Redox Regulation Mechanisms in Inflammatory Macrophages Marina Diotallevi October 2017 A thesis submitted in partial fulfilment of the requirements of the University of Brighton and the University of Sussex for a programme of study undertaken at the Brighton and Sussex Medical School for the degree of Doctor of Philosophy
Transcript
Redox Regulation Mechanisms in
Inflammatory Macrophages
Marina Diotallevi
October 2017
A thesis submitted in partial fulfilment of the requirements of the University of
Brighton and the University of Sussex for a programme of study undertaken at
the Brighton and Sussex Medical School for the degree of Doctor of Philosophy
2
Abstract
Long-lasting activation of inflammation leads to chronic conditions and is particularly
common in autoimmune diseases. The causes of this self-activation in those
conditions are still unknown, but these diseases are often also associated with an
increase in oxidative stress. In fact, reactive oxygen species (ROS) are released
during the inflammatory response and can cause oxidative damage, which in turn can
lead to maintenance of inflammation. However, ROS are not only toxic species but
can also act as signalling molecules to regulate immune responses, for instance via
thiol modification. Thiols present in the cysteine residues of protein are among the
most sensitive targets of ROS. They can undergo many redox changes, including
glutathionylation or disulphide-linked dimerisation, all of which alter the protein and
thus its function, localisation and secretion. This “redox regulation” regulates many
cellular processes such as apoptosis, cell development and differentiation,
homoeostasis and the immune response.
In this project, we hypothesise that changes in thiol oxidation affect the inflammatory
response and two different approaches have been set up to track redox changes in
inflammatory conditions.
Firstly, the role of endogenous glutathione (GSH), the main thiol antioxidant, was
investigated. For this purpose, we used RAW cells, a mouse macrophage cell line.
Cells were depleted of endogenous GSH and then stimulated with a standard
inflammatory stimulus, bacterial lipopolysaccharide (LPS). A microarray analysis was
then performed to identify changes in the gene expression profile. Results indicated
that endogenous GSH does not decrease the inflammatory response but, on the
contrary, favours the host antiviral response as its depletion results in an impaired
LPS-induced increase in gene expression of genes in the interferon pathway,
including oas2, mx2 and irf9. The biological significance of these results was later
confirmed in cells infected with influenza A, showing that the antiviral response elicited
by LPS was inhibited by GSH depletion.
The second approach of this work was the use of a pegylated maleimide (MalPEG –
10kDa) to determine the redox state of three “redoxkines”, protein thiol/disulphide
oxidoreductases with inflammatory properties: Trx, Prx1 and Prx2. MalPEG covalently
binds to free thiols causing a mobility shift that can be detected by Western blot,
leading to differences in the migration of oxidised and reduced proteins. After LPS
stimulation, clear changes in the redox state were detected both intracellularly and in
secreted proteins. To identify potential membrane targets of redoxkines, we set up a
3
technique to identify proteins with redox-sensitive exofacial thiols on the cell surface.
The results of this work show that activation of inflammatory pathway in macrophages
brings about a number of redox changes in protein thiols, some of which may be
related to GSH signalling, which are important in the regulation of both inflammation
cytochrome C oxidase (16/930 proteins), PDI (3/930 proteins).
203
Figure 5.10: Venn diagram of membrane proteins shared by LPS-stimulated and
untreated (NT) RAW cells identified by MS.
204
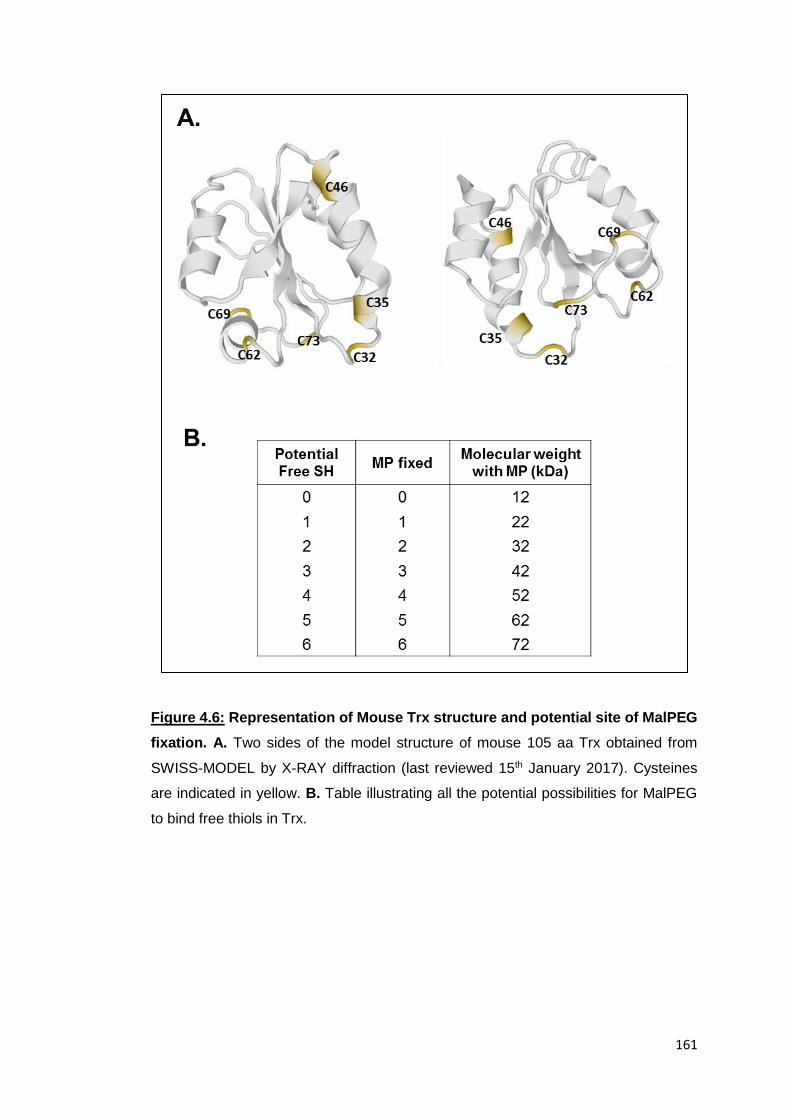
5.8 Investigation of the expression of membrane Trx
Although Trx was not present in the lists of protein identified by MS, some papers
have pointed out its existence at the surface of human cell lines (Sahaf, Soderberg et
al. 1997, Wollman, Kahan et al. 1997). The absence of Trx in the protein identified by
MS could be explained by the fact that membrane Trx is in the oxidized state. In fact,
if a protein was present in the absence of free thiols, the protein would not be labelled
with BIAM and would not attach to the streptavidin beads and consequently the
protein would not be identified by MS.
Therefore, we attempted to detect the protein in the membrane of RAW cells from the
same samples we used for MS identification. As shown in Figure 5.11A, three bands
were detected with the antibody anti-Trx; one at the expected molecular weight (MW)
of 12kDa but also one at a higher MW band (around15kDa) and one at 24kD (all forms
were indicated by black arrows). It has to be noted that no difference was detected
between untreated and LPS-treated samples.
The presence of Trx was confirmed in two other experiments (experiment 2 and
experiment 3) as shown in Figure 5.11. While the form at 24kDa was observed, the
12kDa and 15kDa bands were barely detected (Figure 5.11B and C). Usual controls
of the membrane purification were also made using GAPDH and ATPase marker. This
controls confirmed the low amount of cytosolic protein as illustrated Figure 5.11D, it
also suggests that 12kDa and 15kDa forms of Trx could be due to contamination from
the cytosolic compartment. The 15kDa form could be a Trx in association with a small
chaperone molecule of 3kDa and could belong to the nucleus or other organelles as
cells were lysed differently than in our previous work (Chapter 4).
The form identified at 24kDa could correspond to the molecular weight of the Trx
dimer. To confirm that, β-mercaptoethanol (β-ME) was mixed with the sample in order
to reduce intramolecular disulphide bonds. However, the protein was not reduced
completely as seen Figure 5.12A. DTT, another reducing agent was also tested but
the same result obtained with β-ME was observed (Figure 5.12B). In fact, this dimer
resistance to reductant was recently demonstrated in shrimp Trx as a result of the
disulphide bond Cys73-Cys73 (Campos-Acevedo, Sotelo-Mundo et al. 2017). This
disulphide bonds resist to elevated concentration of DTT higher than 50mM. We
therefore hypothesise that Trx was present at the membrane of RAW cells as a stable
dimer.
205
Figure 5.11: Detection of Trx in membrane RAW cells. A; B; C. Three individual
experiments: Cells were treated or not with 100ng/ml of LPS for 24h, then membranes
were extracted and submitted to electrophoresis. Trx was analysed by Western blot
using a polyclonal antibody anti-mouse Trx antibody. D; Western blot of a marker of
cytosolic protein (GAPDH) and of membrane proteins (ATPase) used as controls for
the membrane extraction in Experiment 2.
206
Figure 5.12: Reduction of the 24kDa form of Trx with β-ME (A) and DTT (B).
Membrane samples were separated by electrophoresis after being mixed with
different concentrations of β-ME or DTT. Trx was then analysed by Western blot using
a polyclonal antibody specific for mouse Trx
207
5.9 Discussion
In the present Chapter, we have successfully set up a protocol for the identification of
proteins with free thiols present at the surface of RAW cells stimulated or not with LPS
for 24h. Despite the limitation that a number of proteins belonging to the nucleus and
cytosolic compartments were identified, suggesting some degree of contamination of
our membrane preparations, a number of observations were made and are discussed
below.
The overall amount of free protein thiols at the surface of 24h LPS-treated cells is
higher than the level of free thiols present in untreated cells. This result is similar to
what was observed in other monocytes cells lines using 100ng/ml of LPS for 24h
(Szabo-Taylor, Toth et al. 2017). This increase in free thiols is also observed in
association with activation of the immune response (Metcalfe, Cresswell et al. 2011).
On the contrary, the levels of surface thiols detected from cells stimulated only 2h with
LPS were similar to the levels observed in untreated cells if not slightly lower. This
may relate to the fact that we detected an increase generation of superoxide anion
(O2.-) at this early time point, as described in Chapter 3 - 3.3. Superoxide, a ROS, is
a highly reactive molecule able to oxidise rapidly proteins and especially free thiols,
and one could have expected an even stronger decrease in the level of free thiols.
Additionally, a number of receptors and signalling proteins were identified by MS
among the membrane proteins bearing a free surface thiol. Integrins were present in
untreated cells but not LPS-treated cells. In fact, integrin α-4, was previously identified
as a redox target in human peripheral blood mononuclear cells and could be oxidised
by oxidoreductases released during LPS stimulation (Laragione, Bonetto et al. 2003).
PDIs were also identified in untreated cells but were less present in LPS-treated cells.
It could be that they actively catalyse thiol disulphide exchange with other molecules
at the surface of the macrophages during LPS stimulation as it was demonstrated in
neutrophils (Hahm, Li et al. 2013). On the other hand, heat shock proteins were only
found in LPS-treated cells and thus correlate with an inflammatory and stress
response.
The fact that some proteins identified in the LPS-treated cells (Oas family; STAT and
Ptgs2) were identified previously as being transcriptionally induced by GSH during
LPS stimulation (as shown in Chapter 3-3.5) strengthens the hypothesis that several
redox changes occur in RAW cells following LPS stimulation, although our results also
suggest some cytosolic contamination.
208
Finally, Trx was detected by Western blot in membrane samples but was not identified
by MS. Although this suggests that membrane Trx is normally in the oxidized state, it
is also possible that its peptides were not identified by MS for technical reasons. In
fact not all peptides ionize to the same extent and some may be difficult to detect by
MS. A better way to confirm one or the other hypothesis would be to use the MalPEG
method; unfortunately Trx detection was not possible potentially due to
conformational changes in the epitopes recognised by the antibody anti-Trx after
attachment of MalPEG molecule(s).
In addition, in our experimental model, Trx was identified as a dimer. This
conformational structure has been suggested in many structural studies but, to our
knowledge, was never detected in the membrane from mammalian cells.
Interestingly, structural studies have suggested that the Trx homodimer plays an
important physiological role due to its high stability, its conserved amino acids
sequence and the fact that it is not a substrate for Trx reductase (Weichsel, Gasdaska
et al. 1996). Possible biological roles of the dimer are still unknown but sensing the
cell redox state has been postulated (Weichsel, Gasdaska et al. 1996). Furthermore,
the oxidized dimer leads to the loss of cytokine-like activity by dimerization of C73
(Gasdaska, Kirkpatrick et al. 1996).
To conclude, the methodology probably needs further refinement including, for
instance, additional solubilisation steps and washes. Nevertheless, we obtained a set
of data pointing at potentially interesting findings. Since the reason why we set up this
technique was to identify potential surface target of extracellular Trx and Prx2, the
next step will consist in inhibiting Trx, to see which protein thiol/disulphides at the cell
surface are among its possible targets.
209
Chapter 6
Redox state of Peroxiredoxin 2 and Thioredoxin
as biomarkers of oxidative stress
210
6.1 Introduction
Coronary artery diseases (CAD) are part of the cardiovascular diseases (CVDs) which
are the main cause of death in the world (World Health Organization n.d.). In 2015,
31% of the deaths worldwide were attributable to CVDs and nearly half of them were
caused by CAD (World Health Organization n.d.). Thus, a decrease of this disease’s
incidence is one of the priorities of the WHO.
Physiologically, CAD occurs when an artery is narrowed, preventing the blood flow to
transport nutrients and oxygen to the heart (also referred as ischemia), and therefore
leading to myocardial infarction, where part of the heart muscle dies. According to the
WHO, risk factors include unhealthy food, alcohol abuse, and tobacco, lack of physical
exercise, diabetes, stress, and ageing. All these factors are linked with oxidation and
inflammation, themselves associated with coronary heart diseases (Hansson 2005,
Libby and Theroux 2005). In fact, CAD is mainly a consequence of years of
atherogenesis, a disorder of the artery walls, which starts as an inflammatory process
through the oxidation of fatty acids activating macrophages and immune response
(Ross and Agius 1992, Libby and Theroux 2005, Hansson and Hermansson 2011).
Essentially, proinflammatory cytokines and chemokines are released in the vessels,
by different processes, attracting monocytes and lymphocytes to the vessels and
leading to their adhesion to endothelium and smooth muscle cells into the intima layer
(just below the endothelium forming the walls of the blood vessels). This forms an
extracellular matrix which binds lipids and lead to the deposit of lipid-laden
macrophages (“foam cells”), differentiated monocytes which ingest lipids. This deposit
undergoes fibrosis and calcification forming an atheroma (Latin for “tumour full of
gruel-like matter”) that stop the blood from correctly supplying the heart (Libby 2002).
This inflammatory process which has become excessive and chronic is defined as
atherosclerosis (Ross and Agius 1992).
Unfortunately, the atheroma cannot be completely removed due to its anchorage in
tissues but can only be controlled by taking drugs such as aspirin in addition of
significantly improving life style with healthy diet and physical activity. However, in
some cases, surgical intervention is the only solution to restore proper blood flow.
This is referred as angioplasty, a common procedure, to flatten the atheroma; a
balloon is inflated directly into the blocked area, pushing the atheroma to the vessels
and placing a stent to prevent it from blocking, again, the blood flow in the vessel.
Ironically, angioplasty can also result in cellular damage due to the reperfusion
ischemia, that is a short period of ischemia followed by reintroduction of molecular
211
oxygen (O2) increasing the formation of ROS and further damage in the vessels
(Kalogeris, Baines et al. 2012 1159).This series of events can initiate inflammatory
responses and aggravate the local injury (Preeshagul, Gharbaran et al. 2013). To
evaluate the potential complication post-injury performed by the insertion of the stent
and to assess patient survival and adapt treatment (Preeshagul, Gharbaran et al.
2013), research has focused on the investigation of damage and inflammation
biomarkers easily accessible in the bloodstream. Ideally a large set of biomarkers is
evaluated for each patient by combining all the data collected leading to different and
specific therapy (Kleber, Goliasch et al. 2014). This required a large pallet of
biomarkers to identify the different facets of the disease; oxidation, inflammation, and
injury.
Currently, the most common biomarkers of CAD are Troponin T, a contractile
component of the heart muscle which is released into the circulation after heart failure,
and C-reactive protein (CRP), an acute protein synthesised in response to
inflammatory cytokines, such as IL-6 and TNF (Saunders, Nambi et al. 2011,
Ikonomidis, Michalakeas et al. 2012, Shrivastava, Singh et al. 2015). More recently,
circulating microRNAs have also been investigated as novel more reliable
biomarkers, easily measured in plasma (Wang, Zhu et al. 2010).
Assessing oxidative damage, an important factor in the progression and complication
of the disease, is however still problematic (Strobel, Fassett et al. 2011). Two main
oxidative biomarkers are currently used: an oxidised lipid, oxidised low-density
lipoprotein (LDL), and myeloperoxidase, a heme peroxidase (Huang, Mai et al. 2008,
Karakas and Koenig 2012). Limitation have been raised in regards to the handling
and standardisation of the samples to keep the same level of oxidative stress
reference (Pastori, Carnevale et al. 2014). Another issue is that the impact of
antioxidant level present in the environment which is not understood, can lead to
different interpretation of these markers (Pastori, Carnevale et al. 2014).
