27
SUMMARY OF SELECTED RESEARCH John M. Mihelcic, Ph.D. 2004 - 2010
| Date post: | 28-Jul-2015 |
| Category: |
Documents |
| Upload: | john-mihelcic |
| View: | 120 times |
| Download: | 1 times |

SUMMARY OF SELECTED RESEARCH
John M. Mihelcic, Ph.D.2004 - 2010

CONTENTS
Progress Toward Hsp90 InhibitorsSlides 3-15
Probing Interactions Between Hsp90
and the Estradiol Receptor Slides 16-20
Total Synthesis of Opiate Analgesics Slides 21-27

PROGRESS TOWARD HSP90 INHIBITORS
Conducted with Dr. Brian S. J. BlaggDepartment of Medicinal ChemistryUniversity of Kansas2008 - 2010

Hsp90 (90 kDa heat shock protein) is a molecular chaperone responsible for the conformational maturation of numerous client proteins.1 Recent studies have demonstrated that Hsp90 multi-protein complexes from tumor cells have higher affinity for ligands than Hsp90 in normal cells; making the Hsp90 multi-protein complex a viable target for cancer therapy.2
Geldanamycin (GDA) and radicicol (RDC) are known inhibitors of Hsp90 and manifest their activity by binding and preventing Hsp90-catalyzed hydrolysis of ATP.3 The IC50 values for GDA and RDC are 49 nM and 23 nM, respectively, when determined in MCF-7 breast cancer cell lines.4 RDC is rapidly metabolized and displays no activity in vivo and GDA displays toxicity issues that are unassociated with Hsp90 inhibition.5
Naturally-Occurring Inhibitors of Hsp90
While derivatives of GDA have entered clinical trials for the treatment of several cancers,6,7 the quinone ring is redox- active. In cells GDA has been shown to generate superoxide radicals which can lead to cell death without interfering directly with Hsp90.8 Consequently, researchers have pursued the development of new Hsp90 inhibitors without these detrimental properties.3

Co-crystal structures of the natural products revealed numerous interactions with the Hsp90 architecture.9 Analysis of the crystal structures revealed that, in addition to other key interactions: 1) the resorcinol moiety of RDC binds in the same location as the adenine ring of ADP and 2) the quinone ring of GDA binds toward the exterior of the pocket and participates in hydrogen bond interactions with the amino acids that normally bind to the diphosphate region of ADP. 10
Interactions with Key Residues of Hsp90
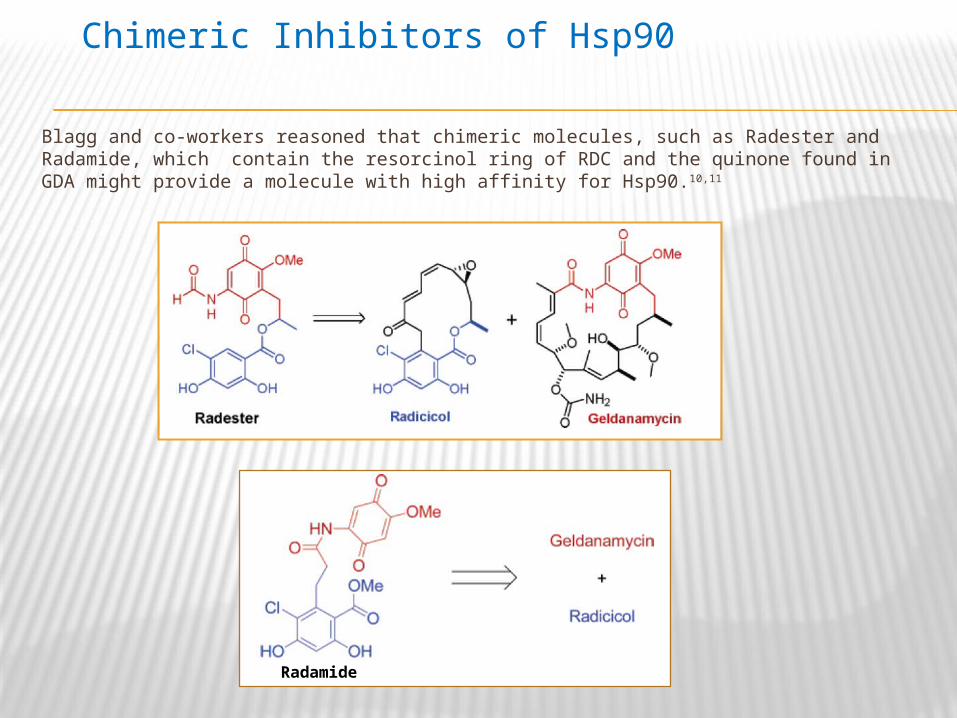
Blagg and co-workers reasoned that chimeric molecules, such as Radester and Radamide, which contain the resorcinol ring of RDC and the quinone found in GDA might provide a molecule with high affinity for Hsp90.10,11
Chimeric Inhibitors of Hsp90
Radamide

