Page 1
REVIEW ARTICLE
Oxidative stress and pathology in muscular dystrophies:focus on protein thiol oxidation and dysferlinopathiesJessica R. Terrill1,2, Hannah G. Radley-Crabb3,1, Tomohito Iwasaki2, Frances A. Lemckert4,Peter G. Arthur2 and Miranda D. Grounds1
1 School of Anatomy, Physiology and Human Biology, University of Western Australia, Perth, Western Australia, Australia
2 School of Biomedical, Biomolecular & Chemical Science, University of Western Australia, Perth, Western Australia, Australia
3 Curtin Health Innovation Research Institute Biosciences Research Precinct, School of Biomedical Sciences, Curtin University,
Western Australia, Australia
4 Institute for Neuroscience and Muscle Research, Children’s Hospital at Westmead, New South Wales, Australia
Keywords
antioxidants; Duchenne muscular dystrophy;
Dysferlinopathies; dystropathology; muscle
necrosis; N-acetylcysteine; oxidative stress;
protein thiol oxidation; skeletal muscle;
reactive oxygen species
Correspondence
M. D. Grounds, School of Anatomy,
Physiology and Human Biology, University
of Western Australia, Perth, Western
Australia 6009, Australia
Fax: +61 8 6488 1051
Tel: +61 8 6488 3486
E–mail: [email protected]
(Received 17 December 2012, revised 14
January 2013, accepted 15 January 2013)
doi:10.1111/febs.12142
The muscular dystrophies comprise more than 30 clinical disorders that are
characterized by progressive skeletal muscle wasting and degeneration.
Although the genetic basis for many of these disorders has been identified,
the exact mechanism for pathogenesis generally remains unknown. It is
considered that disturbed levels of reactive oxygen species (ROS) contrib-
ute to the pathology of many muscular dystrophies. Reactive oxygen spe-
cies and oxidative stress may cause cellular damage by directly and
irreversibly damaging macromolecules such as proteins, membrane lipids
and DNA; another major cellular consequence of reactive oxygen species is
the reversible modification of protein thiol side chains that may affect
many aspects of molecular function. Irreversible oxidative damage of pro-
tein and lipids has been widely studied in Duchenne muscular dystrophy,
and we have recently identified increased protein thiol oxidation in dystro-
phic muscles of the mdx mouse model for Duchenne muscular dystrophy.
This review evaluates the role of elevated oxidative stress in Duchenne
muscular dystrophy and other forms of muscular dystrophies, and presents
new data that show significantly increased protein thiol oxidation and high
levels of lipofuscin (a measure of cumulative oxidative damage) in dysfer-
lin-deficient muscles of A/J mice at various ages. The significance of this
elevated oxidative stress and high levels of reversible thiol oxidation, but
minimal myofibre necrosis, is discussed in the context of the disease mecha-
nism for dysferlinopathies, and compared with the situation for dystro-
phin-deficient mdx mice.
Introduction
Overview of oxidative stress
Reactive oxygen species (ROS) are formed during a
variety of biological processes for all eukaryotes, and
although they are essential for cell signaling, excess
generation of ROS may harm biological components.
This occurs when the action of endogenous defense
mechanisms of the cell, involving various molecules
called antioxidants, is outweighed by the generation of
ROS, a state called oxidative stress [1]. Oxidative stress
Abbreviations
DMD, Duchenne muscular dystrophy; FLM, BODIPY FL–N–(2–aminoethyl) maleimide; NAC, N–acetylcysteine; ROS, reactive oxygen
species; Texas Red, Texas Red C2-maleimide.
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 1
Page 2
is implicated in the pathology of numerous conditions,
including aging, inflammatory disorders, cancer, mus-
cle wasting and muscular dystrophies [2–5]. ROS
causes cellular damage by directly and irreversibly
altering macromolecules such as proteins, membrane
lipids and DNA [6], but another (less studied) major
cellular consequence of ROS exposure is the reversible
modification of protein thiol side chains. Thiols are
organic sulfur derivatives, identified by the presence of
sulfhydryl residues (-SH) at their active site. Biological
thiols include low-molecular-weight free thiols and
protein thiols, the functional group of the amino acid
cysteine. In the presence of ROS, sulfhydryl residues
may undergo reversible modifications, whereby sulfhy-
dryl bonds are broken and disulfides are formed.
These thiol modifications are reversible via the action
of certain antioxidant molecules that reduce disulfides
via thiol/disulfide exchange, including the enzymes thi-
olredoxin and peroxiredoxin, as well as free cysteine
and glutathione. Cysteine availability is a rate-limiting
step in the synthesis of glutathione, which is a ubiqui-
tously expressed tripeptide that is considered to be the
most important cellular antioxidant molecule [7–9].Oxidation of protein thiols may be crucial to the
normal function of a specific protein, and may affect a
vast variety of functions, including protein structure,
protein–protein interactions, catalysis, electron trans-
fer, ion channel modulation, phosphorylation-depen-
dent signal transduction, post-translational protein
modification and transcriptional activation [10,11]. In
skeletal muscle, contractile function and force produc-
tion and the development of fatigue, are directly influ-
enced by the reduction/oxidation (redox) state of
protein thiols of contractile proteins. Contractile (myo-
fibrillar) proteins such as troponin, tropomyosin, myo-
sin and actin contain thiol side chains that are
sensitive to oxidation, and modifications may alter
excitation/contraction coupling and cross-bridge
cycling, and therefore modulate muscle contraction [12
–24]. Excessive oxidative stress, which occurs in condi-
tions such as chronic inflammation, during strenuous
exercise and disease states, may cause muscle weakness
[25], and is implicated in the pathology of numerous
muscular diseases such as muscular dystrophies.
Oxidative stress in muscular dystrophies
The muscular dystrophies comprise more than 30
hereditary clinical disorders that are characterized by
progressive skeletal muscle wasting and degeneration.
They generally share common histological features,
including variation in myofibre size, myofibre degener-
ation and regeneration, and the replacement of muscle
with connective tissue and fat [26]. These conditions
vary in many aspects, including prevalence, age of
onset, severity, the muscles affected and the underlying
gene defect [27]. These disorders are due to mutations
in a wide variety of molecules, including extracellular
matrix, sarcolemmal, cytoskeletal, cytosolic and
nuclear membrane proteins [28,29]. Although the
genetic basis of many of these disorders has been iden-
tified, the exact mechanism for pathogenesis remains
unclear; however, there is evidence that interactions
between the primary genetic defect and elevated levels
of ROS contribute to the pathology of several muscu-
lar dystrophies [30]. Oxidative stress has been investi-
gated extensively in Duchenne muscular dystrophy
(DMD), which is discussed in more detail below, and
is also clearly involved in other dystrophies and myop-
athies [30–35].For example, oxidative stress is strongly implicated
in the SEPN1-related myopathies that are due to defi-
ciency in the protein selenoprotein N (SEPN1): these
comprise four congenital skeletal muscle disorders that
are characterized by severe weakness and wasting of
neck and trunk muscles, leading to scoliosis and respi-
ratory insufficiency [31]. Selenoproteins contain seleno-
cysteine and include many proteins involved in the
regulation of oxidative stress, including glutathione
peroxidases and thioredoxin reductases; selenopro-
tein N has recently been identified as a key protein in
cell protection against oxidative stress and
redox-related calcium homeostasis [32]. Another disor-
der, facioscapulohumeral muscular dystrophy is an
autosomal dominant muscle disease that is character-
ized by progressive weakness and atrophy of facial,
shoulder girdle and upper-arm muscles. Its molecular
pathogenesis is due to deletions that lead to an
increase in function of the DUX4 (double
homeobox 4) protein in muscles [33]. DUX4 is a tran-
scription factor that initiates an extensive gene de-reg-
ulation cascade, and many genes that are differentially
expressed in the muscles of facioscapulohumeral
muscular dystrophy patients are involved in oxidative
stress responses [33]. Altered levels of ROS and a
higher susceptibility to oxidative stress are also a fea-
ture of laminopathies, which result from mutations in
the LMNA gene, encoding A–type lamins, proteins
that are associated with the nuclear membrane [34].
Elevated oxidative stress is also implicated in myopa-
thies due to mutations in the ryanodine receptor
RYR1, an essential component of the excitation/
contraction coupling apparatus; these include several
congenital RYR1-related myopathies that are the most
common non-dystrophic muscle diseases of childhood
[35].
2 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 3
In our laboratory, we have applied a wide range of
quantitative measures to analyze oxidative stress in
two forms of muscular dystrophy, using the mdx
mouse model for DMD and the dysferlin-deficient A/J
mouse model for dysferlinopathies. These two diseases
are discussed in detail below.
