68
Jonathan Tam, MD Clinical Division of Allergy and Immunology Children’s Hospital Los Angeles Recognizing Immunodeficiency & Immune Disorders in Adults

Jonathan Tam, MDClinical Division of Allergy and Immunology
Children’s Hospital Los Angeles
Recognizing Immunodeficiency & Immune Disorders in Adults

October 15, 2020
In accordance with the ACCME policy on relevant financial disclosure, all speakers/planners were asked to reveal any relevant financial relationships.
Research supportRegeneronAstra Zeneca
Disclosures

Learning Objectives
• To recognize signs and symptoms of potential immunodeficiency
• To review immunodeficiencies that can be found in adults
• To review basic evaluation of patients with suspected immunodeficiency

Outline
• Basic immunology
• Primary Immunodeficiency (PIDD)• Newborn Screening
• Example of adult onset immunodeficiency• CVID
• Anti-cytokine autoantibodies


Immunology ReviewJust a Touch

Figure 1-1, Abbas, A. K., et al. "Cellular and molecular immunology, Elsevier Saunders." Eighth Edition. Philadelphia, PA.(2015).
Innate and adaptive immunity


COMPARTMENTS OF IMMUNITYH
UM
OR
AL
CE
LLU
LAR
INNATE ADAPTIVE
Macrophages
Phagocytes
Neutrophils
NK cellsT cells
B cells
Antibody
CD4
CD8
Complement Spleen

Primary Immunodeficiency in ChildrenJust a Touch

PRIMARY IMMUNODEFICIENCES
11
Dosanjh, Amrita. Pediatrics in review 36.11 (2015): 489-94.
PIDs prevalence ~ 1:10,000 to 1:12,000 live births
-vary from common ~1:500 for IgA deficiency to rare ~1:200,000 in CGD to v. rare in LAD ~1:1 million
Antibody deficiencies are the most common
Incidence of Severe combined immunodeficiency (SCID) ~ 1:58,000 live births in the US
Diseases of innate immunity (including NK cells), and complement deficiencies are most rare
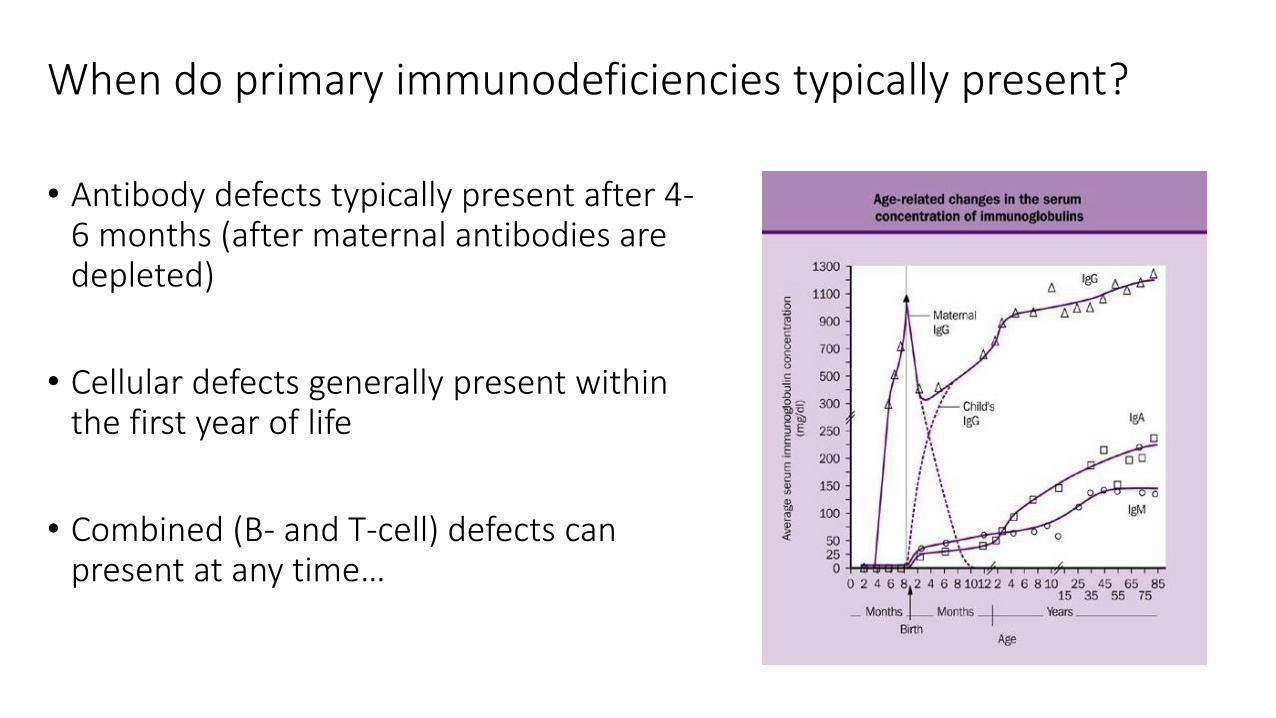
When do primary immunodeficiencies typically present?
• Antibody defects typically present after 4-6 months (after maternal antibodies are depleted)
• Cellular defects generally present within the first year of life
• Combined (B- and T-cell) defects can present at any time…