OHHO
Cl
O
NH
O
OMe
O
O
O
OHHO
Cl
HO
NH
OH
OMe
O
O
O
OH
HN
OH
OMe
CO2Me
OH
OH
Cl
OO
HN
O
OMe
CO2Me
OH
OH
Cl
O
Blagg and co-workers synthesized several chimera that displayed activities in the low mM range in anti-proliferation assays against breast cancer cells.10,11 An unexpected and highly promising result was the enhanced activity that resulted upon replacement of the redox-active quinone ring with a dihydroquinone (1 vs. 2 and 3 vs. 4). The dihydroquinone of radester displayed the highest activity among the four.
Activities of Initial Chimera and Derivatives
3: Radester1: Radamide 2
# IC50 (mM) MCF-71 18.6 +/- 0.912
2 14.0 +/- 1.412
3 13.9 +/- 1.411
4 7.1 +/- 0.311
4

NH
O
HO OH
ClO
RO
OR
X
O
R=H, Me
NH
O
HO OH
ClO
OR
X
O
NH
O
HO OH
ClO
RO
NH2
X
O
NH
O
HO OH
ClO
NH2
X
O
X=CN, Cl
R=H, Me
X=CN, Cl
Proposed Modifications
The dihydroquinone can be metabolized in vivo to the redox-active quinone which displays unwanted toxicity. We sought to remove the dihydroquinone motif through the removal of one oxygen atom or replacement with a nitrogen atom. The phenol was available by default and the aniline was chosen for its excellent hydrogen-bonding characteristics.
We sought to enhance the ability to bind with the Hsp90 architecture by introducing new substituents onto the dihydroquinone ring. Co-crystal structures indicated that replacing the 17-methoxy of GDA with small groups might be well tolerated.13 Chloride and nitrile groups were chosen for their size and hydrogen-bonding characteristics.
The necessity of both hydroxyl or alkoxyl groups of the dihydroquinone would also be investigated.

MeO
HO
NO2H2
Pd/C
MeOH
rt 12 h
MeI
K2CO3
DMF
rt 12 h
tBuC(O)Cl
DMAP pyr.
DCM
0°C 2 hMeO
MeO
NHPv
78% over 3 steps
a) nBuLi, THF
0°C 2 h
b)
THF 0°C to rt
O
85%
MeO
MeO
NHPv
OH
BCl3DCM
-40°C to rtMeO
HO
NHPv
OH80%
conc. HCl
1,4-dioxane
80°C 16 h
30%
a)NaNO2
HCl AcOH
H2O 0°C
b) CuCN
PhH 0°C
HO OH
Cl CO2H
DCC DMAP
THF DMF
50°C 15 hHO OH
ClO
MeO
HO
CN
O
6% over 2 steps
5
6
5
Synthesis of the New Analogs Compounds lacking the formamide were pursued first. A substantial amount of
experimentation was performed on an unsuccessful synthetic approach before establishing the robust route to pivalanalide 5 described below.
Mono-demethylation of 5 was achieved via the directing ability of the amide. Acidic hydrolysis of the amide was complicated by formation of a 7-membered ring by the
proximal alcohol and a low yield was obtained. Sandmeyer reaction to install the nitrile was low-yielding and the coupling reaction to
produced compound 6 suffered from the presence of the free phenol.

