Spectrochimica Acta Part B 57 (2002) 1361–1452 0584-8547/02/$ - see front matter 2002 Elsevier Science B.V. All rights reserved. PII: S0584-8547 Ž 02 . 00069-1 Review Reaction cells and collision cells for ICP-MS: a tutorial review Scott D. Tanner*, Vladimir I. Baranov, Dmitry R. Bandura Perkin Elmer SCIEX, 71 Four Valley Drive, Concord, Ont., Canada L4K 4V8 Received 12 December 2001; accepted 20 June 2002 Abstract This paper reviews the literature published to September 2001 relating to the history, design, operation and application of linear radio-frequency (r.f.)-driven multipole collision cells and reaction cells in combination with inductively coupled plasma mass spectrometry. The available material is supplemented with original experimental data that demonstrates the principles presented. The relation of these devices to collision cells for organic mass spectrometry and to the three-dimensional ion trap is discussed in its historical context. A general tutorial on the fundamentals of ion collision and reaction, including thermochemistry, energy transfer and reaction kinetics, is given. Consideration is given to some of the fundamental aspects of operation and design of linear r.f. devices. This historical and fundamental framework then allows the tutorial to focus on the promotion and control of ion–molecule chemistry in linear r.f.-multipole cells for elemental analysis. Vacuum requirements are considered in some detail, and deal in particular with the issue of contamination of the reaction gas. Special attention is paid to the thermal characteristics of the ions in the cell, as this has important implications for the application of the available databases of thermochemical and thermal kinetic data to the development of analytical methods. Calculation and experimental validation of the efficiency of the ion–molecule chemistry leads to the recognition that secondary, sequential chemistry can play a limiting role in the realization of the potential of the cell method. The two principal means of controlling the analytical impact of the secondary chemistry, through post-cell kinetic energy discrimination and through in-cell mass-bandpassing are discussed and contrasted through spectral data acquired for different reaction gas types and pressures. The available literature on the application of collision cells and reaction cells for the analysis of samples of high purity, environmental, geological and biological materials is critically reviewed. 2002 Elsevier Science B.V. All rights reserved. Keywords: Collision cell; Reaction cell; ICP-MS; Collision induced fragmentation; Ion–molecule chemistry *Corresponding author. Tel.: q1-905-660-9006x262; fax: q1-905-660-2623. E-mail address: [email protected](S.D. Tanner).
Transcript
Spectrochimica Acta Part B 57(2002) 1361–1452
0584-8547/02/$ - see front matter� 2002 Elsevier Science B.V. All rights reserved.PII: S 0 5 8 4 - 8 5 4 7Ž0 2.0 0 0 6 9 - 1
Review
Reaction cells and collision cells for ICP-MS: a tutorial review
Scott D. Tanner*, Vladimir I. Baranov, Dmitry R. Bandura
Perkin Elmer SCIEX, 71 Four Valley Drive, Concord, Ont., Canada L4K 4V8
Received 12 December 2001; accepted 20 June 2002
Abstract
This paper reviews the literature published to September 2001 relating to the history, design, operation andapplication of linear radio-frequency(r.f.)-driven multipole collision cells and reaction cells in combination withinductively coupled plasma mass spectrometry. The available material is supplemented with original experimentaldata that demonstrates the principles presented. The relation of these devices to collision cells for organic massspectrometry and to the three-dimensional ion trap is discussed in its historical context. A general tutorial on thefundamentals of ion collision and reaction, including thermochemistry, energy transfer and reaction kinetics, is given.Consideration is given to some of the fundamental aspects of operation and design of linear r.f. devices. This historicaland fundamental framework then allows the tutorial to focus on the promotion and control of ion–molecule chemistryin linear r.f.-multipole cells for elemental analysis. Vacuum requirements are considered in some detail, and deal inparticular with the issue of contamination of the reaction gas. Special attention is paid to the thermal characteristicsof the ions in the cell, as this has important implications for the application of the available databases ofthermochemical and thermal kinetic data to the development of analytical methods. Calculation and experimentalvalidation of the efficiency of the ion–molecule chemistry leads to the recognition that secondary, sequential chemistrycan play a limiting role in the realization of the potential of the cell method. The two principal means of controllingthe analytical impact of the secondary chemistry, through post-cell kinetic energy discrimination and through in-cellmass-bandpassing are discussed and contrasted through spectral data acquired for different reaction gas types andpressures. The available literature on the application of collision cells and reaction cells for the analysis of samplesof high purity, environmental, geological and biological materials is critically reviewed.� 2002 Elsevier Science B.V. All rights reserved.
1362 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Contents
1. Introduction....................................................................................................................13632. Experimental..................................................................................................................13643. Nomenclature(definitions) ..............................................................................................13644. History ...........................................................................................................................1366
4.1. Tandem mass spectrometry........................................................................................13664.2. Pressurized multipole ion guides for reaction studies....................................................13694.3. The ion trap as a reaction cell....................................................................................13704.4. Pressurized multipole cells for ICP-MS.......................................................................1371
5. Collisional processes.......................................................................................................13755.1. Energy transfer.........................................................................................................13755.2. Collisional fragmentation...........................................................................................1375
6.1.1. Enthalpy of reaction............................................................................................13776.1.2. Specificity of thermal ion–molecule reaction.........................................................1379
6.2. Kinetics ...................................................................................................................13796.2.1. The Langevin collision theory..............................................................................13806.2.2. Measurement of reaction rate constants.................................................................13816.2.3. Ion–molecule reaction profiles in a r.f.-driven reaction cell.....................................1383
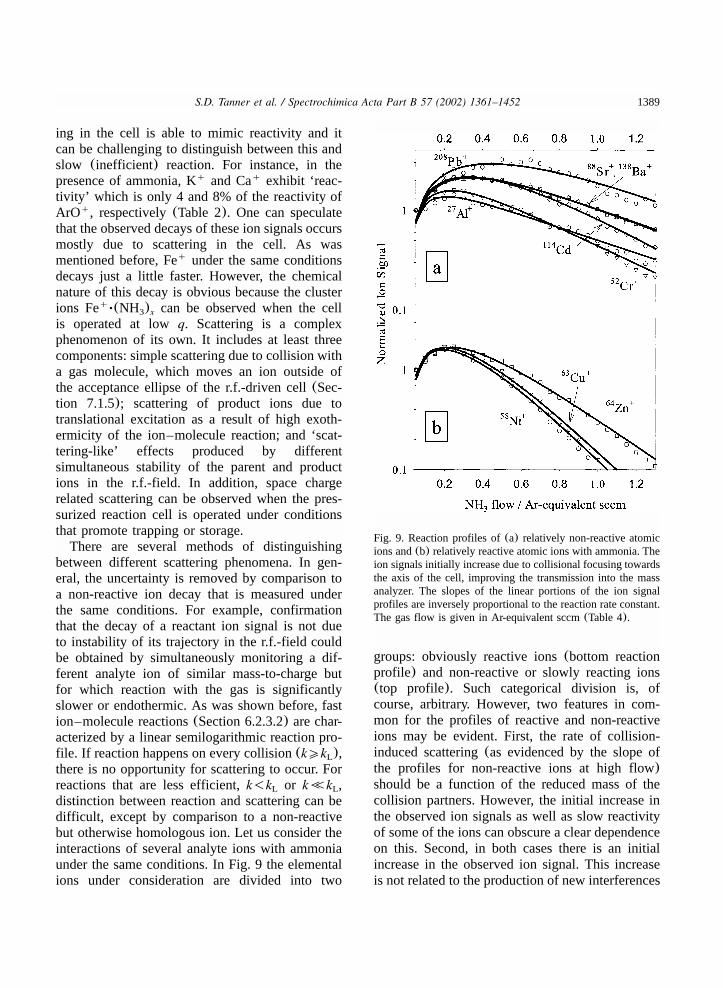
6.2.3.1. Types of ion–molecule reactions....................................................................13836.2.3.2. Plasma ion reactive decay..............................................................................13846.2.3.3. Reactive decay of an interfering plasma ion....................................................13866.2.3.4. Product ions of the primary ion–molecule reaction..........................................13876.2.3.5. Scattering versus reactivity for parent and product ions....................................1388
7. Linear r.f. devices...........................................................................................................13907.1. General characteristics of r.f. multipoles......................................................................1390
7.1.1. Equations of motion............................................................................................13907.1.2. Adiabaticity........................................................................................................13917.1.3. Hyperbolic versus round rods...............................................................................13917.1.4. Auxiliary excitation at the secular frequency of motion..........................................13927.1.5. Acceptance.........................................................................................................13927.1.6. Fringing fields....................................................................................................1392
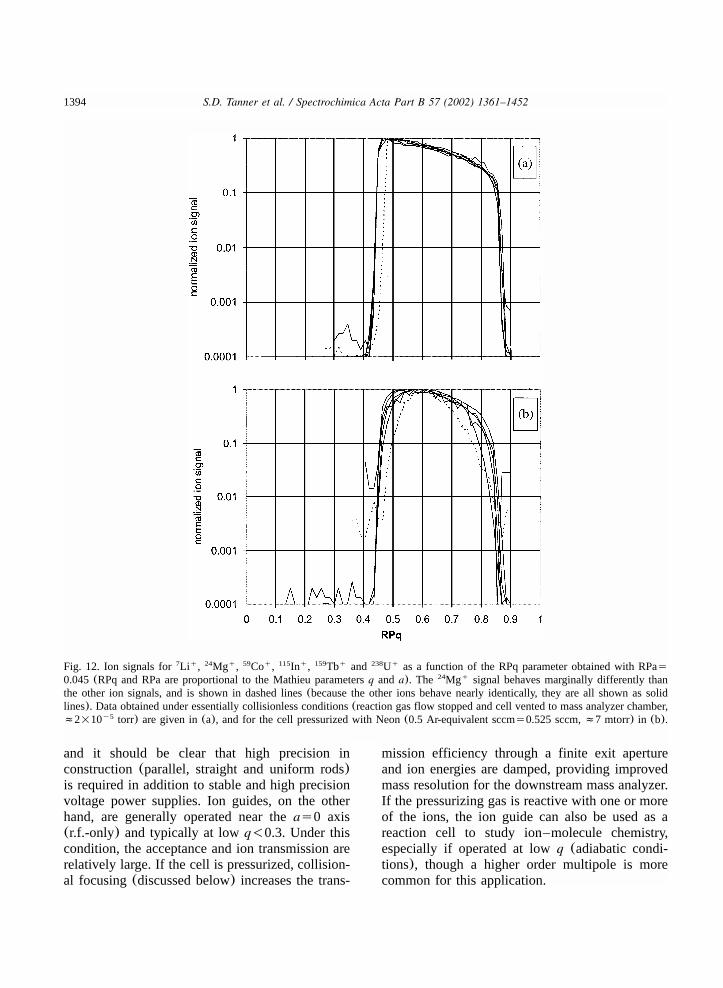
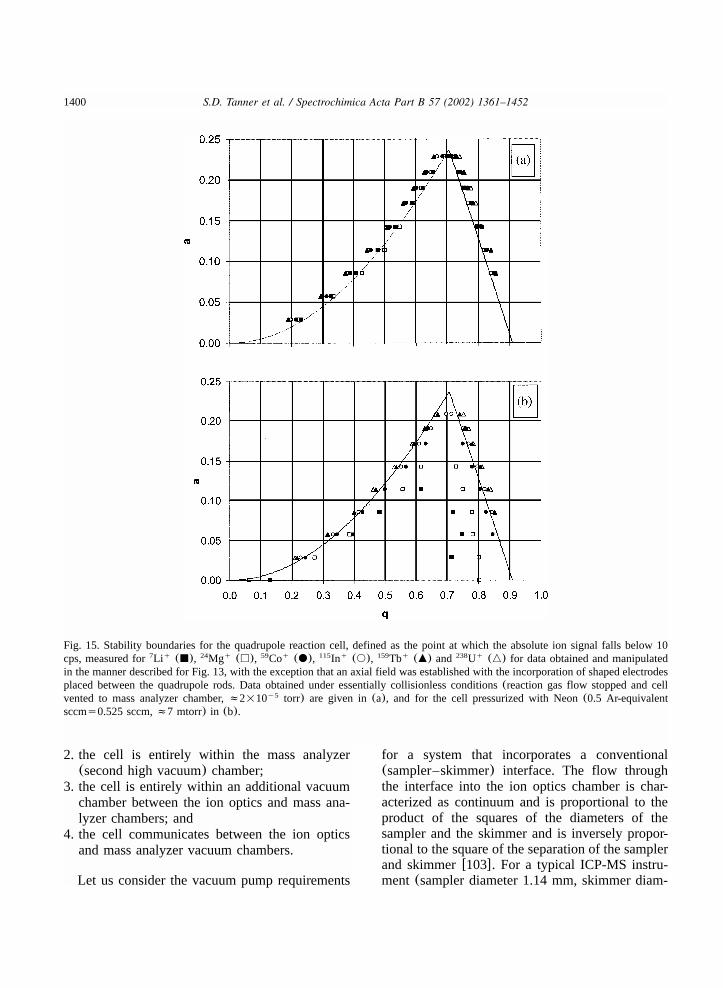
7.2. Quadrupoles.............................................................................................................13927.3. Higher order multipoles.............................................................................................13957.4. Prefilters..................................................................................................................13977.5. Axial fields ..............................................................................................................1398
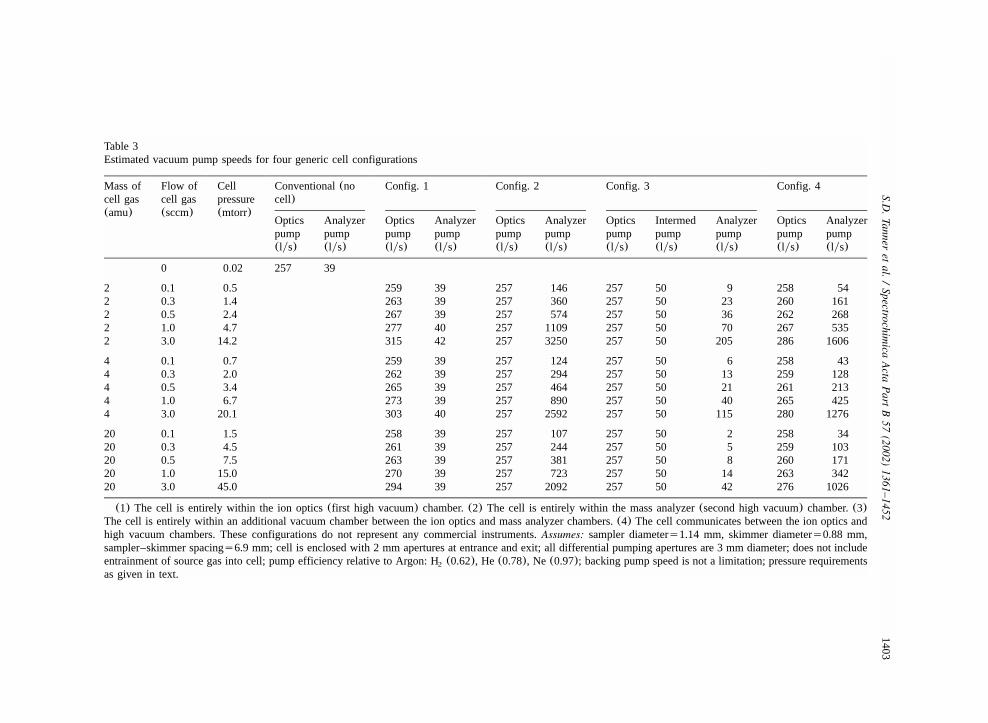
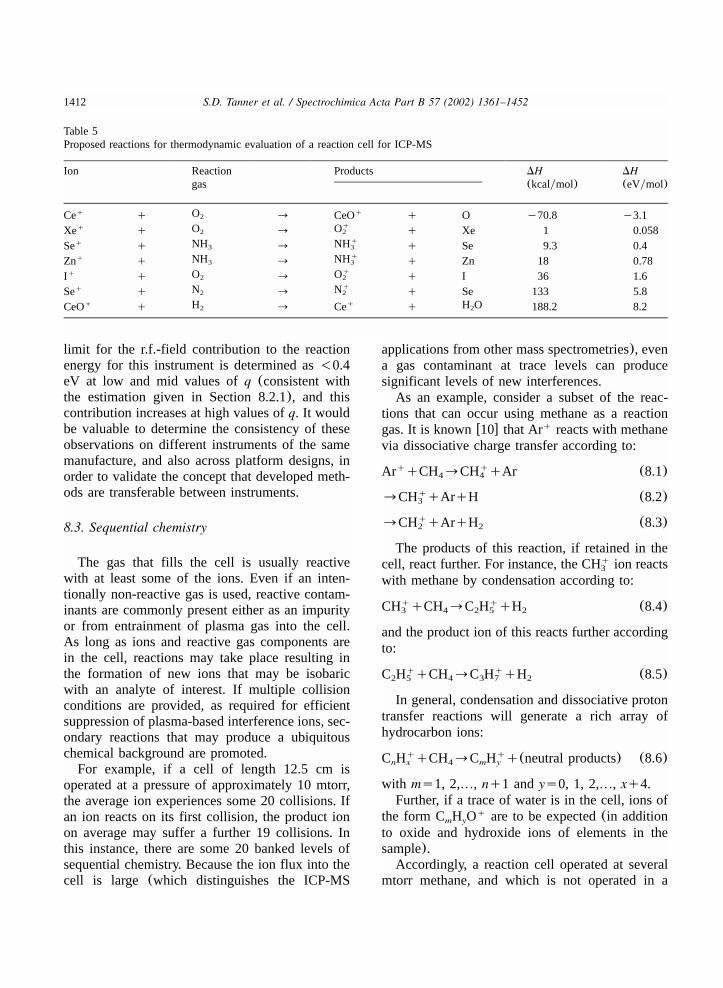
8. Ion chemistry in r.f. devices for analytical ICP-MS............................................................13998.1. Vacuum considerations..............................................................................................13998.2. Reaction energy........................................................................................................1407
8.2.1. Thermalization and collisional focusing.................................................................14078.2.2. Temporal homogenization....................................................................................14098.2.3. r.f. contribution to reaction energy........................................................................14108.2.4. Transferability of methods...................................................................................1411
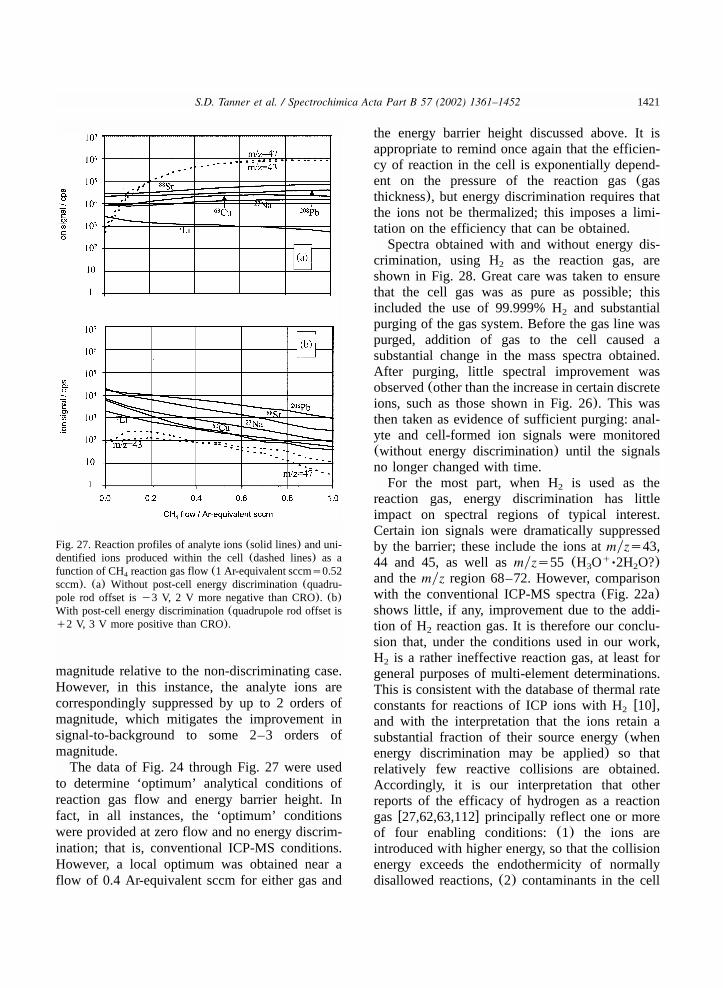
8.4.1. Post-cell kinetic energy discrimination..................................................................1414
1363S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
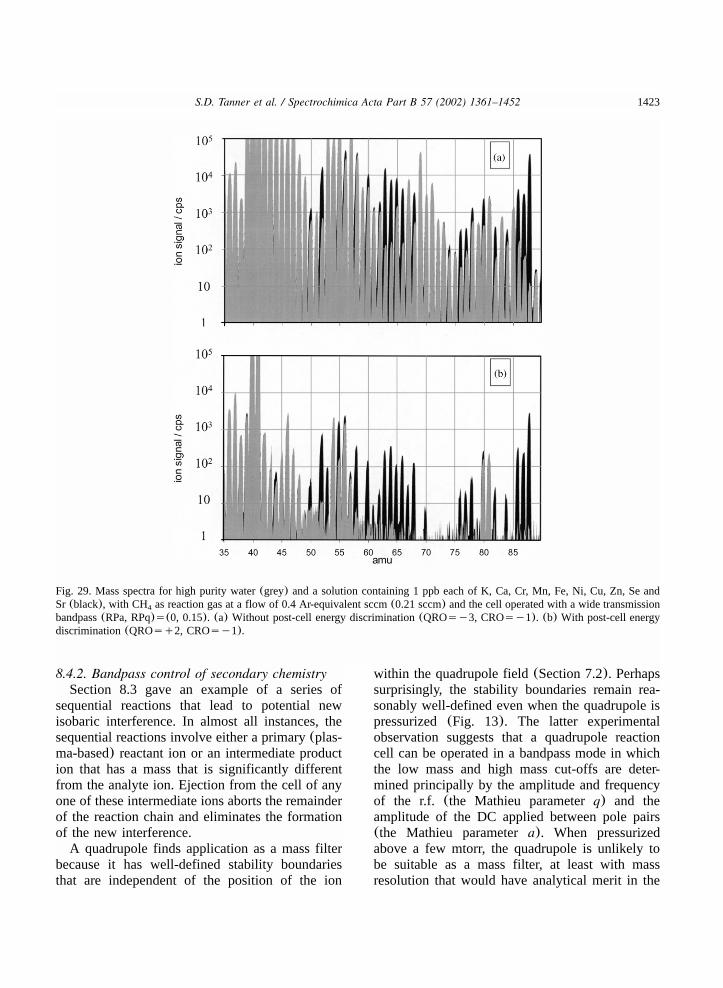
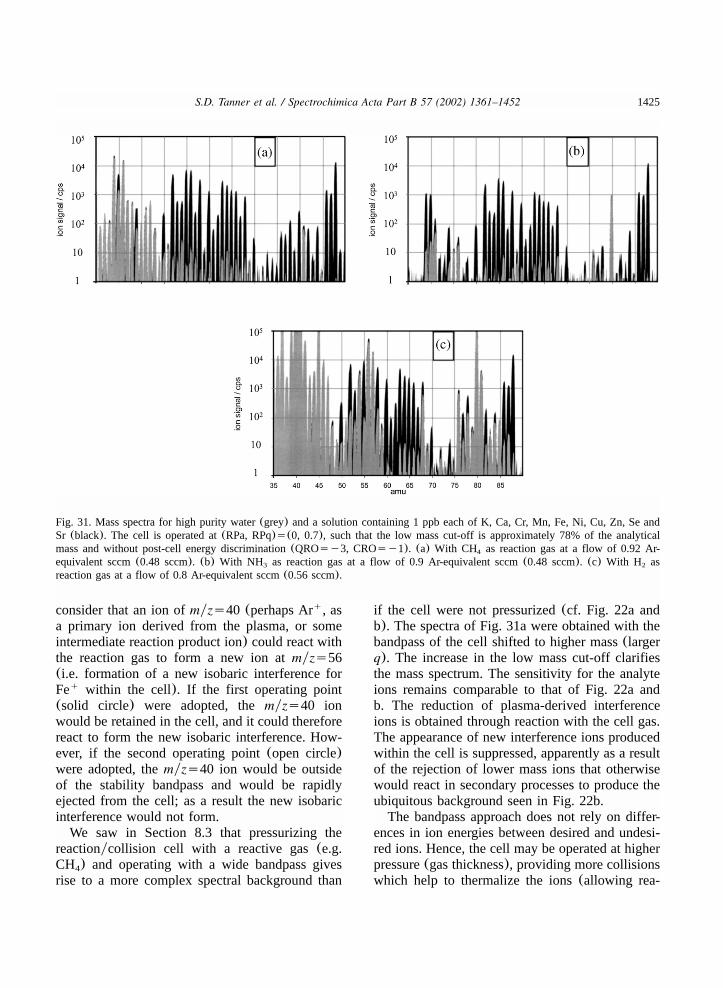
8.4.2. Bandpass control of secondary chemistry..............................................................14238.4.3. Promotion of secondary chemistry........................................................................1429
9. Applications ...................................................................................................................14319.1. Method development.................................................................................................14339.2. Developed methods...................................................................................................1434
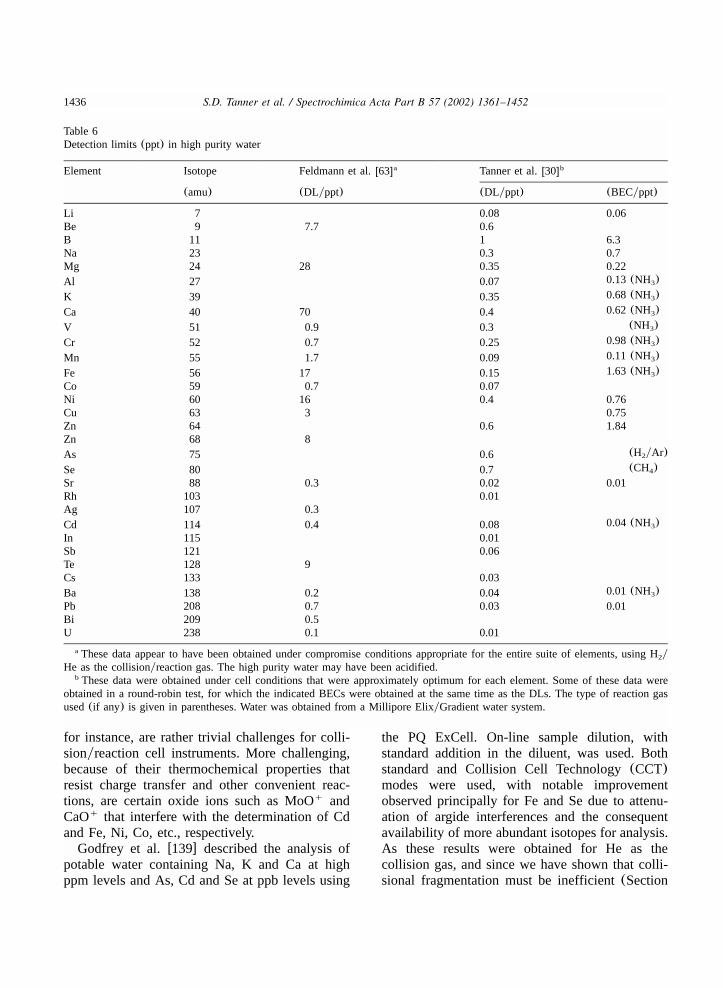
9.2.1. High purity water and process chemicals(semiconductor) ......................................14349.2.2. Environmental....................................................................................................14359.2.3. REE and actinide oxide and hydroxide ions...........................................................14399.2.4. Geological..........................................................................................................14419.2.5. Biological ..........................................................................................................1444
It is our opinion that ion–molecule chemistry,enacted in a radio-frequency(r.f.)-driven multipolethat is pressurized with a reactive gas, will domi-nate high performance elemental analysis byinductively coupled plasma mass spectrometry(ICP-MS) in the near- and mid-future. Ion–mole-cule reactions have been studied for several dec-ades, having their principal application ininterstellar and ionospheric chemistryw1,2x. Thebasic tenets governing the kinetics and reactivityare well understoodw3–11x. The r.f.-only multipolehas found significant application for the study ofnear-thermal or energy-selected ion–moleculereactions, particularly when operated at low r.f.amplitude and high frequencyw12–14x. In addi-tion, the r.f.-only ‘collision cell’ has been impor-tant in organic tandem mass spectrometry for 20years, where it is used to promote collision induceddissociation(CID) and to confine and transportthe resultant fragment ions to a downstream massanalyzerw15–19x. Two important papers appearedin 1989 that discussed the potential of CID andreactivity for the inorganic applicationw20,21x. Itwas shownw20x that CID is an ineffective processfor several polyatomic ions obtained from the ICP,but that ion–molecule chemistry using Ow20x,2
Xe, CH or C H w21x was very specific and4 2 6
efficient. Perhaps because of perceived deficien-cies, difficulties or expense, or because attentionwas focused on other perceived instrument defi-
ciencies, the subject remained dormant for severalyears. The catalyst for the recent resurgence ininterest was the 1994 publication of Barinaga andKoppenaal w22x in which it was observed thatargide ions were nearly absent in the spectraobtained by ICP-IT-MS(ion trap), and this wasimportantly ascribed, at least in part, to chargetransfer reactions of these ions with adventitiouswater molecules. Subsequent work by the samegroupw23–26x showed highly effective applicationof ion–molecule chemistry with other gases(H ,2
O and CH) for general and specific challenges.2 4
The first commercial enactment of a pressurizedmultipole, specific for the ICP-MS application inthe form of a hexapole collision cell, was reportedby Turner et al.w27x, and is now available fromMicromass as the Platform� (a similar technologyis used with a different intent in the MicromassIsoProbe�). Originally, it was claimed in this workthat the effective process was CID using He as thecollision gas. Later, H was used as a reaction gas2
in mixture with He. For reasons now apparent,reactions with H and impurities in the collision2
gas were principally responsible for the observedbenefit. Subsequent commercial entries were madeby Perkin Elmer-SCIEX(ELAN DRC�), Ther-�
mo Elemental(PQ ExCell ), and Agilent(7500c).�
It is the intent of this work to review thetechnology of pressurized r.f.-driven multipolecells and to review the fundamentals of ion–molecule chemistry as it might be applied to the
1364 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
ICP-MS application. The adoption of these tech-nologies is just now taking place, such that insuf-ficient information related to operation or thedevelopment of methods has been published.Accordingly, we will complement our review ofthe existing literature with examples from our ownlaboratory. The resultant manuscript thus becomesa tutorial review, where we provide our owninterpretation and understanding of the processesdiscussed.
2. Experimental
In support of some of the theoretical consider-ations presented in this work, we include somepreviously unpublished demonstrative dataobtained in our laboratory. These data wereobtained using several generations of the proto-types of the ELAN DRC and including the com-mercial instrument itself. The hardware and theoperating modes of these instruments have beendescribedw28,29x. The DRC (Dynamic ReactionCell�) is a high precision quadrupole that isenclosed and may be pressurized. It is operated ata low, user-selected r.f. amplitude, typically 200V measured peak-to-peak. The frequency of therf
r.f. signal is adjusted dynamically in accordancewith the user-selected value of the operatingparameter RPq(related to the Mathieu parameterq, defined below). A DC potential may be appliedbetween pole pairs, having an amplitude definedby the user-selected operating parameter RPa(related to the Mathieu parametera, also definedbelow). The flow of gas into the cell is controlledby a mass flow controller(Model 1479, MKS,Andover, MA). Two principal modes of operationof these systems are supported: the standard mode(which emulates conventional ICP-MS), in whichthe reaction cell is operated at low pressure(f10 torr) obtained by shutting off the reactiony5
gas flow and venting the cell to the high vacuum(mass analyzer) chamber, and the DRC mode,obtained by closing the vent and pressurizing thecell with a reactive(or non-reactive) gas. All ofthe reported data were obtained under normal‘robust’ plasma conditions. Typical operating con-ditions and voltages are given in Ref.w30x.
The data given in Sections 8.3, 8.4 and 9.2 wereobtained using a prototype of the ELAN DRC ,Plus
which differs from the earlier instrument with theaddition of electrodes inserted between the activerods of the reaction cell quadrupole, as describedin Section 7.5, to provide an axial acceleratingfield of approximately 0.2 Vycm.
3. Nomenclature (definitions)
Various terms used in the discussion of reactionenergetics and r.f. multipole theory have properdefinitions, but they are often misused or micro-defined. Therefore, we define our usage of theseterms with specific reference to the pressurizedmultipole cell used for ICP-MS:
A bimolecular process is a reaction(or colli-sion) involving two particles, as is described bythe reaction
AqB™CqD (3.1)
A termolecular reaction involves three particles,where the intermediate transition state of a bimo-lecular process is stabilized by collision with athird particle which removes an amount of energy.These reactions are also calledclustering reactionsor association reactions, and may be described bythe reaction
AqBqM™ABqM (3.2)
which is actually comprised of the two steps:
AqB™AB* (3.3)
AB* qM™ABqM (3.4)
It is convenient to distinguish a bimolecularprocess from a termolecular process on the basisof the pressure dependence of the rate of reaction.The reaction rate of a bimolecular process islinearly dependent on the reaction gas density,whereas that for a termolecular process is depend-ent on the product of the pressure and the reactiongas density. In the instance where a buffer gas isnot used(i.e. the cell is pressurized only by thereaction gas), a bimolecular process is first order,and a termolecular is second order, with respect tothe cell pressure.
Thermochemistry refers to the energy balance ina reaction process. The ‘reaction enthalpy’,DH ,r,T
1365S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
is the difference between the sums of the gas phaseheats of formation,DH , of the reactants andf,T
products at the temperatureT. When the enthalpyis negative, the reaction is said to beexothermic,and when positive it isendothermic. An exother-mic reaction releases energy to the environmentand, thus, isthermodynamically allowed. An endo-thermic reaction absorbs energy from the environ-ment and is thermodynamically disallowed. Anexothermic process may proceed, and is often fast.An endothermic process usually does not proceed,or is very slow (taking place only for thosereactants with energies in the high energy tail ofthe distribution). In fact, the true discriminator ofa thermodynamically allowed gas phase reactionis the ‘free energy of reaction’,DG , whichr,T
includes an entropy term(DG sDH yTDS ),r,T r,T r,T
but in most instances the entropy differencebetween reactants and products of a bimolecularprocess in the gas phase is negligible.
A thermal condition is one under which anensemble of particles has a Maxwell–Boltzmannenergy distribution which has the same temperaturein all degrees of freedom. A thermal ensemble canbe adiabatic(insulated from the container) or canbe in equilibrium with the container walls. There-fore, a ‘thermal ion beam’ is an oxymoron, due toits obvious anisotropy.
A non-thermal condition is one under which anensemble of particles has a non-Maxwell–Boltz-mann energy distribution in one or more degreesof freedom. It can be adiabatic(insulated from thecontainer) or be out of equilibrium with the con-tainer walls. An example of a non-thermal ensem-ble is an ion beam.
A thermal process, with respect to the subjectof this paper, is a reaction that proceeds underthermal conditions. That is, the process is exother-mic and an external source of energy is absent oris irrelevant.
A non-thermal process, with respect to thesubject of this paper, is a reaction that proceedswhen the reactants are not in equilibrium or arenot adiabatically insulated from the container wallsthat are themselves not in equilibrium with thereactants. Such a reaction can be endothermicunder thermal conditions and requires an external
energy source to proceed under non-thermalconditions.
To thermalize a system is to convert a non-thermal ensemble into a thermal ensemble, usuallyby means of spontaneous energy redistribution(equilibration). Thermalization in the case of anion beam injected into a pressurized multipole canbe achieved(to some extent) by means of multiplecollisions with a buffer gas under thermalconditions.
A lab normal condition is one under which athermal ensemble of ions is in equilibrium with abuffer gas under thermal conditions. In most prac-tically important instances, the thermal ensembleof ions should be adiabatically insulated from ther.f. drive of a pressurized multipole.
An adiabatic condition, with special referenceto an r.f. multipole device, is one in which theions do not, on average, gain energy from theapplied r.f. field. This condition is achieved whenthe fundamental frequency of ion motion is essen-tially uncoupled from the applied r.f. frequency,and conveniently describes a case of insulation ofthe ion flow from the r.f. field. It is generallyconsidered that an adiabatic condition is achievedfor r.f.-only multipole devices only when the sta-bility parameterq (see below) is less than approx-imately 0.3w12x.
The parametersa and q describe the regions ofstable or unstable trajectories in an r.f. multipolefield. In general:
eVdcas2n(ny1) (3.5)2 2mv r0
eVrfqsn(ny1) (3.6)2 2mv r0
wheren is the order of the multipole(number ofpairs of poles), e is the elementary charge,V isdc
the DC voltage applied between pole pairs andV is the peak-to-peak r.f. voltage(note that thisrf
definition follows that of Dawsonw31x), m is theion mass,v is the r.f. angular frequency(theapplied r.f. frequency multiplied by 2p), andr is0
the radius of the inscribed circle that is tangentialto the inner surfaces of the rods of the multipolearray.
1366 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Stability, as referred to multipole operation,describes the region in(a, q) parametric spacewhich allows transmission of ions through aninfinitely large theoretical multipole field(fieldonly, no multipole array present). Ions inside ofthe stability region belong to a stable trajectory.
Acceptance, referred to a multipole array,describes the(position, velocity) space(r, drydtin cylindrical coordinates) for which an ionbelonging to a stable trajectory has a maximumradial displacement less thanr . The acceptance0
of the multipole is directly related to the transmis-sion efficiency.
Collisional energy damping and collisionalfocusing are concomitant phenomena(a chickenand egg conundrum) referring to the loss of axialion energy and the reduction of the width of theion energy distribution towards thermalization witha collision gas. This process manifests itself inmigration of ions towards the axis of a multipole.
In this paper, we use the term pressure in twosenses. The term pressure is appropriate in discus-sion of collision frequency, and applies to consid-eration of vacuum system requirements and,amongst other things, to collisional stabilization ofreaction intermediate complexes(termolecularreactions). However, we often refer to pressure asindicative of the number of collisions that an ionsuffers in transit through a device, where we haveassumed a fixed length for the device(12.5 cmfor the cell length, unless otherwise specified). Amore correct term for the latter usage isgasthickness, which is given by the product of the gasdensity and the length of the device. An increasein pressure results in an increase in the number ofcollisions that an ion suffers for a given device,but where devices differ in length, gas thicknessgives a better comparison of the total number ofcollisions.