Oxidative stress in Duchenne muscular
dystrophy
Duchenne muscular dystrophy (DMD) is a lethal
X–chromosome-linked skeletal muscle disease, mani-
fested in children, that is caused by mutations in the
dystrophin gene resulting in the absence or decreased
function of the membrane-associated protein dystro-
phin [27,36]. Other mutations in the same gene result
in a mildly defective dystrophin protein, with a less
severe disease, usually with later onset, called Becker
muscular dystrophy [27]. Skeletal and cardiac myofi-
bres lacking functional dystrophin have an increased
susceptibility to sarcolemma damage after muscle con-
traction, which leads to myofibre necrosis; this results
in inflammation, myogenesis and new muscle forma-
tion to regenerate the tissue [37,38]. However,
repeated cycles of damage and inflammation over
months and years progress to replacement of muscle
by fat and fibrous connective tissues, with severe loss
of muscle function, resulting in premature death,
often due to respiratory or cardiac failure [36,39]. It
has been proposed that growth (as well as muscle size
and mechanical loading) increases the severity of dyst-
ropathology, which is less pronounced in animal
models of DMD, such as mdx mice and Golden
Retriever dogs [40]. The pathology of DMD also
appears to be exacerbated by oxidative stress: pre-
cisely why dystrophin deficiency leads to the genera-
tion of ROS in skeletal muscle is unclear, although
probable reasons include interactions between
excessive intracellular calcium and inflammation
[5,30,41–44]. It is well documented that elevated cyto-
solic calcium concentrations increase mitochondrial
calcium, which is an effector of ATP synthesis, and
an increase in ATP synthesis increases production of
ROS by mitochondria, via higher oxygen consump-
tion and enhanced electron flow through the electron
transport chain [45–47]. Membrane damage stimulates
degranulation of resident mast cells [48] and also
releases intracellular contents that activate the
immune system of the host, further increasing the
inflammatory cell cascade [49,50]. In addition,
immune cells such as neutrophils and macrophages
generate ROS in order to promote phagocytosis [51–53].
A role for oxidative stress in DMD is supported by
a wealth of pre-clinical studies in mdx mice that report
benefits such as improved muscle pathology and
decreased necrosis for many antioxidant drugs and
interventions, such as green tea extracts, resveratrol,
coenzyme Q10 and catalase [54–61]. Many of these
drugs may have broad-based effects in vivo, and the
potential translational benefits of these for clinical
treatment of DMD remain to be substantiated [62,63].
There is a notorious lack of success of antioxidants in
clinical trials [64] that may be due in part to a lack of
understanding of the precise nature of oxidative stress
involved with the particular pathology [5].
While irreversible oxidative damage of protein and
lipids, as measured by the carbonyl and malondiade-
hyde assays, respectively, has been widely studied and
targeted by antioxidant treatment, there has been little
information related to protein thiol oxidation in mus-
cular dystrophies. However, this topic has been a focus
of recent research in our laboratory. The development
of a specific two-tag assay to measure protein thiol
oxidation in skeletal muscle tissues has revealed signifi-
cantly elevated levels of protein thiol oxidation, as well
as elevated protein carbonylation, in dystrophic muscle
of mdx mice [65–68]. In addition, treatment with the
thiol-reducing antioxidant N–acetylcysteine (NAC)
reduces the severity of dystropathology in vivo, as
measured by decreased levels of plasma creatine kinase
and reduced myonecrosis, and this study specifically
demonstrated in vivo that NAC reduced the level of
oxidized protein thiols in dystrophic muscles [67].
NAC has previously been shown to improve force pro-
duction by mdx muscles [69] and the pathology of
mdx hearts [70], and a recent in vivo study further con-
firmed the beneficial effects of NAC on creatine kinase
levels and myonecrosis (using diaphragm muscles), as
well as reduced levels of tumor necrosis factor in mdx
mice [71]. These combined studies implicate protein
thiol oxidation in the dystropathology of DMD, and
support further evaluation of specific thiol antioxidant
drugs for clinical translation.
Oxidative stress in dysferlinopathies
Another group of muscular dystrophies of interest,
that have been far less well studied, are the dysferlin-
opathies. These encompass two disorders, limb-girdle
muscular dystrophy type 2B and Miyoshi myopathy,
which are both rare adult diseases, with weakness in
either distal muscles (Miyoshi myopathy) or proximal
limb-girdle muscles (limb-girdle muscular dystrophy
type 2B) [72,73]. Limb-girdle muscular dystrophy
type 2B and Miyoshi myopathy are considered to
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 3
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 4
result from allelic variations of the same dysferlin
gene; however, the reason why mutations to this gene
give rise to two different phenotypes is unknown [74].
The dysferlin gene encodes dysferlin, a membrane-
associated protein that is involved in membrane vesicle
trafficking and fusion, that is localized to transverse
tubules and the periphery of myofibres and cardiomyo-
cytes and, to a lesser extent, other tissues such as
monocytes, brain and kidney [75–77]. Muscles lacking
dysferlin may have problems in the membrane reseal-
ing that follows mechanical injury of contraction [78],
and an absence of dysferlin reportedly leads to intra-
cellular calcium dysregulation through membrane
modifications and cell signaling dysfunction [78–81].Dysferlin deficiencies are also associated with excessive
inflammation [49,82,83], and, although it has been
hypothesized that this leads to an increased presence
of ROS, very little experimental work on the subject
has been published.
A case study in a single Miyoshi patient identified
increased protein and lipid oxidation and protein thiol
content in affected muscle, suggesting increased ROS
levels in dysferlin-deficient muscle as well as protein
thiol alterations [84]; increased levels of antioxidants,
including glutathione and catalase, were also observed.
A study investigating oxidative damage in DMD, dys-
ferlinopathies and sarcoglycanopathy identified ampli-
fied lipid peroxidation and protein oxidation in all
three human muscular dystrophies [85]. The study also
showed dysregulation of glutathione-recycling antioxi-
dants, such as glutathione reductase and peroxidase,
suggesting perturbations in glutathione metabolism in
the muscle of patients with these dystrophies. Other
studies have used a combination of the antioxidants
coenzyme Q10 and resveratrol in SJL/J dysferlin-defi-
cient mice, and reported reduced inflammation and
muscle pathology, although these results were not
quantified [86,87]. Several strains of mice that lack
dysferlin are available, and include SJL/J, A/J and
Bla/J (A/J mice bred onto a C57Bl/6 background), as
well as genetically engineered null strains such as the
B6.129-Dysf tm1Kcam/J and B10.SJL–Dysf im/AwaJ mice:
all show a late onset but relatively mild pathology
compared with the human condition (http://www.jain-
foundation.org/our-dysferlin-research-institute/research-
tools/mouse-models-dysferlin-deficiency/).
Elevated protein thiol oxidation in
dysferlin-deficient muscles of A/J mice
Here we present new data for protein thiol oxidation,
and other measures of oxidative stress, along with
severity of histopathology, in a range of muscles from
dysferlin-deficient A/J (A/Jdysf�/�) mice, a naturally
occurring dysferlin-deficient strain of mice with a retro-
transposon insertion in dysferlin intron 4 [88]. These
data are compared to those for normal control A/J
mice at various ages, and then critically compared with
data for mdx mice. The A/Jdysf�/� mice exhibit dystro-
phic changes, including necrosis and inflammation, that
are histologically evident by 12 months of age in proxi-
mal muscles, whereas distal muscles appear relatively
unaffected even in late stages of the disease [88]. The
present study examined both distal and proximal mus-
cles, including the psoas and quadriceps (severely
affected) and gastrocnemius, biceps brachii muscles
(mildly affected), at four ages; 3, 8, 12 and 19 months.
The histology of muscles was assessed, and reversible
protein thiol oxidation was quantified using a dual-
labeling technique that indicates the percentage of oxi-
dized to total (reduced and oxidized protein thiols) in
muscles [66,67], and was further analyzed using a quan-
titative method that visualizes the localization of oxi-
dized protein thiols on tissue sections. Other measures
of oxidative stress included determination of irrevers-
ible oxidative damage to proteins using the carbonyl
assay [89] and of cumulative damage by quantification
of lipofuscin granules [90]. All of these measures were
correlated with histopathology. Identifying the targets
of oxidative stress that are affected in dysferlin-deficient
muscle provides insight into the molecular basis for
pathology and may also identify more specific drugs for
possible therapeutic intervention.
Results
Preliminary semi-quantification of necrosis, fat
content and protein thiol oxidation in severely
affected 19-month-old A/Jdysf�/� mice, to select
muscles most affected by pathology
A range of muscles from 19-month-old (severely
affected) A/Jdysf�/� mice were subjected to semi-quan-
titative histological analysis and measurement of total
protein thiol oxidation. The histology results indicated
that all muscles had a low–medium score for necrosis,
apart from the biceps, which scored normal–low(Fig. 1A). For fat content (Fig. 1B), both the psoas
and quadriceps muscles scored medium–high, whilst
the gastrocnemius, biceps and deltoid scored normal–low. Quantification of total protein thiol oxidation of
the muscles of 19-month-old mice (Fig. 2) showed that
both the psoas and quadriceps muscles had signifi-
cantly higher levels of protein thiol oxidation, while
the gastrocnemius, biceps and deltoid had a low level
of oxidation.
4 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 5
As all three parameters were high in psoas and
quadriceps muscles, these muscles were considered the
most affected, and were selected for quantification of
histology and oxidative stress measures in further stud-
ies, together with a relatively unaffected muscle (either
gastrocnemius or deltoid) as an additional control.
Quantification of areas of necrosis and fatty
tissue in 3-, 8-, 12- and 19-month-old A/Jdysf−/−
mice
Quantification of myofibre necrosis (Figs 3A and 4)
and fat content (Figs 3B and 4) for psoas, quadriceps
and gastrocnemius muscles was performed in 3-, 8-,
12- and 19-month-old A/Jdys�/� mice. These data were
compared with age-matched control A/J wild-type
mice (except for 19 months old, as control mice of this
age were not available); data are not shown for wild-
type muscles as all values were very low compared
with A/Jdysf�/� mice, as were A/Jdysf�/� gastrocnemius
*
$ $ $A
B
Fig. 1. Semi-quantitative histological analysis for myofibre necrosis
(A) and fat content (B) in skeletal muscles from 3-, 8-, 12- and 19-
month-old A/Jdysf�/� (dysferlin-null) mice. Means of semi_quantitative
data, where 0 = normal (none in whole tissue area), 1 = < 5%, 2 = 5–
10%, 3 = 10–15%, 4 = > 15%. The asterisk indicates a significant
difference (P < 0.05) between gastrocnemius and quadriceps
muscles (P < 0.05). The dollar symbol ($) indicates a significant
difference (P < 0.05) between biceps and other muscles. Values are
means � SEM (n = 6).
