Review Release factors and their role as decoding proteins: speci¢city and ¢delity for termination of protein synthesis Elizabeth Poole, Warren Tate * Department of Biochemistry and the Centre for Gene Research, University of Otago, P.O. Box 56, Dunedin, New Zealand Received 21 March 2000; received in revised form 13 June 2000; accepted 13 June 2000 Abstract The decoding of stop signals in mRNA requires protein release factors. Two classes of factor are found in both prokaryotes and eukaryotes, a decoding factor and a stimulatory recycling factor. These factors form complexes at the active centre of the ribosome and mimic in overall shape the complexes found at other stages of protein synthesis. The decoding release factor is shaped like a tRNA and has a domain for codon recognition at the decoding site of the ribosome, and a domain for peptidyl-tRNA hydrolysis that is inferred to be near the peptidyltransferase centre. Initial interaction of the decoding factor with the ribosome is a low fidelity event involving multiple contacts with the ribosomal components. A subsequent discrimination step, at present poorly defined, ensures high fidelity of codon recognition. ß 2000 Elsevier Science B.V. All rights reserved. Keywords : Termination ; Protein synthesis ; Release factor ; Molecular mimicry ; Decoding 1. Introduction Decoding of signals in mRNA takes place on the ribo- some and, collectively, many resident and visiting proteins and RNA molecules are directly or indirectly important to the process. For many years now, detailed studies on in- dividual components have accumulated a wealth of knowl- edge that is like pieces of a jigsaw in understanding the overall decoding processes. There have been two ‘key’ breakthroughs that helped to put the jigsaw together. The ¢rst was largely conceptual and was the gradual rec- ognition that the RNAs of the ribosome, rather than the many proteins, were the fundamental molecules critical for function and that the protoribosome was probably simply an RNA molecule [1]. This followed from the idea of an ‘RNA World’, a concept stimulated by the discovery that RNA could catalyse reactions as well as store information [2,3]. From that time in the mid 1980s, there has been a signi¢cant focus on rRNAs and their importance in trans- lation. The fact that the code was contained in an RNA molecule and that the main decoding molecules of trans- lation were tRNAs functioning in initiation and elonga- tion, strongly reinforced this concept. However, the termi- nation mechanism remained an enigma because protein release factors (RFs) rather than RNAs seemed to be the direct participants in decoding and no tRNAs speci¢c for termination have been found. There was the strong possibility that a speci¢c decoding mechanism for this step of protein synthesis was a late ‘add-on’ and previ- ously an unassigned or genuine nonsense codon signalled the complex to ‘fall-o¡’ the ribosome as a default termi- nation mechanism. We now view this process somewhat di¡erently as a result of recent advances in the under- standing of ribosomal structure. 2. Structural mimicry among translational complexes Two technological ‘tours de force’ have provided the second ‘key’ breakthrough. The advent of cryoelectron microscopy giving high resolution structures from the lat- ter part of the 1990’s [4^6] and the very recent X-ray crystallography solutions of the small ribosomal subunit [7,8], large ribosomal subunit [9] and the whole ribosome [10], have brought us much closer to understanding pro- tein synthesis in atomic detail. Firstly, with both tech- niques, individual helices of the RNA and solution struc- tures of individual proteins are being ¢tted into the particle structure. One of the structural features that is now clear, is the active centre that has an inner ‘tRNA- 0167-4781 / 00 / $ ^ see front matter ß 2000 Elsevier Science B.V. All rights reserved. PII:S0167-4781(00)00162-7 * Corresponding author. Fax: +64-3-479-7866; E-mail : [email protected]Biochimica et Biophysica Acta 1493 (2000) 1^11 www.elsevier.com/locate/bba
Transcript
Review
Release factors and their role as decoding proteins: speci¢city and¢delity for termination of protein synthesis
Elizabeth Poole, Warren Tate *Department of Biochemistry and the Centre for Gene Research, University of Otago, P.O. Box 56, Dunedin, New Zealand
Received 21 March 2000; received in revised form 13 June 2000; accepted 13 June 2000
Abstract
The decoding of stop signals in mRNA requires protein release factors. Two classes of factor are found in both prokaryotes andeukaryotes, a decoding factor and a stimulatory recycling factor. These factors form complexes at the active centre of the ribosome andmimic in overall shape the complexes found at other stages of protein synthesis. The decoding release factor is shaped like a tRNA and has adomain for codon recognition at the decoding site of the ribosome, and a domain for peptidyl-tRNA hydrolysis that is inferred to be nearthe peptidyltransferase centre. Initial interaction of the decoding factor with the ribosome is a low fidelity event involving multiple contactswith the ribosomal components. A subsequent discrimination step, at present poorly defined, ensures high fidelity of codonrecognition. ß 2000 Elsevier Science B.V. All rights reserved.
Keywords: Termination; Protein synthesis ; Release factor; Molecular mimicry; Decoding
1. Introduction
Decoding of signals in mRNA takes place on the ribo-some and, collectively, many resident and visiting proteinsand RNA molecules are directly or indirectly important tothe process. For many years now, detailed studies on in-dividual components have accumulated a wealth of knowl-edge that is like pieces of a jigsaw in understanding theoverall decoding processes. There have been two `key'breakthroughs that helped to put the jigsaw together.The ¢rst was largely conceptual and was the gradual rec-ognition that the RNAs of the ribosome, rather than themany proteins, were the fundamental molecules critical forfunction and that the protoribosome was probably simplyan RNA molecule [1]. This followed from the idea of an`RNA World', a concept stimulated by the discovery thatRNA could catalyse reactions as well as store information[2,3]. From that time in the mid 1980s, there has been asigni¢cant focus on rRNAs and their importance in trans-lation. The fact that the code was contained in an RNAmolecule and that the main decoding molecules of trans-lation were tRNAs functioning in initiation and elonga-tion, strongly reinforced this concept. However, the termi-
nation mechanism remained an enigma because proteinrelease factors (RFs) rather than RNAs seemed to bethe direct participants in decoding and no tRNAs speci¢cfor termination have been found. There was the strongpossibility that a speci¢c decoding mechanism for thisstep of protein synthesis was a late `add-on' and previ-ously an unassigned or genuine nonsense codon signalledthe complex to `fall-o¡' the ribosome as a default termi-nation mechanism. We now view this process somewhatdi¡erently as a result of recent advances in the under-standing of ribosomal structure.