PIDD in Children
Children with a normal immune system can average 6-8 upper respiratory
infections (URI) per year
Can be as high as 10-12 URIs per year with various risk factors:
- siblings, day care/preschool, asthma, chronic disease, atopic disease
Mean duration of viral URI symptoms ~8 days, but can extend >2 weeks
-So a “normal” child with >10 URIs per year can potentially have symptoms half the year
‘10 Warning Signs’ of Primary Immunodeficiency (PID) by Jeffrey Model
Foundation
- Can be helpful but just a guide as most signs not very sensitive or specific13

14
http://www.info4pi.org/library/educational-
materials/10-warning-signs

SCID Newborn Screening

Justification for SCID Newborn Screening
SCREENING CRITERIA
• Disease is serious
• Disease is not detectable by routine exam
• Incidence supports screening• (hypothyroidism 1:2,000, galactosemia
1:88,000)
• Simple, reliable test is available
• Confirmation test is available
• Treatment alters disease
• Earlier treatment improves outcomes
SCID MEETS CRITERIAFatal if untreated
SCID babies appear healthy at birth
~1:50,000
TREC
Lymphocyte subset analysis
BMT/HSCT cures SCID
Survival improved if BMT/HSCT done before complicating infections

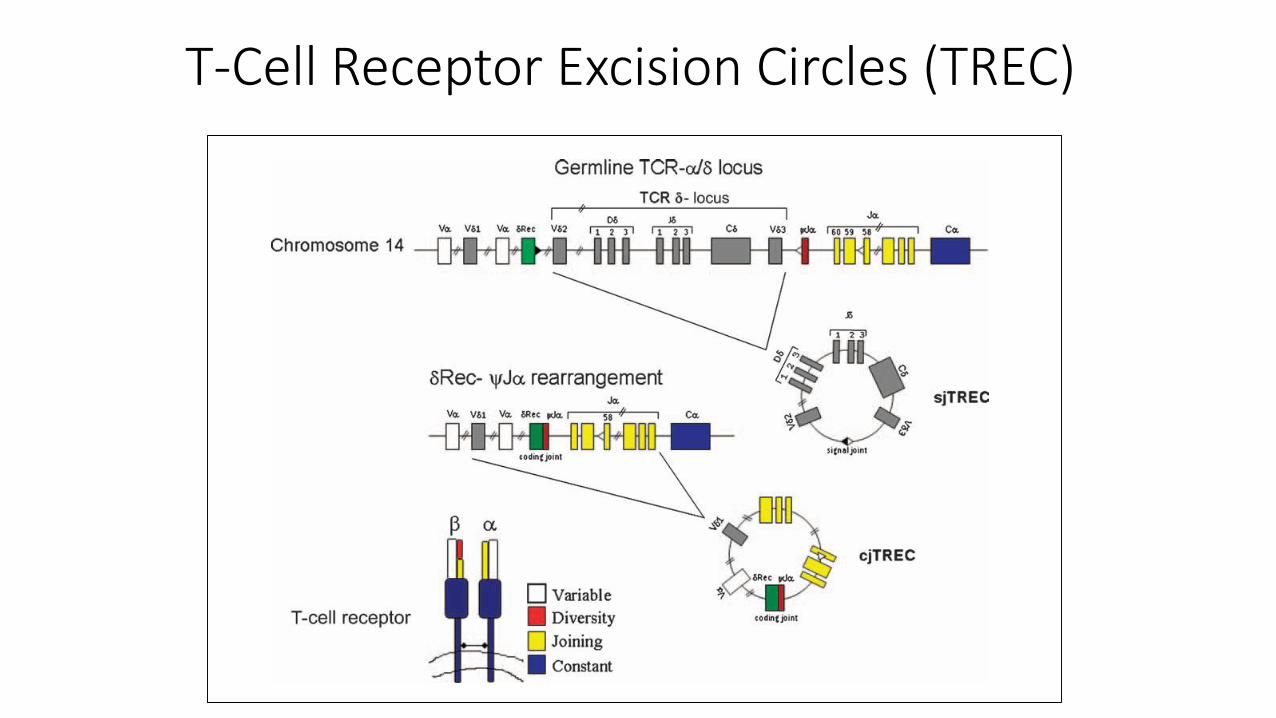
T-Cell Receptor Excision Circles (TREC)
• Small circles of DNA
• By-products of T-cell receptor (TCR) gene rearrangement during generation of antigen specific TCR in the thymus
• Effectively looks for naïve T cells and screens for T cell lymphopenia
• Stable, detectable and quantifiable
• TREC ≈ T-cells
• Actin gene = control
T-Cell Receptor Excision Circles (TREC)