**
Fig. 2. Total protein thiol oxidation in skeletal muscles from 19-
month-old A/Jdysf�/� mice. The asterisk indicates a significant
difference (P < 0.05) from gastrocnemius, biceps and deltoid
muscles. Values are means � SEM (n = 6).
$# $#
$
$
$$
A
B
Fig. 3. Myofibre necrosis (A) and fat (B) (percentage cross-
sectional area) in psoas and quadriceps muscle from 3-, 8-, 12- and
19-month-old A/Jdysf�/� mice. The hash symbol (#) indicates a
significant difference from quadriceps muscle of the same age
(P < 0.05). The dollar symbol ($) indicates a significant difference
from the same muscle at 3 months (P < 0.05). Values are
means � SEM (n = 6).
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 5
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 6
values. Although necrosis was significantly elevated in
the A/Jdysf�/� psoas and quadriceps muscles compared
with normal controls, the extent of necrosis was low
(<1% of the cross-sectional area), and there were
no significant differences in necrosis between these
A/Jdysf�/� muscles at any age.
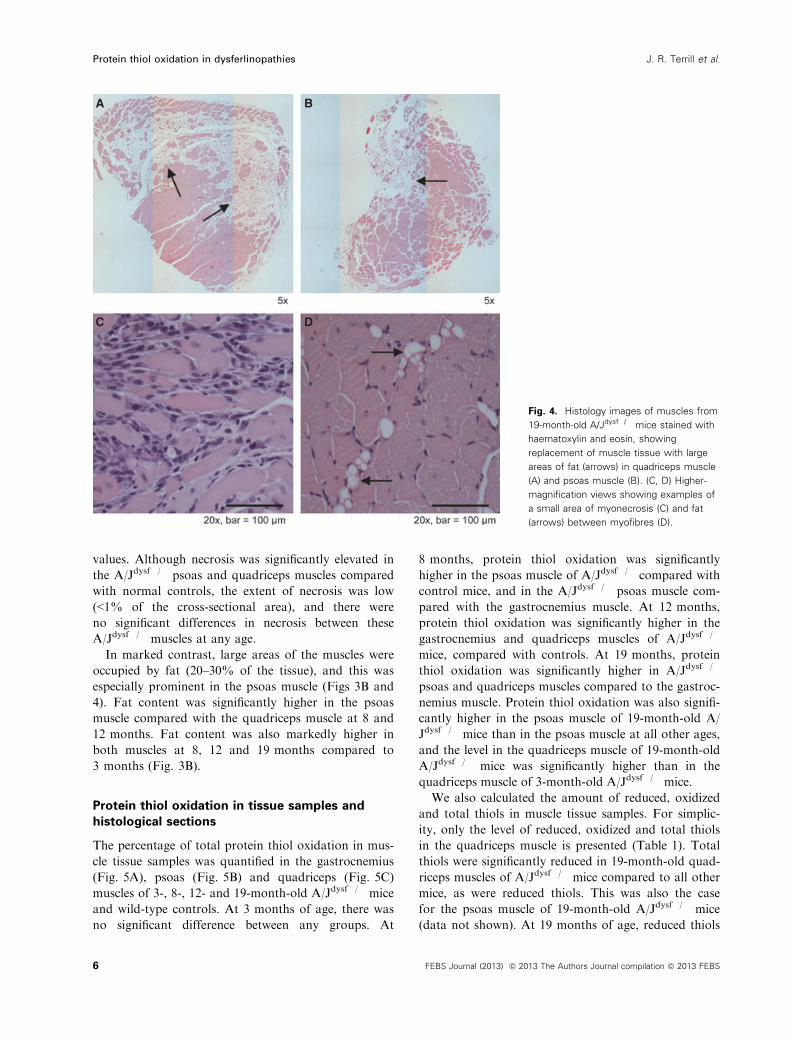
In marked contrast, large areas of the muscles were
occupied by fat (20–30% of the tissue), and this was
especially prominent in the psoas muscle (Figs 3B and
4). Fat content was significantly higher in the psoas
muscle compared with the quadriceps muscle at 8 and
12 months. Fat content was also markedly higher in
both muscles at 8, 12 and 19 months compared to
3 months (Fig. 3B).
Protein thiol oxidation in tissue samples and
histological sections
The percentage of total protein thiol oxidation in mus-
cle tissue samples was quantified in the gastrocnemius
(Fig. 5A), psoas (Fig. 5B) and quadriceps (Fig. 5C)
muscles of 3-, 8-, 12- and 19-month-old A/Jdysf�/� mice
and wild-type controls. At 3 months of age, there was
no significant difference between any groups. At
8 months, protein thiol oxidation was significantly
higher in the psoas muscle of A/Jdysf�/� compared with
control mice, and in the A/Jdysf�/� psoas muscle com-
pared with the gastrocnemius muscle. At 12 months,
protein thiol oxidation was significantly higher in the
gastrocnemius and quadriceps muscles of A/Jdysf�/�
mice, compared with controls. At 19 months, protein
thiol oxidation was significantly higher in A/Jdysf�/�
psoas and quadriceps muscles compared to the gastroc-
nemius muscle. Protein thiol oxidation was also signifi-
cantly higher in the psoas muscle of 19-month-old A/
Jdysf�/� mice than in the psoas muscle at all other ages,
and the level in the quadriceps muscle of 19-month-old
A/Jdysf�/� mice was significantly higher than in the
quadriceps muscle of 3-month-old A/Jdysf�/� mice.
We also calculated the amount of reduced, oxidized
and total thiols in muscle tissue samples. For simplic-
ity, only the level of reduced, oxidized and total thiols
in the quadriceps muscle is presented (Table 1). Total
thiols were significantly reduced in 19-month-old quad-
riceps muscles of A/Jdysf�/� mice compared to all other
mice, as were reduced thiols. This was also the case
for the psoas muscle of 19-month-old A/Jdysf�/� mice
(data not shown). At 19 months of age, reduced thiols
A B
C D
Fig. 4. Histology images of muscles from
19-month-old A/Jdysf�/� mice stained with
haematoxylin and eosin, showing
replacement of muscle tissue with large
areas of fat (arrows) in quadriceps muscle
(A) and psoas muscle (B). (C, D) Higher-
magnification views showing examples of
a small area of myonecrosis (C) and fat
(arrows) between myofibres (D).
6 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 7
were significantly lower in the A/Jdysf�/� psoas muscle
compared to the A/Jdysf�/� gastrocnemius muscle, and
total and reduced thiols were significantly lower in
the A/Jdysf�/� quadriceps muscle compared to the
A/Jdysf�/� gastrocnemius muscle (data not shown).
Even at 8 months of age, the A/Jdysf�/� mice had sig-
nificantly lower levels of reduced thiols than mice at
3 months, while 12-month-old A/Jdysf�/� mice had sig-
nificantly more oxidized thiols than age-matched wild-
type controls.
Protein thiol oxidation was also visualized and quan-
tified on histological tissue sections of 12-month-old
A/Jdysf�/� and wild-type controls (Fig. 6). The total area
of protein thiol oxidation was significantly higher in
A/Jdysf�/� tissue compared with control tissue, and was
evident both in areas with extensive lipid presence and
without (Fig. 6A), being mainly present throughout
intact myofibres (Fig. 6B). This pattern of histological
localisation was compared with mdx muscles, where
pronounced protein thiol oxidation (Fig. 6B) occurs
most dramatically in fragmented myofibres associated
with areas of necrosis, and there are more oxidized thi-
ols in intact myofibres compared to controls.
Oxidative damage to proteins
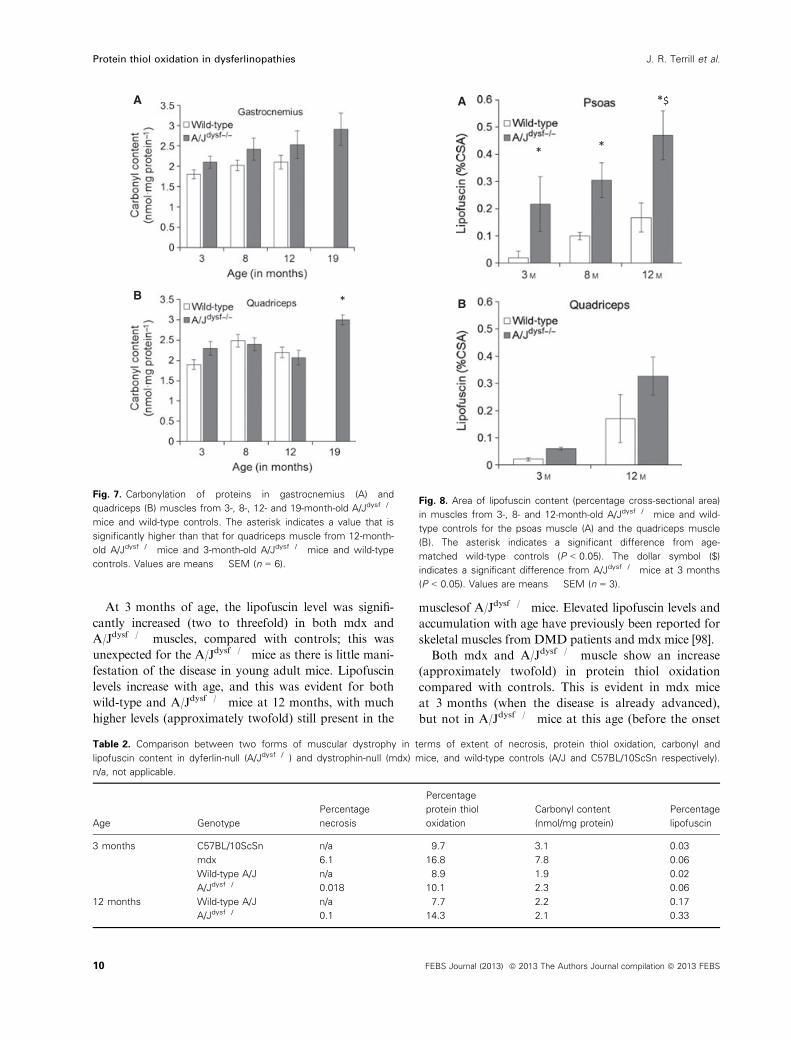
Carbonyl content
As the muscle most affected by oxidative stress (the pso-
as muscle) was too small to measure protein carbonyl
content, the gastrocnemius (Fig. 7A) and quadriceps
(Fig. 7B) muscles were analyzed in 3-, 8-, 12- and
19-month-old A/Jdysf�/� mice and wild-type controls.