Thiol oxidation of blood proteins could be a response to this difficulty by assessing
potential oxidative damage to the vessels through the redox state of a chosen thiol
protein. In fact, blood, in addition to be easily accessible, is a rich environment in
terms of redox reactions and therefore in potential thiol oxidised molecules. As
explained by Butera et al, at least one fifth of all proteins present in blood contain
disulphide bonds which can be involved in processes such as coagulation, thrombosis
and inflammation (Butera, Cook et al. 2014). Human plasma is rich with free cysteine
or cysteine disulphide which are considered as biomarkers of oxidation due to their
sensitivity to smoke, alcohol abuse and high-fat diet all of which are risk factors for
212
CVD (Go and Jones 2011). Red blood cells are also very sensitive to redox
equilibrium and considered as entities with complex antioxidant systems and carrier
of oxidative biomarkers (Pandey and Rizvi 2011).
Interestingly, studies performed on Trx, Prx1 and Prx2 support the importance of
using thiol oxidation biomarkers in assessing oxidative damage in cardiovascular
disease (Choi, Lee et al. 2005, Martinez-Pinna, Lindholt et al. 2010, Martinez-Pinna,
Ramos-Mozo et al. 2011, Madrigal-Matute, Fernandez-Garcia et al. 2015).
A correlation between increase level of Prx1 and Trx and the evolution of the disease
in abdominal aortic aneurysm (AAA), a type of atherosclerotic disease, suggested that
both of them could be biomarkers of its severity (Martinez-Pinna, Lindholt et al. 2010,
Martinez-Pinna, Ramos-Mozo et al. 2011). Furthermore, recently, the same research
group has shown a dependency of their extracellular release, in plasma patient, with
NOX activation suggesting antioxidant response to atherosclerosis (Madrigal-Matute,
Fernandez-Garcia et al. 2015).
Additionally, overoxidation of Prx2 with sulfenic or sulfonic acid could be used as a
biomarker of the thickness of the intima layer in atherosclerosis and its evolution
(Kang, Lee et al. 2013). This overoxidation of Prx2 was observed in rats intima layer
after injury provoked by balloon insertion in the artery, a model of endothelium
damage, but also in human atherosclerotic lesions. Previous studies lead to
hypothesise that overoxidised Prx2 play a role in the healing process of the injury by
mediating the vascular signalling process (Choi, Lee et al. 2005).
Oxidation is present from the beginning of atherosclerosis in CAD patients until
medical intervention via the reintroduction of molecular oxygen in the blood stream.
This inflammatory disease is therefore an ideal condition to study the potential
occurrence in vivo of the redox changes in Trx and Prx2 previously detected in RAW
cells. To study this, in the experiments described in this chapter, we set up a technique
to measure the redox state of Prx and Trx in the blood of patients with CAD
undergoing percutaneous coronary intervention. During this procedure, the stenotic
portion of the artery is dilated with an intracoronary balloon after which a stent is
deployed to provide structural support to the artery.
Aim: Trx and Prx2 go through redox changes specific to LPS stimulation which
can be detected both intracellularly and in the secreted proteins. We wanted to
investigate whether these changes could be used as biomarkers of oxidation in
patient’s blood. Thus, in this chapter, the redox state of Trx and Prx2 will be
213
studied in human blood from healthy donors before to be determined in blood
from patients undergoing angioplasty.
214
6.2 Assessment of the MalPEG technique in Rat blood
Before applying this methodology to studies with human blood, the MalPEG-
technique was tested in rat plasma. In fact, the viscosity of MalPEG, mentioned
previously, added to the viscosity of blood could be problematic by preventing a
correct migration of the proteins in the polyacrylamide gel electrophoresis. As shown
Figure 6.1, both Trx and Prx2 were detectable in normal rat plasma. Two redox states
were observed with Trx: one fully oxidised at 12kDa (= 0 MP) and one more reduced
at 32kDa having two-SH groups (= 2 MP). For Prx2, two forms were also detected in
the MalPEG-treated proteins: an oxidised dimer without any MalPEG attached (= 0
MP), and a more reduced dimer, with two –SH groups, at 64kDa (= 2 MP).
215
Figure 6.1: Measure of the redox state of Trx (A.) and Prx2 (B.) in plasma from
two rats. Plasma from two independent rats (#1 and #2) were treated with 0.5mM of
MalPEG or with 50mM of NEM as described in the methods section. Trx and Prx2
were then analysed by Western blot in non-reducing or reducing conditions. The
arrows indicate the position of reduced or oxidised forms determined by the number
of MP fixed to the protein.
216
6.3 Measurement of the redox state of Prx2 in human plasma
Having shown that this technique is applicable to the detection of the redox state of
proteins in rat plasma, we assessed the redox state of Prx2 in human plasma from
four healthy human donors.
As shown Figure 6.2, Prx2 treated with NEM (which does not modify the molecular
weight) was mainly found as an oligomer (around 250 kDa) but also as two dimeric
forms, both around 44kDa in the plasma. These two dimers were already described
in human serum (Mullen, Hanschmann et al. 2015, Peskin, Pace et al. 2016). The
micro heterogeneity of the dimer, showing as a doublet, is probably due to small
changes in mobility associated with posttranslational modifications such as
glutathionylation. It has been suggested that they correspond to the glutathionylated
form (upper one) and non-glutathionylated dimer (lower one). The MP technique
allowed to discriminate between oxidized dimers, migrating to a putative MW of 44kDa
(=0MP), therefore no free –SH) and reduced dimers at 64kDa (which implies 2 MP
bound and therefore 2 free –SH).
The four healthy donors have a similar redox profile independent of the total amount
of Prx2 present.
217
Figure 6.2: Redox state of Prx2 in plasma from 4 healthy donors (#1 to #4).
Plasma samples were treated with 0.5mM of MalPEG (MP) or 50mM of NEM. Prx2
was then analysed by Western blot following non-reducing conditions. The arrows
indicate the position of reduced or oxidised forms determined by the number of MP
bound to the protein.
218
6.4 Measurement of the redox state of Trx in human red blood cells
In contrast with Prx2, we could not define the redox state of Trx in human plasma.
Indeed, anti-Trx detected aggregates and several bands at different molecular
weights, making the study of its redox state impossible as shown Figure 6.3A. To
overcome this difficulty, we studied Trx in red blood cells (RBCs) lysate. The
advantage of using biomarkers from RBCs is that they do not have a machinery to
synthetize new proteins due the lack of nucleus but RBCs are also the first cell
exposed to redox changes (Pandey and Rizvi 2011). Therefore oxidised protein are
less diluted than in the plasma and are constantly undergoing redox changes. In
addition, RBCs are full of antioxidant proteins such as Prx2, the third most abundant
protein in RBCs (Low, Hampton et al. 2008). The redox state of Trx was measurable
as three bands were observed: one at 12kDa for the completely oxidised form (= 0
MP), one at 32kDa (= 2 MP) and one around 62kDa (= 5 MP) (Figure 6.3B). The
three donors studied had a similar redox profile for Trx.
219
Figure 6.3: Redox state of Trx in plasma (A) or in RBCs lysate (B) (1:10 dilution)
from 3 healthy donors. Plasma or red blood cells samples were treated 0.5mM of
MalPEG (MP), 50mM of NEM, or left untreated (Ctrl). Trx was then analysed by
Western blot following reducing condition. The arrows indicate the position of reduced
or oxidised forms determined by the number of MP fixed to the protein.
220
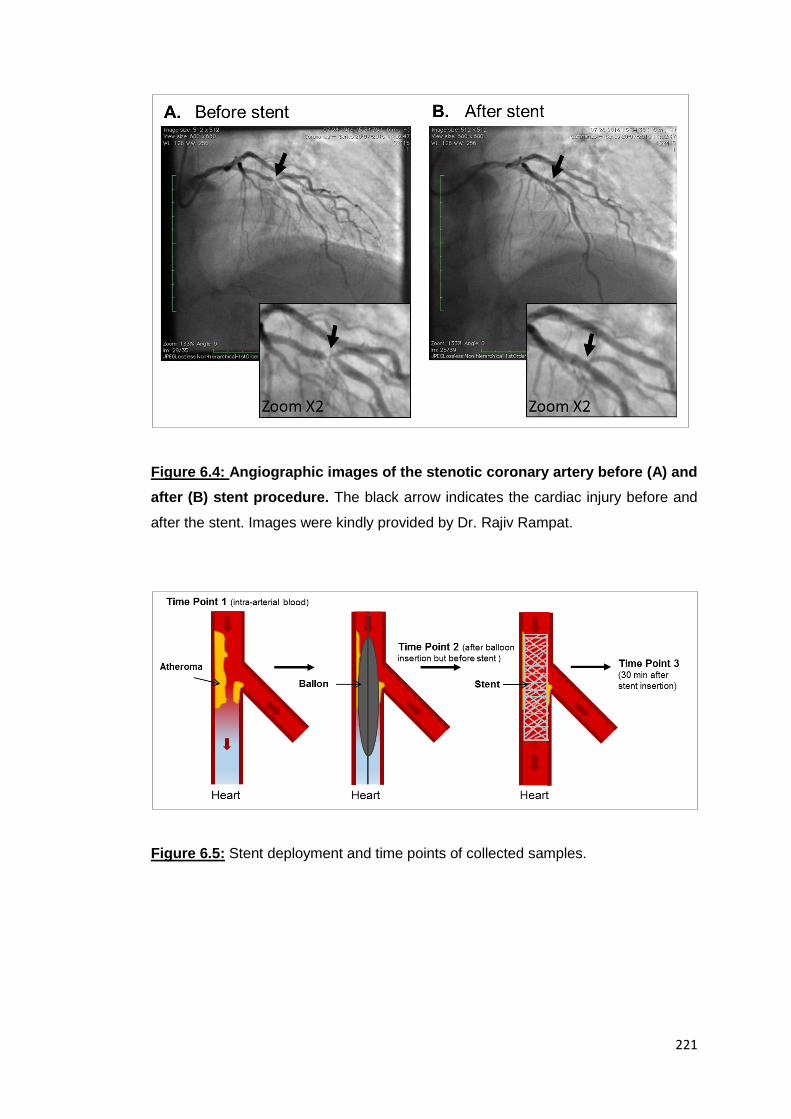
6.5 Assessment of potential redox changes in Trx and Prx2 after stent
insertion in blood samples from CAD patients
Blood was taken from patients suffering from coronary artery disease before and after
angioplasty. During the medical procedure, a biodegradable stent was inserted into
the artery via a catheter and directed by radio-imaging to the injury allowing the blood
to flow freely as visualized in Figure 6.4A before the stent and Figure 6.4B after the
stent. Blood samples were collected at 3 time points (Figure 6.5). Firstly, before the
intervention, an intra-arterial blood sample was taken via peripheral arterial sheath (=
time point 1). Secondly, a sample was collected from the affected coronary artery after
insertion of the ballon but before stent insertion (=time point 2) or, thirdly, 30min after
the insertion of the stent (= time point 3).
As shown in Figure 6.6, the investigation of the redox state of Prx2 in plasma gave
inconsistent results. Indeed, either the Prx2 from samples treated with MalPEG was
not detected or the control Prx2 was not detected.
Therefore, we decided to measure both Prx2 and Trx in RBCs (Figure 6.7 and Figure
6.8). Results were consistent between the two patients where Prx2 was studied and
between the three tested for Trx. As shown in Figure 6.7, two bands were observed
in the MP-treated samples, one at 44kDa which corresponds to the dimer and one
staining stronger at 64kDa (2 MP). In Figure 6.8, MalPEG-Trx migrated as three
bands: one band at 12kDa (=0MP/0SH) which could be either the completely oxidised
form, another band at 32kDa (= 2 MP/2-SH) and a band at 62kDa (= 5 MP/5-SH).
There was no difference detected before and after stent insertion in any of the three
patients tested.
In that we could distinguish the different redox states but they did not change before
and after the stent (time point 1 versus time point 2 or 3). Two hypothesis could explain
the absence of redox changes; neither Trx nor Prx2 underwent redox modifications
under these conditions or because any specifically-oxidized protein would be
immediately washed away and diluted in the total body’s blood volume. However,
due to the difficulty to access blood samples, the study was stopped at that stage.
221
Figure 6.4: Angiographic images of the stenotic coronary artery before (A) and
after (B) stent procedure. The black arrow indicates the cardiac injury before and
after the stent. Images were kindly provided by Dr. Rajiv Rampat.
Figure 6.5: Stent deployment and time points of collected samples.
222
Figure 6.6: Redox state of Prx2 in plasma at different time point before or after
stent insertion. Plasma samples were treated with 0.5mM MalPEG. Prx2 was then
analysed by Western blot in non-reducing condition. The arrows indicate the missing
bands.
Figure 6.7: Redox state of Prx2 in RBCs at different time point before or after
stent insertion. RBCs samples were treated with 0.5mM MalPEG. Prx2 was then
analysed by Western blot in non-reducing condition. The arrows indicate the position
of reduced or oxidised forms determined by the number of MP fixed to the protein.
223
Figure 6.8: Redox state of Trx in RBCs at different time point before or after
stent insertion. RBCs samples were treated with 0.5mM MalPEG. Trx was then
analysed by Western blot in non-reducing condition. The arrows indicate the position
of reduced or oxidised forms determined by the number of MP fixed to the protein.
224
6.6 Oxidation of Trx and Prx2 with diamide
In order to see whether our technique could detect redox changes in Trx/Prx in patient
RBCs lysates, RBCs samples were treated with 10mM diamide, a thiol-oxidising
agent, for 10min (Kosower, Kosower et al. 1969). When treated with diamide, the
most reduced form of Trx disappeared as indicated by the red arrow (Figure 6.9). The
same observation was made for Prx2 (Figure 6.9). Therefore, our methodology can
detect oxidation of Trx and Prx2.
We then studied the applicability of our technique to a clinical setting, where the blood
may not be processed immediately after sampling which is an issue in the
standardisation of oxidative marker reproducibility between studies.
For this purpose, samples were either processed immediately (Control) or left on the
bench at room temperature for one hour. These were compared with samples
oxidised with diamide for 10min. A shown Figure 6.10, Prx2 was not affected by the
length of time of the procedure. Indeed, the two characteristic redox states of Prx2
(i.e. 44kDa and 64kDa) were still present in the same proportion in the sample left in
the bench for an hour and the one processed immediately. On the contrary, the redox
state of Trx was sensitive to the new condition: the most reduced form that is the band
migrating at 62kDa (= 5 MP/5-SH) disappeared similarly to samples exposed to
diamide.
225
Figure 6.9: Effect of diamide on the redox state of Trx and Prx2 in human RBCs.
After 10mM diamide treatment, proteins were treated with 0.5mM MP and analysed
by Western blot. 1 and 2 indicate the time point samples blood were collected. The
red arrows indicate the missing band. The black arrows indicate the different redox
states of Trx and Prx2.
Figure 6.10: Redox state of Trx and Prx2 in human RBCs left 1h at room
temperature. RBCs were tagged with 0.5mM MalPEG after 1h left on the bench or
after being treated 10min with 10mM diamide. Control are samples processed
immediately. Trx and Prx2 were then analysed by Western blot. The arrows indicate
the different redox states of Trx and Prx2.
226
6.7 Discussion
The aim of this chapter was to investigate the redox states of Prx2 and Trx in human
blood and thus measure redox changes in patients undergoing angioplasty due to
stent insertion by percutaneous coronary intervention.
The redox states of Prx2 and Trx were successfully measured in blood using the
MalPEG technique. However, no measurable redox changes were detected in the
blood of patients undergoing stent insertion, despite the fact that the blood was taken
from the injured area. There are two possible explanations for these observations.
First, it is possible that there are no biological redox changes occurring during the
stent insertion. However, it is well known that damage occurs to the artery due to
mechanical rupture of the atheroma by the stent, the presence of a foreign device and
also due to the reintroduction of oxygen in previously deprived blood vessels (Otsuka,
Finn et al. 2012) (Boos, Balakrishnan et al. 2007, Pelliccia, Del Prete et al. 2012).
Similarly, evidence of inflammation and oxidative stress evidences have been
reported during percutaneous coronary intervention (Cordis, Maulik et al. 1998,
Iliodromitis, Kyrzopoulos et al. 2006) (Chao, Li et al. 2004, Berg, Jynge et al. 2005).
In fact, one could hypothesise that the damage perpetuated by the stent insertion will
release DAMPS, and thus activate the TLR4 pathway leading to an inflammatory
response (Lee, Hutchinson et al. 2016). This is also supported by a recent study
demonstrating that hydrocortisone could reduce the inflammatory effects of stent
insertion by reducing TLR4 expression (Bagheri, Sohrabi et al. 2014). Thus, we could
hypothesise that as seen in RAW cells with LPS stimulation of TLR4 pathway, in
Chapter 4, redox changes may occur later than the time points chosen. Indeed, these
changes were measured 24h later from RAW cells, in both intracellular and
extracellular compartments. So it could be that the redox state of the proteins studied
is not affected by those damages yet.
The second possibility is that redox changes were undetectable due to the rapid
speed of blood flow through the systemic circulation. Cardiac output at rest is 5L/min
so one could expect local changes to be quickly diluted by this rapid blood flow.
