22
RNA-Seq Xiaole Shirley Liu STAT115, STAT215, BIO298, BIST520
| Date post: | 19-Dec-2015 |
| Category: |
Documents |
| Upload: | edwin-rodgers |
| View: | 234 times |
| Download: | 0 times |

RNA-Seq
Xiaole Shirley Liu
STAT115, STAT215, BIO298, BIST520

RNA-seq Protocol
2Martin and Wang Nat. Rev. Genet. (2011)

RNA-seq Applications
• Expression levels, differential expression• Alternative splicing, novel isoforms• Novel genes or transcripts, lncRNA• Detect gene fusions• Many different protocols• Can use on any sequenced genome• Better dynamic range, cleaner data
3

Experimental Design
• Assessing biological variation requires biological replicates (no need for technical replicates)
• 3 preferred, 2 OK, 1 only for exploratory assays (not good for publications)
• For differential expression, don’t pool RNA from multiple biological replicates
• Batch effects still exist, try to be consistent or process all samples at the same time
4

Experimental Design
• Ribo-minus (remove too abundant genes)
• PolyA (mRNA, enrich for exons)
• Strand specific (anti-sense lncRNA)
• Sequencing: – PE (resolve redundancy) or SE: expression– PE for splicing, novel transcripts– Depth: 30-50M differential expression, deeper
transcript assembly– Read length: longer for transcript assembly
5

RNA-seq Analysis
6

Alignment
• Prefer splice-aware aligners
• TopHat, BWA, STAR (not DNASTAR)
• Sometimes need to trim the beginning bases
7

Transcript Assembly
8
Reference-based assemblyCufflinks
De novo assemblyTrinity

Quality Control: RSeQC
9

Expression Index
• RPKM (Reads per kilobase of transcript per million reads of library)
– Corrects for coverage, gene length
– 1 RPKM ~ 0.3 -1 transcript / cell
– Comparable between different genes within the same dataset
– TopHat / Cufflinks
• FPKM (Fragments), PE libraries, RPKM/2• TPM (transcripts per million)
– Normalizes to transcript copies instead of reads
– Longer transcripts have more reads
– RSEM, HTSeq 10

Differential Expression
11

Sequencing Read Distribution
• Poisson distribution: – # events within an interval
• Sequencing data is overdispersed Poisson
• Negative binomial– Def: # of successes
before r failures occur, if
Pb(each success) is p
12

Differential Expression
• Negative binomial
for RNA-seq• Variance estimated by
borrowing information from all the genes – hierarchical models
• Test whether μi is the same for gene i between samples j
• FDR?
13

Differential Expression
• Should we do differential expression on RPKM/FPKM or TPM?
• Cufflinks: RPKM/FPKM• LIMMA-VOOM and DESeq: TPM• Power to detect DE is proportional to length• Continued development and updates
14
Gene A (1kb)
Gene B (8kb)

Alternative Splicing
• Assign reads to splice isoforms
15

Isoform Inference
• If given known set of isoforms
• Estimate x to maximize the likelihood of observing n
16

Known Isoform Abundance Inference
17

Isoform Inference
• With known isoform set, sometimes the gene-level expression level inference is great, although isoform abundances have big uncertainty (e.g. known set incomplete)
• De novo isoform inference is a non-identifiable problem if RNA-seq reads are short and gene is long with too many exons
• Algorithm: MATS
18
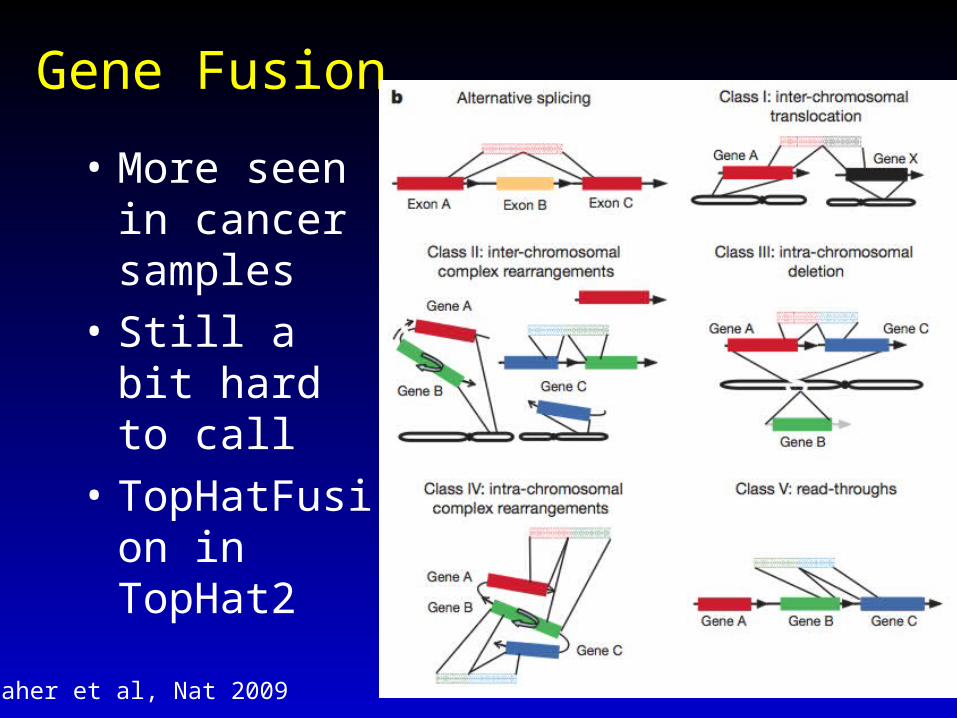
Gene Fusion
• More seen in cancer samples
• Still a bit hard to call
• TopHatFusion in TopHat2
Maher et al, Nat 200919

Other Applications
• RNA editing– Change on RNA sequence after transcription– Most frequent: A to I (behaves like G), C to U– Evolves from mononucleotide deaminases,
might be involved in RNA degradation
• Circular RNA– Mostly arise from splicing– Varying length, abundance, and stability– Possible function: sponge for RBP or miRNA
20

21
Summary• RNA-seq design considerations• Read mapping
– TopHat, BWA, STAR
• De novo transcriptome assembly: TRINITY• Expression index: FPKM and TPM• Differential expression
– Cufflinks: versatile
– LIMMA-VOOM and DESeq: better variance estimates
• Alternative splicing: MATS• Gene fusion, genome editing, circular RNA

Acknowledgement
• Alisha Holloway
• Simon Andrews
• Radhika Khetani
22

















