Page 1
ROLE OF HUMAN HERPESVIRUS 8 VIRAL INTERLEUKIN-6 SIGNALING IN VIRUS BIOLOGY
by Emily M. Cousins
A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy
Baltimore, Maryland December 2014
© 2014 Emily M. Cousins All Rights Reserved
Page 2
ii
ABSTRACT
Human herpesvirus 8 (HHV-8) is associated with B cell and endothelial tumors.
HHV-8 encodes a viral homolog of interleukin 6 (vIL-6), which promotes the proliferation
and survival of virally infected and neighboring cells and induces angiogenesis, suggesting
that vIL-6 contributes to HHV-8 malignant pathogenesis. Furthermore, vIL-6 is expressed
during viral latency and lytic replication, allowing vIL-6 to function in viral latency
maintenance and virus productive replication.
Prior to this dissertation research, little was known regarding the role of vIL-6
interactions with its signal transducer (gp130) within the endoplasmic reticulum (ER),
where vIL-6 predominantly localizes. High levels of phosphorylated STAT3, a product of
vIL-6 signaling, had been observed in HHV-8+ primary effusion lymphomas (PEL), but the
mechanism behind STAT3 activation was unclear. The effects of vIL-6 on virus replication
were limited to one published study, which failed to detect any influence of vIL-6 in a model
culture system. Mechanistically, vIL-6 is distinct from its cellular counterparts because it
can signal from the ER, but the biological functions of ER-localized vIL-6/gp130 signaling
were unknown. Therefore, the goals of this dissertation were to further characterize ER-
localized vIL-6/gp130 interactions, investigate the mechanism(s) responsible for
increased STAT3 activation in PEL cells, decipher the roles of gp130-activated STAT and
ERK signaling in PEL cell maintenance, and evaluate the role of vIL-6 and the vIL-6/gp130
interaction in HHV-8 replication.
Studies performed during this dissertation work demonstrated that ER-localized
vIL-6/gp130 signaling in PEL cells contributes to high levels of active STAT3, that vIL-
6/gp130 signaling also activates STAT1 and ERK1/2 in PEL cells, that ER-localized vIL-
6/gp130 signaling can sustain PEL cell growth and viability, and that STAT3 and ERK1/2
are crucial for cell growth and survival. In addition to promoting the growth and survival of
latently infected PEL cells, vIL-6 and gp130 also promote productive virus replication in
Page 3
iii
PEL and endothelial cells. In PEL cells, vIL-6 pro-replication effects require its interaction
with gp130 in the ER, where vIL-6/gp130-mediated STAT signaling, and not MAPK
signaling, is critical. These studies have expanded our understanding of how vIL-6
functions in normal virus biology and have opened new avenues for targeted HHV-8
therapy.
Readers John Nicholas, PhD (Advisor) Professor of Oncology Johns Hopkins University School of Medicine, Baltimore, MD Gary Ketner, PhD (Committee Member) Professor of Molecular Microbiology and Immunology Johns Hopkins Bloomberg School of Public Health, Baltimore, MD
Page 4
iv
ACKNOWLEDGEMENTS
Many individuals have supported me during my graduate training at the Johns
Hopkins University School of Medicine. First and foremost, I must acknowledge my
mentor, John Nicholas. My development as a scientist can be attributed to John’s patience
and guidance throughout my time in his laboratory. John taught me how to design
experiments with proper controls, how to write scientific articles and grant applications,
and how to develop a story worthy of publication. Additionally, John fostered a
collaborative environment in the laboratory in which techniques, reagents, and expertise
were shared. I would also like to thank the other members of the Nicholas lab for their
guidance, instruction, and friendship: Daming Chen, Young Choi, Gordon (Sandy)
Sandford, Yang Gao, and Kihyun Yoo. Additionally, much of my dissertation research was
funded by a Ruth L. Kirschstein F31 fellowship (5F31CA171933), and I am grateful for the
support of this program.
Prior to joining the Nicholas laboratory, several other scientists taught and
encouraged me. My first foray into research occurred during my undergraduate studies at
Clemson University. My mentor at Clemson, Michael Sehorn, is passionate about science
(and college football), and his continued support and guidance throughout my scientific
training has been incredibly valuable. Additionally, my research rotations with Joel
Pomerantz and Kathryn Wagner were both fun and instructive; Joel and Kathryn are both
gifted scientists, and they have graciously advised me throughout my time at Hopkins.
Furthermore, I owe my gratitude to members of my thesis committee, whose time
and expertise were crucial to the development of my thesis project. Young Choi, Diane
Hayward, Kuan-Teh Jeang, and Gary Ketner were instrumental in the design and
execution of my research projects. Interactions with my thesis committee encouraged me
to ask the right research questions, to determine the best method for answering my
Page 5
v
research questions, and to think about my career after graduation. Their help and support
cannot be overstated.
The training environment within the Graduate Training Program in Cellular and
Molecular Medicine encourages collaboration and friendship from the very beginning. I
would like to thank all of my classmates for making graduate school enjoyable. Afternoon
coffee breaks with friends and fellow students were not only stress-relieving, but these
discussions often yielded new approaches for circumventing experimental failures. Thank
you, Adam Moyer, Laurene Cheung, Laura Gottschalk, Sara Sinclair, deMauri Mackie,
Emily Chang, Kate Laws, and Justin Glenn.
I also want to acknowledge and thank my family for their love and support
throughout this journey. I attribute my love of science to my parents, Shawn and Susan
Gaunt, who have spent their careers in the medical field. They have always encouraged
me and my siblings (Patrick, Benjamin, Christine, and Leah) to study hard, help others,
and explore new things.
Lastly, I could not have completed my PhD without the love, encouragement, and
support of my husband, Matthew Cousins. Having a spouse understand not only the time
commitment of research but also one that you can bounce scientific ideas off of is
incredibly helpful. Learning how to be parents to Mark may be our biggest challenge yet,
but I have no doubt that we will succeed together.
Page 6
vi
TABLE OF CONTENTS
ABSTRACT.................................................................................................................... II
ACKNOWLEDGEMENTS ............................................................................................. IV
TABLE OF CONTENTS ................................................................................................ VI
LIST OF ABBREVIATIONS ........................................................................................ VIII
LIST OF TABLES ....................................................................................................... XIII
LIST OF FIGURES ...................................................................................................... XIV
CHAPTER 1: INTRODUCTION ...................................................................................... 1
CLINICAL SIGNIFICANCE OF HUMAN HERPESVIRUS 8 ................................ 2
HUMAN HERPESVIRUSES ............................................................................... 5
HUMAN HERPESVIRUS GENOME ORGANIZATION ........................................ 7
HISTORY AND UNIQUE FEATURES OF HHV-8 ............................................... 8
HHV-8 LYTIC CYCLE ......................................................................................... 8
HHV-8 LATENCY ...............................................................................................11
VIRAL INTERLEUKIN 6 AND PATHOGENESIS ................................................13
VIRAL INTERLEUKIN 6 CHARACTERISTICS ...................................................16
SIGNIFICANCE OF VIRAL IL-6 AND THIS PROJECT ......................................19
TABLES .............................................................................................................20
FIGURES ...........................................................................................................22
CHAPTER 2: METHODS ..............................................................................................29
INTRODUCTION ...............................................................................................31
RNA EXTRACTION ...........................................................................................31
REVERSE TRANSCRIPTION ............................................................................31
POLYMERASE CHAIN REACTION ...................................................................32
AGAROSE GEL ELECTROPHORESIS .............................................................33
CLONING AND SHORT HAIRPIN RNA DESIGN ..............................................34
MINIPREP DNA EXTRACTIONS .......................................................................36
CELL CULTURE ................................................................................................36
TRANSFECTION OF CULTURED CELLS .........................................................37
WESTERN BLOT ANALYSIS ............................................................................38
IMMUNOFLOURESCENCE STAINING .............................................................39
BRDU INCORPORATION ..................................................................................39
ANNEXIN V-CY3 STAINING ..............................................................................40
LENTIVIRUS PRODUCTION .............................................................................41
LENTIVIRAL TRANSDUCTION OF CULTURED CELLS ...................................41
CELL GROWTH ASSAY ....................................................................................42
HHV-8 R219 GENERATION AND INFECTION OF ENDOTHELIAL CELLS ......42
TITERING OF REACTIVATED HHV-8 ...............................................................43
DEPLETION AND COMPLEMENTATION EXPERIMENTS ...............................44
Page 7
vii
CHAPTER 3: INVOLVEMENT OF GP130 IN VIL-6-MEDIATED PEL CELL GROWTH
AND SURVIVAL .......................................................................................................46
ROLE OF HUMAN HERPESVIRUS 8 INTERLEUKIN-6-ACTIVATED GP130 SIGNAL TRANSDUCER IN PRIMARY EFFUSION LYMPHOMA CELL GROWTH AND VIABILITY ............................................................................49
INTRODUCTION ....................................................................................49
METHODS .............................................................................................51
RESULTS ...............................................................................................55
DISCUSSION .........................................................................................61
FIGURES ...............................................................................................65
CHAPTER 4: ROLE OF GP130 IN VIL-6-MEDIATED VIRUS REPLICATION ..............80
HUMAN HERPESVIRUS 8 VIRAL INTERLEUKIN-6 SIGNALING THROUGH GP130 PROMOTES VIRUS REPLICATION IN PRIMARY EFFUSION LYMPHOMA AND ENDOTHELIAL CELLS....................................................82
INTRODUCTION ....................................................................................82
METHODS .............................................................................................83
RESULTS ...............................................................................................89
DISCUSSION .........................................................................................91
FIGURES ...............................................................................................93
CHAPTER 5: DISCUSSION AND PERSPECTIVES ................................................... 101
DISCUSSION OF MAJOR FINDINGS .................................................. 102
IMPLICATIONS FOR CLINICAL RESEARCH ...................................... 108
FUTURE DIRECTIONS ........................................................................ 110
FIGURES ............................................................................................. 111
REFERENCES ............................................................................................................ 115
CURRICULUM VITAE ................................................................................................. 151
Page 8
viii
LIST OF ABBREVIATIONS
AIDS acquired immunodeficiency syndrome
bFGF basic fibroblast growth factor
BrdU 5-bromo-2-deoxyuridine
BSA bovine serum albumin
CatD cathepsin D
cDNA complementary deoxyribonucleic acid
CTL cytotoxic T lymphocyte
DAPI 4',6-diamidino-2-phenylindole
DHFR dihydrofolate reductase
DMEM Dulbecco’s modified Eagle’s medium
DNA deoxyribonucleic acid
dNTP deoxynucleotide triphosphate
Dox doxycycline
DTT dithiothreitol
EBV Epstein-Barr virus (HHV-4)
EBM-2 endothelial growth basal media
EDTA ethylenediaminetetraacetic acid
ER endoplasmic reticulum
FBS fetal bovine serum
FLICE FADD-like interleukin-1 beta-converting enzyme
gB glycoprotein B
GC guanine-cytosine
GFP green fluorescent protein
GSK3β glycogen synthase kinase-3 beta
Page 9
ix
HAART highly active antiretroviral therapy
HCl hydrochloric acid
HCMV human cytomegalovirus (HHV-5)
HCV hepatitis C virus
MHC major histocompatibility complex
HHV-1 human herpesvirus 1 (HSV-1)
HHV-2 human herpesvirus 2 (HSV-2)
HHV-3 human herpesvirus 3 (VZV)
HHV-4 human herpesvirus 4 (EBV)
HHV-5 human herpesvirus 5 (HCMV)
HHV-6 human herpesvirus 6
HHV-7 human herpesvirus 7
HHV-8 human herpesvirus 8 (KSHV)
hIL-6 human interleukin 6
HIV human immunodeficiency virus type 1 (HIV-1)
HRP horseradish peroxidase
hrs hours
HSV-1 herpes simplex virus 1 (HHV-1)
HSV-2 herpes simplex virus 2 (HHV-2)
IFN interferon
IL-1 interleukin 1
IL-6 interleukin 6
IL6R interleukin 6 receptor (alpha subunit)
JAK Janus kinase
kb kilobase
KICS KSHV inflammatory cytokine syndrome
Page 10
x
KS Kaposi’s sarcoma
KSHV Kaposi’s sarcoma-associated herpesvirus (HHV-8)
LANA latency-associated nuclear antigen
LB Luria broth
MAPK mitogen-activated protein kinase
MCD multicentric Castleman’s disease
MCMV murine cytomegalovirus
MHC major histocompatibility complex
min minute
MIR modulator of immune recognition
miRNA micro ribonucleic acid
mRNA messenger ribonucleic acid
N asparagine
NaB sodium butyrate
NK natural killer
ORF open reading frame
OX-2 orexin receptor 2 (CD 200)
PAN RNA polyadenylated nuclear ribonucleic acid
PBS phosphate-buffered saline
PCR polymerase chain reaction
PDGF platelet-derived growth factor
PEL primary effusion lymphoma
PM plasma membrane
PPMO peptide-conjugated phosphorodiamidate morpholino oligomer
pRb retinoblastoma protein
PS phosphatidylserine
Page 11
xi
RNA ribonucleic acid
Rta replication and transcriptional activator
RT-PCR reverse transcription-polymerase chain reaction
ROS reactive oxygen species
SCID severe combined immunodeficiency
SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel electrophoresis
shRNA short hairpin ribonucleic acid
SOCS3 suppressor of cytokine signaling 3
STAT signal transducer and activator of transcription
STAT3D dominant negative signal transducer and activator of transcription
TAE Tris-acetate-EDTA
TIME telomerase-immortalized microvascular endothelial
TK thymidine kinase
TLR Toll-like receptor
TNFα tumor necrosis factor alpha
TPA 12-O-tetradecanoylphorbol-13-acetate
TR terminal repeat
TS thymidylate synthase
U unit
UTR untranslated region
UV ultraviolet
vBcl-2 viral B-cell lymphoma 2
vCCL-1 viral chemokine (C-C motif) ligand 1
vCCL-2 viral chemokine (C-C motif) ligand 2
vFLIP viral FLICE-inhibitory protein
vGPCR viral G-protein-coupled receptor
Page 12
xii
vIRF viral interferon regulatory factor
VKORC1v1 vitamin K epoxide reductase complex subunit 1 variant 1
VKORC1v2 vitamin K epoxide reductase complex subunit 1 variant 2
VKORC1v3 vitamin K epoxide reductase complex subunit 1 variant 3
VSV/G vesicular stomatitis virus G protein
vIL-6 viral interleukin 6
VZV varicella-zoster virus (HHV-3)
WB western blot
WT wild-type
XBP1 X-box binding protein 1 Y tyrosine
Page 13
xiii
LIST OF TABLES
Table 1.1. Descriptions of HHV-8-specific open reading frames (“K” ORFs) ..................20
Table 1.2. HHV-8 microRNAs ........................................................................................21
Page 14
xiv
LIST OF FIGURES
Figure 1.1. Genomic organization of HHV-8 ..................................................................22
Figure 1.2. vIL-6 and hIL-6 three-dimensional structures ...............................................24
Figure 1.3. Human and viral interleukin 6 signaling complexes ......................................25
Figure 1.4. Known and proposed functions of viral interleukin 6 ....................................27
Figure 3.1. Effects of gp130 depletion on PEL cell growth and viability ..........................65
Figure 3.2. Contribution of gp130 signaling to levels of active STATs and ERKs in PEL
cells ..........................................................................................................................67
Figure 3.3. Contributions of gp130-activated STATs to PEL cell growth ........................69
Figure 3.4. Analysis of culture growth, cell proliferation, and apoptosis rates as a function
of ERK depletion .......................................................................................................71
Figure 3.5. ER-localized vIL-6 signal transduction via gp130 supports PEL cell growth .74
Figure 3.6. Inhibitory targeting of vIL-6/gp130 signaling diminishes PEL cell proliferation
and survival ..............................................................................................................77
Figure 4.1. Involvement of gp130 and vIL-6 in promotion of HHV-8 replication ..............93
Figure 4.2. Importance of ER-localized vIL-6/gp130 interactions for HHV-8 replication .95
Figure 4.3. Contributions of gp130-mediated STAT and ERK signaling to HHV-8
productive replication ................................................................................................97
Figure 4.4. Role of STAT3 in HHV-8 replication in PEL and endothelial cells ................99
Figure 5.1. ER-localized vIL-6 and gp130 signaling in PEL cell growth and survival .... 111
Figure 5.2. Role of vIL-6 and gp130 in HHV-8 replication in PEL and endothelial cells 113
Page 15
1
CHAPTER 1: INTRODUCTION
Page 16
2
CLINICAL SIGNIFICANCE OF HUMAN HERPESVIRUS 8
Human herpesvirus 8 (HHV-8), also known as Kaposi’s sarcoma-associated
herpesvirus (KSHV), has been linked to three diseases in humans: Kaposi’s sarcoma
(KS), primary effusion lymphoma (PEL), and multicentric Castleman’s disease (MCD).