In the experiments using diamide we could demonstrate that the redox state of Prx2
is quite stable and not too sensitive to experimental conditions, particularly when
compared to Trx. Incubation at room temperature for one hour, did not result in any
detectable change in redox forms identified in the case of Prx2, while a similar
incubation of Trx resulted in the loss of the more reduced form. Thus, Trx seems to
be more sensitive to oxidation and could therefore be a better, more sensitive, marker
227
of oxidative conditions. For instance, measuring the redox state of Trx could also help
in the quality control of human blood stored for transfusion as transfusion of oxidised
erythrocytes is often associated with dangerous side effects such as myocardial
infarction which can cause mortality (Bayer, Hampton et al. 2015). Another recent
study also demonstrated the role of Prx2 in erythrocytes as an indicator of cell damage
during storage (Harper, Oh et al. 2015). In fact, stored erythrocytes show increased
oxidative damage due to the lack of oxygenation and Prx2 is known to limit this
damage as long as its function is not altered. In their study, Harper and colleagues
attempted to measure Prx2 dimeric or monomeric forms in RBCs in blood stored in
transfusion bags at 4˚C in the dark from 7 to 35 days in order to control the quality of
erythrocytes as they hypothesised that the Prx2 dimeric form will indicate oxidation of
the blood (Harper, Oh et al. 2015). They then conclude that because Prx2 was present
as a dimer in the RBCs of stored blood that its activity was compromised (Harper, Oh
et al. 2015). The MalPEG technique described here could provide more insight into
whether Prx2 is present as a reduced or a fully oxidised dimer as we believe that there
are different degrees of oxidation of Prx2 (Chapter 4) and thus could provide further
information to stratify the degree of blood oxidation.
It has also to be noted that a previous study has demonstrated that the native redox
state of Prx2 from healthy donors obtained by mixing samples with NEM prior to lysis
is mainly found as a reduced monomer in RBCs whereas the dimer was present due
to overoxidation in lysis buffer and thus this dimeric form is overoxidised (Low,
Hampton et al. 2007). In our study, RBCs were lysed in ice-cold conditions and mixed
directly with MalPEG and Prx2 was found as a dimer. Under these conditions, it is
expected that the oxidation of Prx2 occur at a slow rate. In fact, we could see that our
dimer contained free thiols detected by MalPEG and thus was not overoxidized. This
also demonstrates the importance of defining what it is overoxidised. In our opinion,
overoxidation will mean that the monomers cannot dimerize via disulphide bond
formation and thus cannot catalyse the removal of peroxide substrates, while the
dimer form indicates that the protein is active and can catalyse this removal.
Remarkably, the redox profile of Trx and Prx2 in the RBCs from both patients and
healthy donors were similar to the redox profile obtained in RAW cells lysate: Prx2
with no free -SH or 2-SH and Trx with 0, 2 free -SH or 5 free-SH. This consistency of
the overall redox state detected for Prx2 and Trx suggest that the redox state of these
proteins is maintained both in cultured cells and in human blood and that the redox
state of these proteins is likely to be important for their biological functions.
228
In conclusion, we set up a method for identifying the redox state of Prx2 and Trx in
the blood. Further work will be needed to investigate if redox changes of Prx2 and Trx
occur in disease and if they could have a prognostic or diagnostic value.
229
Chapter 7
Discussion and conclusions
230
7.1 Summary of the study
The main aim of this work was to identify molecular redox mechanisms participating
in the inflammatory response in macrophages. This was achieved by investigating
and measuring redox changes in different ways during the inflammatory response. It
was examined at the transcriptome level by depleting GSH, the main thiol antioxidant
but also a regulator of the redox state of protein thiols and disulphides (Chapter 3),
and in the intracellular and extracellular proteome by measuring the redox state of
redoxkines Prx1, Prx2 and Trx (Chapter 4) as well as at the cell surface of
macrophages by identifying proteins with free thiols and thus potential targets of those
redoxkines (Chapter 5). Redox changes of Prx2 and Trx were also tested as oxidative
biomarkers in human blood from healthy donors and patients undergoing angioplasty
due to coronary artery disease; however, no changes in the redox states of these
proteins were detected (Chapter 6). The main findings of each chapter as well as
further work required are summarised in Table 7.1.
We demonstrated that GSH, the main intracellular antioxidant and ROS scavenger,
also acts as a signalling molecule required for antiviral response but also controls the
amount of Prx1 transcribed. In fact, the microarray data has shown that Prx1
transcription is induced by LPS and its level is regulated by GSH, while both Prx2 and
Trx genes are not affected by either LPS stimulation or GSH depletion. Both of them
are redox-regulated at latter steps, by post-translational modification after LPS
stimulation. In fact, we could determine three redox profiles specific of each of these
redoxkines (Prx1, Prx2 and Trx) depending on their cell location and exposure to
inflammatory stimulation. Prx2 and Trx are both detected as a mixture of reduced and
oxidised forms prior to LPS stimulation. After exposure of the cells to LPS, only the
reduced form is observed intracellularly while the oxidised form seems to be released.
This could be a homeostatic measure to maintain a reduced intracellular environment.
Furthermore, while the oxidised form of Trx is detected extracellularly, Prx2 is found
in the reduced form, suggesting that the oxidised form could have been reduced by
Trx, its main electron donor outside of the cell. Thus, the redox state of both proteins
appears to be in dynamic equilibrium with their catalytic activity dependent on LPS.
On the contrary, Prx1 is only found as a reduced form with 2 free-SH (one for each
monomer) and as a fully oxidised form when released, which could correlate with its
DAMP activity due to inactivation of its catalytic centre.
We then hypothesised that the reduced form of Prx, in cooperation with Trx, could
catalyse thiol disulphide exchanges outside the cell using extracellular substrates
231
such as proteins with free thiols at the cell surface. We thus set up a method to detect
those potential targets by merging alkylation of free thiols at the cell surface with a
biotin tag plus MS identification following streptavidin bead enrichment. Although
preliminary, this technique has allowed the detection of interesting receptors such as
integrin-α previously identified as redox regulated by inhibitors of GSH and by NAC
at the surface of PBMC (Laragione, Bonetto et al. 2003).
Unexpectedly, we could also identify Trx in a dimeric form. It seemed that this form
was only present in the membrane (plasma or exosome) as it was not identified in our
previous work in cell lysates and supernatants. This is in agreement with other studies
suggesting that a Trx dimer could be associated with a redox sensor function
(Weichsel, Gasdaska et al. 1996). At the same time, redox changes detected after
LPS stimulation in macrophages were analysed in the plasma and red blood cells
from healthy donors as well as patients undergoing angioplasty due to coronary artery
disease in order to detect potential redox changes. However, no changes were
observed but the same overall redox state in human red blood cells was detected as
observed in RAW cell lysates demonstrating the stability of Prx2 and Trx redox state
across different cells and under different conditions.
We could then also hypothesise that those redox changes of redoxkines were
dependent on LPS. In fact, this endotoxin only reacts with TLR4 triggering a specific
signalling pathway. Therefore, further work to identify these signalling pathways is
required.
232
Chapter Aim Results Further work
3
Identify genes regulated by GSH during LPS stimulation in RAW cells
GSH acts as a signalling molecule to fine tune the antiviral response
Test this hypothesis at the proteomic level
4
Identify redox state changes of Prx1, Prx2 and Trx in LPS-stimulated RAW cells
Following LPS stimulation in RAW cells, Prx2 and Trx undergo redox changes prior to secretion and seem to have a catalytic activity extracellularly while Prx1 is not sensitive to redox change and seems to act as a DAMP
Identify the signalling pathway of secretion; identify specific biological functions for each redox form detected
5
Identification of surface thiols targets of thiol oxidoreductases released in LPS-stimulated RAW cells
Identification of interesting receptors such as integrin-α; Trx exists as a dimer
Optimisation of the technique of membrane extraction is required as well as inhibiting Trx prior to LPS stimulation
6
Utilisation of the redox state change of Prx2 and Trx as biomarkers of oxidative stress
Redox state of Prx2 and Trx are measurable in human blood; No redox changes in patients undergoing angioplasty
Determination of “use-by date” for stored human blood + use another model or use other tissues than blood
Table 7.1: Findings and further work required for each aim of this study.
233
7.2 Advances in the redox and inflammatory field
Potential for novel antioxidant therapies
It is interesting to note that in our experimental model, endogenous GSH does not
inhibit the inflammatory response and that this is contrary to what has been concluded
by pioneers in this field who used a different experimental approach by adding
exogenously thiols antioxidants (Schreck, Rieber et al. 1991). However, one gene,
CXCL10, was increased when cells were depleted of GSH and then stimulated with
LPS. This chemokine, formerly known as IFNγ-inducible protein 10 (IP-10) has been
recently studied in chronic inflammatory diseases such as diabetes (Antonelli, Ferrari
et al. 2014), autoimmune diseases (Lee, Lee et al. 2009), multiple sclerosis
(Vazirinejad, Ahmadi et al. 2014), and cancer (Lunardi, Jamieson et al. 2014) and
therefore could be linked with the increase of ROS due to GSH depletion. In fact, the
effects of thiol antioxidants and ROS vary in studies depending on the experimental
model used as discussed previously (Chapter 3). The fact that some molecules such
as CXCL10 have been identified in a number of studies strengthens the evidence for
its role in, and as a potential therapeutic target for, chronic inflammatory diseases.
Overall, the microarray analysis also brought new insights in defining how complex
the thiol system antioxidants is, highlighting GSH as a signalling molecule important
for host defence. Our results are consistent with other studies as mentioned
previously in Table 3.5 investigating the effect of GSH depletion on viral replication.
It could also explained why viral infection is often linked with reduced GSH level
(Swietek and Juszczyk 1997) (Herzenberg, De Rosa et al. 1997) (Papi, Contoli et al.
2008) as this could be a viral strategy to increase replication and survival by down-
regulating the host antiviral response.
Prxs and Trx have specific redox states depending on their localisation and
could be used as biomarkers or therapeutic targets
In this study, we identified different redox states of Prx1, Prx2 and Trx specific to their
localisation as well as the condition of the cells. It is possible that these specific redox
states could be used as biomarkers of protein oxidation. The MalPEG technique
developed in this study seems a promising technique to identify different degrees of
oxidation of a protein which could be useful to measure the oxidation and hence
quality of stored blood intended for transfusion (Bayer, Hampton et al. 2015). In
addition, it seems that the redox changes in the proteins studied here were specific to
234
LPS stimulation and thus the TLR4 signalling pathway. This activation of TLR4 has
been described, in many autoimmune diseases (Liu, Yin et al. 2014) such as
inflammatory bowel disease (Oostenbrug, Drenth et al. 2005) or Alzheimer’s disease
(Walter, Letiembre et al. 2007) which could be used as models to determine whether
Prx2 and Trx redox changes could be used for prognostic or diagnostic purposes.
A number of studies have pointed out the potential cytokine- and chemokine- like roles
of Prx2, Trx and Prx1 during the inflammatory response. To summarize, Prx1 acts as
a DAMP (Riddell, Wang et al. 2010), Trx acts as a chemoattractant (Bertini, Howard
et al. 1999) but also as a growth factor (Gasdaska, Berggren et al. 1995) and finally
recent studies have shown that Prx2 can promote release of TNF from cells (Salzano,
Checconi et al. 2014). In addition, and in particular for Trx, these proteins are often
detected in elevated concentrations in autoimmune diseases and cancer compared
to healthy individuals (Nakamura, De Rosa et al. 1996, Lincoln, Ali Emadi et al. 2003)
(Wahlgren and Pekkari 2005) (Riddell, Bshara et al. 2011, Szabo-Taylor, Eggleton et
al. 2012). Thus research to inhibit these proteins has started but has been
inconclusive thus far as illustrated by a recent clinical trial inhibiting Trx from
gastrointestinal cancers patients (Baker, Adab et al. 2013). This failure may be related
to the strategy adopted consisting of completely inhibiting Trx instead of specific
functions of Trx. In fact targeting specific redox states of these proteins could be more
efficient to suppress the pro-inflammatory roles of these proteins. Our study has been
able to identify these specific forms and future work will focus on identifying what
biological roles these forms have and which post-translational modifications are
present. In fact, the development of new therapeutic approaches such as antibodies
targeting specific thiol modifications is being investigated in the field of chronic
diseases (Ryan, Nissim et al. 2014) and one could think that targeting specific redox
states of Prx2 and Trx could help to reduce the inflammatory response in chronic
inflammatory conditions.
7.3 Conclusion
Redox regulation is involved in many cellular processes such as apoptosis, cell
development and differentiation, homoeostasis and the immune response. However
due to the reactivity of ROS, the main effectors, the importance and involvement of
this regulation in signalling functions is difficult to elucidate fully. In this project we
successfully demonstrated the importance of protein and non-protein thiols as targets
235
of redox changes during the inflammatory response directly linked with the TLR4
signalling pathways in macrophages.
236
Bibliography
Abais, J. M., M. Xia, Y. Zhang, K. M. Boini and P. L. Li (2015). "Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector?" Antioxid Redox Signal 22(13): 1111-1129.
Abbas, K., N. Babic and F. Peyrot (2016). "Use of spin traps to detect superoxide production in living cells by electron paramagnetic resonance (EPR) spectroscopy." Methods 109: 31-43.
Abbas, K., J. Breton, C. R. Picot, V. Quesniaux, C. Bouton and J. C. Drapier (2009). "Signaling events leading to peroxiredoxin 5 up-regulation in immunostimulated macrophages." Free Radic Biol Med 47(6): 794-802.
Abbas, K., M. Hardy, F. Poulhes, H. Karoui, P. Tordo, O. Ouari and F. Peyrot (2014). "Detection of superoxide production in stimulated and unstimulated living cells using new cyclic nitrone spin traps." Free Radic Biol Med 71: 281-290.
Abbasi, A., E. Corpeleijn, D. Postmus, R. T. Gansevoort, P. E. de Jong, R. O. Gans, J. Struck, J. Schulte, H. L. Hillege, P. van der Harst, L. M. Peelen, J. W. Beulens, R. P. Stolk, G. Navis and S. J. Bakker (2012). "Peroxiredoxin 4, a novel circulating biomarker for oxidative stress and the risk of incident cardiovascular disease and all-cause mortality." J Am Heart Assoc 1(5): e002956.
Aesif, S. W., V. Anathy, I. Kuipers, A. S. Guala, J. N. Reiss, Y. S. Ho and Y. M. Janssen-Heininger (2011). "Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation." Am J Respir Cell Mol Biol 44(4): 491-499.
Akdis, M., A. Aab, C. Altunbulakli, K. Azkur, R. A. Costa, R. Crameri, S. Duan, T. Eiwegger, A. Eljaszewicz, R. Ferstl, R. Frei, M. Garbani, A. Globinska, L. Hess, C. Huitema, T. Kubo, Z. Komlosi, P. Konieczna, N. Kovacs, U. C. Kucuksezer, N. Meyer, H. Morita, J. Olzhausen, L. O'Mahony, M. Pezer, M. Prati, A. Rebane, C. Rhyner, A. Rinaldi, M. Sokolowska, B. Stanic, K. Sugita, A. Treis, W. van de Veen, K. Wanke, M. Wawrzyniak, P. Wawrzyniak, O. F. Wirz, J. S. Zakzuk and C. A. Akdis (2016). "Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases." J Allergy Clin Immunol 138(4): 984-1010.
Akdis, M., S. Burgler, R. Crameri, T. Eiwegger, H. Fujita, E. Gomez, S. Klunker, N. Meyer, L. O'Mahony, O. Palomares, C. Rhyner, N. Ouaked, A. Schaffartzik, W. Van De Veen, S. Zeller, M. Zimmermann and C. A. Akdis (2011). "Interleukins, from 1 to 37, and interferon-gamma: receptors, functions, and roles in diseases." J Allergy Clin Immunol 127(3): 701-721 e701-770.
Alberts, B. (2002). Molecular biology of the cell. New York, Garland Science.
Alfonso, H., P. Franklin, S. Ching, K. Croft, P. Burcham, N. Olsen, A. Reid, D. Joyce, N. de Klerk and A. B. Musk (2015). "Effect of N-acetylcysteine supplementation on oxidative stress status and alveolar inflammation in people exposed to asbestos: a double-blind, randomized clinical trial." Respirology 20(7): 1102-1107.
Amor, S., F. Puentes, D. Baker and P. van der Valk (2010). "Inflammation in neurodegenerative diseases." Immunology 129(2): 154-169.
237
Andersen, J. F., D. A. Sanders, J. R. Gasdaska, A. Weichsel, G. Powis and W. R. Montfort (1997). "Human thioredoxin homodimers: regulation by pH, role of aspartate 60, and crystal structure of the aspartate 60 --> asparagine mutant." Biochemistry 36(46): 13979-13988.
Antonelli, A., S. M. Ferrari, A. Corrado, E. Ferrannini and P. Fallahi (2014). "CXCR3, CXCL10 and type 1 diabetes." Cytokine Growth Factor Rev 25(1): 57-65.
Arthur, J. S. and S. C. Ley (2013). "Mitogen-activated protein kinases in innate immunity." Nat Rev Immunol 13(9): 679-692.
Babior, B. M., J. T. Curnutte and B. J. McMurrich (1976). "The particulate superoxide-forming system from human neutrophils. Properties of the system and further evidence supporting its participation in the respiratory burst." J Clin Invest 58(4): 989-996.
Babior, B. M., R. S. Kipnes and J. T. Curnutte (1973). "Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent." J Clin Invest 52(3): 741-744.
Bacic, G., A. Pavicevic and F. Peyrot (2016). "In vivo evaluation of different alterations of redox status by studying pharmacokinetics of nitroxides using magnetic resonance techniques." Redox Biol 8: 226-242.
Bagheri, B., B. Sohrabi, A. A. Movassaghpour, S. Mashayekhi, A. Garjani, M. Shokri, M. Pezeshkian and A. Garjani (2014). "Hydrocortisone reduces Toll-like receptor 4 expression on peripheral CD14+ monocytes in patients undergoing percutaneous coronary intervention." Iran Biomed J 18(2): 76-81.