4. History
4.1. Tandem mass spectrometry
The two dimensional r.f.-driven collision cellwas first introduced to mass spectrometry by Yostand Enkew15x in the triple quadrupole configura-tion. A r.f.-only enclosed quadrupole(the collision
cell) was placed between two mass analyzingquadrupoles. Ions of interest, called the ‘parentions’, from the composite of ions from the ionsource are mass-selected in the first quadrupole(Q1). These ions are injected at a selectable energyinto the collision cell(Q2) which has been pres-surized with a target(collision) gas. Upon impactat the collision energy with the collision gas, theions are fragmented. The resultant daughter ionsare confined in the r.f. field and transmitted to thesecond mass analyzer(Q3) where the daughterions are mass analyzed. The process is shownschematically in Fig. 1. The fragmentation processhas been called CID or collision activated disso-ciation. In its initial enactment, the collision cellwas operated at relatively low pressures thatallowed only a few collisions. In this instance, asignificant fraction of the incident ion energy wasretained and each collision occurred at significantenergy. The process of fragmentation may be asingle collision event or a multiple collision eventin which the vibrational degrees of freedom of thepolyatomic ion are pumped in sequential collisionsto the state of fragmentation. Another enactmentof the device used a cross-molecular beam in anopen construction collision cellw18,19x. It is clearthat the fragmentation pathways are a function ofthe incident energy of the ions, the mass ratio ofthe ion-to-target gas, and the number of degreesof freedom of the ion and the target gas. In essence,the daughter ion spectrum may be viewed as ajigsaw puzzle from which the structure of theparent ion may be determined. This mode ofoperation may be termed a daughter ion scan. Withtwo mass analyzers, other modes of operation arealso feasible, including neutral loss scan(wherethe two mass analyzers are scanned in concertwith a fixed mass offset), and parent ion scan(where the Q3 mass analyzer is held to transmit afixed daughter ionmyz and the Q1 analyzer isscanned to determine which parent ions give riseto the daughter ion fragment).
The triple quadrupole is more properly describedas a tandem mass spectrometer, in recognition thatsubsequent iterations of the device included higherorder multipoles(specifically hexapole and octa-pole) as the collision cellw32x. The advantageclaimed for the higher order multipole in this
1367S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 1. Schematic of a tandem quadrupole MSyMS system for organic mass spectrometry. Ions are produced in a corona or elec-trospray source and transmitted through a vacuum interface and r.f.-only multipole ion guide. Ions of interest(parent ions) areselected by a first mass-analyzing quadrupole and are transmitted to a collision cell where they are fragmented in collisions withan inert gas at energies that exceed the bond strength(s) of the parent ion. The daughter ions are confined and transmitted to thesecond mass-analyzing quadrupole. The daughter ions provide structural information regarding the parent ion. Several types of linkedscan modes are possible.(Figure provided by MDS SCIEX.)
application is more efficient confinement andtransport of the daughter ions, due to the deeperand steeper pseudo-potential well and wider sta-bility region of these devices as compared to aquadrupolew12,33–36x. In practice, the r.f.-onlyhexapole and octapole show a practical increasein the mass range of confinement of the order of10% towards high mass and 30% at low massw32x. The higher order multipoles are reported toprovide lower transmission efficiency than thequadrupole at high ion energy, but comparabletransmission at low energies(presumably the con-ditions appropriate for the ICP-MS application)w32x.
Tandem MS has also been enacted in situ in anion trap mass spectrometer, wherein the trap isoperated to confine and then isolate the parent ion,an auxiliary r.f. field is applied to excite the ionand induce CID, and the daughter ions are trappedand mass analyzedw37x. Multiple sequential frag-mentation events may be obtained(MS ) throughn
sequential isolation and excitationw37x. In the 2D
quadrupole, such MS operation requires(2ny1)n
multipole devices in seriesw38,39x. Recent workwith a 2D ion trap has shown the ability toaccomplish MS in a single 2D collision cellw40x.n
Of course, it was recognized early on that,alternative to CID, the tandem MS collision cellcould be used to promote ion–molecule reactions.Hence, the neutral loss scan could also be used asa neutral gain scan, where the ion reacts byaddition or substitution to produce a product ionof higher myz. In some instancesw41,42x, thiscapability is used to facilitate resolution of isobarsby discrimination on the basis of thermochemicalproperties (e.g. on the basis of proton affinityrelative to ammonia as a reaction gas, where onlythe higher proton affinate isobar survives the cell),in a manner similar to conventional chemicalionization ion sources. It is also well known thatthe quadrupole may be operated in a notch filtermode, in which an auxiliary excitation frequencyat the fundamental frequency of motion(which ismass dependent) is applied in order to selectively
1368 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
eject a particularmyz. Watson et al.w43x used thiscapability to delineate the sequence of reactionsthat might take place in a cell operated undermultiple collision conditions. In this instance, theauthors selected a parent ion(Fe from ironq
carbonyl) in Q1 and introduced this ion into thecell operated with 1 mtorr of allyl chloride. Anumber of organic and organometallic ions wereobserved in the daughter ion spectrum, and severalof these were perceived to be due to sequentialreactions of previous daughter ions. Notch filteringat myzs41 caused the suppression of a numberof ions which were thus identified as products ofsubsequent reactions of the C H ion, which itselfq
3 5
is a daughter ion of the Fe reaction with allylq
chloride; other ions were not affected by the notchfiltering at myzs41, indicating that these ionswere not progeny of C H .q
3 5
Tandem MS has found exceptional applicationin organic mass spectrometry, most commonly withoperation of the collision cell for CID. It has beenwidely used for environmental analyses, perhapsmost notably for the determination of dioxins insoils and incineration ashesw44x. With the adventof various incarnations of electrospray ionizationw45x, it has had a profound impact on pharmaceu-tical drug discoveryw46x and related fields suchas proteomicsw47,48x. A good introduction totandem MS, though by now a little dated, is thebook by Busch et al.w49x.
The first application of tandem MS with theICP ion source was reported by Douglasw20x. Theinitial intent was to perform CID on polyatomicions (e.g. Ar , ClO , ArCl and CeO) in orderq q q q
2
to obviate their interference on isobaric atomicions. Clearly, the collision energy must be suffi-ciently large to promote such fragmentation(i.e.the deposited energy must exceed the bondstrength). At 50 eV (in the laboratory frame), itwas observed that the loss cross-section(propor-tional to the loss rate) of atomic ions is comparableto that for the polyatomic ions. This was ascribedto the occurrence of charge transfer reactions ofthe atomic ions with the argon gas target. That is,the collision energy required to successfully frag-ment many metal polyatomic ions exceeds thedifference in the ionization potentials(IPs) of theisobaric atom and the reaction gas. As a result,
conditions which enable CID of the polyatomicions also generally promote charge transfer of theelemental ion of interest, resulting in a correspond-ing loss in sensitivity. This is in addition to thesimple scattering loss of both the polyatomic ionand the atomic ion. Hence, it was recognized thatCID was unlikely to provide large gains in themetal ion to molecular ion ratios. On the otherhand, it was found that some atomic ions rapidlyoxidize in reaction with O , but that the oxide ion2
frequently does not further oxidize to the dioxide.Operation of the triple quadrupole in the neutralgain mode (for Dms16 amu) with O as the2
reaction gas transmits only those ions that add 16amu(O) in the collision cell. Accordingly, Ce isq
transmitted(as CeO ), but CeO is not transmit-q q
ted (as CeO ) since CeO does not furtherq q2
oxidize, as is shown in Fig. 2. The cautiousconclusion that ‘for some special cases«ion–molecule chemistry may provide a way aroundpersistent interferences’ has greater significance inhindsight. Shortly after this was discussed, Rowanand Houk described the direct coupling of the ICPwith a reaction cell and mass analyzerw21x, omit-ting the Q1 mass analyzer. This is the point ofdivergence of the ICP-MS application from theconventional collision cell. We will return to itafter introducing some further relevant art.
An additional important characteristic that hasprofound influence on the use and operation ofpressurized multipole cells for ICP-MS is colli-sional focusing of both the energy and spatialdistributions. It is not clear which is the cause andwhich the effect; these might be viewed as con-comitant phenomena. In the ICP-MS application,the ions are generally introduced into the cell witha relatively high axial translational energy(strong-ly influenced by the ion optical configuration) anda low radial energy(resulting from the transversecooling of the ions in the expansion through theinterface). Collisions with gas molecules in thecell cause retardation of the axial motion and, aswill be seen later, consequent excitation of theradial energy. Subsequent collisions exponentiallydecrease the magnitude and distribution of theseenergies, ultimately yielding a nearly thermal ener-gy distribution in all dimensions(under certainconditions). Coincident with this reduction of the
1369S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 2. Spectra obtained using an ICP source on a tandem quad-rupole MSyMS system, for a sample containing cerium andterbium. (a) The first mass analyzer is operated in r.f.-onlymode and no collision gas is used. Approximately 2%CeO yCe is observed, corresponding to the composition typ-q q
ical for the plasma itself.(b) Both mass analyzers are operatedto provide nominal unit mass resolution, and they are synchro-nously scanned with the second analyzer operating at 16 amuhigher than the first, and the mass transmitted by the first ana-lyzer is shown. Collision gas(air) was introduced to the cellto promote oxidation. Only ions that add 16 mass units in thecollision cell are detected. Ce and Tb are both oxidized andq q
are thus observed. CeO is not efficiently further oxidized andq
is correspondingly significantly suppressed in the detectedspectrum.(From Ref.w20x with permission.)
ion energy, the ions migrate to the axis of themultipole. Since the cell’s extraction aperture ison-axis, this migration causes an increase in theefficiency of transmission of the ions out of thecell. The consequence of the spatial focusing effectis an increase in sensitivity, providing that thecollisional focusing outweighs scattering or reac-tive losses. Energy focusing leads to an improve-ment in the mass resolution of the downstreammass analyzer. The efficiency of these collisional
effects is dependent on the number of degrees offreedom of the ion and neutral, the total numberof collisions, the number of collisions per r.f.cycle, the relative masses of the ion and neutraland the operating point(a, q) of the multipole.These phenomena are described in the general caseby Douglas et al.w50,51x and by Krutchinsky etal. w52x, and in the specific case of the quadrupolereaction cell for ICP-MS by Baranov and Tannerw28x. The simultaneous improvement in transmis-sion efficiency and mass resolution of daughterions of renin substrate has been shown by Thom-son et al.w53x, and of Pb ions(by ICP-MS) byq
Turner et al. w27x. A novel application of thecollision cell, taking advantage principally of itsenergy focusing characteristics, is incorporated inthe Isoprobe from Micromass, which replaces theelectric sector of a double focusing mass spectrom-eter with a hexapole collision cell pressurized withHe w54x. This yields a single focusing magneticsector ICP-MS instrument that has sufficient res-olution for isotope measurement and is now avail-able as either a single collector or multi-collectorsystem.
4.2. Pressurized multipole ion guides for reactionstudies
Multipole ion guides have long been used tostudy ion–molecule reaction chemistry. As anexample, Ervin and Armentroutw13x reported onthe translational energy dependence of the cross-section for the Ar reaction with hydrogen(H ,q
2
D and HD) using a pressurized octapole ion beam2
guide. The subject is thoroughly reviewed, as faras the classic application is concerned, by Gerlichw12x. The principal conclusions(page 62 of Ref.w12x) are that, for the study of near-thermal ion–molecule chemistry, ‘the frequency should be ashigh as possible, the buffer gas should be lightrelative to the ion mass, and the r.f. trap shouldhave a wide field-free region with steep confiningwalls. If collisions play a role, quadrupole iontraps should be avoided.’ In effect, the argumentis that the influence of the r.f. field should beminimized. Accordingly, the r.f. device should beoperated under conditions of adiabaticity(com-monly considered to be forq-0.3), and the
1370 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
collision frequency should be less than the appliedfrequency. A higher order multipole is desirablesince these have a wider region of low r.f. fieldnear the axis, with steeper fields closer to the rods,and because they have a wider stability region(they confine and transmit product ions over awider mass range). Indeed, these characteristicsare desirable when it is intended to study reactionkinetics and product ion distributions under con-ditions of defined energy(including near-thermal).However, we will argue below that other morepractical considerations take precedence in theICP-MS application where it is desired to removeinterferences(which may include the products ofreactions within the cell) rather than trap them.
4.3. The ion trap as a reaction cell
The two dimensions multipole confines ions inonly 2Ds, so that ions can leak out of the cellalong the axis; therefore, there is restricted controlover the time that ions spend in the cell(thetrapping time). In addition, when operated as areaction cell, the pressure is usually higher(1–20mtorr), so that the mean free path is shorter andthe ions gain less energy between collisions fromthe r.f. field. Accordingly, multi-step dissociationof polyatomic ions is relatively inefficient. Forsmall ions with few internal degrees of freedom,scattering loss is more probable than dissociation.
The 3D quadrupole ion trap may be regarded asa 2D quadrupole that is rotated through its trans-verse axis bisecting a pair of rods(according tothe original theory, the trap is asymmetric withr s2z , where 2z is the distance between the2 2
0 0 0
endcaps, though in practice the trap is often‘stretched’ w55x). It is characterized by well-defined stability boundaries similar to those of thequadrupole mass filter. It is commonly pressurizedwith an inert gas, which serves to retard the energyof externally injected ions(thus allowing theirentrapment) and causes the ions to migrate to thecenter of the trap, which provides improved massresolution and sensitivity. While they are in wide-spread use as mass analyzers(and as tandem massanalyzers), they have also been used as near-thermal and non-thermal ion reactors for nearlythree decades. The subject is thoroughly reviewed
in the excellent book by March and Hughesw56xand the more recent set of books edited by Marchand Toddw37x.
The 3D ion trap is usually operated at relativelylow pressure, of the order of 1 mtorr. The efficien-cy of an ion–molecule reaction in the trap, there-fore, is dominated by the trapping time: a longertrapping time leads to more complete reaction.Provided that the r.f. amplitude is low and thefrequency is high(low q), the ions gain only asmall amount of energy between collisions fromthe r.f. field, and the collision energy approximatesthermal energies. In this event, the chemistry ismore-or-less characterized as a thermal process. Athigher q, or when auxiliary excitation at thefundamental frequency of motion of the ion isapplied, the ions can be translationally excited. Inaddition, the internal energy of the ion can beexcited step-by-step in multiple collisions. Accord-ingly, the ion trap can also be used as a super-thermal reactor such that even very endothermicprocesses(such as dissociation of TaOw57x andq
BaOH w58x) can be promoted. Hence, multi-stepq
CID of polyatomic ions is possible with the 3Dion trap.
Much of the recent interest in collision andreaction cells derives from the work of the groupof Koppenaalw22–26x. In recognition that the iontrap has a limited dynamic range of ion confine-ment (of the order of 10 –10 ions before space3 4
charge effects are prominent), the original conceptfor the ICP-IT w22x included a quadrupole ionguide interface that could be used to notch filter(i.e. remove) the Ar ions. Surprisingly, evenq
without the notch filtering capability, few Ar orq
argide polyatomic ions were observed in the spec-trum obtained with the ion trap. It was realizedw22,23x that, at least in part, this was the result ofreactions of these ions(whose corresponding neu-trals have high IPs) with adventitious water in theion trap. This then led to the intentional additionof H to the ion trap in order to promote reactions2
with Ar leading to its consequent efficient remov-q
al from the spectraw24x. The method was shownto be extremely efficient, with 6 orders of magni-tude reduction of the expected Ar ion signal andq
little simultaneous loss of other elemental ions ofanalytical interest. More recently, Furuta et al.
1371S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
w59x, presenting initial results obtained with thefirst commercial ICP-trap-MS(the Hitachi P-5000)and using He as the trapping gas, have shownrelatively efficient conversion of CaO toq
CaOH , presumably due to reaction with impurityq
gases in the ion trap. Eiden et al.w26x havesummarized the attractive performance character-istics of the ICP ion trap for the ICP-MSapplication.
The 2D multipole can act as an analog of the3D ion trap when ions are pulsed into the cell andthe end cap potentials are adjusted to confine theions. Trapping times as long as 5 s with efficiencyapproaching 100% have been reportedw40x. Ionsmay be extracted from the trap either axiallythrough the exit aperture when the potential isdropped, or radially through a slot in one of thepoles. It should be evident that many of the scanmodes available to the 3D trap are also potentiallyapplicable to the 2D configuration, including theapplication of auxiliary r.f. excitation for ion iso-lation or fragmentation. It seems reasonable thatthe 2D trap can be operated at low pressure, hashigher trapping efficiency than the 3D trap, andcan confine more ions because the space chargelimit is higher (approx. proportional to the lengthof the 2D trap). Accordingly, the 2D trap offersintriguing benefits that should be explored. Theincreased space charge limit may be particularlysignificant for the ICP-MS application where theinput ion flux is high.
4.4. Pressurized multipole cells for ICP-MS
To this point we have but briefly noted theimportant 1989 contribution from Rowan andHouk w21x. This work was singular in that itshowed tremendous potential for ion–moleculechemistry enacted in a pressurized multipole cellfor the ICP-MS application, but the work was thenessentially ignored until the introduction of acommercial equivalent 8 years later. The Rowanand Houk instrument contained two quadrupoles,the first of which was operated in the r.f.-onlymode synchronously with the mass filter quadru-pole through capacitive coupling. A schematic isgiven in Fig. 3. The first(r.f.-only) quadrupolecould be pressurized with an external gas(xenon,
methane and ethane were reported). Therefore, theinstrument might be viewed as a triple quadrupolewithout the first mass filter. Accordingly, all ionsthat passed the ion optical region were introducedinto the first quadrupole cell, which could bepressurized. The authors made the further notabledistinction against a triple quadrupole collision cellin that ‘collisions are used to remove unwantedions already present«polyatomic ions must be lostefficiently, relative to analyte ions’w21x. The meanDC potential of the quadrupole arrays(the quad-rupole rod offsets) were independently adjustable.Highly specific and efficient suppression of certainisobaric interferences were reported while retaininga substantial portion of the analyte signal. Inparticular, improvements in the signal-to-back-ground ratio were obtained for Fe(removal ofq
ArO by Xe) and Se (removal of Ar by CH).q q q2 4
Perhaps of the greatest importance, Rowan andHouk recognized that the resultant mass spectrumcontained ions that were formed within the colli-sion cell; obviously these were either primary orsecondary products of reactions of the ionsobtained from the ICP with the reaction gas.Further, it was shown that these product ions,which themselves can act as isobaric interferencesfor other analyte ions, could be discriminatedagainst by application of a potential hill down-stream of the collision cell(which, in this case,was applied simply by making the DC rod offsetof the collision cell slightly more negative thanthat of the mass filter). Accordingly, ions that wereproduced in the cell and, as a result, had lowerkinetic energies than the incompletely thermalizedanalyte ions derived from the source, were pre-vented from entering the mass filter. It should beemphasized that this approach, which we term‘kinetic energy discrimination’, is effective onlywhen there is a discernable difference in the ionenergy distributions of the analyte ions and of theions produced in the cell; that is, it is not effectivewhen the ion population is nearly(or fully) ther-malized in the cell.
Kinetic energy discrimination of analyte andinterference ions has a substantial impact on theefficiency of the ion–molecule chemistry thatmight be used to resolve plasma–ion interferences,and, arguably, is a distinguishing characteristic of
1372 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 3. Schematic of the cell-based ICP-MS instrument of Rowan and Houkw21x, the first reported configuration that introducedthe plasma ion beam into a pressurized multipole for chemical modification before mass analysis.(From Ref.w21x with permission.)
cell operation. In a broad sense, current commer-cial instrumentation may, in part, be distinguishedon this basis. In our opinion, this is one of themajor distinctions between collision cells and reac-tion cells. However, with the commercial introduc-tion of this technology in quadrupole ICP-MSinstruments(the four currently available instru-ments are shown schematically in Fig. 4), desig-nation of the terms ‘collision cell’ and ‘reactioncell’ has created some confusion.
The collision cell has a long history of associa-tion with CID, perhaps principally in the MSyMSconfiguration. The large majority of practitionersare hence familiar with a mode of operation thatis most concerned with only the first few colli-sions; these are at relatively high energies so thatthe center-of-mass energy exceeds at least theweakest chemical bond in the(polyatomic) ionwith the intent to causefragmentation of the parention with trapping of the resultant daughter ions.Certainly, it is common now to operate the colli-sion cell at elevated pressures so as to enhancetransmission of the ions through collisional focus-ing, but this effect is subsequent and supplemen-tary to the initial fragmentation process. Perhapsthe principal distinguishing feature is that thecollision cell, in its historical application, is usedto promote processes that are endothermic underlab normal conditions: usually fragmentation thatis endothermic by at least the weakest bond
strength. It has not been common to introduce ionsinto a collision cell at energies that do not promotefragmentation (other than for purposes of ioncollisional focusing and transmission), and it hascertainly not been common to use a collision cellfor atomic ions.
When the ‘collision cell’ is used to promoteion–molecule chemistry, as we now realize is theprincipal mechanism in the ICP-MS application(Section 5), it has been called a ‘molecule ionreactor’ w60x, ‘ion guide’ w13x, ‘beam guide’w12xor ‘reaction chamber’w43x. It is obvious that ion–molecule collision precedes energy transfer, frag-mentation or chemical reaction. Indeed,fragmentation might be considered an endothermicreaction. Nevertheless, the distinctions in the oper-ating conditions are evident. In our opinion, to calla ‘reaction cell’ a ‘collision cell’ could be consid-ered infringing on the common usage of the latterterm. The confusion in the ICP-MS applicationlikely stems from early misunderstanding or com-mercial interest.
It is clear that Rowan and Houkw21x understoodthat the operative mechanism in their early workthat combined a pressurized cell with an ICP-MSinstrument was ion–molecule chemistry, and alsothat the ions were not thermalized. However, thefirst commercial applicationw27x reverted to infer-ring that the improvements were obtained as aresult of collisional fragmentation. As long as the
1373S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
attention was focused on polyatomic(argide) ionsand the mechanism was understood to be colli-sional fragmentation, the term collision cellseemed to apply. Eiden et al.w25x modified theoctapole ion guide of their ion trap instrument toallow pressurization with reactive gases, and calledthe device a collision cell. They showed at least 4orders of magnitude suppression of Ar using Hq
2
as a reaction gas, and demonstrated specific ion–molecule chemistries for the distinction of otherisobars, notably the elimination of Xe interfer-129 q
ence on I and Y , Zr on Sr with the129 q 90 q 90 q 90 q
addition of O as a reaction gas, and the removal2
of Kr interference on Sr with CHw25x. Bara-q q4
nov and Tannerw28x then described a quadrupolecell operated at relatively high pressure with reac-tive gases(O , NH , N ) and discussed the process2 3 2
and importance of thermalization of the ions. Theypromoted the potential for near-thermal ion–mol-ecule chemistry to provide specific and efficient‘chemical resolution’ w61x, and used the term‘reaction cell’ to distinguish their approach. Theconfusion then began, with various manufacturersand researchers using the terms interchangeably oreven together, as in ‘collision and reaction cell’w62–64x. The distinguishing characteristic thatappears to have been overlooked is that the ‘reac-tion cell’ was intuitively understood to involvethermal chemistry, or at the least near-thermalconditions, so that the reactions are governed bythe thermochemical properties of the ions and thereaction gas. Accordingly, we subsequently pro-posed a distinction based on the thermal charac-teristics of the cellw65x. We justify this distinctionon the premise that a near-thermal energy distri-bution means that the reaction kinetics are gov-erned by the thermal properties of the ions andneutrals, and that the reaction rates correspond tothose that are measured for thermal systems(e.g.selected ion flow tube (SIFT) instrumentsw11,66x). Hence, the distinction is intimately relat-ed to the operating pressure and type of gas usedin the cell, though the measurable characteristic isthe ion energy distributions(or, equivalently, theefficiency of kinetic energy discrimination toimprove the analytical result).
The Micromass Platform and the Thermo Ele-mental PQ ExCell both use an r.f.-only hexapole
as the r.f. device of the cell, and in both instancesthe plasma gas expansion through the sampler–skimmer interface is directed into the on-axisentrance of the cell. To discriminate against plasmaphotons and metastables, the Platform collisioncell is tilted off-axis while the ExCell employs adeflecting chicane lens downstream of the cell.The Agilent 7500c uses an r.f.-only octapole thatis tilted off-axis as the cell r.f. device. An off-axisaperture lens downstream of the skimmer andbefore the collisionyreaction cell serves to simul-taneously disrupt the directed flow of the plasmagas into the cell and blocks the transmission ofplasma photons and metastables. Kinetic energydiscrimination is enabled on all three instrumentsby biasing the pole bias(rod offset) of the collisioncell negative relative to the mass analyzer rodoffset. These instruments are typically operatedwith either He or H or a combination of these as2
the collisionyreaction gas, though the use of Xehas been shown by Masonw67x.
The several generations of the Perkin ElmerSCIEX ELAN DRC use a quadrupole cell thatmay be operated either r.f.-only or r.f.yDC, thefrequency and amplitudes of which are selected onthe basis of the chemistry that is enacted. It istypically operated with relatively heavy reactiongases(NH , CH , O , CH F, N O or others) and3 4 2 3 2
usually at pressures that provide near-thermal con-ditions. Discrimination against ions produced with-in the cell is preferably accomplished byestablishing the appropriate mass bandpass(viathe Mathieu parametersa and q, proportional tothe ELAN parameters RPa and RPq) of the quad-rupolar field (discussed in Section 8.4.2). Kineticenergy discrimination can also be employed whena low mass collision gas is used at relatively lowpressure(i.e. under non-thermal conditions, seeSection 8.4.1). Plasma photons and metastablesare blocked by an on-axis shadow stop, which alsoserves to disrupt the directed motion of the beam.
All four instruments adjust the pressure insidethe cell by controlling the rate of flow of gas intothe cell. At low flow, the cell gas is primarilyplasma gas entrained from the ion optics chamber.In general, this is an undesirable state, since thesampled plasma gas contains as much as 17%oxygen and hydrogenw68x. Increasing the cell
1374S.D
.Tanner
etal.
/Spectrochim
icaA
ctaP
artB
57(2002)
1361–1452
Fig. 4. Schematics of the four cell-based quadrupole analyzer instruments that are presently commercially available: the Micromass Platform(figure courtesy Andrew�
Eaton), the Perkin Elmer SCIEX ELAN DRC� (figure from the authors), the Thermo Elemental PQ ExCell(figure courtesy Jonathan Batey) and the Agilent� �
7500c(figure courtesy Setsuo Muramoto).
1375S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
pressure should eventually achieve a state wherereaction gas flows out from the entrance aperture,thus excluding the plasma gas: the efficiency ofthis exclusion, and its dependence on instrumentdesign, is discussed in Section 8.1. Some of theseinstruments may emulate conventional ICP-MS bystopping the flow of reaction gas and operatingeither at the ambient pressure of the chamber(typically less than 1 mtorr) or by active ventingto the lower pressure mass analyzer chamber, thusto achieve nearly or fully collisionless conditionsso that the cell simply acts as an r.f.-drivenmultipole ion guide.
5. Collisional processes
All interactions of ions with molecules arecollisional processes, and a liberal interpretationmight include energy transfer and collisional frag-mentation as reaction processes. We distinguishthe latter two as collisional events, preferring toconsider reactive processes as those that includetransfer of one or more particles between thereacting partners. For example, fission results inthe transformation of the reactant species, but hasconventionally been considered a physical phe-nomenon. In large measure, the distinction mightbe made on the basis of the energetics of thecollision. The authors tend to consider particletransfer events that take place at, or near, thermalconditions to be chemical reactions, though thispresents a challenge to describe energy-selectedendothermic processes, such as those studied bythe ion guide techniquew12,13x, as the reactionsthat they clearly are. Because we distinguishbetween fragmentation and reaction, we presentthese in separate Sections of this review. Thediscussion presented here of collisional processes,including energy transfer and collisional fragmen-tation, is taken closely from Ref.w65x. The readeris referred to the review by Douglasw51x for amore general and thorough discussion.
5.1. Energy transfer
In an elastic(no internal excitation) non-reactivecollision of an ion of massm and kinetic energy1
E with a stagnant(E s0) neutral of massm ,1 2 2
the energies after collision are given by:
2 2w zm qm1 2x |E9 sE (5.1)1 1 2y ~Ž .m qm1 2
E9 sE yE9 (5.2)2 1 1
As m ™0, E9 ™E , and no energy transfer2 1 1
takes place, with the reactant ion leaving theinteraction with the same energy with which itentered. Ifm sm , the collision partners exit with2 1
equal energy, so that the incident ion loses half ofits initial energy. Multiple collisions of the ionresult in sequential loss of kinetic energy andresults in energy damping(a reduction in the widthand magnitude of the kinetic energy distribution).Thus, the ion loses energy according to the reducedmass of the collision partners: a larger neutralyionmass ratio increases the rate of energy damping ofthe ion. Complete damping to the thermal condi-tion, if possible, means that the ion simply exe-cutes an essentially ‘random walk’ through thecell. A large ion energy at the entrance to the cell(source potential plus expansion energy minus celloffset potential) requires more collisions for energydamping. For a given cell pressure, a higher initialenergy also results in a reduction of efficiencybecause the ion progresses farther into the cellbefore the energy is damped, and hence the numberof collisions is reduced. Further, higher energylowers the probability of reaction during collision,compromises the specificity of the thermal chem-istry, and increases the potential for sputtering cellmaterials.
5.2. Collisional fragmentation
Transfer of energy to internal degrees of freedomduring the collision defines an inelastic collision.The energy that is transferred can be distributedamongst the various internal degrees of freedom:for a polyatomic ion, these include rotational,vibrational and electronic. Subsequent collisionscan transfer (relax) this energy to translation(kinetic energy, heat). If an energy that exceedsthe bond strength accumulates in a single vibra-tional degree of freedom, the chemical bond mayrupture and the polyatomic ion fragments. This
1376 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
process is commonly known as CID. Fragmenta-tion may be successful in a single collision, inwhich case a relatively high collision energy isrequired (to account for the distribution of theenergy in the various degrees of freedom). It canalso occur through multiple collisions, where theinternal energy is accumulated by sequential ener-gy pumping to the dissociation limit. The latter ismost effective in a 3D ion trap(or 2D trap withconfinement using repulsive endcap potentials)because the ion can be confined in three dimen-sions for a long period of time. In these devices,auxiliary excitation may be used to further increasethe collision energy, thus improving the efficiencyof CID. For a given r.f. amplitude and frequency,CID is promoted more efficiently under conditionsof fewer than 1 collision per r.f. cycle(i.e. at lowpressure) because the ion gains higher kineticenergy from the r.f.-field between collisions. In a2D multipole without endcap trapping(i.e. a col-lision cell), multi-collision CID is relatively inef-ficient because the ions are unconstrained in theaxial direction. Though CID is of paramountimportance for organic tandem MS applications,the efficiency required of the process is modest:seldom are more than 90% of the polyatomic ionsfragmented since it is only necessary to producesufficient fragment ions in order to identify theparent polyatomic.
Eqs. (5.1) and (5.2) describe the energetics ofcollisions in the LAB frame(the energies that areapparent to an outside observer). The transfer ofenergy to internal modes of excitation is bestunderstood in the center-of-mass frame of refer-ence. Assuming a stationary neutral molecule, theenergy with which the center-of-mass moves, orthe energyof the center-of-mass,E , is propor-CM
tional to the LAB energy of the ion,E , according1
to:
w zm1x |E sE (5.3)CM 1y ~Ž .m qm1 2
For the same collision pair, the energyin thecenter-of-mass, which is the maximum amount ofenergy that may be converted into internal excita-tion, E , is given by:int,max
w zm2x |E sE yE sE (5.4)int,max 1 CM 1y ~Ž .m qm1 2
For single collision fragmentation, it is thusdesirable to use a neutral having as large a massas possible. Unfortunately, this condition also max-imizes scattering losses. Bandura et al.w65x haveconsidered the energetics of fragmentation ofAr (a particularly favorable case, since the bondq
2
strength is only 1.2 eVw69x), assumed to enter thecell with a LAB energy of 8 eV(3 eV from theplasma potential offset, 4 eV from the supersonicexpansion, and assuming that the ion penetratessufficiently into a cell having an offset potentialof y1 V that the cell potential defines the potentialnear the ion). Recursive use of Eqs.(5.1), (5.3)and (5.4) allows estimation of themaximum pos-sible energy transfer as a function of the numberof collisions under two extreme conditions:
1. prior collisions are elastic(no internal excitationis obtained in prior collisions); and
2. maximum internal excitation in the vibrationalbond is achieved and accumulated on eachcollision.
It is shown that single-collision fragmentationof Ar is possible(but not necessarily obtained)q
2
using neutral Ar as the collision gas, for whicheach of the first two collisions permits energytransfer in excess of the bond strength(the thirdand further collisions provide insufficient energytransfer). Interestingly, this projects a maximumefficiency of 86% for single-collision fragmenta-tion of Ar with Ar. He and H are incapable ofq
2 2
facilitating single-collision fragmentation under theconditions given. Of course, given the above,pumped sequential fragmentation(case 2) with Aris possible, since even the first collision is suffi-cient if maximum excitation is obtained. With Heas the collision gas, a minimum of 4 collisions,each providing maximum transfer of energy to thevibrational bond, are required for pumped frag-mentation. Pumped fragmentation with H as the2
collision gas requires more than 4 collisions, andit should be noted that internal excitation of H2
competes with the fragmentation of the ion.A more effective alternative process for removal
of the argon dimer ion is chemical reaction. There
1377S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
are no exothermic reaction channels for Ar withq2
either Ar or He. However, reaction with H is2
relatively rapid(yielding ArH ), having a thermalq
rate constant of 4.9=10 cm ys w10x, and chargey10 3
transfer is exothermic for reaction with atomic Oand H neutrals(and their molecular combinations)that may be entrained from the plasma gas.Accordingly, it is far more likely that the removalof the argon dimer ion is achieved through chem-ical reaction than through collisional fragmenta-tion. An analytically useful reaction gas is CH ,4
for which Ar reacts by charge transfer with aq2
rate constant of 5.7=10 cm ys, and with whichy10 3
Se has no bimolecular reactionw70x.q
6. Ion–molecule reactions
6.1. Reaction thermochemistry
Enthalpy is a state property, meaning(in part)that it is defined for the set of thermodynamicproperties of a system(which include temperature,pressure, composition, etc.). It is the heat effectdefined as the sum of the internal energy of thesystem plus the expansion work performed on thesurroundings for a constant pressure process. Asnoted by Sussmanw71x, ‘its raison d’etre is con-ˆvenience’. The enthalpy of formation(commonlycalled the heat of formation) of species X,DH (X), is the amount of heat required to pro-f,T
duce X from its standard state components attemperatureT. The ‘standard state’ of an elementis the normal state of aggregation at atmosphericpressure at the specified temperature. Accordingly,at room temperature the standard state of H is H ,2
that of O is O , and that of Ar is Ar. The heats of2
formation of standard states at 298 K are definedas zero. Tabulations of the heats of formation ofions and neutrals, such as Ref.w9x, are valuablebecause they allow determination of the enthalpyof a proposed reaction, which will be shown to bedeterminant of the thermodynamic viability of thatreaction for analytical application(subject only tokinetic validation; see Section 6.2).
6.1.1. Enthalpy of reactionThe change of enthalpy in a reaction:
q qA qB™C qD (6.1)
is (the sum of the heats of formation of theproducts) minus(the sum of the heats of formationof the reactants):
q qDH sDH (C )qDH (D)yDH (A )r f f f
yDH (B) (6.2)f
If the enthalpy of reaction is negative, thereaction is exothermic and might proceed. If theenthalpy of the reaction is positive, the reaction isendothermic and will not take place unless addi-tional energy is contributed to the process(byexcess axial kinetic energy before relaxation in acollisionyreaction cell or by the r.f.).
In reality, the reaction energy is given by thefree energy of reaction:
DG sDH yTDS (6.3)r r r
where T is the temperature(in K) and DS is theentropy change of the reaction(defined the sameas for enthalpy). Few people bother calculatingthe free energy, in part because it is more difficult,the entropy data is not as commonly available, andthe fact that the entropy change of a simple small-particle-transfer reaction(charge transfer, H-atomtransfer or proton transfer) is usually close enoughto zero. The entropy term is important in somecondensation reactions and is particularly impor-tant in association reactions.
In some instances, the enthalpy of formation(heat of formation) of one or more of the speciesis not known or reported, or there may be reasonto doubt the reported value(i.e. different resourceswere used for the same reaction). In some cases,the heat of formation may be calculated from otherinformation. For example, we may wish to deter-mine the reaction energetics of oxidation andhydroxylation of Sr:
q qSr qO ™SrO qO (6.4)2
q qSrO qH O™SrOH qOH (6.5)2
We know the following enthalpies of formation(X, DH (X)), where the enthalpy is given in kcalyf
mol) w9x: Sr (170.6), O (0, by definition),q2
SrO (149), O (59.6), H O (y58), OH (9.3),q2
1378 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
from which we can derive the enthalpy of Eq.(6.4) as
DH (Eq. 6.4)s149q59.6y170.6y0r
s38 kcalymol (6.6)
The reportedw9x heat of formation of SrOH isq
74 kcalymol, which seems questionable. We knowthe heat of formation of SrOH(y49.1 kcalymol),and we know the IP of SrOH(5.1 eV). So we cancalculate the heat of formation of SrOH asq
q ySrOH™SrOH qe (6.7)
The heat of formation of the electron is 0, sothe heat of formation of SrOH isq
q yDH (SrOH )sDH yDH (e )qDH (SrOH)f r f f
s(5.1=23.06)y0q(y49.1)s68.5 kcalymol (6.8)
So the heat of Eq.(6.5) is
Ž . Ž .DH Eq. 6.5s68.5q9.3y149y y58r
sy13.2 kcalymol (6.9)
Of course, this is the same as simply summingreactions, where Eq.(6.5) is the same as the sumof Eqs.(6.10) and(6.11):
q ySrO qH Oqe ™SrOHqOH2
DH s(y49.1)q9.3y149y(y58)y0r
sy130.8 (6.10)q ySrOH™SrOH qe DH sIPr
s5.1 eVs5.1=23.06s117.6 kcalymol (6.11)
where
DH (Eq. 6.5)sDH (Eq. 6.10)r r
qDH (Eq. 6.11)r
sy130.8q117.6sy13.2 kcalymol (6.12)
This is a simple and obvious example, but itwill allow you to combine reactions where neces-sary: the heat of reaction of a sum of reactions isthe sum of the heats of the individual reactions.
In some instances, the only available thermo-chemical data is an ‘appearance potential’, whichis an experimental parameter that can in someinstances be converted to an enthalpy of formation.
In a simple case, the sample is placed in a low-pressure vessel and is subjected to electrons, in anattempt to induce ionization. The electron energyis increased until the product ion appears. This isthen the appearance potential of that ion. If theion is simply the ionized neutral sample, theappearance potential is an approximation to the IPof the neutral sample. If the product ion is adissociation product of the ionization, then theappearance potential is the sum of the IP of thesample and the heat of dissociation reaction. Forexample:
y q y qAqe ™A q2e AP(A );IP(A) (6.13)y q yAqe ™B qCq2e
qAP(B )q;IE(A)qDH (B )qDH (C)f f
qyDH (A ) (6.14)f
where we obtained the AP(B ) through combiningq
the reactions:y q y qŽ . Ž .Aqe ™A q2e AP A ∼IP A (6.15)
q qA ™B qCq qDH sDH (B )qDH (C)yDH (A ) (6.16)r f f f
Clearly, the appearance potential must bedefined with reference to a particular parent neutral(i.e. the appearance potential of CH from CH).q
2 4
A convenient means to gain a qualitative per-spective of reaction energetics is to scatter-plot athermodynamic property against the mass(or iden-tity) of analyte and interference ions and overlayon this horizontal lines that correspond to thevalue of this property for potential reaction gases.Each horizontal line then bisects the scatter-plotinto endothermicyexothermic reactions withrespect to that reaction gas.(Of course, justbecause a particular type of reaction is endothermicdoes not preclude a different reaction channel forthe pair from being exothermic.) An example isgiven in Fig. 5a, which plots the IP of the elementsand possible ICP-MS plasma interferences(argon,argides, oxides and hydroxides of elements)against the masses of the most abundant corre-sponding ions. The elements are indicated by filledcircles and the ‘interferences’ by open circles.Several horizontal lines are given, correspondingto the IPs of potential reaction gases. The(posi-
1379S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
tive) ions of species that are above a line areexothermic for charge transfer with that gas, andthose that are below the line are endothermic forcharge transfer with that gas. For example, Arq
and N are exothermic for charge transfer withq2
H , but all other ions shown are endothermic. On2
the other hand, NO is endothermic for chargetransfer with most analyte ions and is exothermicfor charge transfer with a majority of interferenceions. A similar ‘reaction energetics’ figure is givenin Fig. 5b for O-atom affinities. In this instance,the horizontal lines correspond to the O-atomaffinities of the gas having one less O-atom thanindicated(i.e. the neutrals indicated would be theproduct of an oxidation reaction). Hence, ions thathave a higher affinity than an indicated reactiongas(i.e. that appear above the line) are exothermicto extract an O-atom from the indicated gas, andions that are below(providing that these ionscontain an oxygen atom) are exothermic for dona-tion of an O-atom to a neutral that would formthat gas(i.e. for reaction with the gas having oneless O-atom than indicated).
