Chem. 178 (2005)1452–1463.
[66] S.C. Neumair, S. Vanicek, R. Kaindl, D.M. T€obbens, C. Martineau, F. Taulelle,J. Senker, H. Huppertz, HP-KB3O5 highlights the structural diversity of borates:corner-sharing BO3/BO4 groups in combination with edge-sharing BO4 tetrahedra,Eur. J. Inorg. Chem. 2011 (2011) 4147–4152.
[67] O.L.G. Alderman, D. Iuga, A.P. Howes, K.J. Pike, D. Holland, R. Dupree, Spectralassignments and NMR parameter–structure relationships in borates using high-resolution 11B NMR and density functional theory, Phys. Chem. Chem. Phys.15 (2013) 8208–8221.
[68] K. Seleznyova, N.A. Sergeev, M. Olszewski, P. Stępie�n, S.V. Yagupov,M.B. Strugatsky, J. Kliava, 11B MAS NMR study of Ga1�xFexBO3 mixed crystals,Solid State Nucl. Magn. Reson. 70 (2015) 38–42.
[69] T. Br€auniger, T. Pilz, C.V. Chandran, M. Jansen, Full differentiation and assignmentof boron species in the electrolytes Li2B6O9F2 and Li2B3O4F3 by solid-state
11B NMRspectroscopy, J. Solid State Chem. 194 (2012) 245–249.
[70] B. Zhou, V.K. Michaelis, S. Kroeker, J.E.C. Wren, Y. Yao, B.L. Sherriff, Y. Pan, 11Band 23Na solid-state NMR and density functional theory studies of electric field gra-dients at boron sites in ulexite, CrstEngComm 15 (2013) 8739–8747.
[71] R. Ternane, M.T. Cohen-Adad, G. Panczer, C. Goutaudier, C. Dujardin, G. Boulon,N. Kbir-Ariguib,M. Trabelsi-Ayedi, Structural and luminescent properties of newCe3+
doped calcium borophosphate with apatite structure, Solid State Sci. 4 (2002) 53–59.[72] E. V�eron, M.N. Garaga, D. Pelloquin, S. Cadars, M. Suchomel, E. Suard, D. Massiot,
V. Montouillout, G. Matzen, M. Allix, Synthesis and structure determination ofCaSi1/3B2/3O8/3: a new calcium borosilicate, Inorg. Chem. 52 (2013) 4250–4258.
[73] C. Fild, D.F. Shantz, R.F. Lobo, H. Koller, Cation-induced transformation of boron-coordination in zeolites, Phys. Chem. Chem. Phys. 2 (2000) 3091–3098.
[74] I. Lezcano-Gonzalez, A. Vidal-Moya, M. Boronat, T. Blasco, A. Corma, Modellingactive sites for the Beckmann rearrangement reaction in boron-containing zeolites andtheir interactionwith probemolecules, Phys. Chem.Chem. Phys. 12 (2010) 6396–6403.
[75] P.V.Wiper, J. Amelse, L.Mafra, Multinuclear solid-state NMR characterization of theBrønsted/Lewis acid properties in the BP HAMS-1B (H-[B]-ZSM-5) borosilicatemolecular sieve using adsorbed TMPO and TBPO probe molecules, J. Catal.316 (2014) 240–250.
[76] C. Fild, H. Eckert, H. Koller, Evidence for selective association of tetrahedral BO4
units with Na+ and of trigonal BO3 units with H+ in dehydrated zeolite B-ZSM-5from solid-state NMR spectroscopy, Angew. Chem. Int. Ed. 37 (1998) 2505–2507.
[77] H. Koller, C. Fild, R.F. Lobo, Variable anchoring of boron in zeolite beta, Micropo-rous Mesoporous Mater. 79 (2005) 215–224.
[78] V.R.R. Marthala, W. Wang, J. Jiao, Y. Jiang, J. Huang, M. Hunger, Effect of probemolecules with different proton affinities on the coordination of boron atoms indehydrated zeolite H-[B]ZSM-5, Microporous Mesoporous Mater. 99 (2007) 91–97.
[79] H.T.T. Tong, H. Koller, Control of Al for B framework substitution in zeolite Beta bycounterions, Microporous Mesoporous Mater. 148 (2012) 80–87.
[80] S.-J. Hwang, C.-Y. Chen, S.I. Zones, Boron sites in borosilicate zeolites at variousstages of hydration studied by solid state NMR spectroscopy, J. Phys. Chem. B108 (2004) 18535–18546.