MeO
NHPv
OH
TFAA
NH4NO3
MeCN -10°C
35%
a) NaNO2 HCl
AcOH H2O 0°C
b) CuCN PhH 0°C
9%
H2 Pd/C
MeOH NH3
3 h
90%MeO
HO
NHPv
OH
Tf2NPh
Hunig's base
DMF
75%
HCl
1,4-dioxane
80°C 20 h
80%
33%
HO OH
Cl CO2H
DCC DMAP
THF DMF
50°C 15 h 35%
H2, Pd/C
MeOH 2 h
HO OH
ClO
MeO
NH2
CN
O
MeO
NH2
OH
NO2
MeO
NH2
OH
NO2
7
IC50 (mM) MCF-7
6 28.0 +/- 3.57 45.2 +/- 6.7
The free phenol was replaced with a nitro group in three steps. Yield for the hydrolysis was greatly enhanced in the case of the deactivated aromatic ring.
The sequence just described was used to provide a nitroester that was hydrogenated to give compound 7.
Activity of the new compounds was lower than compound 4 by 4-6 times. The observation that
aniline 7 was less active the phenol 6 suggested that the NH2 was not making significant
interactions with the Hsp90 architecture.
Synthesis and Activity of the New Analogs
HO OH
Cl CO2H
DCC DMAP
THF DMF
50°C 15 h
HO OH
ClO
MeO
OH
CN
O
Isolation of product from either sequence was unsuccessful.
O2N
MeO
HO
CN
OH
O2N
MeO
HO
NH2
OH
O2NHO OH
Cl CO2H
DCC DMAP
THF DMF
50°C
12% HO OH
ClO
MeO
OH
CN
O
O2Na) NaNO2 HCl
AcOH H2O 0°C
b) CuCN PhH 0°C
9%
ca. 25%
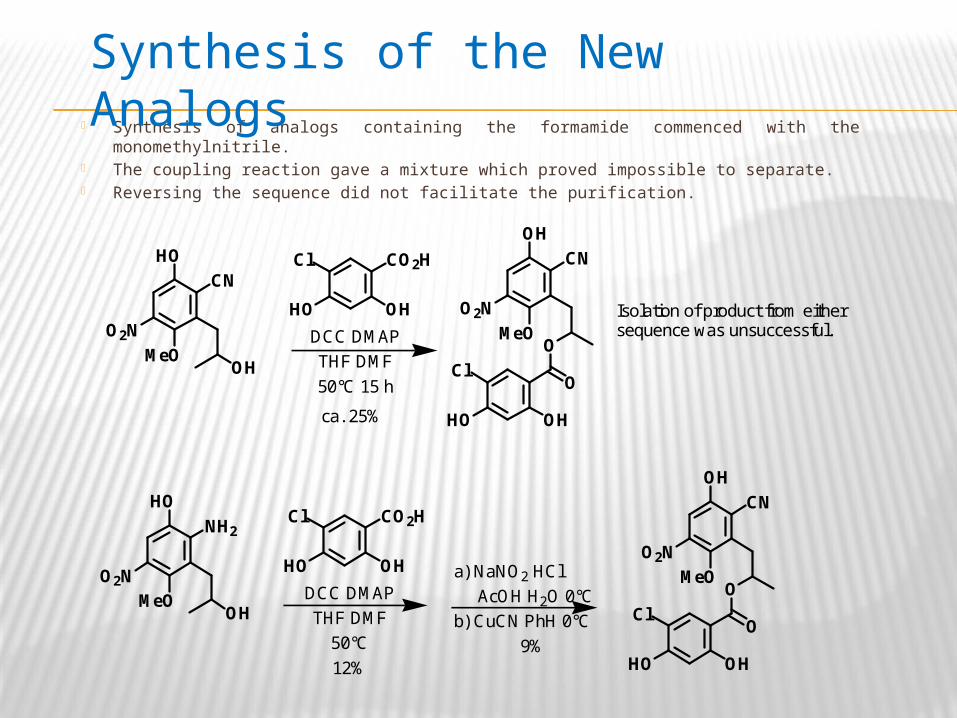
Synthesis of analogs containing the formamide commenced with the monomethylnitrile. The coupling reaction gave a mixture which proved impossible to separate. Reversing the sequence did not facilitate the purification.
Synthesis of the New Analogs