6.1.2. Specificity of thermal ion–molecule reactionThe enthalpy of reaction is, of course, a thermal
property. It is properly defined only when the ionsand neutrals are thermalized. An exothermic reac-tion releases energy to the environment and istermed ‘thermodynamically allowed’. Reactionsleading to polyatomic products(either the production or product neutral) generally release the enthal-py of reaction principally into internal degrees offreedom (vibration and rotation of chemicalbonds). The energy eventually ends up as thermal(kinetic) energy as a result of energy transfer insubsequent collisions. An endothermic reactionabsorbs energy from the environment and, thus, isthermodynamically ‘not allowed’. The enthalpy ofreaction is not correlated with kinetics(the rate ofreaction) except in one very important aspect:under thermal conditions, an exothermic reactionmay take place but an endothermic reaction maynot. If an exothermic small particle transfer reac-tion occurs, it usually occurs with high probability(i.e. close to the collision rate) and is relativelyfast. In principle, this is because such processesdo not generally exhibit an activation energy bar-
rier. In turn, this is due to the relatively strongion-induced dipole electrostatic interaction. Hence,thermochemistry provides a high degree of speci-ficity for ion–molecule reactions. If a reaction gasis chosen such that it has an exothermic channelwith either the analyte ion or the isobaric interfer-ence ion, and thermal conditions prevail, theallowed reaction is likely to take place and thedisallowed reaction is not. This difference provides‘chemical resolution’. An example is the chemicalresolution of Ca from Ar using NH as theq q
3
reaction gas. The IP of Ar(15.76 eV) is greaterthan that of NH (10.16 eV), which in turn is3
greater than that of Ca(6.11 eV). Thus, the IP ofNH is sandwiched between Ar and Ca, and charge3
transfer is allowed for Ar but disallowed forq
Ca . It is observed experimentallyw10x that theq
Ar reaction is fast and the Ca reaction isq q
exceptionally slow(if it proceeds at all). Passingthe ion beam through a cell containing NH as the3
reaction gas therefore promotes reactive loss ofAr while Ca is essentially unaffected, thusq q
achieving chemical resolution. It is important torecognize that the provision of(near) thermalconditions is essential: if the collision energy(inthe center of mass) is sufficiently large to over-come the endothermicity of the disallowed(underlab normal conditions) reaction, the specificity ispotentially forfeit.
6.2. Kinetics
The enthalpy of reaction determines the ther-modynamic viability of a proposed reaction. Theactual value of the reaction for analytical purposesis dependent on the kinetics, by which we meanthe rate of the reaction. The density of a reactantor product ion, which is proportional to the ionsignal observed, is exponentially dependent on therate constant, the density of the reactant neutral,and the reaction time. In addition, the product iondistribution (the identity of the product ions andtheir branching ratio, for multiple products orreaction channels) is of critical importance. In thissection we discuss theoretical aspects of the reac-tion dynamics, compare experimental reaction rateswith theoretical collision rates, review the majortypes of ion–molecule reactions, and consider how
1380 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 5. Some thermochemical properties of some atomic ions and potential atomic and polyatomic interference ions. Solid pointsrepresent the atomic ions usually of interest for inorganic analysis, and the open points represent argon, argide, oxide, hydroxideand other potential interference ions.(a) Ionization potentials. The horizontal lines indicate the IPs of potential reaction gases. Ionsabove a horizontal line are thermodynamically favorable(though not necessarily kinetically favorable) for charge transfer with theindicated neutral.(b) Oxygen-atom affinities. The horizontal lines indicate the O-atom affinities of gases having one less oxygenthan the indicated neutral. Ions above a line are thermodynamically favorable to extract an oxygen atom from the indicated neutral;oxides of the ions below the line a favorable to donate an oxygen atom to a neutral having one less O-atom than the indicatedneutral.
these characteristics affect the observation of ionsignals in the 2D r.f.-driven multipole.
6.2.1. The Langevin collision theoryAccording to the Langevin approach(which has
certain limitations, as will be discussed), the ionis considered as a point charge and all its interac-tions are predetermined by the polarizability,a, ofthe molecule. In this instance, the ion–molecule
potential as a function of the internuclear distance,r, may be taken as:
2 2B E1 e aC FF(r)sy (6.17)44p´ 2rD G0
where ´ is the permittivity of free space. The0
potential is presented in SI units. Accordingly, themolecular polarizability is in J(myV) and the2
elementary charge(e) in Coulombs. In the CGS
1381S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
system of units,a and e would be in cm and3
e.s.u., respectively. An ion–molecule reaction canoccur only when the distance between the centersof the ion and molecule are less than a criticalvalue. Integrating in two dimensions, this deter-mines a cross-section,s, which can be shown todepend on the relative velocityn as:
1y2B Ee aC Fs(v)s (6.18)
2´ v mD G0
wherems(m m )y(m qm ) is the reduced mass1 2 1 2
for the collision of partners having massesm and1
m . Consequently, the cross-section is inversely2
proportional to the relative velocity. This propertyof the cross-section of an ion–molecule reactionin the Langevin theory is very important becausethe rate constantns(n)sk for Eq. (6.1) may beL
deduced from:
q? @d Aqw xw xy s A B vs(v) (6.19)
dt
as
1y2B Ee aC Fk s (6.20)L 2´ mD G0
and is, thus, independent of energy.It is generally accepted that the ion-induced-
dipole model, Eq.(6.17), satisfactorily describesthe interaction at impact energies less than anelectron volt. For higher energies the Langevintheory is not applicable. Moreover, the forcebetween a real molecule and ion is strongly repul-sive at short distances and weakly attractive atlarge distances. Also, the polarizabilities of mole-cules are not scalars but tensors, and not allcollisions are reactive. Given these limitations,kL
can be regarded as the maximum limiting rate ofan ion–molecule reaction(for non-polar mole-cules) and is widely used for comparison with theobserved reaction rate,k. The ratiokyk is gener-L
ally considered as a reaction efficiency of an ion–molecule collision and describes the fraction ofcollisions that lead to reaction.
Assuming that neutral reactant B is present inabundance and its concentration is independent of
the reaction time,Dt, and ion concentration, Eq.(6.19) may be integrated. The result is:
w xq q ykDt B( )w x w xA s A e (6.21)0
where is the initial concentration of ions.qw xA 0
Units for the rate constant,k, are equal to{ 1yDtwBx} , usually given in cmys or more accurately3
cm y(molecule s). The reaction time or concentra-3
tion of the reaction gas may be kept constantduring the ion–molecule reaction, in which casethe logarithm of the ion concentration should decaylinearly as a function ofDt or wBx. In the specialcase of the linear reaction cell, which obviouslyhas a constant length, it is much more convenientto vary the reaction gas flow. A plot of the ionsignal as a function of the reaction gas flow yieldsa reaction profile. Its characteristic feature shouldbe a semilogarithmic linear decay of the parention (A in the case of Eq.(6.1)). As can be seenq
from Eq. (6.21), the slope of the decay of the ionsignal is proportional to the reaction rate constant.A steep decay indicates a fast reaction. If severalions react with the same reactant under the sameconditions and form different reaction profiles, thesteepest one indicates the fastest ion–moleculereaction assuming that other losses are similar.
6.2.2. Measurement of reaction rate constantsRate constants of ion–molecule reactions have
been measured using a variety of techniques,including high pressure ion sourcesw72x, ion trapsw37x, ion cyclotron resonancew73x, flowing after-glows (FAs) w74x and SIFTs w11,66x. In manyinstances, the reported values are remarkably con-sistent, while in others there is substantial varia-tion. The instances of disparate results have incitedvigorous debate. In most instances, argument hasfocused on the validity of the assumption ofthermal conditions(hence, perhaps, the presentauthors’ preoccupation with this characteristic).The rates of certain reactions(e.g. spin-forbiddenreactions and others that show an unusual activa-tion energy) can be very sensitive to the energydistribution of the ions and to residual internalexcitation of the ions. It is our opinion that rateconstants measured with the SIFT technique pro-vide the most reliable thermal data. In this method,ions are produced(by electron impact, thermal
1382 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
ionization, or other means) and mass-filtered toselect the ions of interest. This step, which isadditional to the FA method, ensures that subse-quent concomitant reactions of other source ions,or reactions with source gases, do not interferewith the data analysis. The mass-selected ions arethen extracted into a flight tube where they areentrained in a flow of He(f0.35 torr) and allowedto undergo many()10 ) collisions to ensure5
thermalization at the temperature of the He(whichshould be equilibrated with the flight tube). Areaction gas is added downstream, and the ionsare allowed to react as they flow towards the exitof the tube(having a constant reaction time,Dt),whereupon they are extracted for mass analysisand detection. Knowledge of the flow characteris-tics of the flight tube(mixing, wall effects, veloc-ity distribution, etc.) and of the dependence of theion signals on the flow rate(density) of thereaction gas allows determination of the thermalrate constant with high accuracy and precision. Aswell, the products of the reaction can, in mostinstances, be determined as these are also trans-ported through the tube and are mass-analyzed.The product ions themselves can also react, andthe data can be deconvoluted to yield rate constantsfor each step of the sequential chemistry. Animportant development for the focus of this workis the adaptation of an ICP ion source to a SIFTapparatus in the laboratory of Diethard Bohme atYork University, Canadaw75–77x. With this mod-ification, rate constants of virtually any ion thatthe analytical atomic spectroscopist may encounterwith a variety of neutral reaction gases can bedetermined, and this capability will significantlyenhance method development for the ICP-MSapplication.
It should be noted that some research groups(notably those of Armentroutw14x and Schwarzw78x) have specialized in the measurement of rateconstants of energy-selected ions. The reactions ofions having a known and well-defined energy,sometimes ‘thermalized’ in a pressurized octapoleand accelerated into a collision cell, are studied asa function of the incident ion energy. In mostinstances, the reactions are studied under relativelyrarefied conditions so that, typically, the averageion undergoes only a single collision(or less), and
this ensures that the energy distribution is notsignificantly altered during the promotion of thereaction. In every instance, the ions are ‘non-thermal’, having an excess axial kinetic energy.The dependence of the reaction rate and productson the axial energy is interpreted in terms of thereaction endothermicity(if appropriate) or activa-tion energy.
Anicich has expended considerable effort intabulating thermal bimolecular rate constantsw10x.Two of the adjectives in the preceding sentenceare exceedingly important. The referenced tabula-tion is for thermal reactions and, hence, does notinclude the very many energy-selected rate con-stants that have been reported. Secondly, the tab-ulation includes, for the most part, onlybimolecular processes and, with few exceptions,excludes association(clustering) reactions. Hence,reliance on this database for the ICP-MS applica-tion must be tempered with the recognition thatthree-body reactions may proceed even when thetabulation indicates ‘no reaction’. Anicich hasincluded a variety of measurement techniques inhis compilation without comment on the validityof the assumption of thermalization, and hasincluded in many instances rate constants at tem-peratures other than room temperature. In instanceswhere there are multiple reports of rate constants,Anicich has ‘evaluated’ the data on the basis ofhis considerable experience to recommend a value.
It will become common to report ‘relative rates’measured with cell-based ICP-MS instrumentsw79,80x. These rates are expressed as the numberof orders of magnitude of reaction per unit ofreaction gas flow(i.e. the logarithmic slope of themeasured reaction profile). In many instances,there is good correlation between the absolute rateconstants and the ‘relative rates’w79,80x. However,several important considerations should be noted:
1. By and large, the cell-based ICP-MS instru-ments are not operated under strictly thermalconditions, so the relative rates cannot be con-sidered ‘thermal’.
2. The relative rates of reaction cannot be con-verted to absolute rate constants since the reac-tion time and the pressure are not knownaccurately (though an effective rate constant
1383S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
might be determined through normalization to aknown rate constant).
3. Incursion of plasma or background gas into thecell can significantly affect the apparent relativerate of reaction, especially for relatively non-reactive gases.
4. The apparent relative reaction rates may beaffected by differing rates of scattering loss orcollisional focusing.
5. Care must be taken in the units in which thereaction gas flow is measured. As will bediscussed in Section 8.1, the units of flow thatare reported depend upon the calibration of themass flow controller(if used). It is common touse a mass flow controller calibrated for Argonwith gases other than Argon, and the resultantflow units should be indicated as Ar-equivalentflow units (though in a prior publicationw65xwe have referred to these as ‘arbitrary units’).These may be converted to absolute flow unitsif the calibration correction coefficient is known.
6.2.3. Ion–molecule reaction profiles in a r.f.-driven reaction cell
6.2.3.1. Types of ion–molecule reactions. Thegeneric ion–molecule reaction given in Eq.(6.1)includes several categories or types of processesthat transform the reacting species. It is generallyunderstood that, in the ICP-MS application, themost important, useful and abundant type of reac-tions are charge transfer of the type:
q qA qB™B qAq qŽ .e.g. Ar qNH ™NH qAr (6.22)3 3
because the first applications were to the resolutionof interferences caused by the argide ions(Arq
and ArX , where X may be Ar, O, Cl, C, Na andq
so on). The corresponding neutral argides havehigh IPs and their charge transfer reactions oftenproceed with high efficiency near the collisionrate.
The experimental observation that thermalcharge transfer reactions occur near the collisionrate allows an opportunity to dispel a commonmisunderstanding related to the rates of resonantcharge transfer reactions. For atomic species athigh kinetic energy, where the collision cross-
section approximates the hard sphere limit, inter-action times are short and it is known that chargetransfer processes(in this regime they are some-times called stripping reactions) that are resonanttypically proceed at a higher rate. In effect, thismay be re-stated as: atomic non-resonant chargetransfer processes at high energy are relativelyinefficient. At thermal energies the interaction timeis longer, in part due to the development of theion–dipole attraction, and electron transfer is moreefficient. In the thermal instance, where chargetransfer, whether resonant or not, proceeds on eachcollision, it is not possible to distinguish the rateson the basis of resonance.
With the development of the reaction cell tech-nique, it has become apparent that the scope ofapplication is considerably broader than simplecharge transfer, and it is to be expected that avariety of classes of ion–molecule reactions willbe applied to resolve other interferences. Sincesmall particle transfer reactions are often fast, asecond important class of reactions involves hydro-gen-containing substances. The several types ofthese include:
So-called ‘condensation reactions’ involve trans-fer of atoms other than hydrogen, and sometimesresult in rearrangement to a thermodynamicallystable form. Oxidation reactions are promising dueto their apparent selectivity and speed:
q qA qBO™AO qBq q(e.g. Ce qN O™CeO qN ) (6.26)2 2
This latter type of ion–molecule reaction is verysensitive to the thermodynamic stabilities of theproduct oxide ions and of the residual(neutral)leaving molecule. For example, N O is attractive2
as an oxidizing agent because the correspondingreactions include N as a product molecule. CO2 2
1384 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
is also a promising reactant due to the thermody-namic stability of CO as a leaving group. O has2
a different application because a very strong A –Oq
bond is required for this reaction to proceed.However, as a result, some reactions with oxygenhave very high specificity.
Clustering is a common reaction between manyelectron donor molecules and ions, which can berationalized in terms of the ligand–ion mutualelectron donation. Association or clustering reac-tions of the type:
q q q qA qB™AB (e.g. Ni qNH ™Ni3
ØNH ) (6.27)3
generally play a negative role in reaction cell ionchemistry when applied to the ICP-MS. Ammoniais a good clustering ligand, meaning that it formsadducts readily and hence can be an analyticalcomplication unless steps are taken to control theappearance of cluster ions(Section 8.4.2). Thistype of reaction is often observed with watermolecules, which are present in many reactiongases in trace quantities. Water also facilitatesoxidation, hydroxylation, H-atom transfer and pro-ton transfer reactions.
More complicated multistep ion–molecule reac-tions may be observed in the reaction cell environ-ment but are usually considered as a nuisance.This does not mean that they cannot be used toadvantage; an example is the sequential oxidationof Sr by N O followed by H-atom transfer fromq
2
CH w65,80x.4
6.2.3.2. Plasma ion reactive decay. As was dis-cussed in Section 6.2.2, the r.f.-driven reaction celloperated under multiple collision conditions is anunsuitable environment for accurate determinationof thermal ion–molecule reaction rate constants.Within the validity of certain assumptions regard-ing flow characteristics, the number density of thereaction gas in the cell is directly proportional toits flow rate. If the ions were fully thermalizedthroughout the cell, the reaction time would bedetermined by the macroscopic flow speed of thereaction gas and this depends on the manner inwhich the gas is added and the relative flow ratesthrough the entrance and exit apertures of the cell.However, reaction cells have not been operated
under strictly thermal equilibrium conditions. Usu-ally, the ions are injected into the cell with someenergy, and in addition they experience the r.f.field which alters the ion trajectory(Section 8.2.1).From basic principles, a semilogarithmic lineardecay of the reacting ion should be obtained if theloss process is first order with respect to the gasdensity. A bimolecular reaction is such a process,but it should be realized that other(non-reactive,physical) processes can also emulate the depend-ence with different associated rates. For reactiveloss, first order decay can be observed at therelatively low operating pressure of this techniqueif the number of thermalizing collisions is smallin comparison with the total number of collisionsor if the ion–molecule reaction happens on everycollision.
For example, a semilogarithmic linear decay ofthe parent plasma ion signal is typical for reactioncells operated under conditions close to thermalw28x. As can be seen in Fig. 6, the reaction of theLa , Tb , Ho and Tm ions with CO in ourq q q q
2
laboratory with an ELAN DRC demonstrate lineardecay in the semilogarithmic plot. These analyteions react with CO by formation of the corre-2
sponding oxides:
q qM qCO ™MO qCO (6.28)2
where M is La , Tb , Ho or Tm . Evidently,q q q q q
the response of the parent plasma ion is related tothe probability of reaction during the collision.Broadly, all events of close interaction between anion and a neutral can be divided into two genericgroups: reactive and non-reactive collisions. Non-reactive collisions do not change the chemicalnature of the colliding atoms and molecules. Theusual outcome of such an event is energy exchangebetween different degrees of freedom of the collid-ing partners. Chemical reaction leads to anexchange of mass, charge or energy, which isfollowed by transformation of the reactants intonew chemical entities. From Eq.(6.20) it is clearthat the mean collisional frequencyw of ions is:L
1y2B Ee aw x w x C Fw s B k s B (6.29)L L 2´ mD G0
and that the probability for the ion to traveln
1385S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 6. Ion signals for lanthanide ions as a function of CO2
reaction gas flow(in true sccm). The steepest slope indicatesthe fastest reacting ion with the indicated gas.
times longer than 1yw without collision is equalL
to P se . Assuming that every collision leadsynn
to reaction, this simple dependence allows us toestimate the number of collisions(or the reactiongas pressure) that are required in order to attenuatethe ion signal by a given factor. For example, 10reactive collisions are able to reduce the ion signalby approximately 4 orders of magnitude(1yP s10
2.2=10 ), but 30 reactive collisions will achieve4
13 orders of magnitude(1yP s1.1=10 )! Of1330
course, if the rate of the ion–molecule reaction ishalf the collision rate(reaction efficiency of 50%),the 13 orders of magnitude will require twice asmany collisions: half reactive and half non-reactive.
We know that if the reaction time was inde-pendent of the reaction gas flow(i.e. if the reactioncell was buffered with a non-reactive gas) and theions were thermalized throughout the reaction celland the number density of the reaction gas wasaccurately known, the slope of the logarithmicdecay of the ion signal intensity versus the reactiongas flow would yield the reaction rate constant.All these conditions and many others are notessential for an analytical instrument(though theyare for an instrument intended for kinetic measure-ments), and one should not attempt to obtainaccurate kinetic data from a reaction cell designedfor the ICP-MS analytical application. Therefore,why bother with a discussion of the rate constants?There is important information that can be extract-ed from the reaction dynamics, and this informa-tion can be vital for method development. Forexample, from comparison of the observed reactionprofiles in Fig. 6, one may conclude that the rateconstants for ion–molecule reactions of lanthanideions with CO decay in the orderk(La ))q
2
k(Tb ))k(Ho ))k(Tm ). Therefore, we gainq q q
information on the efficiency of these reactions,and this guides us on the corresponding reactioncell conditions necessary for transformation of theions for analytical purposes. Even some qualitativeinformation can be obtained from this comparativestudy despite the reservations about limitations ofrate determinations using a r.f.-driven reaction cell.Assuming that the fastest reaction(for La ) has aq
relative rate constant equal to 1, it is simple toestimate the other rate constants obtained under
the same conditions(Table 1). It is useful to knowthat, in order to get comparable attenuation ofTm in reaction with CO , the reaction cell pres-q
2
sure should be at least three times higher than inthe case of La reaction. In a relative sense, onlyq
1 reactive collision occurs for every 3 collisionsbetween Tm and CO . Fortunately, the collisionq
2
rate constants,k , for these analytes are veryL
similar and no additional correction is required forthis estimation. This is not exactly true for asimilar assessment of the reactions of Ar , Arq q
2
and ArO with ammonia, even though the effectq
is small or even negligible.One of the most efficient and important practical
reactions in the ICP-MS application is the chargeexchange between Ar and NH :q
3
q qAr qNH ™NH qAr (6.30)3 3
which has been observed under strictly thermoe-quilibrium conditions using a SIFT apparatus toproceed with a reaction rate equal to 1.7=10y9
cm ys w10x. The reaction is considered to be fast3
in comparison with the collision rate(Table 2).Consequently, its efficiency, given by the ratio ofthe experimental reaction rate to the calculatedLangevin collision rate, is more than 100%. Clear-ly, the reaction rate cannot exceed the collisionrate, and this instance demonstrates a significanterror that is introduced by the simplifications ofthe Langevin theory. The average dipole orienta-
1386 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Table 1Comparative rate constants for reaction of the La , Tb , Ho and Tm ions with CO under the DRC operating conditionsq q q q
2
M qCO , Mq2 k (relative) Relative number of k =109
L Relative numberrequired collisions (cm ys)3 of collisions(estimation)a (corrected)
Number of required collisions for equal attenuation of an analyte ion(see text).a
tion (ADO) theory w6x is an improved collisiontheory model that includes the charge-inducedorientation of the molecular dipole moment thatincreases the collision rate for polar molecules.Calculation according to the ADO theory(requiredparameters for ammonia: mass 17 amu, polariza-bilitys2.26=10 cm , dipole momentsy24 3
1.3=10 e.s.u.) yields a collision rate equal to18
2.1=10 cm ys, which puts the efficiency of they9 3
reaction under consideration slightly below 100%.The number of reactive collisions required to
decrease the Ar ion intensity from;10 to 10q 9
cps is close to 20, which under our experimentalconditions corresponds to an NH flow of approx-3
imately 0.53 sccm(;1.0 Ar-equivalent sccm), asseen in Fig. 7a. Chemical conversion of ArO inq
a similar reaction is marginally faster. In addition,the initial ion current of ArO is significantlyq
smaller and, as a result, less flow of ammonia isrequired. Reaction between the argon dimer ionand ammonia is slower, and the initial Ar signalq
2
is intermediate between that of Ar and ArO ;q q
consequently, reduction of the Ar signal to theq2
instrument baseline using this gas is not obtained
even at the highest flows consistent with instru-ment operation(Section 8.1).
6.2.3.3. Reactive decay of an interfering plasmaion. In Fig. 7a a wide range of NH flows in the3
DRC was used, and a curvature in the high flowregion can be observed. Let us consider thiscurvature more closely.
Usually, the ion–molecule reaction profile hastwo slopes if the reactant ionmyz is comprised oftwo (or more) populations that react differently.Using a mass-spectrometer to measure the ionconcentration(number density) one cannot distin-guish between different populations easily. Forinstance, a mixture of an ion in the ground andexcited electronic states might form two slopes inthe reaction profile. Two isobaric ions( Ar and40 q
Ca , e.g.) will possibly react with different40 q
speeds. In both cases, the observed reaction profilewould present an unresolved combination of tworeaction profiles. For instance, ArO interferesq
with iron. Both ions are generated in the plasma,are unresolved in the quadrupole mass spectrum,but have different reactivity toward many reaction
1387S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 7. Reaction profiles formyzs40, 56 and 80 ions withammonia reaction gas(a) for high purity water and(b) for asolution containing 100 ppt Ca and Fe. Ar and ArO reactq q
relatively quickly, and Ar reacts more slowly, with ammonia.q2
The curvature of themyzs40 and 56 signals at high ammoniaflow indicates the presence of other ions at the same mass thatreact much more slowly with ammonia than do Ar andq
ArO ; these other ions become predominant once the majorityq
of the Ar and ArO ions are reacted away. The initial decaysq q
for both (a) and (b) are similar, indicating the predominanceof fast-reacting Ar and ArO . The second linear portions ofq q
the profiles in(b) indicate the relative reaction rates of Caq
and Fe ; the similarity of these slopes with those at high flowq
in (a) provides evidence that the residual signal in the latter isdue to contaminant Ca and Fe in the high purity water. Thegas flow is given in Ar-equivalent sccm(Table 4).
gases. If iron is present in trace quantities(as inDIW), ArO dominates the combined ion signalq
(and the reaction profile) until it is sufficientlyreactively removed that it is no longer the majorcomponent of themyzs56 signal. It is our expe-rience that, even inside a clean room facility,contaminants in the DIW are at the level of severalppt and it is very difficult to exclude them com-pletely. In Fig. 7b it is evident that Fe reactsq
much more slowly with ammonia than doesArO . A similar observation holds for Ar andq q
Ca , which also have very different properties andq
reactivities(Table 2). The difference in reactivitybetween isobaric ions is an essential feature ofmethod development for interference reduction.Because of the difference in the reaction rateconstants, the same extent of signal suppression inreaction with ammonia requires ten times morecollisions for Ca than for Ar . A larger differ-q q
ence in the reaction rate constants provides higherspecificity of the ion–molecule reaction and, as aresult, better detection power of an instrumentbased on the reaction cell technology.
As an additional confirmation that the observedcurvatures in the reaction profiles are due toresidual contamination in DIW, one can comparethe relative reaction rate constants of the contam-ination as evidenced by the slope of the second,high flow part of the reaction profile with thereaction rate(slope) obtained from a solutioncontaining the contaminant(or analyte). As canbe seen in Fig. 7b, the observed slopes(reactionrates) for the Ca or Fe dominated signals(atq q
high flow) are nearly identical to those of thecorresponding background decay rates in Fig. 7a.This gives some confirmation that the limitingsignals in the DIW sample are due to contamina-tion by Ca and Fe. Certainly, such an analysis isconclusive only in the case of a negative result.
6.2.3.4. Product ions of the primary ion–moleculereaction. What happens to the product ion of anion–molecule reaction? Charge should be con-served in a chemical reactor if there is no externalmeans of quenching it. All reaction cells havesome physical boundaries and in a multipole envi-ronment some charged particles can disappear fromthe cell by striking the rods or entranceyexit lenses.
Conservation of charge can be forfeit due tocollisional scattering or to ion rejection accordingto the stability characteristics of the multipole field.Under certain operating conditions(Section 8.4.2),the r.f.yDC field of a quadrupole is able to confineions and successfully deliver them to the exitaperture, but under other conditions is also able toreject them according to their mass-to-charge ratio.In addition, product ions might emerge at the samemass as an analyte ion, which creates a newinterference. For example, the NH ion that is aq
3
1388 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 8. Signals for the product ions of reactions of La and Howith CO (see also Fig. 6). The more rapid increase of the2
LaO signal reflects the faster reaction of La . Further, theq q
approach to unity of the normalization to the total(M qq
MO ) signal indicates that oxidation is the primary reactionq
channel for these ions.
product of Eq.(6.30) might react further if certainstability conditions are met:
q qNH qNH ™NH qNH (6.31)3 3 4 2
Eqs. (6.22) and (6.23) are examples of chargeand proton transfer reactions, respectively, that arecommon under most experimental conditions. Inour instance, the product ions NH and NH ofq q
3 4
charge and proton transfer reactions, respectively,could not be reliably observed. In part, this isbecausemyzs17 and 18 ion signals were alreadylarge (isobaric OH , OH ), resulting from domi-q q
2
nant plasma ions. Other abundant ions(e.g. N ,q2
ArH ) for which the corresponding neutral has aq
higher IP or lower proton affinity than ammoniacan also contribute to their formation, and resolu-tion of the source reactions is not feasible withoutprior mass selection of the reactant ions.
In addition, particularly in the case of a quad-rupole when operated with a restricted bandpass,the large mass-to-charge difference between thereactant and product ions means that both are notnecessarily simultaneously stable. It will be shown(Section 8.4.2) that the DRC is operated in abandpass mode defined by the parametersq andafor the mass that is currently being analyzed bythe mass analyzer. Concomitant ions of other mass-to-charge in the reaction cell are confined accord-ing to the stability parametersq and a
corresponding to their masses:
q m sq m , or q sq m ym (6.32)Ž .1 1 2 2 2 1 1 2
as implied by Eqs.(3.5) and (3.6). Under condi-tions for which the Ar reactant ion is atq)0.39q
(as0), the NH product ion is at highq)0.9q3
and is consequently unstable. Conversely, whenthe (lower mass) product ion is stable, the oper-ating point for the(higher mass) reactant ion is atvery low q, under which conditions the efficiencyof confinement in the quadrupole field is reduceddue to scattering(Section 7.2). Accordingly, theexistence of a charge transfer reaction channel canonly be inferred by observing a reactive loss ofthe reactant ion without observing a concomitantproduct ion whose intensity change accounts forthe loss of the reactant ion.
Determination of the products of atom transfer(hydrogen and oxygen) reactions is more straight-forward because of the small mass shift of theproducts. For example, the products of the Laq
and Ho oxidation reactions with CO can beq2
clearly observed, as shown in Fig. 8. Comparisonwith the decay profiles of Fig. 6 shows that therate of increase of the product oxide ions isproportional to the rate of decay of the parentmetal ions. For a reaction that proceeds throughonly one reaction channel, the magnitude of therates of reactant decay and product appearanceshould be equal. However, there are several com-peting processes that interfere with the observationof the reaction kinetics. Perhaps the most importantof these effects are scattering and collisional focus-ing, both of which are functions of the mass ratioof the ion and neutral, the operating point of themultipole, and the number of degrees of freedomof the ion and neutral.
6.2.3.5. Scattering versus reactivity for parent andproduct ions. Plasma ion decay in the presence ofa reactionycollision gas is not always evidence ofion–molecule reaction. Similarly, product iondecay is not always indicative of a subsequentreaction channel. There was no doubt concerningthe character of ion decays in the case of Eq.(6.28) because the corresponding product oxideions could be easily observed and the rates ofreaction were substantial. However, simple scatter-
1389S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 9. Reaction profiles of(a) relatively non-reactive atomicions and(b) relatively reactive atomic ions with ammonia. Theion signals initially increase due to collisional focusing towardsthe axis of the cell, improving the transmission into the massanalyzer. The slopes of the linear portions of the ion signalprofiles are inversely proportional to the reaction rate constant.The gas flow is given in Ar-equivalent sccm(Table 4).
ing in the cell is able to mimic reactivity and itcan be challenging to distinguish between this andslow (inefficient) reaction. For instance, in thepresence of ammonia, K and Ca exhibit ‘reac-q q
tivity’ which is only 4 and 8% of the reactivity ofArO , respectively(Table 2). One can speculateq
that the observed decays of these ion signals occursmostly due to scattering in the cell. As wasmentioned before, Fe under the same conditionsq
decays just a little faster. However, the chemicalnature of this decay is obvious because the clusterions Fe Ø(NH ) can be observed when the cellq
3 x
is operated at lowq. Scattering is a complexphenomenon of its own. It includes at least threecomponents: simple scattering due to collision witha gas molecule, which moves an ion outside ofthe acceptance ellipse of the r.f.-driven cell(Sec-tion 7.1.5); scattering of product ions due totranslational excitation as a result of high exoth-ermicity of the ion–molecule reaction; and ‘scat-tering-like’ effects produced by differentsimultaneous stability of the parent and productions in the r.f.-field. In addition, space chargerelated scattering can be observed when the pres-surized reaction cell is operated under conditionsthat promote trapping or storage.
There are several methods of distinguishingbetween different scattering phenomena. In gen-eral, the uncertainty is removed by comparison toa non-reactive ion decay that is measured underthe same conditions. For example, confirmationthat the decay of a reactant ion signal is not dueto instability of its trajectory in the r.f.-field couldbe obtained by simultaneously monitoring a dif-ferent analyte ion of similar mass-to-charge butfor which reaction with the gas is significantlyslower or endothermic. As was shown before, fastion–molecule reactions(Section 6.2.3.2) are char-acterized by a linear semilogarithmic reaction pro-file. If reaction happens on every collision(k0k ),L
there is no opportunity for scattering to occur. Forreactions that are less efficient,k-k or k<k ,L L
distinction between reaction and scattering can bedifficult, except by comparison to a non-reactivebut otherwise homologous ion. Let us consider theinteractions of several analyte ions with ammoniaunder the same conditions. In Fig. 9 the elementalions under consideration are divided into two
groups: obviously reactive ions(bottom reactionprofile) and non-reactive or slowly reacting ions(top profile). Such categorical division is, ofcourse, arbitrary. However, two features in com-mon for the profiles of reactive and non-reactiveions may be evident. First, the rate of collision-induced scattering(as evidenced by the slope ofthe profiles for non-reactive ions at high flow)should be a function of the reduced mass of thecollision partners. However, the initial increase inthe observed ion signals as well as slow reactivityof some of the ions can obscure a clear dependenceon this. Second, in both cases there is an initialincrease in the observed ion signal. This increaseis not related to the production of new interferences
1390 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
and it also cannot be attributed to a new source(not associated with the plasma) of the analyteions. Rather, it is a result of collisional focusing,discussion of which we defer to Section 8.2.1.
7. Linear r.f. devices
Consider an array of an even number, 2n, ofparallel rods of lengthl placed evenly about anaxis of symmetry,z, such that an inscribed circleof radiusr contacts the inner surface of each rod.0
Further, suppose that opposing pairs of rods areelectrically connected and supplied with an ACvoltage having radio frequencyf svy2p andamplitudeV , with a phase shift ofpyn radiansrf
between neighboring rods. In addition, all of therods are supplied with an identical DC voltage,V , that defines a rod offset(also called poleRO
bias) potential. An ion that is within this arraythen has an axial kinetic energy,E sKE qz source
PE yV , where KE and PE are thesource RO source source
kinetic energy that the ion has when it exits thesource and the potential at the source, respectively.The ion has a corresponding axial velocityv sz
, where m is the mass of the ion. They2E ymz
(positively charged) ion within the array experi-ences an oscillating field and is at one momentattracted to the nearest pole at negative voltage,and at the next(1y f seconds later) to the neigh-boring rod. If the frequency is sufficiently low, orthe ion mass,m, is sufficiently small, the ion willcollide with the attracting rod before the ACvoltage on that rod reverses polarity. If the fre-quency is sufficiently high, or the ion sufficientlyheavy, its inertia will prevent it from reaching theattractive rod before the polarity changes, and theion will undergo lfyv stable oscillations through-z
out its transit of the array. Operation in this ‘r.f.-only’ mode (V is not considered) provides aRO
high-pass mass filter; only ions having massesabove a ‘low mass cut-off’ are transmitted.
If, in addition, a DC voltage,V , is appliedDC
between pole pairs, the ion will feel a continuousattraction towards the more negative(DC) polepair. If the AC frequency is sufficiently low, orthe ion mass sufficiently heavy, so that the ion’sresponse to the AC field is laggardly, the ion willdrift towards the DC-negative pole, eventually
striking that pole. If the ion mass, AC frequencyand DC voltage are just right, the ion may have astable (though tortuous) trajectory through thearray. Thus, the addition of a DC bias betweenpole pairs provides a high mass cut-off in additionto the low mass cut-off of the r.f. It will be seenthat, to first order approximation, the low masscut-off is essentially determined by the amplitudeof the r.f., V , and the applied AC frequency,v,rf
while the high mass cut-off is essentially deter-mined by the amplitude of the DC,V , and theDC
applied AC frequency,v.
7.1. General characteristics of r.f. multipoles
7.1.1. Equations of motionThis description of ion motion in an r.f.-driven
multipole is overly simplistic. In fact, the ionexecutes a tortuous trajectory in the r.f. field. Inthe theoretical instance, the r.f. and DC fields areapplied to an infinitely long multipole of ordernso as to produce a field of the form:
B E1 y2 2 ny2 C FŽ .FsF x qy cos natan (7.1)0 nr xD G0
whereF sV yV cos(vt).0 dc rf
The ion motion in Cartesian coordinates isdetermined by the classical equations of motion:
2 2≠ x e ≠ y es E ; s E (7.2)x y2 2≠t m ≠t m
The Cartesian components of the electric fieldare obtained as the gradient of potentialF:
≠ ≠E sy F; E sy F (7.3)x y
≠x ≠y
where:
2 2 n1y2w zŽ .x qy≠ x |FsF n0y ~≠x r0
B B EE B B EEy yC C FF C C FFxcos natan qysin natan
x xD D GG D D GG
(7.4)2 2Ž .x qy
1391S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
2 2 n1y2w zŽ .x qy≠ x |FsF n0y ~≠y r0
B B EE B B EEy yC C FF C C FFyycos natan qxsin natan
x xD D GG D D GG
(7.5)2 2Ž .x qy
from which the equations of motion of an ion ina multipole field of ordern can be derived accord-ing to Eq.(7.2). In the special instance thatns2(a quadrupole), it will be seen that Eqs.(7.4) and(7.5) are independent, and this has importantramifications to which we will return.