HHV-8 infection is required for the development of these neoplasias; however, disease
progression is rare in immunocompetent hosts. These diseases are most prevalent in
populations that are immunocompromised, such as those with acquired immunodeficiency
syndrome (AIDS) and those who are immunosuppressed due to organ transplant or
treatment of unrelated disease. It is believed that immunosuppression allows the virus to
replicate and for latently infected cell populations to expand in the absence of host-
immune pressures, leading to more rapid and aggressive disease progression. While KS,
PEL, and MCD share an etiologic cause in HHV-8, they differ in target cell types, clonality
of lesions, and host cell responses to viral infection.
KS is a distinctive condition that inspired one of the alternative names given to
HHV-8, KSHV. Four forms of KS have been described: classical, AIDS-associated,
endemic, and iatrogenic [1-8]. These classifications are based on geographical
prevalence, disease progression, and other factors. Classical KS was first described by
dermatologist Dr. Moritz Kaposi in 1872 [9]. This form of the disease is characterized by
cutaneous lesions of the skin, which are commonly observed in the extremities [9].
Classical KS is most common in elderly men of eastern European and Mediterranean
descent [10], but with the emergence of HIV/AIDS in Africa beginning in the 1980s, the
AIDS-associated form of KS became more prevalent in this geographical region [11]. The
prevalence of AIDS-associated KS has mirrored the prevalence of HIV infection in Africa,
and the advent of highly active antiretroviral therapy (HAART) has led to a rapid decline
in both HIV incidence and AIDS-associated KS [12]. AIDS-associated KS is the most
aggressive form of KS and has the highest prevalence of the four subtypes [11,13].
Page 17
3
Endemic KS is most commonly observed in African children and is characterized by high
mortality rates [14]. Finally, iatrogenic KS is observed in transplant recipients that have
undergone immunosuppressive therapy [15].
Nearly all KS lesions contain cells infected with HHV-8, though the fraction of virus-
positive cells within each lesion varies widely (partially dependent on the stage of disease)
[16,17]. HHV-8 infects endothelial cells, which subsequently become spindloid and
proliferate rapidly [17]. Spindle cells have a distinct morphology and a unique gene
expression profile; these cells express a mixture of blood and lymphatic endothelial cell
markers [18-20]. KS lesions are generally polyclonal in nature, though clonal KS has also
been observed [21,22]. Lesions are reddish, brown, or purple in color due to the induction
of angiogenesis and extravasation of erythrocytes from malformed microvasculature.
Cytokine dysregulation has been implicated as a cause of KS. Pro-inflammatory
factors promote the infiltration of immune cells into infected tissues. Cultured KS cells
require basic fibroblast growth factor (bFGF), interleukins 1 and 6 (IL-1, IL-6), and platelet-
derived growth factor (PDGF) for growth, and cells found in KS lesions express IL-6 and
PDGF receptors [23-27]. Furthermore, cells in KS lesions secrete IL-1β, tumor necrosis
factor alpha (TNFα), and IL-6 [23]. Virus infection drives this cytokine-rich environment,
which promotes virus replication and the infection of new host cells that secrete additional
cellular and viral cytokines, further exacerbating the infection.
HHV-8 is also the causative agent of PEL, a B-cell malignancy that involves the
pleural, peritoneal, and pericardial spaces [28,29]. Infected B-cells express both B-cell
and plasma cell markers (CD138, CD38, and CD23), suggesting that the HHV-8-infected
cells are in fact immature B-cells that were in the process of differentiating into plasma
cells when the virus promoted their proliferation and halted their differentiation [30-33].
PEL tumors are monoclonal, and each infected B-cell contains many (approximately 50)
copies of the viral genome maintained as circular episomes [34,35]. PEL is generally
Page 18
4
diffuse with no detectable mass, but solid tumors have also been reported [36]. Cases of
PEL are most commonly observed in HIV-positive patients or other immunocompromised
individuals; the median survival of patients with this aggressive cancer is only six months
[37]. The initiation of HAART prior to PEL diagnosis is thought to improve clinical prognosis
in patients co-infected with HIV [37].
Finally, HHV-8 is the etiologic agent of MCD, which is characterized by viral
infection of plasmablastic B-cells within the mantle zone of B-cell follicles [38]. Unlike PEL,
MCD is generally polyclonal, though cases of clonal MCD have been reported [39]. MCD
progression in HIV-infected individuals is rapid; the disease is driven by the elevated
expression of pro-inflammatory cytokines, including IL-6, IL-10, and vascular endothelial
growth factor (VEGF) [40-43]. Furthermore, levels of IL-6 in MCD patients were inversely
correlated with patient prognosis [40]. Recently, it was reported that six patients coinfected
with HIV and HHV-8 presented with pathologies and symptoms similar to MCD, but these
patients could not be classified as having MCD and did not develop MCD during follow-up
of 3-60 months [44]. These patients had elevated levels of HHV-8-encoded viral IL-6 (vIL-
6) (similar to levels observed in patients diagnosed with MCD) compared to patients with
mild or severe KS [44]. Furthermore, those with MCD-like inflammatory disease also had
significantly increased levels of serum human IL-6 (hIL-6), serum IL-10, and HHV-8 viral
load compared to control patients with KS [44]. These levels were comparable to serum
hIL-6 levels, IL-10 levels, and HHV-8 viral loads in MCD patients [44]. This MCD-like
disease has since been termed KSHV inflammatory cytokine syndrome (KICS) [45]. In an
AIDS patient presenting with KICS, circulating HHV-8 viral load was calculated as
5,300,000 copies per ml; this HHV-8 viral load is the highest value reported to date [46].
This patient also had high HHV-6A viremia, though the significance of HHV-6A infection
as it relates to KICS is unclear [46]. KICS is thought to arise from the dysregulation of pro-
inflammatory cytokines in the context of increased lytic replication of HHV-8 [45].
Page 19
5
HHV-8 causes neoplasia in the context of an immunocompromised host. This virus
has been linked to three malignancies that complicate the management of infectious and
induced immunodeficiencies. vIL-6 is believed to contribute to all three diseases via its
pro-proliferative, anti-apoptotic, pro-angiogenic, and pro-inflammatory activities. Though
HHV-8 is unique in its possession of vIL-6 and other virus-specific genes, this virus shares
many characteristics with other human herpesviruses.
HUMAN HERPESVIRUSES
Herpesviruses are ancient (approximately 180-220 million years old) [47].
Members of the family Herpesviridae can infect a variety of hosts, including humans, birds,
pigs, mice, elephants, and oysters [47]. Each of the herpesviruses has a linear, double-
stranded, deoxyribonucleic acid (DNA) genome, a lipid bilayer envelope surrounding the
tegument, and an icosahedral capsid architecture (triangulation T=16) [48]. Herpesvirus
genomes range in size from approximately 125-225 kilobases (kb) and contain 70-200
open reading frames (ORFs) [48]. Mature virions measure approximately 120-260
nanometers in diameter [49]. Of the greater than 100 identified members of the
herpesvirus family, eight infect humans [50]. The eight known human herpesviruses fall
into three subfamilies: Alphaherpesvirinae (α), Betaherpesvirinae (β), and
Gammaherpesvirinae (Ɣ) [51].
Alphaherpesviruses include herpes simplex virus 1 (HSV-1; also called HHV-1),
herpes simplex virus 2 (HSV-2; also called HHV-2), and varicella-zoster virus (VZV; also
called HHV-3). HSV-1 and HSV-2 are responsible for cold sores and genital warts,
respectively. During outbreaks, HSV-1 and HSV-2 are reactivated from host neuronal
cells, and virions are shed from the oral or genital mucosa. VZV is associated with both
chicken pox, a primary infection characterized by a vesicular skin rash, and shingles, a
reactivation of latent VZV from neural ganglia. The alphaherpesviruses are characterized
Page 20
6
by accelerated replication in culture, efficient cell lysis of infected host cells, and diverse
cell tropism compared to members of Betaherpesvirinae and Gammaherpesvirinae [48].
The betaherpesvirus subfamily includes human cytomegalovirus (HCMV; formally
HHV-5), human herpesvirus 6 (HHV-6), and human herpesvirus 7 (HHV-7). HCMV infects
cells of the salivary glands, causing characteristic cytomegaly [48]. HCMV has the largest
genome of the eight human herpesviruses at 235 kb and encodes more than 200 ORFs
[52]. HCMV infection is generally asymptomatic in immunocompetent individuals;
however, HCMV infection of immunocompromised individuals can result in pneumonia,
dyspepsia, meningitis, and pericardial effusions [53-56]. Though highly prevalent in
children, HHV-6 (95% seropositive by age 3) and HHV-7 (90% seropositive by age 5) have
minor clinical significance [57,58]. HHV-6 causes roseola infantum, and HHV-7 is often
associated with concurrent HHV-6 infection, though HHV-7 infection alone can also cause
roseola infantum [59-61]. Roseola infantum, also known as exanthema subitum, is
characterized by high fevers and a morbilliform rash that first appears on the trunk and
later on the face and extremities [62]. Viruses within the betaherpesvirinae subfamily
undergo slow replication and spread in culture and have asynchronous lytic cycles [48].
The gammaherpesviruses include Epstein-Barr virus (EBV; formally HHV-4) and
HHV-8 (also called KSHV). According to the 2013 release from the International
Committee on the Taxonomy of Viruses, EBV can be further classified as a member of the
lymphocryptovirus (gamma-1) genus while HHV-8 has been placed within the rhadinovirus
(gamma-2) genus. EBV is associated with infectious mononucleosis, Burkitt’s lymphoma,
Hodgkin’s lymphoma, and nasopharyngeal carcinoma. As noted previously, HHV-8 is the
causative agent of KS, MCD, and PEL [28,38,63]. Gammaherpesviruses exhibit latent
tropism for lymphocytes (with EBV and HHV-8 also infecting epithelial and endothelial
cells, respectively), have transforming capabilities, and are more likely to be oncogenic in
Page 21
7
immunocompromised hosts. HIV-positive individuals and those having received organ
transplants are particularly susceptible to EBV- and HHV-8-associated cancers [64].
HUMAN HERPESVIRUS GENOME ORGANIZATION
Though each of the human herpesviruses has a complement of unique genes,
these viruses also share sets of genes encoded in conserved gene blocks. Though the
relative orientations and genomic locations of the gene blocks differ between the
herpesvirus subfamilies (α, β, and Ɣ), the genes encoded within a specific gene block are
maintained in the same relative arrangement. Many of these conserved genes (>40) are
involved in viral entry, replication, virion packaging, virion maturation, and egress [65].
Enzymes involved in basic metabolism, including thymidine kinase (TK), ribonucleotide
reductase, and uracil-DNA glycosylase, are also encoded within the genomes of all human
herpesviruses. Many viral structural proteins that make up the virion capsid are also
conserved within the human herpesviruses; these capsid proteins include the major capsid
protein, small capsid protein, triplex monomer, portal protein, and portal capping protein
[65].
A number of the human herpesviruses encode “accessory” (non-core) cellular
gene homologs. Frequently, these genes are specific to one or a few members of
Herpesviridae. These genes encode proteins that function as chemokines (CXC
chemokines in HCMV, CC chemokines in HHV-8), cytokines (vIL-10 in EBV and vIL-6 in
HHV-8), and cell cycle regulators (vCyc in HHV-8) [66-73]. Several anti-apoptotic proteins
are also encoded by human herpesviruses (e.g., vBcl-2 in EBV and HHV-8 and viral
FLICE-inhibitory protein [vFLIP] in HHV-8) [74-78]. Additionally, microRNAs (miRNAs) are
encoded by several herpesviruses, including HSV-1, HSV-2, HCMV, HHV-6, EBV, and
HHV-8 [79].
Page 22
8
HISTORY AND UNIQUE FEATURES OF HHV-8
HHV-8, the eighth human herpesvirus to be identified, was discovered in 1994
using representational difference analysis [63]. HHV-8 DNA sequences were found in KS
lesions from an HIV-positive patient but not in adjacent unaffected tissue [63]. Shortly
thereafter, the virus was also causally linked to PEL and MCD [38,80].
HHV-8 encodes at least 90 ORFs within its genome of approximately 170 kb
(Figure 1.1) [34,63]. Guanine-cytosine (GC)-rich terminal repeat (TR) sequences are
present at both ends of the genome and comprise about 30 kb of genomic sequence [81].
The HHV-8 ORFs that are shared with herpesvirus saimiri (the first-sequenced gamma-2
herpesvirus) are designated ORF1 to ORF75 starting at the left end of the genome, and
the HHV-8-unique ORFs are designated K1-K15 and are located at particular regions of
gammaherpesvirus genome divergence (Table 1.1) [82]. The HHV-8-specific ORFs are
also numbered from left to right within the genome. In addition to these protein-encoding
ORFs, 12 microRNAs (miR-K12-1 to miR-K12-12) have recently been identified within the
HHV-8 genome (Tables 1.2). The majority (10) of these miRNAs are located between
ORF71 and K12, while the other two miRNAs are located within the 3’ untranslated region
(UTR) and coding sequence of K12 [83,84]. These miRNAs facilitate host immune system
evasion, cell cycle regulation, latency maintenance, inhibition of apoptosis, promotion of
angiogenesis, growth signaling, and chromatin modification [83,84].