2. Structural mimicry among translational complexes
Two technological `tours de force' have provided thesecond `key' breakthrough. The advent of cryoelectronmicroscopy giving high resolution structures from the lat-ter part of the 1990's [4^6] and the very recent X-raycrystallography solutions of the small ribosomal subunit[7,8], large ribosomal subunit [9] and the whole ribosome[10], have brought us much closer to understanding pro-tein synthesis in atomic detail. Firstly, with both tech-niques, individual helices of the RNA and solution struc-tures of individual proteins are being ¢tted into theparticle structure. One of the structural features that isnow clear, is the active centre that has an inner `tRNA-
0167-4781 / 00 / $ ^ see front matter ß 2000 Elsevier Science B.V. All rights reserved.PII: S 0 1 6 7 - 4 7 8 1 ( 0 0 ) 0 0 1 6 2 - 7
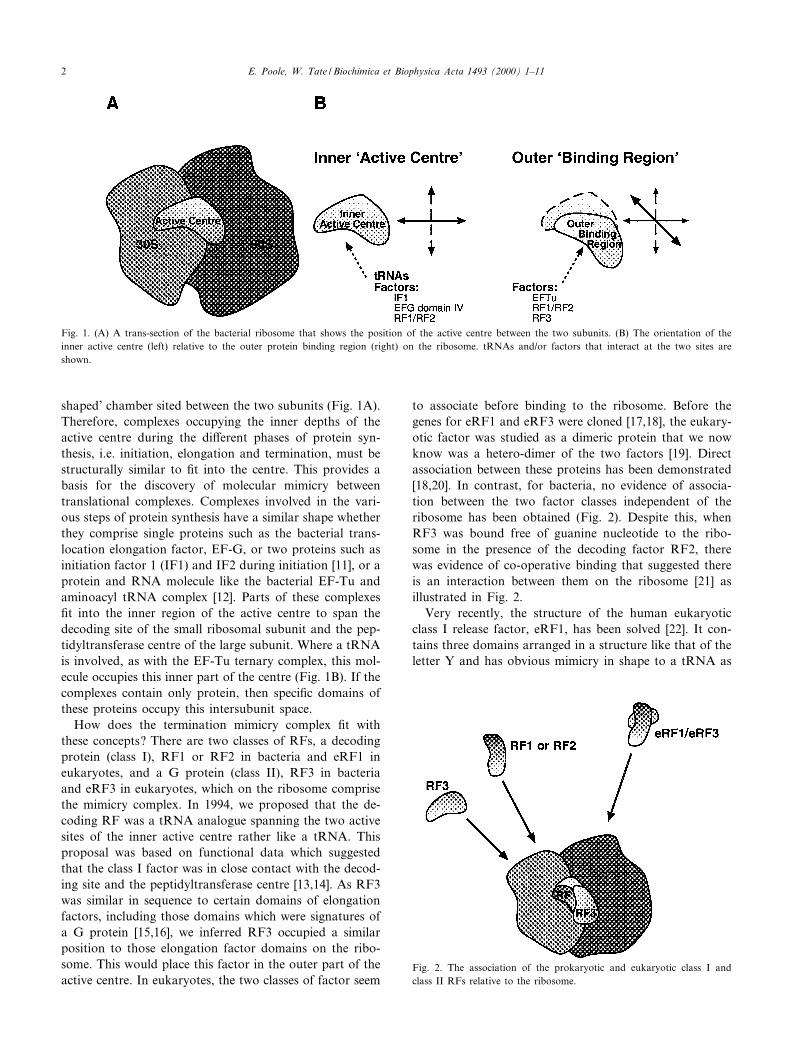
shaped' chamber sited between the two subunits (Fig. 1A).Therefore, complexes occupying the inner depths of theactive centre during the di¡erent phases of protein syn-thesis, i.e. initiation, elongation and termination, must bestructurally similar to ¢t into the centre. This provides abasis for the discovery of molecular mimicry betweentranslational complexes. Complexes involved in the vari-ous steps of protein synthesis have a similar shape whetherthey comprise single proteins such as the bacterial trans-location elongation factor, EF-G, or two proteins such asinitiation factor 1 (IF1) and IF2 during initiation [11], or aprotein and RNA molecule like the bacterial EF-Tu andaminoacyl tRNA complex [12]. Parts of these complexes¢t into the inner region of the active centre to span thedecoding site of the small ribosomal subunit and the pep-tidyltransferase centre of the large subunit. Where a tRNAis involved, as with the EF-Tu ternary complex, this mol-ecule occupies this inner part of the centre (Fig. 1B). If thecomplexes contain only protein, then speci¢c domains ofthese proteins occupy this intersubunit space.
How does the termination mimicry complex ¢t withthese concepts? There are two classes of RFs, a decodingprotein (class I), RF1 or RF2 in bacteria and eRF1 ineukaryotes, and a G protein (class II), RF3 in bacteriaand eRF3 in eukaryotes, which on the ribosome comprisethe mimicry complex. In 1994, we proposed that the de-coding RF was a tRNA analogue spanning the two activesites of the inner active centre rather like a tRNA. Thisproposal was based on functional data which suggestedthat the class I factor was in close contact with the decod-ing site and the peptidyltransferase centre [13,14]. As RF3was similar in sequence to certain domains of elongationfactors, including those domains which were signatures ofa G protein [15,16], we inferred RF3 occupied a similarposition to those elongation factor domains on the ribo-some. This would place this factor in the outer part of theactive centre. In eukaryotes, the two classes of factor seem
to associate before binding to the ribosome. Before thegenes for eRF1 and eRF3 were cloned [17,18], the eukary-otic factor was studied as a dimeric protein that we nowknow was a hetero-dimer of the two factors [19]. Directassociation between these proteins has been demonstrated[18,20]. In contrast, for bacteria, no evidence of associa-tion between the two factor classes independent of theribosome has been obtained (Fig. 2). Despite this, whenRF3 was bound free of guanine nucleotide to the ribo-some in the presence of the decoding factor RF2, therewas evidence of co-operative binding that suggested thereis an interaction between them on the ribosome [21] asillustrated in Fig. 2.
Very recently, the structure of the human eukaryoticclass I release factor, eRF1, has been solved [22]. It con-tains three domains arranged in a structure like that of theletter Y and has obvious mimicry in shape to a tRNA as
Fig. 1. (A) A trans-section of the bacterial ribosome that shows the position of the active centre between the two subunits. (B) The orientation of theinner active centre (left) relative to the outer protein binding region (right) on the ribosome. tRNAs and/or factors that interact at the two sites areshown.
Fig. 2. The association of the prokaryotic and eukaryotic class I andclass II RFs relative to the ribosome.
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^112
predicted in the original tRNA analogue model [14]. TheC-terminal part of the factor that interacts with the classII factor, eRF3, is found in the shorter arm of the Y(equivalent to the hinge region of the tRNA). The authorsexpect the prokaryotic class I RFs to adopt di¡erent pro-tein architectures because the pattern of K-helices and L-strands would be di¡erent to that of eRF1 according tothe secondary structure prediction [22].