Baharom, F., S. Thomas, A. Bieder, M. Hellmer, J. Volz, K. J. Sandgren, G. M. McInerney, G. B. K. Hedestam, I. Mellman and A. Smed-Sorensen (2015). "Protection of Human Myeloid Dendritic Cell Subsets against Influenza A Virus Infection Is Differentially Regulated upon TLR Stimulation." Journal of Immunology 194(9): 4422-4430.
Baker, A. F., K. N. Adab, N. Raghunand, H. H. Chow, S. P. Stratton, S. W. Squire, M. Boice, L. A. Pestano, D. L. Kirkpatrick and T. Dragovich (2013). "A phase IB trial of 24-hour intravenous PX-12, a thioredoxin-1 inhibitor, in patients with advanced gastrointestinal cancers." Invest New Drugs 31(3): 631-641.
Baldridge, C. W. and R. W. Gerard (1933). "The extra respiration of phagocytosis." American Journal of Physiology 103(1): 235-236.
Banerjee, R. (2012). "Redox outside the box: linking extracellular redox remodeling with intracellular redox metabolism." J Biol Chem 287(7): 4397-4402.
Bankar, S. B., M. V. Bule, R. S. Singhal and L. Ananthanarayan (2009). "Glucose oxidase--an overview." Biotechnol Adv 27(4): 489-501.
Barjesteh, N., S. Behboudi, J. T. Brisbin, A. I. Villanueva, E. Nagy and S. Sharif (2014). "TLR Ligands Induce Antiviral Responses in Chicken Macrophages." Plos One 9(8): 11.
Battistuzzi, G., M. Bellei, C. A. Bortolotti and M. Sola (2010). "Redox properties of heme peroxidases." Arch Biochem Biophys 500(1): 21-36.
238
Bayer, S. B., M. B. Hampton and C. C. Winterbourn (2015). "Accumulation of oxidized peroxiredoxin 2 in red blood cells and its prevention." Transfusion 55(8): 1909-1918.
Bedard, K. and K. H. Krause (2007). "The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology." Physiol Rev 87(1): 245-313.
Beedle, A. E., S. Lynham and S. Garcia-Manyes (2016). "Protein S-sulfenylation is a fleeting molecular switch that regulates non-enzymatic oxidative folding." Nat Commun 7: 12490.
Benham, A. M. (2012). "The protein disulfide isomerase family: key players in health and disease." Antioxid Redox Signal 16(8): 781-789.
Berg, K., P. Jynge, K. Bjerve, S. Skarra, S. Basu and R. Wiseth (2005). "Oxidative stress and inflammatory response during and following coronary interventions for acute myocardial infarction." Free Radic Res 39(6): 629-636.
Bernard, G. R., A. P. Wheeler, M. M. Arons, P. E. Morris, H. L. Paz, J. A. Russell and P. E. Wright (1997). "A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group." Chest 112(1): 164-172.
Berndt, C., C. H. Lillig and A. Holmgren (2008). "Thioredoxins and glutaredoxins as facilitators of protein folding." Biochim Biophys Acta 1783(4): 641-650.
Berridge, M. J., M. D. Bootman and H. L. Roderick (2003). "Calcium signalling: dynamics, homeostasis and remodelling." Nat Rev Mol Cell Biol 4(7): 517-529.
Bertini, R., O. M. Howard, H. F. Dong, J. J. Oppenheim, C. Bizzarri, R. Sergi, G. Caselli, S. Pagliei, B. Romines, J. A. Wilshire, M. Mengozzi, H. Nakamura, J. Yodoi, K. Pekkari, R. Gurunath, A. Holmgren, L. A. Herzenberg and P. Ghezzi (1999). "Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells." J Exp Med 189(11): 1783-1789.
Beziere, N., Y. Frapart, A. Rockenbauer, J. L. Boucher, D. Mansuy and F. Peyrot (2010). "Metabolic stability of superoxide and hydroxyl radical adducts of a cyclic nitrone toward rat liver microsomes and cytosol: A stopped-flow ESR spectroscopy study." Free Radic Biol Med 49(3): 437-446.
Bienert, G. P., A. L. Moller, K. A. Kristiansen, A. Schulz, I. M. Moller, J. K. Schjoerring and T. P. Jahn (2007). "Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes." J Biol Chem 282(2): 1183-1192.
Bindoli, A., J. M. Fukuto and H. J. Forman (2008). "Thiol chemistry in peroxidase catalysis and redox signaling." Antioxid Redox Signal 10(9): 1549-1564.
Blackwell, T. S., T. R. Blackwell, E. P. Holden, B. W. Christman and J. W. Christman (1996). "In vivo antioxidant treatment suppresses nuclear factor-kappa B activation and neutrophilic lung inflammation." J Immunol 157(4): 1630-1637.
Blackwell, T. S., J. W. Christman, T. Hagan, P. Price, T. Edens, P. E. Morris, S. N. Wolff, S. A. Goodman and B. W. Christman (2000). "Oxidative stress and NF-kappaB activation: correlation in patients following allogeneic bone marrow transplantation." Antioxid Redox Signal 2(1): 93-102.
239
Blonder, J., K. C. Chan, H. J. Issaq and T. D. Veenstra (2006). "Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS." Nature Protocols 1(6): 2784-2790.
Boos, C. J., B. Balakrishnan, S. Jessani, A. D. Blann and G. Y. Lip (2007). "Effects of percutaneous coronary intervention on peripheral venous blood circulating endothelial cells and plasma indices of endothelial damage/dysfunction." Chest 132(6): 1920-1926.
Boston, U. o. (2017). "NF-kB Target Genes."
Braakman, I. and D. N. Hebert (2013). "Protein folding in the endoplasmic reticulum." Cold Spring Harb Perspect Biol 5(5): a013201.
Brieger, K., S. Schiavone, F. J. Miller, Jr. and K. H. Krause (2012). "Reactive oxygen species: from health to disease." Swiss Med Wkly 142: w13659.
Brigelius-Flohe, R. and M. Maiorino (2013). "Glutathione peroxidases." Biochim Biophys Acta 1830(5): 3289-3303.
Brubaker, S. W., K. S. Bonham, I. Zanoni and J. C. Kagan (2015). "Innate immune pattern recognition: a cell biological perspective." Annu Rev Immunol 33: 257-290.
Brundu, S., L. Palma, G. G. Picceri, D. Ligi, C. Orlandi, L. Galluzzi, L. Chiarantini, A. Casabianca, G. F. Schiavano, M. Santi, F. Mannello, K. Green, M. Smietana, M. Magnani and A. Fraternale (2016). "Glutathione Depletion Is Linked with Th2 Polarization in Mice with a Retrovirus-Induced Immunodeficiency Syndrome, Murine AIDS: Role of Proglutathione Molecules as Immunotherapeutics." J Virol 90(16): 7118-7130.
Brunelle, J. L. and R. Green (2014). "One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE)." Methods Enzymol 541: 151-159.
Buchmann, K. (2014). "Evolution of innate immunity: clues from invertebrates via fish to mammals." Frontiers in Immunology 5.
Buonocore, G., S. Perrone and M. L. Tataranno (2010). "Oxygen toxicity: chemistry and biology of reactive oxygen species." Semin Fetal Neonatal Med 15(4): 186-190.
Bustin, S. A. (2000). "Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays." J Mol Endocrinol 25(2): 169-193.
Butera, D., K. M. Cook, J. Chiu, J. W. Wong and P. J. Hogg (2014). "Control of blood proteins by functional disulfide bonds." Blood 123(13): 2000-2007.
Cadenas, E. and K. J. Davies (2000). "Mitochondrial free radical generation, oxidative stress, and aging." Free Radic Biol Med 29(3-4): 222-230.
Cai, J., Y. Chen, S. Seth, S. Furukawa, R. W. Compans and D. P. Jones (2003). "Inhibition of influenza infection by glutathione." Free Radic Biol Med 34(7): 928-936.
Cai, Z. and L. J. Yan (2013). "Protein Oxidative Modifications: Beneficial Roles in Disease and Health." J Biochem Pharmacol Res 1(1): 15-26.
240
Callewaert, L. and C. W. Michiels (2010). "Lysozymes in the animal kingdom." J Biosci 35(1): 127-160.
Campos-Acevedo, A. A., R. R. Sotelo-Mundo, J. Perez and E. Rudino-Pinera (2017). "Is dimerization a common feature in thioredoxins? The case of thioredoxin from Litopenaeus vannamei." Acta Crystallogr D Struct Biol 73(Pt 4): 326-339.
Carocho, M. and I. C. Ferreira (2013). "A review on antioxidants, prooxidants and related controversy: natural and synthetic compounds, screening and analysis methodologies and future perspectives." Food Chem Toxicol 51: 15-25.
Carvalho, A. N., C. Marques, R. C. Guedes, M. Castro-Caldas, E. Rodrigues, J. van Horssen and M. J. Gama (2016). "S-Glutathionylation of Keap1: a new role for glutathione S-transferase pi in neuronal protection." FEBS Lett 590(10): 1455-1466.
Chantzoura, E., E. Prinarakis, D. Panagopoulos, G. Mosialos and G. Spyrou (2010). "Glutaredoxin-1 regulates TRAF6 activation and the IL-1 receptor/TLR4 signalling." Biochem Biophys Res Commun 403(3-4): 335-339.
Chao, T. H., Y. H. Li, W. C. Tsai, J. H. Chen, P. Y. Liu and L. M. Tsai (2004). "Elevation of the soluble thrombomodulin levels is associated with inflammation after percutaneous coronary interventions." Clin Cardiol 27(7): 407-410.
Chaplin, D. D. (2010). "Overview of the immune response." J Allergy Clin Immunol 125(2 Suppl 2): S3-23.
Checconi, P., S. Salzano, L. Bowler, L. Mullen, M. Mengozzi, E. M. Hanschmann, C. H. Lillig, R. Sgarbanti, S. Panella, L. Nencioni, A. T. Palamara and P. Ghezzi (2015). "Redox proteomics of the inflammatory secretome identifies a common set of redoxins and other glutathionylated proteins released in inflammation, influenza virus infection and oxidative stress." PLoS One 10(5): e0127086.
Chen, X., F. Ren, J. Hesketh, X. Shi, J. Li, F. Gan and K. Huang (2012). "Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-kappaB pathway." Virology 426(1): 66-72.
Chen, Y. and W. G. Junger (2012). "Measurement of oxidative burst in neutrophils." Methods Mol Biol 844: 115-124.
Choi, M. H., I. K. Lee, G. W. Kim, B. U. Kim, Y. H. Han, D. Y. Yu, H. S. Park, K. Y. Kim, J. S. Lee, C. Choi, Y. S. Bae, B. I. Lee, S. G. Rhee and S. W. Kang (2005). "Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II." Nature 435(7040): 347-353.
Cildir, G., K. C. Low and V. Tergaonkar (2016). "Noncanonical NF-kappaB Signaling in Health and Disease." Trends Mol Med 22(5): 414-429.
Cocheme, H. M. and M. P. Murphy (2010). "Can antioxidants be effective therapeutics?" Curr Opin Investig Drugs 11(4): 426-431.
Collet, J. F. and J. Messens (2010). "Structure, function, and mechanism of thioredoxin proteins." Antioxid Redox Signal 13(8): 1205-1216.
241
Compston, A. and A. Coles (2008). "Multiple sclerosis." Lancet 372(9648): 1502-1517.
Cook, G. C. and S. Sherlock (1965). "Results of a Controlled Clinical Trial of Glutathione in Cases of Hepatic Cirrhosis." Gut 6(5): 472-&.
Cooper, A. J., J. T. Pinto and P. S. Callery (2011). "Reversible and irreversible protein glutathionylation: biological and clinical aspects." Expert Opin Drug Metab Toxicol 7(7): 891-910.
Coppock, D. L. and C. Thorpe (2006). "Multidomain flavin-dependent sulfhydryl oxidases." Antioxid Redox Signal 8(3-4): 300-311.
Cordis, G. A., G. Maulik, D. Bagchi, W. Riedel and D. K. Das (1998). "Detection of oxidative DNA damage to ischemic reperfused rat hearts by 8-hydroxydeoxyguanosine formation." J Mol Cell Cardiol 30(10): 1939-1944.
Correa, M. J. U., H. A. Mariz, L. E. C. Andrade and C. Kayser (2014). "Oral N-acetylcysteine in the treatment of Raynaud's phenomenon secondary to systemic sclerosis: a randomized, double-blind, placebo-controlled clinical trial." Revista Brasileira De Reumatologia 54(6): 452-458.
Cox, A. G., C. C. Winterbourn and M. B. Hampton (2010). "Measuring the redox state of cellular peroxiredoxins by immunoblotting." Methods Enzymol 474: 51-66.
Cu, A., Q. Ye, R. Sarria, S. Nakamura, J. Guzman and U. Costabel (2009). "N-acetylcysteine inhibits TNF-alpha, sTNFR, and TGF-beta1 release by alveolar macrophages in idiopathic pulmonary fibrosis in vitro." Sarcoidosis Vasc Diffuse Lung Dis 26(2): 147-154.
Cuadrado, A., Z. Martin-Moldes, J. Ye and I. Lastres-Becker (2014). "Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation." J Biol Chem 289(22): 15244-15258.
Curnutte, J. T., D. M. Whitten and B. M. Babior (1974). "Defective superoxide production by granulocytes from patients with chronic granulomatous disease." N Engl J Med 290(11): 593-597.
D'Autreaux, B. and M. B. Toledano (2007). "ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis." Nat Rev Mol Cell Biol 8(10): 813-824.
Dalle-Donne, I., R. Rossi, G. Colombo, D. Giustarini and A. Milzani (2009). "Protein S-glutathionylation: a regulatory device from bacteria to humans." Trends Biochem Sci 34(2): 85-96.
Debarbieux, L. and J. Beckwith (2000). "On the functional interchangeability, oxidant versus reductant, of members of the thioredoxin superfamily." J Bacteriol 182(3): 723-727.
Demedts, M., J. Behr, R. Buhl, U. Costabel, R. Dekhuijzen, H. M. Jansen, W. MacNee, M. Thomeer, B. Wallaert, F. Laurent, A. G. Nicholson, E. K. Verbeken, J. Verschakelen, C. D. Flower, F. Capron, S. Petruzzelli, P. De Vuyst, J. M. van den Bosch, E. Rodriguez-Becerra, G. Corvasce, I. Lankhorst, M. Sardina, M. Montanari and I. S. Group (2005). "High-dose acetylcysteine in idiopathic pulmonary fibrosis." N Engl J Med 353(21): 2229-2242.
242
Deponte, M. (2013). "Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes." Biochim Biophys Acta 1830(5): 3217-3266.
Deschacht, M., T. Horemans, W. Martinet, H. Bult, L. Maes and P. Cos (2010). "Comparative EPR study of different macrophage types stimulated for superoxide and nitric oxide production." Free Radic Res 44(7): 763-772.
Dhalla, N. S., R. M. Temsah and T. Netticadan (2000). "Role of oxidative stress in cardiovascular diseases." J Hypertens 18(6): 655-673.
Dias, F. F., K. B. Amaral, L. A. Carmo, R. Shamri, A. M. Dvorak, P. F. Weller and R. C. Melo (2014). "Human Eosinophil Leukocytes Express Protein Disulfide Isomerase in Secretory Granules and Vesicles: Ultrastructural Studies." J Histochem Cytochem 62(6): 450-459.
Diaz-Borjon, A., C. M. Weyand and J. J. Goronzy (2006). "Treatment of chronic inflammatory diseases with biologic agents: opportunities and risks for the elderly." Exp Gerontol 41(12): 1250-1255.
Dinarello, C. A. (2007). "Historical insights into cytokines." Eur J Immunol 37 Suppl 1: S34-45.
Dismukes, G. C., V. V. Klimov, S. V. Baranov, Y. N. Kozlov, J. DasGupta and A. Tyryshkin (2001). "The origin of atmospheric oxygen on Earth: the innovation of oxygenic photosynthesis." Proc Natl Acad Sci U S A 98(5): 2170-2175.
Dodd, S., O. Dean, D. L. Copolov, G. S. Malhi and M. Berk (2008). "N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility." Expert Opin Biol Ther 8(12): 1955-1962.
Donath, M. Y. and S. E. Shoelson (2011). "Type 2 diabetes as an inflammatory disease." Nat Rev Immunol 11(2): 98-107.
Drazic, A. and J. Winter (2014). "The physiological role of reversible methionine oxidation." Biochim Biophys Acta 1844(8): 1367-1382.
Dumitriu, I. E., P. Baruah, M. E. Bianchi, A. A. Manfredi and P. Rovere-Querini (2005). "Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells." Eur J Immunol 35(7): 2184-2190.
Dunkelberger, J. R. and W. C. Song (2010). "Complement and its role in innate and adaptive immune responses." Cell Res 20(1): 34-50.
Eaton, P. (2006). "Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures." Free Radic Biol Med 40(11): 1889-1899.
Eisen, M. B., P. T. Spellman, P. O. Brown and D. Botstein (1998). "Cluster analysis and display of genome-wide expression patterns." Proc Natl Acad Sci U S A 95(25): 14863-14868.
Eisner, V., A. Criollo, C. Quiroga, C. Olea-Azar, J. F. Santibanez, R. Troncoso, M. Chiong, G. Diaz-Araya, R. Foncea and S. Lavandero (2006). "Hyperosmotic stress-dependent
243
NFkappaB activation is regulated by reactive oxygen species and IGF-1 in cultured cardiomyocytes." FEBS Lett 580(18): 4495-4500.