7.1.2. AdiabaticityWhen it is of interest to study ion–molecule
interactions under thermal or defined-energy con-ditions using an r.f.-driven ion guide, it is desirableto operate such that the r.f.-field does not contrib-ute significantly to the interaction energy. Underthe influence of the applied r.f., the ion is period-ically accelerated and the amount of r.f. energytransferred into the collision energy is greater forhigher r.f. amplitude for a given operating point(a, q). Hence, it is propitious to operate at arelatively low r.f. voltage consistent with the con-current desire to confine ions within the field. Ina simple view, adiabatic conditions apply whenthe total energy and momentum of the ion ensem-ble are conserved on average. Hence, the termdoes not seem to properly apply when the ionundergoes collisions (with the consequentexchange of energy that eventually ends up asradiated heat), though the concept of adiabaticitystill applies between collisions. It should be evi-dent that operation at pressures that provide morethan 1 collision per r.f. cycle also minimizes thecontribution of the r.f. energy to the collisionenergyw28x. With some liberal interpretation, theconcept of adiabaticity can be used to identifyconditions where the ion collision energetics areat least minimally affected by the applied r.f. It isgenerally accepted that ion motion in a multipoleoperated in the r.f.-only mode at lowq satisfiesthe requirements for conservation of energy andmomentum on average, and that operation at highq or with non-zeroa deviates from this condition.A general rule of thumb, developed by Gerlich
w12x, is that adiabatic conditions hold forq-0.3,though operation outside of this regime does notnecessarily imply substantial deviation from con-stant-energy conditions(depending as it does onthe amplitude of the r.f., the geometry of themultipole, the number of collisions per r.f. cycleand the ratio of ion and neutral masses, the latterapplying before thermalization). Gerlich has rig-orously considered the conditions that provide‘safe operation’ from the point of view of adiabat-icity; the interested reader is referred to that workw12x.
7.1.3. Hyperbolic versus round rodsTo satisfy the detailed theory, the field within
the multipole should be hyperbolic. Accordingly,the rods used for the quadrupole mass filter appli-cation are often produced with a hyperbolic profile.Round profile rods can emulate a hyperbolic fieldif the ratio of the radii of the rods to that of theinscribed circle,ryr , is appropriately chosen on0
the basis of theory, experiment or modeling(MonteCarlo is preferred to SIMIONw81x). For thequadrupole, an incorrect value for this ratio,ryr s1.16, was misquoted by Paul et al.w82x and0
subsequently used by other authors; the morecommonly accepted value is 1.148w31,83x, whichis chosen because it minimizes the dodecapoledistortion. Gibson and Taylorw81x empiricallydetermined that the optimum ratio is in the range1.12-ryr -1.13 through ion trajectory calcula-0
tions. Douglas and Konenkovw84x have showntheoretically that atryr s1.13 the dodecapole0
term is essentially offset by higher order contri-butions, and confirmed the prediction experimen-tally. The quadrupole is often enclosed in aconductive housing that affects the internal fieldand can minimize the higher order distortionsintroduced by the round rod approximationw85x;the calculations of Gibson and Taylorw81x indicatethat this effect is extremely small. Some quadru-pole manufacturers today have experimentallyoptimized theryr ratio in their product configu-0
ration, leading to a substantially different value ofthe ratio, which remains proprietary. A recent studyreports on SIMION calculations of the appropriateratio for hexapole(ns3) and octapole(ns4)
1392 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
arrays, recommendingryr s0.5375 and 0.355,0
respectivelyw86x.Experimental comparison of quadrupole mass
filters having hyperbolic and circular geometriesis difficult, in part because of variations in repro-ducibility of installation and in part because of thedependence of performance on the ion kineticenergy distribution, the ion beam profile at theentrance of the array and fringing fields. Probablythe most thorough study was reported by Brubakerw87,88x, showing that for a given sensitivity thehyperbolic rods can provide up to twice the reso-lution of circular rods, diminishing to comparableperformance at low(nominal unit mass) resolu-tion. The general conclusion is that under thenormal operating conditions(unit mass resolution)there is little difference between round rods andhyperbolic rods.
7.1.4. Auxiliary excitation at the secular frequencyof motion
In addition to ion motion at the frequencyvs2p f of the applied r.f., the ion oscillates at itsfundamental(secular) frequency of motion,v ,:n
B Ev bC Fv sb ; v s 1y v;0 12 2D G
B EbC Fv s 1q v; etc. (7.6)2 2D G
where 0(b(1 and is a complicated function ofthe operating conditions(a, q). This fundamentalmotion usually, and under adiabatic conditions,appears as a dominant low frequency oscillationhaving high frequency secondary motion drivenby the applied r.f. Application of auxiliary r.f. at afrequency which is in resonance with the secularmotion of the ion causes the amplitude of motionto increase, and can therefore be used to excite thespecific ion mass to eject these ions or to increasetheir collision energy in a pressurized multipole.
7.1.5. AcceptanceAn ion that satisfies the stability requirements
of the multipole(recall that stability assumes aninfinitely large and perfect device) is not neces-sarily transmitted through the multipole. The ionmust simultaneously meet the acceptance criteria
of the multipole, which results from the finitedimensions of the device. These criteria relate tothe radial displacement and radial energy(veloci-ty) of the ion and the phase of the r.f. Theacceptance is described as an ellipse in(r, drydt)phase space. In general, the acceptance ellipseconstricts at lower r.f. frequency or higherV orrf
V (higher (a, q)). For a pressurized multipole,DC
the acceptance criteria must be satisfied after eachcollision in order for the ion to remain confined.
7.1.6. Fringing fieldsThe ion must also satisfy the stability and
acceptance criteria as it enters the multipole. Oneof the realistic assumptions often adopted is thatthe magnitude of the r.f.yDC decays outside of thearray as 1yz . Hence,(a, q) is a function of the2
ion position when it is external to the cell. Whenconditions are applied to provide stability for theion within the array, the fringing field is nearlyalways destabilizing. However, rejection of an ionusually requires a few r.f. cycles, and the effect ofthe fringing field can be minimized if the residencetime is short(energy is high). Accordingly, it iscommon to accelerate ions into and out of themultipole. Brubakerw89x has described a means ofmodifying the fringing fields for the r.f.yDC appli-cation, described below.
7.2. Quadrupoles
Eqs.(7.4) and(7.5) described the motion of anion in a multipole of ordern. For the quadrupole(ns2), these equations simplify to:
d 2F0Fs x (7.7)2dx r0
d y2F0Fs y (7.8)2dy r0
and it is clear that motion in theX andY directionsis uncoupled. The trajectory of an ion in a perfectquadrupolar field of infinite length is given by:
2d uµ Ž .∂q a"2qcos2jyj us0 (7.9)02dj
2d zs0 (7.10)2dj
1393S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 10. Stability diagram for an infinitely long and perfectquadrupole in the dimensionless space of(a, q), wherea andq are the Mathieu parameters given by Eqs.(3.5) and(3.6) inthe text. Ions that are within the enclosed region are consideredto be stable, while ions that are outside of this region are con-sidered to be unstable. Also shown are the iso-beta lines thatcan be used in calculating the frequencies of ion oscillation inthe quadrupole.
Fig. 11. Quadrupole stability diagrams as a function of ionmass, plotted inV y f space. Higher masses are stable at2
dc,rf
higher amplitude of the r.f. and DC and at lower operatingfrequency. The mass filter is usually operated by scanning theamplitudes of the voltages such that the tips of the stabilityboundaries are intersected.
whereus(X, Y), js—(vt), j is the initial phase012
of the r.f. field, anda and q are the non-dimen-sional Mathieu parameters defined in Eqs.(3.5)and (3.6), with ns2, and z is defined to be thelengthwise axis of the quadrupole. Because of theindependence of motion inX and Y, the stabilitycharacteristics of a quadrupole are independent ofthe initial position of the ion within the array.Accordingly, a single stability diagram is appro-priate for the quadrupole, as shown in Fig. 10 in(a, q) space. The diagram describes a binary state:the ion is either stable or unstable, and the transi-tion between these is sharply defined at theb s1x
and b s0 boundaries. Recall thata and q arey
inversely proportional to the ion mass. For a givenset of conditions of r.f. and DC amplitude and r.f.frequency, an ion is either stable or not dependingon its mass. Specification of(a, q) for a given ionmass then defines a ‘scan line’ for all other massesthat passes through the origin. The range of ionmasses that are simultaneously stable(the ‘band-pass’) is easily established by determining themasses corresponding to interception of the stabil-ity boundaries. Clearly, the bandpass narrows asthe slope of the scan line increases. Mass filters
operate at the apex of the stability diagram. To seethis more clearly, the mass can be removed fromthe parametersa andq (from the axes of Fig. 10),yielding a series of stability curves for each massas a function of voltage and frequency, as shownin Fig. 11. The scan line that passes through theapex of one stability curve also passes through theapex of the other stability curves for other masses.This describes either of two means of scanningthe mass filter: the amplitudes of the r.f. and DCvoltage may be adjusted in concert(at a ratioV yrf
V that defines the resolution) at fixed frequency,DC
or alternatively the voltages may be kept constantand the frequency scanned. Almost all mass filterstoday operate in the first mode(amplitude-scanned), partially because the mass scale is thenlinearly dependent on the amplitudes and partiallybecause of the high accuracy and stability ofcrystal clocks to define the frequency. Typically,the mass filter is operated at r.f. amplitudes up toapproximately 7 kV and frequencies of the orderof 3 MHz (for the elemental application, for whichthe maximum scan mass isf270 amu).
Quadrupole applications can be considered fortwo general classes: mass filters and ion guides.As noted above, the mass filter application requiresoperation near the apex of the stability diagram,
1394 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 12. Ion signals for Li , Mg , Co , In , Tb and U as a function of the RPq parameter obtained with RPas7 q 24 q 59 q 115 q 159 q 238 q
0.045(RPq and RPa are proportional to the Mathieu parametersq and a). The Mg signal behaves marginally differently than24 q
the other ion signals, and is shown in dashed lines(because the other ions behave nearly identically, they are all shown as solidlines). Data obtained under essentially collisionless conditions(reaction gas flow stopped and cell vented to mass analyzer chamber,f2=10 torr) are given in(a), and for the cell pressurized with Neon(0.5 Ar-equivalent sccms0.525 sccm,f7 mtorr) in (b).y5
and it should be clear that high precision inconstruction(parallel, straight and uniform rods)is required in addition to stable and high precisionvoltage power supplies. Ion guides, on the otherhand, are generally operated near theas0 axis(r.f.-only) and typically at lowq-0.3. Under thiscondition, the acceptance and ion transmission arerelatively large. If the cell is pressurized, collision-al focusing(discussed below) increases the trans-
mission efficiency through a finite exit apertureand ion energies are damped, providing improvedmass resolution for the downstream mass analyzer.If the pressurizing gas is reactive with one or moreof the ions, the ion guide can also be used as areaction cell to study ion–molecule chemistry,especially if operated at lowq (adiabatic condi-tions), though a higher order multipole is morecommon for this application.
1395S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
For various reasons, ion transmission efficiencyis not independent ofa and q. Fig. 12a shows theion signals for severalmyz ions as a function ofthe RPq parameter at RPas0.045, which areproportional toq and a respectively, for a quad-rupole cell of length 12.5 cm having a tubularentrance aperture of 3.2 mm diameter and an exitaperture of 2 mm diameter, under near-collisionlessconditions (pressuref2=10 torr). For thesey5
data, RPq was adjusted by varying the frequencyof the r.f. rather than the more common amplitudescanning. Mean ion energies in the quadrupole cellwere mass-dependent and in the range of 4–11 eV(i.e. cell rod offset, CROs0 V) and were expectedto have an energy distribution of approximatelyone-half the mean energyw90–92x. Under theseconditions, periodic focusing effects are less evi-dent w93,94x. As expected, the normalized signalsas a function of RPq(q) are independent of theion mass. Under r.f.-only(as0) conditions for aquadrupole, ions are normally stable in the range0-q-0.908; for as0.0855 (as appears to beequivalent to RPas0.045; see below), the stabilityrange includes 0.42-q-0.84. The presence ofentrance and exit apertures, which provide bound-ary conditions of fringing fields, distorts theseboundaries somewhat. The decay of the signals asRPq is increased from the maxima near RPqs0.5is believed to result principally from the relativelywide (3.2 mm diameter) ion beam entering theDRC and reflects the constriction of the acceptanceellipse of the quadrupole as RPq increases.
Fig. 12b provides similar data obtained with thecell pressurized to approximately 7 mtorr with Ne.Again, as expected, the dependence of the ionsignals on the RPq(q) parameter are essentiallyindependent of ion mass. The profiles are similarto those of Fig. 12a except for a relative suppres-sion at the low and high RPq sides of the maxi-mum peak(i.e. the profiles are more rounded).The reduced signals at low RPq are likely due toscattering losses because the restoring forces(potential well depth) are insufficient to refocusthe ions back to the axis for transmission throughthe exit aperture. Relative suppression at high RPqis likely due to a different manifestation of scat-tering loss caused by the constriction of the accep-tance of the quadrupole: there is a higher
probability that after the collision(r, drydt) willbe outside of the acceptance.
The data of Fig. 12a and b, and further datarecorded at different values of the RPa parameter,may be converted to stability diagrams by makingan arbitrary definition of ‘stability’: we have cho-sen to consider an ion stable if the ion signal isgreater than 10 cps(i.e. RPq was scanned for agiven RPa, and the stability boundary was definedas that value of RPq for which the ion signal wasabove or below 10 cps). These data appear aspoints in (a, q) space, and are overlaid on theconventional stability diagram defined by Eqs.(3.5) and (3.6) in Fig. 13. The measured stabilitypoints in (RPa, RPq) space have been convertedto (a, q) space according to the(linear) empiricalequationsqs0.95=RPq andas1.9=RPa in orderto obtain congruence of the data with the theoret-ical values. These conversion factors are likelyrequired as a result of capacitive losses inside thevacuum chamber, resulting in approximately 5%lower V than expected, and the RPa conversionrf
includes an additional factor of 2(a simple scalingfactor). With these corrections, the experimentalpoints match the theoretical stability curves quitewell for the instance of near collisionless condi-tions. More importantly, the concept of well-defined stability boundaries holds nearly equallyfor the pressurized condition, and this has impor-tant ramifications for the use of the stabilitycharacteristics of the quadrupole for the suppres-sion of secondary chemistry in a reactive collisioncell (Section 8.4). It appears that the stabilityboundaries are compressed for low mass ionsunder multiple collision conditions. This might beanticipated because of the more severe scatteringfor low values of the ratiom ym . In someion neutral
measure, it is also an artifact of using a 10 cpsthreshold for boundary determination, as the sen-sitivity, particularly for Li , is severely dimin-7 q
ished (mass ratio;0.35) so that, for the 1 ppbconcentration used in these experiments, theboundary threshold is passed ‘early’(closer to theoptimum value ofq).
7.3. Higher order multipoles
The stability of ions in higher order multipoles
1396 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 13. Stability boundaries for the quadrupole reaction cell, defined as the point at which the absolute ion signal falls below 10cps, measured for Li (j), Mg (h), Co (d), In (s), Tb (m) and U (n) for data obtained in the manner of7 q 24 q 59 q 115 q 159 q 238 q
Fig. 12. The theoretical stability boundaries are given by the solid curves. The experimental parameters RPq and RPa were convertedto the Mathieu parametersq anda according to the empirical equationsqs0.95=RPq andas1.9=RPa in order to obtain congru-ence of the data with the theoretical values. Data obtained under essentially collisionless conditions(reaction gas flow stopped andcell vented to mass analyzer chamber,f2=10 torr) are given in(a), and for the cell pressurized with Neon(0.5 Ar-equivalenty5
sccms0.525 sccm,f7 mtorr) in (b).
is a function of the position of the ion in themultipole, and ion motion is non-independent inthe X and Y directions, as evident in Eqs.(7.4)and (7.5) for ns3 (hexapole), 4 (octapole), etc.The stability boundaries have been calculated byHagg and Szabo for a hexapolew34x and anoctapole w36x for different positions of injectioninto the multipole. Fig. 14 gives the resultantstability diagram for the hexapole for the introduc-
tion of ions into a non-pressurized hexapole atrs0.1r . Similar results are obtained for the octapole.0
In contrast to the quadrupole, for which corre-sponding calculations are given in Ref.w34x, thehigher order multipole lacks well-defined stabilityboundaries. Three conditions of stability are shownin Fig. 14: stable(I), partially stable(II) andunstable(III ). Further, different stability character-istics are obtained for different positions of ion
1397S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 14. Stability diagram for a hexapole for ions introducedat radial displacementrs0.1r , obtained by theoretical mod-0
eling. Three regions of relative stability are shown: stable(I),partial stability (II) and unstable(III ). (From Ref.w34x withpermission from Elsevier Science.)
injection. Accordingly, the higher order multipolehas failed to find application as a mass filter.
These data have two important ramifications forthe use of higher order multipoles. It is evidentfrom Fig. 14 that the region of stability of thehexapole(and similarly for the octapole) is widerin (a, q) than for a quadrupole. This means thations having a wider distribution of masses aresimultaneously stable in the higher order multipole(i.e. the bandpass is wider, though less well-defined). Accordingly, the octapole in particularhas found application in MSyMS, for which itprovides a wider range of confinement of daughterion masses than the quadrupole. This is desirablein the CID experiment, where the daughter ionsare used for identification of the parent ion thatwas fragmented. It is not a desirable characteristicwhen a well-defined bandpass is desired for controlof secondary chemistry(Section 8.4). The otherramification derives from the dependence of thestability on the position of the ion in the multipole.In the pressurized cell(collision or reaction cell),the ion must meet the acceptance and stabilitycriteria following each collision. Accordingly, dif-ferent conditions of stability and acceptance applyafter each collision. While this complicates themodeling of the pressurized higher order multipole,
the effect is offset by the wider stability, with thenet result appearing to be somewhat higher effi-ciency of collection over a wider mass range ofdaughteryproduct ions.
The effective potential describes the time-aver-aged radial potential distribution in the multipole.For a multipole of ordern (2n poles), the effectivepotential,V (n), is given byw12x:eff
2 2 2n e V eV2ny2rf dcV (n)s ryr qŽ .eff 02 216mv r 20 0n
= ryr cosnw (7.11)Ž .0where the variables are defined as for Eqs.(3.5)and (3.6) (i.e. V is the peak-to-peak voltage andrf
V is the DC potential between pole pairs). Fordc
multipoles operated in the r.f.-only mode, thesecond term is zero. Thus a higher order multipolehas a wider valley plateau and steeper walls(nearthe rods). For the pressurized multipole, this meansthat there is a larger volume near the axis wherethe restoring forces are weak. Hence the higherorder multipole finds value in applications wherethermal conditions are desirable(e.g. for the studyof thermal kinetics). On the other hand, the morenarrow well of the quadrupole suggests that theions will migrate closer to the axis so that colli-sional focusing(Section 8.2.1) should be morepronounced.
7.4. Prefilters
As discussed in Section 7.1.6, the fringing fieldsat the entrance and exit of the multipole are oftendefocusing. Brubakerw89x realized that the addi-tion of an r.f.-only quadrupole at the entrance andexit of a quadrupole mass filter delays the onsetof the instability because it delays the appearanceof the DC field component. In addition, the accep-tance of an r.f.-only quadrupole is larger than thatof a mass filter. For both reasons, r.f.-only pre-and post-filters improve transmission of ions intoand out of the mass filter. In practice, the post-filter has less benefit, particularly if an attractiveextraction field(such as is common with a close-coupled detector) is present. It has become rela-tively common practice to include only a prefilterin commercial systems. Typically, the prefilter has
1398 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
a length of ;2r . At least for organic mass0
spectrometers, it has become common to signifi-cantly extend the length of the prefilter and operateit at an elevated pressure in order to furtherincrease the transmission efficiency into the massfilter through collisional focusing(Section 8.2.1).To date, it has not been common practice to includepre- and post-filters to a pressurized reactionycollision cell, probably because they are typicallyoperated sufficiently far from the tip of the stabilityboundary(i.e. not as mass filters) that the benefitof delayed DC onset is less apparent.
7.5. Axial fields
Collisions in a pressurized multipole causedamping of the axial ion kinetic energy. If theenergy is sufficiently damped, and ignoring thenet drift of the gas, the ion motion approachesBrownian(essentially a random walk without pref-erence for direction). If this condition applies, thetransmission of ions out of the cell is expected tobe reduced. If the gas(1 sccm) is introducedcoaxially (annular area 0.06 cm) with the ion2
beam at the entrance of the cell(pressure;20mtorr), the gas will have a velocity at the entranceof the cell that is of the order of 10 cmys. While4
the gas flow will also be damped in collisions, theimpulse of the gas at the entrance of the cellinduces a net drift velocity through the cell, andthe ions will be carried with this gas flow. In thiscase, even thermalized ions are encouraged totransit the cell, typically in an average transit timeof a few milliseconds. Hence, the flow of gas canprovide an axial flow field that assists, at the least,with ion penetration into the cell.
Typically, ions are accelerated into the cellthrough an entrance aperture(or tube) at negativevoltage relative to the source. The axial fielddownstream(within the cell) associated with thisentrance potential is retarding, though its impactis marginal after a short penetration(fr ) into0
the cell. Similarly, it is common to apply anextraction potential at the exit of the cell, usuallyby biasing of the exit aperture. This field acceler-ates the ions towards the exit aperture and iseffective over an extraction length of, again, aboutr . Hence, there is a decelerating axial electric0
field at the entrance of the cell, which can bepartially mitigated by an axial flow field, and anaccelerating field at the exit of the cell. For theICP-MS application, a large ion current is typicallyintroduced into the cell, and the resultant chargedensity probably exceeds the space charge limit ofthe cell. Because of the combined effects of thefields at the entrance to the cell, it is expected thatthe maximum charge density occurs at a distanceof approximately 1 to 2r into the cell. Upstream0
of this point, the ions are pushed by their residualentrance kinetic energy and the gas flow stream(if the gas is added coaxially at the entrance ofthe cell). Downstream of this point, the ions feelan accelerating axial field from the space chargefield of the maximum ion density. Hence, thespace charge within the cell may act to introducean axial field gradient which assists with ‘pushing’the ions through the cell.
Thermalization of the ions is associated with abroadening of the distribution of ion transit timesthrough the cell, and hence a degree of homoge-nization of the ion beam takes place within a near-thermal pressurized cell. This has a significantbenefit when the correlation of signals from anoisy ion source(such as the ICP) is desired, asis discussed later in Section 8.2.2. It also inducesan effect known as cross-talk, which is related tothe finite time required for the ion distribution tostabilize after a change in ion population. This isa well-known phenomenon in organic mass spec-trometry, where the pressurized collision cell istypically capacitively coupled to a mass filter.When the mass filter is adjusted to a differentmass, a certain time is required for the ions toredistribute within the collision cell, which causesa lag in the response. If the settling time of themass filterydetector is too short, either or both oftwo observations might be made: the apparent ionsignal corresponding to the new mass may besuppressed because these ions have not yet ‘filled’the collision cell, or ions that were present in theprior state are still apparent. The matter has beendiscussed by several authorsw95,96x for the organ-ic MSyMS application.
Lock and Dyer w96x showed that a voltagegradient along the cell of only 1 V is sufficient toreduce the mean residence time to substantially
1399S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
less than 1 ms, and this alleviates cross-talk.Several means to provide this axial field have beenproposed andyor evaluatedw60,96–99x. A propri-etary electrode configuration for the cell, modeledafter the configuration described by Loboda et al.w100x has recently been introduced under the nameAxial Field Technology in a commercial ICP-MSinstrument(the ELAN DRC from Perkin ElmerPlus
SCIEX). In this instance, the axial field is gener-ated by the addition of electrodes, inserted betweenthe active rods of the cell quadrupole, whichproduce an axial field gradient of approximately0.2 Vycm when a DC potential of 200 V is appliedto the electrodes. Under conditions where there ismore than 1 collisionycm (i.e. typically )2 colli-sionsycm for near-thermal conditions), the fieldtypically adds less than 0.1 eV to the collisionenergy, which corresponds to a temperature of lessthan 1000 K w101x. Accordingly, the axial fielddoes not vigorously accelerate the ions through thecell; rather it ‘herds’ the slower ions towards theexit, thus compressing the arrival time distribution.While these additional electrodes might be expect-ed to introduce an octapolar component to the r.f.field, the quadrupolar stability characteristics aredominant and the stability boundaries remainsharply defined, as shown in Fig. 15. The slightbroadening of the apparent stability boundaries,compared to Fig. 13 likely stems from a possibleapproximately 5% imbalance in the applied axialfield in the prototype instrument which increases(in the experimental configuration used) the valueof the a-parameter.
The principal benefits of incorporating a DCaxial field are obtained for a thermal(or near-thermal) cell and include: compression of thetransit time distribution(allowing for more rapidscanning), increase of the space charge limit(alle-viation of concomitant element effects), improvedsensitivity(particularly with heavier reaction gaseswhere the provision of a directed drift field helpsto overcome scattering losses), and suppression ofclustering reactions(because these reactions arevery sensitive to the effective collision tempera-ture) w102x. Hattendorf and Guntherw79x showed,¨for a DRC cell not provided with an axial field,that the Mg signal could be suppressed using24 q
short dwell times following a mass jump from
U when the bandpass for the latter does not238 q
include the lower mass ion, as shown in Fig. 16a.Increasing the dwell(measurement) time, whichessentially allows longer time for stabilization ofthe ion distribution in the cell following the band-pass adjustment, eliminates the signal suppression.The same instrument was later operated with anaxial field using the original short dwell times,and the need for additional recovery time followinga bandpass jump from high mass was eliminated,as shown in Fig. 16b.
8. Ion chemistry in r.f. devices for analyticalICP-MS
8.1. Vacuum considerations
The gas dynamics of conventional ICP-MS areunderstood well enough for many instrumentdesign purposesw68,92,103–106x. Flow throughthe sampler is characterized as continuum, and thecentral core of this expansion is skimmed at theskimmer. In the absence of distortions, which maybe related to cone geometries, the flow downstreamof the skimmer is supersonic, and the ions gainenergy from the expansion that is proportional totheir mass. This should yield an ion beam char-acterized by mass-dependent kinetic energies witha narrow energy distribution that differs in theaxial and radial directions(high non-thermal ener-gy, but relatively narrow distribution in the axialdirection and low thermal distribution in the radialdirection). Baranov and Tannerw107x have shownthat the true axial energy distribution may bemultimodal due to the effect of rarefaction wavesdownstream of the skimmer, and that the obser-vation of these several energy distributions is afunction of the plasma conditions and ion masses.Niu and Houkw68x have identified a disturbanceat the skimmer that may be related to this. Olneyet al. w106x have studied the beam dispersiondownstream of the skimmer.
At least four configurations of the vacuumsystem for an ICP-MS instrument incorporating acollisionyreaction cell may be envisaged:
1. the cell is entirely within the ion optics(firsthigh vacuum) chamber;
1400 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 15. Stability boundaries for the quadrupole reaction cell, defined as the point at which the absolute ion signal falls below 10cps, measured for Li (j), Mg (h), Co (d), In (s), Tb (m) and U (n) for data obtained and manipulated7 q 24 q 59 q 115 q 159 q 238 q
in the manner described for Fig. 13, with the exception that an axial field was established with the incorporation of shaped electrodesplaced between the quadrupole rods. Data obtained under essentially collisionless conditions(reaction gas flow stopped and cellvented to mass analyzer chamber,f2=10 torr) are given in(a), and for the cell pressurized with Neon(0.5 Ar-equivalenty5
sccms0.525 sccm,f7 mtorr) in (b).
2. the cell is entirely within the mass analyzer(second high vacuum) chamber;
3. the cell is entirely within an additional vacuumchamber between the ion optics and mass ana-lyzer chambers; and
4. the cell communicates between the ion opticsand mass analyzer vacuum chambers.
Let us consider the vacuum pump requirements
for a system that incorporates a conventional(sampler–skimmer) interface. The flow throughthe interface into the ion optics chamber is char-acterized as continuum and is proportional to theproduct of the squares of the diameters of thesampler and the skimmer and is inversely propor-tional to the square of the separation of the samplerand skimmerw103x. For a typical ICP-MS instru-ment (sampler diameter 1.14 mm, skimmer diam-
1401S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 16. Normalized ion signals for Mg (m) and U (h) or Pb (s) as a function of dwell(measurement) time in a24 q 238 q 208 q
pressurized cell. The ions were sequentially measured with a settling time of 3 ms with(RPa, RPq)s(0, 0.45). Accordingly,Mg was rejected from the cell(outside of the bandpass) when the high mass ion was measured.(a) Without axial field, theq
Mg signal is can be suppressed at short dwell times(insufficient time to re-establish the ion distribution within the cell). (b) Withq
an axial field, the ion distribution within the cell is rapidly re-established and the measured signals are largely independent of thedwell time. ((a) Adapted from Ref.w79x with permission.)
eter 0.88 mm, sampler–skimmer separation 6.9mm), the flow of source gas into the ion opticschamber is approximately 10 atomsys. It is19
generally desirable to provide conditions such thatthe ions suffer less than 1 collision with back-ground gas in the ion optics chamber. The numberof collisions that an ion suffers is the length thatthe ion travels in the chamber divided by the meanfree path. For a collision cross-section of 50 A2˚and a path length in the ion optics chamber of 5cm, the required pressure is approximately 1.1mtorr (4=10 moleculesycm ). A vacuum pump13 3
speed of approximately 250 lys is required toprovide this pressure. In the absence of a collisionyreaction cell, the ion optics chamber usually com-
municates with the mass analyzer chamber via adifferential pumping aperture. The type of flowthrough this aperture is determined by the Knudsennumber, Kn, which is defined as the mean freepath divided by the diameter of the aperture.Continuum flow is prescribed for Kn-0.01, effu-sive flow for Kn)10, and transition flow reignsbetween these limits. The pressure required of themass analyzer chamber, in the instance that aquadrupole mass filter is used, is usually definedsuch that the mean free path is substantially greaterthan the path length of the ions within the quad-rupole. Let us assume that the path length is ofthe order of 30 cm(for a 20-cm quadrupole, wherethe additional path length corresponds to the radial
1402 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
excursions of the ions in the r.f. field). If werequire fewer than 0.1 collisions per ion within thequadrupole, the required pressure is then of theorder of 2=10 torr. If the differential pumpingy5
aperture has a diameter of 3 mm, then Kns17and the effusive flow through this aperture is2.7=10 moleculesys. The required pump speed16
for the mass analyzer chamber is then 40 lys. If acollisionyreaction cell is present and gas is addedto the cell, the pumping requirements are increased.While the actual pumping requirements are strong-ly dependent on the instrument configuration(aperture diameters, conductances, etc.), we esti-mate in Table 3 the pump speeds required for thefour generic instrument configurations describedabove as a function of cell gas flow, where wehave assumed for simplicity that the cell isenclosed and has entrance and exit aperture diam-eters of 2 mm.
The collisionyreaction cell entrance is usuallycoaxially aligned with the ion source in order tooptimize transmission of ions into the cell. If thesource flow is not impeded, the pressure at theinlet is greater than the ambient pressure of thechamber with which it communicates. This isbecause the ion source is usually at higher pressurethan the pressure of the chamber in front of thecell. The expansion of gas from the source impartsa directed flow which produces an impact pressureat the cell entrance. If the cell is exposed directlyto the flow from the sampler and skimmer, thesupersonic expansion generates an impact pressureof the order of 10–50 mtorrw106x, though it couldbe as high as several torrw105x for a closelycoupled cell. If the effective source is a bluntaperture downstream of the skimmer, the impactpressure at the cell entrance is a function of theflow characteristics emanating from the effectivesource, as described by Tanner et al.w104,105x,and of the position of the cell relative to theaperture; accordingly, the impact pressure isexpected to be about an order of magnitude lessthan for unimpeded flow, i.e. a few to a few 10sof mtorr w105,106x. Of course, the flow of sourcegas into the cell is a function of the cell pressureand of the impact pressure at the cell entrance. Ifthe cell pressure is lower than the impact pressure(though it may be higher than the ambient pres-
sure), source gas flows into the cell. This isparticularly important as the ICP source gas maycontain up to 17% H and O in the bulk Arw68x.Accordingly, unimpeded exposure of the cellentrance to the directed flow of the source gas isa major source of contamination of the cell gasand causes unintentional chemical modification(e.g. oxidation) of the analyte ions in the cell.Further, substantial penetration of Ar can lead toincreased scattering losses and energy damping(important if kinetic energy discrimination isused). Positioning of the cell at right angles to theexpansion, or disruption of the directed flow byuse of a photon stop, shadow stop or chicane lensin front of the cell, provided that a pressuredifferential across the latter is avoided, reduces theimpact pressure to the ambient pressure and mini-mizes incursion of source gas into the cell(depending, of course, on the pressures of the celland the chamber, and on the Knudsen number atthe cell entrance).
The pressure within the cell is determined bythe net flow into and out of the cell. This flowincludes the intentional flow of collisionyreactiongas, the flow of plasma gas into the cell throughthe entrance aperture(when the cell pressure isless than the impact pressure), and the flow of gasout of the cell through the exit aperture, the ventopening (if present and open) and the entranceaperture(if the cell pressure is greater than thesource gas impact pressure). If the cell communi-cates between the ion optics chamber and the massanalyzer chamber, and no collisionyreaction gas isintroduced and a suitably large vent communicatesbetween the cell and the high vacuum chamber,the pressure in the cell approaches the pressure ofthe mass analyzer chamber(for the ELAN DRC,the cell pressure in this mode is of the order of2=10 torr). In this instance, the mean free pathy5
is of the order of 300 cm. At low r.f. amplitudeand frequency and relatively high ion kineticenergy, the path length of the ion trajectory throughthe cell is approximately the length of the cell. Ifthe cell is 12.5 cm long, the number of collisionssuffered by an ion under the given conditions isapproximately 0.04; that is, 1 ion in 25 suffers asingle collision. Because the cell pressure is lessthan the ambient pressure, the gas composition of
1403S.D
.Tanner
etal.
/Spectrochim
icaA
ctaP
artB
57(2002)
1361–1452
Table 3Estimated vacuum pump speeds for four generic cell configurations
Mass of Flow of Cell Conventional(nocell)
Config. 1 Config. 2 Config. 3 Config. 4cell gas cell gas pressure
(1) The cell is entirely within the ion optics(first high vacuum) chamber.(2) The cell is entirely within the mass analyzer(second high vacuum) chamber.(3)The cell is entirely within an additional vacuum chamber between the ion optics and mass analyzer chambers.(4) The cell communicates between the ion optics andhigh vacuum chambers. These configurations do not represent any commercial instruments.Assumes: sampler diameters1.14 mm, skimmer diameters0.88 mm,sampler–skimmer spacings6.9 mm; cell is enclosed with 2 mm apertures at entrance and exit; all differential pumping apertures are 3 mm diameter; does not includeentrainment of source gas into cell; pump efficiency relative to Argon: H(0.62), He (0.78), Ne (0.97); backing pump speed is not a limitation; pressure requirements2
as given in text.
1404 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
the cell is essentially the plasma gas compositionor condensates thereof. Accordingly, the majorityof the few collisions that occur are with Ar anddo not result in a change in the ionic compositionof the beam. If the plasma gas contains 8% O, assuggested as a maximum by Niu and Houkw68x,8% of the collisions, or 1 in 300 ions, suffers acollision with an oxygen atom. In the unlikelyinstance that every such collision results in theformation of an oxide ion(unlikely because suchprocesses require a second collision to stabilizethe product ion), the oxide ratio should increaseby approximately 0.3%. Accordingly, the ion dis-tribution should remain nearly identical to theplasma source distribution.(If all of the oxygenappears in polyatomic form, such as H O or OH,2
a stabilizing collision for the condensation production is not required and the prediction off0.3%increment in the oxide ratio is more justified.) Theactual distance that an ion travels in its traverse ofthe cell is a function of the ion energy and of ther.f. amplitude and frequency, since the ion respondsto the r.f. field. Assuming that the r.f. amplitude isrelatively large so that the ion responds with asubstantial displacement on each r.f. cycle, that ther.f. frequency is 1 MHz and that the ion energy is20 eV (if there are few collisions, the ion energyis not efficiently damped), the ion experiencesapproximately 13 r.f. cycles. It might be reasonableto assume that the incremental path length duringeach r.f. cycle is of the order of 0.24 cm(thisdepends on the r.f. amplitude and the constructionof the cell), so that the path length traversed bythe ion increases to approximately 15 cm. Theconclusion that the ion distribution is largelyunperturbed from its source distribution remainsvalid. Accordingly, a vented reaction cell operatingat approximately 2=10 torr emulates a conven-y5
tional ICP-MS instrument.If the cell is entirely within the ion optics
chamber, the pressure in the cell will be at leastthe ambient pressure(and greater than this if theimpact pressure is significant). Assuming that thecell pressure is then of the order of 1 mtorr, themean free path decreases to 5.5 cm. Now, for acell of length 12.5 cm, each ion suffers on averageapproximately 2 collisions(actually, more thanthis, because the ion energy is damped after each
collision, so that the residence time increases andthe number of r.f. cycles encountered increaseswith a concomitant increase in the path lengthtraveled). In this instance, 1 in 6 ions will have acollision with an oxygen atom(if present as 8%of the gas in the cell), and since multiple collisionsexist it is more probable that the resultant oxideion will be stabilized. Accordingly, the ion distri-bution should shift significantly toward the for-mation of oxide ions(and other product ions,depending on the composition of the plasma gasentering the cell).
When the cell is operated as a collisionyreactioncell, with the intentional addition of gas to thecell, the pressure is typically of the order of 1–30mtorr. This pressure regime is difficult to measurereproducibly, since there are few types of gaugesthat will read in this range reliably, and those thatdo often suffer from hysteresis following a pressurepulse (for example, when the vacuum system isvented to atmosphere). Accordingly, it is commonto infer the pressure of the cell from the knowledgeof the flow rate of gases into and out of the cell.The flow regime at the entrance and exit lenses islikely in the transition regime between molecular(effusive) and viscous(continuum) flow, but areasonable approximation of the flow through thecell apertures is to be had by assuming effusiveflow. The equations appropriate for effusive floware well known and may be found, e.g. in Ref.w92x. It should be recognized that the effusive flowrate is inversely proportional to the square root ofthe mass of the gas; thus a higher mass gas willyield a higher cell pressure because its flow out ofthe cell is less. This dependence is seen in Fig.17, which approximates the cell pressure for anELAN DRC as a function of the true flow ofreaction gases of different masses.
The flow of collisionyreaction gas into the cellmay be controlled through a critical flow orificeor a needle valve, though it is probably mostcommon to use a mass flow controller. It must beremembered that the calibration of a mass flowcontroller is dependent on the type of gas that isflowing. It is most common to calibrate the massflow controller for either argon or nitrogen, and tomake corrections based on the flow properties ofthe gas. Many such correction factors are available
1405S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 17. Calculated pressure in a reaction cell as a function of flow rate of neutral gases having different molecular weights. Oncesufficient gas is introduced into the cell to exceed the external(ion optics chamber) ambient pressure and the impact pressure atthe entrance to the cell, the pressure may be estimated assuming effusive flow exiting through both the cell entrance and exitapertures. Effusive flow is proportional to the inverse square root of the molecular mass, and hence a higher mass gas establishesa higher pressure. In reality, the flow is at best transitional between effusive and continuum, and the model assumes thin plateapertures of diameter 2.0 and 2.2 mm. Hence, the model does not precisely describe the ELAN DRC, but the result is a reasonableapproximation to expectation and experiment for this instrument.
or can be calculated, and those for some of thecommon gases used for the ICP-MS applicationare given in Table 4.