[81] D.B. Raskar, H. Eckert, B. Ewald, R. Kniep, Characterization of local environmentsin crystalline borophosphates using single and double resonance NMR, Solid StateNucl. Magn. Reson. 34 (2008) 20–31.
[82] N. Feng, Q. Wang, A. Zheng, Z. Zhang, J. Fan, S.-B. Liu, J.-P. Amoureux, F. Deng,Understanding the high photocatalytic activity of (B, Ag)-codoped TiO2 under solar-light irradiation with XPS, solid-state NMR, and DFT calculations, J. Am. Chem.Soc. 135 (2013) 1607–1616.
[83] N. Feng, A. Zheng, Q.Wang, P. Ren, X. Gao, S.-B. Liu, Z. Shen, T. Chen, F. Deng,Boron environments in B-doped and (B, N)-codoped TiO2 photocatalysts: a com-bined solid-state NMR and theoretical calculation study, J. Phys. Chem. C115 (2011) 2709–2719.
[84] S.-J. Hwang, C. Fernandez, J.P. Amoureux, J.-W. Han, J. Cho, S.W. Martin,M. Pruski, Structural study of xNa2S+(1�x)B2S3 glasses and polycrystals by multiple-quantum MAS NMR of 11B and 23Na, J. Am. Chem. Soc. 120 (1998) 7337–7346.
[85] S.-J. Hwang, C. Fernandez, J.P. Amoureux, J. Cho, S.W. Martin, M. Pruski, Quan-titative study of the short range order in B2O3 and B2S3 by MAS and two-dimensionaltriple-quantum MAS 11B NMR, Solid State Nucl. Magn. Reson. 8 (1997) 109–121.
[86] M.A. Beckett, G.C. Strickland, K.S. Varma, D.E. Hibbs, M.B. Hursthouse,K.M.A. Malik, Amine adducts of triarylboroxines: synthesis and characterization ofadducts of tri(2-tolyl) boroxine and crystal structures of (4-MeC6H4)3B3O3 and(4-MeC6H4)3B3O3�4-picoline, J. Organomet. Chem. 535 (1997) 33–41.
[87] J. Brus, J. Czernek, M. Urbanova, L. Kobera, A. Jegorov, An efficient 2D 11B-11Bsolid-state NMR spectroscopy strategy for monitoring covalent self-assembly ofboronic acid-derived compounds: the transformation and unique architecture ofbortezomib molecules in the solid state, Phys. Chem. Chem. Phys. 19 (2017) 487–495.
[88] C. T€onshoff, M. M€uller, T. Kar, F. Latteyer, T. Chass�e, K. Eichele, H.F. Bettinger,B3N3 borazine substitution in hexa-peri-hexabenzocoronene: computational analysisand Scholl reaction of hexaphenylborazine, Chemphyschem 13 (2012) 1173–1181.
[89] R. Conrady-Pigorsch, W. M€uller-Warmuth, G. Schwetlik, M. Wienkenh€over,B. Krebs, Electronic structure and bonding in cyclic B–S and B–Se compounds studiedby solid state 11B NMR, Ber. Bunsen. Phys. Chem 95 (1991) 453–458.
[90] E. Ch�enard, A. Sutrisno, L. Zhu, R.S. Assary, J.A. Kowalski, J.L. Barton, J.A. Bertke,D.L. Gray, F.R. Brushett, L.A. Curtiss, J.S. Moore, Synthesis of pyridine– andpyrazine–BF3 complexes and their characterization in solution and solid state,J. Phys. Chem. C 120 (2016) 8461–8471.
[91] A. Faucher, V.V. Terskikh, R.E. Wasylishen, Spin–spin coupling betweenquadrupolar nuclei in solids: 11B–75As spin pairs in Lewis acid–base adducts,J. Phys. Chem. A 119 (2015) 6949–6960.
[92] C. Rosorius, C.G. Daniliuc, R. Fr€ohlich, G. Kehr, G. Erker, Structural features andreactions of a geminal frustrated phosphane/borane Lewis pair, J. Organomet. Chem.744 (2013) 149–155.
[93] M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert,A.J. Lough, R. Fr€ohlich, C.G. Daniliuc, S. Grimme, D.W. Stephan, G. Kehr,G. Erker, Reactions of phosphorus/boron frustrated Lewis pairs with SO2, Chem.Sci. 4 (2013) 213–219.