MeO
MeO
NH2
OH
O2N
HO OH
Cl CO2H
DCC DMAP
THF DMF
50°C
HO OH
ClO
MeO
MeO
NH2
O
O2N
65%
1.) tBuONO,CuCl22.) H2,Pd/C,MeOH
3.) PhOCHO
40% over 3 steps
HO OH
ClO
MeO
MeO
Cl
O
OHCHN
8
Coupling of the dimethylhydroquinone proceeded in good yield to provide nitroaniline 8 that was used to make the remaining analogs. Formation of the dimethylchloride is shown.
Mono-demethylation was highly problematic because, as anticipated, the amide directing group was no longer present. After significant experimentation only the chloride was selectively demethylated.
Isolation and subsequent testing of the dihydroquinones was accomplished.
Synthesis of the New Analogs

HO OH
ClO
MeO
Cl
O
OHCHN
HO OH
ClO
HO
Cl
O
OHCHN
11 12HO OH
ClO
MeO
CN
O
OHCHN
HO OH
ClO
HO
CN
O
OHCHN
9 10
MCF-7 SKBR3 >100 >100 >100 >100 >100 >100 >100 >100
9101112
IC50 (mM) No activity was observed for compounds 9-12; indicating that the presence only one oxygen atom of the hydroquinone ring is not sufficient to maintain effective binding with Hsp90.
Results of Anti-Proliferation Assays
HO OH
ClO
HO
MeO
Cl
O
OHCHN
HO OH
ClO
MeO
OH
CN
O
HO OH
ClO
MeO
NH2
CN
O
HO OH
ClO
HO
HO
CN
O
OHCHN
HO OH
ClO
HO
HO
Cl
O
OHCHN
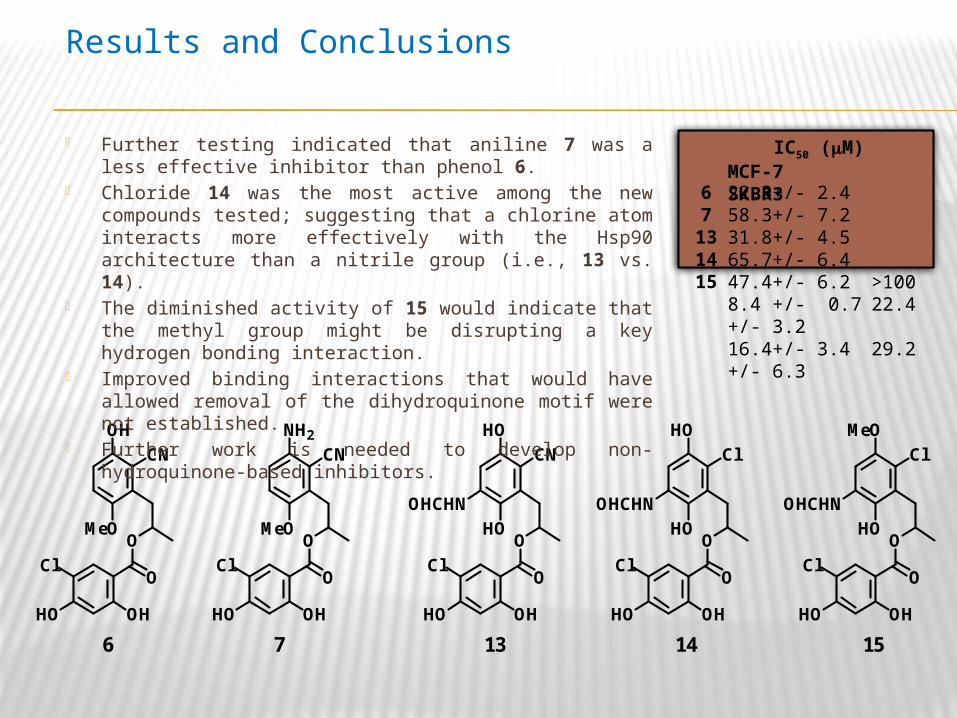
6 7 13 14 15
MCF-7 SKBR322.3+/- 2.4 58.3+/- 7.231.8+/- 4.5 65.7+/- 6.4 47.4+/- 6.2 >1008.4 +/- 0.7 22.4 +/- 3.216.4+/- 3.4 29.2 +/- 6.3
67
131415
IC50 (mM)
Results and Conclusions
Further testing indicated that aniline 7 was a less effective inhibitor than phenol 6.
Chloride 14 was the most active among the new compounds tested; suggesting that a chlorine atom interacts more effectively with the Hsp90 architecture than a nitrile group (i.e., 13 vs. 14).
The diminished activity of 15 would indicate that the methyl group might be disrupting a key hydrogen bonding interaction.
Improved binding interactions that would have allowed removal of the dihydroquinone motif were not established.
Further work is needed to develop non-hydroquinone-based inhibitors.