Ellgaard, L. and L. W. Ruddock (2005). "The human protein disulphide isomerase family: substrate interactions and functional properties." EMBO Rep 6(1): 28-32.
Ellman, G. L. (1959). "Tissue sulfhydryl groups." Arch Biochem Biophys 82(1): 70-77.
Engelman, R., P. Weisman-Shomer, T. Ziv, J. Xu, E. S. Arner and M. Benhar (2013). "Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation." J Biol Chem 288(16): 11312-11324.
Erdil, N., T. Eroglu, B. Akca, O. M. Disli, O. Yetkin, M. C. Colak, F. Erdil and B. Battaloglu (2016). "The effects of N-acetylcysteine on pulmonary functions in patients undergoing on-pump coronary artery surgery: a double blind placebo controlled study." European Review for Medical and Pharmacological Sciences 20(1): 180-187.
Esterbauer, H., R. J. Schaur and H. Zollner (1991). "Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes." Free Radic Biol Med 11(1): 81-128.
Faccio, G., O. Nivala, K. Kruus, J. Buchert and M. Saloheimo (2011). "Sulfhydryl oxidases: sources, properties, production and applications." Appl Microbiol Biotechnol 91(4): 957-966.
Falkowski, P. G. and L. V. Godfrey (2008). "Electrons, life and the evolution of Earth's oxygen cycle." Philos Trans R Soc Lond B Biol Sci 363(1504): 2705-2716.
Fernandes, A. P. and A. Holmgren (2004). "Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system." Antioxid Redox Signal 6(1): 63-74.
Flajnik, M. F. and M. Kasahara (2010). "Origin and evolution of the adaptive immune system: genetic events and selective pressures." Nature Reviews Genetics 11(1): 47-59.
Fomenko, D. E., S. M. Marino and V. N. Gladyshev (2008). "Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases." Molecules and Cells 26(3): 228-235.
Forman, H. J., M. Maiorino and F. Ursini (2010). "Signaling functions of reactive oxygen species." Biochemistry 49(5): 835-842.
Forman, H. J. and M. Torres (2001). "Signaling by the respiratory burst in macrophages." IUBMB Life 51(6): 365-371.
Fratelli, M., H. Demol, M. Puype, S. Casagrande, I. Eberini, M. Salmona, V. Bonetto, M. Mengozzi, F. Duffieux, E. Miclet, A. Bachi, J. Vandekerckhove, E. Gianazza and P. Ghezzi (2002). "Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes." Proc Natl Acad Sci U S A 99(6): 3505-3510.
Fratelli, M., L. O. Goodwin, U. A. Orom, S. Lombardi, R. Tonelli, M. Mengozzi and P. Ghezzi (2005). "Gene expression profiling reveals a signaling role of glutathione in redox regulation." Proc Natl Acad Sci U S A 102(39): 13998-14003.
244
Fukai, T. and M. Ushio-Fukai (2011). "Superoxide dismutases: role in redox signaling, vascular function, and diseases." Antioxid Redox Signal 15(6): 1583-1606.
Garaci, E., A. T. Palamara, M. R. Ciriolo, C. D'Agostini, M. S. Abdel-Latif, S. Aquaro, E. Lafavia and G. Rotilio (1997). "Intracellular GSH content and HIV replication in human macrophages." J Leukoc Biol 62(1): 54-59.
Gardella, S., C. Andrei, D. Ferrera, L. V. Lotti, M. R. Torrisi, M. E. Bianchi and A. Rubartelli (2002). "The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway." EMBO Rep 3(10): 995-1001.
Garlanda, C., C. A. Dinarello and A. Mantovani (2013). "The interleukin-1 family: back to the future." Immunity 39(6): 1003-1018.
Gasdaska, J. R., M. Berggren and G. Powis (1995). "Cell growth stimulation by the redox protein thioredoxin occurs by a novel helper mechanism." Cell Growth Differ 6(12): 1643-1650.
Gasdaska, J. R., D. L. Kirkpatrick, W. Montfort, M. Kuperus, S. R. Hill, M. Berggren and G. Powis (1996). "Oxidative inactivation of thioredoxin as a cellular growth factor and protection by a Cys73-->Ser mutation." Biochem Pharmacol 52(11): 1741-1747.
Geissmann, F., M. G. Manz, S. Jung, M. H. Sieweke, M. Merad and K. Ley (2010). "Development of monocytes, macrophages, and dendritic cells." Science 327(5966): 656-661.
Gelderman, K. A., M. Hultqvist, J. Holmberg, P. Olofsson and R. Holmdahl (2006). "T cell surface redox levels determine T cell reactivity and arthritis susceptibility." Proc Natl Acad Sci U S A 103(34): 12831-12836.
Ghezzi, P. (2013). "Protein glutathionylation in health and disease." Biochim Biophys Acta 1830(5): 3165-3172.
Ghezzi, P., V. Bonetto and M. Fratelli (2005). "Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation." Antioxid Redox Signal 7(7-8): 964-972.
Giacco, F. and M. Brownlee (2010). "Oxidative stress and diabetic complications." Circ Res 107(9): 1058-1070.
Gilgun-Sherki, Y., E. Melamed and D. Offen (2004). "The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy." J Neurol 251(3): 261-268.
Gill, R., A. Tsung and T. Billiar (2010). "Linking oxidative stress to inflammation: Toll-like receptors." Free Radic Biol Med 48(9): 1121-1132.
Gloire, G., S. Legrand-Poels and J. Piette (2006). "NF-kappaB activation by reactive oxygen species: fifteen years later." Biochem Pharmacol 72(11): 1493-1505.
Go, Y. M. and D. P. Jones (2011). "Cysteine/cystine redox signaling in cardiovascular disease." Free Radic Biol Med 50(4): 495-509.
245
Godoy, J. R., M. Funke, W. Ackermann, P. Haunhorst, S. Oesteritz, F. Capani, H. P. Elsasser and C. H. Lillig (2011). "Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse." Biochim Biophys Acta 1810(1): 2-92.
Gorrini, C., P. S. Baniasadi, I. S. Harris, J. Silvester, S. Inoue, B. Snow, P. A. Joshi, A. Wakeham, S. D. Molyneux, B. Martin, P. Bouwman, D. W. Cescon, A. J. Elia, Z. Winterton-Perks, J. Cruickshank, D. Brenner, A. Tseng, M. Musgrave, H. K. Berman, R. Khokha, J. Jonkers, T. W. Mak and M. L. Gauthier (2013). "BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival." J Exp Med 210(8): 1529-1544.
Gosset, P., B. Wallaert, A. B. Tonnel and C. Fourneau (1999). "Thiol regulation of the production of TNF-alpha, IL-6 and IL-8 by human alveolar macrophages." Eur Respir J 14(1): 98-105.
Graham, F. L., J. Smiley, W. C. Russell and R. Nairn (1977). "Characteristics of a human cell line transformed by DNA from human adenovirus type 5." J Gen Virol 36(1): 59-74.
Grek, C. L., J. Zhang, Y. Manevich, D. M. Townsend and K. D. Tew (2013). "Causes and consequences of cysteine S-glutathionylation." J Biol Chem 288(37): 26497-26504.
Grendar, J., J. F. Ouellet, A. McKay, F. R. Sutherland, O. F. Bathe, C. G. Ball and E. Dixon (2016). "Effect of N-acetylcysteine on liver recovery after resection: A randomized clinical trial." Journal of Surgical Oncology 114(4): 446-450.
Griese, M., M. Kappler, C. Eismann, M. Ballmann, S. Junge, E. Rietschel, S. van Koningsbruggen-Rietschel, D. Staab, C. Rolinck-Werninghaus, U. Mellies, T. Kohnlein, T. Wagner, S. Konig, H. Teschler, H. E. Heuer, M. Kopp, S. Heyder, J. Hammermann, P. Kuster, M. Honer, U. Mansmann, I. Beck-Speier, D. Hartl, C. Fuchs, G. Glutathione Study and A. Hector (2013). "Inhalation treatment with glutathione in patients with cystic fibrosis. A randomized clinical trial." Am J Respir Crit Care Med 188(1): 83-89.
Griffith, J. W., C. L. Sokol and A. D. Luster (2014). "Chemokines and chemokine receptors: positioning cells for host defense and immunity." Annu Rev Immunol 32: 659-702.
Griffith, O. W. and A. Meister (1979). "POTENT AND SPECIFIC-INHIBITION OF GLUTATHIONE SYNTHESIS BY BUTHIONINE SULFOXIMINE (S-NORMAL-BUTYL HOMOCYSTEINE SULFOXIMINE)." Journal of Biological Chemistry 254(16): 7558-7560.
Grivennikov, S. I., F. R. Greten and M. Karin (2010). "Immunity, inflammation, and cancer." Cell 140(6): 883-899.
Guo, H., J. B. Callaway and J. P. Ting (2015). "Inflammasomes: mechanism of action, role in disease, and therapeutics." Nat Med 21(7): 677-687.
Guyton, K. Z., Y. Liu, M. Gorospe, Q. Xu and N. J. Holbrook (1996). "Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury." J Biol Chem 271(8): 4138-4142.
Haendeler, J. (2006). "Thioredoxin-1 and posttranslational modifications." Antioxid Redox Signal 8(9-10): 1723-1728.
246
Hahm, E., J. Li, K. Kim, S. Huh, S. Rogelj and J. Cho (2013). "Extracellular protein disulfide isomerase regulates ligand-binding activity of alphaMbeta2 integrin and neutrophil recruitment during vascular inflammation." Blood 121(19): 3789-3800, S3781-3715.
Halliwell, B. (2007). "Biochemistry of oxidative stress." Biochem Soc Trans 35(Pt 5): 1147-1150.
Halliwell, B. (2013). "The antioxidant paradox: less paradoxical now?" Br J Clin Pharmacol 75(3): 637-644.
Hambleton, J., S. L. Weinstein, L. Lem and A. L. DeFranco (1996). "Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages." Proc Natl Acad Sci U S A 93(7): 2774-2778.
Hamzeh, N., L. Li, B. Barkes, J. Huang, B. Canono, M. Gillespie, L. Maier and B. Day (2016). "The effect of an oral anti-oxidant, N-Acetyl-cysteine, on inflammatory and oxidative markers in pulmonary sarcoidosis." Respiratory Medicine 112: 106-111.
Han, R. M., J. P. Zhang and L. H. Skibsted (2012). "Reaction dynamics of flavonoids and carotenoids as antioxidants." Molecules 17(2): 2140-2160.
Han, X., A. Aslanian and J. R. Yates, 3rd (2008). "Mass spectrometry for proteomics." Curr Opin Chem Biol 12(5): 483-490.
Hanschmann, E. M., J. R. Godoy, C. Berndt, C. Hudemann and C. H. Lillig (2013). "Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling." Antioxid Redox Signal 19(13): 1539-1605.
Hansson, G. K. (2005). "Inflammation, atherosclerosis, and coronary artery disease." N Engl J Med 352(16): 1685-1695.
Hansson, G. K. and A. Hermansson (2011). "The immune system in atherosclerosis." Nat Immunol 12(3): 204-212.
Harijith, A., D. L. Ebenezer and V. Natarajan (2014). "Reactive oxygen species at the crossroads of inflammasome and inflammation." Front Physiol 5: 352.
Harper, V. M., J. Y. Oh, R. Stapley, M. B. Marques, L. Wilson, S. Barnes, C. W. Sun, T. Townes and R. P. Patel (2015). "Peroxiredoxin-2 recycling is inhibited during erythrocyte storage." Antioxid Redox Signal 22(4): 294-307.
Harrison, R. (2004). "Physiological roles of xanthine oxidoreductase." Drug Metab Rev 36(2): 363-375.
Hassan, H. M. and I. Fridovich (1979). "Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds." Arch Biochem Biophys 196(2): 385-395.
Hayden, M. S. and S. Ghosh (2008). "Shared principles in NF-kappaB signaling." Cell 132(3): 344-362.
247
Hayden, M. S. and S. Ghosh (2011). "NF-kappaB in immunobiology." Cell Res 21(2): 223-244.
Herzenberg, L. A., S. C. De Rosa, J. G. Dubs, M. Roederer, M. T. Anderson, S. W. Ela, S. C. Deresinski and L. A. Herzenberg (1997). "Glutathione deficiency is associated with impaired survival in HIV disease." Proc Natl Acad Sci U S A 94(5): 1967-1972.
Ho, E., K. Karimi Galougahi, C. C. Liu, R. Bhindi and G. A. Figtree (2013). "Biological markers of oxidative stress: Applications to cardiovascular research and practice." Redox Biol 1: 483-491.
Hoffmann, H. H., W. M. Schneider and C. M. Rice (2015). "Interferons and viruses: an evolutionary arms race of molecular interactions." Trends Immunol 36(3): 124-138.
Hogg, J. C., F. Chu, S. Utokaparch, R. Woods, W. M. Elliott, L. Buzatu, R. M. Cherniack, R. M. Rogers, F. C. Sciurba, H. O. Coxson and P. D. Pare (2004). "The nature of small-airway obstruction in chronic obstructive pulmonary disease." N Engl J Med 350(26): 2645-2653.
Holland, S. M. (2010). "Chronic granulomatous disease." Clin Rev Allergy Immunol 38(1): 3-10.
Holmgren, A. (1985). "Thioredoxin." Annu Rev Biochem 54: 237-271.
Holmgren, A., B. O. Soderberg, H. Eklund and C. I. Branden (1975). "Three-dimensional structure of Escherichia coli thioredoxin-S2 to 2.8 A resolution." Proc Natl Acad Sci U S A 72(6): 2305-2309.
Holmstrom, K. M. and T. Finkel (2014). "Cellular mechanisms and physiological consequences of redox-dependent signalling." Nat Rev Mol Cell Biol 15(6): 411-421.
Hornung, V., F. Bauernfeind, A. Halle, E. O. Samstad, H. Kono, K. L. Rock, K. A. Fitzgerald and E. Latz (2008). "Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization." Nat Immunol 9(8): 847-856.
Huang da, W., B. T. Sherman and R. A. Lempicki (2009). "Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources." Nat Protoc 4(1): 44-57.
Huang, H., W. Mai, D. Liu, Y. Hao, J. Tao and Y. Dong (2008). "The oxidation ratio of LDL: a predictor for coronary artery disease." Dis Markers 24(6): 341-349.
Ikonomidis, I., C. A. Michalakeas, J. Parissis, I. Paraskevaidis, K. Ntai, I. Papadakis, M. Anastasiou-Nana and J. Lekakis (2012). "Inflammatory markers in coronary artery disease." Biofactors 38(5): 320-328.
Iliodromitis, E. K., S. Kyrzopoulos, I. A. Paraskevaidis, K. G. Kolocassides, S. Adamopoulos, G. Karavolias and D. T. Kremastinos (2006). "Increased C reactive protein and cardiac enzyme levels after coronary stent implantation. Is there protection by remote ischaemic preconditioning?" Heart 92(12): 1821-1826.
Iversen, M. B., R. H. Gottfredsen, U. G. Larsen, J. J. Enghild, J. Praetorius, N. Borregaard and S. V. Petersen (2016). "Extracellular superoxide dismutase is present in secretory
248
vesicles of human neutrophils and released upon stimulation." Free Radic Biol Med 97: 478-488.
Iwasaki, A. and R. Medzhitov (2015). "Control of adaptive immunity by the innate immune system." Nat Immunol 16(4): 343-353.
Iyer, G. Y., M. F. Islam and J. H. Quastel (1961). "Biochemical Aspects of Phagocytosis." Nature 192(480): 535-&.
Jacob, C., G. I. Giles, N. M. Giles and H. Sies (2003). "Sulfur and selenium: the role of oxidation state in protein structure and function." Angew Chem Int Ed Engl 42(39): 4742-4758.
Jaramillo, M. and M. Olivier (2002). "Hydrogen peroxide induces murine macrophage chemokine gene transcription via extracellular signal-regulated kinase- and cyclic adenosine 5'-monophosphate (cAMP)-dependent pathways: involvement of NF-kappa B, activator protein 1, and cAMP response element binding protein." J Immunol 169(12): 7026-7038.
Jiang, F., Y. Zhang and G. J. Dusting (2011). "NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair." Pharmacol Rev 63(1): 218-242.
Jiang, Q. (2014). "Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy." Free Radic Biol Med 72: 76-90.
Jiang, X. M., M. Fitzgerald, C. M. Grant and P. J. Hogg (1999). "Redox control of exofacial protein thiols/disulfides by protein disulfide isomerase." J Biol Chem 274(4): 2416-2423.
Johnson, K., C. E. McEvoy, S. Naqvi, C. Wendt, R. A. Reilkoff, K. M. Kunisaki, E. E. Wetherbee, D. Nelson, R. Tirouvanziam and D. E. Niewoehner (2016). "High-dose oral N-acetylcysteine fails to improve respiratory health status in patients with chronic obstructive pulmonary disease and chronic bronchitis: a randomized, placebo-controlled trial." Int J Chron Obstruct Pulmon Dis 11: 799-807.
Jounai, N., K. Kobiyama, F. Takeshita and K. J. Ishii (2012). "Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination." Front Cell Infect Microbiol 2: 168.
Kalinina, E. V., N. N. Chernov and M. D. Novichkova (2014). "Role of glutathione, glutathione transferase, and glutaredoxin in regulation of redox-dependent processes." Biochemistry (Mosc) 79(13): 1562-1583.