[94] M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. P€ottgen,A.J.P. Cardenas, T.H.Warren, R. Fr€ohlich, C.G. Daniliuc, G. Erker, Noninteracting,vicinal frustrated P/B-Lewis pair at the norbornane framework: synthesis, character-ization, and reactions, J. Am. Chem. Soc. 135 (2013) 8882–8895.
[95] T. Wiegand, M. Sajid, G. Kehr, G. Erker, H. Eckert, Solid-state NMR strategies forthe structural characterization of paramagnetic NO adducts of frustrated Lewis pairs(FLPs), Solid State Nucl. Magn. Reson. 61-62 (2014) 19–27.
[96] M. Erdmann, T.Wiegand, J. Blumenberg, H. Eckert, J. Ren, C.G. Daniliuc, G. Kehr,G. Erker, Formation, structural characterization, and reactions of a unique cyclo-trimeric vicinal Lewis pair containing (C6F5)2P-Lewis base and (C6F5)BH-Lewis acidcomponents, Dalton Trans. 43 (2014) 15159–15169.
[97] D.B. Ravnsbæk, Y. Filinchuk, R. �Cerny, M.B. Ley, D. Haase, H.J. Jakobsen,J. Skibsted, T.R. Jensen, Thermal polymorphism and decomposition of Y(BH4)3,Inorg. Chem. 49 (2010) 3801–3809.
[98] L.H. Jepsen, M.B. Ley, R. �Cerny, Y.-S. Lee, Y.W. Cho, D. Ravnsbæk,F. Besenbacher, J. Skibsted, T.R. Jensen, Trends in syntheses, structures, and proper-ties for three series of ammine rare-earth metal borohydrides, M(BH4)3�nNH3 (M ¼Y, Gd, and Dy), Inorg. Chem. 54 (2015) 7402–7414.
[99] E. Roedern, Y.-S. Lee, M.B. Ley, K. Park, Y.W. Cho, J. Skibsted, T.R. Jensen, Solidstate synthesis, structural characterization and ionic conductivity of bimetallic alkali-metal yttrium borohydrides MY(BH4)4 (M ¼ Li and Na), J. Mater. Chem. A4 (2016) 8793–8802.
[100] L.M. Arnbjerg, D.B. Ravnsbæk, Y. Filinchuk, R.T. Vang, Y. Cerenius,F. Besenbacher, J.-E. Jørgensen, H.J. Jakobsen, T.R. Jensen, Structure and dynamicsfor LiBH4�LiCl solid solutions, Chem. Mater. 21 (2009) 5772–5782.
[101] D.B. Ravnsbæk, C. Frommen, D. Reed, Y. Filinchuk, M. Sørby, B.C. Hauback,H.J. Jakobsen, D. Book, F. Besenbacher, J. Skibsted, T.R. Jensen, Structural studiesof lithium zinc borohydride by neutron powder diffraction, Raman and NMR spec-troscopy, J. Alloys Compd. 509S (2011) S698–S704.
[102] B. J€ager, S. Paluch, W. Wolf, P. Herzig, O.J. Zogał, N. Shitsevalova, Y. Paderno,Characterization of the electronic properties of YB4 and YB6 using
11B NMR andfirst-principles calculations, J. Alloys Compd. 383 (2004) 232–238.
[103] B. J€ager, S. Paluch, O.J. Zogał, W. Wolf, P. Herzig, V.B. Filippov, N. Shitsevalova,Y. Paderno, Characterization of the electronic properties of YB12, ZrB12, and LuB12
using 11B NMR and first-principles calculations, J. Phys. Condens. Matter 18 (2006)2525–2535.
[104] D. Koumoulis, C.L. Turner, R.E. Taylor, R.B. Kaner, 11B NMR spectral and nuclearspin–lattice relaxation analyses of ReB2, J. Phys. Chem. C 120 (2016) 2901–2907.
[105] B.J. Suh, X. Zong, Y. Singh, A. Niazi, D.C. Johnston, 11B NMR in the layereddiborides OsB2 and RuB2, Phys. Rev. B 76 (2007) 144511.
[106] S.H. Baek, B.J. Suh, E. Pavarini, F. Borsa, R.G. Barnes, S.L. Bud’ko, P.C. Canfield,NMR spectroscopy of the normal and superconducting states of MgB2 and compar-ison to AlB2, Phys. Rev. B 66 (2002). 104510.
[107] U. Burkhardt, V. Gurin, F. Haarmann, H. Borrmann, W. Schnelle, A. Yaresko,Y. Grin, On the electronic and structural properties of aluminum diboride Al0.9B2,J. Solid State Chem. 177 (2004) 389–394.