The simplest picture of the termination complex wouldhave the functional domains of the class I factor for de-coding, peptidyl-tRNA hydrolysis and ribosome bindingin the inner active centre and a functional domain forinteraction with the G protein factor in the outer part ofthe centre. On the other hand, this G protein in bacteria,RF3, is equivalent to the G, GP and domain II of EF-G,and might be expected to occupy only the outer part of theactive centre (Fig. 3A). As illustrated in Fig. 3B (left im-age), the domains III, IV and V of EF-G mimic a tRNAin shape, and indeed occupy the same position in themimicry complex as does the tRNA of the ternary com-plex. However, a dot plot of RF3 compared with EF-Gshowed a small region of high homology between a C-terminal region of RF3 and domains III and IV of EF-G [23]. A threading analysis of the RF3 sequence against
EF-G gave the possible structure shown in Fig. 3B (rightimage). Not surprisingly, RF3 models well against the G,GP and domain II of EF-G, but, strikingly, also with partof EF-G domain IV, equivalent to the region of the struc-ture that mimics the anticodon stem of a tRNA. Thissuggests that the termination factor mimicry complexmay have a signi¢cant interaction between the decodingRF and the G protein near the decoding site. As RF3 isknown to destabilise the decoding factor and facilitate itsrecycling [24], this point of interaction might be highlysigni¢cant for this function. The functional signi¢canceof the C-terminal region of RF3 that shows homologywith domain IV of EF-G is the subject of current study.
3. Functional domains of the class I RFs
If the decoding RFs are true `mimics' of a tRNA, theymust interact with the decoding site of the small ribosomalsubunit and the peptidyltransferase centre of the large ri-bosomal subunit. In addition, it is likely that they makeother contacts with the ribosomal active centre. The di-mension of the eRF1 structure is such that it could spanthe decoding site and the peptidyltransferase centre like atRNA. Domain 1 is inferred to interact with the stopsignal in the decoding site and domain 2 with the pepti-dyltransferase centre. There are very few interactions be-tween domain 2 and the remainder of the molecule indi-cating a potential for conformational £exibility [22]. Therecently solved structure of the 70S ribosome complexedwith tRNA mini helices [10], indicates that while the P sitetRNA is tightly complexed with elements of the ribosomestructure, the A site tRNA apparently makes only weakcontacts with the ribosome density. This would allow thecodon^anticodon interactions to be exploited for discrim-ination of cognate or non-cognate interactions. In the caseof termination, it is a protein making these contacts, but,by analogy, the contacts between the factor and the ribo-some may be of relatively low a¤nity. Otherwise, therecould be di¤culty in the speci¢city of decoding duringtermination. Indeed, it has not been possible to measurestable RF binding to the ribosome in vitro or to formquantitative termination complexes without the additionof stabilisers such as ethanol or with altered ribosomes.For example, ribosomes lacking protein L11 form stablecomplexes with RF2 [25,26].
When the two classes of bacterial RFs or all bacterial-like RFs including mitochondrial factors are compared, asmall number of highly conserved regions are revealedwithin the linear structure (Fig. 4A). The most conservedand most C-terminal region has been assigned as the pep-tidyl-tRNA hydrolysis domain with putative interactionwith the peptidyltransferase centre. This assignment wasbased on the fact that at the boundary there is a hyper-sensitive proteolytic site that when cleaved, inactivatedpeptidyl-tRNA hydrolysis but still potentiated decoding
Fig. 3. (A) The interrelationship of RF1/2 and RF3 and their probablelocation with respect to the inner active centre and outer binding region.(B) A structure of RF3 (right) compared to the structure of EF-G (left).
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^11 3
[14]. The exchange of a ten-base region in this highly con-served domain between the two factors, RF1 and RF2,also resulted in the loss of peptidyl-tRNA hydrolysis ac-tivity, but not ribosome binding or codon recognition. In-deed, a single residue in Escherichia coli RF2, threonine246, is responsible for this loss of activity and restoring theRF1 residue within this small cassette of residues rein-states activity of the chimeric protein for peptidyl-tRNAhydrolysis [27].
A highly conserved GGQ (position 250^252) motif inthe factors could give a sharp `turn' in the structure.The retention of this small motif in the eukaryotic factoras one of the few points of homology between the pro-karyotic and eukaryotic factors [28], indicates the impor-tance of this region both for tRNA mimicry and for span-ning the ribosomal active centre. The GGQ motif in theeRF1 structure is located on a turn connecting an ex-tended region of KL-strand with the the start of an K-helixand forms the tip of domain 2 of the molecule. This is amini domain believed to be the equivalent of the amino-acyl group attached to the CCA of a tRNA [22]. Residuesof the GGQ motif have been shown to be essential forpeptidyl-tRNA hydrolysis, but not essential for stop signal
recognition or ribosome binding [28]. Moreover, both res-idue 244 in prokaryotic RF2 that when cleaved caused lossof peptidyltransferase activity [14] and residue 246 thatmodulated peptidyltransferase activity [27], are close tothe GGQ motif, suggesting a similar structure in the pro-karyotic family of factors at this site.
The natural threonine in RF2 at position 246 also con-fers a high susceptibility to functional inactivation on RF2expressed from a plasmid, particularly when it is expressedat high concentration in E. coli. This inactivation resultsneither from a loss or gain of a post translational modi¢-cation at the residue, nor from a gross change in thestructure of the protein as determined from proteolyticsensitivity [27]. Rather, the inactivation might resultfrom subtle changes in conformation that could be in-duced by inappropriate protein^protein interactions ifRF2 is present at a high concentration in the bacterialcell. The fact that E. coli RF2 has a unique translationalframeshift mechanism to control precisely its own synthe-sis and, therefore, its concentration in the cell, has beenpuzzling [29]. However, it may relate to the necessity tolimit RF2 concentration to avoid loss of a key functionsuch as that found with the plasmid expressed protein.
Fig. 4. (A) Amino acid positions (vertical black bars) that are identical between all currently available prokaryotic RF1s, RF2s and organellar RFs (allRFs) and IF1s. Probable functions of conserved domains between all RFs are shown in relation to structural domains [30]. (B) The possible location ofthe conserved functional domains within the RF.
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^114
What region(s) of the factors are responsible for decod-ing the translational stop signal? Examination of the se-quences in the RF1 and RF2 families reveals regions thatare factor type-speci¢c. These have been the subject ofmuch interest as codon recognition speci¢city is whatfunctionally di¡erentiates the two classes of factors. Ahighly type-speci¢c region lies within structural domainD, and to a lesser extent type-speci¢c sequences can beseen within structural domains B and F of the theoreticaldomain model shown in Fig. 4 that was developed byNakamura and colleagues after examining twenty twomembers of the RF1 and RF2 families [30]. The regionthat corresponds to domain D, has high homology to theC-terminal half of the small bacterial initiation factor IF1,and there is homology not only with the common sequen-ces within both classes of RF, but also with the sequencesunique to each class [23]. IF1, for which the exact functionhas remained elusive, binds to the small ribosomal subunitand makes exactly the same footprint on the rRNA at thedecoding site of the active centre as an A site-boundtRNA [31,32]. IF1 binds to an empty ribosomal A siteduring initiation of protein synthesis and perhaps preventsthe tRNA from entering the site prematurely or inap-propriately.