HHV-8 LYTIC CYCLE
Immediately upon primary infection of a cell by HHV-8, a burst in lytic gene
expression is triggered and quickly aborted in favor of latency. Under normal conditions,
the virus remains latent until stimulated to reactivate. The mechanisms governing this
latent-to-lytic switch are unclear, though latently infected cultured cells can be
experimentally reactivated by the addition of phorbol esters or sodium butyrate (NaB)
Page 23
9
[35,85-87]. In addition to chemically-induced reactivation, the lytic cycle can be reactivated
by biological stressors, including interferon-Ɣ, toll-like receptor 7 and 8 (TLR7 and TLR8)
agonists, reactive oxygen species (ROS), and the ER stress-activated X-box binding
protein 1 (XBP1) transcription factor [88-91].
Following reactivation or primary infection, lytic genes are transcribed and
expressed. These lytic genes are transcribed in a specific order and are categorized as
immediate-early, delayed-early, and late genes [92]. Immediate-early genes do not require
protein synthesis for their expression, whereas delayed-early genes require protein
synthesis but not DNA replication. Late genes require DNA replication and the expression
of immediate-early and delayed-early gene products for expression. Nine viral genes have
been classified as being expressed very early after butyrate induction, and 27 have been
classified as late genes [93-95].
The major immediate-early gene product, Rta (replication and transcriptional
activator, encoded by ORF50), is required for lytic replication and the induction of
downstream genes (delayed-early and late genes). Rta alone is sufficient to initiate the
lytic cascade, and inhibition of Rta prohibits lytic reactivation [96-99]. Ectopic expression
of Rta in PEL cells leads to the transcription of viral lytic genes [100]. Rta contains both a
DNA-binding domain and a transactivation domain and is detected within 4 hours (hrs) of
n-butyrate induction [92,101]. The DNA-binding domain of Rta allows the viral protein to
bind to viral DNA promoter sequences and to recruit cellular transcription factors, such as
RBP-Jk, via its transactivation domain [101]. Rta can also induce genes lacking Rta-
binding cis sequences via association with promoter-bound transcription factors including
C/EBPα; C/EBPα and Rta cooperatively activate the Rta and PAN promoters [101]. Rta
functions as a tetramer and can be observed as decamers in solution [102]. Genes that
are transcribed soon after Rta expression include ORF45, K3, K5, polyadenylated nuclear
(PAN) ribonucleic acid (RNA), vIL-6, and viral chemokine (C-C motif) ligand 2 (vCCL-
Page 24
10
2/vMIP II); these genes can be detected within 13 hrs of n-butyrate induction [92,93].
Delayed-early genes include TK synthase, vCCL-1/vMIP I, viral G-protein-coupled
receptor (vGPCR), viral B-cell lymphoma 2 (vBcl-2), K12, and dihydrofolate reductase
(DHFR) [92]. The late genes are maximally expressed following viral DNA replication.
Lytic viral DNA replication occurs from two lytic origins (OriLyt-L and OriLyt-R) and
proceeds via a rolling circle mechanism [103]. Like other herpesviruses, HHV-8 encodes
a viral polymerase, polymerase processivity factor, primase, primase-associated factor,
helicase, and single-strand binding protein [82]. When these six gene products were co-
transfected into Vero cells, globular pseudo-replication complexes were observed in the
nucleus [104]. DNA replication is required for the expression of HHV-8 late genes such as
capsid proteins (e.g. ORF65) and other structural proteins found in the tegument [95].
Following expression of late genes, which occurs more than 30 hrs after n-butyrate
induction, viral capsid proteins are assembled, and viral DNA is loaded through the viral
portal protein [92]. Capsid assembly and loading occurs in the host cell nucleus,
necessitating a nuclear escape mechanism for efficient virion release. The nuclear egress
complex, which assists with virion egress from the nucleus, is comprised of the ORF67
and ORF69 gene products [105,106]. The process of virion envelopment and maturation
is complex, and HHV-8 encoded viral glycoprotein B (gB) plays an important role in this
process [107-109]. The virion gains a primary envelope and some tegument proteins as it
traverses the inner nuclear membrane [109]. The viral envelope then fuses with the outer
nuclear membrane as the virus moves across the perinuclear space; the virus particle
then deenvelopes, and the particle traverses the outer nuclear leaflet [109].
Reenvelopment and the acquisition of tegument proteins occur as the virus particle
traverses membranes of the trans-Golgi network [109]. Vesicles containing enveloped and
tegumented virus particles then fuse with the plasma membrane, releasing mature virions
[109]. Studies conducted using wild-type and gB-deleted HHV-8 bacmids demonstrated
Page 25
11
that gB is required for the complete maturation of HHV-8 virions; cells infected with gB-
deleted HHV-8 bacmid did not produce completely enveloped viral particles [107]. After
escaping the cell, progeny virus can infect naïve cells, and the original host cell undergoes
apoptosis.
HHV-8 LATENCY
Viral productive replication is required for the infection of naïve cells and the
expansion of the viral reservoir. However, most infected cells remain latently infected
following primary HHV-8 infection. In B-cells, the virus is nearly completely latent within
hrs of de novo infection, as only a small subset of cells continues to produce virions. In a
study of KS, approximately 3% of cells were found to be lytic, while the remaining infected
cells were latent [92]. The latent virus is maintained as a circular episome within the host
cell nucleus [34,80,110,111]. During latency, a small number of viral genes are expressed,
allowing the virus to evade host immune responses that target viral gene products. Latent
gene products include latency-associated nuclear antigen (LANA), vFLIP, vCyclin, viral
microRNAs, and kaposins A, B, and C [112]. These latency products are encoded in a
cluster within the viral genome known as the latency locus. Two promoters are responsible
for the expression of these latency genes, the LANA promoter (drives expression of LANA,
vCyclin, and vFLIP) and the kaposin promoter (regulates expression of the kaposins and
viral microRNAs) [113,114]. Recently, studies have demonstrated that vIL-6 is also
expressed at low levels by latently infected PEL cells; expression of viral interferon
regulatory factors (vIRFs) 1, 2, and 3 have also been reported in latently infected PEL
cells [115-119].
LANA is a crucial protein of latency that functions to tether the viral episome to the
host chromosome through its interactions with TR regions of the HHV-8 genome and host
cell histone H2A/H2B proteins [120-123]. This tethering allows the HHV-8 genome to be
Page 26
12
replicated during host cell DNA replication and ensures appropriate segregation of viral
genomes to daughter cell nuclei [120,124,125]. Plasmids containing TR regions can be
replicated in the presence of LANA but are not replicated in the absence of LANA,
indicating that LANA is required for episomal replication and maintenance
[123,124,126,127].
LANA expression regulates both host and viral gene transcription. Overexpression
of LANA results in the upregulation of DNA methyltransferase Dnmt3a, which is
responsible for methylating specific promoters, repressing the transcription of these
promoter-driven genes (e.g. cadherin 13) [128]. One study found 186 cellular genes that
were subject to greater than 2-fold positive or negative regulation by LANA; several of
these LANA-responsive genes are involved in the p53 and retinoblastoma protein (pRB)
pathways [129]. Moreover, LANA binds to and inactivates both the p53 and pRb tumor
suppressors, repressing apoptosis and driving E2F-dependent gene transcription
[130,131]. LANA also autoregulates its own transcription and the expression of Rta;
silencing of Rta by LANA inhibits lytic reactivation and promotes latency [132-134].
LANA achieves its pro-growth and survival functions by targeting multiple cellular
pathways in addition to p53 and pRb. LANA enhances levels of β-catenin via sequestration
of glycogen synthase kinase 3β (GSK-3β), ultimately leading to increased expression of
cyclin D and c-Myc [135]. Cellular Myc is an oncogene and a transcription factor
associated with growth, proliferation, and apoptosis [136]. LANA stabilizes c-Myc
expression by repressing the phosphorylation and ubiquitination of the transcription factor
[137,138]. Telomere length is also maintained by LANA, increasing the lifespan of virally-
infected cells [139]. Furthermore, LANA appears to be involved in the initial stages of
lymphogenesis. A transgenic mouse model expressing LANA driven by the LANA
promoter showed increased germinal center formation and an increased incidence of
Page 27
13
lymphoma [140]. Thus, LANA is critical for the maintenance and replication of HHV-8
episomes, the promotion of infected cell growth, and likely contributes to lymphogenesis.
VIRAL INTERLEUKIN 6 AND PATHOGENESIS
Prior to the discovery of HHV-8, elevated levels of IL-6 were observed in KS and
MCD patients, and disease severity was correlated with IL-6 levels [25,40]. Moreover, the
growth of cells derived from an HIV-positive KS patient could be suppressed (reduced by
~67%) by the inhibition of IL-6 via antisense oligomers, indicating potential IL-6
involvement in the pathogenesis of KS [25]. Soon after the discovery of HHV-8, one of the
15 unique ORFs of the virus was found to encode a homolog of interleukin 6 [67,69,141].
Cellular IL-6 is involved in many normal physiologic processes, including hematopoiesis,
inflammation, cell growth, and cell survival. Early studies demonstrated that the vIL-6
homolog could substitute for hIL-6 in some experimental systems. vIL-6 could prevent
murine plasmacytoma B9 cell apoptosis and promote B9 cell proliferation; B9 cells are IL-
6-dependent [67,69]. Furthermore, antibodies specific to the IL-6 receptor inhibited hIL-6-
and vIL-6-mediated growth of B9 cells in culture while control antibodies specific for IL-2
had no effect on cell growth [69]. hIL-6 is also known to induce the expression of acute-
phase proteins in hepatocytes [142]. Thus, cell culture-based methods were employed to
determine whether vIL-6 could similarly promote acute-phase protein production [69]. The
Hep3B hepatocyte cell line was transfected with plasmids encoding vIL-6, hIL-6, or control
sequences, and Northern blots were conducted using harvested RNA from the transfected
cells [69]. Both human and viral cytokines could induce the messenger RNA (mRNA)
expression of α1 acid glycoprotein, a prototypical acute-phase protein [69]. Thus, at the
time of its discovery, many surmised that vIL-6 might play a role in the development and
progression of HHV-8-associated disease [67-69].
Page 28
14
Following the discovery of vIL-6, clinical and other data were reported that
implicated vIL-6 as an important factor in the progression of KS, PEL, and MCD. Elevated
levels of serum vIL-6 were correlated with episodes of acute inflammation in MCD patients
[143]. Additionally, high levels of both hIL-6 and vIL-6 were observed in PEL tumors, and
these cytokines were required for PEL cell growth [115,144]. Depletion of vIL-6 via a short
hairpin RNA (shRNA)-encoding lentiviral vector and inhibition of vIL-6 using neutralizing
antibodies led to decreased PEL growth in culture [115,144]. The engraftment of PEL
tumors in severe combined immunodeficiency (SCID) mice could be inhibited by the
inhibition of vIL-6 via morpholino oligomers specific to vIL-6 [145]. In KS tumors, vIL-6 can
induce the expression of angiogenic factors (including VEGF), which are critical in the
development of KS [146]. In these studies, vIL-6-expressing NIH3T3 fibroblasts were
injected into nude mice, and these mice were compared to control mice that received
injections of NIH3T3 cells expressing control plasmid; vIL-6-expressing mice exhibited
increased hematopoiesis in specific lineages (myeloid, erythroid, and megakaryocytic),
greater vascularization, and polyclonal hypergammaglobulinemia [147]. In NIH3T3 cells
expressing vIL-6, VEGF expression could be detected by immunofluorescence staining;
in control cells, VEGF was not detected [147]. In a mouse model of peritoneal
inflammation, vIL-6 was found in enhance CCL-2 secretion and inhibit CXCL-8 expression,
ultimately blocking the recruitment of neutrophils to the site of inflammation [148]. Finally,
vIL-6 can also enhance the secretion of hIL-6 [149]. Plasmids encoding vIL-6 or the
reverse vIL-6 sequence were transfected into cells, and culture medium was collected for
use in experimental assays [149]. Collected culture medium was then added to several B
and T cell lines, and hIL-6 levels were assessed; the introduction of vIL-6 led to significant
increases (up to 30-fold) in hIL-6 expression in five cell lines (MT-4, THP-1, U937, Raji,
and CESS) [149]. Additionally, vIL-6-containing culture medium was capable of inducing
a 2-fold increase in hIL-6 secretion in cell lines derived from MCD patients [149].
Page 29
15
Other studies have further characterized the role of vIL-6 in HHV-8 pathogenesis.
vIL-6 is expressed maximally during lytic replication, though it is also expressed at low,
though functional, levels during PEL latency [115,116]. vIL-6 transcripts were identified via
limiting dilution reverse transcription-polymerase chain reaction (RT-PCR) and array-
based methods in HHV-8 latently infected SLK cells (KS-derived, HIV-) [116].
Immunofluorescence staining of HHV-8-positive PEL cells for vIL-6 indicates that the viral
cytokine is expressed during latency; vIL-6-specific immunofluorescence staining in PEL
cells was detected above background staining in co-mixed HHV-8-negative Akata cells
[115]. Depletion of vIL-6 via shRNA-mediated lentiviral transduction led to decreased
growth and survival of PEL cells in culture [115]. Following vIL-6 depletion, PEL cell growth
could be rescued by the addition of fully ER-retained, shRNA-resistant vIL-6 [115]. Similar
results were observed when vIL-6 was neutralized using an ER-directed vIL-6-specific
antibody (MAV) or peptide-conjugated phosphorodiamidate morpholino oligomers
(PPMO) directed to vIL-6 [150,151]. These studies are significant because they indicate
that vIL-6 plays an important role in cell growth and viability during latency when the
protein is expressed at much lower levels than during lytic reactivation.
vIL-6 is known to activate both mitogen-activated protein kinase (MAPK) and signal
transducer and activator of transcription (STAT) signaling (discussed in more detail
below), and high levels of active (phosphorylated) STAT3 have been observed in PEL
cells [152]. Several PEL cells lines, including BC-1, BCBL-1, and VG-1, express
constitutively phosphorylated STAT3 [152]. Furthermore, inactivation of STAT3 (via
treatment with JAK2 pharmacological inhibitor AG490 or the overexpression of dominant-
negative STAT3 [STAT3D]) led to decreased PEL cell growth, increased apoptosis, and
decreased expression of survivin, a protein that is required for PEL cell survival [152].
STAT3D contains a mutation within the DNA binding domain of STAT3; thus, STAT3D
can dimerize with wild-type STAT3 but cannot bind STAT3-responsive DNA elements to
Page 30
16
initiate transcription, effectively acting as a competitive inhibitor of wild-type STAT3
[152,153]. Survivin is an inhibitor of apoptosis (IAP) family member, and its inhibition has
been implicated in several cancers [154]. Forced overexpression of survivin in VG-1 PEL
cells was capable of rescuing AG490-mediated apoptosis and STAT3 inhibition,
confirming the importance of STAT3 and survivin for PEL cell viability [152]. Importantly,
STAT3 can also be activated by VEGF in bovine aortic endothelial cells, and VEGF is
upregulated by vIL-6 in other cell culture models [147,155]. Thus, vIL-6 can be linked to
pro-growth and survival as well as pro-angiogenic pathways, further implicating the viral
cytokine in HHV-8 malignant pathogenesis.