[108] R. Schmitt, B. Blaschkowski, K. Eichele, H.-J. Meyer, Calcium tetraboride-does itexist? Synthesis and properties of a carbon-doped calcium tetraboride that is isotypicwith the known rare earth tetraborides, Inorg. Chem. 45 (2006) 3067–3073.
[109] T. Langer, S. Dupke, C. Dippel, M.Winter, H. Eckert, R. P€ottgen, LiBC—synthesis,electrochemical and solid-state NMR investigations, Z. Naturforsch. 67b (2012)1212–1220.
[110] K. Kanehashi, K. Saito, Structural analysis of boron compounds using 11B-3QMASsolid state NMR, J. Mol. Struct. 602-603 (2002) 105–113.
[111] S. Seidel, T. Dierkes, T. J€ustel, C. Benndorf, H. Eckert, R. P€ottgen, Superstructureformation in SrBa8[BN2]6 and EuBa8[BN2]6, Dalton Trans. 45 (2016) 12078–12086.
[112] R.W. Schurko, I. Hung, S. Schauff, C.L.B. Macdonald, A.H. Cowley, Anisotropic11B and 13C NMR interaction tensors in decamethylcyclopentadienyl boron com-plexes, J. Phys. Chem. A 106 (2002) 10096–10107.
[113] R.E. Youngman, J.W. Zwanziger, Network modification in potassium borate glasses:structural studies with NMR and Raman spectroscopies, J. Phys. Chem. 100 (1996)16720–16728.
[114] A. L€otz, J. Voitl€ander, Line intensities in 10B nuclear quadrupole double resonancespectra, J. Magn. Reson. 54 (1983) 427–435.
[115] A. L€otz, J. Voitl€ander, J.A.S. Smith, The electronic structure of borazine as seen by 10B,11B, and 14N nuclear quadrupole double resonance, Z. Naturforsch. 41a (1986)206–207.
[116] S. Grimme, H. Kruse, L. Goerigk, G. Erker, The mechanism of dihydrogen activationby frustrated Lewis pairs revisited, Angew. Chem. Int. Ed. 49 (2010) 1402–1405.
[117] F. Brouwer, J. Alma, H. Valkenier, T.P. Voortman, J. Hillebrand, R.C. Chiechi,J.C. Hummelen, Using bis(pinacolato)diboron to improve the quality of regioregularconjugated co-polymers, J. Mater. Chem. 21 (2011) 1582–1592.
[118] D. Zhou, N.Y. Doumon, M. Abdu-Aguye, D. Bartesaghi, M.A. Loi, L.J. AntonKoster, R.C. Chiechi, J.C. Hummelen, High-quality conjugated polymers via one-pot Suzuki–Miyaura homopolymerization, RSC Adv. 7 (2017) 27762–27769.