Once the true flow of the collisionyreaction gasis known, and the flow into and out of the aperturesof the cell are accounted for, the pressure withinthe cell can be approximated. When the cellpressure is less than the impact pressure, the cellgas must be contaminated by plasma gas. As thecell pressure increases, the incursion of plasma gasdecreases until it is effectively excluded(otherthan for mixing in the aperture region; if theentrance aperture is tubular, the probability ofsource gas migrating against the pressure differ-ential into the source is small). Now, if the cell isentirely within the ion optics chamber, the gas thatsubsequently enters the mass analyzer chamber isa mixture of the plasma gas and the collisionyreaction gas, according to the relative flow ratesof the two. The mass analyzer chamber pressurewill increase only slowly with collisionyreaction
gas flow until the flow of cell gas is comparableto the flow of source gas into the optics chamber.If the cell communicates directly with the massanalyzer chamber(i.e. the only opening is the cellexit aperture), the gas that flows into the massanalyzer chamber is entirely cell gas, and thepressure of the mass analyzer chamber will changein proportion to the pressure of the cell. However,the pressure of the mass analyzer chamber is mostoften measured with a Bayert–Alpert type ioniza-tion gauge, which is sensitive to the compositionof the gas. In the instance that the gas in the massanalyzer chamber is a pure gas, corrections factorsare available. Assuming that the ionization gaugehas been calibrated for N , the correction factors2
for some of the common gases for the presentapplication are given in Table 4.
When the cell is pressurized above the impactpressure, a portion of the cell gas flows into theoptics chamber through the cell entrance aperture.Then the gas within the ion optics chamber con-
1406 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Table 4Gas correction factors for mass flow controller and for Bayert–Alpert gauge
Assumes that MFC is calibrated for Ar; true flowsMFCa
reading=MFC factor.Assumes BAG is calibrated for N2; true pressuresBAGb
reading=BAG factor.
tains a fraction of cell gas in addition to the sourcegas. The fractional composition of the gas in theoptics chamber is determined by the relative flowrates of the source gas(through the skimmer) andthe cell gas(through the cell entrance aperture).For example, for the ELAN DRC pressurized to20 mtorr with ammonia, the optics chamber pres-sure increases by approximately 5%. If the pathlength in the optics chamber(the distance betweenthe skimmer tip and the cell entrance) is 5 cm,and the pressure in the optics chamber is of theorder of 1 mtorr(so that the mean free path isf5cm, and each ion suffers, on average, 1 collision),approximately 1 of each 20 ions will suffer acollision with an ammonia molecule during itstransit between the skimmer and the cell. Thesecollisions are generally non-thermal(the ions areextracted into the cell under a potential difference)and the reaction probability is consequently
reduced, but the potential for a degree of pre-reaction exists, and these pre-reactions may sup-port endothermic reaction channels.
The considerations given here suggest that ifthe cell pressure is less than the impact pressureat the cell entrance, the purity of the collisionyreaction gas is a lesser concern since the cell mustbe contaminated with plasma gas regardless. If thecell pressure is substantially greater than the impactpressure, so that incursion of plasma gas into thecell is less probable, there is value in ensuring thathigh purity cell gases are used. This concept thenallows an opportunity to dispel a misconceptionthat is commonly held: that reactions of ions withthe neutral products of prior reactions in the cellcan be important. For example, it has been saidthat the O atoms produced in the dissociationyreaction of ArO in the cell, or O atoms producedq
in the neutralization of O , might participate inq
further ion–molecule reactions. In fact, this is avery improbable occurrence. The total number ofions measured for a typical ICP-MS mass spectrumis of the order of 10 cps. Assuming that the11
transmission efficiency of the mass analyzer is ofthe order of 10%, it may be concluded that thetotal number of ions entering the collisionyreactioncell is of the order of 10 ys. If every one of these12
ions contained an oxygen atom that was releasedas a neutral O atom following collision in the cell,the total ‘flow’ of O atoms into the cell throughthis route would be 10ys. A flow of reaction gas12
of 1 sccm contributes 4.5=10 moleculesys.17
Therefore, the maximum partial pressure of the Oatoms generated from reaction of ions within thecell is of the order of 2 ppm. That is, one of each500 000 collisions may be with the product of aprior reaction. Even at the highest cell pressures,few ions experience more than 100 collisions.Therefore, even in the extreme case, only 1 ofeach 5000 ions can be expected to experience acollision with a neutral product of a prior reaction.Further, since most of the products of the priorreactions can be expected to be atomic, and theprobability of formation of a stable condensationproduct ion resulting from reaction with an atomin a single collision is small(because a third bodycollision is then required to stabilize the production), the contribution of such reactions must be
1407S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 18. Calculated radial and axial energy as a function oftime for an ion in an r.f.-only quadrupole. The upper figureshows the radial energy in the absence of collisions. The mid-dle figure shows the initial increase and then decay of radialenergy resulting from conversion of axial energy to radial ener-gy, followed by an approach to equilibrium, caused by colli-sions with the gas. The lower figure shows the decay of theaxial energy caused by collisions with the gas.(From Ref.w28xwith permission.)
small. Therefore, in most instances, reactions ofions with neutrals derived from source ions isinconsequential, and the observation of unexpectedoxide ions most probably reflects contaminationof the cell gas by other means. The exception isthe instance when the precursor ion is abundantand the background at the product ion is low suchthat the maximum contribution of 0.02% of theprecursor ion through this route might be observed.
8.2. Reaction energy
8.2.1. Thermalization and collisional focusingUnder usual operating conditions, ions must be
accelerated(using an entrance aperture bias poten-tial) through a counter flow of gas in order toenter the cell. In this instance, the ions enter thecell with a relatively large axial kinetic energy,and the initial collisions with the cell gas areenergetic. If the energy in the center of mass(Section 5.2) exceeds the endothermicity of afeasible reaction channel, that reaction process maybe enabled. This mechanism has been invoked byDouglas w20x to explain the relatively large losscross-sections for small ions introduced into acollision cell at 50 eV(i.e. promotion of endo-thermic charge transfer).
In most collisions(Section 5.1), the ion transfersa fraction of its kinetic energy to the collisionneutral, and the ion energy is damped. In the usualinstance for ICP-MS, the axial kinetic energy ofthe ion is significantly greater than the radialenergy, and the first step in the collisional dampingof the ion’s axial energy is transfer to(excitationof) the ion’s radial energy. In an electrostaticcollision cell (without r.f.), this radial excitationwould likely lead to an increased probability ofion loss. This is one of the advantages of the r.f.-driven collision cell: the restoring force providedby the r.f. drives the radially excited ions backtoward the cell axis. Transfer of energy from axialto radial excitation continues until the two modesapproach equilibrium, and then both translationaldegrees of freedom relax together. Eventually, theaxial energy should be completely relaxed to near-thermal, under which conditions the ion motion inthe axial direction is close to Brownian. However,the radial energy does not completely relax, as it
continues to be excited to a degree by the r.f. field.At pseudo-equilibrium, the total ion energy isdistributed between the radial, azimuthal and axialdegrees of freedom, with radial being dominantdue to the r.f. excitation. The magnitude of thetotal pseudo-equilibrium energy is a function ofthe operating parameters(a, q), and of the numberof collisions per r.f. cycle(and is thus dependenton the pressure and r.f. frequency). For typicaloperating conditions(q-0.6), the total energy atpseudo-equilibrium may be of the order of 0.1 eV,which is near-thermal. A model describing theprocess of thermalization for the ICP-MS applica-tion has been reportedw28x. Fig. 18 shows theenergy transfer and relaxation due to collisions inan r.f. device.
1408 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 19. Calculated trajectories with(lighter shaded) and with-out (darker shaded) collisions for an ion in a quadrupole r.f.field at (a, q)s(0, 0.4). The same initial modeling conditionswere used, and the initial displacement is indicated by thearrow. Without collisions, the ion trajectory is bound by anoutside and an inside surface, as a result of the requirementfor conservation of the total energy and the angular momen-tum, respectively. Collisions involve transfer of energy(axialto radial, and ion to neutral), and the ion displacement initiallyincreases then collapses towards the quadrupole axis.(FromRef. w28x with permission.)
Fig. 9 showed an initial increase in ion signalsat low flows of gas that was ascribed to collisionalfocusingw50,51x. The effect is significant for non-reactive ions(k<k ). It is less pronounced forL
moderately reactive ions because the gain in sen-sitivity is offset by the reactive loss. For highlyreactive ions(k0k ), collisional focusing cannotL
occur because each collision is successful at con-verting the ion. The effect is intimately connectedwith energy damping and thermalization, wherethe reduction of ion energy causes the ions tomigrate towards the minimum of the effectivepotential well (Section 7.3) near the multipoleaxis. The effect is shown by trajectory modelingin Fig. 19. Under collisionless and approximatelyadiabatic conditions, ion motion in the multipoleis bound by the requirements for conservation ofenergy and momentum. Collisions, transferringaxial energy into radial energy, first cause anincrease in the magnitude of the radial excursions,
and then the ions collapse towards the axis as theaxial and radial energies relax together. Collisionalfocusing can be used in order to increase theanalyte ion intensity by factors of 2–5. Thisincrease directly improves the detection limitbecause the corresponding continuum(photon)background is not increased; even if the back-ground is spectral, an improvement of the detectionlimit on the order of the square root of the intensitygain is obtained because of counting statistics(ormore, if temporal homogenization is improved, asdiscussed in Section 8.2.2).
The benefits of collisional focusing are not yetwidely adopted in the ICP-MS application partiallydue to the novelty of the technique and perhapsbecause of a perception that the reaction cell inthe pressurized mode should be used only forinterfered analytes. Several aspects should be con-sidered if the benefits of collisional focusing areto be employed. To improve sensitivity, non-reac-tive gases ought to be carefully selected. The massof the collision gas should be below that of theanalyte ion in order to minimize scattering; thegas should be thoroughly dried to avoid anyreactions with contaminant water vapors; and thegas flow control manifold should be sufficientlystable (with respect to flow and composition ofthe gas in the cell) to allow quantitative analysison the top of the optimization mountain.
The thermalization of the ions has at least fourimportant ramifications.(1) As the ion energiesare relaxed, collisional focusing improves thetransmission efficiency through an on-axis exitaperture of the cell. The improvement in sensitivityfor non-reactive ions is a function of severalparameters(number of degrees of freedom of ionand neutral, mass ratio, operating point, etc.). It isimportant also to determine that the backgrounddoes not increase disproportionately, as in someinstances new interferences may limit the improve-ment in analytical performance(Sections 8.3 and8.4). (2) The collision cross-section increases withreduction of ion energy, leading to a higher numberof collisions and, thus, a higher efficiency ofreaction. At energies above approximately 1 eV,the interaction of the ion with the neutral issufficiently short that polarization of the neutraldoes not occur to an appreciable extent. Under this
1409S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 20. Calculated collision cross-sections as a function of ion kinetic energy. Above approximately 1 eV, the cross-section isdominated by the ‘hard sphere’ physical sizes of the collision partners. Below approximately 0.5 eV, electrostatic interaction effectsincrease the cross-section.a is the neutral polarizability andm is the permanent dipole moment of the neutral.(From Ref.w65xD
with permission,�Springer-Verlag.)
condition, the collision cross-section is essentiallythe hard-sphere cross-section determined by thesum of the physical radii of the collision partners.As the ion energy approaches thermal, the cross-section increases due to interaction with theinduced and permanent(if any) dipole momentsof the neutral. Fig. 20 shows the calculated cross-sections for interaction of Ar ions with neutralsq
having different polarizabilities and dipolemoments. A lower mass collision gas is lesseffective at damping the axial energy, as impliedby Eq.(5.1). Bandura et al.w65x have shown that,at relatively high energy, the number of collisionsexperienced by the ion increases only slowly(linearly) with cell pressure, reflecting the domi-nance of the(energy-independent) hard-sphere col-lision cross-section(see also Fig. 20). The numberof collisions as a function of cell pressure increasesrapidly at energies lower than 1 eV due to thesignificant increase in the collision cross-section.Since the extent of reaction is exponentiallydependent on the number of reactive collisions(given by kDtwBx in Eq. (6.21)), the efficiency ofthe cell increases dramatically when near-thermalconditions are obtained.(3) Under thermal condi-
tions, the specificity inherent in thermal chemistrymay be used to improve chemical resolution ofisobars.(4) The selection of an appropriate reac-tion gas is immensely simplified by reference tothe existing (and growing) database of thermalrate constantsw10,75–77x. These rate constants,and the product branching ratios, have for the mostpart been measured under assiduously thermalconditions, using techniques such as the FAw74xand SIFTw11,66x. Provision of a controlled thermalenvironment in the reaction cell, achieved throughcollisional thermalization, allows direct applicationof these data in the development of new methods.
8.2.2. Temporal homogenizationCollisional focusing is also accompanied by
collisional cooling, wherein substantial reductionof the average ion kinetic energy and of the widthof the energy distribution is achieved. It can beexpected that collisions in the pressurized deviceshould also modify the arrival time distribution ofions. The fact that short transient signals arebroadened in the pressurized multipole was recent-ly used in a collisional damping ion transmissiondevice for decoupling a pulsed MALDI ion source
1410 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
from a continuous beam orthogonal-accelerationtime-of-flight mass-analyzer, as described byKrutchinsky et al.w108x. Collisional broadening ofthe ion pulses allowed, in this case, the conversionof a pulsed ion beam into a quasi-continuous beam,with average arrival times for MALDI ions ofDalargin, Substance P, Melittin and Insulin rangingfrom 9 to 26 ms and half-width of the arrival timedistributions of the same order. It was observedthat the average arrival time depends on the pres-sure in the devicew108x and on the DC potentialdifference between the entrance and exit apertureplates and the quadrupole rodsw52x. Work per-formed in our laboratory for elemental ions froman ICP suggests that the degree of the collisionalbroadening in a pressurized r.f. quadrupole alsodepends on the target gas(dipole moment andpolarizability, as well as the number of vibrationaldegrees of freedom) and on the Mathieu parameterq of the quadrupolew109x. Arrival time distributionprofiles for Ag ion pulses transmitted through107 q
a r.f.-only quadrupole pressurized with differentgases is given in Fig. 1 of Ref.w109x. Polyatomicgases, even if lighter, produce more significantbroadening. This can be attributed to more rapidthermalization of the ions due to a larger fractionof the translational energy being converted intointernal energy of the polyatomic target, and alsoto the higher number of collisions with the polya-tomic target gases due to their higher polarizabilityand thus higher(thermal) collisional cross-section.The dependence of the arrival time distribution onthe operating parameterq is attributed to a com-bination of r.f.-heating, the depth of the pseudo-potential well, and the dependence of the stabilitycharacteristics of the dominant ion population inthe quadrupole onq. As plasma matrix ions enterthe pressurized quadrupole at a rate off1012
ionsys, the stability of the dominant ions and thedevelopment of a space-charge hill can play amajor role in defining the residence time of otherions in the device.
Collisional damping has been used to homoge-nize the fluctuations of the ion beam extractedfrom an ICPw110x in order to improve the preci-sion of the isotope ratios measured by a quadrupoleanalyzer. As collisional broadening occurs on atime scale of several milliseconds, fluctuations of
the ion current at high and intermediate frequenciesare smoothed, resulting in better correlationbetween sequentially sampled ion populations. Itwas shown that the collisional homogenizationallows achievement of the counting statistics limitfor the isotope ratio internal precision, with dem-onstrated precision of 0.02–0.03% R.S.D. for
at sample concentrations of 40 ngyml. Conven-tional methods for reduction of plasma noise, suchas free sample aspiration or use of a torch bonnet,were not used. It was also shown that the externalprecision of Pb isotope ratios, measured for 7samples atns9 replicates per sample, was approx-imately6n times lower than the internal precision,which indicated that the external precision wasalso defined by counting statistics only. Whenatomic gases are used for collisional homogeniza-tion, the number of collisions needed to achievesufficient (on a millisecond scale) temporalhomogenization can be enough to cause scatteringlosses of ion signal, so that the absolute value ofratio precision(for a given sample concentration),although being at its statistical limit, may notimprove compared to non-pressurized cell opera-tion. As polyatomic gases produce more significantdamping of fast ion density fluctuationsw108x, alesser number of collisions is required, and theabsolute value of the ratio precision is improved.The most practical case of collisional homogeni-zation is with use of a polyatomic gas for chemicalresolution of isobaric interferences. Use of a reac-tive gas such as NH allows simultaneous reactive3
removal of isobaric interferences and collisionalhomogenization of the analyte ion populations. Ithas been shownw109x that the ratios of thenormally interfered isotopes Fe, Fe and Fe54 56 57
can be measured by quadrupole ICP-MS at thecounting statistics limited internal precision of0.05–0.1% for a sample concentration of 50 ngyml.
8.2.3. r.f. contribution to reaction energyIt should be evident that the r.f. contributes to
the reaction energy through the radial excitationof the ions. The energy contributed in this fashionis essentially the pseudo-equilibrium energy dis-cussed above. Accordingly, a higher reaction ener-
1411S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 21. Ion signals for Mn and Zn as a function of ammonia gas flow at several operating points,q, in the quadrupole stabilityq q
diagram. Both ions are unreactive(or slowly reactive, perhaps scattered) at low and mid values ofq. Reaction is promoted forZn at highq, while Mn remains unreactive. Charge transfer for Zn and Mn with ammonia is endothermic(at room temperature)q q q q
by 0.8 and 2.8 eV, respectively. The results suggest that the ion energies are less than 0.8 eV at low and mid values ofq, but areabove 0.8 eV due to the r.f. contribution to the ion energy under non-adiabatic conditions at highq. (From Ref.w28x with permission.)
gy is provided by a higher r.f. amplitude or byfewer collisions per r.f. cycle(i.e. a lower pressureor higher frequency). Competing with the latter isthe breakdown of the adiabatic approximation athigherq, which appears to dominate. For example,Mn has an IP of 7.4 eV and Mn is unreactiveq
with NH since charge transfer is 2.76 eV endo-3
thermic. Zn is endothermic for charge transferq
with NH by only 0.78 eV. As shown in Fig. 21,3
neither Mn nor Zn are seen to react withq q
ammonia at q-0.7. However, asq is furtherincreased(by reducing the r.f. frequency; the r.f.amplitude was constant through this experiment),reaction of Zn is facilitated, presumably byq
charge transfer, while Mn remains unreactiveq
(and serves as a normalizing signal to correct forpotential scattering losses). It is concluded that thereaction cell employed contributes less than 0.8eV to the reaction energy atq-0.7, but that ther.f. contribution to the reaction energy can beadjusted through the operating parameterq.
8.2.4. Transferability of methodsIf methods are to be transferable from one
instrument to another, and the methods take advan-tage of near-thermal chemistry, the instrumentsmust provide similar thermal environments and amethod must be available to validate this assump-tion. One means to determine the thermal charac-teristics of a reaction cell is to establish athermodynamic ladder of reactions. A proposal forsuch a scheme has been madew28x, and is givenin Table 5. It is proposed that these reactions beinvestigated by measuring the reactant ion signalas a function of reactant neutral flow, in order todetermine the lower limit of endothermicity sup-ported by the instrument. For example, the reactionof Se with NH is observed to proceed(thoughq
3
the product ion was not observed, it is presumedto react by charge transfer) over a wide range ofthe operating parameterq w28x with an ELANDRC. As noted above, the reaction of Zn withq
NH is observed only atq)0.7. Therefore, a lower3
1412 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Table 5Proposed reactions for thermodynamic evaluation of a reaction cell for ICP-MS
Ion Reaction Products DH DHgas (kcalymol) (eVymol)
Ceq q O2 ™ CeOq q O y70.8 y3.1Xeq q O2 ™ Oq
2 q Xe 1 0.058Seq q NH3 ™ NHq
3 q Se 9.3 0.4Znq q NH3 ™ NHq
3 q Zn 18 0.78Iq q O2 ™ Oq
2 q I 36 1.6Seq q N2 ™ Nq
2 q Se 133 5.8CeOq q H2 ™ Ceq q H O2 188.2 8.2
limit for the r.f.-field contribution to the reactionenergy for this instrument is determined as-0.4eV at low and mid values ofq (consistent withthe estimation given in Section 8.2.1), and thiscontribution increases at high values ofq. It wouldbe valuable to determine the consistency of theseobservations on different instruments of the samemanufacture, and also across platform designs, inorder to validate the concept that developed meth-ods are transferable between instruments.
8.3. Sequential chemistry
The gas that fills the cell is usually reactivewith at least some of the ions. Even if an inten-tionally non-reactive gas is used, reactive contam-inants are commonly present either as an impurityor from entrainment of plasma gas into the cell.As long as ions and reactive gas components arein the cell, reactions may take place resulting inthe formation of new ions that may be isobaricwith an analyte of interest. If multiple collisionconditions are provided, as required for efficientsuppression of plasma-based interference ions, sec-ondary reactions that may produce a ubiquitouschemical background are promoted.
For example, if a cell of length 12.5 cm isoperated at a pressure of approximately 10 mtorr,the average ion experiences some 20 collisions. Ifan ion reacts on its first collision, the product ionon average may suffer a further 19 collisions. Inthis instance, there are some 20 banked levels ofsequential chemistry. Because the ion flux into thecell is large (which distinguishes the ICP-MS
applications from other mass spectrometries), evena gas contaminant at trace levels can producesignificant levels of new interferences.
As an example, consider a subset of the reac-tions that can occur using methane as a reactiongas. It is knownw10x that Ar reacts with methaneq
via dissociative charge transfer according to:
q qAr qCH ™CH qAr (8.1)4 4
q™CH qArqH (8.2)3
q™CH qArqH (8.3)2 2
The products of this reaction, if retained in thecell, react further. For instance, the CH ion reactsq
3
with methane by condensation according to:
q qCH qCH ™C H qH (8.4)3 4 2 5 2
and the product ion of this reacts further accordingto:
q qC H qCH ™C H qH (8.5)2 5 4 3 7 2
In general, condensation and dissociative protontransfer reactions will generate a rich array ofhydrocarbon ions:
q qC H qCH ™C H q(neutral products) (8.6)n x 4 m y
with ms1, 2,«, nq1 andys0, 1, 2,«, xq4.Further, if a trace of water is in the cell, ions of
the form C H O are to be expected(in additionqm y
to oxide and hydroxide ions of elements in thesample).
Accordingly, a reaction cell operated at severalmtorr methane, and which is not operated in a
1413S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 22. Spectra obtained using an ELAN DRC with an axial field applied, for high purity water(grey) and a solution containingPlus
1 ppb each of K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, Se and Sr(black). (a) Standard mode(no gas added to cell, cell vented to massanalyzer vacuum chamber), with (RPa, RPq)s(0, 0.2), CPV (entrance and exit lenses of cell)sy16, CRO(cell rod offset)sy12 and QRO(mass analyzer quad rod offset)sq2. (b) DRC mode with CH reaction gas flows0.92 Ar-equivalent sccm(0.484
sccm,f6 mtorr), with (RPa, RPq)s(0, 0.15), CPVsy18, CROsy1 and QROsy3.
manner to suppress either the formation or trans-mission of product ions, should be expected toshow an elevated spectral background at manymasses. Fig. 22a shows overlaid mass spectra fora DIW blank and a mixture of analyte elementseach at 1 ppb recorded without reaction gas in thecell (i.e. ‘standard mode’ to emulate conventionalICP-MS). The relatively low spectral backgroundin important regions of the spectrum allows sen-sitive determination of many elements(e.g. Ni (as
Ni ), Cu, Zn and Sr are readily observed), while60 q
K, Ca, Cr, Mn, Fe, Ni and Se are obfuscated by58
argide ions(ArH , Ar , ArC , ArN , ArO ,q q q q q
Ar O and Ar , respectively). By contrast, the18 q q2
same spectra are shown in Fig. 22b with methaneas a reaction gas under conditions where a broadrange of ion masses are simultaneously stable inthe cell. While some of the argide ion signals aresubstantially reduced in intensity, as a result ofreaction with CH , a generally elevated spectral4
background is observed. This background is prin-cipally the hydrocarbon ions produced in sequen-tial reactions, as discussed above. The resultantdetection power of the instrument for elements
1414 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
that are not normally interfered is seriously degrad-ed by the secondary chemistry.
It should be evident that a reaction cell can beoperated in a manner to efficiently reactivelyremove plasma-based interference ions(e.g.though the scales used make it less evident, the
Ar and Ar signals in Fig. 22b are reduced40 q 40 q2
by nearly 4 orders of magnitude relative to Fig.22a), but this improvement is more than offset bythe formation of new interferences at many masses.This then raises the issue of the three forms ofefficiency that are required of the reaction cell:(1) efficient removal of plasma-based interferenceions, (2) efficient transmission of analyte ions ofinterest, and (3) efficient suppression of theappearance of new interference ions produced insecondary chemistry in the reaction cell. As wehave discussed above, the first two efficiencies areaddressed by the selection of the reaction gas(reactive with one of the interference or the anal-yte) and the pressure of the gas(gas thickness) inthe cell, and may be influenced also by electricalconditions under which the cell is operated(e.g.the end cap voltages and the r.f. amplitude). Wenow must address means of achieving the thirddimension of efficiency: suppression of unwantednew ions produced in the cell. To make thingsmore difficult, there are also instances in which itmay be desirable to determine an analyte at adifferent mass, as the product of an atom transferreaction; examples, including oxidation of V,hydroxylation of Sr and fluorination of Sr, aregiven in Section 8.4.3. When this is desired, it isalso desirable to suppress the formation of otherions that might interfere at the same mass. Hence,a degree of selectability in the promotion andsuppression of product ions in the cell would bevaluable.
8.4. Secondary chemistry control
There are a number of possible means to controlthe appearance of secondary reaction product ionsin the mass spectrum. Two of these dominate intoday’s instruments. Kinetic energy discriminationafter the cell allows operation of the cell as apassive device(in the sense that the chemistry isallowed to proceed unhindered). In this method,
resolution of the plasma ions from cell-producedions is based on the kinetic energy differencebetween them. A potential energy barrier is estab-lished downstream of the cell and provides adegree of resolution of plasma polyatomic ionsfrom atomic ions. Alternatively, the cell may beoperated in a bandpass mode to establish a windowof ion masses that are simultaneously stable withinthe cell and that rapidly ejects ions from the cellthat are outside of this window in order to restrictthe occurrence of secondary chemistry within thecell itself.
8.4.1. Post-cell kinetic energy discriminationWe earlier noted that ions are generally intro-
duced into the cell with excess axial energy. Thisis because the cell is typically operated at higherpressure than the optics chamber that precedes it;therefore, the ions must be accelerated through thecounterflow of gas emanating from the cellentrance aperture. Under conditions of high colli-sion number, the ion loses axial kinetic energysequentially in subsequent collisions until a near-thermal distribution of energies is obtained. Thegas can be considered as essentially stagnant inthese collisions. If the ion is substantially moreheavy than the gas molecule and reacts with thegas by small charged particle transfer(an electronor proton), the resultant product ion remains nearlyas stagnant as the reactant gas molecule was(essentially,E9 in Eq. (5.2)). Reaction of an ion2
by transfer of a heavy atom(e.g. O) also leads toa product ion having a lower kinetic energy, inpart because at least a part of the product ion isderived from the stagnant neutral and, perhapsmore importantly, because such reactions typicallyrequire a relatively long interaction time betweenthe ion and neutral during which the kinetic energyof the partners is redistributed. Polyatomic ions,either as the reactant ion(perhaps sampled fromthe ion source) or as the product ion, present aspecial case. In this instance, some of the energythat is delivered to the collision complex from theion’s pre-collision kinetic energy can be redistrib-uted into the internal degrees of freedom of theproduct(or original ion that has undergone colli-sion without reaction) polyatomic ion. As a result,its post-collision kinetic energy can be lower than
1415S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
the kinetic energy of an atomic ion of the samemass-to-charge ratio. Moreover, the polyatomicions, due to their relatively large size, may havesignificantly larger hard-sphere collision cross-sec-tions than those of atomic ions. As a result, atcollision energies above approximately 1 eV, theyexperience a larger number of collisions and thuslose more kinetic energy per unit path length thanatomic ions do. The difference in collision cross-sections generally diminishes at lower energies, asthey are then defined principally by the ion–dipoleinteraction(which is a function of the neutral andnot substantially of the ion).
Therefore, prior to thermalization of the incidentplasma ions, the product ions might be discrimi-nated from the plasma ions on the basis of theirkinetic energy. Placement of a potential barrierdownstream of the cell(e.g. an aperture lens orthe mass analyzer quadrupole bias voltage at apotential more positive than the cell pole biaspotential) discriminates against ions that haveenergies less than the barrier potential. If thebarrier height is between the axial energies of theplasma ions and the energies of ions that areproduced within the cell, a degree of discriminationagainst the latter is afforded. If the energy distri-butions of the plasma ions and cell-formed ionsoverlap, the potential barrier cannot be used effi-ciently and the transmission of both analyte andinterference ions is suppressed.
As noted in Section 5.1, the rate of energydamping is proportional to the ratio of the massof the ion to the mass of the gas molecule. Fig.23 gives the results of calculations of energydamping as a function of ion and neutral massassuming elastic(no internal excitation) interac-tion. As assumed for the fragmentation calculationsof Section 5.2, the ion energy at the entrance ofthe cell is taken as 8 eV. Relatively realistic casesare considered, though the ions and neutrals aretreated as elastic particles and hence we disregardvibrational modes of energy damping and ignoreion–dipole interactions(i.e. we consider the hardsphere case). To a first approximation the examplescould be considered to represent the collision ofAr with H (m ym s40), Ar with Heq q
2 2 ion neutral
(m ym s10) and Ar with CH (m yqion neutral 4 ion
m s2.5). The cell pressure is assumed to beneutral
20 mtorr and r.f. effects are ignored. Fig. 23a givesthe residual ion energy as a function of the numberof collisions suffered. Clearly, a higher mass ratiois less efficient at damping the ion energy; after agiven number of collisions, the ion retains moreof its initial energy, and hence is more easilydistinguished from ions produced within the cellon the basis of the kinetic energy difference. Asan example, we have indicated a horizontal line at1.6 eV which is intended to represent the kineticenergy barrier height that might be used. Onaverage, a collision pair having a high mass ratio(e.g. Ar yH s40) allows discrimination(assum-q
2 2
ing the unrealistically favorable case that the ionenergy distributions are extremely narrow) up to35 collisions, whereas the lower mass ratio pairsbecome indistinguishable(on the basis of kineticenergy alone) at as few as 3 collisions. These dataare displayed differently in Fig. 23b, where theabscissas axis has been converted to distancetraveled through the cell. Because the higher massratio case leaves the ion with greater residualenergy after each collision, that ion also travelsthe furthest for a given number of collisions. Sincethe energy discrimination device is applied at ordownstream of the exit of the cell, the ions musttransit the cell retaining sufficient energy to sur-mount the potential energy barrier. Therefore, themaximum pressure(which is interchangeable withdistance in this example) that allows for kineticenergy discrimination is proportional to the ionyneutral mass ratio.
As has been emphasized earlier, the efficiencyof removing the plasma-based interference ion isexponentially dependent on the number of reactivecollisions. If each collision considered in Fig. 23is effective at reacting the isobar, the efficiency ofthe cell as a function of ionymass ratio can beinferred from the data given. However, conditionsmust be applied that ensure that the majority ofions retain an energy above the potential barrierheight, and thus these ion–molecule collisions areenergetic(non-thermal). As shown earlier, the rateof reaction typically decreases as energy(temper-ature) increases, and so it may be concluded thatthese energetic collisions may be less effective inleading to reaction, resulting in lesser efficiencyof isobar removal than the number of collisions
1416 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 23. Calculated residual kinetic energy as a function of the number of collisions(a) or distance traveled through the cell(b)for an ion (Ar or Ar ) with H , He or CH , corresponding to ionyneutral mass ratios of 40, 10 and 2.5. The horizontal dashedq q
2 2 4
line is intended to indicate the height of a possible kinetic energy barrier placed downstream of the cell: ions that have energiesabove the barrier height may be transmitted to the mass analyzer, while ions having energies below the barrier height are discrim-inated against.
alone implies. Further, because the ion kineticenergy adds to the reaction energy, endothermicprocesses are facilitated and may diminish thespecificity provided by thermal chemistry by pro-moting unwanted reaction of the analyte ion orallowing alternate reaction channels yielding unex-pected product ions.
The kinetic energy barrier may be applied as arelatively repulsive ion optic component down-stream of the cell(e.g. an aperture lens), or evenat the exit end cap of the cell. It is more commonto discriminate the ions using the rod offset of themass analyzing filter(or, equivalently, the bias
potential of the cell rods). Under thermalizedconditions, the ion energy is defined by the cellbias potential, as the ions ‘forget’ the energy thatthey had at the ion source. If the mass analyzerrod offset (QRO in the instance of the ELANDRC) is more negative than the CRO, exiting ionsare accelerated and energy discrimination is notapplied; energy discrimination may be effectedwhen QRO is more positive than CRO providedthat the ion energies are thermalized to near CRO.Fig. 24 shows the ion signals for three analyteions (Na , Cu and Pb) and an ion that isq q q
principally formed in the cell(myzs45, not iden-
1417S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 24. Ion signals for several ions as a function of quadrupole rod offset potential for various H gas flows. The gas flow, in Ar-2
equivalent sccm(1 units0.7 sccm H), is given beside the arrows. The CRO wasy1.0 V (indicated by the grey vertical line).2
The ion atmyzs45 (not identified) is primarily produced within the cell. Ion energies tend towards the in-cell ion energy(nearthe CRO potential) at higher H flows and lower ion masses. Note that the vertical scales differ.2
tified) as a function of the mass analyzer rod offsetpotential(QRO) and as a function of the flow ofH into the cell. For these experiments, the CRO2
was y1.0 V. The pressure within the cell is notdirectly monitored for logistical reasons; providedthat the flow of the gas and the conductance ofthe cell apertures is known, the cell pressure canbe calculated with reasonable accuracy. Our bestattempt at these calculations for our instrumentwas given in Fig. 17, where the flow of Hydrogen(2 amu) is corrected according to the factors givenin Table 4. Because the ion transmission efficiencythrough the mass filter is a function of the ionenergy in the mass filter, which discriminatesagainst low energy ions, these ‘stopping curves’may not be directly interpreted as ion kineticenergy curves(though these are related, and wewill discuss them as though they reflect the ion
energy distribution). If there were no bias in theenergy filtering, the stopping curves could bedifferentiated to yield ion energy distributions.There are two important characteristics of theenergy distribution that the stopping curve pro-vides: the ‘most probable energy’ and the energydistribution width. These may be interpreted todescribe a stream having a flow velocity corre-sponding to the most probable energy and aneffective temperature given by the width of theenergy distribution. If the energy distribution isapproximately thermal, the ‘most probable energy’typically falls near the point where the normalizedstopping curve falls below 1ye; we will use this‘definition’ as a marker for ion energy. Secondly,it should be clear that a steeper slope correspondsto a lower ‘temperature’ and a broader slope to ahigher ‘temperature’. We will use this interpreta-
1418 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
tion to compare sets of data below. The zero pointof the energy is approximately the energy withwhich the ions leave the vacuum interface(plasmapotential plus the energy corresponding to theexpansion velocity plus the interface potential if itis positive) at low cell pressure and shifts to thecell bias potential(CRO) when many collisionsthermalize the ions. In fact, prior to thermalization,the flow velocity may have a wide distribution(poorly defined most probable energy) and two ormore simultaneous temperature distributions.
When no gas is added to the cell(and in thisinstance the cell is vented to the high vacuumchamber so that the mean free path is very long),the stopping curves reflect the energies that theions gain in the supersonic expansion to vacuum.When gas is added to the cell, the most probableenergies of the source-derived analyte ions(Na ,q
Cu and Pb ) decrease, but notably the tempera-q q
ture of these ions increase. This is completely inaccord with the description given in Section 8.2.1,whereby the axial kinetic energy of the ions is firstconverted into radial excitation and then both theaxial and radial energies(temperatures) relaxtogether towards an asymptotic temperature distri-bution. Before relaxation is complete, the temper-ature gradient(i.e. the rate of change of the widthof the energy distribution) can be quite dramatic,and different ions can demonstrate different ‘tem-peratures’. In the data shown here, the Na andq
Cu ions relax somewhat more than do the higherq
mass Pb ions, though all of these source-derivedq
ions remain well above room temperature even atthe highest H flow. Clearly, H is ineffective at2 2
thermalizing the ions, though the temperature andmost probable energy of the ions is inverselyproportional to the ionyneutral mass ratio. On theother hand, themyzs45 ions are cool relative tothe atomic analyte ions, reflecting their formationwithin the cell (evidenced by nearly 4 orders ofmagnitude increase in signal intensity with addi-tion of H to the cell).2
On the other hand, the use of a heavier andpolyatomic gas such as methane induces a rapidcooling of the ions and reduction of the flowvelocity, as seen in Fig. 25. At a flow of 0.2 Ar-equivalent sccm(0.10 sccm,f2 mtorr, estimated),the most probable energy has decreased signifi-
cantly and the ‘temperatures’ of many of the ionshas increased somewhat. With 0.4 Ar-equivalentsccm flow (0.21 sccm, f3 mtorr), the flowvelocity is quite low (the most probable energyfor most of the ions is near the CROsy1.0 V)and the majority of the ions have a relatively lowtemperature. Further increases in methane flowtend to thermalize the ions in the tail of the energydistribution.
Kinetic energy discrimination nearly equallydiscriminates against desired product ions(e.g.oxidation or fluorination, in the instances wherethese are promoted for analytical benefit) and theunwanted product ions of the sequential secondarychemistry. That is, kinetic energy discriminationdoes not facilitate measurement of an analyteshifted in mass by the intentional addition of anatom while simultaneously suppressing the appear-ance of background ions produced in the cell inthe sequential chemistry.
In order to properly demonstrate the efficiencyof the cell at improving the signalybackgroundratio by plotting reaction profiles either as afunction of reaction gas flow or potential barrierheight, it is necessary to show data for both amatrix blank and an analyte sample. Measurementof the ion signal as a function of the kinetic energydiscrimination barrier height measures two phe-nomena simultaneously: ion loss due to scattering,reaction or fragmentation, and signal loss due toenergy damping(the ion energies shift below thebarrier height). Only the former results in animprovement in the signalybackground ratio unlessthere is a substantial difference in the rate ofenergy loss of the isobaric ions. Because of theiradditional degrees of freedom in which collisionenergy can be deposited, polyatomic ions loseenergy somewhat more rapidly than do atomicions. Therefore, kinetic energy discrimination pro-vides a degree of resolution of atomic(analyte)ions from polyatomic(plasma-based interference)ions, but this distinction is generally small and isachieved through differential signal loss(i.e. boththe atomic ion and polyatomic ion signals arereduced, with the latter somewhat more signifi-cantly). Therefore, observation of suppression ofa plasma-based interference ion using kinetic ener-gy discrimination(either by scanning the height
1419S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 25. Ion signals for several ions as a function of quadrupole rod offset potential for various CH gas flows. The gas flow, in4
Ar-equivalent sccm(1 units0.52 sccm CH), is given beside the arrows. The CRO wasy1.0 V (indicated by the grey vertical4
line). The ion atmyzs43 (not identified) is primarily produced within the cell. Ion energies tend towards the in-cell ion energy(near the CRO potential) at higher CH flows and lower ion masses. Note that the vertical scales differ.4
of the barrier or by varying the gas density at agiven barrier height) might infer a concomitantimprovement in analyte signal-to-background, butthis must be confirmed by similar measurement ofthe analyte ion signal. For example, Boulyga andBecker w111x have reported that the Ar signalq
2
can be suppressed by 5 orders of magnitude usingH yHe and a potential barrier height of 1.6 eV2
(the difference between the Hexapole Pole Biasand the mass analyzer pole bias), but only by 2orders of magnitude with the barrier removed. Atthe same time, they report that the ratio of theSe ion signal to background(Ar ) signal, meas-q q
2
ured with the potential barrier, improves only by2 orders of magnitude. The implication is that 3of the 5 orders of magnitude suppression of Arq
2
results from damping the ion energy below thebarrier height, and that Se is similarly suppressed.q
To select an appropriate set of cell conditions todemonstrate the efficacy of post-cell kinetic energydiscrimination, we recorded reaction profiles(ionsignal vs. gas flow) at various barrier heights(applied as the mass filter rod offset, QRO, relativeto the CRO). As will become evident shortly, wewere unable to select amyz for an analyte ion thatwas interfered by a cell-formed ion for which theinterference ion could be efficiently removed byreaction and for which energy discrimination pro-vided a substantial improvement in detectability.Therefore, despite the paragraph above, we record-ed reaction profile data for several generally-not-interfered analyte ions and several ions thatappeared to be formed within the cell. The selec-tion of the ‘optimum’ kinetic energy barrier height(QRO) was taken from the data of Figs. 24 and25, where the ratio of the Cu signal to an63 q
1420 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 26. Reaction profiles of analyte ions(solid lines) and uni-dentified ions produced within the cell(dashed lines) as afunction of H reaction gas flow(1 Ar-equivalent sccms0.72
sccm). (a) Without post-cell energy discrimination(quadru-pole rod offset isy3 V, 2 V more negative than CRO). (b)With post-cell energy discrimination(quadrupole rod offset isq2 V, 3 V more positive than CRO).