VIRAL INTERLEUKIN 6 CHARACTERISTICS
hIL-6 and vIL-6 share approximately 25% amino acid sequence identity and 63%
similarity [67-69]. While the primary sequences of the homologs are quite divergent, both
cytokines fold into similar three-dimensional structures consisting of 4-alpha helical
bundles (Figure 1.2) [156]. This structural similarity allows both cytokines to engage the
IL-6 α receptor (IL6R or gp80) and gp130 (β subunit) and initiate signaling, leading to the
induction of MAPK and STAT signaling (Figure 1.3) [156-160]. Phosphorylation of tyrosine
(Y) 759 (Y759) of gp130 results in MAPK signaling, while the phosphorylation of Y767,
Y804, Y905, or Y915 leads to the initiation of STAT signaling [156,161-163]. Ultimately,
hIL-6 and vIL-6 signaling lead to cell growth and survival, and both cytokines can sustain
IL-6-dependent B9 cells in culture [67,141]. While both proteins can initiate signaling via
interactions with gp80 and gp130, hIL-6 and vIL-6 have different requirements for signaling
induction. hIL-6-containing signaling complexes comprise two molecules each of hIL-6,
gp80, and the gp130 signal transducer (hIL-62-gp802-gp1302) [156-160]. vIL-6 can also
form identical hexameric complexes but is unique among IL-6 proteins in that it can signal
in the absence of gp80 via tetrameric complexes (vIL-62-gp1302) [158,164-166]. While
Page 31
17
gp80 is not required for vIL-6-initiated signaling, the presence of gp80 facilitates a more
sustained signaling event [159,167,168]. Hexameric (gp80-containing) signaling
complexes are found at the plasma membrane (PM), while (unique) vIL-6 signaling from
the ER occurs exclusively through tetrameric (gp80-devoid) complexes [115,169]. vIL-6-
containing tetrameric complexes may also form at the cell surface. Thus, hIL-6 can signal
at the cell surface, while vIL-6 can signal from the cell surface and intracellularly within the
ER. These distinct signaling mechanisms allow vIL-6 to signal in both a paracrine and
strictly autocrine (intracellular) manner.
In addition to differences in signaling complex composition, vIL-6 is mostly retained
within the ER compartment, while hIL-6, like other cellular cytokines, is efficiently secreted
from the cell [115,170,171]. Secretion kinetics indicate that vIL-6 is secreted approximately
8-fold slower than hIL-6 [170]. Within the ER, vIL-6 interacts with calnexin, which is
involved in proper folding of the cytokine [171]. Depletion of calnexin led to reduced levels
of intracellular vIL-6 but did not alter levels of secreted vIL-6; vIL-6 levels could be rescued
by overexpression of gp130 in calnexin-depleted cells, but the underlying mechanism is
unclear [171]. Furthermore, vIL-6 contains two potential asparagine glycosylation sites
(asparagines [N] 78 and 89), and secreted vIL-6 is completely glycosylated [170,172].
Glycosylation of vIL-6 N89 is required for the induction of vIL-6-initiated signaling but not
for its interaction with calnexin, for which N78 glycosylation is sufficient [171,172]. Thus,
posttranslational modification differences between the human and viral cytokines may
partially explain differences in secretion kinetics and interactions with ER-resident
proteins.
vIL-6, but not hIL-6, was found to bind directly to a previously uncharacterized
protein called vitamin K epoxide reductase complex subunit 1 variant 2 (VKORC1v2) in
addition to calnexin [173]. The major isoform, VKORC1v1 (vitamin K epoxide reductase
complex subunit 1 variant 1), plays a role in the vitamin K cycle and is the target of the
Page 32
18
anticoagulant warfarin [174]. Evidence demonstrating that VKORC1v2 interacts with vIL-
6 was the first reported function of VKORC1v2. VKORC1 variants 1 and 2 share the first
58 amino acids, and splicing gives rise to variant 2 [173]. Only VKORC1v2 interacts with
vIL-6, though the interaction domain (vIL-6 binding domain [vBD], residues 31-39) of
VKORC1v2 is shared with VKORC1v1 [173]. The difference in vIL-6-binding between the
two variants can be explained by the opposing orientations of the two proteins in the ER
membrane. Both VKORC1 variants contain a transmembrane domain between amino
acids 11 to 29 [173]. For VKORC1v1, the N-terminus of the protein is located in the ER
lumen, and amino acids 31-39 (vBD) are located in the cytoplasm [173]. Conversely, the
N-terminus of VKORC1v2 is located in the cytoplasm, and amino acids 31-39 are located
within the ER lumen and are accessible to vIL-6 [173]. A third VKORC1 variant,
VKORC1v3, is similar to VKORC1v1 but has a deletion of amino acids 58-73 due to
alternative splicing, and VKORC1v3 is also incapable of binding to vIL-6 [175]. Studies
have shown that depletion of VKORC1v2, but not VKORC1v1, leads to reduced rates of
PEL cell growth and increased rates of apoptosis in these cells, identifying the importance
of VKORC1v2 for PEL cell viability [173]. Growth of VKORC1v2-depleted cells could be
rescued by the addition of shRNA-resistant wild-type but not vBD-mutated VKORC1v2
[173]. Similarly, overexpression of vBD led to reduced growth and increased apoptosis in
PEL cultures, presumably due to disruption of the vIL-6/VKORC1v2 interaction [173].
These findings indicate the importance of the vIL-6/VKORC1v2 interaction for PEL cell
viability.
The interaction between vIL-6 and VKORC1v2 can enhance VKORC1v2
interactions with the proenzyme form of cathepsin D (CatD) [175]. CatD, in its mature,
lysosomally localized form, is an aspartate protease, which can be released into the
cytoplasm in response to certain stress signals [176]. Depletion of vIL-6 results in
increased levels of ER-localized/transiting pro-CatD and mature CatD, leading to
Page 33
19
decreased PEL cell viability in culture [175]. Introduction of vIL-6/VKORC1v2-disrupting
vBD also led to increased levels of CatD and reduced PEL cell viability [175]. PEL cell
apoptosis could be induced with the overexpression of CatD, suggesting that vIL-6 may
suppress CatD function to maintain cell viability [175]. PEL cell viability is critical for
efficient virus replication, and depletion of vIL-6 or introduction of vIL-6/VKORC1v2-
disrupting vBD in lytically reactivated BCBL-1 and JSC-1 PEL cells led to increased levels
of CatD and decreased HHV-8 viral titers [175]. In contrast, depletion of CatD in these
PEL cell lines led to increased viral titers [175]. These results suggest a role for vIL-6 in
HHV-8 replication via its interactions with VKORC1v2 and regulation of CatD.
SIGNIFICANCE OF VIRAL IL-6 AND THIS PROJECT
In order to develop therapeutic agents for the treatment of HHV-8-associated
disease, it will be crucial to develop a better understanding of the mechanisms governing
vIL-6-mediated pathogenesis. Several research questions must be addressed before
beginning the process of selecting therapeutic targets. First, the role of ER-localized vIL-
6/gp130 interactions must be investigated. vIL-6/gp130-mediated signaling likely activates
STAT and MAPK pathways, though this has not been shown in the context of PEL.
Furthermore, the mechanism(s) responsible for elevated levels of active STAT3 in PEL
tumors should be elucidated. In addition to its proposed roles in HHV-8 latency, functions
of the vIL-6/gp130 interaction in the context of virus replication are unclear. The focus of
this dissertation project was to functionally characterize ER-localized vIL-6/gp130
interactions in the context of both viral latency and virus productive replication.
Page 34
20
TABLES
Table 1.1. Descriptions of HHV-8-specific open reading frames (“K” ORFs).
ORF Encoded Protein Protein Function Reference
K1 VIPa activation of NFĸB; pro-angiogenic; downregulation of B-cell
receptor; cytokine induction
[177-182]
K2 vIL-6 pro-growth; pro-survival; pro-angiogenic; pro-virus replication [67-69,115,152,169,175,183]
K3 K3 protein downregulation of MHCb class I; NKc cell and CTLd evasion [94,184-186]
K4 vCCL-2 pro-angiogenic; pro-survival; pro-virus replication; Th2
polarization
[187-193]
K4.1 vCCL-3 pro-angiogenic; Th2 polarization [193,194]
K5 K5 protein downregulation of MHC class I; NK and CTL evasion [94,184-186,195]
K6 vCCL-1 pro-angiogenic; pro-survival; pro-virus replication; Th2
polarization
[187,196]
K7 vIAPe pro-survival [197-199]
K8 K8.1A/K8.1B target cell recognition; heparin binding [200,201]
K9 vIRF1 pro-survival; inhibition of interferon signaling [202-211]
K10 vIRF4 p53 destabilization [202,212,213]
K10.5 vIRF3 pro-angiogenic; pro-survival; inhibition of IFN signaling [117,202,214-217]
K11 vIRF2 inhibition of interferon signaling [119,202,218]
K12 kaposin A, B, & C prevention of cytokine mRNA decay; activation of MAPK
signaling; PROX1 stabilization
[219-223]
K13 vFLIPf anti-apoptotic via inhibiting caspase-8 activation; pro-survival
via NFĸB activation
[78,224-226]
K14 K14 cell surface protein
interacts with CD200 receptor; downregulation of TNFα and myeloid cell production
[227]
K15 K15 membrane protein
Pro-growth; pro-survival; pro-angiogenic
[228-231]
a Variable immunoreceptor tyrosine-activation motif (ITAM) protein; b major histocompatibility complex; c natural killer; d cytotoxic T lymphocyte; e viral inhibitor of apoptosis; f viral FADD-like interleukin-1 beta-converting enzyme (FLICE) inhibitory protein
Page 35
21
Table 1.2. HHV-8 microRNAs.
microRNA Targets Function Reference
miR-K12-1 p21, caspase 3, THBS1, IkBα
inhibition of cell-cycle arrest; anti-apoptotic; pro-angiogenic; suppression of viral replication; induction of MAPK signaling [232-235]
miR-K12-2 unknown upregulated in KS lesions [236]
miR-K12-3 caspase 3, NFIB, THBS1 anti-apoptotic; pro-latency; pro-angiogenic [234,235,237]
miR-K12-4 caspase 3, Rbl2 anti-apoptotic; epigenetic modifications [234,238]
miR-K12-5 BCLAF1, Rta, MyD88 anti-apoptotic; pro-latency; suppression of inflammatory cytokine production [238-240]
miR-K12-6 MAF, THBS1 cell fate reprogramming; pro-angiogenic [20,235]
miR-K12-7 Rta, MICB pro-latency; NK evasion [241,242]
miR-K12-8 unknown upregulated in KS lesions [236]
miR-K12-9 BCLAF1, Rta, IRAK1 anti-apoptotic; pro-latency; decreased inflammatory cytokine production [239,240,243]
miR-K12-10 TWEAKR, BCLAF1 repression of caspase activation; anti-apoptotic [239,244]
miR-K12-11 BACH1, SMAD5, MAF, THBS1
pro-survival; suppression of TGF-β signaling; cell fate reprogramming; pro-angiogenic; induction of MAPK signaling
[20,235,245-248]
miR-K12-12 unknown upregulated during lytic reactivation [249,250]
Page 36
22
FIGURES
Figure 1.1. Genomic organization of HHV-8. Gammaherpesvirus-conserved “core” gene
blocks are shown in blue. Divergent loci (DL-A through DL-D and IRF locus) outside of the
conserved blocks are labelled. Signaling membrane receptors (pink), cytokines (blue),
interferon regulatory factors (orange), and latency genes (green) are highlighted. The K1,
vOX2/vCD200 (K14), and K15 receptors, the viral cytokines, and the viral IRFs have thus
far been identified only in HHV-8 and closely-related macaque rhadinoviruses [251-253].
K7 (survivin) and K12 (kaposin) are unique to HHV-8. Latency genes encoding LANA,
vCyclin, and vFLIP are present in and characteristic of other gamma-2 herpesviruses. .
Abbreviations: see Table 1.1; modulator of immune recognition 2 (MIR2), thymidylate
synthase (TS), modulator of immune recognition 1 (MIR1), viral B-cell lymphoma 2 (vBcl-
2), viral chemokine ligand (vCCL), orexin receptor 2 (OX-2).
Page 38
24
Figure 1.2. vIL-6 and hIL-6 three-dimensional structures. hIL-6 and vIL-6 are composed
of four alpha helices (A-D) and fold into similar three-dimensional structures. This
structural similarity allows both cytokines to initiate signaling through complexes
containing the IL-6α receptor and gp130 signal transducer.
Figure 1.2
The above figure has been published previously and is reproduced here with permission from the American Society for Microbiology (Copyright © American Society for Microbiology, [Journal of Virology, 2012, 86(3):1577-1588, doi: 10.1128/JVI.05782-11]). Chen D, Cousins E, Sandford G, and Nicholas J (2012). Human herpesvirus 8 viral interleukin-6 interacts with splice variant 2 of vitamin K epoxide reductase complex subunit 1. 86(3):1577-1588.
Page 39
25
Figure 1.3. Human and viral interleukin 6 signaling complexes. Human IL-6 (hIL-6) initiates
signaling through hexameric signaling complexes containing two molecules each of hIL-
6, gp80, and gp130; these complexes form exclusively at the plasma membrane. Viral IL-
6 (vIL-6) is capable of signaling via identical hexameric complexes, though vIL-6 is unique
in its ability to signal via tetrameric complexes (vIL-62-gp1302) within the endoplasmic
reticulum (ER). Following complex formation, tyrosine (Y) residues within the cytoplasmic
tail of gp130 are phosphorylated. Depending on the specific tyrosine phosphorylation
event, signal transducer and activator of transcription (STAT) 1 or 3 or mitogen-activated
protein kinase (MAPK) signaling cascades are activated downstream of gp130.
Page 41
27
Figure 1.4. Known and proposed functions of viral interleukin 6. Viral interleukin 6 (vIL-6)
is a multifunctional viral protein and is active during both the latent and lytic phases of the
virus life cycle. During latency in PEL cells, vIL-6 acts as an autocrine factor to promote
cell growth and survival; it may perform a similar role in other cell types. During the lytic
phase, vIL-6 is thought to promote pathogenically significant angiogenesis and
inflammation through induction of cellular factors, including VEGF and hIL-6. vIL-6 inhibits
neutrophil recruitment via downregulation of CCL-2. These activities are believed to
promote virus replication in vivo. The hypotheses that vIL-6 autocrine signaling via gp130
can contribute to latently infected cell viability and to productive replication form the basis
of the thesis project.
Page 43
29
CHAPTER 2: METHODS
Page 44
30
Summary
Many laboratory techniques were utilized to conduct the studies described in this
dissertation. Detailed methods are provided in the chapters describing the studies in
which they were used.
