Relativistic hybrid density functional calculations of indirect nuclear spin–spin coupling tensors — Comparison with experiment for diatomic alkali metal halides 1,2 David L. Bryce and Jochen Autschbach Abstract: The accurate calculation of the isotropic (Jiso) and anisotropic (DJ) parts of indirect nuclear spin–spin coupling tensors is a stringent test for quantum chemistry, particularly for couplings involving heavy isotopes where relativistic ef- fects and relativity – electron correlation cross terms are expected to play an important role. Experimental measurements on diatomic molecules in the gas phase offer ideal data for testing the success of computational approaches, since the data are essentially free from intermolecular effects, and precise coupling anisotropies may be reliably extracted in favourable cases. On the basis of available experimental molecular-beam coupling-tensor parameters for diatomic alkali metal halides, we tabulate known values of Jiso and, taking rotational–vibrational corrections to the direct dipolar coupling constant into account, precise values of DJ are determined for the ground rovibrational state. First-principles calculations of the cou- pling tensors were performed using a recently developed program based on hybrid density functional theory using the two- component relativistic zeroth-order regular approximation (ZORA). Experimental trends in Jiso and DJ are reproduced with correlation coefficients of 0.993 and 0.977, respectively. Periodic trends in the coupling constants and their depend- ence on the product of the atomic numbers of the coupled nuclei are discussed. Finally, the hybrid functional method is also successfully tested against experimental data for a series of polyatomic xenon fluorides and group-17 fluorides. Key words: density functional theory, relativistic effects, spin–spin coupling constant, dipolar coupling, rovibrational aver- aging, alkali metal halides, group-17 fluorides, xenon fluorides. Re ´sume ´: Le calcul pre ´cis des parties isotropes (J iso ) et anisotropes (DJ) des tenseurs de couplage spin–spin nucle ´aire indi- rect est un test rigoureux pour la chimie quantique, en particulier pour les couplages impliquant des isotopes lourds pour lesquels on peut s’attendre a ` ce qu’il y ait un ro ˆle important de jouer par les effets relativistes et les effets dus aux termes croise ´s de la relativite ´ et la corre ´lation e ´lectronique. Des mesures expe ´rimentales sur des mole ´cules diatomiques, en phase gazeuse, fournissent les donne ´es ide ´ales pour ve ´rifier les re ´sultats des approches the ´oriques puisque les donne ´es ne compor- tent pratiquement aucun effet intermole ´culaire et que dans les cas favorables il est possible d’en extraire des anisotropies de couplage fiables. Sur la base des parame `tres de tenseur de couplage obtenus a ` partir d’une expe ´rience faisceau mole ´cu- laire pour des haloge ´nures diatomiques de me ´taux alcalins, on a pre ´pare ´ un tableau des valeurs connues de Jiso et, prenant en compte les corrections rotationnelles-vibrationnelles pour la constante de couplage dipolaire directe, on a de ´termine ´ des valeurs pre ´cises de DJ pour l’e ´tat vibrationel fondamental. On a aussi effectue ´ des calculs a ` base de principes premiers des tenseurs de couplage en faisant appel a ` un programme de ´veloppe ´ re ´cemment et base ´ sur une the ´orie hybride de la fonction- nelle de la densite ´ utilisant une approximation re ´gulie `re d’ordre ze ´ro relativiste a ` deux composantes « ZORA ». Les valeurs expe ´rimentales de Jiso et de DJ sont ainsi reproduites avec des coefficients de corre ´lation respectifs de 0,993 et 0,977. On discute des tendances pe ´riodiques des constantes de couplage et de leur corre ´lation avec le produit des nume ´ros atomiques de noyaux participants au couplage. Enfin, la me ´thode fonctionnelle hybride a aussi e ´te ´ teste ´e avec succe `s avec des donne ´es expe ´rimentales pour une se ´rie de fluorures de xe ´non polyatomiques et un groupe de fluorures du groupe 17. Mots-cle ´s : the ´orie de la fonctionnelle de la densite ´, effets relativistes, constante de couplage spin-spin, couplage dipolaire, moyenne rovibrationnelle, haloge ´nures de me ´taux alcalins, fluorures du groupe 17, fluorures de xe ´non. [Traduit par la Re ´daction] Introduction Indirect nuclear spin–spin coupling (J-coupling) tensors are widely used, in the form of their isotropic average (J iso ), for structural elucidation in diverse areas of the chemical, 3–7 materials, 8,9 and biochemical sciences. 10–14 The direct dipo- lar coupling constant (R DD ), which depends on the motional average of the inverse cube of the distance between two spins, is the key NMR parameter for providing internuclear distance information in solids, 15 and highly precise bond vector orientations relative to an overall molecular align- Received 21 December 2008. Accepted 6 March 2009. Published on the NRC Research Press Web site at canjchem.nrc.ca on 26 May 2009. D.L. Bryce. 1 Department of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, Ottawa, ON K1N 6N5, Canada. J. Autschbach. 2 Department of Chemistry, State University of New York at Buffalo, New York 14260-3000, USA. 1 Corresponding author (e-mail: [email protected]). 2 Corresponding author (e-mail: [email protected]). 927 Can. J. Chem. 87: 927–941 (2009) doi:10.1139/V09-040 Published by NRC Research Press
Transcript
Relativistic hybrid density functional calculationsof indirect nuclear spin–spin coupling tensors —Comparison with experiment for diatomic alkalimetal halides1,2
David L. Bryce and Jochen Autschbach
Abstract: The accurate calculation of the isotropic (Jiso) and anisotropic (DJ) parts of indirect nuclear spin–spin couplingtensors is a stringent test for quantum chemistry, particularly for couplings involving heavy isotopes where relativistic ef-fects and relativity – electron correlation cross terms are expected to play an important role. Experimental measurementson diatomic molecules in the gas phase offer ideal data for testing the success of computational approaches, since the dataare essentially free from intermolecular effects, and precise coupling anisotropies may be reliably extracted in favourablecases. On the basis of available experimental molecular-beam coupling-tensor parameters for diatomic alkali metal halides,we tabulate known values of Jiso and, taking rotational–vibrational corrections to the direct dipolar coupling constant intoaccount, precise values of DJ are determined for the ground rovibrational state. First-principles calculations of the cou-pling tensors were performed using a recently developed program based on hybrid density functional theory using the two-component relativistic zeroth-order regular approximation (ZORA). Experimental trends in Jiso and DJ are reproducedwith correlation coefficients of 0.993 and 0.977, respectively. Periodic trends in the coupling constants and their depend-ence on the product of the atomic numbers of the coupled nuclei are discussed. Finally, the hybrid functional method isalso successfully tested against experimental data for a series of polyatomic xenon fluorides and group-17 fluorides.
Resume : Le calcul precis des parties isotropes (Jiso) et anisotropes (DJ) des tenseurs de couplage spin–spin nucleaire indi-rect est un test rigoureux pour la chimie quantique, en particulier pour les couplages impliquant des isotopes lourds pourlesquels on peut s’attendre a ce qu’il y ait un role important de jouer par les effets relativistes et les effets dus aux termescroises de la relativite et la correlation electronique. Des mesures experimentales sur des molecules diatomiques, en phasegazeuse, fournissent les donnees ideales pour verifier les resultats des approches theoriques puisque les donnees ne compor-tent pratiquement aucun effet intermoleculaire et que dans les cas favorables il est possible d’en extraire des anisotropiesde couplage fiables. Sur la base des parametres de tenseur de couplage obtenus a partir d’une experience faisceau molecu-laire pour des halogenures diatomiques de metaux alcalins, on a prepare un tableau des valeurs connues de Jiso et, prenanten compte les corrections rotationnelles-vibrationnelles pour la constante de couplage dipolaire directe, on a determine desvaleurs precises de DJ pour l’etat vibrationel fondamental. On a aussi effectue des calculs a base de principes premiers destenseurs de couplage en faisant appel a un programme developpe recemment et base sur une theorie hybride de la fonction-nelle de la densite utilisant une approximation reguliere d’ordre zero relativiste a deux composantes « ZORA ». Les valeursexperimentales de Jiso et de DJ sont ainsi reproduites avec des coefficients de correlation respectifs de 0,993 et 0,977. Ondiscute des tendances periodiques des constantes de couplage et de leur correlation avec le produit des numeros atomiquesde noyaux participants au couplage. Enfin, la methode fonctionnelle hybride a aussi ete testee avec succes avec des donneesexperimentales pour une serie de fluorures de xenon polyatomiques et un groupe de fluorures du groupe 17.
Mots-cles : theorie de la fonctionnelle de la densite, effets relativistes, constante de couplage spin-spin, couplage dipolaire,moyenne rovibrationnelle, halogenures de metaux alcalins, fluorures du groupe 17, fluorures de xenon.
are widely used, in the form of their isotropic average (Jiso),for structural elucidation in diverse areas of the chemical,3–7
materials,8,9 and biochemical sciences.10–14 The direct dipo-lar coupling constant (RDD), which depends on the motionalaverage of the inverse cube of the distance between twospins, is the key NMR parameter for providing internucleardistance information in solids,15 and highly precise bondvector orientations relative to an overall molecular align-
Received 21 December 2008. Accepted 6 March 2009.Published on the NRC Research Press Web site atcanjchem.nrc.ca on 26 May 2009.
D.L. Bryce.1 Department of Chemistry and Centre for CatalysisResearch and Innovation, University of Ottawa, Ottawa, ONK1N 6N5, Canada.J. Autschbach.2 Department of Chemistry, State University ofNew York at Buffalo, New York 14260-3000, USA.
Can. J. Chem. 87: 927–941 (2009) doi:10.1139/V09-040 Published by NRC Research Press
ment frame in liquid-crystal NMR of proteins and nucleicacids.16–18 The orientation-dependent anisotropic part of theJ-coupling (DJ) is also important, not only from a funda-mental perspective, but also due to the fact that any meas-ured ‘‘effective’’ dipolar coupling also contains acontribution from DJ, i.e., Reff = RDD – DJ/3. Consequently,RDD and DJ cannot be measured via NMR independently ofone another. Although RDD is often much larger than DJ/3,particularly for light nuclei,19,20 there are several caseswhere the opposite is true,20,21 e.g., in I2, the value of DJ ismore than 14 times that of RDD.22 Also, available theoreticaldata on J-coupling tensors in molecules with heavy elementshave for a long time indicated that DJ can be of the sameorder of magnitude or larger than Jiso.23 Thus, an a priori as-sumption of a small DJ/Jiso ratio in a molecule should notbe made without theoretical information or experimentaldata for related systems.
Experimental values of Jiso measured in solids and solu-tions, while plentiful in the literature, are influenced bycrystal-packing effects or solvent effects. These effects addan extra layer of complexity when attempting to interpretcoupling-tensor information in a quantitative fashion. Reli-able experimental values of DJ measured in solids orliquid-crystal solutions are scarce for several reasons,15,20
e.g., DJ may be much smaller than RDD, the need to havean independent measure of RDD to extract DJ, the need toaccount for how molecular motion will affect RDD, and sym-metry considerations may simply increase the number oftensor elements to the point where these are underdeter-mined by the experimental data.