As the class I RFs also enter the A site to decode thetranslational stop signal, there was an implication that thestructural domain D region with high sequence homologyto IF1 was a good candidate for the `anticodon region'(previously called the tRNA anticodon mimicry region[30]) of the protein. Subsequently, a detailed structuraldomain exchange study has identi¢ed a small number ofresidues within domain D as the key determinants of co-don discrimination [33]. These residues may be the equiv-alent of the nucleotides of the anticodon that actuallyinteract with the translational stop signal in the mRNA.The RF1 family has a `prolineWalanineWthreonine' motif andthe RF2 family a `serineWprolineWphenylalanine' motif in theequivalent position. Exchanging these key residues be-tween RF1 and RF2 has been shown to change the spe-ci¢city of codon recognition, and the ¢rst and third resi-dues of the motif may recognise the second and thirdnucleotides of the stop codon where discrimination mustoccur. These residues (RF2 205^207) are in functional do-main 1A illustrated in Fig. 4B, toward the C-terminal endof the IF1 homology region. It is of particular interest thatthe eukaryotic class 1 factor that recognises all three stopcodons contains a motif RVNRLSV, similar to the highlyconserved prokaryotic RF1 motif RVQRVPA adjacent toand partly including the PAT tripeptide `anticodon' [33].The motif RVNRLSV may comprise the equivalent eu-karyotic factor anticodon site as it is located in the K3helix of domain 1 in the structure postulated to be thestop codon interaction site [22].
In structural domain C, another residue also in£uencesthe speci¢city of codon recognition. A selected RF2 mu-tant that suppressed a temperature sensitive phenotype of
a variant RF1 was able to recognise UAG as well asretaining UGA and UAA recognition properties, albeitsomewhat more weakly [34]. This RF2 mutant had achange at residue 167 in a region showing little homologybetween the two families of factors. E. coli RF1 and RF2have di¡erent residues at this position. Therefore, it islikely that there could have been a topological change inthe variant protein that modi¢ed its discrimination. In-deed, it was reported that other codons were also recog-nised.
Strong evidence that the RFs are in close contact withthe translational stop signal comes from site-directedcrosslinks formed from the ¢rst position (+1) of the stopcodon (where the +1 U was replaced by photoactive 4-thio-U) [13] and from the three positions downstream ofthe stop codon (+4 to +6) [35]. Beyond the +6 base in themRNA, no crosslinks were formed to the protein. Thecrosslink patterns were consistent with the discovery thatthe +4 base is a major determinant of the in vivo e¤ciencyof the stop signal [36], and that the +6 nucleotide can alsoin£uence stop signal strength in vivo [37]. This has led to aproposal that the translational stop signal could be de-scribed as a sequence element rather than a simple tripletcodon [38]. The technically di¤cult task of determiningwhich RF residue(s) form crosslinks with the +1 positionof the stop codon has been attempted using HPLC peptideanalysis and peptide antibodies targeted against key re-gions in the functional domains 1A, 1B and 2. As indi-cated in Fig. 5, these functional domains relate to struc-tural domains C, D and E, respectively. A crosslink wasidenti¢ed in structural domain D (containing functionaldomain 1A) near to or within the peptide `anticodon' re-gion described by Nakamura and colleagues [33]. A sec-ond crosslink was found within functional domain 1B.
4. Fidelity of polypeptide release
The RF can bind to the ribosome as a result of manyinteractions between parts of its structure and rRNA nu-cleotides and residues of the ribosomal proteins. To date,there has been no detailed analysis of the points of closecontact with rRNA, although a RF footprint has beenfound to cover nucleotides of the K-sarcin loop of thelarge subunit rRNA (C.M. Brown and W.P. Tate, unpub-lished data). In addition, some nucleotides in rRNA havebeen shown to in£uence termination [39]. On the otherhand, there are several studies that indicate that the L7/L12 and L11 ribosomal proteins are essential for higha¤nity factor binding [40,25]. These interactions are inaddition to those that determine codon speci¢city withthe mRNA positioned in the decoding site. Indeed, a ri-bosomal particle lacking one or both of the importantproteins is still able to bind RFs, but with much lowera¤nity, implying that multiple interactions occur. Thecritical question, given the multiple interactions of the
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^11 5
factor with the ribosome, is what prevents inappropriatepeptidyl-tRNA hydrolysis when this interaction is non-cognate, and, speci¢cally, when it is near-cognate (for ex-ample, when the codon has two of the three correct nu-cleotides as with the codon UGG)?
Site-directed crosslinks from the stop signal of designedRNAs has been used as a tool to detect functional bindingof the RF into the ribosomal A site [13]. A number ofexperiments have revealed that the ability to form cross-links is a sensitive indicator and is very dependent on theorientation of the mRNA to the RF. The crosslink e¤-ciency can be in£uenced by the upstream sequence in themRNA, the P site codon and the tRNA [41]. Tradition-ally, the best in vitro bu¡er to detect crosslinks has con-tained high salt and also ethanol to stabilise the complex.For studies to determine whether the discriminating codonalone could determine the formation of a functional ter-mination complex, a more physiological bu¡er was used(low salt, polyamines) [42]. However, the inclusion of etha-nol was still necessary for stabilisation. These experimentsshowed whether the factor was entering the ribosomalactive centre in the correct orientation with cognate ornear-cognate stop signals. A summary of these studies isshown in Table 1 with the variations in the +2 and +3base positions of the stop codon (I) and also the key +4base position (II). What was clear from these experiments,was that poor discrimination resulted when the codon inthe A site was near-cognate (the +1 U and one of the tworemaining nucleotides cognate). In these cases, site-di-rected crosslinks between the codon and RF still formed.For example, UCA supported a +1 crosslink with RF2,U(A/G)U formed crosslinks with both factors, and UGAformed a complex with RF1 and UAG with RF2 despiteUAG and UGA being discriminatory codons. The +4 basealso a¡ected the orientation of the factor within the com-plex. For example, UAGG allowed crosslinks to form
with RF2, but not RF1, and UAGU formed crosslinksto RF1 but not RF2.
The functional domains described in Fig. 4 are clusteredin the central to C-terminal region of the factors, with less
Fig. 5. The conserved amino acid positions (vertical black bars) in the prokaryotic RF2 family, indicating the structural domains [30], the position ofthe conserved functional domains between all RFs, and the amino acid locality of the antipeptides used to locate crosslinked residues. The position ofthe exposed cleavage site for chymotrypsin is shown.