There was no significant difference in either muscle for
protein carbonylation between strains or ages, apart
from the level in the quadriceps muscle of 19-month-old
A/Jdysf�/� mice, which was significantly higher than that
in the quadriceps muscle of 12-month-old A/Jdysf�/�
mice, and also that in the quadriceps muscle of
3-month-old A/Jdysf�/� and wild-type control mice.
Lipofuscin content
The presence of lipofuscin (also known as ceroid) indi-
cates the extent to which a tissue has been exposed
over time to irreversible oxidative stress [91], although
decreased degradation through impaired autophagy
may also increase the accumulation of lipofuscin [92].
Lipofuscin was evident in the psoas and quadriceps
muscles of A/Jdysf�/� mice as autofluorescent granules
at all ages (Fig. 8A). The amount of lipofuscin was
significantly higher in the psoas muscles of A/Jdysf�/�
mice at all ages (3, 8 and 12 months) compared with
age-matched controls, and was also higher in the psoas
muscle of 12-month-old A/Jdysf�/� mice compared
with 3-month-old A/Jdysf�/� mice (Fig. 8A). Although
there was a trend for increased lipofuscin content in
$
*
*#
*#&
A
B
C
Fig. 5. Total protein thiol oxidation in muscles from 3-, 8-, 12- and
19-month-old A/Jdysf�/� mice and wild-type controls. The asterisk
indicates a significant difference from age-matched wild-type
control (P < 0.05). The hash symbol (#) indicates a significant
difference from gastrocnemius muscle of the same age (P < 0.05).
The ampersand (&) indicates a significant difference from 3-month-
old A/Jdysf�/� mice (P < 0.05). The dollar symbol ($) indicates a
significant difference from A/Jdysf�/� and wild-type at all other ages
(P < 0.05). Values are means � SEM (n = 6).
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 7
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 8
quadriceps muscle (Fig. 8B), this was not statistically
significant, possibly due to low group numbers (n = 3).
Discussion
Correlation between pathology and thiol
oxidation in affected dysferlin-null muscles
While the level of myonecrosis was very low (< 1%) in
dysferlin-deficient muscles at all ages, the replacement
of muscle with fat was a very striking feature of the
affected muscles (psoas and quadriceps) in A/Jdysf�/�
mice. In diseases such as DMD, the increased adipos-
ity of muscle is a result of progressive replacement of
myofibres in response to repeated cycles of myofibre
necrosis [39,93], but this does not appear to be the
explanation for dysferlin-deficient muscles, in which
myofibre necrosis is relatively low: instead it must be
due to another pathogenic mechanism. An increase in
adiposity in skeletal muscle is associated with modifi-
cations to adipogenic genes and cell signaling path-
ways [94], and this may occur in dysferlin-deficient
muscle as a direct cellular consequence of dysferlin
deficiency, or indirectly as a secondary consequence of
this gene defect. Further experimental work is required
to understand the reasons for increased fat content in
dysferlin-deficient muscles and why only some muscles
are so severely affected.
Oxidative stress may lead to cellular dysfunction
through permanent damage to proteins, lipids and
DNA, as well as causing reversible modifications to
thiol side chains on cysteine residues, leading to altera-
tions in protein function [6]. The protein carbonyl
assay is commonly used as an indicator of irreversible
oxidative damage to proteins, and increased carbonyl
levels have been reported in human muscle lacking
dysferlin [84,85]. The fact that protein carbonylation
was not significantly increased in the muscles of
A/Jdysf�/� mice (compared to age-matched controls)
may reflect the relatively mild phenotype of this mouse
model up to 12 months of age, as carbonyl content
was significantly increased in the severely affected
quadriceps muscles of 19-month-old A/Jdysf�/� mice
compared with younger A/Jdysf�/� and control mice
(Fig. 7).
A more striking result in A/Jdysf�/� mice was the
reversible modification of protein thiol side chains,
with significantly elevated protein thiol oxidation in
the most severely affected psoas and quadriceps
muscles as early as 8 and 12 months, respectively (com-
pared with controls), and a significant further increase
by 19 months. This pattern of protein thiol oxidation
correlates with disease manifestation in specific muscles
and progression with age. Visualization of protein thiol
oxidation in tissue sections indicated that affected
dysferlin-deficient myofibres had high levels of protein
thiol oxidation: this was not evident in the interstitial
tissue and was not particularly pronounced in areas of
severe pathology where fat was present.
Another measure of oxidative stress that was
increased in both dystrophin- and dysferlin-deficient
muscle is the accumulation of lipofuscin: lipofuscin lev-
els are already significantly elevated in the psoas muscles
of A/Jdysf�/� mice at 3 months of age and increase fur-
ther with age and disease progression (Fig. 8). This ele-
vated lipofuscin level at the young age of 3 months
(when no striking pathology is evident in A/Jdysf�/�
muscles) appears to be of particular importance as it
indicates that there are already significant disturbances
to oxidative stress levels and implies an early role for
oxidative stress in subsequent disease manifestation.
Is the pattern of oxidative stress similar in DMD
and dysferlinopathies?
There is clearly a very different pattern in the time of
onset and the nature of pathology for DMD and
dysferlinopathies, and it is of interest to compare the
Table 1. Redox state of protein thiols in quadriceps muscle from A/Jdysf�/� mice and wild-type controls. The asterisk indicates a significant
difference from age-matched wild-type controls (P < 0.05). The ampersand (&) indicates a significant difference from 3-month-old A/Jdysf�/�
and wild-type mice (P < 0.05). The dollar symbol ($) indicates a significant difference from A/Jdysf�/� and wild-type mice at all other ages
(P < 0.05). Values are means � SEM (n = 6).
Age Genotype
Reduced thiols
(nmol�mg protein�1)
Oxidized thiols
(nmol�mg protein�1)
Total thiols
(nmol�mg protein�1)
Percentage
oxidized (%)
3 months Wild-type 36.4 � 1.8 4.6 � 0.4 41.0 � 2.1 8.9 � 1.1
A/Jdysf�/� 38.3 � 0.7 5.2 � 0.1 38.2 � 1.6 10.1 � 1.1
8 months Wild-type 31.7 � 1.5 5.2 � 0.5 38.1 � 1.7 11.5 � 0.3
A/Jdysf�/� 30.6 � 2.1& 4.8 � 0.4 35.4 � 2.2 14.2 � 1.4
12 months Wild-type 36.7 � 1.0 3.1 � 0.3 39.8 � 1.2 7.7 � 0.5
A/Jdysf�/� 33.6 � 1.7 4.5 � 0.3* 36.9 � 1.1 14.3 � 1.6*
19 months A/Jdysf�/� 24.1 � 0.9$ 4.9 � 0.3 29.1 � 0.8$ 17.6 � 1.2$
8 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 9
parameters of oxidative stress for the mouse models of
these diseases. Myofibre necrosis and oxidative stress
measures (protein thiol oxidation, carbonyl content
and lipofuscin accumulation) are compared in Table 2
for mdx and A/Jdysf�/� mice and their respective con-
trol strains (C57BL/10ScSn and A/J) at 3 months,
with additional data at 12 months for A/Jdysf�/� mice
as the disease was evident by this later age. At
3 months, active myofibre necrosis occupies approxi-
mately 6% of the muscle area for mdx mice, whereas
necrosis is very low in the muscles of A/Jdysf�/� mice.
This correlates with protein carbonyl oxidation, which
is elevated in mdx mice but not in A/Jdysf�/� mice at
this young age. Increased protein carbonyl content has
been reported in both DMD and mdx muscle [65,85,95
–97], but whether this is a cause or consequence of the
myonecrosis (and associated inflammation) is unclear.
Protein carbonylation is also evident in dysferlinopa-
thies after disease manifestation in humans [84,85] and
for very old A/Jdysf�/� mice at 19 months, with this
late onset suggesting that this is more likely to be a
consequence rather than a cause of the pathology.