J-coupling tensor data available for diatomics from high-resolution rotational24,25 or molecular beam electric reso-nance (MBER)26 spectroscopies are beautiful examples ofthe highly precise benchmark information which can bemeasured experimentally.21,27 Since the experiments are car-ried out at low pressure on essentially isolated molecules ofknown structure, all of the usual problems associated withdetermining DJ (vide supra) are no longer an issue. The ex-perimental data thus provide an excellent test for computa-tional methods. Very good agreement between benchmarkexperimental values extracted from molecular beam data forselect diatomic molecules has been obtained through multi-configurational self-consistent field21 or DFT22 calculations.Recently, Cederberg and co-workers reported coupling-tensor parameters for gaseous KBr and KI, making the po-tassium halides the first series of alkali metal halides forwhich a complete set of isotropic and anisotropic J-couplingsare available with high precision.28
On the theoretical front, quantum chemists have long had agreat interest in the computation of NMR parameters, such asspin–spin coupling tensors. For example, see Pople’s andSantry’s early work on J-coupling.29,30 Because of the sensi-tivity of NMR parameters to the electronic structure of a mol-ecule, including effects from approximations made, and thepotentially formidable computational cost of high-accuracycorrelated-wave-function-based methods, applications offirst-principles-correlated theoretical methods have only be-come widespread after the development of efficient DFT im-plementations for molecules.31 The availability of generallyapplicable DFT programs was soon followed by develop-ments for DFT computations of NMR parameters, including
methods to compute J-coupling.23,32,33 The application toheavy atomic systems has met with particular challenges be-cause of the need for a relativistic theoretical framework.23
Indeed, relativistic effects on J-coupling can be spectacularlylarge due to the strong effects on the hyperfine integrals34
and other relativistic influences.35 In the DFT field, one ofthe first steps to incorporate relativistic effects in J-couplingswas taken by Khandogin and Ziegler36 with a Pauli-operator-based method designed to obtain relativistically scaled hyper-fine integrals. The method met with limited success becauseof the variational instability of the Pauli operator and wassoon superseded by a variationally stable method based onthe Zeroth-order regular approximate (ZORA) two-componentHamiltonian.37,38 Other recent two-component relativisticDFT developments for J-coupling include a modified infin-ite-order regular approximation (IORAmm) approach39 anda second-order Douglas–Kroll–Hess method reported byMelo et al.40 The ZORA code originally developed byAutschbach and Ziegler has been recently extended to al-low for analytic-derivative two-component relativistic hy-brid DFT calculations of J-coupling.41 The method isapplicable to atoms across the periodic table, including‘‘strongly relativistic’’ ones such as Pt, Hg, Pb, or Tl, andsupports a variety of orbital-based analysis methods, whichcan be used to determine chemically intuitive contributionsto the coupling tensor.42–44
The recent report of the development of the two-componenthybrid ZORA DFT method for J-coupling41 has been ac-companied by benchmark computations of the reduced iso-tropic coupling Kiso and the reduced coupling anisotropyDK for interhalogen diatomics and for the series TlXwhere X = F, Cl, Br, I. For these systems, experimentaldata for Kiso and DK are available from microwave orMBER spectroscopy.21,45–51 For the TlX series, computa-tions with the PBE0 hybrid functional52 (25% Hartree–Fock exchange, this functional is a typical representativeof the class of non-empirical hybrid functionals) yieldedclear improvements over nonhybrid functionals for Kisoand in particular for DK. The availability of this hybridDFT method and the availability of experimental data fornumerous other molecules20,53 (usually from MBER spec-troscopy) have therefore motivated us to perform furthercomputational studies of isotropic spin–spin coupling andcoupling anisotropies with the aim of extending the avail-able body of benchmark data, providing further validationof the theoretical method, its range of applicability, and itslimitations, and re-evaluating some additional polyatomicsystems studied previously where the application of an im-proved functional might be beneficial. We also use a com-bination of computational and experimental data to predictthe J-coupling for the RbBr diatomic.
This paper is organized as follows: following a brief pre-sentation of the computational details, hybrid DFT resultsfor polyatomic xenon fluorides and group-17 fluorides arepresented and assessed compared with previous pure DFTresults and experiment;54 a detailed discussion of extractingall known available experimental spin–spin coupling-tensordata for alkali metal halides (MX; M = Li, Na, K, Rb, Cs;X = F, Cl, Br, I) is then presented, including a discussionof rovibrational averaging effects; relativistic hybrid DFTcalculations of the coupling tensors for the MX series are
928 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
presented and discussed in the context of the experimentaldata as well as pure DFT and Hartree–Fock results.
Computational detailsAll calculations were carried out with a developer’s ver-
sion of the ADF code.55–57 Indirect nuclear spin–spin cou-pling tensors were calculated using the CPL module33,37,38,58
in its most recent incarnation where the scalar and spin–or-bit functionalities were extended to hybrid functionals.41,52
The computation of the Hartree–Fock exchange-relatedterms for the unperturbed ground state and the coupling ten-sor make use of the density fitting machinery implementedin the ADF package. A ‘‘bug’’ that was present in the imple-mentation has been corrected; details will be provided in anerratum to ref. 41. Five sets of calculations were performedfor the alkali metal halides: non-relativistic, ZORA scalarrelativistic, ZORA spin–orbit relativistic, ZORA spin–orbitrelativistic using a PBE0 hybrid functional with 25% HF ex-change,41,52 and a Hartree–Fock ZORA spin–orbit relativis-tic calculation. The first three sets of pure DFT calculationsused the VWN59 local density approximation along with theBecke88-Perdew86 (BP86) generalized gradient approxi-mation functional, and the VWN response kernel in theJ-coupling calculations. These settings are similar to pre-vious J-coupling studies that we have carried out 22, 37,38
and were chosen for comparison with this previous work.For the PBE0 calculations, PBE60 terms in the J-couplingcalculations were included as detailed in ref. 61 (in addi-tion to the HF exchange terms). Doubly polarized valencetriple-z all-electron Slater-type basis sets optimized forZORA calculations (TZ2P from the ADF basis set library)were used for all atoms as a starting point to derive thebasis sets used here for the J-coupling computations: Func-tions with an exponent much higher than the nuclearcharge Z were removed, if present, then four additional s-functions with exponents 2Z, 3Z, 4Z, and 100Z were addedalong with an additional set of density-fit functions similarto the approach taken in ref. 41. High-exponent s-functionsare required for accurate calculation of spin–spin couplingtensors as was shown explicitly for relativistic computa-tions in ref. 32 (note that, for elements heavier than thoseinvestigated here, a significantly more extended set of s-and p-functions should be added32). The J-coupling imple-mentation used here is based on point nuclear charges andpoint nuclear magnetic dipoles. An extension of the pro-gram to include finite nucleus corrections will be reportedin a forthcoming publication. All contributions to the cou-pling tensors were calculated for all molecules. In the two-component relativistic framework adopted here, these con-tributions are the relativistic analogs of the diamagneticspin–orbit (DSO), paramagnetic spin–orbit (PSO), Fermi-contact (FC), and spin–dipolar (SD) terms, the FC–SDcross term, as well as spin–orbit PSO–FC and PSO–SDcross terms. For details, we refer to some of the availablereview articles on relativistic NMR calculations23,62,63 andthe Discussion in this work. The hybrid DFT extension ofthe program is described in detail in ref. 41. The computa-tions yield the reduced indirect coupling tensor K alongwith its isotropic average. The molecules were orientedalong the z axis (Kzz = K||; Kxx = Kyy = K?) and the tensoranisotropy DK obtained, as usual, as K|| – K?.
The reduced coupling tensor, K, which does not dependon the magnetogyric ratios of the coupled nuclei, is relatedto the J tensor as follows:
½1� J ¼ hg1g2
4p2K
K tensors and J tensors were interconverted using themagnetogyric ratios found in ref. 64.
Atomic coordinates for the structures of the xenon fluo-rides and group-17 fluorides were obtained from refs. 65–78; further details on the structures used are given in ref.54. Equilibrium structures of the diatomic alkali metal hal-ides were obtained from the NIST Chemistry Webbook.79
Results and discussion
The PBE0 hybrid functional. Results for xenon fluoridesand group-17 fluorides
In this section, we test the performance of the PBE0 func-tional and the augmented basis set on a series of polyatomicxenon, chlorine, bromine, and iodine fluoride molecules forwhich experimental isotropic coupling data are available.Results of ZORA-DFT spin–orbit relativistic calculations ofthe coupling tensors with a nonhybrid GGA functional arealso available,54 and enable an assessment of the differencesin the results obtained through inclusion of a portion of exactexchange. One of the reasons why it is interesting to reinves-tigate these systems with a hybrid functional approach is thattheir coupling constants tend to have significant contribu-tions from the PSO mechanism. In a spin-free non-hybridDFT approach, the paramagnetic mechanisms of J-couplingand nuclear magnetic shielding require a set of so-called‘‘uncoupled’’ perturbed Kohn–Sham equations to be solved.The lack of ‘‘coupling’’ in these equations was for sometime thought to be a DFT problem that requires specificcures80 (see, however, ref. 81). Although a spin–orbit relativ-istic framework generally leads to a set of coupled-perturbedKohn–Sham equations, there are still no orbital current-density contributions in the perturbed Kohn–Sham potentialto evaluate. This situation changes when a hybrid DFTframework is adopted (or within a formalism based on thecurrent-density): already at the spin-free level of theoryself-consistent contributions from the perturbed Fock opera-tor need to be evaluated while solving the coupled per-turbed Kohn–Sham equations for the PSO perturbation(likewise for the paramagnetic shielding). With spin–orbitcoupling present, one also needs to pay attention to thetreatment of the spin-density perturbation. Already in theinitial implementation,38 a so-called non-collinear approachwas adopted, which has also been used for the hybrid func-tional computations performed in this work and in ref. 41.