This work was supported by funding from NIH/NCI grant CA109265.
1. Zhang, H.; Burrows, F. J. Mol. Med. 2004, 82, 488.2. Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M. F.; Fritz, L. C.; Burrows, F. J. Nature 2003, 425, 4073. Chiosis, G.; Vilenchik, M.; Kim, J.; Solit, D. Drug Discovery Today 2004, 9, 881.4. Yamamoto, K.; Garbaccio, R. M.; Stachel, S. J.; Solit, D. B.; Chiosis, G.; Rosen, N.; Danishefsky, S. J. Angew. Chem.,
Int. Ed. 2003, 42,1280.5. Geng, X.; Yang, Z.-Q.; Danishefsky, S. J. Synlett 2004, 8, 1325.6. Sausville, E. A.; Tomaszewski, J. E.; Ivy, P. Curr. Cancer Drug Targets 2003, 3, 377.7. (a) Adams, J.; Elliott, P. J. Oncogene 2000, 19, 6687. (b) Neckers, L. Trends Mol. Med. 2002, 8, S55.8. Dikalov, S.; Landmesser, U.; Harrison, D. G. J. Biol. Chem. 2002, 277, 25480.9. Roe, M.S.; Prodromou, C.; O’Brien, R.; Ladbury, J.; Piper, P.; Pearl, L. J. Med Chem. 1999, 26010.Clevenger, R.S.; and Blagg, B.S.J. Org.Lett. 2004, 24, 445911.Shen, G.; Blagg, B.S.J. Org.Lett. 2005, 25, 215712.Hadden, K.; Blagg, B.S.J. J.Org.Chem. 2009, 74, 469713.Immormino, R.; Metzger, L.; Reardon, P.; Dollins, D.E.; Blagg, B.S.J; Gewirth, D. J. Mol. Biol. 2009, 388, 1033
References and Acknowledgement

PROBING PROXIMAL INTERACTIONS BETWEEN HSP90 AND THE ESTRADIOL RECEPTORConducted with Dr. Brian S. J. BlaggDepartment of Medicinal ChemistryUniversity of Kansas2008 - 2010

O
NH
HO
O
OMeOMe
O
OH2N
O
OOH
HO
Linker
The aim of this novel application is to determine the proximity of the estradiol receptor to the Hsp90 chaperone complex while the two are bound in an activated state.
A molecule of GDA would be tethered to one component of a “click chemistry” partner and an estradiol derivative would contain the complementary “click” appendage.
Probes with various length tethers would first be allowed to bind to their respective targets in vivo. The hypothesis is that tethers of appropriate length would meet and enter into a click reaction to form a covalently bound macromolecule.
Isolation of the macromolecule and determination of the tether length would provide an estimate of the distance between Hsp 90 and the estradiol receptor.
Background