3 mm hole punched from blood spot
~3 ul blood
Measure TRECs by PCR
Extract DNA
50 ul blood/drop
Guthrie Card
TREC DRIED BLOOD SPOT ASSAY

General Warning SignsBack to Including Adults

Symptoms of Immunodeficiency
• Infections• Frequent, severe, unusual, resistant organisms
• Often common infections that are hard to eradicate
• Autoimmune disease• Immune system no longer able to properly distinguish “self” from “non-self”
• Immune dysregulation• Unchecked immune cell proliferation
• Hematopoietic malignancy
• Impaired tumor surveillance

Mimickers of Immunodeficiency
• Infection• HIV – destroys T-cells (CD4+)
• Malnutrition• Vitamin deficiencies, thymic involution
• Smoking • Ciliary dysfunction
• Loss of barrier function• Burns, influenza with superinfection (secondary pneumonia)
• Medications• Corticosteroids, immunosuppressants
• Genetic Causes• Cystic fibrosis, primary ciliary dyskinesia• Splenic dysfunction (Sickle Cell disease, congenital asplenia)

COMPARTMENTS OF IMMUNITYH
UM
OR
AL
CE
LLU
LAR
INNATE ADAPTIVE
Macrophages
Phagocytes
Neutrophils
NK cells T cells B cells
Antibody
CD4
CD8
Complement Spleen
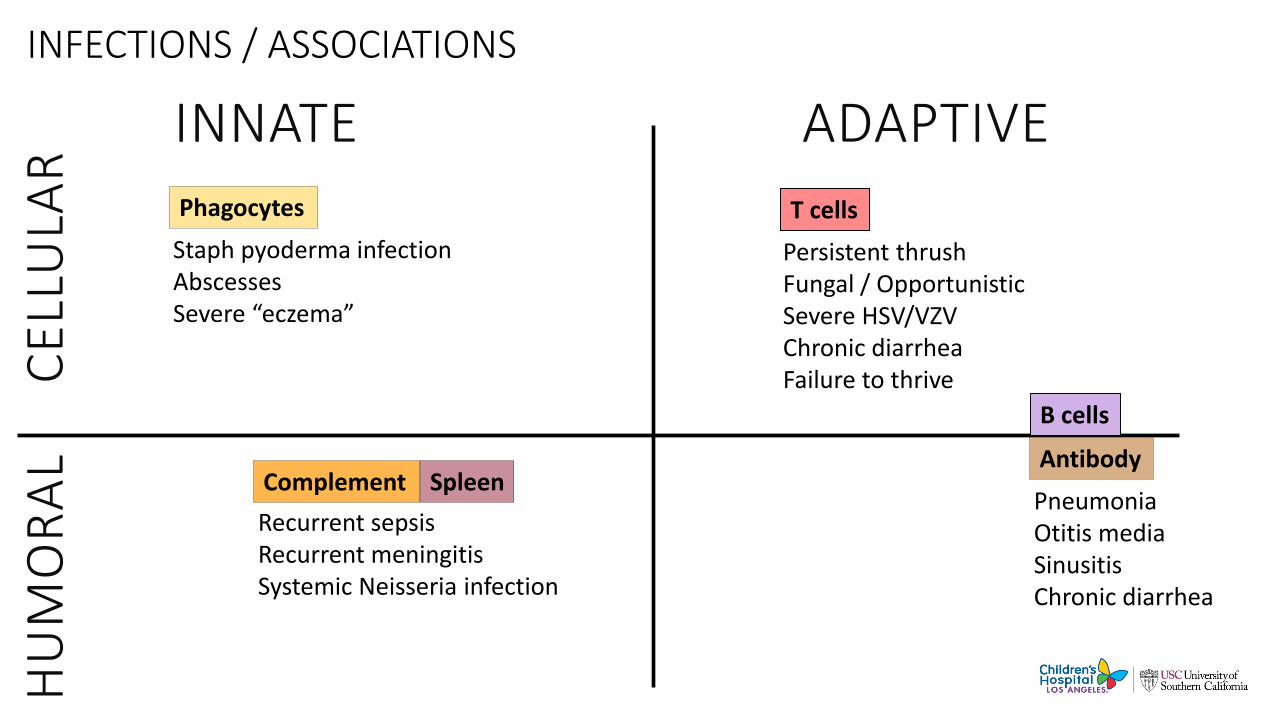
INFECTIONS / ASSOCIATIONS
24HU
MO
RA
L
CE
LLU
LAR
INNATE ADAPTIVEPhagocytes
Complement
T cells
Staph pyoderma infectionAbscessesSevere “eczema”
Persistent thrushFungal / OpportunisticSevere HSV/VZVChronic diarrheaFailure to thrive
PneumoniaOtitis mediaSinusitisChronic diarrhea
Recurrent sepsisRecurrent meningitisSystemic Neisseria infection
Antibody
B cells
Spleen