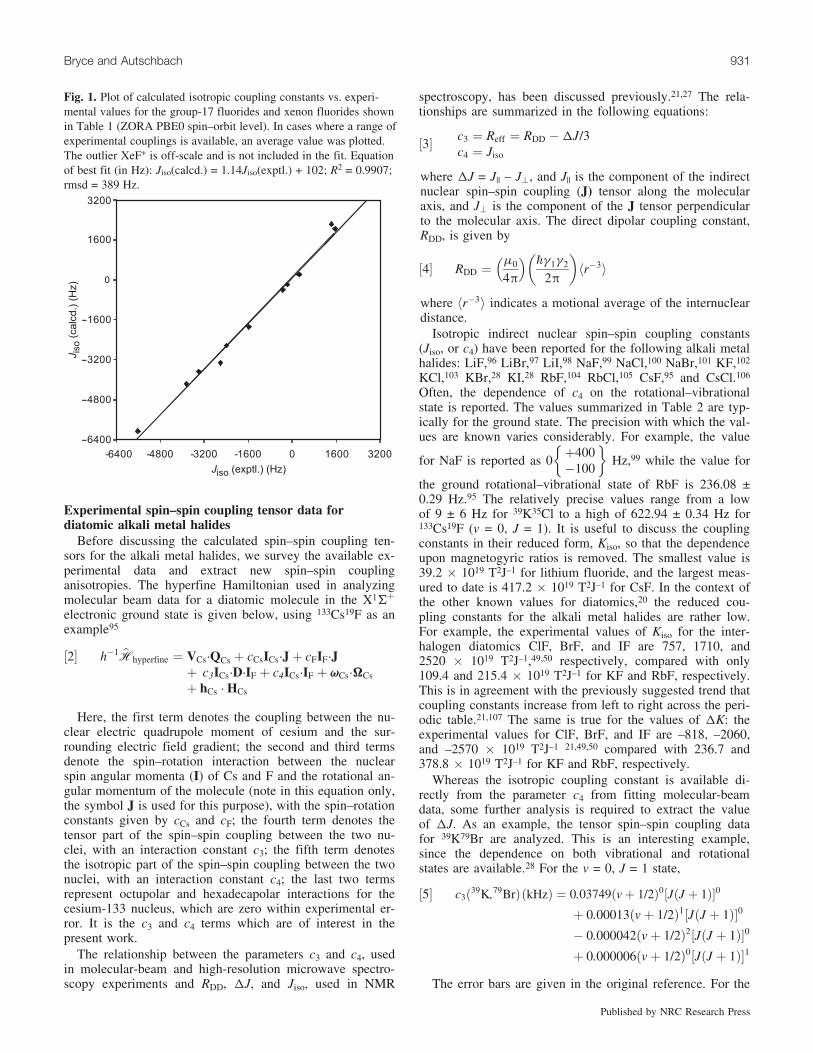
The hybrid functional results (with spin–orbit relativisticcorrections) are presented in Table 1, along with the experi-mental data for comparison. Analogously to the previouslyreported non-hybrid DFT results, good agreement is ob-tained in comparison with experiment (Jiso). This is demon-strated in Fig. 1, where a plot of the calculated couplingconstants vs. the experimental data yields a slope of 1.14and a correlation coefficient, R2, of 0.99. Furthermore, a cal-culation of the root-mean-square-deviation (rmsd) betweenthe computed results and the experimental data (where an
Bryce and Autschbach 929
Published by NRC Research Press
average value has been used in the cases where a range of J-couplings are available for a particular species) yields389 Hz with the PBE0 functional and the augmented basis.Considering that the range in experimental coupling con-stants considered is on the order of 7200 Hz (the outlierXeF+ was not included in the analysis), the rmsd representsonly about 5% of this total range. Regarding the outlier, ithas been pointed out by Bagno et al. that the experimentalcoupling constant for this system was obtained in supera-cids, thus there are good reasons to assume that the speciespresent in solution have NMR parameters that might be sig-nificantly different from those of the isolated XeF+ ion.82
The contributions from the PSO and the FC + SD mecha-nisms to the total isotropic coupling constant for these mol-ecules are consistent with the results obtained previously inthat the PSO contribution is seen to increase with the num-ber of lone pairs on the central heavy atom involved in thecoupling.54 There are some variations in the exact percent-
age contributions to Kiso for the hybrid DFT results com-pared with the pure DFT results; however, overall, the twosets of results are consistent with each other.
The results obtained presently for xenon fluorides andgroup-17 fluorides demonstrate the utility of the relativistichybrid DFT implementation for spin–spin coupling in ob-taining the correct trends and magnitudes of calculated cou-pling tensors when compared with experimental data. Theagreement with experiment is similar to the previously pub-lished non-hybrid DFT results,54 if slightly worse. It shouldbe kept in mind that both functional and basis set have beenchanged with respect to ref. 54. Further improvement to theoverall rmsd is likely to require consideration of the fact thatmost of the experimental data were recorded in solutionwhile the calculations are for isolated molecules. In the fol-lowing sections, the application of the theoretical approachto the challenging case of the alkali metal halides will bediscussed.
Table 1. One-bond isotropic and anisotropic coupling constants for a series of group-17 fluorides and xenonfluorides.
Jiso (Hz)
Experimentala Calculatedb Kiso DK PSO (%) FC + SD (%)ClF2
aReferences for experimental coupling constants: ClF3: 83; ClF6þ: 84; BrF6
þ: 85; BrF6�: 86; XeFþ: 87, 88; XeF2: 89, 90;
XeF3þ: 91, 92; XeF4: 89, 93; XeF5
þ: 94. See also ref. 54 for further discussion of the choice of experimental data.bCalculated using the PBE0 hybrid functional with relativistic spin–orbit terms.cLarge values of opposite sign.
930 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
Experimental spin–spin coupling tensor data fordiatomic alkali metal halides
Before discussing the calculated spin–spin coupling ten-sors for the alkali metal halides, we survey the available ex-perimental data and extract new spin–spin couplinganisotropies. The hyperfine Hamiltonian used in analyzingmolecular beam data for a diatomic molecule in the X1Sþ
electronic ground state is given below, using 133Cs19F as anexample95
Here, the first term denotes the coupling between the nu-clear electric quadrupole moment of cesium and the sur-rounding electric field gradient; the second and third termsdenote the spin–rotation interaction between the nuclearspin angular momenta (I) of Cs and F and the rotational an-gular momentum of the molecule (note in this equation only,the symbol J is used for this purpose), with the spin–rotationconstants given by cCs and cF; the fourth term denotes thetensor part of the spin–spin coupling between the two nu-clei, with an interaction constant c3; the fifth term denotesthe isotropic part of the spin–spin coupling between the twonuclei, with an interaction constant c4; the last two termsrepresent octupolar and hexadecapolar interactions for thecesium-133 nucleus, which are zero within experimental er-ror. It is the c3 and c4 terms which are of interest in thepresent work.
The relationship between the parameters c3 and c4, usedin molecular-beam and high-resolution microwave spectro-scopy experiments and RDD, DJ, and Jiso, used in NMR
spectroscopy, has been discussed previously.21,27 The rela-tionships are summarized in the following equations:
½3� c3 ¼ Reff ¼ RDD �DJ=3
c4 ¼ Jiso
where DJ = J|| – J?, and J|| is the component of the indirectnuclear spin–spin coupling (J) tensor along the molecularaxis, and J? is the component of the J tensor perpendicularto the molecular axis. The direct dipolar coupling constant,RDD, is given by
½4� RDD ¼m0
4p
� � Zg1g2
2p
� �hr�3i
where hr�3i indicates a motional average of the internucleardistance.
Isotropic indirect nuclear spin–spin coupling constants(Jiso, or c4) have been reported for the following alkali metalhalides: LiF,96 LiBr,97 LiI,98 NaF,99 NaCl,100 NaBr,101 KF,102
KCl,103 KBr,28 KI,28 RbF,104 RbCl,105 CsF,95 and CsCl.106
Often, the dependence of c4 on the rotational–vibrationalstate is reported. The values summarized in Table 2 are typ-ically for the ground state. The precision with which the val-ues are known varies considerably. For example, the value
for NaF is reported as 0þ400
�100
� �Hz,99 while the value for
the ground rotational–vibrational state of RbF is 236.08 ±0.29 Hz.95 The relatively precise values range from a lowof 9 ± 6 Hz for 39K35Cl to a high of 622.94 ± 0.34 Hz for133Cs19F (v = 0, J = 1). It is useful to discuss the couplingconstants in their reduced form, Kiso, so that the dependenceupon magnetogyric ratios is removed. The smallest value is39.2 � 1019 T2J–1 for lithium fluoride, and the largest meas-ured to date is 417.2 � 1019 T2J–1 for CsF. In the context ofthe other known values for diatomics,20 the reduced cou-pling constants for the alkali metal halides are rather low.For example, the experimental values of Kiso for the inter-halogen diatomics ClF, BrF, and IF are 757, 1710, and2520 � 1019 T2J–1,49,50 respectively, compared with only109.4 and 215.4 � 1019 T2J–1 for KF and RbF, respectively.This is in agreement with the previously suggested trend thatcoupling constants increase from left to right across the peri-odic table.21,107 The same is true for the values of DK: theexperimental values for ClF, BrF, and IF are –818, –2060,and –2570 � 1019 T2J–1 21,49,50 compared with 236.7 and378.8 � 1019 T2J–1 for KF and RbF, respectively.
Whereas the isotropic coupling constant is available di-rectly from the parameter c4 from fitting molecular-beamdata, some further analysis is required to extract the valueof DJ. As an example, the tensor spin–spin coupling datafor 39K79Br are analyzed. This is an interesting example,since the dependence on both vibrational and rotationalstates are available.28 For the v = 0, J = 1 state,
The error bars are given in the original reference. For the
Fig. 1. Plot of calculated isotropic coupling constants vs. experi-mental values for the group-17 fluorides and xenon fluorides shownin Table 1 (ZORA PBE0 spin–orbit level). In cases where a range ofexperimental couplings is available, an average value was plotted.The outlier XeF+ is off-scale and is not included in the fit. Equationof best fit (in Hz): Jiso(calcd.) = 1.14Jiso(exptl.) + 102; R2 = 0.9907;rmsd = 389 Hz.
Bryce and Autschbach 931
Published by NRC Research Press
Table 2. Comparison of experimental and calculated isotropic coupling constants for alkali metal halides.
aHybrid functional with scalar and SO relativistic terms. Coupling constants are reported for the following isotopes:7Li,23Na,39K, 85Rb, 133Cs, 19F,35Cl, 79Br, and 127I.
bThe coupling constant has been averaged according to the procedure described in the text.cFor the v = 0 vibrational state.dNot available to our knowledge.eFor the v = 0, J = 1 rotational–vibrational state.
Table 3. Equilibrium and rovibrationally corrected direct dipolar coupling constants, experimental values of c3 and DJ, and rovi-brationally averaged calculated values of DJ for the alkali metal halides.
Note: See the text for explanation of the averaging procedure. Hybrid DFT calculations, which include scalar and spin–orbit relativistic effects.aNot available to our knowledge.