Page 45
31
INTRODUCTION
In order to answer the research questions posed in this body of work, many
techniques were employed. Several of the techniques have been well-described by others,
but other protocols were specifically developed in the course of this project. Classic and
novel techniques are described in general terms below; more detailed methods are
provided in the relevant chapters that follow.
RNA EXTRACTION
HHV-8 and cellular mRNA were extracted from cell cultures in order to quantify
gene expression levels. To extract RNA, cells were washed with phosphate-buffered
saline (PBS) and suspended in TRizol (Thermo Fisher Scientific, Inc., Waltham, MA).
Following a 3 minute (min) incubation in TRizol, chloroform was added to the mixture, and
the samples were vortexed briefly and incubated for 10 min at room temperature. Samples
were then centrifuged in a microcentrifuge (10 min at maximum speed at room
temperature), and the top layer was removed and placed in a clean Eppendorf tube.
Isopropanol was added to precipitate RNA, and samples were centrifuged in a
microcentrifuge (10 min at maximum speed at 4°C) to pellet the RNA. Finally, RNA pellets
were washed with 70% ethanol and resuspended in RNase- and DNase-free water.
Concentrations of RNA samples were determined via spectrophotometer.
REVERSE TRANSCRIPTION
After viral and cellular RNA were extracted from cultured cells, the RNA was
reverse-transcribed to complementary deoxyribonucleic acid (cDNA) to allow amplification
of a gene of interest. The reverse-transcription reaction requires a pure RNA template
devoid of contaminating DNA molecules. As a result, the extracted RNA was treated with
DNase I (45 min incubation at room temperature) to destroy contaminating DNA
Page 46
32
molecules; ethylenediaminetetraacetic acid (EDTA) was subsequently used to inactivate
the DNase I enzyme by chelating magnesium and calcium required for enzyme activity.
Random hexamer primers and deoxynucleotide triphosphates (dNTPs) were added to
DNase-treated samples. Samples were then heated (65°C for 5 min) and cooled on ice (5
min); this process facilitated hexamer primer annealing. Finally, dithiothreitol (DTT;
Invitrogen, Carlsbad, CA), reverse transcriptase buffer (5X First Strand Buffer; Invitrogen),
RNase inhibitor (RNaseOUT; Invitrogen), and reverse transcriptase (SuperScript II RT;
Invitrogen) were added to the samples. RNA samples were then reverse-transcribed into
cDNA by incubating at 25°C for 10 min, 42°C for 50 min, and 70°C for 15 min. cDNA
samples were stored at -20°C until further processing.
POLYMERASE CHAIN REACTION
Polymerase chain reaction (PCR) was used to amplify a DNA template in order to
create vast numbers of copies of a given amplicon. PCR products are more stable than
cDNA derived from reverse-transcription and can be further analyzed or manipulated.
PCR reactions included a DNA template, dNTPs, forward and reverse primers,
polymerase buffer, and DNA polymerase. PCR amplification times depended on the length
of the PCR product and the polymerase used. Generally, PCR reactions included
approximately 50 nanograms of DNA template, 4 μl of dNTPs (2.5 mM), 1 μl each of
forward and reverse primers (10 μM each), 1X polymerase buffer, 1.5 μl of MgCl2 (50 mM),
and 1 unit (U) of Taq polymerase isolated from Thermus aquaticus (Invitrogen) in 1X
polymerase buffer. For cloning purposes, 2.5 U of PFU Ultra High-Fidelity polymerase
(Stratagene, La Jolla, CA) was used, and MgCl2 was not added. PFU Ultra High-Fidelity
polymerase is less error-prone than Taq polymerase and is therefore more suitable for
cloning.
Page 47
33
Amplification using Taq polymerase was conducted as follows: 94°C for 2 min
(denaturing); 25-35 cycles of 94°C for 30 sec (denaturing), 55°C for 30 sec (annealing),
and 72°C for 1 min per kb of DNA template (extension); and a final extension of 72°C for
10 min. Samples were then cooled to 4°C and stored at -20°C until they were used for
further analysis. A similar protocol was employed for reactions containing PFU Ultra High-
Fidelity enzyme: 95°C for 2 min; 25-35 cycles of 95°C for 30 sec, 50-55°C for 30 sec, 72°C
for 1 min per kb of template DNA (for vector templates <10 kb); followed by a final
extension of 72°C for 10 min. For vector templates greater than 10 kb, an extension time
of 2 min per kb and an extension temperature of 68°C were used. PCR amplification was
confirmed and visualized by agarose gel electrophoresis (described below).
AGAROSE GEL ELECTROPHORESIS
PCR amplification was verified by running PCR products on an agarose gel with
an appropriate DNA size marker. PCR samples were mixed with DNA loading buffer (40%
sucrose and 0.25% bromophenol blue) and loaded into agarose gel lanes. DNA size
marker was loaded into a gel lane as a reference to determine the size of PCR products.
As DNA is negatively charged, PCR products migrated from the negatively charged pole
toward the positively charged pole through the gel matrix. Ethidium bromide was included
in the agarose gel and intercalated into the DNA duplexes, allowing for the detection of
labeled DNA fragments by ultraviolet (UV) light. Smaller DNA fragments migrated through
the gel more rapidly than larger DNA fragments; thus, DNA was separated based on size.
While agarose gel electrophoresis was used to determine PCR product size,
relative abundance was also assessed in some instances. For example, RT-PCR was
often used to determine whether a specific shRNA adequately repressed its target gene
expression. In this case, RT-PCR-derived DNA samples from control and shRNA-treated
cells were compared on an agarose gel. Amplified target gene DNA should be less
Page 48
34
abundant than amplified target gene DNA from control cells if the shRNA used was
effective. Thus, target gene expression was semi-quantified by agarose gel
electrophoresis.
CLONING AND SHORT HAIRPIN RNA DESIGN
Several plasmid constructions were prepared in order to address questions related
to vIL-6 functions in cell growth, apoptosis, and virus replication. Some genes were cloned
with tags (e.g. FLAG) to facilitate detection, while others were cloned with localization
sequences (e.g. KDEL motif) to restrict the gene product to specific cellular locations.
Gene segments were also cloned (e.g. soluble gp130 [sgp130]) and introduced into cells
to inhibit the interaction between vIL-6 and gp130. Additionally, shRNAs were cloned into
lentiviral vectors to deplete endogenous cellular or viral proteins in cultured cells. Finally,
shRNA-resistant genes were cloned into lentiviral vectors for use in depletion-
complementation experiments. Specific details regarding the primers, restriction enzyme
sites, and vectors utilized for these constructions are provided in the relevant chapters.
In general, genes were amplified from various vector templates using PFU Ultra
High-Fidelity polymerase and then purified. In brief, PCR products were precipitated from
PCR reaction mixtures with sodium acetate and ethanol to remove primers, excess
dNTPs, polymerase, and reaction buffer components. Purified PCR products were then
digested with restriction enzymes to create appropriate 5’ and 3’ overhangs (or left
undigested in the event of a blunt-ended ligation). Restriction enzyme-digested PCR
products and vectors were then separated on an agarose gel, and the appropriate DNA
bands were excised from the gel under the guidance of UV light. DNA was purified from
the agarose gel using a DNA gel extraction kit (QIAquick Gel Extraction Kit; Qiagen,
Valencia, CA). Gel-purified DNA vector and PCR product insert were then ligated using
Page 49
35
T4 DNA ligase (New England Biolabs; Ipswitch, MA) for 1 hr at room temperature or
overnight at 16°C.
Ligated vector-insert constructions were transformed into competent DH5α E. coli
cells via a chemical transformation technique. Briefly, ligated DNA constructions were
incubated with competent bacterial cells on ice for 30 min prior to heat shocking (42°C)
for 30-60 seconds. Ligation product-bacterial mixtures were then placed on ice briefly and
plated on Luria broth (LB) agar plates with appropriate antibiotics for selection of
transformed bacteria. Depending on the plasmid used in the transformation, agar plates
were incubated at 30°C or 37°C overnight to allow for the growth of transformed bacteria.
Individual bacterial colonies were picked from plates, amplified in liquid LB, and
miniprepped to extract bacterial DNA. Colony-derived DNA was used for PCR-based or
restriction enzyme-based techniques to validate cloning constructions. Bacterial-derived
DNA was PCR amplified using construct-specific primers to verify the presence of the
DNA insert, DNA signal sequence, or sequence tag. Restriction enzyme digestion was
also used to verify the insertion of restriction enzyme-containing DNA inserts, signal
sequences, or tags. Prior to use in experiments, all constructions were commercially
sequenced to verify the modified DNA sequence.
Several experiments utilized shRNA-encoded lentiviral vectors to deplete
endogenous viral or host proteins. Proteins were targeted by 19 nucleotide sequences
embedded in a characteristic hairpin structure. Targeting sequences were designed with
the aid of iRNAi (version 2.1; Nucleobytes, The Netherlands). Complementary
oligonucleotides encoding candidate target sequences were annealed, phosphorylated,
and ligated into the pYNC352 lentiviral vector between the BamHI and MluI restriction
sites. shRNA-encoded lentiviral vectors were then sequenced. To assess depletion
efficiency of specific shRNA-encoded lentiviral vectors, vectors were co-transfected into
293T cultured cells with overexpressed target protein. Twenty-four hrs post-transfection,
Page 50
36
cell lysates were harvested and subjected to western blot analysis to determine protein
levels in control and shRNA-overexpressing cells (described below). In some cases, target
mRNA depletion was assessed via RT-PCR following shRNA-encoding lentiviral
overexpression (described above).
MINIPREP DNA EXTRACTIONS
Transformed bacteria were amplified in liquid LB overnight at 30°C or 37°C
depending on the transformed plasmid and harvested for the extraction of plasmid DNA.
Bacterial cells were pelleted via centrifugation, and cell pellets were resuspended in STET
buffer (8% sucrose, 5% Triton X-100, and 50 mM EDTA, Tris [pH 8.0]). Samples were
vortexed prior to the addition of lysozyme (100 μg/ml) and then boiled for 1 min and
centrifuged at maximum speed for 10 min at room temperature. Supernatants were
transferred to clean tubes, and an equal volume of isopropanol was added to each tube
to precipitate DNA. Tubes containing sample mixtures were chilled at -20°C for 10 min
and then centrifuged at maximum speed for 10 min at 4°C to pellet DNA. DNA pellets were
washed in 70% ethanol and finally resuspended in DNase-free water containing RNase A
to eliminate contaminating RNA.
CELL CULTURE
HEK293T cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM)
supplemented with 10% heat-inactivated fetal bovine serum (FBS). BCBL-1,
TRExBCBL1-RTA, JSC-1, BJAB, BC-1, and BC-3 cells were maintained in RPMI 1640
media supplemented with 10% FBS. Telomerase-immortalized microvascular endothelial
(TIME) cells and TIME-TRE/RTA cells were grown in endothelial growth basal media
(EBM-2) containing FBS and supplements (Lonza; Walkersville, MD). All cells were grown
in a 5% CO2 humidified chamber maintained at 37°C.
Page 51
37
TRANSFECTION OF CULTURED CELLS
Transfection of cultured HEK293T cells was utilized for functional experiments
assessing viral and cellular protein-mediated signaling, shRNA depletion efficiency,
protein-protein interactions, and lentivirus production. Transfections of HEK293T cells
were conducted using standard calcium-phosphate protocols. Briefly, HEK293T cells were
plated such that they would achieve 50% confluence at the time of transfection. DNA
plasmids were then mixed with calcium phosphate (2.5 M) and 2X HEPES-buffered saline
(HBS; 12 mM dextrose, 50 mM HEPES, 10 mM KCl, 280 mM NaCl, and 1.5 mM
Na2HPO4·2H2O, pH 7.05), vortexed, incubated at room temperature for 5 min, and added
to HEK293T cell culture media. Following 8 hr of incubation, culture medium was replaced
with fresh DMEM containing 10% FBS. Transfection efficiency was determined for
plasmids encoding green fluorescent protein (GFP) by visualizing GFP fluorescence. To
assess the transfection efficiency of plasmids not encoding fluorescent markers, a subset
of transfected cells were fixed and stained for transfected proteins via
immunohistochemical methods (described below).
Transfected cells were harvested 24-48 hr post-transfection for either RNA or cell
lysates. RNA was harvested using the TRizol method described above. For preparation
of cell lysates, cells were washed with PBS, scraped from the surface of the well or flask,
and incubated in lysis buffer (20 mM Tris [pH 8.0], 5 mM EDTA, 50 mM NaCl, 0.2% NP40,
and 1X protease and phosphatase inhibitors [Sigma; St. Louis, MO]) for 30 min on ice.
Samples were then centrifuged (maximum speed for 5 min at 4°C in a microcentrifuge) to
pellet cellular debris, and supernatants (lysates) were collected and stored at -20°C until
western blot analysis.
Page 52
38
WESTERN BLOT ANALYSIS
Western blot analysis was conducted to observe signaling events in transfected
HEK293T cells or transduced PEL cell cultures. Western blot analysis was also performed
to determine the efficiency of shRNA-mediated depletion in HEK293T, PEL, and TIME cell
cultures. Preparation of cell lysates used for western blot analysis was described above.
Specific antibodies and product numbers are provided in the methods section of the
relevant chapters.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was
performed to separate lysate proteins via electrophoresis. Similar to agarose gel
electrophoresis, proteins were separated based on size. Smaller proteins migrated
through the gel matrix more rapidly than larger proteins. Depending on the size of the
target protein, 8-12% polyacrylamide gels were used. Smaller target proteins were more
efficiently separated on higher percentage (e.g. 12%) polyacrylamide gels, whereas larger
proteins were better separated on lower percentage (e.g. 8%) gels. SDS loading buffer
(188 mM Tris-HCl [pH 6.8], 3% SDS, 30% glycerol, 0.01% bromophenol blue, and 15% ß-
mercaptoethanol) was added to each protein sample prior to denaturation via boiling (5
min), and a protein size marker was loaded onto the gel as a reference. Following gel
electrophoresis, proteins were electrophoretically transferred onto nitrocellulose
membranes using standard protocols. Membranes were then washed in TBS-T (50 mM
Tris-HCl [pH 7.4], 150 mM NaCl, and 0.1% Tween 20) and blocked with 5% nonfat milk in
TBS-T for 1 hr at room temperature. After briefly washing blocked membranes in TBS-T,
primary antibodies (diluted in SuperBlock Blocking Buffer in TBS; Thermo Scientific,
Rockford, IL) were incubated with membranes for 1 hr at room temperature or overnight
at 4°C. After primary antibody incubation, membranes were washed three times with TBS-
T and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody
(diluted in 5% non-fat milk in TBS-T) for 1 hr at room temperature. Membranes were
Page 53
39
continuously rocked during both antibody incubations. Again, membranes were washed
three times in TBS-T and then developed using standard chemiluminescence procedures
with ECL reagents (GE Healthcare Bio-Sciences; Pittsburgh, PA). Images were captured
digitally on a GeneGnomeXRQ machine (Syngene; Frederick, MD).