Table 1The e¡ect of stop signal +2, +3 and +4 base identity on release factordiscrimination
Signal RF2 RF1
I. E¡ect of the +2 and +3 baseUNAG
A 4 1G 4 3C 3 0UNGG
A 4 0G 3 1C 0 0UNCG
A 0 0G 3 0UNUG
A 3 2G 5 2
II. E¡ect of the +4 baseUAAN
G 4 1U 0 2
UGAN
G 4 3U 5 4C 4 3
UAGN
G 4 0U 0 2
UGGN
G 3 1U 4 3
The results of di¡erent experiments represent a relative score of densito-metric measurements of the crosslink signals detected by autoradiogra-phy. 0, no signal; 1^5, very weak signalCvery strong signal.
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^116
homology toward the N-terminus. However, truncatedRF1 and RF2 proteins containing C-terminal deletionscan suppress nonsense mutations [43] and this has led tothe suggestion that the unassigned N-terminal homologyregion might be primarily responsible for ribosome bind-ing. This would explain why termination complexes areformed with non-cognate codons as an initial bindingstep. The proteins L7/L12 and L11 are at the entranceto the active centre and it has been shown by immuno-electron microscopy that the RF occupies a similar positionto that for elongation factor EF-Tu at this entrance [44].Certainly, factor interactions at the entrance to the activecentre, mediated through proteins L7/L12 and L11 and asyet unidenti¢ed rRNA nucleotides and structures, couldrepresent initial binding prior to codon recognition anddiscrimination. During elongation, non-cognate EF-TuWaminoacyl-tRNAWGTP ternary complexes that bind intothe active centre before delivering the aminoacyl-tRNA tothe A site must be discriminated against. An attractivehypothesis has been suggested that postulates an initialweak a¤nity of the site for the tRNA and correct co-don-anticodon interaction is necessary to trigger a confor-mational change in the K-sarcin region of the large subunitrRNA before other interactions of the complex with theribosome become signi¢cant [45]. Our site-directed cross-link studies have not yet provided evidence that a mecha-nism involving a ribosomal conformational change canexplain codon-dependent discrimination during termina-tion of protein synthesis at the level of factor^codon in-teraction. Such factor-codon interaction may co-ordinatethe discrimination of this recognition function togetherwith a productive hydrolyis event. Certainly, whilethe cognate stop signal can signi¢cantly enhance stableRF binding to the ribosome, the factor can still beobserved in the ribosomal complex in the absence of co-don when such complexes are analysed by SDS^PAGE[46].
These studies suggest that termination complexes canform on the ribosome with non-cognate or near-cognatecodons. Therefore, there must be another mechanism thatdetermines the ¢delity of the termination event to sensewhether correct codon discrimination has or has not oc-curred. Indeed, a near physiological in vitro translationsystem developed by Ehrenberg and colleagues [47] hasbeen used with di¡erent codons that suggested peptiderelease occurs with a high degree of ¢delity [48]. Usingthis system, the discrimination between cognate andnear-cognate codons was found to be between three andsix orders of magnitude, apparently without an energy-de-pendent proofreading mechanism. Two near-cognate co-dons, UGG and UAU were identi¢ed as potential siteswhere premature chain termination might occur. Both ofthese codons contained within mRNAs were found in oursite-directed crosslink assay to support relatively strongtermination complexes (RF1, UAU and RF2, UAU andUGG, see Table 1). In our studies, the base following the
stop codon has a signi¢cant in£uence on the strength ofthe crosslink with a particular codon.
What might be the likely mechanism that precludes thepremature release of the growing polypeptide as the ribo-some is processing along the mRNA?
5. Possible mechanisms for translation termination ¢delity
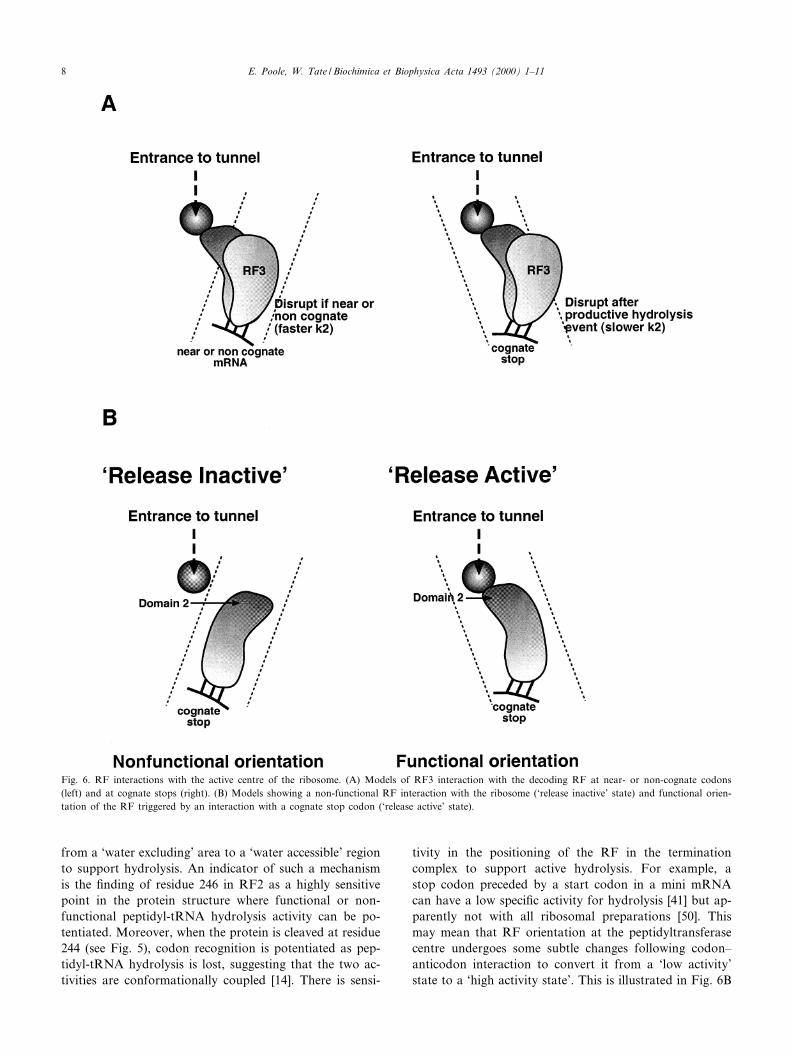
In reality, in the bacterial cell, the EF ternary complexesare of su¤cient concentration to compete e¡ectively andexclude RFs from the ribosomal active centre at non-cog-nate or near-cognate codons. However, this alone is un-likely to be su¤cient to account for the high degree of¢delity measured in vivo [49]. Also, it cannot explain thediscrimination between the stop signals themselves by thetwo bacterial factors, RF1 and RF2. There is more thanone feasible explanation that could account for the highdegree of ¢delity. One reason is that a functional termi-nation complex may form with either a cognate codon ora near-cognate codon and have the potential to release agrowing peptide. In this case, a kinetic mechanism mightact as the discriminator, mediating factor release beforeproductive peptidyl-tRNA hydrolysis. Such an event islikely to be functioning near the codon/factor recognitionsite. The intriguing indication that part of the structure ofthe dissociating factor, RF3, may interleave with the RFat the decoding site (Fig. 6A) might provide an importantclue. This is likely to correspond to the ribosomal sitewhere the key RF residues of structural domain D interactwith the stop codon. RF3 may be able to disrupt thisinteraction if it is not su¤ciently strong (for example, atnear-cognate codons) and cause dissociation of the decod-ing RF before a productive peptide hydrolysis event oc-curs. Although such a situation would be di¤cult to sim-ulate in vitro, site-directed crosslink experiments with thetwo factors have indicated that RF3 does disturb thecrosslink patterns in di¡erent ways with the two factorsand with di¡erent stop codons (E.S. Poole, unpublisheddata). Such a mechanism may also provide an explanationfor puzzling in vivo experiments with weak and strongcognate UAG-containing stop signals where high concen-trations of RF3 can either enhance the e¤ciency of termi-nation as expected, or, surprisingly, with some signals de-crease their e¤ciency (L.L. Major, unpublished data). Inthe latter cases, the decoding factor may be dissociated byRF3 before a productive hydrolysis event can occur atsome of the ribosomal passages through the signal.