* *
A
B
Fig. 6. (A) Protein thiol oxidation on muscle sections from 12-month-old A/Jdysf�/� mice and wild-type controls, in areas with and without
extensive fat. The asterisk indicates a significant difference from age-matched wild-type control (P < 0.05). Values are means � SEM (n = 3).
(B) Comparison of protein thiol oxidation on muscle tissue sections in dysferlin-null (A/Jdysf�/�) and dystrophin-null (mdx) mice, and wild-type
controls (A/J and C57BL/10ScSn respectively). Red., reduced thiols; Ox., oxidized thiols. Protein thiol oxidation is confined to intracellular
proteins of myofibres in tissue sections of skeletal muscles, and is increased in both A/Jdysf�/� and mdx muscle compared with controls.
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 9
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 10
At 3 months of age, the lipofuscin level was signifi-
cantly increased (two to threefold) in both mdx and
A/Jdysf�/� muscles, compared with controls; this was
unexpected for the A/Jdysf�/� mice as there is little mani-
festation of the disease in young adult mice. Lipofuscin
levels increase with age, and this was evident for both
wild-type and A/Jdysf�/� mice at 12 months, with much
higher levels (approximately twofold) still present in the
musclesof A/Jdysf�/� mice. Elevated lipofuscin levels and
accumulation with age have previously been reported for
skeletal muscles from DMD patients and mdx mice [98].
Both mdx and A/Jdysf�/� muscle show an increase
(approximately twofold) in protein thiol oxidation
compared with controls. This is evident in mdx mice
at 3 months (when the disease is already advanced),
but not in A/Jdysf�/� mice at this age (before the onset
*
A
B
Fig. 7. Carbonylation of proteins in gastrocnemius (A) and
quadriceps (B) muscles from 3-, 8-, 12- and 19-month-old A/Jdysf�/�
mice and wild-type controls. The asterisk indicates a value that is
significantly higher than that for quadriceps muscle from 12-month-
old A/Jdysf�/� mice and 3-month-old A/Jdysf�/� mice and wild-type
controls. Values are means � SEM (n = 6).
* *
*$A
B
Fig. 8. Area of lipofuscin content (percentage cross-sectional area)
in muscles from 3-, 8- and 12-month-old A/Jdysf�/� mice and wild-
type controls for the psoas muscle (A) and the quadriceps muscle
(B). The asterisk indicates a significant difference from age-
matched wild-type controls (P < 0.05). The dollar symbol ($)
indicates a significant difference from A/Jdysf�/� mice at 3 months
(P < 0.05). Values are means � SEM (n = 3).
Table 2. Comparison between two forms of muscular dystrophy in terms of extent of necrosis, protein thiol oxidation, carbonyl and
lipofuscin content in dyferlin-null (A/Jdysf�/�) and dystrophin-null (mdx) mice, and wild-type controls (A/J and C57BL/10ScSn respectively).
n/a, not applicable.
Age Genotype
Percentage
necrosis
Percentage
protein thiol
oxidation
Carbonyl content
(nmol/mg protein)
Percentage
lipofuscin
3 months C57BL/10ScSn n/a 9.7 3.1 0.03
mdx 6.1 16.8 7.8 0.06
Wild-type A/J n/a 8.9 1.9 0.02
A/Jdysf�/� 0.018 10.1 2.3 0.06
12 months Wild-type A/J n/a 7.7 2.2 0.17
A/Jdysf�/� 0.1 14.3 2.1 0.33
10 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 11
of pathology), although it is evident by 12 months in
A/Jdysf�/� mice when the pathology has already mani-
fested. In the mdx muscles, pronounced protein thiol
oxidation (Fig. 6B) occurs most dramatically in frag-
mented myofibres associated with areas of necrosis,
and, like the muscles of A/Jdysf�/� mice, there are also
significantly more oxidized thiols inside intact myofi-
bres (also evident as lower levels of reduced protein
thiols in Fig. 6B). This suggests that skeletal muscle
proteins are undergoing protein thiol oxidation in rela-
tively ‘unaffected’ myofibres of the dystrophic muscles,
potentially causing changes in protein function and
thus contributing to the resulting pathology and
altered muscle performance. There are a wide range of
target proteins for thiol oxidation and modulation of
function that include many contractile proteins, and
we have recently identified significant changes in the
thiol oxidation state of such proteins, specifically myo-
sin and tropomyosin in the quadriceps muscle of
3-month-old mdx mice (J. Terrill, M. Grounds,
P. Arthur, unpublished data).
The A/Jdysf�/� mouse is a useful model to study dysf-
erinopathies, with a strong correlation between the onset
and severity of pathology in specific muscles and the
incidence of various measures of oxidative stress. The
early elevation of protein thiol oxidation in affected mus-
cles (compared with the late increases in carbonylation)
suggests a potentially important role for such reversible
oxidation of key muscle proteins in the manifestation of
pathology in dysferlinopathies, and presents the opportu-
nity to assess the effects of specific thiol-reducing antioxi-
dants such as NAC in the A/Jdysf�/� mice. In mdx mice,
NAC has various benefits and reduces the severity of
pathology [67,69,71], providing further evidence for the
probable important role of protein thiol oxidation in the
pathology of DMD. It is also of interest to identify the
specific proteins that are affected by thiol oxidation in
the muscles of A/Jdysf�/� mice to determine their poten-
tial role in the mechanism of the disease. The fact that
lipofuscin is already significantly elevated by 3 months
of age in the muscles of A/Jdysf�/� mice, before the
disease is obvious, indicates that this is a particularly
sensitive measure and strongly supports an early and key
role for altered oxidative stress prior to disease manifes-
tation. Further investigations into such aspects of
oxidative stress in dysferlinopathies (as have already
been initiated for the mdx mouse and DMD) appear
warranted.
Experimental procedures
All reagents were obtained from Sigma-Aldrich (St. Louis,
MO, USA) unless otherwise specified.
Animal procedures
A/J control mice were obtained from the Animal Resources
Centre (Murdoch, Western Australia). A/Jdysf�/� (dysferlin
null) mice were obtained from the Institute for Neurosci-
ence and Musclemuscular Research, Children’s Hospital at
Westmead, (Sydney, Australia). Mice were transported to
the University of Western Australia and maintained on a
12 h light/dark cycle under standard conditions, with free
access to food and drinking water. All animal experi-
ments were performed in strict accordance with the
guidelines of the National Health and Medical Research
Council of Australia code of practice for the care and
use of animals for scientific purposes (2004), and the
Animal Welfare Act of Western Australia (2002), and
were approved by the Animal Ethics Committee at the
University of Western Australia.
Tissue collection, histology and image
acquisition
All mice were killed by complete cervical dislocation while
under terminal anesthesia (2% v/v AttaneTMisoflurane,
Bomac, Hornsby, NSW, Australia). Muscles were collected
from wild-type control mice at 3, 8 and 12 months of age,
and from A/Jdysf�/� mice at 3, 8, 12 and 19 months
(19-month-old wild-type controls were omitted as these
were not available for analysis), and immediately snap-fro-
zen in liquid nitrogen for biochemical analysis. For histol-
ogy, one whole upper limb and one lower limb were
immersed in 4% paraformaldehyde and fixed for 1 week;
samples were then processed for paraffin histology. Trans-
verse muscle sections (5 lm) were cut through the mid-
region of each muscle on a Leica microtome, as previously
described [99], and sections were stained with haematoxylin
and eosin for morphological analysis.
Groups of three control and three A/Jdysf�/� mice at
three ages (3, 8 and 12 months old) were sampled, and mus-
cles were obtained for frozen histology for the analysis of
lipofuscin and protein thiol oxidation. The fresh muscles
were bisected transversely and longitudinally and mounted
on cork squares using tragacanth gum. The muscles were
frozen in a slurry of isopentane cooled in liquid nitrogen.
Cryostat sections (8 lm) were cut directly onto silinised
glass slides, and stored at �20 °C until stained or analyzed.
Histological image analysis
Myofibre necrosis was identified as areas of myofibres with
fragmented sarcoplasms and/or increased inflammatory cell
infiltration. Fat content was identified as areas of many
large circular cells unstained by haematoxylin and eosin.
Both were assessed semi-quantitatively in all skeletal muscles
(psoas, biceps brachii, quadriceps, gastrocnemius, deltoid)
from 8-, 12- and 19-month-old A/Jdysf�/� mice. In brief,
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 11
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 12
skeletal muscles were prepared for paraffin histology,
stained with haematoxylin and eosin [67,68,99], and all mus-
cles (n = 6 per group) were examined by light microscopy at
109 magnification. For the preliminary semi-quantitative
analysis, each parameter was given a score of 0–4; where
0 = normal, 1 = < 5%, 2 = 5–10%, 3 = 10–15%, 4 => 15%. These scores for six samples were averaged to pro-
vide an overall score. After the semi-quantitative analysis
had revealed that psoas and quadriceps muscles were the
most affected, these muscles were quantitatively assessed for
necrosis and fat content using non-overlapping tiled images
of transverse muscle sections that provided a picture of the
entire muscl’e cross-section. Digital images were acquired
using a Leica Microsystems (Wetzlar, Germany) DM RBE
microscope, a Hitachi (Tokyo, Japan) HVC2OM digital
camera, IMAGE PRO PLUS 4.5.1 software (Media Cybernetics,
Rockville, MD, USA) and VEXTA STAGE MOVEMENT software
(Oriental Motor Co, Tokyo, Japan). Tiled images were
taken at 109 magnification. Muscle morphology was
drawn manually by the researcher using IMAGE PRO PLUS
4.5.1 software. The area occupied by necrotic myofibres
(i.e. myofibres with fragmented sarcoplasm and/or areas of
inflammatory cells) or fat was measured as a percentage of
the whole muscle section area. Histological analysis was
completed according to the TREAT-NMD recommended
standard protocol ‘Histological Measurements of
Dystrophic Muscle – M.1.2_007’ (http://www.treat-nmd.
eu/research/preclinical/dmd-sops/).