Key DISORDERS to considerH
UM
OR
AL
C
ELL
ULA
R
INNATE ADAPTIVEPhagocytes
Complement
T cells
Chronic Granulomatous Disease Hyper IgE
SCID or other CID DiGeorge syndrome HIV / AIDS
25
Terminal Complement deficiency (C5-C9)
Antibody
B cells
SIGAD THI SAD CVID XLA
Spleen
Hyposplenism

Initial outpatient screenH
UM
OR
AL
C
ELL
ULA
RINNATE ADAPTIVE
Phagocytes
Complement
T cells
CBC w/diff Phagocyte oxidase activity
Dihydrorhodamine (DHR) or
Nitroblue-tetrazolium (NBT)
CBC w/diff IgG, IgA, IgM, IgE Tetanus and Hib antibody levels HIV antibody or PCR
26
CH50 CBC w/diff with smear
for Howell-Jolly bodies
Antibody
B cells
Spleen
CBC w/diff IgG, IgA, IgM, IgE Tetanus and Hib antibody levels HIV antibody or PCR

Two Examples

Common Variable Immunodeficiency (CVID)

CVID
• Primary immunodeficiency of unknown etiology*.• Mutations in TACI in approximately 10%• Increased risk of CVID—sibs with identical mutations but
without disease• Most cases not inherited (family member with IgA deficiency in 8-
10%, some linkage to MHC)• Incidence is uncertain: estimated 1:30,000 to 1:50,000 (under
diagnosed!!!)
* Small numbers of patients dx with CVID have mutations in BTK, SH2DIA (SLP), TACI and ICOS.

J Allergy Clin Immunol Pract 2016;4(1):38-59
CVID (ICON) 2016 Consensus Definition1. At least one of the characteristic clinical manifestations (infection,
autoimmunity, lymphoproliferation)
2. Hypogammaglobulinemia according to the age-adjusted reference range in at least 2 measurements more than 3 weeks apart.
3. IgA or IgM level must also be low.
4. Decreased/absent ability to make specific Abs (polysaccharide Ags> protein Ags)
5. Other causes of hypogammaglobulinemia must be excluded.
6. Genetic studies to investigate monogenic forms of CVID

CVID
• Delay in dx common--4 to 8.9 years—(Clin Exp Immunol. 147: 306-12, 2007; J Clin Immunol. 27:308-316, 2007, J. Ped. 154:888-94, 2009)
• Index of suspicion needs to be MUCH higher.
• Minimal workup: Quantitative immunoglobulins are indicated in any person with 2 or more CXR-documented pneumonias requiring Abx

CVID: Clinical Presentation
• Recurrent Upper/Lower RTI-most common presenting symptom
• Think Ab deficiency in patient >1 pneumonia, intractable sinusitis, recurrent otitis media
• GI Tract Infections also common (Giardia, enteroviruses)
• Think Ab deficiency with intractable or recurrent GI infections with giardia
• Less common: meningitis, septicemia, osteomylelitis
• Autoimmunity, increased malignancies in some forms of Ab deficiency

CVID: Common Bacterial Pathogens
• Encapsulated bacteria (S. pneumoniae, H. influenza)• GNR in patients repeatedly Rxd with antimicrobials
• Atypical bacteria (Mycoplasma sp., Ureaplasma sp.)• Unique susceptibility—must cover these organisms when Rx
URTI (acute sinusitis) or LRTI

Inflammatory or autoimmune manifestations of New York CVID cohort
Ann Allergy Asthma Immunol. 2019 Nov;123(5):454-460.