‘interference ion’(myzs45 for H , myzs43 for2
CH ) was near maximum and the analyte signal4
intensity was simultaneously as large as possible(i.e. on the basis of background equivalent con-centration, BEC). Accordingly, data were recordedfor QROsy3 (non-discriminating) and QROsq2 (energy discriminating).
Reaction profiles for H as the reaction gas and2
withywithout energy discrimination are given inFig. 26. In the absence of energy discrimination,analyte ion signals are insensitive to the H flow.2
On the other hand, the dramatic(orders of mag-nitude) increases in the not-identified ion signalsat myzs37 and 45 evidences their formation by
reaction within the cell. When kinetic energydiscrimination is applied, only a slight loss of theanalyte signals is obtained, corresponding to thefraction of these ions that have been cooled belowthe barrier height. However, the ‘interference ions’(e.g. myzs45) are suppressed by up to 3 ordersof magnitude(relative to the non-discriminatingcase). As much as these ‘interference ions’(albeitat a differentmyz than the analyte ions) reflectthe behavior of same-mass interference ions, theresultant improvement in the signal-to-backgroundis approximately 2 orders of magnitude.
It is important to realize that the application ofthe kinetic energy barrier does not affect theformation of the ‘interference ions’ within the cell:they are formed at the same rate independent ofthe QRO voltage. Only the transmission of theseions through the mass filter, and hence theirdetection, is affected by the energy barrier: thisreduces the interference by 1–2 orders ofmagnitude.
Reaction profiles for CH as the reaction gas,4
obtained under similar conditions, are given inFig. 27. In the instance where energy discrimina-tion is not applied, the analyte ion signals otherthan Li actually increase with methane flow,7 q
presumably a result of collisional focusing whichis more effective for the heavier, polyatomic col-lisionyreaction gas. The production of new ‘inter-ference ions’(e.g. myzs43) is again evidencedby a 3 order ofmagnitude increase of these signals.However, the application of an energy barriercauses a substantial loss of analyte signal. Thisreflects the more efficient cooling of the ions bythe more heavy, polyatomic reaction gas, so thatthe energies of a large fraction of the analyte ionsfall below the barrier. It could be noted, then, thatcollisional focusing is not commensurate withenergy discrimination: the increase in signals dueto collisional focusing is coincident with the reduc-tion of their ion energies, and these lower energyions are those that are discriminated against by theenergy barrier. Accordingly, the ions that aredetected tend to be those that have had the fewestcollisions, and these have consequently not beenfocused to the axis of the cell. Even more so thanin the instance of H , the ‘interference ions’ are2
highly discriminated against, by up to 5 orders of
1421S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 27. Reaction profiles of analyte ions(solid lines) and uni-dentified ions produced within the cell(dashed lines) as afunction of CH reaction gas flow(1 Ar-equivalent sccms0.524
sccm). (a) Without post-cell energy discrimination(quadru-pole rod offset isy3 V, 2 V more negative than CRO). (b)With post-cell energy discrimination(quadrupole rod offset isq2 V, 3 V more positive than CRO).
magnitude relative to the non-discriminating case.However, in this instance, the analyte ions arecorrespondingly suppressed by up to 2 orders ofmagnitude, which mitigates the improvement insignal-to-background to some 2–3 orders ofmagnitude.
The data of Fig. 24 through Fig. 27 were usedto determine ‘optimum’ analytical conditions ofreaction gas flow and energy barrier height. Infact, in all instances, the ‘optimum’ conditionswere provided at zero flow and no energy discrim-ination; that is, conventional ICP-MS conditions.However, a local optimum was obtained near aflow of 0.4 Ar-equivalent sccm for either gas and
the energy barrier height discussed above. It isappropriate to remind once again that the efficien-cy of reaction in the cell is exponentially depend-ent on the pressure of the reaction gas(gasthickness), but energy discrimination requires thatthe ions not be thermalized; this imposes a limi-tation on the efficiency that can be obtained.
Spectra obtained with and without energy dis-crimination, using H as the reaction gas, are2
shown in Fig. 28. Great care was taken to ensurethat the cell gas was as pure as possible; thisincluded the use of 99.999% H and substantial2
purging of the gas system. Before the gas line waspurged, addition of gas to the cell caused asubstantial change in the mass spectra obtained.After purging, little spectral improvement wasobserved(other than the increase in certain discreteions, such as those shown in Fig. 26). This wasthen taken as evidence of sufficient purging: anal-yte and cell-formed ion signals were monitored(without energy discrimination) until the signalsno longer changed with time.
For the most part, when H is used as the2
reaction gas, energy discrimination has littleimpact on spectral regions of typical interest.Certain ion signals were dramatically suppressedby the barrier; these include the ions atmyzs43,44 and 45, as well asmyzs55 (H O Ø2H O?)q
3 2
and themyz region 68–72. However, comparisonwith the conventional ICP-MS spectra(Fig. 22a)shows little, if any, improvement due to the addi-tion of H reaction gas. It is therefore our conclu-2
sion that, under the conditions used in our work,H is a rather ineffective reaction gas, at least for2
general purposes of multi-element determinations.This is consistent with the database of thermal rateconstants for reactions of ICP ions with Hw10x,2
and with the interpretation that the ions retain asubstantial fraction of their source energy(whenenergy discrimination may be applied) so thatrelatively few reactive collisions are obtained.Accordingly, it is our interpretation that otherreports of the efficacy of hydrogen as a reactiongasw27,62,63,112x principally reflect one or moreof four enabling conditions:(1) the ions areintroduced with higher energy, so that the collisionenergy exceeds the endothermicity of normallydisallowed reactions,(2) contaminants in the cell
1422 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 28. Mass spectra for high purity water(grey) and a solution containing 1 ppb each of K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, Se andSr (black), with H as reaction gas at a flow of 0.4 Ar-equivalent sccm(0.28 sccm) and the cell operated with a wide transmission2
bandpass(RPa, RPq)s(0, 0.15). (a) Without post-cell energy discrimination(QROsy3, CROsy1). (b) With post-cell energydiscrimination(QROsq2, CROsy1).
gas, introduced either with the gas or inadvertentlyfrom the external vacuum chamber or plasma gasentrainment, are the effective reaction neutrals,(3)in some instances(notably Se yAr ), differentialq q
2
energy loss rates may allow a degree of energydiscrimination against the polyatomic ion,(4)some collisional fragmentation of weakly boundpolyatomic ions(e.g. Ar ) occurs.q
2
Fig. 29 provides spectra using methane as thereaction gas with and without energy discrimina-tion. In this instance, the methane substantiallyreduces the argide interferences on Se , Fe80 q 56 q
and Cr , but introduces a number of new and52 q
significant interference ions(especially atmyzs43–48, 53–55, 57 and 60–75) when energy dis-
crimination is not applied. These new interferencesare dramatically suppressed with energy discrimi-nation, but, as noted above, the analyte signals arealso substantially reduced. Accordingly, it is evi-dent that CH has some desirable characteristics4
as a reaction gas in removing certain argide inter-ferences, but the efficiency of cooling the ionsmeans that low pressures are necessary for energydiscrimination, with concomitant reduction in reac-tion efficiency. Because of these antagonistic char-acteristics, methane is not viable as a reaction gaswhen energy discrimination is required, and thiscan almost certainly be projected to also includeother relatively heavy and polyatomic reactiongases.
1423S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 29. Mass spectra for high purity water(grey) and a solution containing 1 ppb each of K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, Se andSr (black), with CH as reaction gas at a flow of 0.4 Ar-equivalent sccm(0.21 sccm) and the cell operated with a wide transmission4
bandpass(RPa, RPq)s(0, 0.15). (a) Without post-cell energy discrimination(QROsy3, CROsy1). (b) With post-cell energydiscrimination(QROsq2, CROsy1).
8.4.2. Bandpass control of secondary chemistrySection 8.3 gave an example of a series of
sequential reactions that lead to potential newisobaric interference. In almost all instances, thesequential reactions involve either a primary(plas-ma-based) reactant ion or an intermediate production that has a mass that is significantly differentfrom the analyte ion. Ejection from the cell of anyone of these intermediate ions aborts the remainderof the reaction chain and eliminates the formationof the new interference.
A quadrupole finds application as a mass filterbecause it has well-defined stability boundariesthat are independent of the position of the ion
within the quadrupole field(Section 7.2). Perhapssurprisingly, the stability boundaries remain rea-sonably well-defined even when the quadrupole ispressurized(Fig. 13). The latter experimentalobservation suggests that a quadrupole reactioncell can be operated in a bandpass mode in whichthe low mass and high mass cut-offs are deter-mined principally by the amplitude and frequencyof the r.f. (the Mathieu parameterq) and theamplitude of the DC applied between pole pairs(the Mathieu parametera). When pressurizedabove a few mtorr, the quadrupole is unlikely tobe suitable as a mass filter, at least with massresolution that would have analytical merit in the
1424 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 30. Quadrupole stability diagram with hypothetical scan line.(m) Denotes the high mass cut-off, and(j) denotes the lowmass cut-off. The bandpass includes a lower mass range when operated at(d) than at(s), and this can affect the formation ofsecondary interferences if the precursor ion is includedyexcluded under these conditions.
ICP-MS application. However, even a relativelywide bandpass(a few amu to 10s of amu) candramatically suppress the secondary chemistry. Inthe instance where it is desired to remove anexisting isobaric interference, the bandpass isestablished in order to make the analyte ion(andthe isobaric interference) stable. This provides acondition for efficient transmission of the analyteion and an environment which permits the isobarto react with the reaction gas. Provided that thebandpass excludes at least one of the intermediateproduct ions of the sequential chemistry that wouldotherwise create a new interference, that sequentialchemistry is aborted and the new interference isnot formed.
The position of the stability boundary relativeto the analyte ion is conveniently selected byadjustment of either the r.f. voltage, as is typicalfor the mass filter, or the frequency of operation.Because the r.f. contribution to the reaction energy,which is important in the establishment of thermalconditions, is a strong function of the r.f. ampli-
tude, it is convenient to use the frequency for thispurpose in the reaction cell. The impact of theselection of the operating point(a, q) on thebandpass mass limits is considered in reference toFig. 30. In this figure, the low mass cut-off isindicated by the solid square and the high masscut-off by the solid triangle, and either of twooperating points is indicated by the circles. Thesolid circle indicates an operating point of(a, q)s(0.05, 0.5). If the analyte mass is 56 amu, thebandpass extends from approximately 33 to 147amu (calculated for an ideal quadrupole undercollisionless conditions). If, instead, the operatingpoint was chosen as indicated by the open circle(a, q)s(0.07, 0.7), the bandpass would includeapproximately 47–205 amu. In both instances, theanalyte ion (e.g. Fe ) is within the stabilityq
bandpass and is expected to be retained and trans-mitted. An isobaric plasma-based ion(e.g. ArO )q
is stable with respect to the quadrupole field, butmay be reactively removed if the reaction gas isappropriately chosen. For the sake of argument,
1425S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 31. Mass spectra for high purity water(grey) and a solution containing 1 ppb each of K, Ca, Cr, Mn, Fe, Ni, Cu, Zn, Se andSr (black). The cell is operated at(RPa, RPq)s(0, 0.7), such that the low mass cut-off is approximately 78% of the analyticalmass and without post-cell energy discrimination(QROsy3, CROsy1). (a) With CH as reaction gas at a flow of 0.92 Ar-4
equivalent sccm(0.48 sccm). (b) With NH as reaction gas at a flow of 0.9 Ar-equivalent sccm(0.48 sccm). (c) With H as3 2
reaction gas at a flow of 0.8 Ar-equivalent sccm(0.56 sccm).
consider that an ion ofmyzs40 (perhaps Ar , asq
a primary ion derived from the plasma, or someintermediate reaction product ion) could react withthe reaction gas to form a new ion atmyzs56(i.e. formation of a new isobaric interference forFe within the cell). If the first operating pointq
(solid circle) were adopted, themyzs40 ionwould be retained in the cell, and it could thereforereact to form the new isobaric interference. How-ever, if the second operating point(open circle)were adopted, themyzs40 ion would be outsideof the stability bandpass and would be rapidlyejected from the cell; as a result the new isobaricinterference would not form.
We saw in Section 8.3 that pressurizing thereactionycollision cell with a reactive gas(e.g.CH ) and operating with a wide bandpass gives4
rise to a more complex spectral background than
if the cell were not pressurized(cf. Fig. 22a andb). The spectra of Fig. 31a were obtained with thebandpass of the cell shifted to higher mass(largerq). The increase in the low mass cut-off clarifiesthe mass spectrum. The sensitivity for the analyteions remains comparable to that of Fig. 22a andb. The reduction of plasma-derived interferenceions is obtained through reaction with the cell gas.The appearance of new interference ions producedwithin the cell is suppressed, apparently as a resultof the rejection of lower mass ions that otherwisewould react in secondary processes to produce theubiquitous background seen in Fig. 22b.
The bandpass approach does not rely on differ-ences in ion energies between desired and undesi-red ions. Hence, the cell may be operated at higherpressure(gas thickness), providing more collisionswhich help to thermalize the ions(allowing rea-
1426 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 32. (a) Optimization of the RPq parameter for Cu as63 q
an analyte ion and three unidentified ions that are producedwithin the cell. CH was used as reaction gas at 0.92 Ar-equiv-4
alent sccm(0.48 sccm), and post-cell energy discriminationwas not employed(QROsy3, CROsy1). (b) The samedata is presented as a function of the low mass cut-off, assum-ing a cut-off near RPqs0.89. In the latter case, extrapolationof the decay of the signal with increasing cut-off mass(shownby the dashed lines) corresponds roughly to the mass of theprecursor ion that gives rise to the observed signal. Two suchprecursor ions are evident for themyzs57 ion.
gent selection on the basis of the thermal databaseand providing the specificity of the thermal chem-istry) and improve the efficiency of the reactiveremoval of plasma-based interference ions. This isevident in a comparison of Fig. 29 with Fig. 31a.The former data were obtained with a methaneflow of 0.4 Ar-equivalent sccm(0.21 sccm,f3mtorr) whereas the latter were obtained at 0.92Ar-equivalent sccm(0.48 sccm,f6 mtorr). Adramatic reduction in the signal intensities of theplasma-based argide interference ions(e.g. Ar ,q
ArH , ArC , ArO and Ar ) is obtained, a directq q q q2
result of the exponential dependence of the effi-ciency of these reactions on the reaction gaspressure, and the formation of new interferencesin the cell(most of the ions in the DIW spectrumof Fig. 29) are suppressed by the application ofthe bandpass. It should be recognized that, whereaspost-cell energy discrimination allows the second-ary chemistry to proceed and discriminates theions downstream of the cell, the bandpass approachsuppresses the secondary chemistry as it occursthrough removal of the precursor ions. It shouldalso be noted that the bandpass of the DRC cell isadjusted in concert with themyz set for thedownstream mass analyzer; the bandpass is thusdynamically adjusted as opposed to being static ata given r.f. amplitude and frequency.
The effect of the bandpass on the secondarychemistry, and some insight into the sequence ofthe chemistry, can be obtained by recording theion signal as a function of the bandpass parameters.Fig. 32a shows the ion signals for Cu and63 q
several not-identified ions that are formed withinthe cell with methane as the reaction gas as afunction of the RPq parameter(the Mathieu qparameter) in r.f.-only mode. If all of these ionswere derived from the plasma, they should showa similar dependence on RPq, as shown in Fig.12b. Clearly this is not the case, and the implica-tion is that the not-identified ions are created insidethe cell; this also agrees with the increasing signalsfor these ions as the methane flow is increased(the myzs47 profile is seen in Fig. 27). While itis preferred, in order to determine the optimumvalue of RPq, to measure the RPq-profiles at thesamemyz for a standard and a blank, the presentresult suggests that operation at RPq)0.6 should
alleviate many interferences. Of course, a differentvalue of RPq may be optimum for each mass andfor each gas and pressure, but the data presentedsuggests that a reasonable compromise can beadopted for multi-element measurement.
As noted earlier(Section 7.2), the experimentaldefinition of the stability boundary is a matter ofinterpretation. If we now accept that the RPq-profile for Cu is representative of a non-reactive63 q
ion, and define that this ion becomes unstable
1427S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
when its signal falls below 1% of its maximum,near RPqs0.89sRPq (recall thatqs0.908 ismax
the theoretical r.f.-only stability boundary for aninfinitely long and accurate quadrupole), the RPqscale of Fig. 32a can be converted to a low masscut-off scale (using Eq. (6.32) with q and m1 1
referring to the RPq and mass of the monitoredion, q sRPq s0.89 andm being the cut-off2 max 2
mass), as in Fig. 32b. Here, the sequential natureof the chemistry leading to the formation of themyzs47, 57 and 69 is evident. Both themyzs57and 69 ions apparently derive from sequentialchemistry involving a precursor ion having amyzof approximately 17 amu(probably CH , whereq
x
extrapolation of the first decay region to 1%suggests thatx is probably 5), and a secondprecursor in the vicinity of myzs27 (eitherC H or C H , by the same logic and on theq q
2 3 2 5
basis of the known chemistry of hydrocarbon ionswith methane). Therefore, the chemistry formingthese ions involves at least three steps: formationof CH , which reacts to form C H , whichq q
x 2 x
subsequently reacts either directly or indirectly toform the ions atmyzs57 and 69. It will berecognized that there are at least two ions havingmyzs57 in this example. The chemistry leadingto the dominant ion has just been described, andaccounts for the RPq-profile below approximatelyRPqs0.4 (low mass cut-offs27 amu). However,either Fig. 32a or b show that there is also arelatively stable ion atmyzs57. This is evidentfrom the recognizable RPq-dependence of the sig-nal above RPqs0.4, which emulates that of the
Cu ion. Indeed, these data were recorded for a63 q
sample that contained Fe, and the high-RPq con-tribution comes from the unreactive Fe isotope.57 q
The myzs47 ion has a different chemistry thanthe others. Clearly, it is not a primary stable ionderived from the plasma(it is suppressed at toolow a RPq value for this to be true). Also, it doesnot appear to be a pure hydrocarbon ion, as it doesnot involve intermediates of the ‘normal’ hydro-carbon ions C H . Rather, it appears to deriveq
n x
from reaction of an ion having amyz of approxi-mately 31. It may well be a methane cluster ofHNO , known to be a prominent ICP ion inq
aqueous sampling.
The important point to note is that at lowq(RPq), the secondary chemistry is enabled becausethe precursor ions are stable, and the product ionsappear in the mass spectrum. In most instances,this is undesirable; the exception is discussed inSection 8.4.3. When it is desired to suppress theappearance of these ions, a higher operating point(larger RPq) may be sufficient. If the chemistryrather derives from a higher mass ion(fragmen-tation or dissociative reaction), a non-zero valuefor the Mathieu a parameter(RPa in the ELANDRC) may be effective, as this institutes a highmass cut-off; an example of this for an interferencefor Al was shown in our early work with27 q
‘unclean’ hydrogenw29x but has not been pub-lished elsewhere. In addition, there is some evi-dence that a non-zero value for the Mathieu aparameter provides more sharply defined stabilityboundaries(for both the low- and high-mass) inthe pressurized quadrupole, and this will be thesubject of continuing investigation.
For the mass range below 100 amu, methaneappears to be a generally suitable reaction gas. Afew atomic ions, including As and Ti reactq q
with methane, perhaps making it not a universalselection for ultratrace multi-element determina-tions including these ions(though it will be shownin Section 8.4.3 that there is a solution for theseas well). However, CH reacts too slowly with4
Ar and ArH to allow ppt determinations of Kq q
and Ca. An alternate gas of relatively generalutility, but that is particularly well suited for thedetermination of K, Ca and Fe, is ammonia.Spectra similar to those shown for H and CH2 4
above, using NH as the reaction gas, are given in3
Fig. 31b. The NH gas flow used to obtain these3
spectra was on the high side of common usage,and was selected to optimize determination ofCa at myzs40. Such a high pressure promotesq
the reaction of even slowly reacting atomic ions,and hence the signature isotopes of Se are miss-q
ing (the products have not yet been determined; itis possible that charge transfer, endothermic by 0.4eV, may be promoted at the relatively high RPqused for these data). Simply to provide a referencefor other workers, we include the correspondingspectra using H as the reaction gas in Fig. 31c.2
As noted earlier, these data were obtained under
1428 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 33. Mass spectra in the vicinity of Fe and its ammonia clusters, recorded with an NH flow of 1.5 Ar-equivalent sccm(0.8q3
sccm). The upper spectra were recorded at lowq (RPa, RPq)s(0, 0.2), and the lower spectra at higherq (RPa, RPq)s(0, 0.5).Cluster ions of Fe are observed when the low mass cut-off is below the mass of Fe(so that the ‘naked’ ion and its clusters areq q
simultaneously stable). Notably, the first cluster is relatively absent, reflecting its fast rate of adding a second ammonia cluster.When the bandpass is set to exclude Fe when determining masses corresponding to the higher cluster ions, the clusters are virtuallyq
absent, because they cannot be formed when the precursor ion is removed early in the cell.
as clean conditions as possible, and little differencefrom the conventional spectra of Fig. 22a is to beseen(other than increased background signals atmyzs43, 45, 46, 47 and 69).
We were not able to observe the products of theCa and K ion–molecule reactions with ammo-q q
nia. Although the Fe decay is just slightly fasterq
(Table 2), the products of the ion–molecule reac-tion can be observed under conditions where boththe Fe ion and its product ions are stable(Fig.q
33).It has been observed that Fe reacts with ammo-q
nia by termolecular association and sequentiallyforms the ligated cations Fe(NH ) , wherexs1–q
3 x
4:q qŽ . Ž .Fe NH qNH ™Fe NH (8.7)xy1 x3 3 3
These reactions have been observed by othersin thermalized systemsw113,114x. NH plays the3
role of a stabilizing third body. Mass spectra of
the mass region including the product ions arepresented in Fig. 33 forqs0.2 and 0.5(as0 inboth cases), where it is seen that the cluster ionsare present at lowq but are largely absent at highq. The progress of the reaction is apparent fromthe sequence of the intermediate product ions.
The ability to support several consecutive stepsof ion–molecule reactions is related to the numberof reactive and scattering collisions and to theconfinement properties of the r.f.-field. Reflectingthe conditions that allow production and confine-ment of product ions in a quadrupole cell, it wasobserved that every consecutive step of ion–molecule reaction is discriminated with respect tothe precursor ion. Assuming that all reaction chan-nels and product ions have been monitored, it isfeasible to correct the product ion intensities inorder to conserve the overall charge, but we willnot do so here.
1429S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Selection of suitable ligands and operation atrelatively high pressure is very important to enablethe utilization of ternary association reactions forthe potential determination of an analyte as itscluster ion. For example, M –O or M –N bond-q q
2 2
dissociation energies are perhaps large enough toexpect observable addition, but the lifetime of theintermediate complex involving diatomic ligandsis relatively short because of the small number ofinternal degrees of freedom expected to be effec-tive in the energy dispersal. Hence, these clusterions are unlikely to be prominent. In contrast,ammonia is a good electron donor ligand and hasaccessible internal degrees of freedom for energydispersal, so that at higher pressures its collisioncomplex may have a sufficiently long lifetime toallow collisional stabilization. In some instances,its cluster ions might have analytical value. Thistype of reaction rarely proceeds at the collisionrate. Most often, the intermediate ligated ion mustbe collisionally stabilized before unimoleculardecomposition back to the separated reactants. Ahigher pressure provides a higher probability of astabilizing collision.
The chemical stability of products must beaccompanied by the stability of their trajectoriesin the DRC. In Fig. 33, it is apparent that operatingat highq suppresses the appearance of cluster ionsnearly completely. The most important reason forthis is the rejection of the parent ions in thequadrupole field due to their instability simulta-neously with the product ions. A lesser contribut-ing cause is that a higher value ofq provides alarger contribution to the thermal energy, therebypromoting the unimolecular decomposition of thecollision complex and interrupting the chain ofsuccessive reactions. The rate of formation of thefirst adduct, in particular, depends on the operatingpoint because it has a limited number of degreesof freedom. Successive clustering has a distinctivefeature: on every reaction step the product ion hasa higher mass-to-charge ratio than the precursorion. In our experimental arrangement, the DRC isinterposed between the ICP ion source and quad-rupole mass analyzer. The DRC has dynamic low-and high-mass cut-offs, which are adjusted inconcert with the analyzed mass and the requestedq and a at that mass. Therefore, considering this
tandem arrangement, the products of the additionreaction between Fe and ammonia are dynami-q
cally scanned. Under the DRC operating condi-tions, parent and daughter ions are alwayspositioned in different places on the stability dia-gram. For example, when the third adduct isanalyzed at(a, q)s(0, 0.5), the parent Fe is atq
(a, q)s(0, 0.96), which is in the unstable regionof the stability diagram, and is rejected from thecell before it can react. In order to see the thirdadduct, all of the precursor ions throughout thechain of successive reactions must be stable in thecell. Therefore, favorable and unfavorable condi-tions for clustering of many plasma ions can bechosen in the DRC by selecting the appropriatestability boundaries of the r.f.-field.
8.4.3. Promotion of secondary chemistryIt was noted in Section 8.3 that the reaction cell
should be efficient both at removing the isobaricinterference ion and at suppressing the secondarychemistry that would otherwise create new inter-ferences. However, there are instances in which itis more convenient to react the analyte ion(gen-erally by transfer of an atom) than to reactivelyremove the interference ion. Most often, this situ-ation will arise when the thermochemistry is unfa-vorable for charge transfer(i.e. the interferenceion has a similar or lower IP than the analyte).Such an instance arises when it is desired tochemically resolve certain isobaric atomic ions(e.g. Rb and Sr). If heavy atom transfer(e.g.87 87
oxidation) is to be a viable option, the product ionmyz should be relatively free of interference, orthe interference should also react at a sufficientrate that it no longer interferes with determinationof the analyte as the MX ion. If this is the case,q
the efficiency required of the reaction chemistryneed not be exceptional: it may be sufficient toconvert only a fraction of the atomic analyte ionto achieve a substantial improvement in detectionability. Also, it is clearly necessary to enable thesecondary chemistry. Therefore, kinetic energy dis-crimination is of little value, as this would discrim-inate against the desired product analyte ion. If thebandpass approach to secondary chemistry controlis used, the bandpass must include both the react-ing (atomic) analyte ion and the product(molec-
1430 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
ular) ion. If the bandpass is established withreference to the product ionmyz, the favorablevalues of(a, q) will be less than for an atomic(not transformed) ion. If the atomic analyte ionhas amyzsm and the product ion hasmyzsm ,1 2
then the product ion will be observed only for:
B Em1C Fq $ 0.908 (8.8)m2 mD G2
More typically, if the ion signal maximizes atqsq (typically ;0.5) under pressurized condi-opt
tions, the approximately optimum value ofq form , will be:2
Ž .m qm1 2q s q (8.9)m opt2 2m2
For example, in chloride matrices V is inter-q
fered by ClO . ClO should be expected to reactq q
with ammonia by charge transfer(0.79 eV exo-thermic). It was originally reportedw76x that reac-tion does not occur, but reexaminationw115xconfirms the relatively fast charge transfer reactionrate constant of 6=10 cm ys. V reacts slowlyy10 3 q
to form VNH (ks2.2=10 cm ys) w10x. Theq y12 3
300-fold difference in reaction rate should makeNH an attractive reaction gas for this application.3
However, at low values of RPq ammonia isobserved to be ineffective at resolving V fromq
ClO w116x. This appears to be due to a minorq
reaction channel of Cl forming NH Cl (exo-q q2
thermic by 25 kcalymol) at the samemyz, whichis consistent with unreported work from our lab-oratory with ND as a reaction gas indicating the3
formation of both ND Cl and ClØND at lowq q2 3
RPq. For the production of the NH Cl ion to beq2
suppressed,q must be greater than the valuem2
calculated according to Eq.(8.8), or 35y51=0.908s0.62. Indeed, it is observed that theinterference atmyzs51 in 1% HCl solution isreduced to 3 ppt BECw116x or to the instrumentbackground levelw117x for operation at RPqs0.65 or higher.
An alternative approach is to oxidize the V toq
VO (myzs67) using N O as the reaction gas.q2
Unfortunately, ClO also reacts, albeit slowly, toq
form ClO at the samemyz. However, VO canq q2
be readily further oxidized to VO (myzs83)q2
whereas ClO does not. Observation of V asq2
VO requires that V , VO and the final productq q q2
VO be simultaneously stable, and according toq2
Eq. (8.9) the optimum should occur nearq sm2
(51q83)y(2=83)=0.5s0.4. Results presentedby Bandura et al.w65x show that the operatingpoint is optimized as predicted.
It is well known that Se is interfered inq
conventional ICP-MS by the argon dimer ion, andAs is interfered by ArCl in samples containingq q
chlorine. H has been used to allow the determi-2
nation of As in Cl matricesw30,118–121x, but isineffective for the determination of Se at tracelevels (Fig. 31c). On the other hand, CH is4
effective for the determination of Se(Fig. 31a,and Sloth and Larsenw122x), but As reacts toq
form AsCH (at myzs89). It is often of interestq2
to determine As and Se nearly simultaneously, e.g.in LC-ICP-MS where the time resolution of theAs and Se species are not sufficient to allowchange of the reaction gas. In the absence of Y89
in the sample, methane may be used for thesimultaneous determination of Se and As, whereSe is measured as the atomic ion and As as theproduct ion, as shown in Fig. 34.
Beta decay of Rb to Sr allows age determi-87 87
nation of rocks through the isochrone obtained bythe measurement of the Sry Sr isotope ratio87 86
together with the RbySr element ratiow118,123,124x. Because of the overlap of themyzs87 isobars, chemical separation is usuallyrequired before analytical measurement. However,Sr is readily oxidized(by N O) whereas Rb isq q
2
not; if this reaction could be efficiently promoted,chemical separation could be performed in situ inthe reaction cell, with measurement of the Srisotope ratio as the oxide ions, eliminating the riskof contamination and ratio distortion in an addi-tional separation step. The reaction profiles forSr and Rb with N O are given by Bandura etq q
2
al. w65x. While it is clear that the concept hasmerit, the experimental observation was that theproduct ion SrO reacted further with an unknownq
impurity (water in the N O?) to form SrOH , andq2
the partitioning between the oxide and the hydrox-ide complicates the measurement of the Sr isotoperatio. However, it was also shown that SrO reactsq
rapidly with methane to form the hydroxide, and
1431S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 34. Mass spectra for a high purity water(grey) and asolution containing 1 ppb of As and Se(black). (a) Standardconditions(no reaction gas, cell vented to mass analyzer cham-ber). As is observed at its atomic mass(the solution did notq
contain chloride), and Se is largely hidden by the Ar back-q q2
ground. (b) Cell pressurized with CH at a flow of 0.7 Ar-4
equivalent sccm(0.36 sccm), and operated at(RPa, RPq)s(0, 0.5). Approximately 80% of the As is reacted toq
AsCH atmyzs89, and the Se isotopes are readily apparentq q2
at their atomic masses.
Fig. 35. Mass spectra for 50 ppb Rb and Sr, measured sepa-rately and overlaid to show the contributions of the isotopes tothe combined spectrum, with CH F as reaction gas. Sr reactsq
3
to form SrF (F is monoisotopic, so that the isotope ratio ofq
SrF reflects the isotope ratio of Sr), and Rb is unreactive.q q q
(From Ref. w118x with permission of The Royal Society ofChemistry.)
this can be used to force the secondary reaction(forming the hydroxide) to near completion. Con-sequently, a mixed reaction gas of N O and CH2 4
(which must be mixed in the low-pressure gasmanifold, because they are combustible at higherpressure) allows rapid sequential oxidation andhydroxylation with high efficiency. This was ourfirst experience with the promotion of sequentialchemistry using two reaction gases.
Superior results for the geochronological appli-cation have been reported by Moens et al.w118xusing CH F in a Ne buffer. Sr reacts with methylq
3
fluoride by F-atom transfer, forming SrF , and Rbq
is unreactive. An advantage is had using thischemistry because F is monoisotopic and hencemeasurement of the Sr isotope ratios is not con-voluted by further corrections. Fig. 35 provides asuperimposition of spectra for two solutions, onecontaining Rb and the other Sr, so that the separatecontributions to the combined mass spectrum canbe observed. The Sr signal is largely shifted toq
SrF , and the Rb signal is obtained at its atomicq q
mass. It might be noted, as mentioned earlier, thatexceptional efficiency of conversion of the Srisotopes is not required for this application, as themyzs85 corresponds only to Rb and can be usedfor the construction of the isochrone. Neon bufferwas used to homogenize the ion temporal distri-bution (Section 8.2.2), and yielded isotope andelement ratios that approximated the counting sta-tistics limit. The resultant isochrone, expressed inthe usual isotope configuration, is shown in Fig.36, where the ICP-DRC-MS result using in situchemical element resolution compared very favor-ably with the accepted TIMS result that used priorchemical separationw118x.
9. Applications
In the 5 years since their first commercialimplementation, publications relating to the use of
1432 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 36. SryRb isochrone for an intrusive rock formation,obtained by dissolution and direct nebulization without elementseparation, using CH F to convert the Sr to SrF , with Neonq q
3
buffer to improve temporal homogenization. The derived ageof the rock is in agreement with the TIMS resultw161x and hascomparable precision.(From Ref. w118x with permission ofThe Royal Society of Chemistry.)
collisionyreaction cells in ICP-MS have been dom-inated by fundamental studies and phenomenolog-ical characterization rather than practicalapplication. It should be expected, and recenttrends justify this, that acceptance of the techniquehas begun an explosion in the number ofpublications dealing with the solution of real ana-lytical challenges. We review here the reports ofapplications of which we were aware as of Septem-ber 2001.
The 3D ion trap inherently lends itself to oper-ation as a collisionyreaction cell device. Variablestorageyreaction time can be optimized for speedof analysis or efficiency of reaction, and extendsthe dynamic range of the instrument which isotherwise severely limited by the space charge ofthe accumulated ions. It has historically beenoperated in a bandpass mode(even single amu)using filtered waveforms or broadband excitation,principally to allow for sensitivity enhancement byselective ion trapping, and this capability can beused to suppress the appearance of secondaryreactions, though at the expense of duty cycle.Some applications of the ion trap were reviewedin Section 4.3 and are not repeated here. We refer
to the paper of Eiden et al.w26x for a summary ofthe performance characteristics of the ion trap thatmake it attractive for elemental analysis.
Though a collisionyreaction cell appears to offersignificant advantage for sector-based ICP-MS, andprincipally for multicollector detection, adoptionin this configuration has been slow. The MicromassIsoProbe has been available for some time, andtakes advantage of collisional energy damping toeliminate the need for an electrostatic sector, andthe Thermo Elemental Axiom(having both mag-netic and electrostatic sectors) has been offered ina cell-configuration(but is no longer). Both ofthese instruments are(were) available with multi-collector detection, and should allow the determi-nation of isotope ratios of interfered elements. Anearly report w125x indicates that high precisionisotope ratios of Fe are indeed possible, but alsothat the use of H(or impurity H O) as the reaction2 2
gas can lead to the formation of hydroxides suchas ArOH that may lead to inaccurate(thoughq
precise) results.Guzowski and Hieftjew126x have suggested the
potential analytical benefits of incorporating acollisionyreaction cell in an orthogonal Time-of-Flight ICP-MS, including the improvement of res-olution and sensitivity as a result of the narrowingof the ion energy distribution before acceleration.While significant improvement in the determina-tion of elements interfered by the argide ions isobtained, it seems that further work is necessaryto realize the TOF-specific advantages. Perhaps anotable exception is the reduction of continuumbackground, probably resulting from more efficientexclusion of argon from the flight tube with acorresponding reduction in the production of argonmetastables that may, in part, be responsible forthis background. Further, it was shown that theresolution is sufficiently improved to allow spectraldiscrimination of hydrocarbon ions produced inunwanted secondary reactions. Similar conclusionsare reported by Leach and Hieftjew127x for anaxial-acceleration ICP-TOF-MS.
By far, the most common application of colli-sionyreaction cells has been in quadrupole massfilter instruments. Of course, this is partly a resultof the general acceptance of these instruments inthe conventional ICP-MS configuration because of
1433S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
their relative ease of use, but it is also a reflectionthat the cell offers relief of the principal disadvan-tage of the low (unit mass) resolution of thequadrupole. The sequential scanning characteristicof the quadrupole is also compatible with bandpassoperation of the cell. Further, the quadrupole hasless-demanding vacuum requirements than either asector or TOF, and so the cell may be operated athigher pressures(higher reaction efficiency), high-er conductance to the mass analyzer, andyor withless expensive vacuum pumping.
9.1. Method development
It is convenient if the instrument may be oper-ated either with or without collisionyreaction gas.Operation as a conventional ICP-MS allows optim-ization of the sample introduction, plasma and ionoptics in the familiar manner and permits approx-imately independent optimization of the cell para-meters(recognizing that certain parameters, suchas cell and analyzer rod offsets, are mode-depend-ent). This recognizes that, for the most part,operation of the cell is independent of the plasmaand sampling characteristics. The latter statementis not entirely true, but simplifies the first-orderoptimization of the instrument. Of course, different‘interfering’ plasma ions, or the intensities of theseions, are obtained under different sampling andplasma conditions, and the operating conditions ofthe cell should be adjusted to account for this(i.e.higher or lower efficiency depending on the rela-tive intensities of the analyte and interference,which prescribes the type or pressure of gas to beused in the cell). Where satisfactory performanceis obtained using conventional(non-pressurizedcell) conditions, this is almost certainly preferred.Of course, an experienced user will realize thatimproved sensitivity (detection limit) can beobtained by taking advantage of collisional focus-ing with a pressurized cell(even for a non-interfered ion, where a non-reactive gas may bepreferred) w111,112,128,129x. However, where thisadditional performance is not required for theapplication at hand, it seems propitious to takeadvantage of the familiar convenience of thismode.