[119] R. K€oppe, H. Schn€ockel, The boron–boron triple bond? A thermodynamic and forcefield based interpretation of the N-heterocyclic carbene (NHC) stabilization proce-dure, Chem. Sci. 6 (2015) 1199–1205.











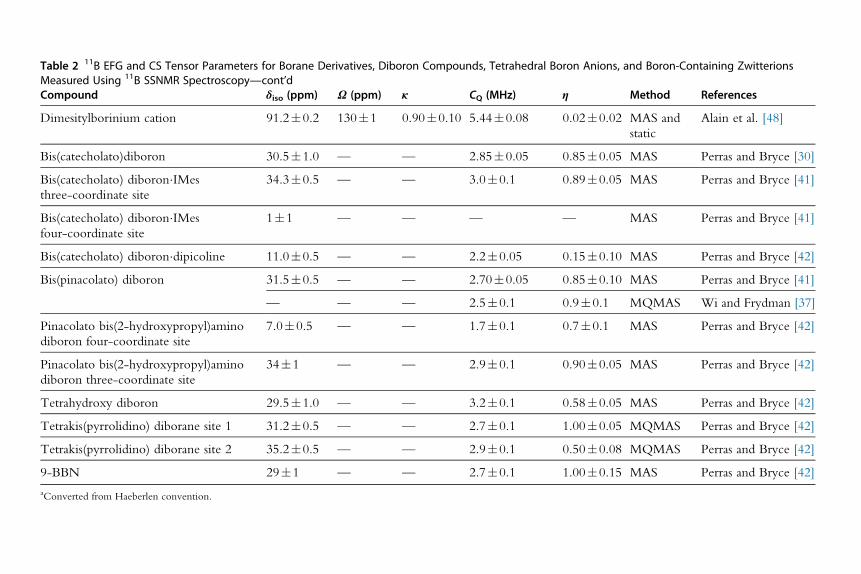








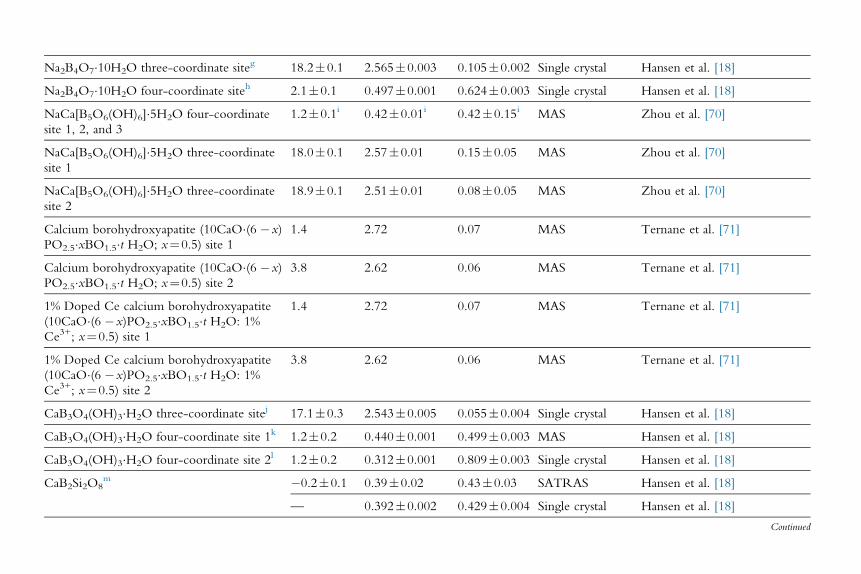




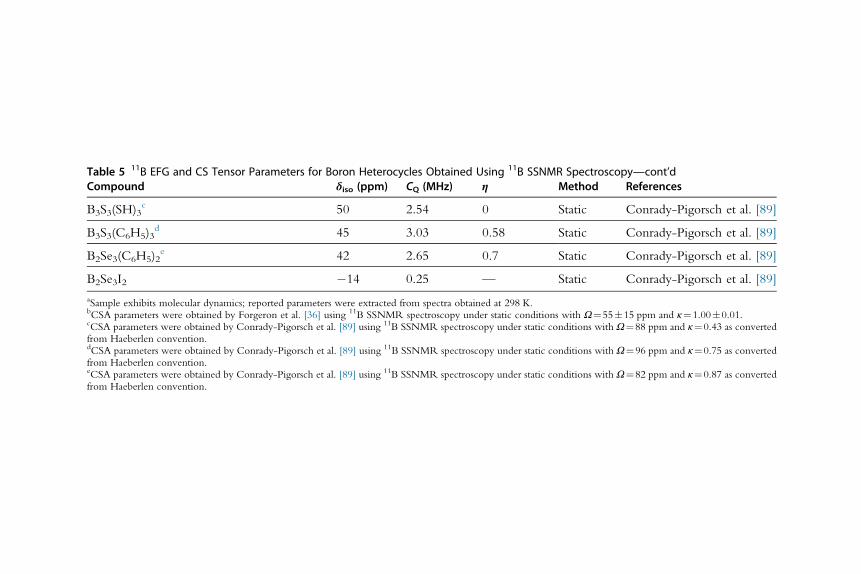

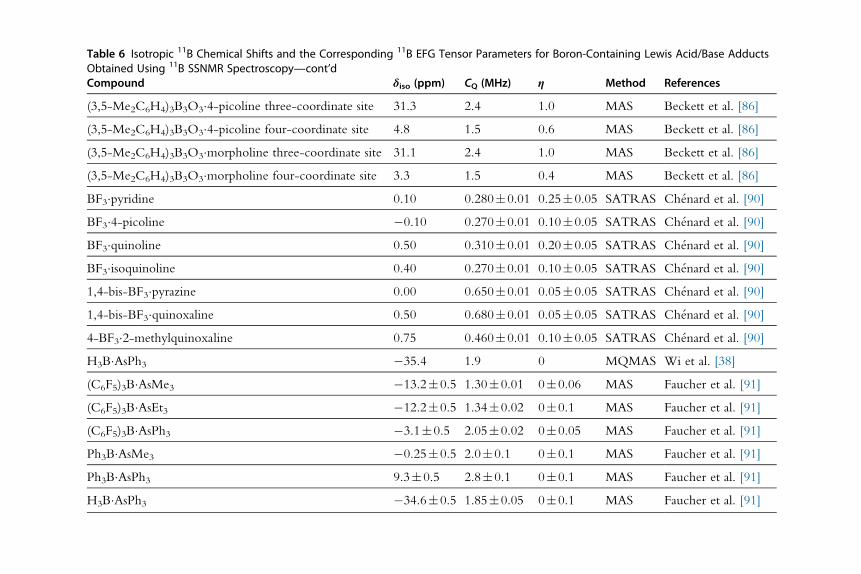

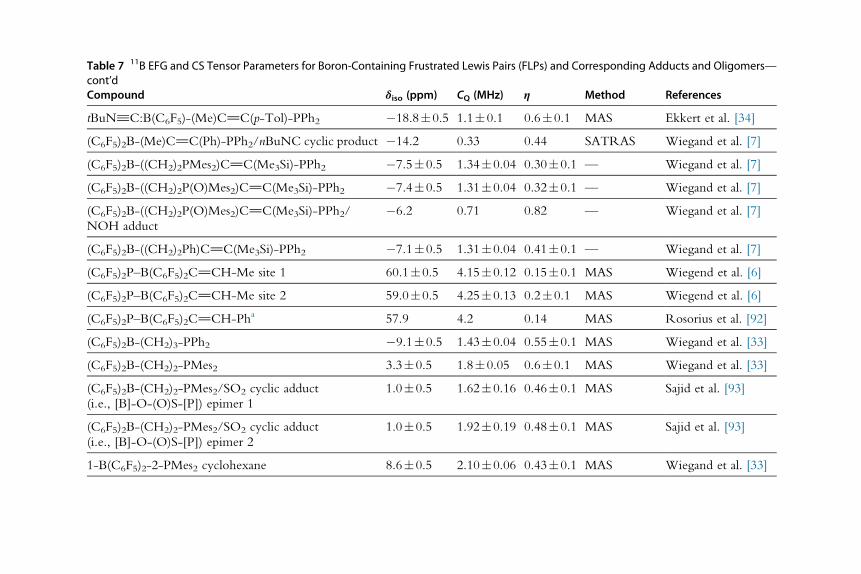










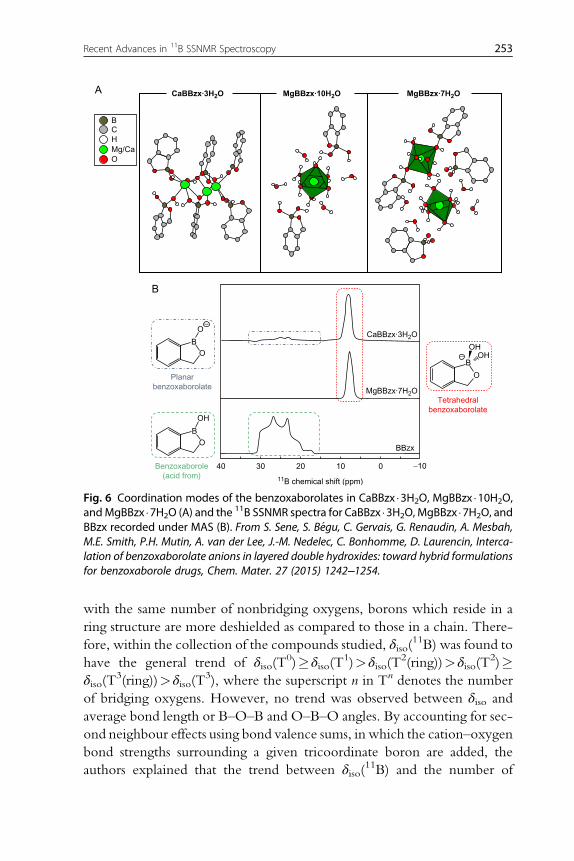
































![Areviewofoxygen-17solid-stateNMRoforganic materials ... · to spin-1 2 nuclei and in solids it is the residual second-order quadrupolar broadening for half-integer spin ... (MQMAS)[31].MQMAS](https://static.documents.pub/doc/80x56/5aed63f07f8b9a66258fd835/areviewofoxygen-17solid-statenmroforganic-materials-spin-1-2-nuclei-and-in-solids.jpg)