Another possible mechanism would involve a conforma-tional change in the decoding RF following cognate co-don^anticodon recognition that might transmit a signalfrom the codon recognition domain of the factor (domains1A and B) to the part of the protein in contact with thepeptidyltransferase centre (domain 2 ^ see Fig. 6B). Thiscould involve some repositioning of the elements of thepeptidyltransferase centre that would convert the centre
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^11 7
from a `water excluding' area to a `water accessible' regionto support hydrolysis. An indicator of such a mechanismis the ¢nding of residue 246 in RF2 as a highly sensitivepoint in the protein structure where functional or non-functional peptidyl-tRNA hydrolysis activity can be po-tentiated. Moreover, when the protein is cleaved at residue244 (see Fig. 5), codon recognition is potentiated as pep-tidyl-tRNA hydrolysis is lost, suggesting that the two ac-tivities are conformationally coupled [14]. There is sensi-
tivity in the positioning of the RF in the terminationcomplex to support active hydrolysis. For example, astop codon preceded by a start codon in a mini mRNAcan have a low speci¢c activity for hydrolysis [41] but ap-parently not with all ribosomal preparations [50]. Thismay mean that RF orientation at the peptidyltransferasecentre undergoes some subtle changes following codon^anticodon interaction to convert it from a `low activity'state to a `high activity state'. This is illustrated in Fig. 6B
Fig. 6. RF interactions with the active centre of the ribosome. (A) Models of RF3 interaction with the decoding RF at near- or non-cognate codons(left) and at cognate stops (right). (B) Models showing a non-functional RF interaction with the ribosome (`release inactive' state) and functional orien-tation of the RF triggered by an interaction with a cognate stop codon (`release active' state).
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^118
as a bending of domain 2 of the factor towards the en-trance of the exit tunnel of the large ribosomal subunitwhere the terminal amino acid will be positioned awaitingcleavage from the tRNA. In this model, dissociation of thedecoding RF by RF3 may not be accelerated, but stilloccur before a productive hydrolysis event. Murgola andcolleagues [51,52] have provided genetic evidence that aninteraction between the GTPase centre of domain II of thelarge ribosomal subunit rRNA, and the entrance to do-main V (the peptidyltransferase ring) occurs during termi-nation and that may be part of an activation process sup-porting hydrolysis. They speculate that the RF maymediate this interaction. Failure to switch the peptidyl-transferase centre from peptide bond forming mode tohydrolysis mode may be the consequence of non- ornear-cognate interaction between codons and RF.
RF-mediated termination can be summarised in a pos-sible kinetic scheme as shown in Fig. 7. An initial bindingstep may occur with cognate or non-cognate codons (A).If the interaction is non-cognate, then the rate of thisassociation (k1) may be su¤ciently slow to be competedout in most instances by the rate of association of theelongation factor ternary complex (k1P). Where a func-tional recognition event has occurred, either with cognate,near-cognate or non-cognate codons, two kinds of com-plex are possible; a functionally active complex potentiallycompetent in peptide release (B), or a non-competent com-plex for release (C). In the former case, an active dissoci-ation mechanism (mechanism I), perhaps involving RF3,
may allow non-cognate complexes to be distinguishedfrom cognate complexes. This rate of dissociation (k2P)could be high when the mRNA a¤nity with the proteinis low (no bases, or only one or two bases recognised outof three). As shown for mechanism II, if there is a non-competent release complex formed with cognate or non-cognate codons, then this must be activated, perhaps by asubtle conformational change in the factor a¡ecting theribosomal peptidyltransferase centre, to reach a competentstate (k4P). If this activation does not occur, as would bethe case with non-cognate codon recognition, then disso-ciation of the complex would follow (k2). This means thatthere are several potential places where ¢delity could bedetermined. Fidelity could involve codon recognitioncoupled with dissociation of the complex, or codon recog-nition coupled with a conformational change in either orboth the factor and the ribosomal active centre itself. Atpresent, there is some evidence supporting both mecha-nisms I and II although further studies will be requiredto determine which of these best describes termination¢delity during protein synthesis.
Acknowledgements
The work of our own laboratory described here hasbeen supported by the Marsden Fund of New Zealand,the Health Research Council of New Zealand, the HumanFrontier Science Program (Grants RG 416/93 awarded to
Fig. 7. A kinetic scheme of the RF interaction with the ribosome. (A) The initial binding step. (B) A functionally active complex potentially competentin peptide release. (C) A non-competent complex unable to release a product with a conformational change in the RF.
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^11 9
Y. Nakamura and WPT and RG32/97 awarded to Y. Na-kamura, L. Kisselev, M. Philippe and W.P.T.) and a How-ard Hughes International Investigator award to W.P.T.
References
[1] P.B. Moore, The ribosome returns, Nature 331 (1988) 223^227.[2] K. Kruger, P.J. Grabowski, A.J. Zaug, J. Sands, D.E. Gottschling,
T.R. Cech, Self-splicing RNA: autoexcision and autocyclization ofthe ribosomal RNA intervening sequence of Tetrahymena, Cell 31(1982) 147^157.