Autoimmunity in CVID cohort of USIDNET
J Clin Immunol, 38 (2018), pp. 28-34

REMINDER: Diagnostic Studies in Patients with CVID and antibody defects (or people on IVIg)
• DO NOT use serological assays for dx in pts. with CVID, XLA or other forms of panhypogammaglobulinemia
• Serological assays: measure antibodies in gammaglobulin in patients receiving IVIG
• Dx of infectious disease MUST be done by culture, PCR or other direct methods to directly test the presence of the pathogen

Anti-cytokine AutoantibodyA class of disorders

The Story of NTMWhen it looks like Primary Immunodeficiency, but…
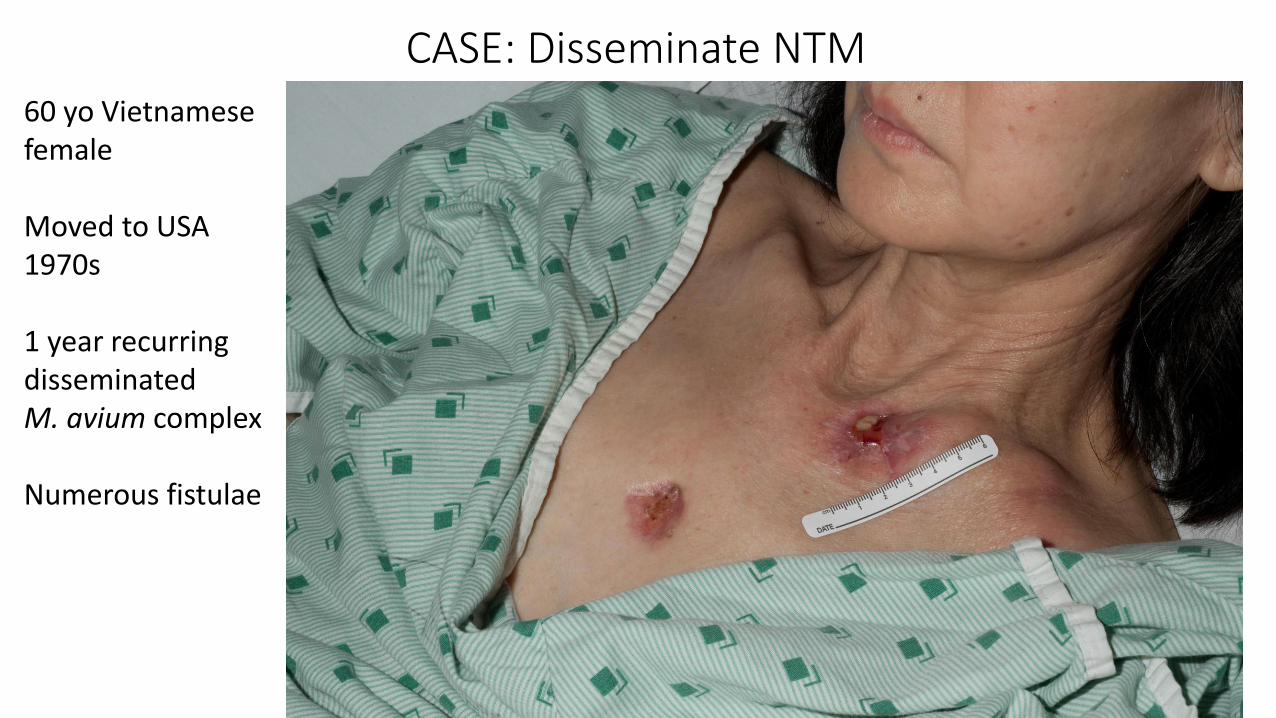
60 yo Vietnamesefemale
Moved to USA 1970s
1 year recurringdisseminated M. avium complex
Numerous fistulae
CASE: Disseminate NTM


Mycobacteria
Relative Virulence
M. tuberculosis
M. kansasii
M. leprae
M. avium complex
M. abscessus
M. gordonae
M. smegmatis
Bacille Calmette-Guerin
M. fortuitum
Koch, 1882
NTM

Clinical Spectrum of NTM Infections
Disseminated
Severe, Young
Immune defects
Pulmonary
Chronic, Older
Bronchiectasis
Cystic fibrosis (CF)
Ciliary dyskinesia (PCD)
SkinExposure
Inoculation

T/NK Cell
Macrophage
IFN γ R
IL-12R
Mycobacterial Control
IFN γ
IL-12

Mendelian Susceptibility to Mycobacterial Disease (MSMD)
Pathogens in human IFN γ /IL-12 defects: Intracellular Pathogens
Nontuberculous mycobacteria (NTM)
Bacille Calmette Guerin (BCG)
M. tuberculosis (MTB)
Salmonella
Burkholderia
Listeria
Histoplasma
Coccidioides
VZV, CMV, HSV, HHV-8
RSV

What is Causing this Late Onset Immunodeficiency?
The infections suggest IFNg/IL-12 pathway.
The age is late for a typical primary immunodeficiency.
Genetic testing did not find a cause.