Kalliolias, G. D. and L. B. Ivashkiv (2016). "TNF biology, pathogenic mechanisms and emerging therapeutic strategies." Nat Rev Rheumatol 12(1): 49-62.
Kalogeris, T., C. P. Baines, M. Krenz and R. J. Korthuis (2012). "Cell biology of ischemia/reperfusion injury." Int Rev Cell Mol Biol 298: 229-317.
Kang, D. H., D. J. Lee, J. Kim, J. Y. Lee, H. W. Kim, K. Kwon, W. R. Taylor, H. Jo and S. W. Kang (2013). "Vascular injury involves the overoxidation of peroxiredoxin type II and is recovered by the peroxiredoxin activity mimetic that induces reendothelialization." Circulation 128(8): 834-844.
249
Karakas, M. and W. Koenig (2012). "Myeloperoxidase production by macrophage and risk of atherosclerosis." Curr Atheroscler Rep 14(3): 277-283.
Kawasaki, T. and T. Kawai (2014). "Toll-like receptor signaling pathways." Front Immunol 5: 461.
Kehrer, J. P. (2000). "The Haber-Weiss reaction and mechanisms of toxicity." Toxicology 149(1): 43-50.
Kellett-Clarke, H., M. Stegmann, A. N. Barclay and C. Metcalfe (2015). "CD44 Binding to Hyaluronic Acid Is Redox Regulated by a Labile Disulfide Bond in the Hyaluronic Acid Binding Site." PLoS One 10(9): e0138137.
Khan, A. A., A. H. Rahmani, Y. H. Aldebasi and S. M. Aly (2014). "Biochemical and pathological studies on peroxidases -an updated review." Glob J Health Sci 6(5): 87-98.
Kimbrell, D. A. and B. Beutler (2001). "The evolution and genetics of innate immunity." Nat Rev Genet 2(4): 256-267.
Kirkman, H. N. and G. F. Gaetani (2007). "Mammalian catalase: a venerable enzyme with new mysteries." Trends Biochem Sci 32(1): 44-50.
Kirkpatrick, D. L., M. Kuperus, M. Dowdeswell, N. Potier, L. J. Donald, M. Kunkel, M. Berggren, M. Angulo and G. Powis (1998). "Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides." Biochem Pharmacol 55(7): 987-994.
Klebanoff, S. J. (1967). "Iodination of bacteria: a bactericidal mechanism." J Exp Med 126(6): 1063-1078.
Klebanoff, S. J. (1975). "Antimicrobial mechanisms in neutrophilic polymorphonuclear leukocytes." Semin Hematol 12(2): 117-142.
Kleber, M. E., G. Goliasch, T. B. Grammer, S. Pilz, A. Tomaschitz, G. Silbernagel, G. Maurer, W. Marz and A. Niessner (2014). "Evolving biomarkers improve prediction of long-term mortality in patients with stable coronary artery disease: the BIO-VILCAD score." J Intern Med 276(2): 184-194.
Klein, E. A., I. M. Thompson, Jr., C. M. Tangen, J. J. Crowley, M. S. Lucia, P. J. Goodman, L. M. Minasian, L. G. Ford, H. L. Parnes, J. M. Gaziano, D. D. Karp, M. M. Lieber, P. J. Walther, L. Klotz, J. K. Parsons, J. L. Chin, A. K. Darke, S. M. Lippman, G. E. Goodman, F. L. Meyskens, Jr. and L. H. Baker (2011). "Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT)." JAMA 306(14): 1549-1556.
Klotz, L. O., C. Pellieux, K. Briviba, C. Pierlot, J. M. Aubry and H. Sies (1999). "Mitogen-activated protein kinase (p38-, JNK-, ERK-) activation pattern induced by extracellular and intracellular singlet oxygen and UVA." Eur J Biochem 260(3): 917-922.
Koenderman, L., W. Buurman and M. R. Daha (2014). "The innate immune response." Immunol Lett 162(2 Pt B): 95-102.
250
Kosek, D., S. Kylarova, K. Psenakova, L. Rezabkova, P. Herman, J. Vecer, V. Obsilova and T. Obsil (2014). "Biophysical and structural characterization of the thioredoxin-binding domain of protein kinase ASK1 and its interaction with reduced thioredoxin." J Biol Chem 289(35): 24463-24474.
Kosower, N. S., E. M. Kosower, B. Wertheim and W. S. Correa (1969). "Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide." Biochemical and Biophysical Research Communications 37(4): 593-596.
Kowaltowski, A. J., N. C. de Souza-Pinto, R. F. Castilho and A. E. Vercesi (2009). "Mitochondria and reactive oxygen species." Free Radic Biol Med 47(4): 333-343.
Kvietys, P. R. and D. N. Granger (2012). "Role of reactive oxygen and nitrogen species in the vascular responses to inflammation." Free Radic Biol Med 52(3): 556-592.
Kwon, A. T., D. J. Arenillas, R. Worsley Hunt and W. W. Wasserman (2012). "oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets." G3 (Bethesda) 2(9): 987-1002.
Lambeth, J. D. (2004). "NOX enzymes and the biology of reactive oxygen." Nat Rev Immunol 4(3): 181-189.
Landmann, R., F. Scherer, R. Schumann, S. Link, S. Sansano and W. Zimmerli (1995). "LPS directly induces oxygen radical production in human monocytes via LPS binding protein and CD14." J Leukoc Biol 57(3): 440-449.
Laragione, T., V. Bonetto, F. Casoni, T. Massignan, G. Bianchi, E. Gianazza and P. Ghezzi (2003). "Redox regulation of surface protein thiols: identification of integrin alpha-4 as a molecular target by using redox proteomics." Proc Natl Acad Sci U S A 100(25): 14737-14741.
Laurent, T. C., E. C. Moore and P. Reichard (1964). "Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B." J Biol Chem 239: 3436-3444.
Laurindo, F. R., L. A. Pescatore and C. Fernandes Dde (2012). "Protein disulfide isomerase in redox cell signaling and homeostasis." Free Radic Biol Med 52(9): 1954-1969.
Lawrence, T. and D. W. Gilroy (2007). "Chronic inflammation: a failure of resolution?" Int J Exp Pathol 88(2): 85-94.
Lee, D. H., J. H. Park, S. B. Han, D. Y. Yoon, Y. Y. Jung and J. T. Hong (2017). "Peroxiredoxin 6 overexpression attenuates lipopolysaccharide-induced acute kidney injury." Oncotarget.
Lee, E. Y., Z. H. Lee and Y. W. Song (2009). "CXCL10 and autoimmune diseases." Autoimmun Rev 8(5): 379-383.
Lee, S. M., M. Hutchinson and D. A. Saint (2016). "The role of Toll-like receptor 4 (TLR4) in cardiac ischaemic-reperfusion injury, cardioprotection and preconditioning." Clin Exp Pharmacol Physiol 43(9): 864-871.
251
Lee, W., K. S. Choi, J. Riddell, C. Ip, D. Ghosh, J. H. Park and Y. M. Park (2007). "Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2." J Biol Chem 282(30): 22011-22022.
Leonard, S. E. and K. S. Carroll (2011). "Chemical 'omics' approaches for understanding protein cysteine oxidation in biology." Curr Opin Chem Biol 15(1): 88-102.
Lerner, A., P. Jeremias and T. Matthias (2015). "The World Incidence and Prevalence of Autoimmune Diseases is Increasing." International Journal of Celiac Disease 3(4): 151-155.
Libby, P. (2002). "Inflammation in atherosclerosis." Nature 420(6917): 868-874.
Libby, P. (2007). "Inflammatory mechanisms: the molecular basis of inflammation and disease." Nutr Rev 65(12 Pt 2): S140-146.
Libby, P. and P. Theroux (2005). "Pathophysiology of coronary artery disease." Circulation 111(25): 3481-3488.
Lillig, C. H. and C. Berndt (2013). "Glutaredoxins in thiol/disulfide exchange." Antioxid Redox Signal 18(13): 1654-1665.
Lincoln, D. T., E. M. Ali Emadi, K. F. Tonissen and F. M. Clarke (2003). "The thioredoxin-thioredoxin reductase system: over-expression in human cancer." Anticancer Res 23(3B): 2425-2433.
Liu, Y., H. Yin, M. Zhao and Q. Lu (2014). "TLR2 and TLR4 in autoimmune diseases: a comprehensive review." Clin Rev Allergy Immunol 47(2): 136-147.
Liu, Z. G. (2005). "Molecular mechanism of TNF signaling and beyond." Cell Res 15(1): 24-27.
Livak, K. J. and T. D. Schmittgen (2001). "Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method." Methods 25(4): 402-408.
Lo Conte, M. and K. S. Carroll (2013). "The redox biochemistry of protein sulfenylation and sulfinylation." J Biol Chem 288(37): 26480-26488.
Lo Conte, M., J. S. Lin, M. A. Wllson and K. S. Carroll (2015). "A Chemical Approach for the Detection of Protein Sulfinylation." Acs Chemical Biology 10(8): 1825-1830.
Low, F. M., M. B. Hampton, A. V. Peskin and C. C. Winterbourn (2007). "Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte." Blood 109(6): 2611-2617.
Low, F. M., M. B. Hampton and C. C. Winterbourn (2008). "Peroxiredoxin 2 and peroxide metabolism in the erythrocyte." Antioxid Redox Signal 10(9): 1621-1630.
Lu, J. and A. Holmgren (2014). "The thioredoxin antioxidant system." Free Radic Biol Med 66: 75-87.
252
Lu, S. C. (2013). "Glutathione synthesis." Biochim Biophys Acta 1830(5): 3143-3153.
Lunardi, S., N. B. Jamieson, S. Y. Lim, K. L. Griffiths, M. Carvalho-Gaspar, O. Al-Assar, S. Yameen, R. C. Carter, C. J. McKay, G. Spoletini, S. D'Ugo, M. A. Silva, O. J. Sansom, K. P. Janssen, R. J. Muschel and T. B. Brunner (2014). "IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival." Oncotarget 5(22): 11064-11080.
Luu, K., C. J. Greenhill, A. Majoros, T. Decker, B. J. Jenkins and A. Mansell (2014). "STAT1 plays a role in TLR signal transduction and inflammatory responses." Immunol Cell Biol 92(9): 761-769.
Macchia, I., A. T. Palamara, C. Bue, P. Savini, M. Ciriolo, R. Gaziano and P. di Francesco (1999). "Increased replication of Sendai virus in morphine-treated epithelial cells: evidence for the involvement of the intracellular levels of glutathione." Int J Immunopharmacol 21(3): 185-193.
Madrigal-Matute, J., C. E. Fernandez-Garcia, L. M. Blanco-Colio, E. Burillo, A. Fortuno, R. Martinez-Pinna, P. Llamas-Granda, O. Beloqui, J. Egido, G. Zalba and J. L. Martin-Ventura (2015). "Thioredoxin-1/peroxiredoxin-1 as sensors of oxidative stress mediated by NADPH oxidase activity in atherosclerosis." Free Radic Biol Med 86: 352-361.
Marino, S. M. and V. N. Gladyshev (2009). "A structure-based approach for detection of thiol oxidoreductases and their catalytic redox-active cysteine residues." PLoS Comput Biol 5(5): e1000383.
Marino, S. M. and V. N. Gladyshev (2010). "Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces." J Mol Biol 404(5): 902-916.
Martina, V., A. Masha, V. R. Gigliardi, L. Brocato, E. Manzato, A. Berchio, P. Massarenti, F. Settanni, L. Della Casa, S. Bergamini and A. Iannone (2008). "Long-term N-acetylcysteine and L-arginine administration reduces endothelial activation and systolic blood pressure in hypertensive patients with type 2 diabetes." Diabetes Care 31(5): 940-944.
Martinez-Pinna, R., J. S. Lindholt, L. M. Blanco-Colio, T. Dejouvencel, J. Madrigal-Matute, P. Ramos-Mozo, M. Vega de Ceniga, J. B. Michel, J. Egido, O. Meilhac and J. L. Martin-Ventura (2010). "Increased levels of thioredoxin in patients with abdominal aortic aneurysms (AAAs). A potential link of oxidative stress with AAA evolution." Atherosclerosis 212(1): 333-338.
Martinez-Pinna, R., P. Ramos-Mozo, J. Madrigal-Matute, L. M. Blanco-Colio, J. A. Lopez, E. Calvo, E. Camafeita, J. S. Lindholt, O. Meilhac, S. Delbosc, J. B. Michel, M. Vega de Ceniga, J. Egido and J. L. Martin-Ventura (2011). "Identification of peroxiredoxin-1 as a novel biomarker of abdominal aortic aneurysm." Arterioscler Thromb Vasc Biol 31(4): 935-943.
Martinon, F., V. Petrilli, A. Mayor, A. Tardivel and J. Tschopp (2006). "Gout-associated uric acid crystals activate the NALP3 inflammasome." Nature 440(7081): 237-241.
McInnes, I. B. and G. Schett (2007). "Cytokines in the pathogenesis of rheumatoid arthritis." Nat Rev Immunol 7(6): 429-442.
Medzhitov, R. (2008). "Origin and physiological roles of inflammation." Nature 454(7203): 428-435.
253
Medzhitov, R. and T. Horng (2009). "Transcriptional control of the inflammatory response." Nat Rev Immunol 9(10): 692-703.
Meister, A. and M. E. Anderson (1983). "Glutathione." Annu Rev Biochem 52: 711-760.
Meng, T. C., T. Fukada and N. K. Tonks (2002). "Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo." Mol Cell 9(2): 387-399.
Messmer, D., H. Yang, G. Telusma, F. Knoll, J. Li, B. Messmer, K. J. Tracey and N. Chiorazzi (2004). "High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization." J Immunol 173(1): 307-313.
Metcalfe, C., P. Cresswell and A. N. Barclay (2012). "Interleukin-2 signalling is modulated by a labile disulfide bond in the CD132 chain of its receptor." Open Biol 2(1): 110036.
Metcalfe, C., P. Cresswell, L. Ciaccia, B. Thomas and A. N. Barclay (2011). "Labile disulfide bonds are common at the leucocyte cell surface." Open Biol 1(3): 110010.
Meyer, A., R. Buhl and H. Magnussen (1994). "The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis." Eur Respir J 7(3): 431-436.
Michigami, T. (2013). "Extracellular phosphate as a signaling molecule." Contrib Nephrol 180: 14-24.
Mihm, S., D. Galter and W. Droge (1995). "Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply." FASEB J 9(2): 246-252.
Mikami, T., N. Satoh, I. Hatayama and A. Nakane (2004). "Buthionine sulfoximine inhibits cytopathic effect and apoptosis induced by infection with human echovirus 9." Arch Virol 149(6): 1117-1128.
Mills, K. H. (2011). "TLR-dependent T cell activation in autoimmunity." Nat Rev Immunol 11(12): 807-822.
Mogensen, T. H. (2009). "Pathogen recognition and inflammatory signaling in innate immune defenses." Clin Microbiol Rev 22(2): 240-273, Table of Contents.
Morgan, M. J. and Z. G. Liu (2011). "Crosstalk of reactive oxygen species and NF-kappaB signaling." Cell Res 21(1): 103-115.
Mosmann, T. (1983). "Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays." J Immunol Methods 65(1-2): 55-63.
Mullen, L., E. M. Hanschmann, C. H. Lillig, L. A. Herzenberg and P. Ghezzi (2015). "Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion." Mol Med 21: 98-108.
Mullen, L., M. Seavill, R. Hammouz, B. Bottazzi, P. Chan, D. Vaudry and P. Ghezzi (2015). "Development of 'Redox Arrays' for identifying novel glutathionylated proteins in the secretome." Sci Rep 5: 14630.
254
Murphy, M. P. (2009). "How mitochondria produce reactive oxygen species." Biochem J 417(1): 1-13.
Murray, P. J. and T. A. Wynn (2011). "Protective and pathogenic functions of macrophage subsets." Nat Rev Immunol 11(11): 723-737.
Nagy, P. (2013). "Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways." Antioxid Redox Signal 18(13): 1623-1641.
Nakamura, H., S. De Rosa, M. Roederer, M. T. Anderson, J. G. Dubs, J. Yodoi, A. Holmgren, L. A. Herzenberg and L. A. Herzenberg (1996). "Elevation of plasma thioredoxin levels in HIV-infected individuals." Int Immunol 8(4): 603-611.
Napetschnig, J. and H. Wu (2013). "Molecular basis of NF-kappaB signaling." Annu Rev Biophys 42: 443-468.
Naqui, A., B. Chance and E. Cadenas (1986). "Reactive oxygen intermediates in biochemistry." Annu Rev Biochem 55: 137-166.
Narayanan, A., M. Amaya, K. Voss, M. Chung, A. Benedict, G. Sampey, K. Kehn-Hall, A. Luchini, L. Liotta, C. Bailey, A. Kumar, S. Bavari, R. M. Hakami and F. Kashanchi (2014). "Reactive oxygen species activate NFkappaB (p65) and p53 and induce apoptosis in RVFV infected liver cells." Virology 449: 270-286.
Nasef, N. A., S. Mehta and L. R. Ferguson (2017). "Susceptibility to chronic inflammation: an update." Arch Toxicol 91(3): 1131-1141.
Nathan, C. and A. Ding (2010). "Nonresolving inflammation." Cell 140(6): 871-882.
Neurath, M. F. (2014). "Cytokines in inflammatory bowel disease." Nat Rev Immunol 14(5): 329-342.
Newton, K. and V. M. Dixit (2012). "Signaling in innate immunity and inflammation." Cold Spring Harb Perspect Biol 4(3).
Nickel, W. (2003). "The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes." Eur J Biochem 270(10): 2109-2119.