932 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
v = 0, J = 1 state, we obtain c3 = 37.56 ± 0.50 Hz. To extractDJ(v = 0, J = 1) from this value, we need the rovibrationallyaveraged value of RDD (cf. eqs. [3] and [4]). The equilibriumvalue of RDD is simple to calculate; we obtain 62.87 Hz usingthe equilibrium bond length of 2.82078 A. The next step is tofind the rovibrationally averaged value of RDD, since c3 isknown for the v = 0, J = 1 state. RDD may be corrected tofourth order as follows:
½6� RDDðv; JÞ ¼m0
4p
� � Zg1g2
2p
� �1
r3e
½1� 3hxi þ 6hx2i
� 10hx3i þ 15hx4i�
where hxi and higher order terms result from an expansionof the term (1 + hxi)–3. Here, x = (r – re)/re. The lengthyexpressions for hxin are available in the literature, and thesedepend on Dunham coefficients (an). We use the experimen-tal values for a1, a2, and a3 summarized by Noorizadeh andNazari108 for all alkali metal halides, and the values for a4and a5 determined by the same authors through a recursionformula. The result for 39K79Br is that RDD(v = 0, J = 1) is62.67 Hz, i.e., a reduction in the equilibrium value of ap-proximately 0.5%. Larger corrections on the order of severalHz are obtained for other molecules, particularly the fluor-ides (see Table 3). The largest percentage correction, about0.9%, is not surprisingly obtained for the lightest molecule,LiF. We note that slightly different values of RDD(v = 0, J =1) and DJ(v = 0, J = 1) are reported here for LiF comparedwith ref. 21 due to updated magnetogyric ratios64 used inthe calculation RDD(eq) and updated Dunham parametersused in the rovibrational correction.108
Using the relationship between c3, RDD, and DJ (eq. [3],the experimental value of DJ(v = 0, J = 1) for 39K79Br isfound to be 75.33 ± 1.49 Hz. Data obtained for all other al-kali metal halides are reported in Table 3. Although the ro-vibrational corrections to RDD are all less than 1% in thiscase, it is very important to perform such corrections to getaccurate values of DJ, particularly due to the error propaga-tion which is introduced into the values obtained due to thefactor of three in eq. [3]. The precision in DJ which is ob-tainable in some cases is remarkable. For example, DJ forthe ground rovibrational state in 133Cs19F is 690.48 ±2.57 Hz and that for 85Rb19F is 415.12 ± 1.22 Hz, i.e., theseerrors of one standard deviation are as little as 3% of the to-tal value of DJ. Clearly, such data represent a formidablechallenge for theoretical chemistry methods of calculatingNMR parameters. The data for these molecules are verywell-suited for comparison to theoretical results, since theexperiments are performed at low pressures, thereby ap-proaching the idealized case of an isolated molecule treatedin the calculations.
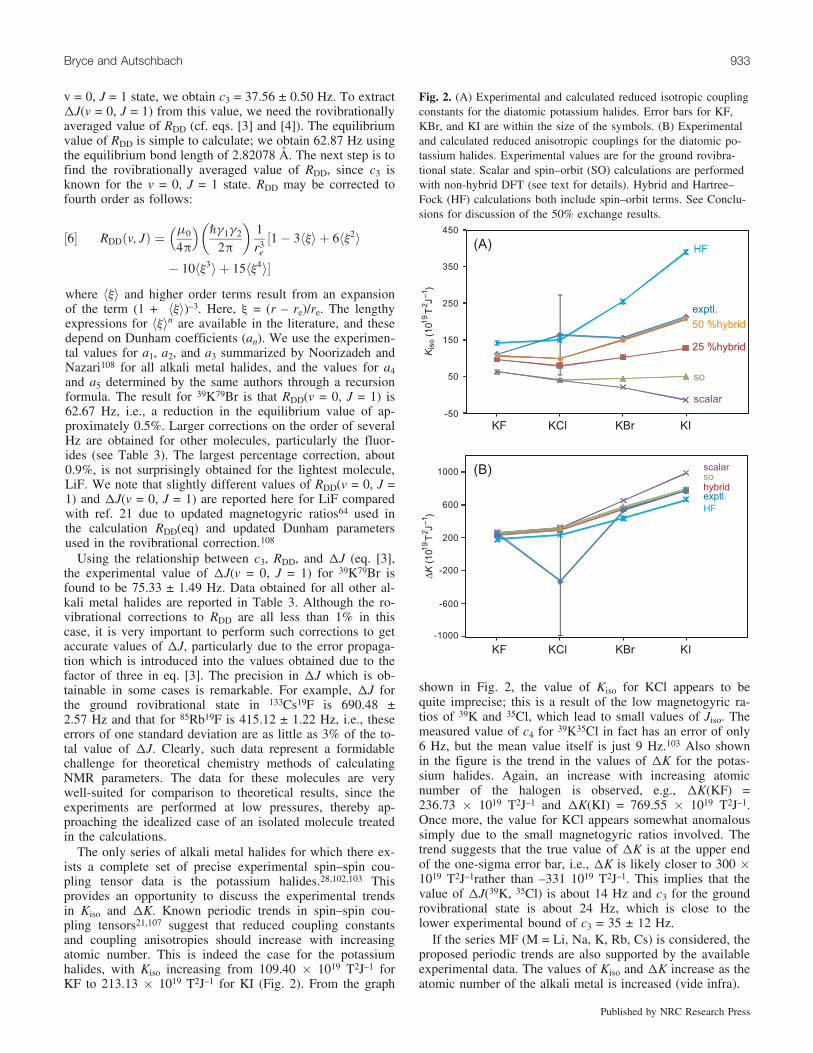
The only series of alkali metal halides for which there ex-ists a complete set of precise experimental spin–spin cou-pling tensor data is the potassium halides.28,102,103 Thisprovides an opportunity to discuss the experimental trendsin Kiso and DK. Known periodic trends in spin–spin cou-pling tensors21,107 suggest that reduced coupling constantsand coupling anisotropies should increase with increasingatomic number. This is indeed the case for the potassiumhalides, with Kiso increasing from 109.40 � 1019 T2J–1 forKF to 213.13 � 1019 T2J–1 for KI (Fig. 2). From the graph
shown in Fig. 2, the value of Kiso for KCl appears to bequite imprecise; this is a result of the low magnetogyric ra-tios of 39K and 35Cl, which lead to small values of Jiso. Themeasured value of c4 for 39K35Cl in fact has an error of only6 Hz, but the mean value itself is just 9 Hz.103 Also shownin the figure is the trend in the values of DK for the potas-sium halides. Again, an increase with increasing atomicnumber of the halogen is observed, e.g., DK(KF) =236.73 � 1019 T2J–1 and DK(KI) = 769.55 � 1019 T2J–1.Once more, the value for KCl appears somewhat anomaloussimply due to the small magnetogyric ratios involved. Thetrend suggests that the true value of DK is at the upper endof the one-sigma error bar, i.e., DK is likely closer to 300 �1019 T2J–1rather than –331 1019 T2J–1. This implies that thevalue of DJ(39K, 35Cl) is about 14 Hz and c3 for the groundrovibrational state is about 24 Hz, which is close to thelower experimental bound of c3 = 35 ± 12 Hz.
If the series MF (M = Li, Na, K, Rb, Cs) is considered, theproposed periodic trends are also supported by the availableexperimental data. The values of Kiso and DK increase as theatomic number of the alkali metal is increased (vide infra).
Fig. 2. (A) Experimental and calculated reduced isotropic couplingconstants for the diatomic potassium halides. Error bars for KF,KBr, and KI are within the size of the symbols. (B) Experimentaland calculated reduced anisotropic couplings for the diatomic po-tassium halides. Experimental values are for the ground rovibra-tional state. Scalar and spin–orbit (SO) calculations are performedwith non-hybrid DFT (see text for details). Hybrid and Hartree–Fock (HF) calculations both include spin–orbit terms. See Conclu-sions for discussion of the 50% exchange results.
Bryce and Autschbach 933
Published by NRC Research Press
The relative anisotropy ratios, DJ/Jiso, range from 111%in CsF to 361% in KI. The size of the indirect contribution(DJ) to c3 relative to the averaged direct dipolar contribu-tion (RDD (v = 0, J = 1)) ranges from 1.7% in LiF to 219%in KI. The trends are in agreement with the result observedfor diatomic fluorides that the ratio DJ/RDD increases as onemoves down a group in the periodic table.21 It is thereforeanticipated that the heaviest molecules, i.e., RbI, CsBr, andCsI, will exhibit the largest experimental DJ/RDD ratios.
Alkali metal halides: calculated coupling tensorsPresented in Table 4 are calculated values of Kiso and DK
for the twenty molecules. Results are shown for non-hybridDFT calculations incorporating scalar relativistic effects,non-hybrid DFT calculations incorporating scalar and spin–orbit relativistic effects, hybrid DFT calculations incorporatingscalar and spin–orbit relativistic effects, and, for compari-son to the limit of ‘‘100% exact exchange but no electroncorrelation’’, Hartree–Fock calculations with scalar andspin–orbit relativistic effects. If we consider some of theheaviest molecules in the set, it is noted that the effectsfrom spin–orbit coupling in the coupling tensor can bequite large relative to the scalar relativistic values of Kisoand DK. The spin–orbit effect (SO) on the coupling tensormay be analyzed by considering hyperfine matrix elementsinvolving an s1/2 atomic orbital (AO) on one atom and ap1/2 AO on the other atom, as was done already a longtime ago by Pyykko and Wiesenfeld.109 For instance, SOcoupling causes an increasingly pronounced mixing be-tween local s and p symmetries in bonding and antibond-
ing orbitals as the nuclear charges increase. In addition,relativistic effects increase the hyperfine matrix elementsfor s1/2 and p1/2 AOs to a similar degree. As a conse-quence, the importance of the SO terms in the couplingtensor strongly increases as the atoms involved in the cou-pling become heavier. Further aspects of SO effects on J-coupling tensors will be discussed below. For some sys-tems, in particular when involving heavy p-block elements,the SO effect may not just be a small correction but a ma-jor influence that should not be ignored even when discus-sing qualitative trends. For the set of alkali halides, thespin–orbit effects for some of the heaviest systems causelarge relative changes in the isotropic coupling constants.For RbI, where the SO effect is particularly large, thespin-free isotropic coupling is balanced by FC and PSOmechanisms of roughly equal magnitude but opposite sign,while in the SO case, the two mechanisms reinforce eachother. Changes observed when comparing the coupling ten-sors obtained using the hybrid functional relative to non-hybrid DFT are also significant for most of the molecules.For example, in the case of CsBr, the value of Kisochanges from 277.9 to 471.6 � 1019 T2J–1 and DK changesfrom 1715.4 to 1546.3 � 1019 T2J–1. In general, for themolecules studied here, the effect of including some exactexchange in the functional is to increase the magnitude ofKiso and decrease the magnitude of DK. The same trendswere previously also found for spin–spin coupling tensorsobtained for thallium halide diatomics.41
There are significant differences between the results ob-tained using relativistic Hartree–Fock (HF) calculations and
Table 4. Calculated reduced indirect nuclear spin–spin coupling tensors for the diatomic alkali metal halides (units are 1019 T2J–1).
Scalara Spin–orbitb 25% Hybrid–SOc 50% Hybrid–SOe Hartree–Fock SO Hybrid–SOd
aNon-hybrid DFT calculation, which includes scalar relativistic effects.bNon-hybrid DFT calculation, which includes scalar and spin–orbit relativistic effects.cHybrid DFT (PBE0 functional, 25% exchange) calculation, which includes scalar and spin–orbit relativistic effects.dPercentage contributions to Kiso for the hybrid SO (25% exchange) calculations. The DSO contribution is less than 1% in all cases.ePBE-based hybrid with 50% HF exchange. See Conclusions section for a discussion.
934 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
those obtained using DFT (Table 4). For Kiso, the HF resultsare consistently higher than the hybrid DFT results. For theanisotropies, DK, the HF results are systematically lowerthan the hybrid results for all molecules studied. Althoughnot necessarily expected, since the transition from PBE0 toHF entails the loss of all electron correlation contributions,this behaviour appears to represent mainly an extrapolationof the trend seen when going from PBE to PBE0.