Clin Infect Dis. 2000 Jan;30(1):29-34.
59 Adult Thai Cases of NTM with Coinfections
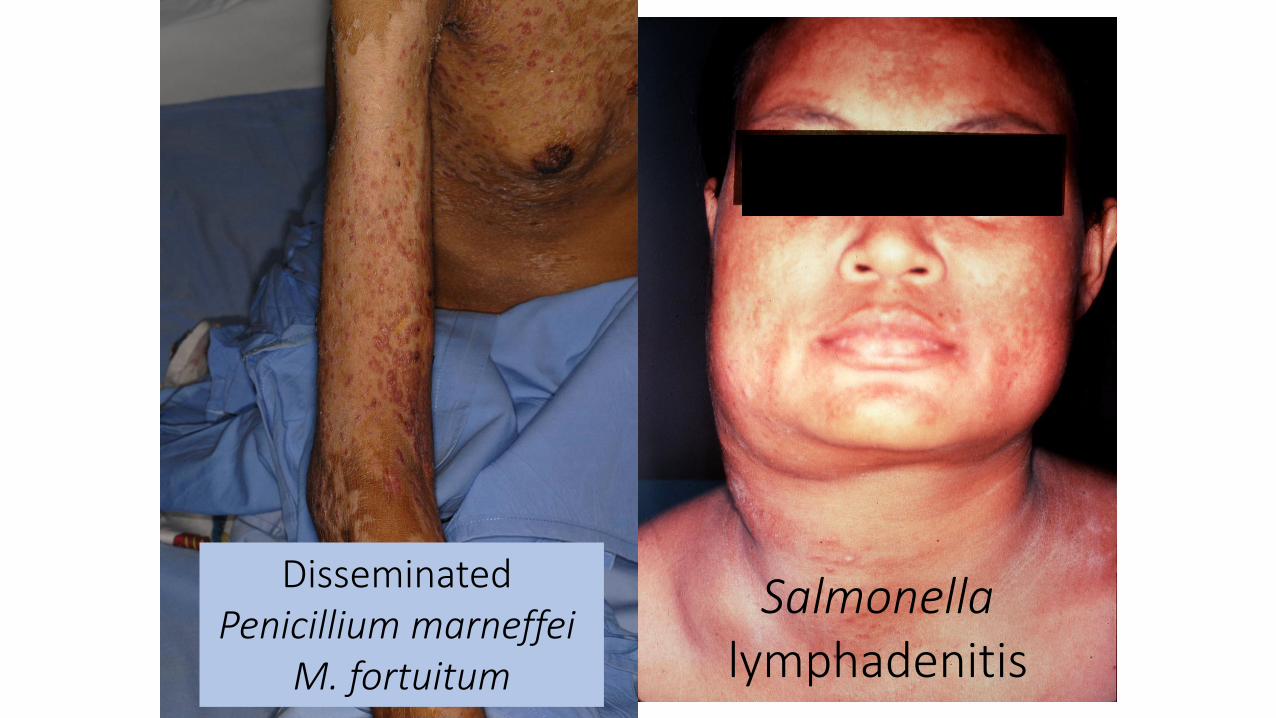
Disseminated Penicillium marneffei
M. fortuitum
Salmonella lymphadenitis

Cryptococcosis
M. abscessus
Neutrophilic pustulosis

Sweet Syndrome (acute febrile neutrophilic dermatosis)

NEJM 2012;367:72590% of Thai patients with severe NTM infection had neutralizing anti-IFNγ autoantibodies

7
6
5
4
3
2
Only anti-IFNg Autoantibodies AreAssociated with Disseminated NTM
41 autoantibodies tested by LIPS (Luciferase)
Anti-IFNg

T/NK Cell
Macrophage
IFN γ R
IL-12R
Mycobacterial Control
IFN γ
IL-12

Autoantibodies IFN γ
• Mostly adults from age 40-70
• Susceptible to severe or disseminated NTM, salmonella (29-40%), VZV
• Almost all patients with anti-IFNγ mostly in Southeast Asia and Japan• HLA association?
Human Genetics volume 139, pages783–794(2020)

Autoantibodies to CytokinesIn sufficient concentration, anti-cytokine autoantibodies could block the signaling or neutralize the biological function of target cytokines via:
preventing the direct binding to its receptor and/or
depleting the cytokine through forming a cytokine/autoantibodies complex
Unknown stimulus, external exposure leading to cross-reactive antigens or multi-step breakdown of tolerance?