Niedzielska, E., I. Smaga, M. Gawlik, A. Moniczewski, P. Stankowicz, J. Pera and M. Filip (2016). "Oxidative Stress in Neurodegenerative Diseases." Mol Neurobiol 53(6): 4094-4125.
Ning, J. L., L. W. Mo and X. N. Lai (2010). "Low- and high-dose hydrogen peroxide regulation of transcription factor NF-E2-related factor 2." Chin Med J (Engl) 123(8): 1063-1069.
Nussbaum, C., A. Klinke, M. Adam, S. Baldus and M. Sperandio (2013). "Myeloperoxidase: a leukocyte-derived protagonist of inflammation and cardiovascular disease." Antioxid Redox Signal 18(6): 692-713.
O'Brien, P. J. (2000). "Peroxidases." Chem Biol Interact 129(1-2): 113-139.
255
O'Neill, L. A. J. (2008). "'Fine tuning' TLR signaling." Nature Immunology 9(5): 459-461.
Oka, S., H. Kamata, K. Kamata, H. Yagisawa and H. Hirata (2000). "N-acetylcysteine suppresses TNF-induced NF-kappaB activation through inhibition of IkappaB kinases." FEBS Lett 472(2-3): 196-202.
Oliveira-Marques, V., H. S. Marinho, L. Cyrne and F. Antunes (2009). "Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator." Antioxid Redox Signal 11(9): 2223-2243.
Oostenbrug, L. E., J. P. Drenth, D. J. de Jong, I. M. Nolte, E. Oosterom, H. M. van Dullemen, K. van der Linde, G. J. te Meerman, G. van der Steege, J. H. Kleibeuker and P. L. Jansen (2005). "Association between Toll-like receptor 4 and inflammatory bowel disease." Inflamm Bowel Dis 11(6): 567-575.
Oosting, M., S. C. Cheng, J. M. Bolscher, R. Vestering-Stenger, T. S. Plantinga, I. C. Verschueren, P. Arts, A. Garritsen, H. van Eenennaam, P. Sturm, B. J. Kullberg, A. Hoischen, G. J. Adema, J. W. van der Meer, M. G. Netea and L. A. Joosten (2014). "Human TLR10 is an anti-inflammatory pattern-recognition receptor." Proc Natl Acad Sci U S A 111(42): E4478-4484.
Otsuka, F., A. V. Finn, S. K. Yazdani, M. Nakano, F. D. Kolodgie and R. Virmani (2012). "The importance of the endothelium in atherothrombosis and coronary stenting." Nat Rev Cardiol 9(8): 439-453.
Pacher, P., J. S. Beckman and L. Liaudet (2007). "Nitric oxide and peroxynitrite in health and disease." Physiol Rev 87(1): 315-424.
Paget, M. S. and M. J. Buttner (2003). "Thiol-based regulatory switches." Annu Rev Genet 37: 91-121.
Palamara, A. T., C. F. Perno, M. R. Ciriolo, L. Dini, E. Balestra, C. D'Agostini, P. Di Francesco, C. Favalli, G. Rotilio and E. Garaci (1995). "Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication." Antiviral Res 27(3): 237-253.
Panday, A., M. K. Sahoo, D. Osorio and S. Batra (2015). "NADPH oxidases: an overview from structure to innate immunity-associated pathologies." Cell Mol Immunol 12(1): 5-23.
Pandey, K. B. and S. I. Rizvi (2011). "Biomarkers of oxidative stress in red blood cells." Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 155(2): 131-136.
Papi, A., M. Contoli, P. Gasparini, L. Bristot, M. R. Edwards, M. Chicca, M. Leis, A. Ciaccia, G. Caramori, S. L. Johnston and S. Pinamonti (2008). "Role of xanthine oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells." J Biol Chem 283(42): 28595-28606.
Park, H. S., J. N. Chun, H. Y. Jung, C. Choi and Y. S. Bae (2006). "Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells." Cardiovasc Res 72(3): 447-455.
Pastori, D., R. Carnevale and P. Pignatelli (2014). "Is there a clinical role for oxidative stress biomarkers in atherosclerotic diseases?" Intern Emerg Med 9(2): 123-131.
256
Pedone, E., B. Ren, R. Ladenstein, M. Rossi and S. Bartolucci (2004). "Functional properties of the protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus: a member of a novel protein family related to protein disulfide-isomerase." Eur J Biochem 271(16): 3437-3448.
Pelliccia, F., G. Del Prete, A. Del Prete, C. Greco and C. Gaudio (2012). "Effects of percutaneous coronary intervention and stenting with different drug-eluting coatings and platforms on endothelial damage and inflammation." Int J Cardiol 156(2): 242-243.
Pellom, S. T., R. D. Michalek, K. E. Crump, P. K. Langston, D. G. Juneau and J. M. Grayson (2013). "Increased cell surface free thiols identify effector CD8+ T cells undergoing T cell receptor stimulation." PLoS One 8(11): e81134.
Perez-Perez, M. E., A. Mata-Cabana, A. M. Sanchez-Riego, M. Lindahl and F. J. Florencio (2009). "A comprehensive analysis of the peroxiredoxin reduction system in the Cyanobacterium Synechocystis sp. strain PCC 6803 reveals that all five peroxiredoxins are thioredoxin dependent." J Bacteriol 191(24): 7477-7489.
Peristeris, P., B. D. Clark, S. Gatti, R. Faggioni, A. Mantovani, M. Mengozzi, S. F. Orencole, M. Sironi and P. Ghezzi (1992). "N-acetylcysteine and glutathione as inhibitors of tumor necrosis factor production." Cell Immunol 140(2): 390-399.
Perkins, A., K. J. Nelson, D. Parsonage, L. B. Poole and P. A. Karplus (2015). "Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling." Trends Biochem Sci 40(8): 435-445.
Peskin, A. V., P. E. Pace, J. B. Behring, L. N. Paton, M. Soethoudt, M. M. Bachschmid and C. C. Winterbourn (2016). "Glutathionylation of the Active Site Cysteines of Peroxiredoxin 2 and Recycling by Glutaredoxin." J Biol Chem 291(6): 3053-3062.
PETER W. RIDDLES, R. L. B., AND BURT ZERNER (1978). "Ellman’s Reagent: 5,5’-Dithiobis(2-nitrobenzoic
Acid) -a Reexamination " ANALYTICAL BIOCHEMISTRY 94.
Phaniendra, A., D. B. Jestadi and L. Periyasamy (2015). "Free radicals: properties, sources, targets, and their implication in various diseases." Indian J Clin Biochem 30(1): 11-26.
Pick, E., T. Kroizman and A. Abo (1989). "Activation of the superoxide-forming NADPH oxidase of macrophages requires two cytosolic components--one of them is also present in certain nonphagocytic cells." J Immunol 143(12): 4180-4187.
Pieragostino, D., P. Del Boccio, M. Di Ioia, L. Pieroni, V. Greco, G. De Luca, S. D'Aguanno, C. Rossi, D. Franciotta, D. Centonze, P. Sacchetta, C. Di Ilio, A. Lugaresi and A. Urbani (2013). "Oxidative modifications of cerebral transthyretin are associated with multiple sclerosis." Proteomics 13(6): 1002-1009.
Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton and B. Beutler (1998). "Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene." Science 282(5396): 2085-2088.
Poole, L. B. (2015). "The basics of thiols and cysteines in redox biology and chemistry." Free Radic Biol Med 80: 148-157.
257
Preeshagul, I., R. Gharbaran, K. H. Jeong, A. Abdel-Razek, L. Y. Lee, E. Elman and K. S. Suh (2013). "Potential biomarkers for predicting outcomes in CABG cardiothoracic surgeries." J Cardiothorac Surg 8: 176.
Qiao, S., Q. Luo, Y. Zhao, X. C. Zhang and Y. Huang (2014). "Structural basis for lipopolysaccharide insertion in the bacterial outer membrane." Nature 511(7507): 108-111.
Radi, R. (2013). "Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects." Acc Chem Res 46(2): 550-559.
Rahman, I. and W. MacNee (2000). "Oxidative stress and regulation of glutathione in lung inflammation." Eur Respir J 16(3): 534-554.
Rao, Z., S. Wang and J. Wang (2017). "Peroxiredoxin 4 inhibits IL-1beta-induced chondrocyte apoptosis via PI3K/AKT signaling." Biomed Pharmacother 90: 414-420.
Raschke, W. C., S. Baird, P. Ralph and I. Nakoinz (1978). "Functional macrophage cell lines transformed by Abelson leukemia virus." Cell 15(1): 261-267.
Rayman, M. P. (2000). "The importance of selenium to human health." Lancet 356(9225): 233-241.
Remer, K. A., M. Brcic and T. W. Jungi (2003). "Toll-like receptor-4 is involved in eliciting an LPS-induced oxidative burst in neutrophils." Immunol Lett 85(1): 75-80.
Reuter, S., S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal (2010). "Oxidative stress, inflammation, and cancer: how are they linked?" Free Radic Biol Med 49(11): 1603-1616.
Rhee, S. G. (2016). "Overview on Peroxiredoxin." Mol Cells 39(1): 1-5.
Rhee, S. G., S. W. Kang, T. S. Chang, W. Jeong and K. Kim (2001). "Peroxiredoxin, a novel family of peroxidases." IUBMB Life 52(1-2): 35-41.
Riddell, J. R., W. Bshara, M. T. Moser, J. A. Spernyak, B. A. Foster and S. O. Gollnick (2011). "Peroxiredoxin 1 Controls Prostate Cancer Growth through Toll-Like Receptor 4-Dependent Regulation of Tumor Vasculature." Cancer Research 71(5): 1637-1646.
Riddell, J. R., X. Y. Wang, H. Minderman and S. O. Gollnick (2010). "Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4." J Immunol 184(2): 1022-1030.
Riederer, B. M. (2009). "Oxidation Proteomics: The Role of Thiol Modifications." Current Proteomics 6(1): 51-62.
Romo, M. R., D. Perez-Martinez and C. C. Ferrer (2016). "Innate immunity in vertebrates: an overview." Immunology 148(2): 125-139.
Ross, R. and L. Agius (1992). "The process of atherogenesis--cellular and molecular interaction: from experimental animal models to humans." Diabetologia 35 Suppl 2: S34-40.
258
Rossi, F. and M. Zatti (1964). "Biochemical Aspects of Phagocytosis in Polymorphonuclear Leucocytes . Nadh + Nadph Oxidation by Granules of Resting + Phagocytizing Cells." Experientia 20(1): 21-&.
Rotruck, J. T., A. L. Pope, H. E. Ganther, A. B. Swanson, D. G. Hafeman and W. G. Hoekstra (1973). "Selenium: biochemical role as a component of glutathione peroxidase." Science 179(4073): 588-590.
Rubartelli, A., A. Bajetto, G. Allavena, E. Wollman and R. Sitia (1992). "Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway." J Biol Chem 267(34): 24161-24164.
Rudyk, O. and P. Eaton (2014). "Biochemical methods for monitoring protein thiol redox states in biological systems." Redox Biol 2: 803-813.
Rusinova, I., S. Forster, S. Yu, A. Kannan, M. Masse, H. Cumming, R. Chapman and P. J. Hertzog (2013). "Interferome v2.0: an updated database of annotated interferon-regulated genes." Nucleic Acids Res 41(Database issue): D1040-1046.
Ryan, B. J., A. Nissim and P. G. Winyard (2014). "Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases." Redox Biology 2: 715-724.
Sadler, A. J. and B. R. Williams (2008). "Interferon-inducible antiviral effectors." Nat Rev Immunol 8(7): 559-568.
Sagara, M., J. Satoh, X. P. Zhu, K. Takahashi, M. Fukuzawa, G. Muto, Y. Muto and T. Toyota (1994). "Inhibition with N-acetylcysteine of enhanced production of tumor necrosis factor in streptozotocin-induced diabetic rats." Clin Immunol Immunopathol 71(3): 333-337.
Sahaf, B., A. Soderberg, G. Spyrou, A. M. Barral, K. Pekkari, A. Holmgren and A. Rosen (1997). "Thioredoxin expression and localization in human cell lines: detection of full-length and truncated species." Exp Cell Res 236(1): 181-192.
Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis and H. A. Erlich (1988). "Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase." Science 239(4839): 487-491.
Saitoh, M., H. Nishitoh, M. Fujii, K. Takeda, K. Tobiume, Y. Sawada, M. Kawabata, K. Miyazono and H. Ichijo (1998). "Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1." EMBO J 17(9): 2596-2606.
Salzano, S., P. Checconi, E. M. Hanschmann, C. H. Lillig, L. D. Bowler, P. Chan, D. Vaudry, M. Mengozzi, L. Coppo, S. Sacre, K. R. Atkuri, B. Sahaf, L. A. Herzenberg, L. A. Herzenberg, L. Mullen and P. Ghezzi (2014). "Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal." Proc Natl Acad Sci U S A 111(33): 12157-12162.
Samuni, Y., S. Goldstein, O. M. Dean and M. Berk (2013). "The chemistry and biological activities of N-acetylcysteine." Biochim Biophys Acta 1830(8): 4117-4129.
259
Sandalio, L. M., M. Rodriguez-Serrano, M. C. Romero-Puertas and L. A. del Rio (2013). "Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules." Subcell Biochem 69: 231-255.
Sanjabi, S., L. A. Zenewicz, M. Kamanaka and R. A. Flavell (2009). "Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity." Curr Opin Pharmacol 9(4): 447-453.
Satriano, J. and D. Schlondorff (1994). "Activation and attenuation of transcription factor NF-kB in mouse glomerular mesangial cells in response to tumor necrosis factor-alpha, immunoglobulin G, and adenosine 3':5'-cyclic monophosphate. Evidence for involvement of reactive oxygen species." J Clin Invest 94(4): 1629-1636.
Saunders, J. T., V. Nambi, J. A. de Lemos, L. E. Chambless, S. S. Virani, E. Boerwinkle, R. C. Hoogeveen, X. Liu, B. C. Astor, T. H. Mosley, A. R. Folsom, G. Heiss, J. Coresh and C. M. Ballantyne (2011). "Cardiac troponin T measured by a highly sensitive assay predicts coronary heart disease, heart failure, and mortality in the Atherosclerosis Risk in Communities Study." Circulation 123(13): 1367-1376.
Sbarra, A. J. and M. L. Karnovsky (1959). "The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes." J Biol Chem 234(6): 1355-1362.
Schena, M., D. Shalon, R. W. Davis and P. O. Brown (1995). "Quantitative monitoring of gene expression patterns with a complementary DNA microarray." Science 270(5235): 467-470.
Schmidt, K. N., P. Amstad, P. Cerutti and P. A. Baeuerle (1995). "The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B." Chem Biol 2(1): 13-22.
Schmitt, B., M. Vicenzi, C. Garrel and F. M. Denis (2015). "Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study." Redox Biology 6: 198-205.
Schreck, R., P. Rieber and P. A. Baeuerle (1991). "Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1." EMBO J 10(8): 2247-2258.
Sedlak, J. and R. H. Lindsay (1968). "Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent." Anal Biochem 25(1): 192-205.
Shabab, M., S. A. Khan, H. Vogel, D. G. Heckel and W. Boland (2014). "OPDA isomerase GST16 is involved in phytohormone detoxification and insect development." FEBS J 281(12): 2769-2783.
Shah, B. and L. Mayer (2010). "Current status of monoclonal antibody therapy for the treatment of inflammatory bowel disease." Expert Rev Clin Immunol 6(4): 607-620.
Shen, H., D. Kreisel and D. R. Goldstein (2013). "Processes of sterile inflammation." J Immunol 191(6): 2857-2863.
260
Shohrati, M., F. Dermanaki, F. Babaei and S. M. Alavian (2010). "Evaluation of the effects of oral N-acetylcysteine and a placebo in paraclinical and oxidative stress parameters of patients with chronic hepatitis B." Hepat Mon 10(2): 95-100.
Shrivastava, A. K., H. V. Singh, A. Raizada and S. K. Singh (2015). "C-reactive protein, inflammation and coronary heart disease." The Egyptian Heart Journal 67(2): 89-97.
Sies, H. (2014). "Role of metabolic H2O2 generation: redox signaling and oxidative stress." J Biol Chem 289(13): 8735-8741.
Sikorski, K., S. Chmielewski, L. Przybyl, U. Heemann, J. Wesoly, M. Baumann and H. A. Bluyssen (2011). "STAT1-mediated signal integration between IFNgamma and LPS leads to increased EC and SMC activation and monocyte adhesion." Am J Physiol Cell Physiol 300(6): C1337-1344.
Simon, A. R., U. Rai, B. L. Fanburg and B. H. Cochran (1998). "Activation of the JAK-STAT pathway by reactive oxygen species." Am J Physiol 275(6 Pt 1): C1640-1652.
Smith, A. D. and H. Dawson (2006). "Glutathione is required for efficient production of infectious picornavirus virions." Virology 353(2): 258-267.
Sobotta, M. C., W. Liou, S. Stocker, D. Talwar, M. Oehler, T. Ruppert, A. N. Scharf and T. P. Dick (2015). "Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling." Nat Chem Biol 11(1): 64-70.
Soriano, F. X., P. Baxter, L. M. Murray, M. B. Sporn, T. H. Gillingwater and G. E. Hardingham (2009). "Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin." Mol Cells 27(3): 279-282.
Staal, F. J., M. Roederer, L. A. Herzenberg and L. A. Herzenberg (1990). "Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus." Proc Natl Acad Sci U S A 87(24): 9943-9947.