According to Ramsey’s non-relativistic theory,26,110 thecoupling tensor may be considered to result from a sum ofcontributions from various coupling mechanisms: the Fermi-contact mechanism (FC), spin–dipolar mechanism (SD), dia-magnetic spin–orbital mechanism (DSO), and paramagneticspin–orbital mechanism (PSO). Within the two-componentZORA formalism, the formal separation between thesemechanisms may be kept the same. Therefore, couplingmechanisms analogous to Ramsey’s theory have been for-mulated, and we report the ZORA PSO and combinedZORA FC + SD terms, which contribute to Kiso, in Table 4.In addition, there are cross terms of the type PSO � (FC +SD).113 The contributions listed in Table 4 incorporate thesespin–orbit cross terms. For reasons of efficiency of the pro-gram implementation, the FC and SD terms are usually notcomputed separately. Since both terms arise from the sameelectron spin – nuclear spin coupling mechanism, the divi-sion is somewhat arbitrary to begin with. We note that anactual ‘‘contact’’ term is absent in the more general relativis-tic picture, but it is formally possible to separate the spin-mechanism operator into a generalized FC and SDterm.38,114 In most cases, the couplings of the alkali halidesare dominated by a positive FC + SD contribution, with anon-negligible negative PSO contribution. For LiF and KF,the breakdown of contributing mechanisms is in agreementwith previous multiconfigurational self-consistent field(MCSCF) calculations.21 It is reassuring that the presentDFT results yield an interpretation of the spin–spin couplingmechanism that is similar to a correlated-wave-function-based ab initio theory. We note in passing that the domi-nance of the electron-spin mechanism (FC + SD) is not al-ways assured. For instance, non-hybrid ZORA DFT resultspreviously reported for diatomic interhalogen compoundsshowed that in these systems, the coupling is more stronglydominated by the PSO mechanism.22 Table 4 also includescases where the PSO mechanism is as large or even largerin magnitude as the electron-spin mechanism (FC + SD).
Alkali metal halides: comparison of calculated resultswith experimental data
We have already emphasized that spin–spin coupling ten-sor results from molecular-beam experiments are excellentfor comparison with calculated values because the experi-mental data are for essentially isolated molecules in the gasphase, and therefore, intermolecular effects are negligible.One of the issues to be addressed in comparing experimentaland calculated spin–spin coupling tensor data is that of rovi-brational averaging. The relationship between the value of amolecular property at the equilibrium geometry for a dia-tomic molecule, P(eq), and the value of the same propertyfor a particular rotational–vibrational state, P(v,J), is to alowest-order anharmonic expansion of the potential-energy
surface and a quadratic expansion of the property (P) sur-face around the equilibrium geometry given by
½7� Pðv;JÞ ¼ PðeqÞ þ ðvþ 1=2Þ Be
ue
� �� @2P
@x2
� �x¼0
� 3a@P
@x
� �x¼0
" #
þ 4ðJ2 þ JÞ Be
ue
� �2@P
@x
� �x¼0
Here, x is the reduced displacement, (r – re)/re; Be and ueare spectroscopic constants specific to the molecule understudy; a is the cubic force constant, equal to�1� ðaeue=6B
2eÞ;
v is the vibrational quantum number, and J is the rotationalquantum number. Derivatives of the coupling constants
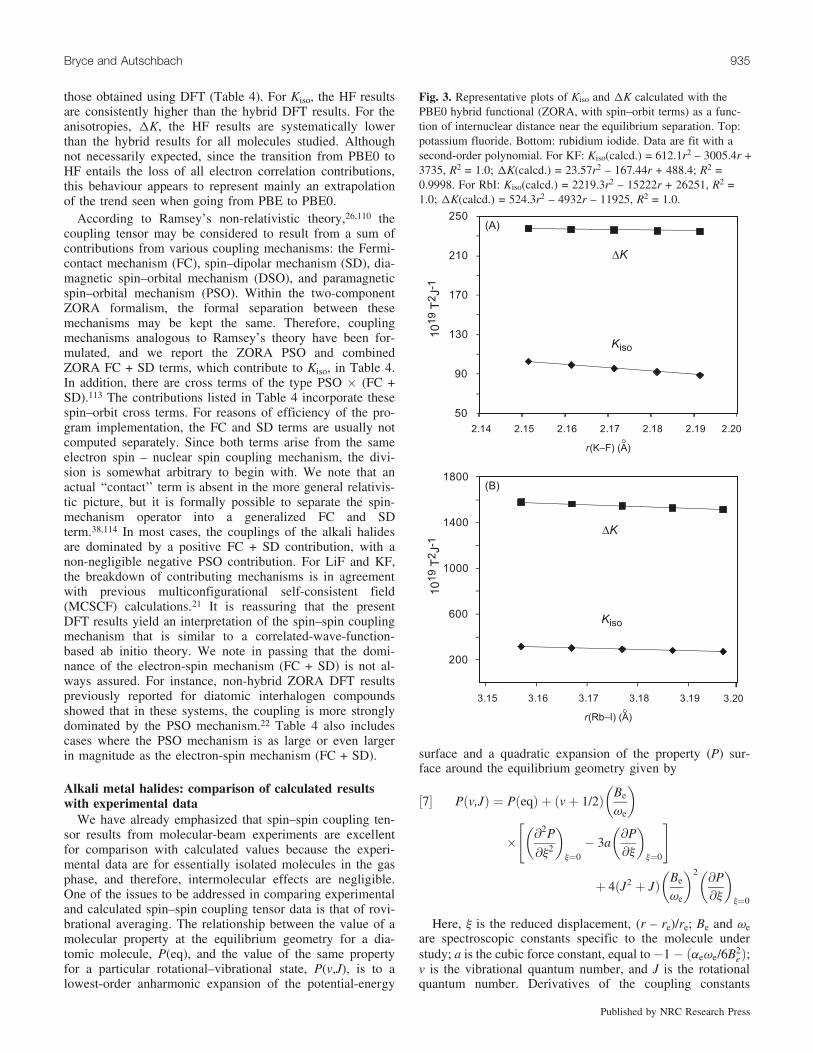
Fig. 3. Representative plots of Kiso and DK calculated with thePBE0 hybrid functional (ZORA, with spin–orbit terms) as a func-tion of internuclear distance near the equilibrium separation. Top:potassium fluoride. Bottom: rubidium iodide. Data are fit with asecond-order polynomial. For KF: Kiso(calcd.) = 612.1r2 – 3005.4r +3735, R2 = 1.0; DK(calcd.) = 23.57r2 – 167.44r + 488.4; R2 =0.9998. For RbI: Kiso(calcd.) = 2219.3r2 – 15222r + 26251, R2 =1.0; DK(calcd.) = 524.3r2 – 4932r – 11925, R2 = 1.0.
Bryce and Autschbach 935
Published by NRC Research Press
were estimated by carrying out hybrid DFT calculationswith the bond length set to re ± 0.01, ± 0.02 A, and fittingthe results to quadratic functions. Examples of the depend-ence of the calculated reduced isotropic and anisotropiccoupling constants on bond length are presented in Fig. 3.First and second derivatives of the reduced isotropic cou-pling constants are summarized in Table 5.
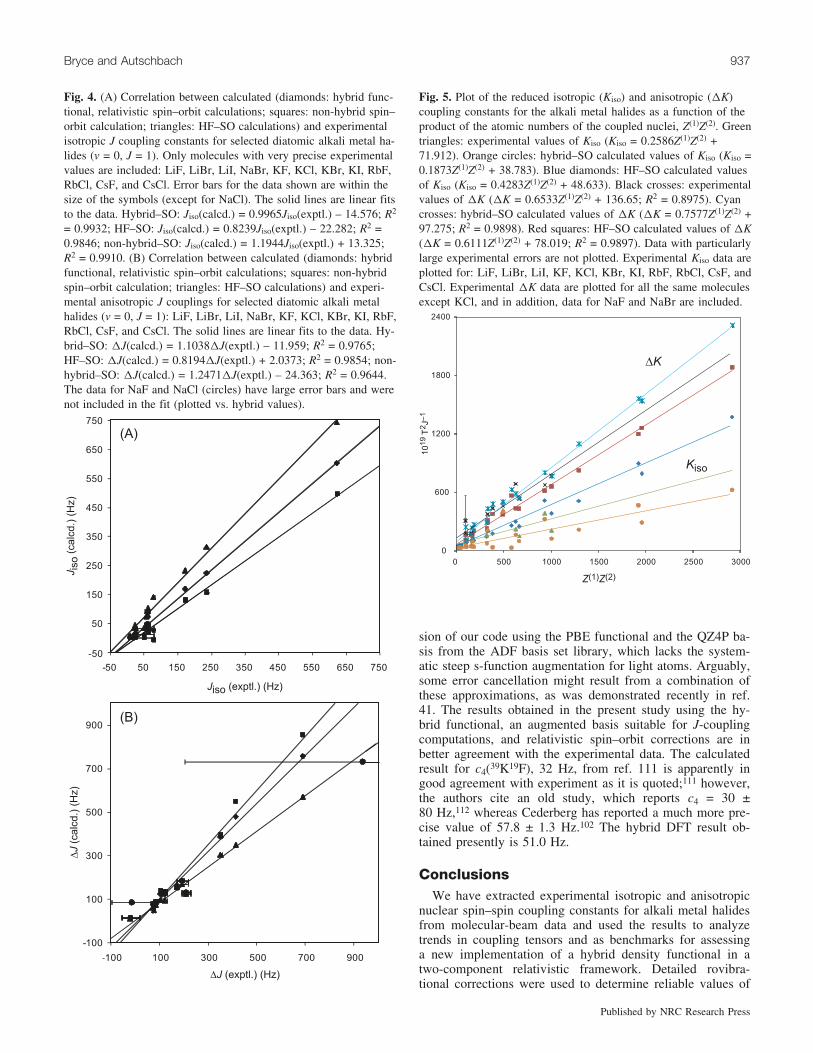
The hybrid DFT calculated values of Jiso were rovibra-tionally averaged according to eq. [6] and plotted againstthe experimental values (Table 2 and Fig. 4). The averagingresults in changes of at most 3.8% (for NaI), but in the ma-jority of cases, the change in Jiso is in the order of 1%. Theplot shows an excellent correlation with the experimentaldata (R2 = 0.9932) and a slope of unity (i.e., Jiso(calcd.) =0.9965Jiso(exptl.) – 14.576). The HF–SO (SO = spin–orbit)results underestimate the experimental data. If we consideragain the data for the potassium halides (Fig. 2), it is seenthat the best agreement with experiment across the wholeseries is achieved with the hybrid–SO calculations. TheHF–SO results follow approximately the correct trend; how-ever, the results for KBr and KI in particular are overesti-mated.
The hybrid DFT calculations also reproduce the couplinganisotropies very well, as demonstrated in Fig. 4B for allavailable experimental data, and for the subset of the potas-sium halides in Fig. 2B. Considering all the molecules forwhich precise data are available, the fit obtained is describedby DJ (calcd.) = 1.1038DJ(exptl.) – 11.959; R2 = 0.9765.The fact that the correct trend observed for Kiso using theHF–SO results is fortuitous is emphasized here, where it isseen that this method does poorly at reproducing the cou-pling anisotropies compared with the hybrid DFT results.