IMMUNOFLOURESCENCE STAINING
Immunofluorescence staining was performed to determine transfected or
transduced protein expression, to visualize cellular localization of target proteins, and to
calculate the percentage of apoptotic cells, proliferating cells, and LANA-positive cells. To
prepare cells for immunofluorescence staining, cells were washed in PBS, plated on glass
slides, and allowed to air dry. Once dry, slides were fixed in ice cold methanol for 10 min.
Slides were then allowed to air dry completely.
Following fixation, primary antibody (diluted 1:100 in SuperBlock Blocking Buffer
in PBS; Thermo Scientific) was added to slides and incubated in a humidified chamber at
37°C for 1 hr. Primary antibody was then removed, and the slide was washed three times
with PBS. Secondary antibody (diluted 1:200 in SuperBlock Blocking Buffer in PBS) was
then applied to the slide, and the slide was incubated for 1 hr as before. After washing
three times with PBS, nuclei were stained with 4’,6’-diamidino-2-phenylindole (DAPI).
Finally, coverslips were mounted on slides prior to visualization on a UV microscope.
BRDU INCORPORATION
To determine the percentage of dividing (S-phase) cells, 5-bromo-2-deoxyuridine
(BrdU) incorporation was assessed. BrdU is a thymidine analog and is incorporated into
replicating DNA; thus, BrdU incorporation occurs exclusively in actively replicating cells.
BrdU (0.01 M diluted in culture media, 1000X) was added to cell cultures, and the
cultures were incubated for 2 hr at 37°C; cells were then washed in PBS, spotted on glass
Page 54
40
slides, and allowed to air dry. Slides were fixed in methanol as described above and
washed in PBS. Cells were then permeabilized with 2M hydrochloric acid (HCl) for 20 min
at room temperature, and sodium borate (0.1 M) was then added to the slides for 2 min to
neutralize HCl. Permeabilized cells were then washed with PBS containing 3% bovine
serum albumin (BSA) and incubated with an Alexa Fluor 488-conjugated anti-BrdU
antibody for 1 hr at 37°C. After washing the slide in PBS, nuclei were stained with DAPI
prior to mounting of cover slips. Finally, slides were visualized on a fluorescence
microscope. The percentage of BrdU-positive cells was calculated as the number of BrdU-
positive cells in the DAPI-positive population. Multiple random fields were assessed to
generate an average number of BrdU-positive cells in the sample.
ANNEXIN V-CY3 STAINING
Cells undergoing apoptosis express phosphatidyl serine (PS) on the outer leaflet
of the plasma membrane lipid bilayer. In healthy cells, PS is localized to the inner leaflet
of the lipid bilayer. Annexin A5 (or annexin V) binds with high affinity to PS, which is
accessible only with PS is localized to the outer leaflet. Thus, annexin V can be used as
a marker of apoptotic cells.
To calculate the percentage of cells undergoing apoptosis, cells were first washed
with PBS and resuspended in annexin V binding buffer (10 mM HEPES [pH 7.4], 140 mM
NaCl, 2.5 mM CaCl2) containing stock Annexin V-Cy3 (BioVision; Mountain View, CA).
Following a 5 min incubation at room temperature in the dark, cells were pelleted and
washed with fresh annexin V binding buffer. Washed cell pellets were resuspended in 4%
paraformaldehyde and incubated at room temperature for 20 min. Paraformaldehyde-fixed
cells were spotted on glass slides, permeabilized with 0.25% Triton-X 100 in PBS, and
stained with DAPI prior to visualization by fluorescence microscopy. The percentage of
annexin V-Cy3-positive cells was calculated as the number of annexin V-Cy3-positive cells
Page 55
41
in the total DAPI-positive population. Multiple random fields were assessed to obtain an
average value of the percentage of annexin V-positive cells.
LENTIVIRUS PRODUCTION
After lentiviral vectors were cloned and assessed for efficiency via HEK293T
transfection and western blot or RT-PCR analysis, lentivirus was produced for use in cell-
based experiments. To produce lentivirus, HEK293T cells were plated in T75 flasks such
that they achieved 50% confluence at the time of transfection. For each T75 flask, 12 μg of
lentiviral vector, 9 μg of Pax2 gag/pol, and 3 μg of vesicular stomatitis virus G protein
(VSV/G) were mixed with 1 ml CaCl2 (2.5 M) and 1 ml 2X HBS. Transfections were
conducted as described above. Two days post-transfection, culture medium was collected
and passed through a 0.45 μm filter. Filtered medium was then centrifuged at 49,000 x g
in a SW28 rotor (Beckman Coulter, Inc; Palo Alto, CA) for 2 hr at 4°C in an ultracentrifuge.
Lentivirus pellets were resuspended in RPMI 1640 medium, aliquoted, and stored at
-80°C. Titering of lentivirus was assessed by GFP fluorescence (for pYNC352-derived
lentiviral vectors) or immunofluorescent staining of cells transduced with limiting dilutions
of aliquoted lentivirus.
LENTIVIRAL TRANSDUCTION OF CULTURED CELLS
Transduction experiments were conducted following lentivirus titration. For
adherent cells (e.g. HEK293T or TIME cells), culture medium was removed. Then, RPMI
1640 medium containing lentivirus and polybrene (1 μg/ml) was added to cells. After 8 hr
of incubation at 37°C, medium containing lentivirus was removed and replaced with fresh
cell-appropriate culture medium. For suspension cells (e.g. PEL cells), cells were pelleted,
and culture medium was removed. Cell pellets were resuspended in lentivirus- and
polybrene-containing RPMI 1640 medium and incubated for 8 hr as described above.
Page 56
42
Following incubation with lentivirus, PEL cells were pelleted and resuspended in fresh
RPMI 1640 medium containing 10% FBS and allowed to recover for 48 hr prior to initiation
of cell growth assays or HHV-8 reactivation.
CELL GROWTH ASSAY
To determine the role of vIL-6 and cellular proteins on the growth of PEL cells in
culture, PEL cells were first transduced with vIL-6- or cellular protein-specific shRNA-
encoding lentivirus. Two days post-transduction, trypan blue-excluding (viable) cells were
counted using a hemocytometer. Equivalent numbers (1 x 105 cells/well) of control (non-
silencing shRNA-encoding lentivirus) and experimental (e.g. vIL-6-specific shRNA-
encoding lentivirus) cells were plated in duplicate wells of a 24-well cell culture plate.
Plated cells were then counted daily for three additional days to generate growth curves.
At the end of the growth experiment, remaining cells were harvested for preparation of cell
lysates for western blot analysis.
HHV-8 R219 GENERATION AND INFECTION OF ENDOTHELIAL CELLS
HHV-8 r219 virus was designed such that GFP expression is driven by the human
elongation factor 1 alpha (EF-1α) promoter, and RFP expression is driven by the viral PAN
RNA promoter [254]. GFP is expressed constitutively, while RFP is expressed exclusively
in lytic cells as PAN RNA is expressed only during the viral lytic cycle. Therefore, GFP
and RFP fluorescence can be used as markers to determine whether cells have been
infected with HHV-8 r218 (GFP+) or have been reactivated (RFP+, lytic).
HHV-8 r219 virus was generated in Vero-r219 cells infected with baculovirus-
encoded Rta (a gift from Prashant Dasai). Briefly, Vero-r219 cells were grown in DMEM
supplemented with 10% FBS and puromycin (5 μg/ml). Medium was removed from Vero-
r219 cells, and baculovirus-encoded Rta was added to the cell culture medium. The cells
Page 57
43
and virus were incubated at 37°C for 2 hr with continuous rocking. Cells were then washed
with fresh DMEM and incubated with DMEM containing 10% FBS and NaB (1 mM) for 24
hr. Medium was then replaced with DMEM supplemented with 10% FBS, and cells were
incubated for an additional four days. Finally, culture medium was harvested and
centrifuged briefly at 400 x g for 5 min to remove cellular debris. The supernatant was then
centrifuged at 16,000 rpm (SW41 rotor, Beckman Coulter, Inc) for 2 hr at 4°C in an
ultracentrifuge to pellet the virus. Viral pellets were resuspended in EBM-2 without serum
or supplements, aliquoted, titered on naïve TIME-TRE/RTA cells, and frozen at -80°C until
needed.
TIME-TRE/RTA endothelial cells were infected with HHV-8 r219 virus for use in
virus reactivation experiments. Prior to infection, TIME-TRE/RTA cells were plated such
that they would achieve approximately 90% confluence at the time of infection. Culture
medium was removed, and HHV-8 r219 virus was added to cells. Cell cultures containing
virus were centrifuged clockwise at 400 x g for 30 min at 37°C and then counterclockwise
for an additional 30 min at the same speed and temperature. Following an additional 1 hr
incubation at 37°C at 5% CO2, medium containing virus was removed and replaced with
fresh EBM-2 containing supplements and FBS. Two days post-infection, cells were
visualized for GFP expression via fluorescence microscopy to determine infectious titers.
For reactivation experiments in endothelial cells, HHV-8 r219-infected TIME-TRE/RTA
cells were then transduced with lentivirus targeting viral or host proteins and reactivated
as described below.
TITERING OF REACTIVATED HHV-8
To assess the role of viral and host proteins in HHV-8 productive replication,
protein-depleted TIME and PEL cells were reactivated, and viral titers were calculated.
Transduced JSC-1 PEL cells were reactivated with NaB (0.5 μM) and 12-O-
Page 58
44
tetradecanoylphorbol-13-acetate (TPA, 20 nM), and medium was harvested four days
post-induction. Transduced TRExBCBL1-RTA and HHV-8 r219-infected TIME-TRE/RTA
cells were induced with doxycycline (Dox, 1 μg/ml) and harvested at four (TRExBCBL1-
RTA) or six (TIME-TRE/RTA) days post-induction. Harvested culture medium containing
virus was passed through a 0.45 μm filter and centrifuged at 16,000 rpm (SW41 rotor,
Beckman Coulter, Inc) in an ultracentrifuge for 2 hr at 4°C to pellet virus. Viral pellets were
resuspended in EBM-2, and aliquots of harvested virus were applied to naïve TIME-
TRE/RTA cells for 2 hrs. Cells and virus-containing medium were centrifuged clockwise
at 400 x g for 30 min at 37°C and then counterclockwise using the same conditions.
Following a 1 hr incubation at 37°C in a 5% CO2 chamber, medium containing virus was
removed, and fresh EBM-2 containing FBS and supplements was added. The following
day, cells were trypsinized, washed with PBS, and plated on glass chamber slides. One
day after the cells were plated, slides were fixed in methanol as described above and
stained using an anti-LANA primary antibody and a Cy3-conjugated fluorescent secondary
antibody. Nuclei were stained with DAPI, and slides were visualized via fluorescence
microscopy. The percentage of LANA-positive cells was calculated as the number of
LANA-positive cells in the total DAPI-positive cell population. Multiple random fields were
assessed.
DEPLETION AND COMPLEMENTATION EXPERIMENTS
Depletion and complementation experiments were utilized to address several
research questions, including whether an ER-localized vIL-6 signaling mutant could
rescue PEL cell growth in vIL-6 depleted cells and whether ER-localized vIL-6/gp130-
mediated STAT and MAPK signaling played a role in virus replication. These experiments
required that cells be transduced with multiple lentiviruses.
Page 59
45
For experiments assessing the rescue of vIL-6 depletion with an ER-localized
vIL-6 signaling mutant, cells were first transduced with lentivirus encoding an ER-localized
and shRNA-resistant vIL-6 signaling mutant. After allowing time for the transduced cells
to recover, cultures were selected in puromycin (1 μg/ml) to eliminate untransduced cells.
Then, puromycin-selected cells were transduced with lentivirus encoding vIL-6-specific or
non-silencing shRNAs. Cells were allowed to recover for two days following the second
lentiviral transduction. Then, cells were normalized and plated for cell growth assays as
described above.
For depletion-complementation assays addressing virus replication, TRExBCBL1-
RTA PEL cells were transduced with the first lentivirus (overexpressed vIL-6 variants or
gp130-signaling mutants) and allowed to recover for two days prior to being transduced
with the second (shRNA-encoding) lentivirus. GFP fluorescence was used as a marker of
transduction efficiency of the shRNA-encoded lentivirus. A subset of doubly transduced
TRExBCBL1-RTA cells were harvested in TRIzol for RT-PCR analysis to verify the
expression of vIL-6 and gp130 signaling mutants. As before, cells were allowed to recover
for two days prior to cell count normalization and reactivation with Dox. Virus was
harvested from culture media and applied to naïve TIME-TRE/RTA cells for LANA staining
as described above. Specific procedures are detailed in the methods sections of the
appropriate chapters.
Page 60
46
CHAPTER 3: INVOLVEMENT OF GP130 IN VIL-6-MEDIATED PEL CELL GROWTH
AND SURVIVAL
Page 61
47
Summary
Prior to initiating this dissertation work, vIL-6 had been implicated in the
pathogenesis of HHV-8-associated diseases via pro-proliferative, anti-apoptotic, and pro-
angiogenic activities. In PEL cells, vIL-6 is produced in functional amounts during viral
latency and promotes the growth of these cells, mediating its activity from the ER, where
it is predominantly localized. This vIL-6 activity is dependent, in part, on its interaction with
a splice variant of VKORC1, termed VKORC1v2. However, the roles of gp130 and ER-
localized vIL-6/gp130 interactions in PEL cells were unknown. Thus, this study sought to
determine the influence of gp130 on PEL cell viability and growth, the role of ER-localized
vIL-6/gp130 signaling in PEL cell maintenance, and the contributions of gp130-mediated
MAPK and STAT signaling to PEL cell proliferation and survival.
Targeted depletion of gp130, vIL-6, ERKs 1 and 2, STAT1, and STAT3 was
conducted to determine the role of these proteins in PEL cell growth and survival.
Additionally, gp130-activated MAPK and STAT signaling was assessed following gp130
depletion to determine the contribution of gp130 to activation of these signaling pathways
in PEL cells. Finally, two approaches were utilized to establish the significance of ER-
localized vIL-6/gp130 signaling on the proliferation and viability of PEL cells.
This study found that the IL-6 signal transducer, gp130, is required for optimal PEL
cell growth and viability. Levels of activated ERKs 1 and 2 and STATs 1 and 3,
phosphorylated following gp130 stimulation, were reduced in gp130-depleted BCBL-1 and
BC-1 PEL cells. Diminished STAT activation was also detected in two other PEL cell lines,
JSC-1 and BC3. Growth effects of gp130 depletion could be mimicked by shRNA targeting
of ERKs 1 and 2 or by depletion of STAT3. Finally, inhibition of vIL-6/gp130 interactions
specifically within the ER compartment suppressed cell proliferation and viability, mirroring
the effects of gp130 depletion. Combined, these data demonstrate that gp130, in addition
to VKORC1v2, is essential for normal PEL cell growth and survival and that ER-localized
Page 62
48
vIL-6/gp130 interactions are critical for these activities. Targeting of intracellular vIL-
6/gp130 interactions could potentially provide a means of PEL therapy.