Steinhubl, S. R. (2008). "Why have antioxidants failed in clinical trials?" Am J Cardiol 101(10A): 14D-19D.
Stockert, J. C., A. Blazquez-Castro, M. Canete, R. W. Horobin and A. Villanueva (2012). "MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets." Acta Histochem 114(8): 785-796.
Stone, J. R. and S. Yang (2006). "Hydrogen peroxide: a signaling messenger." Antioxid Redox Signal 8(3-4): 243-270.
Stottmeier, B. and T. P. Dick (2016). "Redox sensitivity of the MyD88 immune signaling adapter." Free Radic Biol Med 101: 93-101.
Strobel, N. A., R. G. Fassett, S. A. Marsh and J. S. Coombes (2011). "Oxidative stress biomarkers as predictors of cardiovascular disease." Int J Cardiol 147(2): 191-201.
Sturn, A., J. Quackenbush and Z. Trajanoski (2002). "Genesis: cluster analysis of microarray data." Bioinformatics 18(1): 207-208.
261
Sun, S., H. Zhang, B. Xue, Y. Wu, J. Wang, Z. Yin and L. Luo (2006). "Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats." Inflamm Res 55(11): 504-510.
Sundaresan, M., Z. X. Yu, V. J. Ferrans, K. Irani and T. Finkel (1995). "Requirement for generation of H2O2 for platelet-derived growth factor signal transduction." Science 270(5234): 296-299.
Suzuki, Y. J., M. Carini and D. A. Butterfield (2010). "Protein carbonylation." Antioxid Redox Signal 12(3): 323-325.
Swietek, K. and J. Juszczyk (1997). "Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis." J Viral Hepat 4(2): 139-141.
Szabo-Taylor, K. E., P. Eggleton, C. A. Turner, M. L. Faro, J. M. Tarr, S. Toth, M. Whiteman, R. C. Haigh, J. A. Littlechild and P. G. Winyard (2012). "Lymphocytes from rheumatoid arthritis patients have elevated levels of intracellular peroxiredoxin 2, and a greater frequency of cells with exofacial peroxiredoxin 2, compared with healthy human lymphocytes." Int J Biochem Cell Biol 44(8): 1223-1231.
Szabo-Taylor, K. E., E. A. Toth, A. M. Balogh, B. W. Sodar, L. Kadar, K. Paloczi, N. Fekete, A. Nemeth, X. Osteikoetxea, K. V. Vukman, M. Holub, E. Pallinger, G. Nagy, P. G. Winyard and E. I. Buzas (2017). "Monocyte activation drives preservation of membrane thiols by promoting release of oxidised membrane moieties via extracellular vesicles." Free Radic Biol Med 108: 56-65.
Szkudlinska, M. A., A. D. von Frankenberg and K. M. Utzschneider (2016). "The antioxidant N-Acetylcysteine does not improve glucose tolerance or beta-cell function in type 2 diabetes." Journal of Diabetes and Its Complications 30(4): 618-622.
Takeuchi, O. and S. Akira (2010). "Pattern Recognition Receptors and Inflammation." Cell 140(6): 805-820.
Talasaz, A. H., H. Khalili, F. Fahimi, Y. Jenab, M. A. Broumand, M. Salarifar and F. Darabi (2014). "Effects of N-Acetylcysteine on the Cardiac Remodeling Biomarkers and Major Adverse Events Following Acute Myocardial Infarction: A Randomized Clinical Trial." American Journal of Cardiovascular Drugs 14(1): 51-61.
Tang, D., R. Kang, C. B. Coyne, H. J. Zeh and M. T. Lotze (2012). "PAMPs and DAMPs: signal 0s that spur autophagy and immunity." Immunol Rev 249(1): 158-175.
Tanudji, M., S. Hevi and S. L. Chuck (2003). "The nonclassic secretion of thioredoxin is not sensitive to redox state." Am J Physiol Cell Physiol 284(5): C1272-1279.
Tarca, A. L., R. Romero and S. Draghici (2006). "Analysis of microarray experiments of gene expression profiling." Am J Obstet Gynecol 195(2): 373-388.
Tarpey, M. M. and I. Fridovich (2001). "Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite." Circ Res 89(3): 224-236.
Thomas, P. and T. G. Smart (2005). "HEK293 cell line: a vehicle for the expression of recombinant proteins." J Pharmacol Toxicol Methods 51(3): 187-200.
262
Thorpe, C., K. L. Hoober, S. Raje, N. M. Glynn, J. Burnside, G. K. Turi and D. L. Coppock (2002). "Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes." Arch Biochem Biophys 405(1): 1-12.
Tian, Y., W. Jiang, N. Gao, J. Zhang, W. Chen, D. Fan, D. Zhou and J. An (2010). "Inhibitory effects of glutathione on dengue virus production." Biochem Biophys Res Commun 397(3): 420-424.
Tien, A. C., A. Rajan, K. L. Schulze, H. D. Ryoo, M. Acar, H. Steller and H. J. Bellen (2008). "Ero1L, a thiol oxidase, is required for Notch signaling through cysteine bridge formation of the Lin12-Notch repeats in Drosophila melanogaster." Journal of Cell Biology 182(6): 1113-1125.
Tousoulis, D., E. Oikonomou, E. K. Economou, F. Crea and J. C. Kaski (2016). "Inflammatory cytokines in atherosclerosis: current therapeutic approaches." Eur Heart J 37(22): 1723-1732.
Towbin, H., T. Staehelin and J. Gordon (1979). "Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications." Proc Natl Acad Sci U S A 76(9): 4350-4354.
Turner, M. D., B. Nedjai, T. Hurst and D. J. Pennington (2014). "Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease." Biochim Biophys Acta 1843(11): 2563-2582.
Turpaev, K. T. (2013). "Keap1-Nrf2 signaling pathway: mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles." Biochemistry (Mosc) 78(2): 111-126.
Turvey, S. E. and D. H. Broide (2010). "Innate immunity." J Allergy Clin Immunol 125(2 Suppl 2): S24-32.
Valko, M., D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser (2007). "Free radicals and antioxidants in normal physiological functions and human disease." Int J Biochem Cell Biol 39(1): 44-84.
Varlamova, E. G., M. V. Gol'tiaev, S. V. Novoselov, V. I. Novoselov and E. E. Fecenko (2013). "[Characterization of some thiol oxidoreductase family members]." Mol Biol (Mosk) 47(4): 568-582.
Vazirinejad, R., Z. Ahmadi, M. Kazemi Arababadi, G. Hassanshahi and D. Kennedy (2014). "The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis." Neuroimmunomodulation 21(6): 322-330.
Venereau, E., M. Casalgrandi, M. Schiraldi, D. J. Antoine, A. Cattaneo, F. De Marchis, J. Liu, A. Antonelli, A. Preti, L. Raeli, S. S. Shams, H. Yang, L. Varani, U. Andersson, K. J. Tracey, A. Bachi, M. Uguccioni and M. E. Bianchi (2012). "Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release." J Exp Med 209(9): 1519-1528.
Venereau, E., C. Ceriotti and M. E. Bianchi (2015). "DAMPs from Cell Death to New Life." Front Immunol 6: 422.
263
Voigt, D., U. Scheidt, T. Derfuss, W. Bruck and A. Junker (2017). "Expression of the Antioxidative Enzyme Peroxiredoxin 2 in Multiple Sclerosis Lesions in Relation to Inflammation." Int J Mol Sci 18(4).
Vossen, R. C., M. C. Persoons, M. E. Slobbe-van Drunen, C. A. Bruggeman and M. C. van Dam-Mieras (1997). "Intracellular thiol redox status affects rat cytomegalovirus infection of vascular cells." Virus Res 48(2): 173-183.
Wahlgren, C. M. and K. Pekkari (2005). "Elevated thioredoxin after angioplasty in peripheral arterial disease." Eur J Vasc Endovasc Surg 29(3): 281-286.
Wajant, H., K. Pfizenmaier and P. Scheurich (0000). "Tumor necrosis factor signaling." Cell Death Differ 10(1): 45-65.
Walter, S., M. Letiembre, Y. Liu, H. Heine, B. Penke, W. Hao, B. Bode, N. Manietta, J. Walter, W. Schulz-Schuffer and K. Fassbender (2007). "Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease." Cell Physiol Biochem 20(6): 947-956.
Wanders, R. J. and H. R. Waterham (2006). "Biochemistry of mammalian peroxisomes revisited." Annu Rev Biochem 75: 295-332.
Wang, G. K., J. Q. Zhu, J. T. Zhang, Q. Li, Y. Li, J. He, Y. W. Qin and Q. Jing (2010). "Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans." Eur Heart J 31(6): 659-666.
Wang, Y., X. Huang, H. Cang, F. Gao, T. Yamamoto, T. Osaki and J. Yi (2007). "The endogenous reactive oxygen species promote NF-kappaB activation by targeting on activation of NF-kappaB-inducing kinase in oral squamous carcinoma cells." Free Radic Res 41(9): 963-971.
Wani, R., A. Nagata and B. W. Murray (2014). "Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology." Front Pharmacol 5: 224.
Waters, K. M., J. G. Pounds and B. D. Thrall (2006). "Data merging for integrated microarray and proteomic analysis." Brief Funct Genomic Proteomic 5(4): 261-272.
Watowich, S. S., D. J. Hilton and H. F. Lodish (1994). "Activation and inhibition of erythropoietin receptor function: role of receptor dimerization." Mol Cell Biol 14(6): 3535-3549.
Weichsel, A., J. R. Gasdaska, G. Powis and W. R. Montfort (1996). "Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer." Structure 4(6): 735-751.
Weiss, G. and U. E. Schaible (2015). "Macrophage defense mechanisms against intracellular bacteria." Immunol Rev 264(1): 182-203.
Weiss, S. J., R. Klein, A. Slivka and M. Wei (1982). "Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation." J Clin Invest 70(3): 598-607.
Weiss, U. (2008). "Inflammation." Nature 454(7203): 427.
264
Wilkinson, B. and H. F. Gilbert (2004). "Protein disulfide isomerase." Biochim Biophys Acta 1699(1-2): 35-44.
Williams, L. T. and E. G. Janzen (1970). "Spin Trapping of Alkyl and Aryl Radicals Produced in Reactions of Organometallic Compiunds - Orgn." Abstracts of Papers of the American Chemical Society(Feb): 20-&.
Wink, L. K., R. Adams, Z. M. Wang, J. E. Klaunig, M. H. Plawecki, D. J. Posey, C. J. McDougle and C. A. Erickson (2016). "A randomized placebo-controlled pilot study of N-acetylcysteine in youth with autism spectrum disorder." Molecular Autism 7.
Winterbourn, C. C. and M. B. Hampton (2008). "Thiol chemistry and specificity in redox signaling." Free Radic Biol Med 45(5): 549-561.
Witschi, A., S. Reddy, B. Stofer and B. H. Lauterburg (1992). "The Systemic Availability of Oral Glutathione." European Journal of Clinical Pharmacology 43(6): 667-669.
Wollman, E. E., A. Kahan and D. Fradelizi (1997). "Detection of membrane associated thioredoxin on human cell lines." Biochem Biophys Res Commun 230(3): 602-606.
Wood, Z. A., E. Schroder, J. Robin Harris and L. B. Poole (2003). "Structure, mechanism and regulation of peroxiredoxins." Trends Biochem Sci 28(1): 32-40.
World Health Organization. (n.d., May 2017). "Cardiovascular diseases (CVDs)." Retrieved 17 June, 2017, from http://www.who.int/mediacentre/factsheets/fs317/en/.
Wu, C., M. R. Jain, Q. Li, S. Oka, W. Li, A. N. Kong, N. Nagarajan, J. Sadoshima, W. J. Simmons and H. Li (2014). "Identification of novel nuclear targets of human thioredoxin 1." Mol Cell Proteomics 13(12): 3507-3518.
Wu, C., A. M. Parrott, C. Fu, T. Liu, S. M. Marino, V. N. Gladyshev, M. R. Jain, A. T. Baykal, Q. Li, S. Oka, J. Sadoshima, A. Beuve, W. J. Simmons and H. Li (2011). "Thioredoxin 1-mediated post-translational modifications: reduction, transnitrosylation, denitrosylation, and related proteomics methodologies." Antioxid Redox Signal 15(9): 2565-2604.
Wu, H. H., J. A. Thomas and J. Momand (2000). "p53 protein oxidation in cultured cells in response to pyrrolidine dithiocarbamate: a novel method for relating the amount of p53 oxidation in vivo to the regulation of p53-responsive genes." Biochem J 351(Pt 1): 87-93.
Wynn, T. A. (2008). "Cellular and molecular mechanisms of fibrosis." J Pathol 214(2): 199-210.
Xie, Y., S. Kole, P. Precht, M. J. Pazin and M. Bernier (2009). "S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling." Endocrinology 150(3): 1122-1131.
Yamamoto, M. and K. Takeda (2010). "Current views of toll-like receptor signaling pathways." Gastroenterol Res Pract 2010: 240365.
Yang, H., H. S. Hreggvidsdottir, K. Palmblad, H. Wang, M. Ochani, J. Li, B. Lu, S. Chavan, M. Rosas-Ballina, Y. Al-Abed, S. Akira, A. Bierhaus, H. Erlandsson-Harris, U. Andersson and K. J. Tracey (2010). "A critical cysteine is required for HMGB1 binding to Toll-like receptor 4
and activation of macrophage cytokine release." Proc Natl Acad Sci U S A 107(26): 11942-11947.
Yang, H., P. Lundback, L. Ottosson, H. Erlandsson-Harris, E. Venereau, M. E. Bianchi, Y. Al-Abed, U. Andersson, K. J. Tracey and D. J. Antoine (2012). "Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1)." Mol Med 18: 250-259.
Yu, M., H. Wang, A. Ding, D. T. Golenbock, E. Latz, C. J. Czura, M. J. Fenton, K. J. Tracey and H. Yang (2006). "HMGB1 signals through toll-like receptor (TLR) 4 and TLR2." Shock 26(2): 174-179.
Yun, H. M., K. R. Park, E. C. Kim and J. T. Hong (2015). "PRDX6 controls multiple sclerosis by suppressing inflammation and blood brain barrier disruption." Oncotarget 6(25): 20875-20884.
Zhang, C., Y. Liu, C. Feng, Q. Wang, H. Shi, D. Zhao, R. Yu and Z. Su (2015). "Loss of PEG chain in routine SDS-PAGE analysis of PEG-maleimide modified protein." Electrophoresis 36(2): 371-374.
Zhang, W., W. Ji, L. Yang, L. Yao, G. Wang, A. Xuan and Z. Zhuang (2013). "The involvement of epigenetic silencing of Foxa2 in cellular replicative and premature senescence induced by hydrogen peroxide." Free Radic Res 47(4): 325-332.
Zhao, H., J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman (2001). "Synthesis and biochemical applications of a solid cyclic nitrone spin trap: a relatively superior trap for detecting superoxide anions and glutathiyl radicals." Free Radic Biol Med 31(5): 599-606.
266
Appendix
Appendix 1: Transcripts induced by LPS at 2h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05). Only the top 50 transcripts most affected by LPS (sorted by fold
change) are shown.
Appendix 2: Transcripts down-regulated by LPS at 2h. Transcripts were selected
for their differential expression between the two groups LPS vs Control (cut-off was:
fold change, 1.5; P<0.05). Only the top 50 transcripts most affected by LPS (sorted
by fold change) are shown.
Appendix 3: Transcripts induced by LPS at 6h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05). Only the top 50 transcripts most affected by LPS (sorted by fold
change) are shown.
Appendix 4: Transcripts down-regulated by LPS at 6h. Transcripts were selected
for their differential expression between the two groups LPS vs Control (cut-off was:
fold change, 1.5; P<0.05). Only the top 50 transcripts most affected by LPS (sorted
by fold change) are shown.
Appendix 5: Expression images of cluster 1, 2, 3 and 4 at 2h. The median of the
expression for a cluster is in pink. The level of expression is indicated by the ratio of
each condition versus the average of the control (+4;-4).
Appendix 6: Expression images of cluster 1, 2, 3 and 4 at 6h. The median of the
expression for a cluster is in pink. The level of expression is indicated by the ratio of
each condition versus the average of the control (+4;-4).
Appendix 7: List of genes in Group 1 at 2h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
267
Appendix 8: List of genes in Group 2 at 2h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 9: List of genes in Group 3 at 2h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 10: List of genes in Group 4 at 2h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 11: List of genes in Group 1 at 6h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 12: List of genes in Group 2 at 6h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 13: List of genes in Group 3 at 6h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 14: List of genes in Group 4 at 6h. Transcripts were selected for their
differential expression between the two groups LPS vs Control (cut-off was: fold
change, 1.5; P<0.05) and the two groups BSO+LPS vs LPS alone (cut-off was: fold
change, 1.5; P<0.05).
Appendix 15: Functional categories of Group 2. Functional categories were
obtained by DAVID using the Functional Annotation Chart tool with GO TERM BP and
268
KEGG pathway for each group independently of the time point and were ordered by
EASE score a modified Fisher’s Test (P value <0.5).
Appendix 16: List of the 80 first proteins out of 1871 from which most peptides
have been identified by MS in untreated cells. Proteins were previously selected
with a FDR<1 (=-10logP) and were ordered by their number of peptides identified by
MS.
Appendix 17: List of the 80 first proteins out of 2427 from which most peptides
have been identified by MS in LPS-treated cells. Proteins were previously selected
with a FDR<1 (=-10logP) and were ordered by their number of peptides identified by