CASE: CNS Nocardia
43-year-old male
Acute seizure
Brain biopsy: pus
Culture: Nocardia
Chest CT: Rare ground glass
opacities
Pulmonary function: normal

J Immunol 2013, 190 (8) 3959-3966

mBio 2014;5:e00912-14
Anti-GM-CSFAutoantibodies
ONLYin C. gattii

Anti-GM-CSF in Plasma of CNS Nocardia Patients
Flu
ore
scence I
nte
nsity
GM-CSF
G-CSFIFNαIFNβ
IFNωIFNγ
IFNλ1
IFNλ2
IFNλ3
IL-1αIL-4IL-6IL-7
IL-10
IL-12p70
IL-15
IL-17A
IL-17F
IL-22
IP-10
TNFα
TNFβ
0
5000
10000
15000
20000
25000

Autoantibodies to GM-CSF
• Anti-GM-CSF autoantibodies in pulmonary and meningeal cryptococcosis and Nocardia

Anti-cytokine Autoantibody Treatment
• Antibiotics/antifungals
• Rituximab

Both groups lack effective immune responses that depend on type I interferon.

Interferons
• Interferon type I: The type I interferons present in humans are IFN-α, IFN-β, IFN-ε, IFN-κ and IFN-ω. In general, type I interferons are produced when the body recognizes a virus that has invaded it. They are produced by fibroblasts and monocytes.
• Interferon type II (IFN-γ in humans): This is also known as immune interferon and is activated by Interleukin-12.
• Interferon type III: Discovered more recently than type I and type II IFNs, important in some types of virus or fungal infections.