N3
CO2R
F
F
Bertozzi Cycloalkyne CO2R
F
F+
NN
N
The Bertozzi cycloalkyne could be used as one of the probes in conjugation with an azide tether.
“Click reaction” would provide a fused bicyclic triazine macromolecule.
Background
Bertozzi, et. al., JACS, 2008, 11486–11493
TMSClNa
toluenereflux
CO2Et
CO2Et
8 g scale 85-90%
OTMS
OTMS
O
O
Et2ZnCH2I2
toluene0°C to rt, 12 h
95%
SelectfluorCs2CO3MeCN
0°C to rt, 1.5 h40%
HIO4EtOH
0°C to rt, 3 h42%
O
O
DBU THF0°C to rt, 12 h
60%
CO2Me
Br,PPh3Pd/C, H2MeOH
0°C to rt, 3 h95%
O
CO2Me
F
F
Tf2NPhKHMDS
THF -78°C to rt, 15 h
58%
OTf
CO2Me
F
F
CO2H
F
FLDA THF-20°C to rt
35%
O
CO2Me
F
F LiOHH2O dioxane
55°C95%
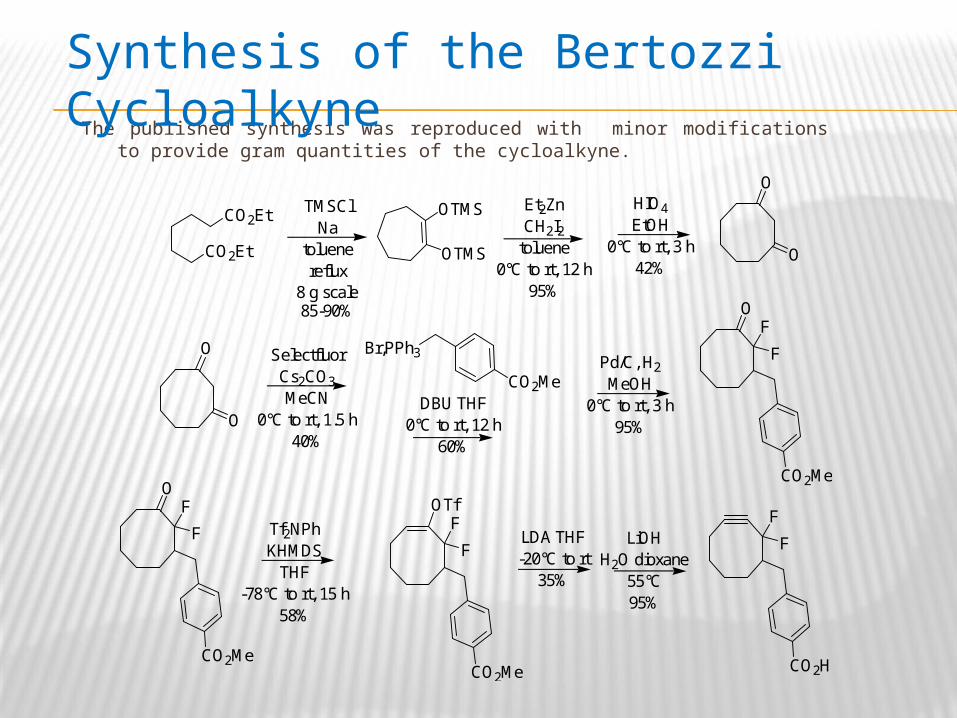
The published synthesis was reproduced with minor modifications to provide gram quantities of the cycloalkyne.
Synthesis of the Bertozzi Cycloalkyne

Proof of Concept for the GDA-EA Tether
O
NH
HO
O
OMeOMe
O
OH2N
O
HN
HN
O
F
FO
NH
HO
O
OMeOMe
O
OH2N
O
OMe H2N NH21.
DCM 2.
C(O)Cl
F
F
Geldanamycin
AH 7-30
N3O
OH
HO
+
O
NH
HO
O
OMeOMe
O
OH2N
O
HN
HN
O
FF
N
NNO
OH
HO
IC50=15.2 +/- 3.0 mM MCF-7
Initial proof of concept was achieved by first coupling GDA with 1,5-diaminopentane. The Bertozzi alkyne was then converted to an acid chloride and used to form an amide bond with the GDA amino tether.
Click reaction of the tethered alkyne was conducted in the lab of Dr. Robert Hanson at Northeastern University in Boston.
The GDA-estradiol AH 7-30 displayed moderate activity in the anti-proliferation assay against MCF-7 breast cancer cells.
The ability of the macromolecule to permeate the cell walls and exhibit a cytotoxic effect has been demonstrated. The next phase will be to determine if AH 7-30 is binding to the HSP90 chaperone complex or if cell death is occurring by another pathway.
For a related approach see: Danishefsky, et. al., BMCL, 1999, 1233