Where an application requires the use of a cellto improve the performance, it is very importantto understand the analytical objectives and theconstraints that these impose. Where an increasein sensitivity by as much as a factor or 5 or 10 isthe principal goal, collisional focusing with a non-reactive gas may be sufficientw111,112,128x. Thegas should have a mass that is a significant fractionof the mass of the analyte ions of interest, andyorthe cell should be operated at relatively highpressure. It is important that the gas be as pure aspossible and that other gases(plasma or ion opticschamber gases) are excluded as much as possibleto minimize reaction. Where collisional focusingis obtained, a significant improvement in abun-dance sensitivity may also be achieved because ofthe concomitant narrowing of the ion energy dis-tribution which improves the resolution of thedownstream quadrupole mass filterw27,53x. Con-comitantly, such conditions lead to temporalhomogenization of rapid variations of ion flux,such as high frequency plasma source noise. If themass filter is scanned sufficiently rapidly toaccount for low frequency fluctuations, the colli-sional broadening within the cell(on the order of5 ms, or homogenization of fluctuations faster than200 Hz) leads to improved signal correlation andprecision of isotope ratiosw109x.
Where suppression of a background interferenceis required, a reactive gas is prescribed. It isimportant to recognize whether the application isspecific to a few elements or is generic for a widerange of elements. In general, a multi-elementmethod requires some compromise of performancerelative to a specific method. In this instance, aless reactive gas, such as H , may be appropriate,2
as few of the atomic analyte ions react. However,our experience has been that high purity hydrogendoes not provide high efficiency of argide ionsuppression(cf. Fig. 31c). This is in contrast tothe conclusions of other reportsw27,79,121x but isconsistent with the TOF results of the Hieftjegroup w126,127x. Careful review of the resultsreported by Feldmann et al.,w62,63x and Boulygaand Beckerw112x indicates that, while improve-ments in signal-to-background(or BEC) of 2–4orders of magnitude are obtained for some of theargide ions, the background is far from eliminated
1434 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
(which requires improvements of up to 9 ordersof magnitude). Further study to determine thereasons for the disparate results reported for H as2
a reaction gas are required and should probablyfocus on the role of impurities in the chemistry orthe importance of concomitant damping gasesw30,79x to facilitate thermalization of the ions.Other gases, such as methane, ethane, ethylene, oreven ammonia may be more effective ‘generalpurpose’ gases, though reaction of some analyteions with the gas(such as As with CH or NH)q
4 3
must be accounted for(and in these instances, itmay be appropriate to determine these reactiveions at the masses of their product ions).
Where the analytical challenge is limited to afew elements, exceedingly high efficiency ofimprovement in the BEC can be obtained withappropriate choice of the reaction gas. Kineticw10xand thermochemicalw9x databases provide thebasis for selection of the appropriate gas. Optimi-zation of the cell pressure(flow rate of gas) andmeans of suppression of the secondary chemistry(kinetic energy discrimination or bandpass para-meters) should be performed by comparison of amethod blank and a spiked sample. Where a trueblank is available(most environmental, clinical,geological samples), the criterion of optimizationmight be the achievable detection limit. Wheretemporal homogenization leads to background sig-nal noise that is characterized by counting statis-tics, it is convenient to determine the ‘estimateddetection limit’ defined as 3 times the square rootof the blank signal divided by the net sensitivity(for optimization purposes it is not essential todefine the measurement time). Many elements willoptimize at similar cell pressure, though some(especially Ca) will optimize at a higher pres-40 q
sure because of the higher efficiency required.Some compromise for this parameter may then berequired, but this usually results in detection limitsthat are within a factor of a few of the ultimateelement-specific values. Where a true blank is notavailable (e.g. for semiconductor pure water orhigh purity acid analyses), it is our contention thata preferred basis of optimization is the BEC, asthe blank itself in this instance is also the sample,and the minimum BEC must correspond to the
upper limit concentration of the analyte in thesample.
To be of greatest value, a method should betransferable between instruments and laboratories.Certainly, variations in achievable results will beinevitable due to differences in skill levels insample preparation(notably in contamination), butthe method itself should be applicable with aslittle variation as possible. This is possible wherethe different instruments provide comparable ther-mal characteristics, consistent ion optic optimiza-tion, and uniform gas flow calibration. Indeed, itis to be expected that the majority of practitionerswill operate under the conditions of methods devel-oped in other laboratories, and that ‘cookbook’recipes will be provided for many common appli-cations. Nonetheless, the development of specificapplications can be a challenging exercise, andwill be facilitated with the promulgation of ageneric, knowledge-based approach to methoddevelopment.
9.2. Developed methods
9.2.1. High purity water and process chemicals(semiconductor)
Because reaction cell methods provide a rathersimple and effective means of suppressing Arq
and argide polyatomic interferences on Ca , Kq q
and Fe , principally because the argide ions haveq
relatively high electron affinities(the correspond-ing neutrals have high IPs) and hence react bycharge transfer with a variety of neutral gases andproduce few secondary interferences, initial atten-tion focused on its use as an alternative to coolplasma methodsw130,131x. In general, comparableresults are obtained for high purity water analysis,but the reaction cell approach has an advantagefor the analysis of high purity acids and othercomplex samples where the cool plasma approachsuffers from concomitant element effects(easilyionized element effects) in the plasma whichdiminish sensitivityw131x. Even for the high puritywater application, the reaction cell approach hasan advantage in that the method allows identifi-cation of the residual argide signal through theelement-specific slope of the reaction profilew30x,
1435S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
whereas this contribution to the signal in the coolplasma method may be uncertain and variable.
The suppression of the argide ions was clearlyshown in the early work of Rowan and Houkw21xand the ion trap results of Koppenaal et al.w22–26x, though the application to semiconductor mate-rials analysis was not explicitly noted. WhileTurner et al.w27x claimed high efficiency for H2as a collision gas, the role of impurities was notrecognized and the quantitative benefits were notobvious. Feldmann et al.w63x showed low-pptdetection limits, using H as a reaction gas, though2
several of the important analyte elements(Ca, Fe,Ni) were detectable at levels higher than requiredfor the semiconductor application, and a value forK was notably absent. Boulyga and Beckerw111xshowed that up to 5 orders of magnitude suppres-sion of Ar , ArO and Ar could be obtainedq q q
2
with H using kinetic energy discrimination, with2
a concomitant, though lesser, effect on the sensi-tivity to atomic analyte ions. Tanner et al.w30,119,132x showed greater than 8 orders ofmagnitude suppression of Ar , while retaining theq
sensitivity to Ca , using NH as a reaction gas,q3
providing sub-ppt detection limits for a suite ofelements including Ca, K and Fe in high puritywater. It was also shown that As reacts withq
NH , not recognizing the potential for determina-3
tion of As as AsNH , and that satisfactory resultsq2
for As were obtained using H in a buffer gas of2
Ar. The NH results are consistent with those3
reported by Bollinger and Schleismanw133,134x.The latter work also showed the notable potentialfor determination of As as AsO , though theq
mechanism of this conversion was not clearlyproven and appeared to be a result of reaction withadventitious contaminants. Olesik et al.w135xshowed oxidation of As with O , and correspond-q
2
ing oxidation of Se . Recent work by Bandura etq
al. w65,80x has shown the efficacy of oxidationusing O as the reaction gas; results given in Fig.2
34 demonstrate a similar potential for determina-tion of As as AsCH with CH reaction gas. Tableq
2 4
6 summarizes the reported detection limits in highpurity water.
Vollkopf et al. w136x met semiconductor guide-lines requiring"25% recovery at the 10 ppt levelin 31% hydrogen peroxide, even for Fe which56
was determined at 3.3 ppt in repetitive analysesover a 5 day period with 95% mean spike(11.25ppt) recovery. Bollinger and Schleismanw133xreport detection limits for 41 elements in highpurity water, nitric acid and hydrochloric acid.
Kishi and Kawabataw137x show that the effi-ciency of chemical resolution in a reaction cellallows the determinations to be conducted underrobust plasma conditions where even relativelyconcentrated acids(nitric and sulfuric) do notcause plasma suppression effects; exemplary datacomparing the sensitivity for K as a function ofq
nitric acid concentration and plasma power aregiven in Fig. 37. Table 7 gives the results ofPorche et al.w138x showing spike recoveries ofbetter than"20% for a suite elements in 2000ppm Si and determination of these at the low pptlevel.
9.2.2. EnvironmentalEnvironmental samples present a spectrum of
challenges: high salt content requires a ruggedinterface and optics, high concentrations of con-comitant elements present spectral interferences,and in some instances exceptional detection limitsare required(e.g. determination of actinide con-tamination). The first of these presents a particularchallenge for cell-based instruments, as it is to beexpected that direct exposure of the cell to theplasma source risks contamination of the cell withthe major elements of the sample both throughdeposition of atomic ions(which can be expectedto ‘stick’ once neutralized on a surface) and bydirect impact of incompletely vaporized particles.Contamination of the cell can lead to memoryeffects, as deposited material can be sputtered andreionized within the cell, and can cause surfacecharging of sensitive optical components(particu-larly the multipole rods) resulting in unstablesignals. Common high concentration elements,such as Na, form argides readily; fortunately, mostof the argide ions, including the metal argides,have relatively high electron affinities(the corre-sponding neutrals have high IPs) and can bechemically resolved in a manner entirely analogousto that for ArO , ArH , etc. Hence, these histor-q q
ically challenging interferences, that have limitedthe application of ICP-MS for seawater analysis
1436 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Table 6Detection limits(ppt) in high purity water
Element Isotope Feldmann et al.w63xa Tanner et al.w30xb
These data appear to have been obtained under compromise conditions appropriate for the entire suite of elements, using Hya2
He as the collisionyreaction gas. The high purity water may have been acidified.These data were obtained under cell conditions that were approximately optimum for each element. Some of these data wereb
obtained in a round-robin test, for which the indicated BECs were obtained at the same time as the DLs. The type of reaction gasused(if any) is given in parentheses. Water was obtained from a Millipore ElixyGradient water system.
for instance, are rather trivial challenges for colli-sionyreaction cell instruments. More challenging,because of their thermochemical properties thatresist charge transfer and other convenient reac-tions, are certain oxide ions such as MoO andq
CaO that interfere with the determination of Cdq
and Fe, Ni, Co, etc., respectively.Godfrey et al.w139x described the analysis of
potable water containing Na, K and Ca at highppm levels and As, Cd and Se at ppb levels using
the PQ ExCell. On-line sample dilution, withstandard addition in the diluent, was used. Bothstandard and Collision Cell Technology(CCT)modes were used, with notable improvementobserved principally for Fe and Se due to attenu-ation of argide interferences and the consequentavailability of more abundant isotopes for analysis.As these results were obtained for He as thecollision gas, and since we have shown that colli-sional fragmentation must be inefficient(Section
1437S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 37. Potassium ion signal( K ), normalized to the signal39 q
for pure water, as a function of nitric acid concentration at threeplasma powers. Under cool plasma conditions(800 W), theacid causes matrix suppression of the K signal. The suppres-q
sion is less severe under ‘normal’(1280 W) plasma conditions,but is still substantial. Little dependence on the nitric acid con-centration is observed under ‘hot’ plasma conditions(1600W). Data were obtained using ammonia reaction gas to removethe ArH interference.(From Ref.w137x with permission.)q
Table 7Spike recovery and determination in 2000 ppm Si(from Ref. w138x)
5.2), it must be concluded that the beneficialeffects result from contaminants derived eitherfrom the gas(the purity was not indicated) or,
more likely, from incursion of plasma gases intothe cell.
Leonhard et al.w121x provide the first reportusing the Octapole Reaction System with theAgilent 7500c, showing multi-element determina-tion in 1:10 diluted seawater and in 0.3% NaClsolution. Three modes of analysis are described:standard(no cell gas), H -mode and He-mode. A2
lesser abundant isotope of Se was used for analy-sis, though dramatic improvement in the determi-nation of Fe is shown at its major isotope atmyzs56. The ArCl interference on As isq q
reportedly resolved in He-mode, though theauthors note that the role of contaminants in thegas is unknown. Reaction profiles are providedshowing more rapid suppression of argide andoxide interferences than of analyte ion signals,where the latter appears to be ascribed to energydiscrimination as the cell pressure is increased.The result, nonetheless, is a substantial improve-ment of more than 2 orders of magnitude in BECsfor Cr, Fe, Co, Cu and As. Detection limits for asuite of elements are sufficient for multi-elementdetermination in diluted seawater, though half ofthe elements are determined at or near their detec-tion limits. No mention is made of means to
1438 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 38. Reaction profiles for Mo , MoO and Cd with O reaction gas, obtained for a solution containing 20 ppb Mo and 2 ppbq q q2
Cd, at the operating point(RPa, RPq)s(0, 0.75) with a high accelerating field. Mo is sequentially oxidized to MoO andq q
MoO . Operation with a high low mass cut-off(high q) minimizes formation of MoO within the cell by rejecting the precursorq q2
Mo , which in turn facilitates quantitative conversion of the monoxide ion to the dioxide ion.q
account for the difficult oxide interferencesCaO and MoO on Fe and Cd, respectively.q q
Bandura et al.w65,80x investigated ion–mole-cule chemistries that may be of value for thedetermination of Fe in high Ca matrices, for whichthe interference imposed by CaO is a significantq
challenge. Because of its high thermodynamicstability (low corresponding IP and O-atom affin-ity), the CaO is resistant to many convenientq
reaction gases. Two potential strategies were pre-sented: oxidation of Fe with N O to remove theq
2
analyte ion to FeO for mass analysis atmyzs72q
(further oxidation of CaO is slow), or O-atomq
abstraction from CaO to CO(Fe is non-reactiveq q
with CO). Neither approach will eliminate theinterference at very high CayFe ratios, but thelatter in particular offers the potential for an order-of-magnitude improvement in the BEC for Fe(perhaps limited by concomitant re-oxidation ofCa by impurities in the gas).q
Recent unreported work from our laboratorysuggests that the MoO interference on Cd mightq q
be resolved by promotion of oxidation with O as2
the reaction gas. The O-atom affinity of Cd isq
not known (but reaction with O is apparently2
endothermic) while oxidation of Mo to MoOq q
is nearly thermoneutral(according to the NISTdatabase of Ref.w9x, the reaction is exothermic by
2.7 kcalymol, which is consistent with the obser-vation that the reaction is observed to proceed,albeit slowly, in the SIFT apparatusw140x, as isthe subsequent oxidation to MoO). For thisq
2
scheme to be effective, relatively high O pressures2
are required, since it is necessary to achievequantitative conversion of plasma-producedMoO to its dioxide. The formation of MoO inq q
the cell, which would exacerbate the challenge, issuppressed by operation at relatively highq (toremove the precursor Mo from the cell). Provi-q
sion of an axial field within the cell to promotetransport of the ions through the relatively heavyand high-pressure gas is essential. Reaction profilesfor Mo , MoO and Cd are given in Fig. 38.q q q
The increment of the MoO interference on Cdq q
at low flow is a result of the primary oxidation ofMo , but at higher O flows the monoxide isq
2
nearly quantitatively converted to MoO . Concur-q2
rently, Cd is transported without reaction. Spectraq
of 10 ppb Mo in high purity water with andwithout 10 ppt Cd are given in Fig. 39 underconditions optimized for Cd determination in aMo-matrix.
Exceptional sensitivity is required for the anal-ysis of actinides in the environment, such as thedetermination of uranium isotope ratios for iden-tification of spent fuels. Boulyga et al.w128x
1439S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 39. Mass spectrum of a solution containing 10 ppb Mo with(black) and without(grey) 10 ppt Cd, obtained with O reaction2
gas(1.6 Ar-equivalent sccms1.14 sccm), (RPa, RPq)s(0, 0.75). Oxidation of MoO to MoO relieves the interference of theq q2
monoxide ion on Cd .q
analyzed soils in the vicinity of the Chernobylincident, and were able to identify fallout from theaccident through the unnatural abundance of Uy235
U and Uy U ratios. Combination of ultra-238 236 238
sonic nebulization with collisional focusingprovided by He in the collision cell of the Platformprovided astounding sensitivity of up to 27 GHzyppm, though it is not clear how the analog outputof the Daly detector is converted to ion count rate.Nonetheless, the sensitivity was sufficient to pro-vide for single digit ppq detection of U, and to236
identify the unnatural isotope distribution in thesoil samples. Because of the relative abundance ofthe isotopes, care was required to minimize for-mation of hydrides, and a membrane desolvatorwas effective in this regard though at the cost ofsome sensitivity.
9.2.3. REE and actinide oxide and hydroxide ionsThe atomic ions of the rare earth elements
(REEs) have relatively high O-atom affinities.Several REE-oxide ions persist even at the plasmatemperature at levels sufficient to convolute thedetermination of the suite of REEs. Du and Houkw141x have shown that the collisionyreaction gasesHeyH significantly reduce the signals for metal2
oxide ions relative to atomic ions. The effect isobserved only when the mass analyzer bias poten-tial is positive with respect to the collision cell
bias potential(i.e. when kinetic energy discrimi-nation is employed). A reaction mechanism forthe relative suppression of the oxide ions is notobvious. A reasonable interpretation is that theoxide ions are discriminated against relative to thecorresponding atomic ions on the basis of theirpost-collision kinetic energies. If a substantial frac-tion of the oxide ions observed were in factproduced within the cell, due to reaction withimpurities introduced either with the gas or byentrainment of plasma gas, the product ions wouldhave lower kinetic energy than the atomic ionssince the product ion of the reaction is retardedsomewhat by addition of the oxygen atom fromthe relatively stagnant gas. In addition, polyatomicions have larger collision cross-sections than thecorresponding atomic ions and, hence, will suffermore collisions with resultant increased energydamping. Finally, the polyatomic ions have addi-tional internal degrees of freedom in which todissipate the collision energy, so that the post-collision translational energy is, on average, lessthan for a corresponding atomic ion. Together,these effects suggest that the metal oxide ions willhave a lower average kinetic energy than theatomic ions, and hence are more efficiently dis-criminated against by the potential barrier estab-lished at the mass filter. This mechanism does notaccount for the observed element-specific variation
1440 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
of the efficiency of oxide-suppression noted by Duand Houk, unless there are differences in theproportion of the oxides that are formed withinthe cell, the cross-sections are substantially differ-ent, or the efficiencies of energy conversion intointernal excitation differ because of the differentbond strengths.
As an aside, Du and Houkw141x present anexpression for the mean free path in a collisioncell, ls( psn) , that differs from that used toy1y2describe the mean free path in, for instance, theion optics, ls(sn) . The 62 term is properlyy1
used when the ion velocity is comparable to theneutral velocity; that is, for a thermalized condi-tion, appropriate for a thermalized cell. When theion velocity is substantially greater than the neutralvelocity, as in the ion optics or in a cell prior tothermalization, the62 term is omitted. However,the p term is appropriate only when the cross-section,s, is given as the square of the impactparameter. As used by Du and Houk, and usedhere, the cross-section is given as the integral areaof interaction(i.e. p(r qr ) for the hard sphere2
1 2
case), in which case thep term is inherentlyincluded. Accordingly, Du and Houk have under-estimated the mean free path, and over-estimatedthe number of collisions, by approximately a factorof 3.
Simpson et al.w142x propose the use of O as a2
reaction gas to suppress the appearance of oxideions that interfere with the determination of thenoble metals. The noble metals are relativelyresistant to oxidation, whereas potential interfer-ences(such as the oxides of Zr, Nb and Hf) appearto react with O , presumably by further oxidation.2
The result is that the noble metals can be deter-mined with much reduced(or eliminated) interfer-ence. Two mechanisms are suggested for thisobservation. The first is that the oxide ions, ortheir further oxidation products, are more efficient-ly scattered from the cell due to their more efficientthermalization with the gas. However, relativelyhigh flow rates of O were used, implying a2
relatively large number of collisions, and the gasitself is relatively heavy. Hence, it is expected thatall of the ions are nearly uniformly thermalized,so that scattering losses of the polyatomic andatomic ions should be comparable. The alternative
suggestion is that the refractory interferences aresequentially oxidized to high levels(e.g. HfO )q
4
which removes the interfering ions from the spec-tral region of interest. These higher oxides(andhydroxides, presumably derived from reaction withimpurity water in the gas) were observed for Hf,and it is suggested that yet further oxidationremoves the interfering ions completely beyondthe mass range of the instrument. A further possi-bility might be that sequential oxidation eventuallyleads to an ion that might be unstable(i.e. thateliminates O as a product ion), in a mannerq
x
analogous to NO that hydrates three timesq
(NO (H O) ) before subsequently producingq2 3
H O (H O) w143x, hence removing the interfer-q3 2 2
ence ion and product ions which include it fromthe spectrum.
Though CO is an effective reductant(its O-atom affinity is sufficiently large to extract an Oatom from CaO , as discussed above), CO willq
2
donate an oxygen atom to most of the REE ions(except Eu and Yb). As shown by Baranov etq q
al. w144x, CO as a reaction gas nearly quantita-2
tively converts most of the REE ions to theiroxides. While there are stronger oxidant reactiongases that will allow oxidation of all of thelanthanide ions, the advantage of CO is that the2
O-atom affinities of most of the REE-oxide ionsappear to be less than that of CO, so that theoxides do not further oxidize to the dioxide. Hence,the REE pattern is simply reproduced 16 amuhigher than the atomic spectrum(with the additionof the O contribution, which can be corrected18
for using the natural abundance of the oxygenisotopes), alleviating the spectral overlap of theoxides.
Hattendorf and Guntherw145x used a reaction¨cell in an innovative study of the formation ofdoubly charged oxide and hydroxide ions of Th.These ThO and ThOH are typically present2q 2q
(in various ICP-MS instruments) at approximately1% of the Th signal level, which is itself 1–3%2q
of the singly charged Th signal. All of the doublyq
charged ions optimize in a similar manner withrespect to the plasma conditions, which distin-guishes them from ThO which optimizes underq
cooler plasma conditions. They studied the relativeoxidation and hydroxylation rates of the doubly
1441S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
charged Th ion in the reaction cell, showing2q
that doubly charged ion reaction rates are typicallygreater than those of the corresponding singlycharged ions. This allowed the preliminary conclu-sion that the doubly charged oxides and hydroxidesare likely formed in reactive collisions with O-containing species either in the expansion throughthe interface or within the ion optics chamber,while singly charged polyatomics are more prob-ably characteristic of the plasma source itself.Interestingly, the reaction study was performedwith high purity neon as the reaction gas, takingadvantage of the oxide impurities in the gas(whichwas used directly and not processed through agetter).
The doubly charged ion reactivity report allowscomment on an interesting but separate issue. Thepresent authors originally expected that the use ofa reaction cell should cause the doubly chargedions to be under-represented in the spectrum. Thiswas based on the recognition that the second IPof most species is relatively large, so that chargetransfer is in many instances exothermic, and thatthe collision rate should be larger than for singlycharged ions because of the enhanced charge-induced-dipole potential, leading to more collisionsand hence higher reaction efficiency. The effect isgenerally not observed, at least for atomic neutralreactants. This is a result of the surprisingly lowrate constant(efficiency) for charge transfer reac-tions of many doubly charged atomic ions withatomic neutral speciesw146x. The interaction of adoubly charged ion and a neutral is attractive atlarge internuclear separation and repulsive at smallseparation, but the products are two singly chargedions that are mutually repulsive. It might besimplistically viewed that the repulsion of theproduct ions causes separation of the transitionstate before the electron is completely transferred.In fact, there is a ‘reaction window’ based on theenergetics of the reaction(for DH ;y4"1 eV)r
for which the reaction typically proceeds with ratescomparable to efficient singly charged ion–mole-cule reactionsw146–148x, which makes this typeof reaction unique in being ‘thermodynamicallydriven’ (the rate constant depends on the heat ofreaction) w148x. In many reactions with molecularneutrals, but not all, the multitude of electronic
and vibrational states cause curve crossings in thereaction coordinate system that facilitate fast(fre-quently dissociative) charge transfer reactionw146x.
It may be opportune here to dispel yet a furtherfrequent misunderstanding. It is common to deter-mine the ‘oxide ratio’, as a significant performancecharacteristic for conventional ICP-MS, throughmeasurement of the CeOyCe ratio. The presentq q
authors have often been told that this ratio isdramatically increased when NH is used as a3
reaction gas, with the interpretation that the oxideratio is increased, presumably resulting from reac-tion with impurities in the gas. However, Ceq
reacts with ammonia forming CeNH andq
CeNH , the latter being isobaric with CeO , andq q2
the increase in this measured ratio simply reflectsthe reaction efficiency. Spectra are given in Fig.40 for reaction of Ce with NH and ND .q
3 3
Deuterated ammonia causes a shift of the production masses that is consistent with the formation ofCeNH and CeNH , with little, if any, evidenceq q
2
for concomitant formation of an oxide ion(thoughthe spectra require correction for the contributionsof the Ce isotopes, for the DyH purity of theND , indicated by the manufacturer to be 99%,3
and for the possible persistence of thef3%CeO that derives from the plasma as its reactivityq
with NH is not known). If it is desired to know3
the oxide ratio as an indication of plasma condi-tions, it should be measured without gas in thecell. If it is necessary to use a pressurized cell, theplasma conditions are reflected only if an elementwhose atomic and oxide ions are either non-reactive (a possibility may be BayBaO , forq q
which at least the atomic ion appears to be unreac-tive with NH ) or similarly reactive; otherwise the3
observed ratio is reflective of the relative reactivitywithin the cell, even with high purity inert gases,as shown by Hattendorf and Guntherw145x.¨
9.2.4. GeologicalAlleviation of the interferences caused by REE
oxides, discussed above, is probably most relevantto geological applications. Other applications ofcollisionyreaction cells in this area include theresolution of interelement isobaric interferences,improvement of isotope ratio precision through
1442 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
Fig. 40. Mass spectra obtained for a solution containing 5 ppb Ce, using NH(grey) or ND (black) as reaction gas. Reaction gas3 3
flow was 0.3 Ar-equivalent sccm(0.16 sccm) yielding approximately 50% reaction of the Ce , and the operating point was(RPa,q
RPq)s(0, 0.5). The principal reaction product ion is CeNH(CeND ), with a minor (f20%) reaction channel leading toq q
CeNH (CeND ). (Ion signals below 140 amu are dominated by washout of Ba from an earlier experiment: the ND experimentq q2 2 3
was performed first, so the Ba signals appear enhanced but these are artefactual.)q
collisional homogenization, and laser ablation ofinhomogeneous materials.
Mason w149x has recently reviewed the use ofcollision and reaction cells for laser ablation. Adescription of available instrumentation is given,together with a brief review of some of the relevantfundamentals. Applications for the analysis of avariety of geological matrices, including chromite,carbonate and carbon-rich minerals, inclusions andsulphides are reviewed, and the potential for bio-geochemical determinations is suggested. It isnoted that the ‘dry’ plasma typically used in thisapplication is characterized by different polyatomicinterferences than are encountered during solutionnebulization. Notably, the oxides are less of achallenge because the oxygen content of the plas-ma is less; the problem does not disappear, but isreduced in magnitude. The argide ions cause themost interference, including the argide ions ofconcomitant elements(ArC , ArNa , ArCr ,q q q
etc.). Fortunately, as noted earlier, these argideions have high electron affinities and are normallyresolved through using the same chemistries asappropriate for Ar , ArH , ArO , etc. Multi-q q q
element conditions, which may require a degree ofcompromise in the overall chemistry, is particularlyimportant for transient samples, such as rasterscanning of inhomogeneous materials, or the anal-ysis of fluid inclusions. Mason’s identification ofa potential problem in rapid scanning over atransient has been discussed in Section 7.5, whereit is shown that an axial field minimizes the settlingtime for ion signal stabilization.
Hattendorf and Guntherw79x, however, note that¨this is not the only deficiency of using a reactivegas, such as ammonia, for multi-element determi-nation. While noting the limitation of the iontransit time for the original DRC instrument, theyalso note that certain important elements are reac-tive with ammonia and hence are suppressed dueto their conversion to other species. While it maybe possible to determine these elements at theirproduct ions, this may not be consistent with full-elemental coverage, as there may be a concomitantneed to suppress the observation of condensationproducts that are interferences and to promotesimilar processes for the measurement of the reac-tive ions. That is, a gas is not appropriate if it
1443S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
reacts with an element in the sample to form aninterference for another element if the product ioncannot be, or is desired not to be, suppressed. Inthis instance, the efficiency of the chemistry mustbe compromised by selection of a less reactivegas. The latter work compared the efficiency ofH and NH for the suppression of common2 3
interferences, noting that the latter is approximate-ly 1 order of magnitude favored, consistent withliterature values of the reaction rate constants.Addition of a relatively heavy buffer gas, such asNe or Xe, enhances the thermalization of the ionsand improves the efficiency of H as a reaction2
gas, allowing 3 orders of magnitude suppressionof the Ar signal (by contrast, buffering with40 q
He provided only approx. 1 order of magnitudereaction). It was found that buffered H was2
sufficient for the analytical purpose, consistentwith the precept noted in Section 9.1 that thereaction gas is to be selected on the basis of theanalytical objectives and the constraints that theseimpose. Ca was determined in high purity quartzusing the most abundantmyzs40 isotope, provid-ing an improvement in detection limit of 2 ordersof magnitude over prior work(f1 mgyg in theglass). It was also shown that the ArCr interfer-q
ence on Nb and Zr could also be relieved, allowingthe determination of these elements in pure chro-mium metal. In related work using solution nebu-lization, Hattendorf et al.w150x determined Zr andNb in chromium matrix solutions. Suppression ofthe ArCr interference was obtained as above, butq
a new interference related to the use of solutionnebulization, formation of CrO , was observed.q
x
These ions apparently derive from reactions ofCr with oxygen species in the cell and areq
suppressed by operating with a bandpass having arelatively high low mass cut-off(RPqs0.7). Thedetection limits obtained for Nb and Zr, estimatedto be 2 and 5 ngyg, respectively, in the pure Crmetal, appear to be limited by Nb and Zr contam-ination in the Cr stock solution(as determined bythe terminal slope of the reaction profile, as dis-cussed in Section 6.2.3.3).
Gunther et al.w151x show equivalence of the¨standard mode(unpressurized) and DRC mode(pressurized with HyNe) for major, minor and2
trace element determinations in fluid inclusions by
LA-ICP-MS, concluding that the reaction cell iswell-suited for fast transient multi-element analy-sis. They note that most(uninterfered) elementsare unaffected by the reaction cell conditionsemployed(though the sensitivity for the refractoryelements Ce and Th is reduced due to oxidationwith impurities in the gas), and that substantialimprovement in detectability for normally inter-fered elements(a factor of 250 in detection limitfor Ca and 20 for Fe) is obtained. They also showthat the temporal response for fast transients(onthe order of a second) is unaffected by pressurizingthe cell, which is consistent with a temporalhomogenization period on the millisecondtimescale.
Isotope ratio measurements are of particularimportance in some geological applications. Sectorinstruments equipped with multicollector detectorsprovide excellent ratio precision, and the incorpo-ration of a collisionyreaction cell should allowaccurate measurements for normally interfered ele-mentsw125x. Feldmann et al.w63x report Pb isotoperatios, measured with a quadrupole(sequentialscanning) equipped with hexapole collision cell,with precisions better than 0.1%. The measurementof U isotope ratios in contaminated soilsw128x hasbeen discussed earlier, and measurement of the
Uy U ratio at 0.07% R.S.D. has been reported235 238
w129x. Measurement of the Pb and U ratios are notusually convoluted by spectral interferences requir-ing chemical resolution, but use of the collisioncell to provide enhanced sensitivity through colli-sional focusing is advantageous. Boulyga andBeckerw111x discuss the use of a collision cell forthe measurement of isotope ratios of Ca, Fe andSe where a hydrogenyhelium mixture is used toeliminate the common spectral interferences thathitherto have prevented accurate measurements atlow (sub-ppm) concentrations. Optimizationresults with respect to helium flow(providingcollisional focusing), hydrogen flow (providingchemical resolution) and hexapole bias(kineticenergy discrimination) are provided. Mass discrim-ination factors across the mass range are deter-mined and compare favorably to conventionalquadrupole instruments. Combined uncertainty forthe Ca and Fe(10 ppb) and Se(100 ppb) isotopesis generally better than 0.7%, and the accuracy is
1444 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
generally better than 1%. Improved precision ofisotope ratio measurements have also been reportedby Bandura et al.w109,110x, as discussed inSection 7.5, and by Moens et al.w118x, as dis-cussed in Section 8.4.3.
Mason et al.w67x have discussed optimizationfor the determination of Sy S ratios in lake34 32
water which is affected by biogenic redox reac-tions. The interference of O on sulfur is shownq
2
to be reduced by reaction with Xe(with He bufferand enhanced with the addition of H) in a2
collisionyreaction cell. The BEC is reduced byabout an order of magnitude to approximately 1ppm, which is sufficient for the application at hand(determination of S isotope ratios at 10–50 ppm),and detection limits in the range 20–50 ppb areshown. Precision in the isotope ratio measurements(for 10–50 ppm S in water and acidic solutions)approaches the counting statistics limit(f0.2–0.4% R.S.D.). The method is applied to the deter-mination of sulfur concentration and isotope ratiosin crater-lake acid water and spring water and isin good agreement with other measurements.
9.2.5. BiologicalWe consider biological applications in the wider
sense to include speciated determination of drugs,nutrients and poisons and their metabolites, clinicaldiagnostics and proteomicygenomic analyses. Withparticular reference to the latter, Jakubowskiw152xhas said ‘in the new millennium, the largest impacton organic mass spectrometry will be from inor-ganic mass spectrometry’. While this may havebeen made as a provocative statement, we concurthat ICP-MS will make important contributions toproteomic applications, wherein the particularadvantages of the method over organic MS meth-ods(relative independence of sensitivity on speciesand sample matrix, large dynamic range, lowdetection limits, accurate quantitation) will becapitalized despite the lack of specific mass spec-tral identification of the species.
Sloth and Larsenw122x showed effective sup-pression of the Ar interference on Se isotopesq
2
using CH as a reaction gas, and then used this to4
speciate selenoamino acids using cation exchangechromatography. Detection limits for these specieson the order of 20 pgyml (as selenium) is said to
allow determination at physiologically significantlevels. Because of the removal of the argideinterference, isotopic ratio determination allowsconfirmation of the presence of Se, although itwas shown that a correction for the formation ofSeH (at f9.6%) is necessary. Interestingly,q
experiments with deuterated methane indicate thatthe hydride does not derive from the reaction gas,suggesting the role of contaminants in the cell.
A thorough comparison of different nebulizers(Meinhard, HHPN and MCN) and chromatograph-ic methods(reversed-phase and ion-pair) for thespeciation of selenium in nutritional supplements,using H yHe as a reactionycollision gas, has been2
reported by Marchante-Gayon et al.w64x. Whilethe HHPN in combination with either chromatog-raphy offered the best relative detection limits(35–90 pgyml), the lowest absolute detection limit(500 femtogram range) was obtained with MCNcoupled to reversed-phase chromatography. Sele-nium species were hot-water-extracted from com-mercial nutritional supplements, and several wereidentified through their retention times. The frac-tion of organic and inorganic selenium variesdramatically with supplement type and brand.
The determination of V and Cr in serum andurine are reported by Nixon et al.w153,154x. Thesamples were diluted 1:10 and analyzed directlyby aspirationynebulization. Ammonia reaction gasprovided effective suppression of the ClO andq
ArC interferences, yielding detection limits inq
the serum and urine in the range of 12 ngyl (V)and 55–80 ngyl (Cr). These detection limits aresubstantially better than those obtained usingGFAAS, and equivalence for determination ofthese elements in real patient samples by the twomethods is shown. Similarly, determination of Sein serum and urine using methane as reaction gasis capable of detection limits on the order of 50ngyl in the undiluted samplew155x. The determi-nation of Fe and Cu in liver tissue, with detectionlimits of 20 and 2 ngyg (in the tissue) respectively,is shown by Nixon et al.w156x, using ammonia tosuppress the ArO , ArOH and ArNaq q q
interferences.The degree of phosphorylation of isolated or
chromatographically separated proteins is animportant diagnostic in proteomics. The usual
1445S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
analytical method involves radioisotope labelingwith P w157x. Wind et al.w158x have shown the32
power of using ICP-MS for the determination ofnatural P. Both a quadrupole ICP-MS with a31
hexapole collisionyreaction cell, operated with Hy2
He, and a high mass resolution instrument wereused to resolve the N O and N O H inter-15 16 q 14 16 1 q
ferences. The collision cell approach required theconcomitant use of desolvation, which appeared toresult in some suppression of the signals, and itwas concluded that the high resolution approachis preferred. In a very recent reportw159x, theynote that the concomitant measurement of Sq
(with high mass resolution) allows effective deter-mination of the degree of phosphorylation throughthe ratio of P yS for homologous proteins thatq q
contain cysteine or methionine. Bandura et al.w160x show that P and S may be efficientlyq q
oxidized using O as a reaction gas, allowing2
determination of these elements at less-interferedmasses. The BECs under optimum conditions inDIW are 0.5 and 5 ppb, respectively, and detectionlimits of 0.1 and 0.2 ppb are reported. The BECsof the oxide ions do not appear to be affected bya sample matrix containing 5% acetonitrile, 5%formic acid and 1 mM ammonium bicarbonate,whereas one would expect these to impact at theatomic ion masses. The method was shown tocorrectly yield the number of P-atoms and the PyS ratio fora-casein, and the residual phosphate ofdephosphorylateda-casein was determined.
10. Summary
The r.f.-driven reactionycollision cell in combi-nation with a quadrupole mass filter has becomean essential tool in many analytical laboratories. Itprovides an effective means for the chemical res-olution of plasma-based isobaric interferences aswell as complementary benefits such as improvedsensitivity (through collisional focusing),improved mass resolution and abundance sensitiv-ity (through collisional energy damping) andimproved precision(through temporal homogeni-zation of high frequency signal fluctuationsderived from the plasma source).
Collisional fragmentation is a relatively ineffec-tive process for the ICP-MS application. Ion–
molecule chemistry, used either to remove anisobaric interference or to shift the analyte to aless-interfered mass, can be exceptionally efficient.Our preference for chemical reaction derives fromthe specificity of thermal ion–molecule reactions,the large rate constants resulting from the ion–dipole interaction, and the general lack of activa-tion energy barriers that allow prediction of therelevant chemistry from thermochemical informa-tion. An extensive database of thermal ion–mole-cule reaction rate constants facilitates methoddevelopment. In order to best take advantage ofthese characteristics, the reaction cell should beoperated under near-thermal conditions. Accord-ingly, we distinguish collision cells from reactioncells on the basis of the thermal characteristics ofthe ions extracted from the cell.
The r.f.-driven reaction cell necessarily operatesunder conditions that deviate from strictly thermal.The relatively large axial energy of the ions at theentrance, required in order to accelerate ions intothe cell that is commonly operated above theambient pressure, is initially converted into radialexcitation. After a number of collisions, the radialand axial energies relax together, with a residualdegree of radial excitation derived from the secularand applied drive frequencies. This r.f. contributionto the collision energy is a function of the operat-ing conditions including the amplitudes of the r.f.and DC voltages applied between pole pairs, thedrive frequency and the number of collisions perr.f. cycle. The rate of thermalization of the ions isa function of the operating conditions and the ratioof the ion and neutral masses. The energy andspatial focusing that derives from these collisionalprocesses results in improved transmission of ionsthrough an on-axis exit aperture(collisional focus-ing) and an improvement in the resolution of thedownstream mass filter.