Shown in Fig. 5 is a plot of experimental and calculatedvalues of Kiso and DK as a function of the product of theatomic numbers of the coupled nuclei. Such correlations
have been demonstrated previously, and are expected on thebasis that all of the non-relativistic coupling mechanisms ex-cept FC depend on the expectation value of the electron–nu-clear distance, hr�3i. In the present case, the couplings aredominated by the FC + SD term, and this is likely one ofthe reasons for the increased scatter in the plot. Another rea-son, which has been discussed previously, is the somewhatanomalous value of hr�3i for fluorine relative to the otherhalogens.22,41 Although there is more scatter in the correla-tion, even for calculated values, than has been observed pre-viously for other series of diatomics,21,22 the overall trend ofincreasing Kiso and DK roughly with Z(1)Z(2) is clear. This isparticularly true for DK, where a stronger correlation is ob-served compared with Kiso, along with somewhat less scatter.
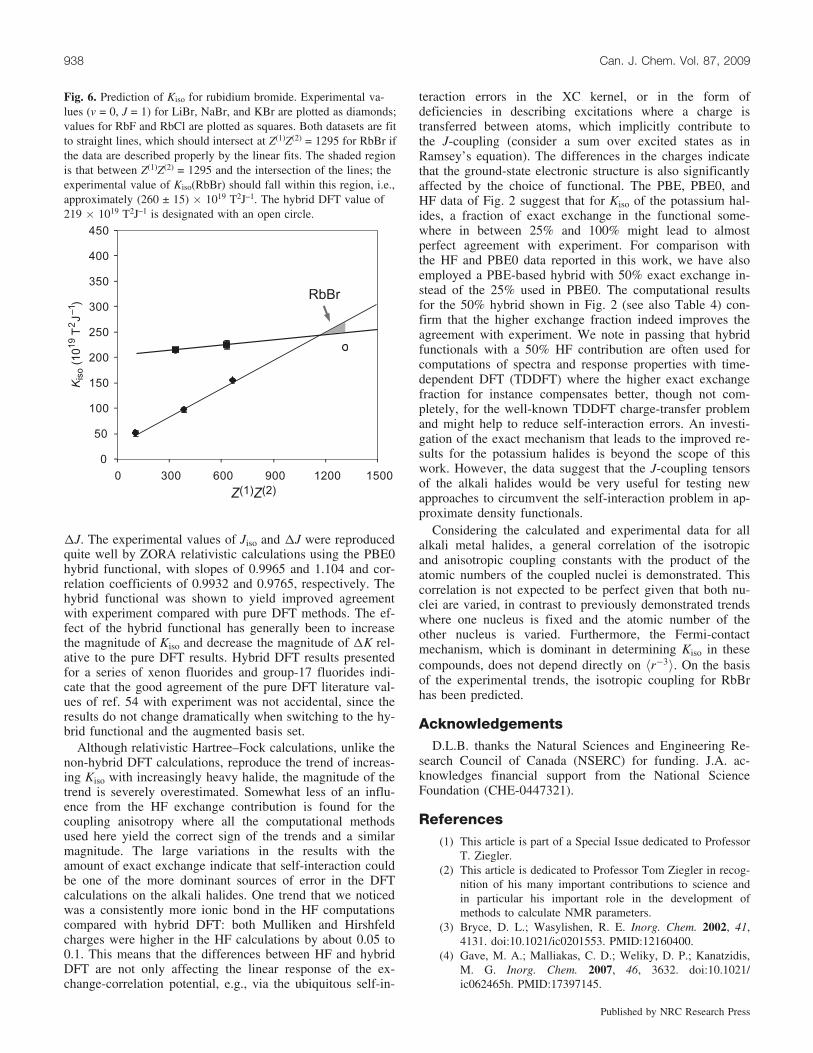
On the basis of the available data, some estimates may bemade of the coupling parameters for alkali metal halides,which are lacking experimental data. For example, shown inFig. 6 is a plot of the available values of Kiso for the alkalimetal bromides (LiBr, NaBr, and KBr) as a function ofZ(1)Z(2), and also the available values for the rubidium hal-ides (RbF and RbCl). Linear extrapolation of both datasetsshould converge to give the coupling constant for RbBr. Ifthe data are ideal and the linear fits properly describe thedata, the two lines should also converge to the correct valueof Z(1)Z(2) for RbBr, i.e., 1295. As shown, the result is quitegood and allows us to estimate the experimental value ofKiso(RbBr) as (260 ± 15) � 1019 T2J–1, or c4(85Rb79Br) =76.1 ± 4.4 Hz for the ground rotational–vibrational state.
Finally, we mention briefly the results of Aldegunde etal., who recently reported DFT calculations of spin–spincoupling parameters for 39K19F, 85Rb19F, 85Rb35Cl, and133Cs19F.111 They obtained some agreement with experimentfor c3 and c4 for these molecules. However, rovibrationalcorrections were not performed in their study, and the J-cou-pling computations were performed with a non-hybrid ver-
Table 5. Calculated derivatives and averaged values of the isotropic reduced spin–spin coupling constants for the alkali metal halides.
Note: Hybrid DFT calculations, which include scalar and spin–orbit relativistic effects.aEquilibrium bond lengths.
936 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
sion of our code using the PBE functional and the QZ4P ba-sis from the ADF basis set library, which lacks the system-atic steep s-function augmentation for light atoms. Arguably,some error cancellation might result from a combination ofthese approximations, as was demonstrated recently in ref.41. The results obtained in the present study using the hy-brid functional, an augmented basis suitable for J-couplingcomputations, and relativistic spin–orbit corrections are inbetter agreement with the experimental data. The calculatedresult for c4(39K19F), 32 Hz, from ref. 111 is apparently ingood agreement with experiment as it is quoted;111 however,the authors cite an old study, which reports c4 = 30 ±80 Hz,112 whereas Cederberg has reported a much more pre-cise value of 57.8 ± 1.3 Hz.102 The hybrid DFT result ob-tained presently is 51.0 Hz.
ConclusionsWe have extracted experimental isotropic and anisotropic
nuclear spin–spin coupling constants for alkali metal halidesfrom molecular-beam data and used the results to analyzetrends in coupling tensors and as benchmarks for assessinga new implementation of a hybrid density functional in atwo-component relativistic framework. Detailed rovibra-tional corrections were used to determine reliable values of
Fig. 4. (A) Correlation between calculated (diamonds: hybrid func-tional, relativistic spin–orbit calculations; squares: non-hybrid spin–orbit calculation; triangles: HF–SO calculations) and experimentalisotropic J coupling constants for selected diatomic alkali metal ha-lides (v = 0, J = 1). Only molecules with very precise experimentalvalues are included: LiF, LiBr, LiI, NaBr, KF, KCl, KBr, KI, RbF,RbCl, CsF, and CsCl. Error bars for the data shown are within thesize of the symbols (except for NaCl). The solid lines are linear fitsto the data. Hybrid–SO: Jiso(calcd.) = 0.9965Jiso(exptl.) – 14.576; R2
= 0.9932; HF–SO: Jiso(calcd.) = 0.8239Jiso(exptl.) – 22.282; R2 =0.9846; non-hybrid–SO: Jiso(calcd.) = 1.1944Jiso(exptl.) + 13.325;R2 = 0.9910. (B) Correlation between calculated (diamonds: hybridfunctional, relativistic spin–orbit calculations; squares: non-hybridspin–orbit calculation; triangles: HF–SO calculations) and experi-mental anisotropic J couplings for selected diatomic alkali metalhalides (v = 0, J = 1): LiF, LiBr, LiI, NaBr, KF, KCl, KBr, KI, RbF,RbCl, CsF, and CsCl. The solid lines are linear fits to the data. Hy-brid–SO: DJ(calcd.) = 1.1038DJ(exptl.) – 11.959; R2 = 0.9765;HF–SO: DJ(calcd.) = 0.8194DJ(exptl.) + 2.0373; R2 = 0.9854; non-hybrid–SO: DJ(calcd.) = 1.2471DJ(exptl.) – 24.363; R2 = 0.9644.The data for NaF and NaCl (circles) have large error bars and werenot included in the fit (plotted vs. hybrid values).
Fig. 5. Plot of the reduced isotropic (Kiso) and anisotropic (DK)coupling constants for the alkali metal halides as a function of theproduct of the atomic numbers of the coupled nuclei, Z(1)Z(2). Greentriangles: experimental values of Kiso (Kiso = 0.2586Z(1)Z(2) +71.912). Orange circles: hybrid–SO calculated values of Kiso (Kiso =0.1873Z(1)Z(2) + 38.783). Blue diamonds: HF–SO calculated valuesof Kiso (Kiso = 0.4283Z(1)Z(2) + 48.633). Black crosses: experimentalvalues of DK (DK = 0.6533Z(1)Z(2) + 136.65; R2 = 0.8975). Cyancrosses: hybrid–SO calculated values of DK (DK = 0.7577Z(1)Z(2) +97.275; R2 = 0.9898). Red squares: HF–SO calculated values of DK(DK = 0.6111Z(1)Z(2) + 78.019; R2 = 0.9897). Data with particularlylarge experimental errors are not plotted. Experimental Kiso data areplotted for: LiF, LiBr, LiI, KF, KCl, KBr, KI, RbF, RbCl, CsF, andCsCl. Experimental DK data are plotted for all the same moleculesexcept KCl, and in addition, data for NaF and NaBr are included.
Bryce and Autschbach 937
Published by NRC Research Press
DJ. The experimental values of Jiso and DJ were reproducedquite well by ZORA relativistic calculations using the PBE0hybrid functional, with slopes of 0.9965 and 1.104 and cor-relation coefficients of 0.9932 and 0.9765, respectively. Thehybrid functional was shown to yield improved agreementwith experiment compared with pure DFT methods. The ef-fect of the hybrid functional has generally been to increasethe magnitude of Kiso and decrease the magnitude of DK rel-ative to the pure DFT results. Hybrid DFT results presentedfor a series of xenon fluorides and group-17 fluorides indi-cate that the good agreement of the pure DFT literature val-ues of ref. 54 with experiment was not accidental, since theresults do not change dramatically when switching to the hy-brid functional and the augmented basis set.