Page 63
49
ROLE OF HUMAN HERPESVIRUS 8 INTERLEUKIN-6-ACTIVATED GP130 SIGNAL
TRANSDUCER IN PRIMARY EFFUSION LYMPHOMA CELL GROWTH AND
VIABILITY
This work has been published and is reproduced here (with modifications) with permission from the American Society for Microbiology (Copyright © American Society for Microbiology, [Journal of Virology, 2013, 87(19):10816-10827, doi: 10.1128/JVI.02047-13]). Cousins E, Nicholas J (2013) Role of human herpesvirus 8 interleukin-6-activated gp130 signal transducer in primary effusion lymphoma cell growth and viability. 87(19):10816-10827.
INTRODUCTION
HHV-8 encodes several proteins that are believed to contribute to the onset and/or
progression of endothelial KS and B-cell malignancies PEL and MCD [255-258]. vIL-6,
like its cellular counterparts, is a growth factor for B-cells and other cell types and promotes
inflammatory and angiogenic responses. These activities implicate the viral cytokine as a
contributory factor in HHV-8 associated neoplasias [144,259]. In PEL cells, true latent
expression of vIL-6 suggests that the viral protein can contribute to this disease in a direct,
autocrine fashion by promoting PEL cell proliferation and survival, in addition to possibly
maintaining viral latent reservoirs during normal (disease-free) infection [115,116].
While the three-dimensional structures of vIL-6 and hIL-6 are similar and both can
bind to and induce dimerization of the gp130 signal transducer, vIL-6 is unique in that it is
“pre-conformed” to mediate gp130 dimerization without first binding the non-signaling
gp80 IL-6 receptor subunit [165,260,261]. However, vIL-6 can bind gp80 and form
hexameric complexes (vIL-62:gp1302:gp802) in addition to tetrameric (gp80-devoid)
complexes [159,165]. Hexameric and tetrameric complexes have distinguishable
signaling properties [262], likely mediated in part by gp80 stabilization of vIL-6-induced
gp130 dimerization [159,165]. Within the ER, vIL-6 induces the formation of tetrameric
complexes exclusively [115,170]. ER-directed hIL-6 is unable to induce gp130 complexing
Page 64
50
and signal transduction. vIL-6, hIL-6, and other cellular IL-6 proteins activate STATs 1 and
3 via gp130-associated Janus kinase (JAK)-mediated tyrosine phosphorylation of the
transcription factors [156]. MAPK signaling is activated following SHP2 recruitment to
gp130 and phosphorylation by JAK, which leads to downstream phosphorylation and
activation of ERKs 1 and 2 [156]. In addition to differences in gp80-dependency of ligand-
induced gp130 dimerization and the ability of vIL-6 to signal from the ER, inefficient
secretion of vIL-6 distinguishes it from its cellular counterparts [170]. Thus, vIL-6 is found
largely intracellularly, specifically within the ER, and its ability to signal from this
compartment suggests that this may be functionally important for both virus biology and
viral pathogenesis. Indeed, vIL-6 depletion-mediated inhibition of PEL cell growth in
culture can be reversed by transduction of ER-retained (KDEL motif-tagged) vIL-6 [115].
Also, vIL-6 support of PEL cell growth can be inhibited by an ER-localized single-chain
antibody specific to vIL-6 [150]. It is reasonable to hypothesize that vIL-6 may contribute
to PEL pathogenesis via gp130 signaling. STAT3, a major target of such signaling and a
transcription factor implicated in many human cancers [263-265], is activated in PEL cells
and appears to be important for their viability, in part via the STAT3-induced pro-survival
protein survivin [152].
However, demonstration of vIL-6-mediated signal transduction via gp130 in PEL
cells and the role of gp130 in PEL cell biology have not been reported. Recently, vIL-6
was found to interact with the ER membrane protein VKORC1v2, a splice variant of
warfarin target VKORC1 (variant 1) [174], and this interaction was shown to be important
for the pro-growth and anti-apoptotic activities of vIL-6 in PEL cells [173]. Interaction of
vIL-6 with VKORC1v2 occurs via a transmembrane-proximal region of the ER-lumenal
domain; its precise mapping enabled the development of a vIL-6 refractory variant and an
interaction-inhibitory peptide that helped reveal the functional relevance of the vIL-6-
Page 65
51
VKORC1v2 interaction [173]. The mechanism of vIL-6 activity via VKORC1v2 appears to
be independent of gp130.
In the present study, we have investigated directly the influence of gp130 on PEL
cell proliferation and viability, the role of the vIL-6-gp130 interaction in gp130-mediated
activities, and the contributions of gp130-activated MAPK and STAT signaling to PEL cell
growth. Our data indicate that gp130 is essential for normal proliferation and viability of
PEL cells and that ER-localized vIL-6/gp130-activated ERK and STAT3 signaling are
critically important. These findings suggest that inhibitory targeting of vIL-6 interactions
with the signal transducer within the ER may provide a useful strategy for PEL therapy or
for targeting HHV-8 latency in normal B-cells should vIL-6 be expressed in this latent
reservoir.
METHODS
Cell culture, transfections, and lentivirus production
Cell cultures were maintained as described in Chapter 2. HEK293T cells were
transfected with plasmids using standard calcium-phosphate-DNA co-precipitation as
explained in Chapter 2. Cells were harvested 24 to 48 hrs post-transfection. Cell lysates
were prepared as outlined in Chapter 2, and lysates were analyzed directly or stored at
-20°C prior to Western blot analysis. Lentivirus was produced by transfecting HEK293T
cells with shRNA-, vIL-6-, or sgp130-encoding or empty lentivirus plasmids (pYNC352 and
pDUET001) together with gag/pol and VSV/G expression vectors, as previously described
and in Chapter 2 [173]. Infectious titers were established by using GFP-based or
immunofluorescence microscopy to identify vector-transduced cells in PEL and other
cultures prior to experimental utilization of lentiviral vectors (see Chapter 2). For vIL-6
“rescue” experiments, PEL cells were first transduced with pYNC419-vIL-6.KDEL or
pYNC419-vIL-6.W167G.KDEL, allowed to recover for 5 days, selected in puromycin (1.0
Page 66
52
μg/ml) for 2 weeks, and then transduced with either NS or vIL-6-specific shRNA encoded
by pYNC352-derived lentiviruses. For shRNA and sgp130-transduced cultures, cells were
allowed to recover for 2 days prior to normalizing cell numbers for cell growth assays.
Oligonucleotides and plasmids
shRNAs were chosen or designed to selectively deplete gp130, STAT1, STAT3,
ERK1, or ERK2. Complementary oligonucleotides encoding 19-nucleotide short hairpin
regions were annealed, phosphorylated, and cloned into the BamHI and MluI sites of the
pYNC352 lentivirus vector. The following, pre-validated shRNA sequences were used:
STAT1 (5’-GCGTAATCTTCAGGATAAT-3’ [266]), STAT3 (CATCTGCCTAGATCGGCTA
[266,267] used independently or together with AGTCAGGTTGCTGGTCAAA [266]), ERK1
(GCCATGAGAGATGTCTACA [268]), and ERK2 (GAGGATTGAAGTAGAACAG [268]).
Three shRNA sequences were designed and tested for gp130 depletion:
GTGGGATCACCTATGAAGA (sh1), CTGTCCAAGACCTTAAACC (sh2), and
TGCCCTTGGGAAGGTTACA (sh3); sh1 was used in subsequent functional analyses,
alone or together with sh3. vIL-6 shRNA oligonucleotides and non-silencing (NS) control
shRNAs were described previously [115]. For generation of a lentivirus vector expressing
KDEL motif-tagged vIL-6 variant W167G [166], the corresponding vIL-6 coding sequences
were first cloned between the BamHI and SmaI sites of pSG5.KDEL [166] to generate
contiguous vIL-6.W167G-KDEL coding sequences, and these sequences were then PCR
amplified and inserted between the NotI and XhoI sites of pTYB6-derived lentivirus vector
pYNC419 [166,269]. Lentiviral vector-cloned vIL-6 and vIL-6-KDEL and pSG5-cloned vIL-
6, vIL-6-KDEL, vIL-6.W167G, hIL-6, hIL-6-KDEL, gp130-Fc, and chitin binding domain
(CBD)-fused VKORC1v2 have been described previously [69,115,165,166,173]. The
soluble gp130 (sgp130) expression vector was made by amplifying the first 972
nucleotides of gp130 (encoding domains 1-3) and inserting this sequence between the
Page 67
53
EcoRI and BamHI sites within the pSG5.KDEL vector, generating pSG5-sgp130.KDEL. A
sequence encoding the FLAG epitope was then inserted at the BamHI site using
appropriate complementary oligonucleotides, generating pSG5-sgp130.FLAG.KDEL. The
sgp130.FLAG.KDEL coding region was then amplified and ligated into the pDUET001
lentiviral vector [270] between the BamHI and XhoI sites. The integrities of all
constructions were verified by sequencing.
Immunoblotting and co-precipitation assays
For western blotting, heat-denatured and reduced proteins in cell lysate samples
were size fractionated by SDS-PAGE and electrophoretically transferred to nitrocellulose
membranes using standard procedures (see Chapter 2). Membranes were blocked,
probed with primary antibodies, washed, incubated with appropriate HRP-conjugated
secondary antibodies, and exposed as described in Chapter 2. For co-precipitation
assays, cell lysates (prepared as described above) were added to either chitin beads (for
precipitating VKORC1v2-CBD) or protein-A agarose beads (for precipitating gp130-Fc)
and incubated with continuous rotation overnight at 4°C. Beads were then washed 3 times
with lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 0.1% Tween 20) prior to
boiling and loading onto SDS-PAGE gels. Untreated lysate samples were also separated
on SDS-PAGE gels to provide controls for protein expression, extraction, and transfer to
immunoblotting membranes.
Antibodies
The following commercially available antibodies were used for western blotting:
BrdU (BD Biosciences, Rockville, MD; catalog number 558599); pSTAT3 and pSTAT1
(Cell Signaling Technology, Beverly, MA; catalog numbers 9131 and 9171); β-actin
(Sigma, St. Louis, MO; catalog number A2228); pERK, ERK1, STAT3, p-tyrosine, and
Page 68
54
gp130 (Santa Cruz Biotechnology, Santa Cruz, CA; catalog numbers sc-7383, sc-94, sc-
482, sc-508, and sc-655); hIL-6 (R&D Systems, Minneapolis, MN; catalog number MAB-
206); and CBD (New England Biolabs, Ipswich, MA; catalog number E8034S). Rabbit
antiserum to vIL-6 was generated in our laboratory and reported previously [166]. The
gp130 (BD Biosciences, Rockville, MD; catalog number 555756) and FLAG (Sigma;
catalog number F1804) primary antibodies and the Alexa Fluor 594 donkey anti-mouse
IgG (H+L) secondary antibody (Invitrogen, Carlsbad, CA; catalog number A21203) were
used for immunofluorescence staining.
Apoptosis, proliferation, and cell growth assays
Apoptosis was assessed via annexin V-Cy3 staining of cells as outlined in Chapter
2. Several fields were used to calculate the percentage of annexin V-Cy3-positive cells in
the total DAPI-positive cell population. Cell proliferation rates were determined by BrdU-
incorporation assay, which was detailed in Chapter 2. Slides were visualized under UV
microscopy to assess the percentage of BrdU-positive cells in the total cell population
(DAPI+). Multiple fields were assessed to determine an average percentage of BrdU-
positive cells. For cell growth assays, appropriate, high-efficiency infection with the GFP-
positive lentiviral vectors was verified by >90% GFP fluorescence in the cultures, which
was sustained over the course of the 3-day growth experiment. Cell growth assays were
initiated following cell density normalization and seeding of cells (105) in fresh media 48
hrs after lentiviral infection. Growth of the cultures was quantified by daily counting of
trypan blue-excluding (viable) cells per ml using a hemocytometer. Cultures transduced
with equivalent titers of lentiviral vector expressing NS shRNA were used as controls.
Page 69
55
RESULTS
Contributions of gp130 to PEL cell growth. In previous studies, we found that
depletion of vIL-6 in BCBL-1 and JSC-1 PEL cells led to autocrine-mediated reductions in
cell proliferation and survival and that ER-localized vIL-6 activity was sufficient for these
functions [115]. Subsequently, we determined that expression of VKORC1v2 and its
interaction with vIL-6 were important gp130-independent contributors to the pro-growth
and anti-apoptotic activities of vIL-6 in these cells [173]. However, the possibility remained
that vIL-6 signaling through gp130, in the ER and/or at the cell surface, also contributes
to vIL-6 effects on PEL cells. The potential contribution of gp130 to PEL cell growth was
tested using lentiviral-delivered gp130-specific shRNA to deplete endogenous expression
of the signal transducer. Three shRNAs for gp130 depletion were designed (Methods) and
tested in lentivirus-shRNA vector-transfected HEK293T cells, and each significantly
depleted overexpressed gp130 levels relative to vector-expressed gp130 observed in NS
shRNA-transfected control cultures (data not shown). Two of these gp130 shRNA-
expressing lentivirus vectors were used for single or dual transduction (see Methods) for
subsequent experiments in PEL cells. The control NS shRNA-encoding lentiviral vector
was used for comparison. Equivalent inoculating doses of pre-titered lentiviruses were
used, sufficient to provide ~90% transduction efficiency, as determined by lentivirus-
expressed GFP fluorescence. After a post-infection incubation period of two days, growth
of cell density-normalized PEL cultures was monitored for an additional three days. For all
PEL cell lines tested (BCBL-1, JSC-1, BC-1, and BC-3), gp130-targeted shRNAs led to
significantly reduced cell growth relative to NS shRNA-transduced controls (Figure 3.1A).
Potential off-target, generally cytotoxic effects of gp130-directed shRNA were controlled
for by equivalent experiments in HHV-8-negative BJAB cells, which were essentially
unaffected by gp130 shRNA transduction relative to NS shRNA-transduced controls.
The observed reductions in PEL cell growth following gp130 depletion could be
Page 70
56
due to decreased cell survival and/or decreased rates of proliferation. This was
investigated utilizing annexin V-Cy3 staining assays to detect apoptotic cells and BrdU
incorporation analysis to identify S-phase cells in BCBL-1 and JSC-1 cultures transduced
with either NS or gp130-specific shRNA. The data from these experiments revealed a
correlation between gp130 depletion and both increased rates of apoptosis (Figure 3.1B)
and decreased rates of DNA synthesis (Figure 3.1C). The combined results indicate that
gp130 signaling supports PEL cell growth by promoting both cell proliferation and cell
survival.