Efficient primary ion–molecule chemistry thatdistinguishes analyte ions from plasma-based inter-ference ions is accompanied by efficient secondaryreactions that create new isobaric interferenceswithin the cell. Either of two methods is commonlyused to suppress the appearance of these secondaryinterference ions: post-cell kinetic energy discrim-ination and in-cell bandpassing. The former maybe applied when the cell conditions are such as to
1446 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
retain a fraction of the initial kinetic energy of theions introduced into the cell; that is, under non-thermal conditions. When the number of collisionssuffered by the ions is sufficiently small to allowdistinction of the energies of the primary ions fromthose of the secondary ions produced within thecell, kinetic energy discrimination does providehigh efficiency of suppression of the secondaryions. The efficiency of post-cell kinetic energydiscrimination is antagonistic to efficiency of theprimary reaction chemistry: the former is favoredunder conditions of few collisions and the latter isfavored under conditions of many collisions. Oper-ation of the cell with a bandpass that excludes atleast one of the precursor ions of the secondarychemistry suppresses the formation of the second-ary interference ions and allows operation of thecell under many-collision conditions that optimizethe efficiency of the primary chemistry.
Under multiple collision conditions, the purityof the reaction gas is very important: relativelysmall traces of impurities can have a dispropor-tionate effect on the overall chemistry if theirreaction rate constants with the ions are large.Therefore, when high efficiency of the primarychemistry is required, it is necessary to use thehighest purity gases available and to minimizeincursion of other gases(plasma gas, ambientvacuum chamber gas) into the cell.
While originally thought to be of most value forthe determination of interfered elements in highpurity materials, especially those used in the semi-conductor industry, the cell has found wide accep-tance in other areas already adapted to the ICP-MSmethod. Many commercial and industrial labora-tories have already adopted the method for qualityassurance of reagents and products, and for directdeterminations in complex samples such as sea-water, urine and serum. New opportunities alsoarise, for instance in geochronology where theion–molecule chemistry can be used for in situisobar resolution and the temporal homogenizationcharacteristics of the cell provide isotope ratioprecision limited by counting statistics. It is to beanticipated that new applications will continue todevelop and these will yet further broaden thescope of the ICP-MS method. It is also obvious
that an outstanding opportunity for fundamentalresearch is facilitated.
Acknowledgments
The authors appreciate the stimulating discus-sions and observations of John Olesik(Ohio StateUniversity), Sam Houk(Iowa State University),Bodo Hattendorf(ETH, Zurich) and other practi-tioners that have inspired us to this work. Thepatience and encouragement of the editor, RalphSturgeon, is much appreciated. We thank Drs KenNeubauer and Ruth Wolf(Perkin Elmer, Connect-icut) for their thorough review of the manuscript.
References
w1x H.I. Schiff, D.K. Bohme, An ion–molecule scheme forthe synthesis of hydrocarbon-chain and organonitrogenmolecules in dense interstellar clouds, Astrophysical J.232 (1979) 740–746.
w2x D. Smith, The ion chemistry of interstellar clouds,Chem. Rev. 92(1992) 1473–1485.
w3x S.G. Lias, P. Ausloos, Ion–Molecule Reactions: TheirRole in Radiation Chemistry, American Chemical Soci-ety, Washington, 1975.
w4x G. Gioumousis, D.P. Stevenson, Reactions of gaseousmolecule ions with gaseous molecules V. theory, J.Chem. Phys. 29(1958) 294–299.
w6x L. Bass, T. Su, W.J. Chesnavich, M.T. Bowers, Ion–polar molecule collisions. A modification of the averagedipole orientation theory: the cos model, Chem. Phys.Lett. 34 (1975) 119–122.
w7x T. Su, W.J. Chesnavich, Parameterization of the ion–polar molecule collision rate constant by trajectorycalculations, J. Chem. Phys. 76(1982) 5183–5185.
w8x T. Su, E.C.F. Su, M.T. Bowers, Ion–polar moleculecollisions. Conservation of angular momentum in theaverage dipole orientation theory. The AADO theory, J.Chem. Phys. 69(1978) 2243–2250.
w9x S.G. Lias, J.E. Bartmess, J.F. Liebman, J.L. Holmes,R.D. Levin, W.G. Mallard, Gas-phase ion and neutralthermochemistry, J. Phys. Chem. Ref. Data 17 1(Suppl.1) (1988) 1–861, Available from http:yyweb-book.nist.govychemistryy.
w10x V.G. Anicich, A survey of bimolecular ion–moleculereactions for use in modeling the chemistry of planetaryatmospheres, cometary comae and interstellar clouds,Astrophysical J. Suppl. Series 84(1993) 215, Availablefrom http:yyastrochem.jpl.nasa.govyaschy.
1447S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
w11x D.K. Bohme, Experimental studies of positive ion chem-istry with flow-tube mass spectrometry: birth, evolution,and achievements in the 20th century, Int. J. MassSpectrom. 200(2000) 97–136.
w12x D. Gerlich, Inhomogeneous RF fields: a versatile toolfor the study of processes with slow ions, in: C.-Y. Ng,M. Baer (Eds.), State-Selected and State-to-State Ion–Molecule Reaction Dynamics Part l. Experiment, Adv.Chem. Physics Series, vol. LXXXII, Wiley, 1992, pp.1–176.
w13x K.M. Ervin, P.B. Armentrout, Translational energydependence of ArqXY™ArX qY (XYsH , D ,q q
2 2
HD) from thermal to 30 eV cm, J. Chem. Phys. 83(1985) 166–189.
w14x P.B. Armentrout, Kinetic energy dependence of ion–molecule reactions: guided ion beams and thresholdmeasurement, Int. J. Mass Spectrom. 200(2000)219–241.
w15x R.A. Yost, C.G. Enke, Selected ion fragmentation witha tandem quadrupole mass spectrometer, J. Amer. Chem.Soc. 100(1978) 2274–2275.
w16x R.A. Yost, C.G. Enke, Triple quadrupole mass spectrom-etry for direct mixture analysis and structure elucidation,Anal. Chem. 51(1979) 1251A–1264A.
w17x R.A. Yost, C.G. Enke, D.C. McGilvery, D. Smith, J.D.Morrison, High efficiency collision-induced dissociationin an rf-only quadrupole, Int. J. Mass Spectrom. IonPhys. 30(1979) 127–136.
w18x P.H. Dawson, J.B. French, J.A. Buckley, D.J. Douglas,D. Simmons, The use of triple quadrupoles for sequen-tial mass spectrometry: 1 the instrument parameters,Org. Mass Spectrom. 17(1982) 205–211.
w19x P.H. Dawson, J.B. French, J.A. Buckley, D.J. Douglas,D. Simmons, The use of triple quadrupoles for sequen-tial mass spectrometry: 2 a detailed case study, Org.Mass Spectrom.(1982) 212–217.
w20x D.J. Douglas, Some current perspectives on ICP-MS,Can. J. Spectroscopy 34(1989) 38–49.
w21x J.T. Rowan, R.S. Houk, Attenuation of polyatomic ioninterferences in inductively coupled plasma mass spec-trometry by gas-phase collisions, Appl. Spectroscopy43 (1989) 976–980.
w22x C.J. Barinaga, D.W. Koppenaal, Ion-trap mass spectrom-etry with an inductively coupled plasma source, RapidCommun. Mass Spectrom. 8(1994) 71–76.
w23x D.W. Koppenaal, C.J. Barinaga, M.R. Smith, Perform-ance of an inductively coupled plasma source ion trapmass spectrometer, J. Anal. At. Spectrom. 9(1994)1053–1058.
w24x G.C. Eiden, C.J. Barinaga, D.W. Koppenaal, Selectiveremoval of plasma matrix ions in plasma source massspectrometry, J. Anal. At. Spectrom. 11(1996)317–322.
w25x G.C. Eiden, C.J. Barinaga, D.W. Koppenaal, Beneficialionymolecule reactions in elemental mass spectrometry,Rapid Commun. Mass Spectrom. 11(1997) 37–42.
w26x G.C. Eiden, C.J. Barinaga, D.W. Koppenaal, Analyticalperformance of the plasma source RF quadrupole iontrap in elemental and isotopic MS, J. Anal. At. Spec-trom. 14(1999) 1129–1132.
w27x P. Turner, T. Merren, J. Speakman, C. Haines, Interfacestudies in the ICP-mass spectrometer, in: G. Holland,S.D. Tanner(Eds.), Plasma Source Mass Spectrometry:Developments and Applications, The Royal Society ofChemistry, Cambridge, 1997, pp. 28–34.
w28x V.I. Baranov, S.D. Tanner, A dynamic reaction cell forinductively coupled plasma mass spectrometry(ICP-DRC-MS). I. The RF-field energy contribution in ther-modynamics of ion–molecule reactions, J. Anal. At.Spectrom. 14(1999) 1133–1142.
w29x S.D. Tanner, V.I. Baranov, A dynamic reaction cell forinductively coupled plasma mass spectrometry(ICP-DRC-MS). II. Reduction of interferences produced with-in the cell, J. Am. Soc. Mass Spectrom. 10(1999)1083–1094.
w30x S.D. Tanner, V.I. Baranov, U. Vollkopf, A dynamicreaction cell for inductively coupled plasma mass spec-trometry (ICP-DRC-MS). III. Analytical performance,J. Anal. At. Spectrom. 15(2000) 1261–1269.
w31x P.H. Dawson, Quadrupole Mass Spectrometry and ItsApplications, American Institute of Physics, Woodbury,New York, 1995.
w32x J.E.P. Syka, Characteristics of multipole collision cells,presented at the 36th ASMS Conference on MassSpectrometry and Allied Topics, San Francisco, 1988,Book of abstracts, 1328–1329.
w33x I. Szabo, New ion-optical devices utilizing oscillatoryelectric fields. I. Principle of operation and analyticaltheory of multipole devices with two-dimensional elec-tric fields, Int. J. Mass Spectrom. Ion Processes 73(1986) 197–235.
w34x C. Hagg, I. Szabo, New ion-optical devices utilizingoscillatory electric fields. II. Stability of motion in atwo-dimensional hexapole field, Int. J. Mass Spectrom.Ion Processes 73(1986) 237–275.
w35x C. Hagg, I. Szabo, New ion-optical devices utilizingoscillatory electric fields. IV. Computer simulations oftransport of an ion beam through an ideal quadrupole,hexapole, and octapole operating in the rf-only mode,Int. J. Mass Spectrom. Ion Processes 73(1986)295–312.
w36x C. Hagg, I. Szabo, New ion-optical devices utilizingoscillatory electric fields. III. Stability of ion motion ina two-dimentional octapole field, Int. J. Mass Spectrom.Ion Processes 73(1986) 277–294.
w37x R.E. March, J.F.J. Todd, Practical Aspects of Ion TrapMass Spectrometry, 1. Fundamentals of Ion Trap MassSpectrometry; 2. Ion Trap Instrumentation; 3. Chemical,Environmental and Biomedical Applications, CRCSeries, Modern Mass Spectrometry, Boca Raton, FL,1995.
w38x J.D. Morrison, K.A. Stanney, J. Tedder, The design anddevelopment of a quinquequadrupole mass spectrometer,
1448 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
presented at the 34th Annual Conference on MassSpectrometry and Allied Topics, Ohio, 1986, Book ofabstracts, 222–223.
w39x C. Beaugrand, G. Devant, S.N. Nermag, C. Rolando,D. Jaouen, A multiquad triple analyzer MSyMSyMS,presented at the 34th Annual Conference on MassSpectrometry and Allied Topics, Cincinnati, 1986, Bookof abstracts, 220–221.
w40x B.A. Collings, J.M. Campbell, D. Mao, D.J. Douglas,A combined linear ion trap time-of-flight system withimproved performance and MS capabilities, Rapidn
Commun. Mass Spectrom. 15(2001) 1777–1795.w41x C.G. Enke, R.A. Yost, J.D. Morrison, Tandem quadru-
pole mass spectrometer for selected ion fragmentationstudies and low energy collision induced dissociatortherefore, US patent 4,234,791(1980).
w42x M. Morris, P. Thibault, R.K. Boyd, Low-energy ionymolecule products from collisions with ammonia, RapidCommun. Mass Spectrom. 7(1993) 1136–1140.
w43x J.T. Watson, C. Jaouen, H. Mestdagh, C. Rolando, Atechnique for mass selective ion rejection in a quadru-pole reaction chamber, Int. J. Mass Spectrom. IonProcesses 93(1989) 225–235.
w44x T. Sakuma, N. Gurprasad, S.D. Tanner, A. Ngo, W.R.Davidson, H.A. McLeod, B.P.-Y. Lau, J.J. Ryan, Theapplication of rapid gas chromatography–tandem massspectrometry in the analysis of complex samples forchlorinated dioxins and furans, in: L.H. Keith, C. Rappe,G. Choudhary(Eds.), Chlorinated Dioxins and Diben-zofurans in the Total Environment II, Butterworth Pub-lishers, Boston, 1985, pp. 139–152.
w45x I.I. Stewart, Electrospray mass spectrometry: a tool forelemental speciation, Spectrochim. Acta Part B 54(1999) 1649–1695.
w46x M.S. Lee, E.H. Kerns, LCyMS applications in drugdevelopment, Mass Spectrom. Rev. 18(1999) 187–279.
w47x R. Aebersold, D.R. Goodlett, Mass spectrometry inproteomics, Chem. Rev. 101(2001) 269–295.
w48x J. Godovac-Zimmerman, L.R. Brown, Perspectives formass spectrometry and functional proteomics, MassSpectrom. Rev. 20(2001) 1–57.
w49x K.L. Busch, G.L. Glish, S.A. McLuckey, Mass Spec-trometryyMass Spectrometry: Techniques and Applica-tions of Tandem Mass Spectrometry, VCH, New York,1988.
w50x D.J. Douglas, J.B. French, Collisional focusing effectsin radio frequency quadrupoles, J. Am. Soc. MassSpectrom. 3(1992) 398–408.
w51x D.J. Douglas, Applications of collision dynamics inquadrupole mass spectrometry, J. Am. Soc. Mass Spec-trom. 9 (1998) 101–113.
w52x A.N. Krutchinsky, I.V. Chernushevich, V.L. Spicer, W.Ens, K.G. Standing, Collisionally damping interface foran electrospray time-of-flight mass spectrometer, J. Am.Soc. Mass Spectrom. 9(1998) 569–579.
efficiency and mass resolution on a triple quadrupolemass spectrometer system, Anal. Chem. 67(1995)1696–1704.
w54x M. Nicholls, Z. Palacz, P. Turner, High precision isotoperatio measurement by multicollector ICP-MS, presentedat Winter Conference on Plasma Spectrochemistry,Scottsdale, Arizona, 1998, Book of abstracts, 413.
w55x M. Splendore, F.A. Londry, R.E. March, R.J.D. Morri-sion, P. Perrier, J. Andre, A simulation study of ionkinetic energies during resonant excitation in a stretchedion trap, Int. J. Mass Spectrom. Ion Processes 156(1996) 11–29.
w56x R.E. March, R.J. Hughes, Quadrupole Storage MassSpectrometry, Wiley, New York, 1989.
w57x D. Duckworth, D.E. Goering, G.V. Van Berkel, S.A.McLuckey, Gas-phase metal ion chemistry investiga-tions using quadrupole ion traps: Unimolecular disso-ciation, ionymolecule and ionyion reactions, presentedat Federation of Analytical Chemistry and SpectroscopySocieties (FACSS), 1999, Book of abstracts, papernumber 57.
w59x N. Furuta, A. Takeda, J. Zheng, Evaluation of induc-tively coupled plasma–ion trap mass spectrometry, in:G. Holland, S.D. Tanner(Eds.), Plasma Source MassSpectrometry: The New Millennium, The Royal Societyof Chemistry, Cambridge, 2001, pp. 90–96.
w60x A. Dodonov, V. Kozlovski, A. Loboda, V. Raznikov, I.Soulimenkov, A. Tolmachev, A. Kraft, H. Wollnik, Anew technique for decomposition of selected ions inmolecule ion reactor coupled with ortho-time-of-flightmass spectrometry, Rapid Commun. Mass Spectrom. 11(1997) 1649–1656.
w61x E.R. Denoyer, S.D. Tanner, U. Vollkopf, A new dynamicreaction cell for reducing ICP-MS interferences usingchemical resolution, Spectroscopy 14(1999) 43–54.
w62x I. Feldmann, N. Jakubowski, D. Steuwer, Applicationof a hexapole collision and reaction cell in ICP-MS partI: instrumental aspects and operational optimization,Fresenius’ J. Anal. Chem. 365(1999) 415–421.
w63x I. Feldmann, N. Jakubowski, C. Thomas, D. Steuwer,Application of a hexapole collision and reaction cell inICP-MS part II: analytical figures of merit and firstapplications, Fresenius’ J. Anal. Chem. 365(1999)422–428.
w64x J.M. Marchante-Gayon, C. Thomas, I. Feldmann, N.Jakubowski, Comparison of different nebulisers andchromatographic techniques for the speciation of sele-nium in nutritional commercial supplements by hexa-pole collision and reaction cell ICP-MS, J. Anal. At.Spectrom. 15(2000) 1093–1102.
w65x D.R. Bandura, V.I. Baranov, S.D. Tanner, Reactionchemistry and collisional processes in multipole devicesfor resolving isobaric interferences in ICP-MS, Fresen-ius’ J. Anal. Chem. 370(2001) 454–470.
1449S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
w66x N.G. Adams, D. Smith, The selected ion flow tube(SIFT); a technique for studying ion-neutral reactions,Int. J. Mass Spectrom. Ion Phys. 21(1976) 349–359.
w67x P.R.D. Mason, K. Kaspers, M.J. vanBergen, Determi-nation of sulfur isotope ratios and concentrations inwater samples using ICP-MS incorporating hexapoleion optics, J. Anal. At. Spectrom. 14(1999) 1067–1074.
w68x H. Niu, R.S. Houk, Fundamental aspects of ion extrac-tion in inductively coupled plasma mass spectrometry,Spectrochim. Acta Part B 51(1996) 779–815.
w69x R.S. Houk, N. Praphairaksit, Dissociation of polyatomicions in the inductively coupled plasma, Spectrochim.Acta Part B 56(2001) 1069–1096.
w70x V.V. Lavrov, D.K. Bohme, Personal communication,2000.
w71x M.V. Sussman, Elementary General Thermodynamics,Addison-Wesley Publishing, Toronto, Canada, 1972.
w72x P. Kebarle, Gas phase ion thermochemistry based onion-equilibria: from the ionosphere to the reactive cen-ters of enzymes, Int. J. Mass Spectrom. 200(2000)313–330.
w73x J.L. Beauchamp, M.C. Caserio, T.B. McMahon, Ion–molecule reactions oftert-butyl alcohol by ion cyclotronresonance spectroscopy, J. Amer. Chem. Soc. 96(1974)6243–6251.
w74x F.C. Fehsenfeld, A.L. Schmeltekopf, P.D. Goldan, H.I.Schiff, E.E. Ferguson, Thermal energy ion-neutral reac-tion rates. 1. Some reactions of helium ions, J. Chem.Phys. 44(1966) 4078–4094.
w75x G.K. Koyanagi, V.V. Lavrov, V. Baranov, D. Bandura,S.D. Tanner, J.W. McLaren, D.K. Bohme, A novel ICPySIFT mass spectrometer for the study of reactions ofatomic and atomic oxide ions, Int. J. Mass Spectrom.194 (2000) L1–L5.
w76x G.K. Koyanagi, V.I. Baranov, S.D. Tanner, D.K. Bohme,An inductively coupled plasmayselected-ion flow tubemass spectrometric study of the chemical resolution ofisobaric interferences, J. Anal. At. Spectrom. 15(2000)1207–1210.
w77x G.K. Koyanagi, D.K. Bohme, Oxidation reactions oflanthanide cations with N O and O : periodicities in2 2
reactivity, J. Phys. Chem. 105(2001) 8964–8968.w78x C. Rue, P.B. Armentrout, I. Kretzschmar, D. Schroder,
H. Schwarz, Guided ion beam studies of the reactionsof Fe and Co with CS and COS, J. Phys. Chem. Aq q
2
105 (2001) 8456–8464.w79x B. Hattendorf, D. Gunther, Characteristics and capabil-¨
ities of an ICP-MS with a dynamic reaction cell for dryaerosols and laser ablation, J. Anal. At. Spectrom. 15(2000) 1125–1131.
w80x D.R. Bandura, S.D. Tanner, V.I. Baranov, G.K. Koyan-agi, V.V. Lavrov, D.K. Bohme, Ion–molecule chemistrysolutions to the ICP-MS analytical challenge, in: G.Holland, S.D. Tanner(Eds.), Plasma Source Mass Spec-trometry: The New Millennium, The Royal Society ofChemistry, Cambridge, 2001, pp. 130–147.
w81x J.R. Gibson, S. Taylor, Numerical investigation of theeffect of electrode size on the behaviour of quadrupolemass filters, Rapid Commun. Mass Spectrom. 15(2001)1960–1964.
w82x W. Paul, H.P. Reinhard, U. von Zahn, Das elektrischemassenfilter als massenspektrometer und isotopentren-ner, Z. Physik. 152(1958) 143–182.
w83x I.E. Dayton, F.C. Shoemaker, R.F. Mozley, The meas-urement of two-dimensional fields. Part II. Study of aquadrupole magnet, Rev. Sci. Instru. 25(1954)485–489.
w84x D.J. Douglas, N.V. Konenkov, Influence of the sixthand tenth spatial harmonics on the peak shape ofquadrupole mass filter with round rods’, Rapid Com-mun. Mass Spectrom. 16(2002) 1425–1431.
w85x D.R. Denison, Operating parameters of a quadrupole ina grounded cylindrical housing, J. Vac. Sci. Technol. 8(1971) 266–269.
w86x V.V.K.R. Rao, A. Bhutani, Electric hexapoles and octa-poles with optimized circular section rods, Int. J. MassSpectrom. 202(2000) 31–36.
w87x W.M. Brubaker, Comparison of quadrupole mass spec-trometers with round and hyperbolic rods, J. Vac. Sci.Technol. 4(1967) 326, abstract only.
w88x W.M. Brubaker, W.S. Chamberlin, Theoretical andexperimental comparison of quadrupole mass analyzerswith round and hyperbolic field-forming surfaces, pre-sented at Recent Developments in Mass Spectrometry,Proceedings International Conference Mass Spectros-copy, University Park Press, Baltimore, MD, 1970, pp.98–103.
w89x W.M. Brubaker, An improved quadrupole mass analyzer,Adv. Mass Spectrom. 4(1968) 293–299.
w91x S.D. Tanner, Experimental studies of ion kinetic energiesin ICP-MS, in: G. Holland, A.N. Eaton(Eds.), Appli-cation of Plasma Source Mass Spectrometry II, TheRoyal Society of Chemistry, Cambridge, 1993, pp.222–234.
w92x D.J. Douglas, S.D. Tanner, Fundamental considerationsin ICP-MS, in: A. Montaser(Ed.), Inductively CoupledPlasma Mass Spectrometry, VCH, 1998, pp. 615–679.
w93x F. Muntean, Transmission study for rf-only quadrupolesby computer simulation, Int. J. Mass Spectrom. IonProcesses 151(1995) 197–206.
w94x K. Blaum, Ch. Geppert, P. Muller, K. Nortershauser, K.Wendt, B.A. Bushaw, Peak shape for a quadrupole massspectrometer: comparison of computer simulation andexperiment, Int. J. Mass Spectrom. 202(2000) 81–89.
w95x M. Morris, P. Thibault, R.K. Boyd, Characterization ofa high-pressure quadrupole collision cell for low-energycollision-induced dissociation, J. Am. Soc. Mass Spec-trom. 5 (1994) 1042–1063.
w96x C.M. Lock, E. Dyer, Characterisation of high pressurequadrupole collision cells possessing direct current axial
1450 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
fields, Rapid Commun. Mass Spectrom. 13(1999)432–448.
w97x G. Javahery, B. Thomson, A segmented radiofrequen-cy—only quadrupole collision cell for measurements ofion collision cross section on a triple quadrupole massspectrometer, J. Am. Soc. Mass Spectrom. 8(1997)697–702.
w98x B.A. Thomson, C.L. Jolliffe, Spectrometer with axialfield, US patent 5,847,386(1998).
w99x A. Loboda, A. Krutchnisky, J. McNabb, V. Spicer, W.Ens, K. Standing, Novel LINAC II electrode geometryto create an axial field in a multipole ioon guide,presented at the 48th Am. Soc. Mass Spectrom. Meeting,Long Beach, California, 2000, Book of abstracts, posternumber WPA016.
w100x A. Loboda, A. Krutchnisky, O. Loboda, J. McNabb, V.Spicer, W. Ens, K. Standing, Novel Linac II electrodegeometry for creating an axial field in a multipole ionguide, Eur. J. Mass Spectrom. 6(2000) 531–536.
w101x D.R. Bandura, V.I. Baranov, S.D. Tanner, Inductivelycoupled plasma mass spectrometer with an axial fieldin a quadrupole reaction cell, J. Am. Soc. Mass Spec-trom. (2002), in press.
w102x D.R. Bandura, V. Baranov, S.D. Tanner, Dynamic reac-tion cell with axial field, presented at the 47th Interna-tional Conference on Analytical Sciences andSpectroscopy, Toronto, 2001.
w103x D.J. Douglas, J.B. French, Gas dynamics of the induc-tively coupled plasma mass spectrometry interface, J.Anal. At. Spectrom. 3(1988) 743–747.
w104x S.D. Tanner, L.M. Cousins, D.J. Douglas, Reduction ofspace charge effects using a three-aperture gas dynamicvacuum interface for inductively coupled plasma-massspectrometry, Appl. Spectroscopy 48(1994)1367–1372.
w105x S.D. Tanner, D.J. Douglas, J.B. French, Gas and iondynamics of a three-aperture vacuum interface for induc-tively coupled plasma-mass spectrometry, Appl. Spec-troscopy 48(1994) 1373–1378.
w106x T.N. Olney, W. Chen, D.J. Douglas, Gas dynamics ofthe ICP-MS interface: impact pressure probe measure-ments of gas flow profiles, J. Anal. At. Spectrom. 14(1999) 9–17.
w107x V.I. Baranov, S.D. Tanner, Multi-thermal plasma distri-butions from ion kinetic energy measurements, present-ed at the 24th Federation of Analytical Chemistry andSpectroscopy Societies, Providence, Rhode Island, 1997,Book of abstracts, 133.
w108x A.N. Krutchinsky, A.V. Loboda, V.L. Spicer, R. Dwor-schak, W. Ens, K.G. Standing, Orthogonal injection ofmatrix-assisted laser desorptionyionization ions into atime-of-flight spectrometer through a collisional damp-ing interface, Rapid Commun. Mass Spectrom. 12(1998) 508–518.
w109x D.R. Bandura, V.I. Baranov, S.D. Tanner, Effect ofcollisional damping and reactions in a dynamic reaction
cell on the precision of isotope ratio measurements, J.Anal. At. Spectrom. 15(2000) 921–928.
w110x D.R. Bandura, S.D. Tanner, Effect of collisional damp-ing in the dynamic reaction cell on the precision ofisotope ratio measurements, At. Spectroscopy 20(1999)69–72.
w111x S.F. Boulyga, J.S. Becker, ICP-MS with hexapole col-lision cell for isotope ratio measurements of Ca, Fe andSe, Fresenius’ J. Anal. Chem. 370(2001) 618–623.
w112x S.F. Boulyga, J.S. Becker, Inductively coupled plasmamass spectrometry with hexapole collision cell: figuresof merit and applications, Entwicklung und Adwendungmassenspektrometrischer Methoden zur Spuren-, Ultras-puren,-Isotopen- und Oberflachenanalytik fur Forchung-saufgaben des Forschungszentrums Julich, Tei 12(2000) 13–44.
w113x V. Baranov, G. Javahery, A.C. Hopkinson, D.K. Bohme,Intrinsic coordination properties of iron in FeO : kinet-q
ics at 294"3 K for gas-phase reactions of the groundstates of Fe and FeO with inorganic ligands contain-q q
ing hydrogen, nitrogen, and oxygen, J. Am. Chem. Soc.117 (1995) 12801–12809.
w114x R.K. Milburn, V.I. Baranov, A.C. Hopkinson, D.K.Bohme, Sequential ligation of Mg , Fe ,(c-q q
C H )Mg , and(c-C H )Fe with ammonia in the gasq q5 5 5 5
phase: transition from coordination to solvation in thesequential ligation of Mg , J. Phys. Chem. A 102q
(1998) 9803–9810.w115x G.K. Koyanagi, D.K. Bohme, Personal communication,
2001.w116x J.W. Olesik, Personal communication, 2001.w117x K. Neubauer, Personal communication, 2001.w118x L.J. Moens, F.F. Vanhaecke, D.R. Bandura, V.I. Baranov,
S.D. Tanner, Elimination of isobaric interferences inICP-MS, using ion–molecule reaction chemistry: RbySr age determination of magmatic rocks, a case study,J. Anal. At. Spectrom. 16(2001) 991–994.
w119x U. Vollkopf, V.I. Baranov, S.D. Tanner, ICP-MS multie-lement analysis at sub-ppt levels applying new instru-mental design concepts, in: G. Holland, S.D. Tanner(Eds.), Plasma Source Mass Spectrometry: New Devel-opments and Applications, The Royal Society of Chem-istry, Cambridge, 1999, pp. 63–79.
w120x F. Abou-Shakra, The determination of arsenic in sea-water by platform ICP-HEX-MS, in: G. Holland, S.D.Tanner(Eds.), Plasma Source Mass Spectrometry: NewDevelopments and Applications, The Royal Society ofChemistry, Cambridge, 1999, pp. 120–131.
w121x P. Leonhard, R. Pepelnik, A. Prange, N. Yamada, T.Yamada, Analysis of diluted seawater at the ng l levely1
using an ICP-MS with an octapole reaction cell, J. Anal.At. Spectrom. 17(2002) 189–196.
w122x J.J. Sloth, E.H. Larsen, The application of inductivelycoupled plasma dynamic reaction cell mass spectrometryfor measurement of selenium isotopes, isotope ratiosand chromatographic detection of selenoamino acids, J.Anal. At. Spectrom. 15(2000) 669–672.
1451S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
w123x J.R. deLaeter, Mass spectrometry and geochronology,Mass Spectrom. Rev. 17(1998) 97–125.
w124x F. Vanhaecke, G. De Wannemacker, L. Moens, J. Her-togen, The determination of strontium isotope ratios bymeans of quadrupole-based ICP mass spectrometry: ageochronological case study, J. Anal. At. Spectrom. 14(1999) 1691–1696.
w125x P.J. Turner, T.O. Merren, J. Speakman, R.C. Haines, Z.Palacz, Ion optics of multi-collector ICP-MS systemsfor precise and accurate isotope ratio measurement,presented at Winter Conference on Plasma Spectro-chemistry, Ft. Lauderdale, 2000, Book of abstracts, 330–331.
w126x J.P. Guzowski, G.M. Hieftje, Characteristics of a hexa-pole ion-guide interface for plasma-source time-of-flightmass spectrometry, J. Anal. At. Spectrom. 16(2001)781–792.
w127x A.M. Leach, G.M. Hieftje, Use of an ion guide collisioncell to improve the analytical performance of an induc-tively coupled plasma time-of-flight mass spectrometer’,Int. J. Mass Spectrom. 212(2001) 49–63.
w128x S.F. Boulyga, J.S. Becker, J.L. Matusevitch, H.-J. Dietze,Isotope ratio measurements of spent reactor uranium inenvironmental samples by using inductively coupledplasma mass spectrometry, Int. J. Mass Spectrom. 203(2000) 143–154.
w129x J.S. Becker, H.-J. Dietze, Precise and accurate isotoperatio measurements by ICP-MS, Fresenius’ J. Anal.Chem. 368(2000) 23–30.
w130x K. Sakata, K. Kawabata, Reduction of fundamentalpolyatomic ions in inductively coupled plasma massspectrometry, Spectrochim. Acta Part B 49(1994)1027–1038.
w131x S.D. Tanner, Characterization of ionization and matrixsuppression in inductively coupled ‘cold’ plasma massspectrometry, J. Anal. At. Spectrom. 10(1995)905–921.
w132x S.D. Tanner, V.I. Baranov, Fundamental processesimpacting performance of an ICP-MS dynamic reactioncell, in: G. Holland, S.D. Tanner(Eds.), Plasma SourceMass Spectrometry: New Development and Applica-tions, The Royal Society of Chemistry, Cambridge,1999, pp. 46–62.
w133x D.S. Bollinger, A.J. Schleisman, Analysis of high purityacids using a dynamic reaction cell ICP-MS, At. Spec-troscopy 20(1999) 60–63.
w134x D.S. Bollinger, A.J. Schleisman, Initial experiences withthe dynamic reaction cell ICP-MS, in: G. Holland, S.D.Tanner(Eds.), Plasma Source Mass Spectrometry: NewDevelopments and Applications, The Royal Society ofChemistry, Cambridge, 1999, pp. 80–92.
w135x J.W. Olesik, C. Hensman, S. Rabb, D. Rago, Sampleintroduction, plasma-sample interactions, ion transportand ion–molecule reactions: fundamental understandingand practical improvements in ICP-MS, in: G. Holland,S.D. Tanner(Eds.), Plasma Source Mass Spectrometry:
The New Millennium, The Royal Society of Chemistry,Cambridge, 2001, pp. 3–16.
w136x U. Vollkopf, K. Klemm, M. Pfluger, The analysis ofhigh purity hydrogen peroxide by dynamic reaction cellICP-MS, At. Spectroscopy 20(1999) 53–59.
w137x Y. Kishi, K. Kawabata, Effect of Plasma Parameters onthe Analysis of Semiconductor Process Chemicals byICP-MS, D-6482, Perkin Elmer Instruments, Shelton,CT, 2001.
w138x A.F. Porche, Y. Kishi, R.E. Wolf, Analysis of SiliconWafers Using the ELAN DRC ICP-MS, ApplicationNote D-6444, Perkin Elmer Instruments, Shelton, CT,2001.
w139x J. Godfrey, J. Cantle, P. Sigsworth, Multi-element anal-ysis of potable water using the VG PQ ExCell ICP-MSincorporating Collision Cell Technology(CCT), pre-sented at ninth ISMAS-WS, 2000, 114–123.
w140x Available from http:yywww.chem.yorku.cayprofsyboh-meyresearchyresearch.html(last accessed June 4, 2002).
w141x Z. Du, R.S. Houk, Attenuation of metal oxide ions ininductively coupled plasma mass spectrometry withhydrogen in a hexapole collision cell, J. Anal. At.Spectrom. 15(2000) 383–388.
w142x L.A. Simpson, M. Thomsen, B.J. Alloway, A reactionmechanism for solving the oxide problem in ICP-MSanalysis of the noble metals, in: G. Holland, S.D. Tanner(Eds.), Plasma Source Mass Spectrometry: The NewMillennium, The Royal Society of Chemistry, Cam-bridge, 2001, pp. 148–161.
w143x M.L. Huertas, J. Fontan, Evolution times of troposphericpositive ions, Atmos. Environ. 9(1975) 1018–1026.
w144x V.I. Baranov, D.R. Bandura, S.D. Tanner, Reaction cellapproach to the oxide problem, presented at WinterConference on Plasma Spectrochemistry, Ft. Lauderdale,Florida, 2000, Book of abstracts, 375.
w145x B. Hattendorf, D. Gunther, Experimental evidence for¨the formation of doubly charged oxide and hydroxideions in inductively coupled plasma mass spectrometry,Fresenius’ J. Anal. Chem. 370(2001) 483–487.
w146x W. Lindinger, Reactions of doubly charged ions at nearthermal energies, Physica Scripta T3(1983) 115–119.
w147x Z. Herman, Dynamics of charge transfer and chemicalreactions of doubly-charged ions at low collision ener-gies, Int. Rev. Phys. Chem. 15(1996) 299–324.
w148x W. Lindinger, A. Hansel, Z. Herman, Ion–moleculereactions, Adv. Atomic Mol. Optical Phys. 43(2000)243–294.
w149x P.R.D. Mason, Expanding the capabilities of laser abla-tion ICP-MS with collision and reaction cells, in: P.Sylvester (Ed.), Laser-Ablation-ICPMS in the EarthSciences—Principles and Applications, The Mineralog-ical Association of Canada, Ottawa, Canada, 2001, pp.63–81.
w150x B. Hattendorf, D. Gunter, M. Schonbachler, A. Halliday,Simultaneous ultra trace determination of Zr and Nb inchromium matrices with ICP Dynamic Reaction CellMS, Anal. Chem. 73(2001) 5494–5498.
1452 S.D. Tanner et al. / Spectrochimica Acta Part B 57 (2002) 1361–1452
w151x D. Gunther, B. Hattendorf, A. Audetat, Multi-element¨analysis of melt and fluid inclusions with improveddetection capabilities for Ca and Fe using laser ablationwith a dynamic reaction cell ICP-MS, J. Anal. At.Spectrom. 16(2001) 1085–1090.
w152x N. Jakubowski, Instrumental concepts in ICP-MS tocope with spectral interferences, presented at Interna-tional Congress of Pacific Basic Societies, 2000, papernumber ANAL 0384.
w153x D.E. Nixon, J. Butz, S. Eckdahl, M.F. Burritt, K.R.Neubauer, Determination of Vanadium in Serum andUrine Using the ELAN DRC ICP-MS, Application NoteD-6456, Perkin Elmer Instruments, Shelton, CT, 2001.
w154x D.E. Nixon, J. Butz, S.J. Eckdahl, M.F. Burritt, K.R.Neubauer, Determination of Chromium in Serum andUrine, Application Note D6356, Perkin Elmer Instru-ments, Shelton, CT, 2000.
w155x D.E. Nixon, J. Butz, S.J. Eckdahl, M.F. Burritt, K.R.Neubauer, R.E. Wolf, Determination of Selenium inSerum and Urine Using the ELAND DRC ICP-MS,Application Note D-6420, Perkin Elmer Instruments,Shelton, CT, 2000.
w156x D.E. Nixon, J. Butz, S.J. Eckdahl, M.F. Burritt, K.R.Neubauer, R.E. Wolf, Determination of Copper and Iron
in Liver Tissue Using the ELAN DRC ICP-MS, Appli-cation Note D6419, Perkin Elmer Instruments, Shelton,CT, 2000.
w157x K. Wilson, J. Walker, Principals and Techniques ofPractical Biochemistry, Cambridge University Press,2000, pp. 319–322.
w158x M. Wind, M. Edler, N. Jakubowski, M. Linsheid, H.Wesch, W.D. Lehmann, Analysis of protein phosphory-lation by capillary liquid chromatography coupled toelement mass spectrometry with P detection and to31
electrospray mass spectrometry, Anal. Chem. 73(2001)29–35.
w159x M. Wind, H. Wesch, W.D. Lehmann, Protein phospho-rylation degree: determination by capillary liquid chro-matography and inductively coupled plasma massspectrometry, Anal. Chem. 73(2001) 3006–3010.
w160x D.R. Bandura, V.I. Baranov, S.D. Tanner, Detection ofultra-trace phosphorous and sulfur by quadrupole ICP-MS with Dynamic Reaction Cell’, Anal. Chem. 74(2002) 1497–1502.
w161x K.R. Ludwig, Using IsoplotyEx (version 1.00): A Geo-chronological Toolkit for Microsoft Excel, BerkeleyGeochronology Center, Berkeley, CA, 1998, SpecialPublication No. 1.