Although relativistic Hartree–Fock calculations, unlike thenon-hybrid DFT calculations, reproduce the trend of increas-ing Kiso with increasingly heavy halide, the magnitude of thetrend is severely overestimated. Somewhat less of an influ-ence from the HF exchange contribution is found for thecoupling anisotropy where all the computational methodsused here yield the correct sign of the trends and a similarmagnitude. The large variations in the results with theamount of exact exchange indicate that self-interaction couldbe one of the more dominant sources of error in the DFTcalculations on the alkali halides. One trend that we noticedwas a consistently more ionic bond in the HF computationscompared with hybrid DFT: both Mulliken and Hirshfeldcharges were higher in the HF calculations by about 0.05 to0.1. This means that the differences between HF and hybridDFT are not only affecting the linear response of the ex-change-correlation potential, e.g., via the ubiquitous self-in-
teraction errors in the XC kernel, or in the form ofdeficiencies in describing excitations where a charge istransferred between atoms, which implicitly contribute tothe J-coupling (consider a sum over excited states as inRamsey’s equation). The differences in the charges indicatethat the ground-state electronic structure is also significantlyaffected by the choice of functional. The PBE, PBE0, andHF data of Fig. 2 suggest that for Kiso of the potassium hal-ides, a fraction of exact exchange in the functional some-where in between 25% and 100% might lead to almostperfect agreement with experiment. For comparison withthe HF and PBE0 data reported in this work, we have alsoemployed a PBE-based hybrid with 50% exact exchange in-stead of the 25% used in PBE0. The computational resultsfor the 50% hybrid shown in Fig. 2 (see also Table 4) con-firm that the higher exchange fraction indeed improves theagreement with experiment. We note in passing that hybridfunctionals with a 50% HF contribution are often used forcomputations of spectra and response properties with time-dependent DFT (TDDFT) where the higher exact exchangefraction for instance compensates better, though not com-pletely, for the well-known TDDFT charge-transfer problemand might help to reduce self-interaction errors. An investi-gation of the exact mechanism that leads to the improved re-sults for the potassium halides is beyond the scope of thiswork. However, the data suggest that the J-coupling tensorsof the alkali halides would be very useful for testing newapproaches to circumvent the self-interaction problem in ap-proximate density functionals.
Considering the calculated and experimental data for allalkali metal halides, a general correlation of the isotropicand anisotropic coupling constants with the product of theatomic numbers of the coupled nuclei is demonstrated. Thiscorrelation is not expected to be perfect given that both nu-clei are varied, in contrast to previously demonstrated trendswhere one nucleus is fixed and the atomic number of theother nucleus is varied. Furthermore, the Fermi-contactmechanism, which is dominant in determining Kiso in thesecompounds, does not depend directly on hr�3i. On the basisof the experimental trends, the isotropic coupling for RbBrhas been predicted.
AcknowledgementsD.L.B. thanks the Natural Sciences and Engineering Re-
search Council of Canada (NSERC) for funding. J.A. ac-knowledges financial support from the National ScienceFoundation (CHE-0447321).
References(1) This article is part of a Special Issue dedicated to Professor
T. Ziegler.(2) This article is dedicated to Professor Tom Ziegler in recog-
nition of his many important contributions to science andin particular his important role in the development ofmethods to calculate NMR parameters.
(3) Bryce, D. L.; Wasylishen, R. E. Inorg. Chem. 2002, 41,4131. doi:10.1021/ic0201553. PMID:12160400.
(4) Gave, M. A.; Malliakas, C. D.; Weliky, D. P.; Kanatzidis,M. G. Inorg. Chem. 2007, 46, 3632. doi:10.1021/ic062465h. PMID:17397145.
Fig. 6. Prediction of Kiso for rubidium bromide. Experimental va-lues (v = 0, J = 1) for LiBr, NaBr, and KBr are plotted as diamonds;values for RbF and RbCl are plotted as squares. Both datasets are fitto straight lines, which should intersect at Z(1)Z(2) = 1295 for RbBr ifthe data are described properly by the linear fits. The shaded regionis that between Z(1)Z(2) = 1295 and the intersection of the lines; theexperimental value of Kiso(RbBr) should fall within this region, i.e.,approximately (260 ± 15) � 1019 T2J–1. The hybrid DFT value of219 � 1019 T2J–1 is designated with an open circle.
938 Can. J. Chem. Vol. 87, 2009
Published by NRC Research Press
(5) Thomas, W. A. Prog. Nucl. Magn. Reson. Spectrosc. 1997,30, 183. doi:10.1016/S0079-6565(96)01033-3.
(6) Gee, M.; Wasylishen, R. E.; Ragogna, P. J.; Burford, N.;McDonald, R. Can. J. Chem. 2002, 80, 1488. doi:10.1139/v02-178.
(7) Foucault, H. M.; Bryce, D. L.; Fogg, D. E. Inorg. Chem.2006, 45, 10293. doi:10.1021/ic061021i. PMID:17140238.
(8) Tomaselli, M.; deGraw, D.; Yarger, J. L.; Augustine, M.P.; Pines, A. Phys. Rev. B 1998, 58, 8627. doi:10.1103/PhysRevB.58.8627.
(9) Martineau, C.; Fayon, F.; Legein, C.; Buzare, J. Y.; Silly,G.; Massiot, D. Chem. Commun. 2007, 2720. doi:10.1039/b703321d. PMID:17594032.
(11) Chou, J. J.; Case, D. A.; Bax, A. J. Am. Chem. Soc. 2003,125, 8959. doi:10.1021/ja029972s. PMID:12862493.
(12) Bystrov, V. F. Prog. Nucl. Magn. Reson. Spectrosc. 1976,10, 41. doi:10.1016/0079-6565(76)80001-5.
(13) Vuister, G. W.; Wang, A. C.; Bax, A. J. Am. Chem. Soc.1993, 115, 5334. doi:10.1021/ja00065a071.
(14) Vuister, G. W.; Delaglio, F.; Bax, A. J. Biomol. NMR1993, 3, 67. PMID:8448436.
(15) Wasylishen, R. E. In Encyclopedia of nuclear magnetic re-sonance; Grant, D. M., Harris, R. K., Eds.; John Wiley &Sons: Chichester, 1996; pp. 1685–1695.
(18) Prestegard, J. H.; Mayer, K. L.; Valafar, H.; Benison, G.C. Methods Enzymol. 2005, 394, 175. doi:10.1016/S0076-6879(05)94007-X. PMID:15808221.
(19) Bryce, D. L.; Wasylishen, R. E. J. Biomol. NMR 2004, 28,273. doi:10.1023/B:JNMR.0000013701.16162.0c. PMID:14752260.
(20) Vaara, J.; Jokisaari, J.; Wasylishen, R. E.; Bryce, D. L.Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41, 233.doi:10.1016/S0079-6565(02)00050-X.
(21) Bryce, D. L.; Wasylishen, R. E. J. Am. Chem. Soc. 2000,122, 3197. doi:10.1021/ja9942134.
(22) Bryce, D. L.; Wasylishen, R. E.; Autschbach, J.; Ziegler,T. J. Am. Chem. Soc. 2002, 124, 4894. doi:10.1021/ja012596b. PMID:11971740.
(23) Autschbach, J.; Ziegler, T. In Calculation of NMR andEPR parameters: Theory and applications; Kaupp, M.,Buehl, M., Malkin, V. G., Eds.; Wiley-VCH: Weinheim.2004.
(24) Townes, C. H.; Schawlow, A. L. Microwave spectroscopy;McGraw-Hill: New York. 1955.
(25) Winnewisser, G. Vib. Spectrosc. 1995, 8, 241. doi:10.1016/0924-2031(94)00053-J.
(26) Ramsey, N. F. Molecular Beams 1956, 162, 208.(27) Bryce, D. L.; Wasylishen, R. E. Acc. Chem. Res. 2003, 36,
(36) Khandogin, J.; Ziegler, T. J. Phys. Chem. A 2000, 104,113. doi:10.1021/jp992571n.
(37) Autschbach, J.; Ziegler, T. J. Chem. Phys. 2000, 113, 936.doi:10.1063/1.481874.
(38) Autschbach, J.; Ziegler, T. J. Chem. Phys. 2000, 113,9410. doi:10.1063/1.1321310.
(39) Filatov, M.; Cremer, D. J. Chem. Phys. 2004, 120, 11407.doi:10.1063/1.1752876. PMID:15268175.
(40) Melo, J. I.; Ruiz de Azua, M. C.; Peralta, J. E.; Scuseria,G. E. J. Chem. Phys. 2005, 123, 204112. doi:10.1063/1.2133730. PMID:16351245.
(41) Autschbach, J. J. Chem. Phys. 2008, 129, 094105. doi:10.1063/1.2969100. PMID:19044863.
(42) Autschbach, J. J. Chem. Phys. 2007, 127, 124106. doi:10.1063/1.2768363. PMID:17902892.
(43) Autschbach, J.; Le Guennic, B. J. Chem. Educ. 2007, 84,156.
(44) Le Guennic, B.; Matsumoto, K.; Autschbach, J. Magn. Re-son. Chem. 2004, 42, S99. doi:10.1002/mrc.1450. PMID:15366046.
(45) Boeckh, R. v.; Graff, G.; Ley, R. Z. Physik 1964, 179, 285.doi:10.1007/BF01381648.
(46) Hammerle, R. H.; Dickinson, J. T.; VanAusdal, R. G.; Ste-phenson, D. A.; Zorn, J. C. J. Chem. Phys. 1969, 50, 2086.doi:10.1063/1.1671337.
(47) Stephenson, D. A.; Dickinson, J. T.; Zorn, J. C. J. Chem.Phys. 1970, 53, 1529. doi:10.1063/1.1674207.
(48) Dickinson, J. T.; Stephenson, D. A.; Zorn, J. C. J. Chem.Phys. 1970, 53, 1525. doi:10.1063/1.1674206.
(49) Fabricant, B.; Muenter, J. S. J. Chem. Phys. 1977, 66,5274. doi:10.1063/1.433908.
(50) Muller, H. S. P.; Gerry, M. C. L. J. Chem. Phys. 1995,103, 577. doi:10.1063/1.470092.
(51) Wallerand, J.-P.; du Burck, F.; Mercier, B.; Goncharov, A.N.; Himbert, M.; Borde, C. J. Eur. Phys. J. D 1999, 6, 63.
(52) Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158.doi:10.1063/1.478522.
(53) Widdifield, C. M.; Chapman, R. P.; Bryce, D. L. Annu.Rep. Nucl. Magn. Reson. Spectrosc. 2009, in press.
(54) Bryce, D. L.; Wasylishen, R. E. Inorg. Chem. 2002, 41,3091. doi:10.1021/ic020025u. PMID:12054987.
(55) te Velde, G.; Bickelhaupt, F. M.; van Gisbergen, S. J. A.;Fonseca Guerra, C.; Baerends, E. J.; Snijders, J. G.; Ziegler,T. J. Comput. Chem. 2001, 22, 931. doi:10.1002/jcc.1056.
(56) Fonseca Guerra, C.; Snijders, J. G.; te Velde, G.; Baerends,E. J. Theor. Chem. Acc. 1998, 99, 391. doi:10.1007/s002140050021.