Carbonylated protein
Oxidative damage to proteins in muscles was determined
by measuring the carbonyl content using 2,4–dini-
trophenylhydrazine as previously described [65,100,101].
Frozen muscles were crushed under liquid nitrogen, and
protein was extracted using 20% trichloroacetic acid/
acetone. The protein pellets were washed in acetone and
ethanol, precipitated, dried, re-suspended in 10 mM
2,4–dinitrophenylhydrazine in 2 M HCl, and incubated for
30 min at room temperature in the dark. Proteins were
washed with ethyl acetate/ethanol (1 : 1) for one hour at
room temperature, dissolved in 6 M guanidine, and absor-
bance was measured at 370 nm. Protein concentration
(mg�mL�1) was determined using the Bio–Rad (Hercules,
CA, USA) Bradford protein assay. Carbonyl concentra-
tions are expressed as nmol carbonyl per mg protein.
Lipofuscin quantification in muscle
Lipofuscin is composed of autofluorescent granules that
accumulate in tissue and are generated as a consequence
of irreversible oxidative stress [91]. The granules are visible
on unstained frozen tissue sections using fluorescent
microscopy. The amount of lipofuscin in frozen muscle
sections was measured by the non-subjective boot strap-
ping method [90]. Images were captured using a fluores-
cent Nikon (Tokyo, Japan) Eclipse Ti microscope
equipped with a Roper Industries (Sarasota, FL, USA)
CoolSNAP-HQ2 camera, a 450–490 nm excitation filter, a
515 nm emission barrier filter, and Nikon NIS-Elements
software. A 409 magnification was used, and sections
were scanned using an automatic stage control setting that
generated a grid structure of images in a set area. For
each section scanned by fluorescent microscopy, eight
images were selected for lipofuscin analysis: images with
tissue edges or obvious artefacts were discarded. Quantifi-
cation of lipofuscin granules was performed using IMAGEJ
version 1.44 (http://imagej.nih.gov/ij/download/), and the
green channel of an eight-bit RBG image was analyzed.
The area occupied by fluorescent granules was expressed
as a percentage of the total image area.
Protein thiol oxidation
Reduced and oxidized protein thiol levels were measured
using a dual-labeling technique [66–68]. In brief, snap-frozen
quadriceps muscle was crushed under liquid nitrogen, and
protein was extracted using 20% trichloroacetic acid/ace-
tone. Protein was solubilized in 0.5% SDS/0.5 M Tris at pH
7.3 (SDS buffer), and protein thiols were labeled with the
first tag, the fluorescent dye BODIPY FL-N–(2-aminoethyl)
maleimide (FLM) (Invitrogen, Mulgrave, VIC, Australia).
After removal of the unbound dye using ethanol, protein
was re-solubilized in SDS buffer, pH 7, and oxidized thiols
were reduced using Tris(2–carboxyethyl)phosphine, before
labeling of the resultant unlabeled reduced thiols with a sec-
ond tag, the fluorescent dye Texas Red C2-maleimide (Texas
Red) (Invitrogen). The sample was washed in ethanol and
resuspended in SDS buffer. Samples were read using a fluo-
rescent plate reader (Fluostar Optima, BMG Labtech, Oten-
berg, Germany) with wavelengths of 485 nm for excitation
and 520 nm for emission for FLM, and 595 nm for excita-
tion and 610 nm for emission for Texas Red. A standard
curve for each dye was generated using ovalbumin, and the
results are expressed per mg of protein, quantified using a
Detergent Compatible Protein Assay (Bio–Rad).
Protein thiol oxidation on tissue sections
Reduced and oxidized protein thiols on frozen tissue sec-
tions were measured using an adaptation of the dual-label-
ing technique described above (T. Iwasaki, J. Terrill,
M. Grounds and P. Arthur, unpublished). Serial muscle
sections (9 lm) used for detecting reduced thiols were trea-
ted immediately in FLM. After washing in NaCl/Pi, sec-
tions were fixed with 4% paraformaldehyde in 0.1 M
phosphate buffer, and immersed in NaCl/Pi overnight. For
detection of oxidized thiols, frozen sections from each
muscle were treated with N–ethylmaleimide to block
free thiols. After washing in NaCl/Pi to remove unreacted
12 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 13
N–ethylmaleimide, sections were fixed with 4% paraformal-
dehyde in 0.1 M phosphate buffer. The fixed sections were
washed again with NaCl/Pi. Oxidized thiols were reduced
with Tris(2–carboxyethyl)phosphine); this was omitted in
negative control sections. The reduced thiols were washed
twice in NaCl/Pi to remove Tris(2–carboxyethyl)phosphine
before labeling with FLM. All sections were mounted with
polyvinyl acetate mounting medium for microscopy.
Another serial section from each muscle was stained with
haematoxylin and eosin for morphometric observation.
Fluorescence images were acquired as per lipofuscin
quantification. Sections were scanned using an automatic
stage control setting that generated a grid structure of
images covering a set area. The level of oxidized thiols in
muscle sections was estimated by image analysis using
IMAGEJ version 1.44. For each section scanned by fluores-
cence microscopy, three images were selected for all
image analysis: images with tissue edges or obvious arte-
facts were discarded. The selected fluorescence images
were used for quantification of the mean fluorescence
intensity (arbitrary units) of the section.
Statistics
Significant differences between groups were determined
using one-way ANOVA with post hoc tests, and all data
are presented as means � standard error of the mean.
Significance was set at P < 0.05.
Acknowledgements
We thank Sandra Cooper (Institute for Neuroscience
and Musclemuscular Research, Children’s Hospital at
Westmead, Sydney, Australia) for generously provid-
ing the dysferlin-deficient A/J mice. This research was
supported by funding from the Jain Foundation, the
National Health and Medical Research Council of
Australia, and an Australian Postgraduate Award
Scholarship to J.R.T.
References
1 Davies KJA (2000) Oxidative stress, antioxidant
defenses, and damage removal, repair, and replacement
systems. IUBMB Life 50, 279–289.
2 Dr€oge W (2003) Oxidative stress and aging. Adv Exp
Med Biol 543, 191–200.
3 Valko M, Rhodes C, Moncol J, Izakovic M & Mazur
M (2006) Free radicals, metals and antioxidants in
oxidative stress-induced cancer. Chem Biol Interact
160, 1–40.
4 Rando TA (2002) Oxidative stress and the
pathogenesis of muscular dystrophies. Am J Phys Med
Rehabil 81, S175–S186.
5 Arthur PG, Grounds MD & Shavlakadze T (2008)
Oxidative stress as a therapeutic target during muscle
wasting: considering the complex interactions. Curr
Opin Clin Nutr Metab Care 11, 408–416.
6 Halliwell B & Gutteridge JM (2007) Free Radicals in
Biology and Medicine, Vol. 4. Oxford University Press,
New York.
7 Ferreira LF & Reid MB (2008) Muscle-derived ROS
and thiol regulation in muscle fatigue. J Appl Physiol
104, 853–860.
8 Medved I, Brown MJ, Bjorksten AR, Murphy KT,
Petersen AC, Sostaric S, Gong X & McKenna MJ
(2004) N–acetylcysteine enhances muscle cysteine and
glutathione availability and attenuates fatigue during
prolonged exercise in endurance-trained individuals.
J Appl Physiol 97, 1477–1485.
9 Dilger RN & Baker DH (2007) Oral N–acetyl-L–
cysteine is a safe and effective precursor of cysteine.
J Anim Sci 85, 1712–1718.
10 Biswas S, Chida AS & Rahman I (2006) Redox
modifications of protein-thiols: emerging roles in cell
signaling. Biochem Pharmacol 71, 551–564.
11 Paulsen CE & Carroll KS (2009) Orchestrating redox
signaling networks through regulatory cysteine
switches. ACS Chem Biol 5, 47–62.
12 Andrade FH, Reid MB, Allen DG & Westerblad H
(1998) Effect of hydrogen peroxide and dithiothreitol
on contractile function of single skeletal muscle fibres
from the mouse. J Physiol 509, 565–575.
13 Liu D, Wang D & Stracher A (1990) The accessibility
of the thiol groups on G- and F–actin of rabbit
muscle. Biochem J 266, 453–459.
14 Crowder M & Cooke R (1984) The effect of myosin
sulphydryl modification on the mechanics of fibre
contraction. J Muscle Res Cell Motil 5, 131–146.
15 Dalle-Donne I, Giustarini D, Rossi R, Colombo R &
Milzani A (2003) Reversible S–glutathionylation of
Cys374 regulates actin filament formation by inducing
structural changes in the actin molecule. Free Radic
Biol Med 34, 23–32.
16 Hertelendi Z, T�oth A, Borb�ely A, Galajda Z, van der
Velden J, Stienen GJM, �Edes I & Papp Z (2008)
Oxidation of myofilament protein sulfhydryl groups
reduces the contractile force and its Ca2+ sensitivity in
human cardiomyocytes. Antioxid Redox Signal 10,
1175–1184.
17 Prochniewicz E, Spakowicz D & Thomas DD (2008)
Changes in actin structural transitions associated with
oxidative inhibition of muscle contraction.