[3] C. Guerrier-Takada, K. Gardiner, T. Marsh, N. Pace, S. Altman,The RNA moiety of ribonuclease P is the catalytic subunit of theenzyme, Cell 35 (1983) 849^857.
[4] H. Stark, E.V. Orlova, J. Rinke-Appel, N. Junke, F. Mueller, M.Rodnina, W. Wintermeyer, R. Brimacombe, M. van Heel, Arrange-ment of tRNAs in pre- and posttranslocational ribosomes revealed byelectron cryomicroscopy, Cell 88 (1997) 19^28.
[5] R.K. Agrawal, P. Penczek, R.A. Grassucci, J. Frank, Vizualisation ofelongation factor G on the Escherichia coli 70S ribosome: the mech-anism of translocation, Proc. Natl. Acad. Sci. USA 95 (1998) 6134^6138.
[6] I.S. Gabashvili, R.K. Agrawal, C.M.T. Spahn, R.A. Grassucci, D.I.Svergun, J. Frank, P. Penczek, Solution structure of the E. coli 70Sribosome at 11.5 Aî resolution, Cell 100 (2000) 537^549.
[7] W.M. Clemons Jr., J.L.C. May, B.T. Wimberly, J.P. McCutcheon,M.S. Capel, V. Ramakrishnan, Structure of a bacterial 30S ribosomalsubunit at 5.5 Aî resolution, Nature 400 (1999) 833^840.
[8] A. Tocilj, F. Schlu«nzen, D. Janell, M. Glu«hmann, H.A.S. Hansen, J.Harms, A. Bashan, H. Bartels, I. Agmon, F. Franceschi, A. Yonath,The small ribosomal subunit from Thermus thermophilus at 4.5 Aî
resolution: pattern ¢ttings and the identi¢cation of a functionalsite, Proc. Natl. Acad. Sci. USA 96 (1999) 14252^14257.
[9] N. Ban, P. Nissen, J. Hanson, M. Cappel, P.B. Moore, T.A. Steitz,Placement of protein and RNA structures into a 5 Aî -resolution mapof the 50S ribosomal subunit, Nature 400 (1999) 841^847.
[11] S. Brock, K. Szkaradkiewicz, M. Sprinzl, Initiation factors of proteinbiosynthesis in bacteria and their structural relationship to elongationand termination factors, Mol. Microbiol. 29 (1998) 409^417.
[12] P. Nissen, M. Kjeldgaard, S. Thirup, G. Polekhina, L. Reshetnikova,B.F.C. Clark, J. Nyborg, Crystal structure of the ternary complex ofPhe-tRNAPhe, EF-Tu, and a GTP analog, Science 270 (1995) 1464^1472.
[13] C.M. Brown, W.P. Tate, Direct recognition of mRNA stop signals byEscherichia coli polypeptide chain release factor 2, J. Biol. Chem. 269(1994) 33164^33170.
[14] J.G. Mo¡at, W.P. Tate, A single proteolytic cleavage in release factor2 stabilizes ribosome binding and abolishes peptidyl-tRNA hydrolysisactivity, J. Biol. Chem. 269 (1994) 18899^18903.
[15] O. Mikuni, K. Ito, J. Mo¡at, K. Matsumura, K. McCaughan, T.Nobukuni, W. Tate, Y. Nakamura, Identi¢cation of the prfC gene,which encodes peptide-chain-release factor 3 of Escherichia coli, Proc.Natl. Acad. Sci. USA 91 (1994) 5798^5802.
[16] G. Grentzmann, D. Brechemier-Baey, V. Heugue-Hamard, R.H.Buckingham, Function of polypeptide chain release factor 3 in Es-cherichia coli, J. Biol. Chem. 270 (1995) 10595^10600.
[17] L. Frolova, X. Le Go¡, H.H. Rasmussen, S. Cheperegin, G. Dru-geon, M. Kress, I. Arman, A-L. Haenni, J.E. Cells, M. Philippe, J.Justesen, L. Kisselev, A highly conserved eukaryotic protein familypossessing properties of polypeptide chain release factor, Nature 372(1994) 701^703.
[18] G. Zhouravleva, L. Frolova, X. Le Go¡, R. Le Guellec, S. Inge-Vechtomov, L. Kisselev, M. Philippe, Termination of translation ineukaryotes is governed by two interacting polypeptide chain releasefactors, eRF1 and eRF3, EMBO J. 14 (1995) 4065^4072.
[20] I. Stans¢eld, K.M. Jones, V.V. Kushnirov, A.R. Dagkesamanskaya,A.I. Poznyakovski, S.V. Paushkin, C.R. Nierras, B.S. Cox, M.D.Ter-Avanesyan, M.F. Tuite, The products of SUP45 (eRF1) andSUP35 genes interact to mediate translation termination in Saccha-romyces cerevisiae, EMBO J. 14 (1995) 4365^4373.
[21] H.J. Pel, J.G. Mo¡at, K. Ito, Y. Nakamura, W.P. Tate, Escherichiacoli release factor 3: Resolving the paradox of a typical G proteinstructure and atypical function with guanine nucleotides, RNA 4(1998) 47^54.
[22] H. Song, P. Mugnier, A.K. Das, H.M. Webb, D.R. Evans, M.F.Tuite, B.A. Hemmings, D. Barford, The crystal structure of humaneukaryotic release factor eRF1-Mechanism of stop codon recognitionand peptidyl-tRNA hydrolysis, Cell 100 (2000) 311^321.
[23] D.N. Wilson, M.E. Dalphin, H.J. Pel, L.L. Major, J.B. Mansell,W.P. Tate, Factor-mediated termination of protein synthesis: awelcome return to the mainstream of translation, in: R.A. Garrett,S.R. Douthwaite, A. Liljas, A.T. Matheson, P.B. Moore, H.F.Noller (Eds.), The Ribosome: Structure, Function, Antibiotics, andCellular Interactions, ASM Press, Washington, DC, 2000, pp. 495^508.
[24] D.V. Freistro¡er, M.Yu. Pavlov, J. MacDougall, R.H. Buckingham,M. Ehrenberg, Release factor RF3 in E. coli accelerates the dissoci-ation of release factors RF1 and RF2 from the ribosome in a GTP-dependent manner, EMBO J. 16 (1997) 4126^4133.
[25] W.P. Tate, H. Schulze, K. Nierhaus, The Escherichia coli ribosomalprotein L11 supresses release factor 2 but promotes release factor 1activities in peptide chain termination, J. Biol. Chem. 258 (1983)12816^12820.
[26] W.P. Tate, M.J. Dognin, M. Noah, M. Sto«¥er-Meilicke, G. Sto«¥er,The NH2-terminal domain of Escherichia coli ribosomal protein L11,J. Biol. Chem. 259 (1984) 7317^7324.
[27] D.N. Wilson, D. Guevremont, W.P. Tate, Residue 246 links theribosome binding and peptidyl-tRNA hydrolysis functions of releasefactor 2, EMBO J., submitted.