TOWARD A MORE EFFICIENT SYNTHESIS OF OPIATE ANALGESICSConducted at Mallinckrodt – A Division of Covidien, formerly a division of Tyco Healthcare2003 - 2004

Background
MeO
ONCH3
Thebaine
MeO
O
O
NCH3
Hydrocodone
MeO
O
O
NCH3
Oxycodone
OH
MeO
O
HO
NCH3
CodeineMeO
TotalSynthesis
2 steps4 steps2 steps
H H
Mallinckrodt refines the morphine, codeine and thebaine that are present in poppy resin. The retrosynthetic analysis illustrates how thebaine is converted to the popular analgesic
oxycodone. Codeine is a commercial product and can also be converted to hydrocodone which is a precursor to thebaine.
Supply of oxycodone is limited by the naturally occurring raw materials. . Mallinckrodt licensed the synthesis developed by Kenner Rice and conducted extensive
process research to establish a scalable route to hydrocodone.

NR
O
MeO Br
HO Br
OMe
HO
O
NR
"H+"
Br
OMe
HO
O
NROH
OMe
O
O
NROH
4 steps
12 steps
H
Is an advanced intermediate available via oxidative cyclization?
Oxycodone16 17
18
Intramolecular Cyclization Strategies
Intermediate 16 was prepared in 10 steps and converted into tetracyclic intermediate 17 with anhydrous acidic catalysis. Oxycodone is then produced from 17 in 12 more steps. We envisioned that oxidative cyclization of 16 could produce intermediate 18 which could be quickly transformed into oxycodone.

Preparation of the Cyclization Substrate
MeO
MeO NH2
CO2H
NCHO
O
MeO Br
HO
N
MeO
MeO
HO
MeO
CO2HHO
NH
MeO
MeO
HO
1.Br2, AcOH, 95%2.NaOH, CuSO4 H2O, reflux then HCl, 90%
Xylene, DST reflux, 85%
1. Ru cat. NEt3-HCO2H ACN 95%, 90%ee2.Birch Reduction 90%
1. iPrCHO2. MeSO3H, CHCl3 ethylene glycol then NBA, -20°C 3. 88% HCO2H, DMF 70%
1. POCl3, ACN, reflux then NaOH/H2O reflux2. NH4OH, H2O/MeOH, 85%
16
This chemistry was performed on scales between 200 g and 1 kg.

NCHO
O
MeO Br
HOBr
OMe
HO
O
NCHO
"H+" NH4F-HF in TfOH: 60%TfOH: 30%
TfOH/TfO2: 70-85%
16 17
Anhydrous Acidic Catalysis
Trace amounts of water adversely affected the yield of the cyclization reaction conducted with triflic acid, TfOH.
Refluxing triflic acid with triflic anhydride prior to adding the substrate provided extremely anhydrous conditions that led to highly improved yield.

Proposed Oxidative Cyclization
NCHO
O
MeO Br
HO
BrMeO
HO
O
NCHONCHO
O
MeO Br
HO
-e- -H+
-e-
H
BrMeO
HO
O
NCHO
H
The proposed oxidative cyclization would be conducted in an electrolysis cell. Loss of one electron to the anode would produce a radical cation that could undergo
attack by the aromatic ring. Re-aromatization via deprotonation followed by loss of a second electron would produce
the tertiary cation shown. The cation could by trapped by methanol to give methyl-protected oxycodone.

NCHO
O
MeO Br
RO
NCHO
MeO
MeO Br
RO
R= H or Ac
NR
MeO
MeO Br
HO
Li/TMSClTHF
then aq. HCl
NR
O
OMe
Br
OH
TMS
Anodic Oxidation
Anodic Oxidation
Anodic Oxidation
R= H or Ac
Product not observed
Product not observed
Product not observed
Attempts at Oxidative Cyclization
The majority of effort was devoted to making 0.25 kg of intermediate 16.
A new electrolysis system was purchased, validated and calibrated.
The attempted cyclization reactions shown are representative of the numerous unsuccessful trials that were attempted.
Further study is needed.

