Biochemistry 47, 11811–11817.
18 Root DD & Reisler E (1992) Cooperativity of thiol-
modified myosin filaments. ATPase and motility assays
of myosin function. Biophys J 63, 730–740.
19 Tiago T, Sim~ao S, Aureliano M, Mart�ın-Romero FJ &
Guti�errez-Merino C (2006) Inhibition of skeletal
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 13
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 14
muscle S1–myosin ATPase by peroxynitrite.
Biochemistry 45, 3794–3804.
20 Putkey J, Dotson D & Mouawad P (1993) Formation
of inter-and intramolecular disulfide bonds can
activate cardiac troponin C. J Biol Chem 268,
6827–6830.
21 Pinto JR, de Sousa VP & Sorenson MM (2010)
Redox state of troponin C cysteine in the D/E helix
alters the C–domain affinity for the thin filament of
vertebrate striated muscle. Biochim Biophys Acta
1810, 391–397.
22 Williams DL Jr & Swenson CA (1982) Disulfide
bridges in tropomyosin. Eur J Biochem 127, 495–499.
23 Andrade FH, Reid MB & Westerblad H (2001)
Contractile response of skeletal muscle to low peroxide
concentrations: myofibrillar calcium sensitivity as a
likely target for redox-modulation. FASEB J 15,
309–311.
24 Mollica JP, Dutka TL, Merry T, Lamboley C,
McConell GK, McKenna MJ, Murphy RM & Lamb
GD (2012) S–glutathionylation of troponin I (fast)
increases contractile apparatus Ca2+ sensitivity in fast-
twitch muscle fibres of rats and humans. J Physiol 590,
1443–1463.
25 Smith MA & Reid MB (2006) Redox modulation of
contractile function in respiratory and limb skeletal
muscle. Respir Physiol Neurobiol 151, 229–241.
26 Manzur AY & Muntoni F (2009) Diagnosis and new
treatments in muscular dystrophies. J Neurol
Neurosurg Psychiatry 80, 706–714.
27 Emery AEH (2002) The muscular dystrophies. Lancet
359, 687–695.
28 Cohn RD & Campbell KP (2000) Molecular basis of
muscular dystrophies. Muscle Nerve 23, 1456–1471.
29 Mercuri E & Muntoni F (2012) The ever-expanding
spectrum of congenital muscular dystrophies. Ann
Neurol 72, 9–17.
30 Tidball JG & Wehling-Henricks M (2007) The role of
free radicals in the pathophysiology of muscular
dystrophy. J Appl Physiol 102, 1677–1686.
31 Arbogast S, Beuvin M, Fraysse B, Zhou H, Muntoni
F & Ferreiro A (2009) Oxidative stress in SEPN1-
related myopathy: from pathophysiology to treatment.
Ann Neurol 65, 677–686.
32 Arbogast S & Ferreiro A (2010) Selenoproteins and
protection against oxidative stress: selenoprotein N as
a novel player at the crossroads of redox signaling
and calcium homeostasis. Antioxid Redox Signal 12,
893–904.
33 Turki A, Hayot M, Carnac G, Pillard F, Passerieux E,
Bommart S, de Mauverger ER, Hugon G, Pincemail J
& Pietri S (2012) Functional muscle impairment in
facioscapulohumeral muscular dystrophy is correlated
with oxidative stress and mitochondrial dysfunction.
Free Radic Biol Med 53, 1068–1079.
34 Sieprath T, Darwiche R & De Vos WH (2012) Lamins
as mediators of oxidative stress. Biochem Biophys Res
Commun 421, 635–639.
35 Dowling JJ, Arbogast S, Hur J, Nelson DD, McEvoy
A, Waugh T, Marty I, Lunardi J, Brooks SV &
Kuwada JY (2012) Oxidative stress and successful
antioxidant treatment in models of RYR1-related
myopathy. Brain 135, 1115–1127.
36 Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens
PR, Cripe L, Kaul A, Kinnett K, McDonald C &
Pandya S (2010) Diagnosis and management of
Duchenne muscular dystrophy, part 1: diagnosis, and
pharmacological and psychosocial management.
Lancet Neurol 9, 77–93.
37 Petrof BJ, Stedman HH, Shrager JB, Eby J, Sweeney
HL & Kelly AM (1993) Adaptations in myosin heavy
chain expression and contractile function in dystrophic
mouse diaphragm. Am J Physiol 265, C834–C841.38 Lapidos KA, Kakkar R & McNally EM (2004) The
dystrophin glycoprotein complex – signaling strength and
integrity for the sarcolemma. Circ Res 94, 1023–1031.
39 Biggar W (2006) Duchenne muscular dystrophy.
Pediatr Rev 27, 83–88.
40 Grounds MD & Shavlakadze T (2011) Growing
muscle has different sarcolemmal properties from adult
muscle: a proposal with scientific and clinical
implications. BioEssays 33, 458–468.
41 Chang WJ, Iannaccone ST, Lau KS, Masters BS,
McCabe TJ, McMillan K, Padre RC, Spencer MJ,
Tidball JG & Stull JT (1996) Neuronal nitric oxide
synthase and dystrophin-deficient muscular dystrophy.
Proc Natl Acad Sci USA 93, 9142–9147.
42 Brenman JE, Chao DS, Xia H, Aldape K & Bredt DS
(1995) Nitric oxide synthase complexed with dystrophin
and absent from skeletal muscle sarcolemma in
Duchenne muscular dystrophy. Cell 82, 743–752.
43 Wehling M, Spencer MJ & Tidball JG (2001) A nitric
oxide synthase transgene ameliorates muscular
dystrophy in mdx mice. J Cell Biol 155, 123–132.
44 Whitehead NP, Yeung EW & Allen DG (2006) Muscle
damage in mdx (dystrophic) mice: role of calcium and
reactive oxygen species. Clin Exp Pharmacol Physiol
33, 657–662.
45 Brookes PS, Yoon Y, Robotham JL, Anders MW &
Sheu SS (2004) Calcium, ATP, and ROS: a
mitochondrial love–hate triangle. Am J Physiol 287,
C817–C833.
46 Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ &
Camello PJ (2006) Mitochondrial reactive oxygen species
and Ca2+ signaling. Am J Physiol 291, C1082–C1088.
47 Feissner RF, Skalska J, Gaum WE & Sheu SS (2009)
Crosstalk signaling between mitochondrial Ca2+ and
ROS. Front Biosci 14, 1197–1218.
48 Radley H & Grounds M (2006) Cromolyn
administration (to block mast cell degranulation)
14 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.
Page 15
reduces necrosis of dystrophic muscle in mdx mice.
Neurobiol Dis 23, 387–397.
49 Han R (2011) Muscle membrane repair and
inflammatory attack in dysferlinopathy. Skelet Muscle
1, 10.
50 Andrews NW (2005) Membrane repair and
immunological danger. EMBO Rep 6, 826–830.
51 Tidball JG (2005) Inflammatory processes in muscle
injury and repair. Am J Physiol 288, R345–R353.
52 Halliwell B (1991) Reactive oxygen species in living
systems: source, biochemistry, and role in human
disease. Am J Med 91, S14–S22.
53 Schraufst€atter I, Browne K, Harris A, Hyslop PA,
Jackson JH, Quehenberger O & Cochrane C (1990)
Mechanisms of hypochlorite injury of target cells.
J Clin Invest 85, 554–562.
54 Dorchies O, Wagner S, Vuadens O, Waldhauser K,
Buetler T, Kucera P & Ruegg U (2006) Green tea
extract and its major polyphenol (–)–epigallocatechin
gallate improve muscle function in a mouse model for
Duchenne muscular dystrophy. Am J Physiol 290,
C616–C625.
55 Buetler T, Renard M, Offord E, Schneider H & Ruegg
U (2002) Green tea extract decreases muscle necrosis
in mdx mice and protects against reactive oxygen
species. Am J Clin Nutr 75, 749–753.
56 Call J, Voelker K, Wolff A, McMillan R, Evans N,
Hulver M, Talmadge R & Grange R (2008) Endurance
capacity in maturing mdx mice is markedly enhanced
by combined voluntary wheel running and green tea
extract. J Appl Physiol 105, 923–932.
57 Nakae Y, Hirasaka K, Goto J, Nikawa T, Shono M,
Yoshida M & Stoward PJ (2008) Subcutaneous
injection, from birth, of epigallocatechin-3–gallate, a
component of green tea, limits the onset of muscular
dystrophy in mdx mice: a quantitative histological,
immunohistochemical and electrophysiological study.
Histochem Cell Biol 129, 489–501.
58 Evans NP, Call JA, Bassaganya-Riera J, Robertson JL
& Grange RW (2010) Green tea extract decreases
muscle pathology and NF–jB immunostaining in
regenerating muscle fibers of mdx mice. Clin Nutr 29,
391–398.
59 Hori YS, Kuno A, Hosoda R, Tanno M, Miura T,
Shimamoto K & Horio Y (2011) Resveratrol
ameliorates muscular pathology in the dystrophic mdx
mouse, a model for Duchenne muscular dystrophy.
J Pharmacol Exp Ther 338, 784–794.
60 Selsby JT (2011) Increased catalase expression
improves muscle function in mdx mice. Exp Physiol
96, 194–202.
61 Radley HG, De Luca A, Lynch GS & Grounds MD
(2007) Duchenne muscular dystrophy: focus on
pharmaceutical and nutritional interventions. Int J
Biochem Cell Biol 39, 469–477.