(57) Baerends, E. J.; Autschbach, J.; Berces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P. M.; Cavallo, L.; Chong, D. P.;Deng, L.; Dickson, R. M.; Ellis, D. E.; van Faassen, M.;Fan, L.; Fischer, T. H.; Fonseca Guerra, C.; van Gisbergen,S. J. A.; Gotz, A. W.; Groeneveld, J. A.; Gritsenko, O. V.;Gruning, M.; Harris, F. E.; van den Hoek, P.; Jacob, C. R.;Jacobsen, H.; Jensen, L.; van Kessel, G.; Kootstra, F.; Kry-
Bryce and Autschbach 939
Published by NRC Research Press
kunov, M. V.; van Lenthe, E.; McCormack, D. A.; Micha-lak, A.; Neugebauer, J.; Nicu, V. P.; Osinga, V. P.; Patch-kovskii, S.; Philipsen, P. H. T.; Post, D.; Pye, C. C.;Ravenek, W.; Rodriguez, J. I.; Ros, P.; Schipper, P. R. T.;Schreckenbach, G.; Snijders, J. G.; Sola, M.; Swart, M.;Swerhone, D.; te Velde, G.; Vernooijs, P.; Versluis, L.;Visscher, L.; Visser, O.; Wang, F.; Wesolowski, T. A.;van Wezenbeek, E. M.; Wiesenekker, G.; Wolff, S. K.;Woo, T. K.; Yakovlev, A. L.; Ziegler, T. ADF2008.01,SCM, Theoretical Chemistry. Vrije Universiteit, Amster-dam, The Netherlands. Available from http://www.scm.com. 2008.
(59) Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58,1200.
(60) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett.1996, 77, 3865. doi:10.1103/PhysRevLett.77.3865. PMID:10062328.
(61) Le Guennic, B.; Patchkovskii, S.; Autschbach, J. J. Chem.Theor. Comput. 2005, 1, 601. doi:10.1021/ct050042j.
(62) Autschbach, J.; Ziegler, T. In Encyclopedia of nuclearmagnetic resonance; Grant, D. M., Harris, R. K., Eds.;John Wiley & Sons: Chichester, 2002; Vol. 9, pp. 306–323.
(63) Autschbach, J.; Zheng, S. Annu. Rep. Nucl. Magn. Reson.Spectrosc. 2009, in press.
(64) Harris, R. K.; Becker, E. D.; Cabral de Menezes, S. M.;Goodfellow, R.; Granger, P. Pure Appl. Chem. 2001, 73,1795. doi:10.1351/pac200173111795.
(65) Bougon, R.; Cicha, W. V.; Lance, M.; Meublat, L.; Nier-lich, M.; Vigner, J. Inorg. Chem. 1991, 30, 102. doi:10.1021/ic00001a019.
(66) Smith, D. F. J. Chem. Phys. 1953, 21, 609. doi:10.1063/1.1698976.
(67) Haubrich, S. T.; Roehrig, M. A.; Kukolich, S. G. J. Chem.Phys. 1990, 93, 121. doi:10.1063/1.459611.
(68) Christe, K. O.; Zhang, X.; Sheehy, J. A.; Bau, R. J. Am.Chem. Soc. 2001, 123, 6338. doi:10.1021/ja003347a.PMID:11427058.
(69) Edwards, A. J.; Taylor, P. J. Chem. Soc., Dalton Trans.1975, 2174. doi:10.1039/dt9750002174.
(70) Lehmann, J. F.; Schrobilgen, G. J.; Christe, K. O.; Kor-nath, A.; Suontamo, R. J. Inorg. Chem. 2004, 43, 6905.doi:10.1021/ic040015o. PMID:15500329.
(71) Magnuson, D. W. J. Chem. Phys. 1957, 27, 223. doi:10.1063/1.1743675.
(72) Mahjoub, A. R.; Hoser, A.; Fuchs, J.; Seppelt, K. Angew.Chem. Int. Ed. Engl. 1989, 28, 1526. doi:10.1002/anie.198915261.
(73) Brassington, N. J.; Edwards, H. G. M.; Long, D. A. J.Chem. Soc., Faraday Trans. II 1978, 74, 1208. doi:10.1039/f29787401208.
(74) Boldrini, P.; Gillespie, R. J.; Ireland, P. R.; Schrobilgen, G.J. Inorg. Chem. 1974, 13, 1690. doi:10.1021/ic50137a030.
(75) Christe, K. O.; Curtis, E. C.; Dixon, D. A.; Mercier, H. P.;Sanders, J. C. P.; Schrobilgen, G. J. J. Am. Chem. Soc.1991, 113, 3351. doi:10.1021/ja00009a021.
(76) Leary, K.; Templeton, D. H.; Zalkin, A.; Bartlett, N. Inorg.Chem. 1973, 12, 1726. doi:10.1021/ic50126a004.
(77) Christe, K. O.; Dixon, D. A.; Sanders, J. C. P.; Schrobil-gen, G. J.; Wilson, W. W. J. Am. Chem. Soc. 1993, 115,9461. doi:10.1021/ja00074a011.
(78) Huis, T. J. v.; Galbraith, J. M.; Schaefer, H. F., III. Mol.Phys. 1996, 89, 607. doi:10.1080/002689796173949.
(79) Huber, K. P.; Herzberg, G. In Constants of diatomic mole-cules (data prepared by J.W. Gallagher and R.D. Johnson,III); Linstrom, P. J., Mallard, W. G., Eds.; NIST ChemistryWebBook, NIST Standard Reference Database Number 69,National Institute of Standards and Technology: Gaithers-burg MD. Available from http://webbook.nist.gov (re-trieved November 24, 2008).
(80) Malkin, V. G.; Malkina, O. L.; Salahub, D. R. Chem. Phys.Lett. 1993, 204, 87. doi:10.1016/0009-2614(93)85609-R.
(81) Autschbach, J.; Seth, M.; Ziegler, T. J. Chem. Phys. 2007,126, 174103. doi:10.1063/1.2735301. PMID:17492853.
(82) Bagno, A.; Saielli, G. Chem. Eur. J. 2003, 9, 1486. doi:10.1002/chem.200390168.
(83) Alexandre, M.; Rigny, P. Can. J. Chem. 1974, 52, 3676.doi:10.1139/v74-549.
(84) Christe, K. O.; Hon, J. F.; Pilipovich, D. Inorg. Chem.1973, 12, 84. doi:10.1021/ic50119a022.
(85) Gillespie, R. J.; Schrobilgen, G. J. Inorg. Chem. 1974, 13,1230. doi:10.1021/ic50135a043.
(86) Mahjoub, A. R.; Zhang, X.; Seppelt, K. Chem. Eur. J.1995, 1, 261. doi:10.1002/chem.19950010410.
(87) Keller, N.; Schrobilgen, G. J. Inorg. Chem. 1981, 20, 2118.doi:10.1021/ic50221a036.
(88) Holloway, J. H.; Schrobilgen, G. J.; Granger, P.; Brevard,C. C. R. Acad. Sci. Paris Ser. C 1976, 282, 519.
(89) Birchall, T.; Myers, R. D.; de Waard, H.; Schrobilgen, G.J. Inorg. Chem. 1982, 21, 1068. doi:10.1021/ic00133a039.
(90) Gillespie, R. J.; Netzer, A.; Schrobilgen, G. J. Inorg.Chem. 1974, 13, 1455. doi:10.1021/ic50136a040.
(91) Schrobilgen, G. J.; Holloway, J. H.; Granger, P.; Brevard,C. Inorg. Chem. 1978, 17, 980. doi:10.1021/ic50182a037.
(92) Gillespie, R. J.; Schrobilgen, G. J. Inorg. Chem. 1974, 13,2370. doi:10.1021/ic50140a015.
(93) Gerken, M.; Schrobilgen, G. J. Coord. Chem. Rev. 2000,197, 335. doi:10.1016/S0010-8545(99)00188-5.
(94) Gillespie, R. J.; Schrobilgen, G. J. Inorg. Chem. 1974, 13,765. doi:10.1021/ic50134a001.
(95) Cederberg, J.; Ward, J.; McAlister, G.; Hilk, G.; Beall, E.;Olson, D. J. Chem. Phys. 1999, 111, 8396. doi:10.1063/1.480213.
(96) Cederberg, J.; Olson, D.; Soulen, P.; Urberg, K.; Ton, H.;Steinbach, T.; Mock, B.; Jarausch, K.; Haertel, P.; Bresna-han, M. J. Mol. Spectrosc. 1992, 154, 43. doi:10.1016/0022-2852(92)90027-L.
(97) Hilborn, R. C.; Gallagher, J. T. F.; Ramsey, N. F. J. Chem.Phys. 1972, 56, 855. doi:10.1063/1.1677242.
(98) Cederberg, J.; Olson, D.; Nelson, A.; Laine, D.; Zimmer,P.; Welge, M.; Feig, M.; Hoft, T.; London, N. J. Chem.Phys. 1999, 110, 2431. doi:10.1063/1.477972.
(99) Hollowell, C. D.; Hebert, A. J.; Street, K., Jr. J. Chem.Phys. 1964, 41, 3540. doi:10.1063/1.1725764.
(100) De Leeuw, F. H.; van Wachem, R.; Dymanus, A. J. Chem.Phys. 1970, 53, 981. doi:10.1063/1.1674166.
(101) Cederberg, J.; Nitz, D.; Kolan, A.; Rasmusson, T.; Hoff-man, K.; Tufte, S. J. Mol. Spectrosc. 1987, 122, 171.doi:10.1016/0022-2852(87)90227-X.
Bongard, M.; Randolph, J.; Nitz, D. J. Chem. Phys. 2006,124, 244304. doi:10.1063/1.2212414. PMID:16821974.
(105) Cederberg, J.; Fortman, S.; Porter, B.; Etten, M.; Feig, M.;Bongard, M.; Langer, L. J. Chem. Phys. 2006, 124,244305. doi:10.1063/1.2212413. PMID:16821975.
(106) Cederberg, J. J. Chem. Phys. 1977, 66, 5247. doi:10.1063/1.433756.
(107) Jameson, C. J.; Gutowsky, H. S. J. Chem. Phys. 1969, 51,2790. doi:10.1063/1.1672415.
(108) Noorizadeh, S.; Nazari, F. J. Mol. Struct. 2004, 673, 99.(109) Pyykko, P.; Wiesenfeld, L. Mol. Phys. 1981, 43, 557.
doi:10.1080/00268978100101511.(110) Ramsey, N. F. Phys. Rev. 1953, 91, 303. doi:10.1103/
PhysRev.91.303.(111) Aldegunde, J.; Rivington, B. A.; Zuchowski, P. S.; Hutson,
J. M. Phys. Rev. A 2008, 78, 033434. doi:10.1103/PhysRevA.78.033434.
(112) Bonczyk, P. A.; Hughes, V. W. Phys. Rev. 1967, 161, 15.doi:10.1103/PhysRev.161.15.
(113) These cross terms are also derived at the non-relativisticlevel, but the contributions from alpha and beta orbitals tothese terms cancel identically. Therefore, the cross termsdo not need to be evaluated in computations that excludespin–orbit coupling.
(114) They can be computed separately in separate runs of theprogram, which has the same computational expense as se-parating the terms in the implementation. We decided notto perform this separation and focus on the influence ofelectron spin versus electron–orbital terms instead.





















![M. Billaud-Friess ,A.Nouyand O. Zahm€¦ · canonical tensors, Tucker tensors, Tensor Train tensors [27,40], Hierarchical Tucker tensors [25] or more general tree-based Hierarchical](https://static.documents.pub/doc/80x56/606a2ea8ed4bc80bc83876de/m-billaud-friess-anouyand-o-zahm-canonical-tensors-tucker-tensors-tensor-train.jpg)