Science. 2020 Sep 24;eabd4585. doi: 10.1126/science.abd4585.
More than 10% of people who develop severe disease have neutralizing autoantibodies against type I interferon.
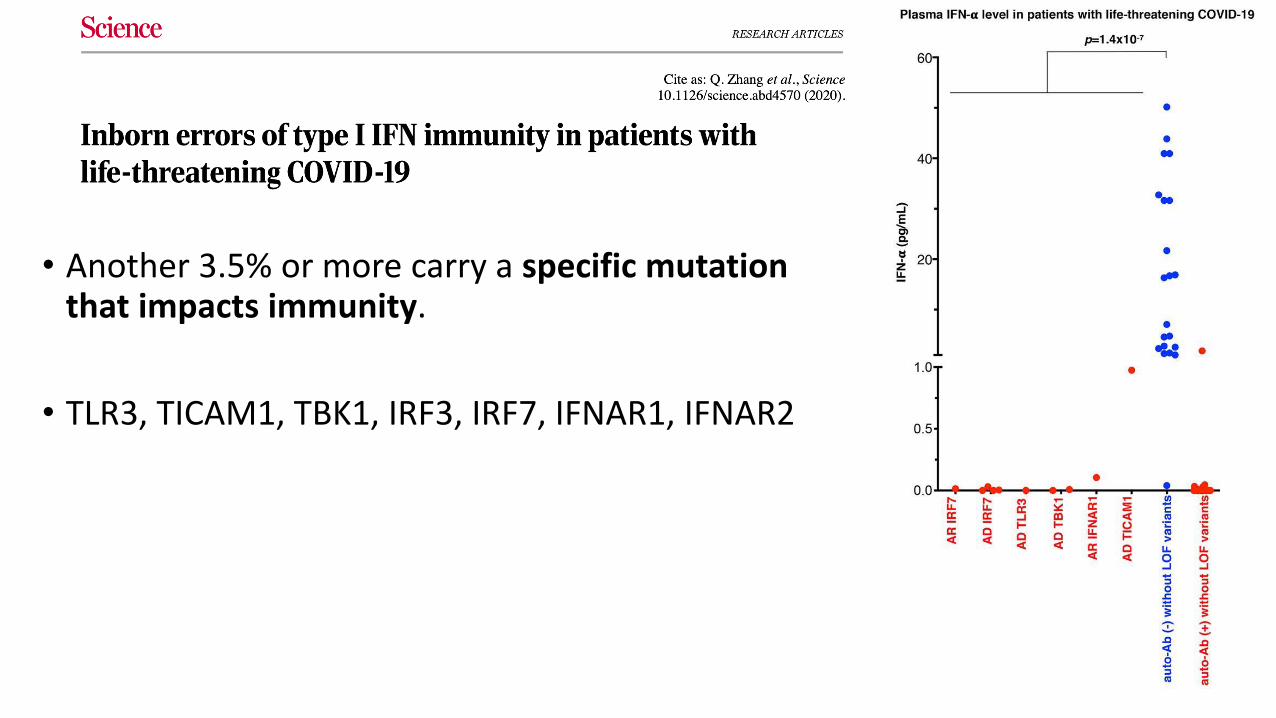
• Another 3.5% or more carry a specific mutation that impacts immunity.
• TLR3, TICAM1, TBK1, IRF3, IRF7, IFNAR1, IFNAR2
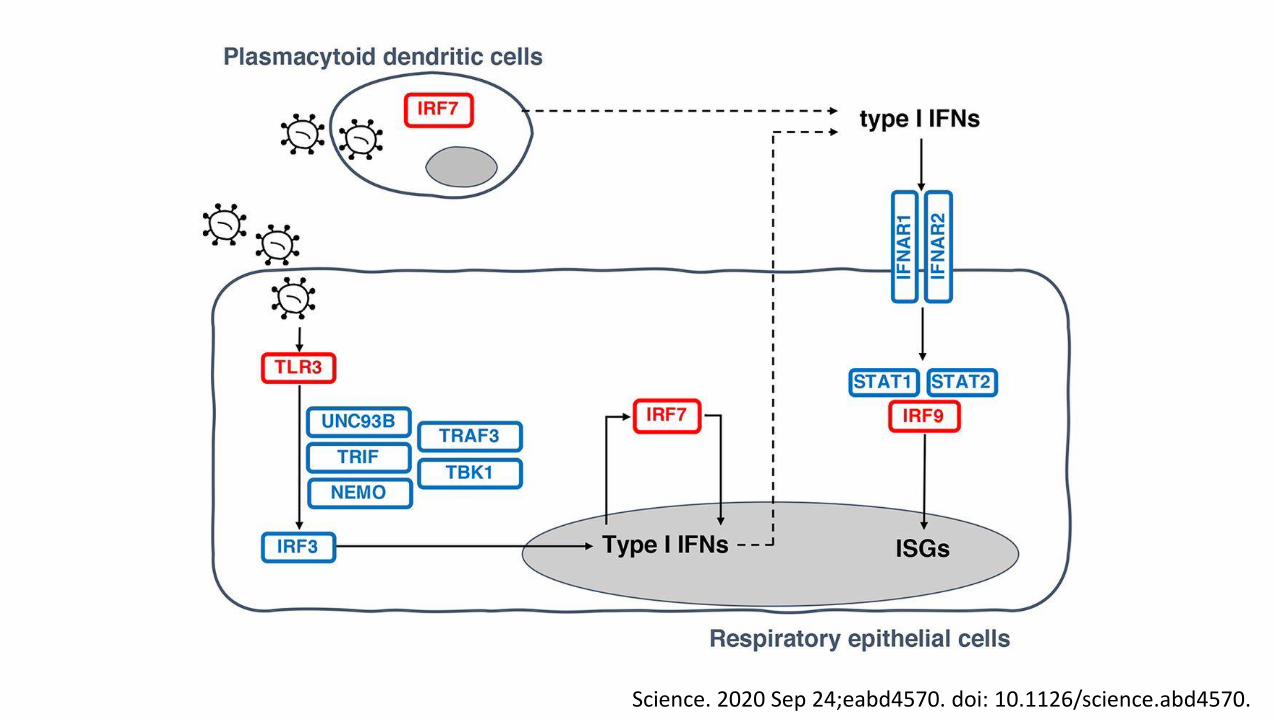
Science. 2020 Sep 24;eabd4570. doi: 10.1126/science.abd4570.

Cytokine Autoantibodies Summary
Cytokine Susceptibility Note
Type 1 Interferon (IFNa, IFN⍵)
SARS-CoV2, Influenza
Type 2 Interferon (IFNγ)
Nontuberculous mycobacteria
GMCSF Pulmonary and meningeal cryptococcosis (gattii) and Nocardia
pulmonary alveolar proteinosis
IL6 Severe bacterial infection, pyogenic infection low CRP despite infection
TH17 cytokines (IL17A, IL17F, IL22, IL23)
Fungal infections, mucocutaneous candidiasis

Conclusions
• Immunodeficiencies can present in adulthood
• Keep a high index of suspicion
• CVID may present with inflammatory of autoimmune manifestations
• Anti-cytokine autoantibodies may explain poor response to specific infectious diseases
• New diagnostic testing may open new therapeutic options

Thank youQuestions?

