62 Malik V, Rodino-Klapac LR & Mendell JR (2012)
Emerging drugs for Duchenne muscular dystrophy.
Expert Opin Emerg Drugs 17, 261–277.
63 De Luca A (2012) Pre-clinical drug tests in the mdx
mouse as a model of dystrophinopathies: an overview.
Acta Myol 31, 40–47.
64 Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG
& Gluud C (2012) Antioxidant supplements for
prevention of mortality in healthy participants and
patients with various diseases. Cochrane Database Syst
Rev 3, CD007176.
65 El-Shafey A, Armstrong A, Terrill J, Grounds M &
Arthur P (2011) Screening for increased protein thiol
oxidation in oxidatively stressed muscle tissue. Free
Radic Res 45, 991–999.
66 Armstrong AE, Zerbes R, Fournier PA & Arthur PG
(2010) A fluorescent dual labeling technique for the
quantitative measurement of reduced and oxidized
protein thiols in tissue samples. Free Radic Biol Med
50, 510–517.
67 Terrill JR, Radley-Crabb HG, Grounds MD & Arthur
PG (2012) N–acetylcysteine treatment of dystrophic
mdx mice results in protein thiol modifications and
inhibition of exercise induced myofibre necrosis.
Neuromuscul Disord 22, 422–434.
68 Radley-Crabb H, Terrill J, Shavlakadze T, Tonkin J,
Arthur P & Grounds MD (2012) A single 30 min
treadmill exercise session is suitable for ‘proof-of
concept studies’ in adult mdx mice: a comparison of
the early consequences of two different treadmill
protocols. Neuromuscul Disord 22, 170–182.
69 Whitehead NP, Pham C, Gervasio OL & Allen DG
(2008) N–acetylcysteine ameliorates skeletal
muscle pathophysiology in mdx mice. J Physiol 585,
2003–2014.
70 Williams IA & Allen DG (2007) The role of reactive
oxygen species in the hearts of dystrophin-deficient
mdx mice. Am J Physiol 293, H1969–H1977.
71 de Senzi Moraes Pinto R, Ferretti R, Moraes LHR,
Neto HS, Marques MJ & Minatel E (2012)
N–Acetylcysteine treatment reduces TNF–a levels and
myonecrosis in diaphragm muscle of mdx mice. Clin
Nutr, doi:org/10.1016/j.clnu.2012.06.001.
72 Tesi Rocha C & Hoffman EP (2010) Limb-girdle and
congenital muscular dystrophies: current diagnostics,
management, and emerging technologies. Curr Neurol
Neurosci Rep 10, 267–276.73 Laval S & Bushby K (2004) Limb-girdle muscular
dystrophies – from genetics to molecular pathology.
Neuropathol Appl Neurobiol 30, 91–105.
74 Ueyama H, Kumamoto T, Nagao S, Masuda T,
Horinouchi H, Fujimoto S & Tsuda T (2001) A new
dysferlin gene mutation in two Japanese families with
limb-girdle muscular dystrophy 2B and Miyoshi
myopathy. Neuromuscul Disord 11, 139–145.
FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS 15
J. R. Terrill et al. Protein thiol oxidation in dysferlinopathies
Page 16
75 Anderson LVB, Davison K, Moss JA, Young C,
Cullen MJ, Walsh J, Johnson MA, Bashir R, Britton S
& Keers S (1999) Dysferlin is a plasma membrane
protein and is expressed early in human development.
Hum Mol Genet 8, 855–861.
76 Glover L & Brown RH (2007) Dysferlin in membrane
trafficking and patch repair. Traffic 8, 785–794.
77 Lek A, Evesson F, Sutton B, North K & Cooper T
(2012) Ferlins: regulators of vesicle fusion for auditory
neurotransmission, recpetor trafficking and membrane
repair. Traffic 13, 185–194.
78 Bansal D, Miyake K, Vogel SS, Groh S, Chen CC,
Williamson R, McNeil PL & Campbell KP (2003)
Defective membrane repair in dysferlin-deficient
muscular dystrophy. Nature 423, 168–172.
79 de Morr�ee A, Hensbergen PJ, van Haagen HH, Dragan
I, Deelder AM, ‘t Hoen PAC, Frants RR & van der
Maarel SM (2010) Proteomic analysis of the dysferlin
protein complex unveils its importance for sarcolemmal
maintenance and integrity. PLoS One 5, e13854.
80 McNeil PL & Kirchhausen T (2005) An emergency
response team for membrane repair. Nat Rev Mol Cell
Biol 6, 499–505.
81 Covian-Nares JF, Koushik SV, Puhl HL & Vogel SS
(2010) Membrane wounding triggers ATP release and
dysferlin-mediated intercellular calcium signaling.
J Cell Sci 123, 1884–1893.
82 Rawat R, Cohen TV, Ampong B, Francia D,
Henriques-Pons A, Hoffman EP & Nagaraju K (2010)
Inflammasome up-regulation and activation in dysferlin-
deficient skeletal muscle. Am J Pathol 176, 2891–2900.
83 Nagaraju K, Rawat R, Veszelovszky E, Thapliyal R,
Kesari A, Sparks S, Raben N, Plotz P & Hoffman EP
(2008) Dysferlin deficiency enhances monocyte
phagocytosis: a model for the inflammatory onset of
limb-girdle muscular dystrophy 2B. Am J Pathol 172,
774–785.
84 Dhanarajan R, Patil AB, Alexander M, Chacko G &
Oommen A (2011) Degradation of myofibrillar
proteins and inadequate antioxidants in selective
muscle wasting of limb girdle muscular dystrophy. Int
J Case Rep Images 2, 6–11.
85 Renjini R, Gayathri N, Nalini A & Srinivas Bharath
M (2012) Oxidative damage in muscular dystrophy
correlates with the severity of the pathology: role of
glutathione metabolism. Neurochem Res 37, 885–898.
86 Potgieter M, Pretorius E, Van der Merwe C, Beukes
M, Vieira W, Auer R, Auer M & Meyer S (2011)
Histological assessment of SJL/J mice treated with the
antioxidants coenzyme Q10 and resveratrol. Micron
42, 275–282.
87 van der Spuy WJ & Pretorius E (2011) The qualitative
effects of resveratrol and coenzyme Q10 administration
on the gluteus complex muscle morphology of SJL/J
mice with dysferlinopathy. Int J Morphol 29, 876–884.
88 Ho M, Post CM, Donahue LR, Lidov HGW, Bronson
RT, Goolsby H, Watkins SC, Cox GA & Brown RH
Jr (2004) Disruption of muscle membrane and
phenotype divergence in two novel mouse models of
dysferlin deficiency. Hum Mol Genet 13, 1999–2010.
89 Halliwell B & Whiteman M (2004) Measuring reactive
species and oxidative damage in vivo and in cell
culture: how should you do it and what do the results
mean? Br J Pharmacol 142, 231–255.
90 Tohma H, Hepworth AR, Shavlakadze T, Grounds
MD & Arthur PG (2011) Quantification of ceroid and
lipofuscin in skeletal muscle. J Histochem Cytochem
59, 769–779.
91 Sohal R & Brunk U (1989) Lipofuscin as an indicator
of oxidative stress and aging. Adv Exp Med Biol 266,
17–26.
92 Stroikin Y, Dalen H, L€o€of S & Terman A (2004)
Inhibition of autophagy with 3–methyladenine results
in impaired turnover of lysosomes and accumulation
of lipofuscin-like material. Eur J Cell Biol 83, 583–590.
93 Deconinck N & Dan B (2007) Pathophysiology of
Duchenne muscular dystrophy: current hypotheses.
Pediatr Neurol 36, 1–7.
94 Shavlakadze T & Grounds M (2006) Of bears, frogs,
meat, mice and men: complexity of factors affecting
skeletal muscle mass and fat. BioEssays 28, 994–
1009.
95 Haycock JW, Neil SM, Jones P, Harris JB & Mantle D
(1996) Oxidative damage to muscle protein in Duchenne
muscular dystrophy. NeuroReport 8, 357–361.
96 Kaczor JJ, Hall JE, Payne E & Tarnopolsky MA
(2007) Low intensity training decreases markers of
oxidative stress in skeletal muscle of mdx mice. Free
Radic Biol Med 43, 145–154.
97 Disatnik MH, Chamberlain JS & Rando TA (2000)
Dystrophin mutations predict cellular susceptibility to
oxidative stress. Muscle Nerve 23, 784–792.
98 Nakae Y, Stoward PJ, Kashiyama T, Shono M, Akagi
A, Matsuzaki T & Nonaka I (2004) Early onset of
lipofuscin accumulation in dystrophin-deficient skeletal
muscles of DMD patients and mdx mice. J Mol Histol
35, 489–499.
99 Grounds MD, Radley HG, Lynch GS, Nagaraju K &
De Luca A (2008) Towards developing standard
operating procedures for pre-clinical testing in the mdx
mouse model of Duchenne muscular dystrophy.
Neurobiol Dis 31, 1–19.
100 Levine RL, Garland D, Oliver CN, Amici A, Climent
I, Lenz AG, Ahn BW, Shaltiel S & Stadtman ER
(1990) Determination of carbonyl content in
oxidatively modified proteins. Methods Enzymol 186,
464–478.101 Hawkins CL, Morgan PE & Davies MJ (2009)
Quantification of protein modification by oxidants.
Free Radic Biol Med 46, 965–988.
16 FEBS Journal (2013) ª 2013 The Authors Journal compilation ª 2013 FEBS
Protein thiol oxidation in dysferlinopathies J. R. Terrill et al.