[28] L.Yu. Frolova, R.Yu. Tsivkovskii, G.F. Sivolobova, N.Yu. Oparina,O.I. Serpinsky, V.M. Blinov, S.I. Tatkov, L.L. Kisselev, Mutations inthe highly conserved GGQ motif of class 1 polypeptide release factorsabolish the ability of human eRF1 to trigger peptidyl-tRNA hydro-lysis, RNA 5 (1999) 1014^1020.
[29] W.J. Craigen, R.G. Cook, W.P. Tate, C.T. Caskey, Bacterial peptidechain release factors: conserved primary structure and possibleframeshift regulation of relase factor 2, Proc. Natl. Acad. Sci. USA82 (1985) 3616^3620.
[30] K. Ito, K. Ibihara, M. Uno, Y. Nakamura, Conserved motifs inprokaryotic and eukaryotic polypeptide release factors: tRNA-pro-tein mimicry hypothesis, Proc. Natl. Acad. Sci. USA 93 (1996) 5443^5448.
[31] D. Moazed, R.R. Samaha, C. Gualerzi, H.F. Noller, Speci¢c protec-tion of 16S rRNA by translational initiation factors, J. Mol. Biol. 248(1995) 207^210.
[32] K. Dahlquist, J.D. Puglisi, Investigating the structure and function oftranslation initiation factor 1 in Escherichia coli, Nucleic Acids Symp.Ser. 33 (1995) 170^171.
[33] K. Ito, M. Uno, Y. Nakamura, A tripeptide `anticodon' deciphersstop codons in messenger RNA, Nature 403 (2000) 680^684.
[34] K. Ito, M. Uno, Y. Nakamura, Single amino acid substitution inprokaryote polypeptide release factor 2 permits it to terminate trans-lation at all three stop codons, Proc. Natl. Acad. Sci. USA 95 (1998)8165^8169.
[35] E.S. Poole, L.L. Major, S.A. Mannering, W.P. Tate, Translationaltermination in Escherichia coli : three bases following the stop codon
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^1110
crosslink to release factor 2 and a¡ect the decoding e¤ciency ofUGA-containing signals, Nucleic Acids Res. 26 (1998) 954^960.
[36] E.S. Poole, C.M. Brown, W.P. Tate, The identity of the base follow-ing the stop codon determines the e¤ciency of in vivo translationaltermination in Escherichia coli, EMBO J. 14 (1995) 151^158.
[37] L.L. Major, E.S. Poole, M.E. Dalphin, S.A. Mannering, W.P. Tate,Is the in-frame termination signal of the Escherichia coli release fac-tor-2 frameshift site weakened by a particularly poor context?, Nu-cleic Acids Res. 24 (1996) 2673^2678.
[38] W.P. Tate, E.S. Poole, M.E. Dalphin, L.L. Major, D.J.G. Crawford,S.A. Mannering, The translational stop signal : codon with a contextor extended factor recognition element?, Biochimie 78 (1996) 945^952.
[39] A.L. Arkov, D.V. Freistro¡er, M. Ehrenberg, E.J. Murgola, Muta-tions in RNAs of both ribosomal subunits cause defects in translationtermination, EMBO J. 17 (1998) 1507^1514.
[40] I.L. Armstrong, W.P. Tate, The requirement for the Escherichia coliribosomal proteins L7 and L12 in release factor-dependent peptidechain termination, FEBS Lett. 109 (1980) 228^232.
[41] K.K. McCaughan, E.S. Poole, H.J. Pel, J.B. Mansell, S.A. Manner-ing, W.P. Tate, E¤cient in vitro translational termination in Esche-richia coli is constrained by the orientations of the release factor, stopsignal and peptidyl-tRNA within the complex, Biol. Chem. 379(1998) 857^866.
[42] S. Schilling-Bartetzko, F. Franceschi, H. Sternbach, K.H. Nierhaus,Apparent association constants of tRNAs for the ribosomal A, P,and E sites, J. Biol. Chem. 267 (1992) 4693^4702.
[43] K. Yoshimura, K. Ito, Y. Nakamura, Amber (UAG) suppressors inUGA/UAA- speci¢c polypeptide release factor 2 of bacteria: geneticprediction of initial binding to ribosome preceding stop codon rec-ognition, Genes Cells 4 (1999) 253^266.
[44] B. Kastner, C.N.A. Trotman, W.P. Tate, Localization of the releasefactor-2 binding site on 70S ribosomes by immuno-electron micros-copy, J. Mol. Biol. 212 (1990) 241^245.
[45] K.H. Nierhaus, Solution of the ribosome riddle how the ribosomeselects the correct aminoacyl-tRNA out of 41 similar contestants,Mol. Microbiol. 9 (1993) 661^669.
[46] M.E. Askarian-Amiri, H.J. Pel, D. Guevremont, K.K. McCaughan,E.S. Poole, V.G. Sumpter, W.P. Tate, Functional characterization ofyeast mitochondrial release factor 1, J. Biol. Chem. 275 (2000) 17241^17248.
[47] M.Yu. Pavlov, M. Ehrenberg, Rate of translation of natural mRNAsin an optimised in vitro system, Arch. Biochem. Biophys. 328 (1996)9^16.
[48] D.V. Freistro¡er, M. Kwiatkowski, R.H. Buckingham, M. Ehren-berg, The accuracy of codon recognition by polypeptide release fac-tors, Proc. Natl. Acad. Sci. USA 97 (2000) 2046^2051.
[49] F. JÖrgensen, F.M. Adamski, W.P. Tate, C.G. Kurland, Releasefactor-dependent false stops are infrequent in Escherichia coli,J. Mol. Biol. 230 (1993) 41^50.
[50] G. Grentzmann, P.J. Kelly, Ribosomal binding site of release factorsRF1 and RF2, J. Biol. Chem. 272 (1997) 12300^12304.
[51] A.L. Arkov, E.J. Murgola, Ribosomal RNAs in translation termina-tion: facts and hypotheses, Biochemistry (Moscow) 64 (1999) 1354^1359.
[52] E.J. Murgola, A.L. Arkov, N.S. Chernyaeva, K.O.F. Hedenstierna,F.T. Pagel, rRNA functional sites and structures for peptide chaintermination, in: R.A. Garrett, S.R. Douthwaite, A. Liljas, A.T.Matheson, P.B. Moore, H.F. Noller (Eds.), The Ribosome: Struc-ture, Function, Antibiotics, and Cellular Interactions, ASM Press,Washington, DC, 2000, pp. 509^518.
BBAEXP 93432 25-8-00 Cyaan Magenta Geel Zwart
E. Poole, W. Tate / Biochimica et Biophysica Acta 1493 (2000) 1^11 11