Influence of gp130 on STAT and ERK signaling in PEL cells. Signal
transduction by gp130 involves activation of STAT and MAPK signaling. This proceeds
via JAK-mediated tyrosine phosphorylation of gp130-recruited STATs 1 and 3 and tyrosine
and threonine phosphorylation of ERKs 1 and 2 following the activation of the MAPK
signaling cascade, which is triggered by JAK phosphorylation of gp130-associating SHP2
[156]. Active STAT3 has been reported previously to be expressed at constitutively high
levels in PEL cells and to be important for PEL cell viability in culture [152]. However, the
mechanism of constitutive STAT3 activation in PEL cells and the biological significance of
ERK signaling in these cells are unknown. To address the role of gp130 in the activation
of STAT3 and ERK in the context of PEL cells, we again utilized gp130 depletion via
lentiviral vector-mediated shRNA transduction. Targeting of gp130 resulted in detectably
reduced levels of phospho-STAT3 (pSTAT3, active) and pSTAT1 in JSC-1, BCBL-1,
BC-1, and BC-3 cells (Figure 3.2A,B) and a marked decrease in phospho-ERK (pERK)
levels in BCBL-1 and BC-1 cells but not in JSC-1 and BC-3 cells (Figure 3.2C). The data
from these experiments identify, for the first time, the contribution of gp130 to constitutively
high pSTAT3 and pSTAT1 levels in PEL cells and provide evidence that gp130 can
significantly influence ERK signaling in at least some PEL cell types (shown here for
BCBL-1 and BC-1).
Page 71
57
Contributions of STATs to PEL cell growth. It has been reported by others that
STAT3 is essential for the viability of PEL cells (BC-1, BCBL-1, and VG-1) and that this
activity involves STAT3-mediated induction of caspase-inhibitory and anti-apoptotic
protein survivin [152]. These studies utilized transduced dominant-negative STAT3 and
the JAK pharmacological inhibitor AG490 to suppress levels of pSTAT3. Others have
documented the importance of JAK-mediated, STAT-activating signaling in several PEL
cell lines via the experimental use of JAK1-inhibitory curcumin; this was found to inhibit
cell proliferation, promote apoptosis, and suppress IAP family proteins, including survivin
[271]. To independently assess the role of STAT3 in PEL cell growth, the transcription
factor was targeted for depletion by shRNA-encoding lentiviruses. Two previously reported
STAT3-targeted shRNAs [266,267] were first tested in transfected HEK293T cells and
verified as effective (data not shown). These shRNAs were each cloned into a lentiviral
vector for introduction into PEL cells. Initial experiments indicated that single shRNA
transduction was insufficient to achieve strong STAT3 depletion in PEL cultures, whereas
co-infection of PEL cells with both lentiviral vectors was effective. For growth assays,
BCBL-1 and JSC-1 cells were simultaneously infected with both STAT3-targeting lentiviral
vectors or with an equivalent infectious dose of NS shRNA-encoding control lentivirus.
Cell growth of post-transduction cultures was monitored by daily counting of trypan blue-
excluding (viable) cells to determine cell densities (Figure 3.3A). Suppression of STAT3
relative to NS shRNA-transduced cultures was verified in terminal (day 3) cultures by
western blotting of cell extracts (Figure 3.3A, right panels). The data revealed that STAT3
depletion substantially diminished growth of these PEL cells, consistent with previously
published studies [152], and identified JSC-1 PEL cells (not previously examined) as
dependent on STAT3 for normal growth.
Further analyses were undertaken to test the contributions of STAT3 to
proliferation and survival of BCBL-1 and JSC-1 cells. These experiments involved
Page 72
58
immunofluorescence detection of BrdU incorporation and annexin V-Cy3 staining,
respectively, as outlined above and for the equivalent studies following gp130 depletion.
Cells were harvested 5 days following lentiviral transduction (3 days post-normalization
and culture) for BrdU and annexin V analyses. The results mirrored the effects of gp130
depletion, demonstrating that STAT3 depletion inhibited cell proliferation (proportion of
cells in S phase) (Figure 3.3B) and increased the rate of apoptosis (Figure 3.3C). Thus,
both of these components of cell biology are influenced by STAT3, which in turn is
activated by gp130 in these PEL cell lines.
As gp130 signaling involves activation of STAT1 in addition to STAT3, we also
explored the potential contribution of STAT1 activity to BCBL-1 and JSC-1 cell growth.
Lentiviral-delivered shRNA [266] was used to deplete STAT1 in the respective cultures,
and cell growth was compared to that of control (NS) shRNA-transduced cultures. The
data revealed no significant effects of STAT1 depletion on BCBL-1 or JSC-1 cell growth,
despite achieving robust depletion of pSTAT1 in BCBL-1 cells and a nearly 2-fold
reduction of pSTAT1 in JSC-1 cells, as demonstrated by western blotting of terminal
culture extracts (Figure 3.3D). Although STAT1 depletion was somewhat limited in JSC-1
cells, these data suggest that STAT1 activity is not critical for PEL cell growth.
Contributions of ERKs 1 and 2 to PEL cell growth. As gp130 signaling involves
the activation (phosphorylation) of ERKs 1 and 2 in addition to STATs, we undertook
experiments to investigate the influence of ERK signaling on PEL cell growth. ERKs 1 and
2 were simultaneously targeted for depletion in BCBL-1 and JSC-1 cells using techniques
analogous to those employed for STAT depletion. The NS- or ERK-shRNA lentiviral
vector-transduced cells were monitored for growth over a three-day period by daily
calculation of viable (trypan blue-excluding) cell densities from triplicate cultures for each
cell type. For both PEL cell lines, ERK depletion led to dramatic reductions in cell growth
relative to control (NS) shRNA-transduced cultures (Figure 3.4A). Therefore, ERKs 1 and
Page 73
59
2, in addition to STAT3, are essential for normal growth of these cells.
The shRNA-transduced JSC-1 and BCBL-1 cultures were further analyzed to
assess the effects of ERK depletion on cell proliferation and survival, specifically. Rates
of proliferation and apoptosis in these cells following ERK1 and ERK2 depletion were
examined by BrdU incorporation and annexin V-Cy3 staining, respectively, as outlined
above. These analyses revealed that both proliferation and cell survival were reduced
appreciably (between two- and three-fold) by ERK depletion (Figure 3.4B,C).
Analysis of the effects of ERK depletion in two additional PEL cell lines, BC-1 and
BC-3, indicated the general importance of ERKs 1 and 2 for PEL cell growth (Figure 3.4D).
Effects were not as marked in these cell lines as they were for BCBL-1 and JSC-1 cells,
but it is possible that this was due to reduced efficacy of ERK depletion (Figure 3.4D,
bottom panels). In contrast, ERK depletion in the HHV-8-negative BJAB B-cell line,
reported previously to be insensitive to inhibition of ERK kinase (MEK) by PD98059 [272],
were unaffected by ERK shRNA transduction, verifying lack of general cytotoxic effects of
lentiviral infection and ERK shRNA transduction (Figure 3.4D, right). Combined, the data
from our ERK depletion studies reveal the importance of ERK signaling, sustained in part
via gp130 signal transduction in at least some PEL cell lines (BCBL-1 and BC-1, shown
here), for PEL cell growth and viability.
ER-localized gp130/vIL-6 interaction contributes to vIL-6 and gp130
activities. We have previously reported that ER-localized vIL-6 and its interaction with the
ER protein VKORC1v2 are important for PEL cell proliferation and viability [173]. A
previously characterized vIL-6 variant, vIL-6.W167G, was used to determine the
contribution of ER-localized vIL-6/gp130 interactions to PEL cell growth and survival. This
altered vIL-6 protein is mutated in a region (“site III”) that interacts with gp130 domain 1
and is unable to induce dimerization of gp130 absent gp80 co-expression and hexameric
complexing [159,166]. As ER-localized vIL-6 signaling occurs exclusively through
Page 74
60
tetrameric (vIL-62:gp1302) complexes [171], vIL-6.W167G should be unable to induce
gp130 dimerization and signaling from the ER compartment. We first verified that ER-
retained, KDEL motif-tagged vIL-6.W167G was unable to induce gp130-mediated signaling
in transfected HEK293T cells. Unlike wild-type vIL-6-KDEL, vIL-6.W167G-KDEL was
unable to induce tyrosine phosphorylation of gp130 or STAT3 (Figure 3.5A). Co-
precipitation assays from transfected cell lysates confirmed that the W167G mutation did
not affect the interaction between vIL-6 and VKORC1v2, which was tagged with CBD to
facilitate precipitation and enable immune-detection with a CBD-specific antibody (Figure
3.5B). KDEL-tagged, shRNA-resistant versions of wild-type and W167G-mutated vIL-6
were then used to supplement vIL-6-depleted PEL cells to determine if the gp130
dimerization-defective but VKORC1v2 binding-competent variant could rescue cell
growth. These experiments were analogous to those undertaken previously with vIL-6-
KDEL, which demonstrated sufficiency of ER-localized vIL-6 for maintenance of BCBL-1
and JSC-1 cell growth and viability [171]. In contrast to vIL-6-KDEL, vIL-6.W167G-KDEL
was unable to complement endogenous vIL-6 depletion (Figure 3.5C), indicating that vIL-
6-induced dimerization of gp130 and its downstream signaling is critically important for
pro-proliferative activity of ER-localized vIL-6 in PEL cells.
The importance of intracellular gp130 signaling was further tested by utilizing
lentiviral-transduced soluble gp130 (sgp130), which was KDEL motif-fused for ER
retention. Initial experiments in transfected HEK293T cells were undertaken to verify the
ability of this protein to inhibit ER-localized (vIL-6.KDEL), but not extracellular (hIL-6),
signal transduction via endogenously expressed gp130 (Figure 3.6A). Additionally, the
intracellular localization of sgp130.KDEL (Flag-tagged) was tested and compared to the
distribution of full-length gp130 by α-Flag and α-gp130 antibody-based
immunofluorescence assays in transfected cells; cells were co-transfected with GFP-
KDEL, which provided an ER marker [115]. In contrast to native gp130, sgp130.KDEL was
Page 75
61
present at high levels intracellularly with little, if any, surface fluorescence detectable
(Figure 3.6B). Having confirmed the expression and appropriate ER localization of
lentiviral vector-expressed sgp130.KDEL, this vector, or an empty vector control, was then
used to transduce PEL cells to determine the effects of ER-localized gp130 inhibition on
cell growth and apoptosis. The data revealed significant effects of sgp130.KDEL, relative
to the empty vector, on the growth of both BCBL-1 and JSC-1 PEL cells (Figure 3.6C).
BrdU incorporation assays using cells harvested at day 2 from the growth experiment
confirmed inhibition of proliferation (i.e., percentage of cells in S-phase) by introduced
sgp130.KDEL (Figure 3.6D). In a separate experiment, transduction of sgp130.KDEL into
BCBL-1 and JSC-1 cells led to substantial increases in rates of apoptosis, as determined
by annexin V-Cy3 staining (Figure 3.6E). As vIL-6 is the only known cytokine to signal
through gp130 from the ER compartment (and ER-retained hIL-6 has been demonstrated
to be unable to do so [115]), these data suggest strongly that vIL-6/gp130 signaling is of
biological significance and contributes, wholly or in part, to the pro-growth and pro-survival
effects of gp130 in these cells.
DISCUSSION
The work reported here addresses the contribution of gp130 signaling to the
constitutively high levels of active STAT3 reported in PEL cells, the influence of gp130
activity on STAT1 and ERK activation in these cells, and the functional relevance of vIL-
6-mediated gp130 activity to PEL cell growth. To our knowledge, our data are the first to
document a mechanism for the constitutively high levels of STAT3 in PEL cells, to
establish a critical role of the gp130 signal transducer in supporting PEL cell proliferation
and viability, and to identify the relevance of gp130 targeting by vIL-6 to these activities.
Thus, in addition to the important association of vIL-6 with VKORC1v2 for PEL cell growth
and survival [115], activity of the viral cytokine through its well-established signal
Page 76
62
transducer is also critical. This activity is mediated from the ER, where vIL-6 is
predominantly localized. A VKORC1v2 binding-competent, gp130 dimerization-defective
vIL-6 variant (W167G) that was restricted to the ER was unable to rescue the effects of vIL-
6 depletion, in contrast to KDEL motif-tagged wild-type vIL-6. Furthermore, ER-localized
sgp130, used as a competitive inhibitor of gp130/vIL-6 interactions in the ER
compartment, enabled identification of the specific role of vIL-6 as a mediator of gp130
function in the context of PEL. Thus, gp130 and ER-localized vIL-6 signaling via gp130
have been shown for the first time to support PEL cell proliferation and viability. Therefore,
gp130 and its interaction with vIL-6 have been identified as potential targets for therapeutic
intervention.
Another important finding from this study is that ERK activation contributes
significantly to the growth and survival of PEL cells. Depletion of ERK led to severe
abrogation of cell proliferation (3-fold reductions in BrdU incorporation) and to greatly
increased rates of apoptosis (2- to 3-fold) for BCBL-1 and JSC-1 cultures, effectively
halting culture growth. Significant effects of ERK depletion on the growth of BC-1 and BC-
3 cells were also identified. At least for BCBL-1 and BC-1 cells (included in this study), the
high levels of active, tyrosine-phosphorylated ERK (pERK) are dependent on gp130.
However, gp130 depletion did not diminish the levels of pERK in JSC-1 and BC-3 cells;
pERK was actually increased in both cell lines. These patterns were observed in multiple
repeat experiments in these PEL cell lines. In contrast to the effects of ERK depletion on
PEL cell growth, ERK depletion in HHV-8-negative BJAB cells did not affect cell growth at
all. The role of ERK signaling in HHV-8 biology has previously been noted only in
connection with de novo infection, establishment of latency, and lytic reactivation. Thus,
pharmacological inhibition of ERK activation suppresses TPA-induced lytic gene
expression and replication in PEL cultures [273-276]. Additionally, activation of ERK-
targeting signaling pathways and their requirement for de novo infection, post-entry lytic
Page 77
63
gene expression, and latency establishment have been reported in endothelial cell
systems [275,277,278]. Our results now add to this body of knowledge regarding the role
of ERK signaling in HHV-8 biology by identifying ERK as an important factor for
maintenance of PEL cell viability and by establishing gp130 signaling as a mechanism
contributing to high levels of active ERK in at least some PEL cell types.
Previous studies by Aoki and colleagues [152] demonstrated that the JAK inhibitor
AG490 and ectopic expression of a dominant-negative STAT3 protein (STAT3D, DNA
binding-refractory) led to dramatically reduced PEL cell viability. The PEL cell lines
investigated were BCBL-1, BC-1 and VG-1. Our experiments investigating the role of
STAT3 utilized BCBL-1 and JSC-1 cells, characterized in depth previously with respect to
latent expression and pro-growth and pro-survival activities of ER-localized vIL-6 [115]. A
caveat of the experiments utilizing AG490 is that this pharmacological inhibitor can affect
JAK-activated receptors other than gp130 and receptor-mediated activation of STATs
other than STAT3; AG490 may also have non-specific effects on other pathways. Of
additional note is that experimental use of the STAT3D protein, while very effective at
blocking endogenous STAT3 activity, also has the potential to inhibit STAT1 and other
STAT3-hetrodimerizing proteins; thus it may affect STAT3-independent mechanisms.
However, the previously reported data from Aoki et al. [152] combined with our
independently-derived and concordant results from STAT3 depletion experiments provide
very strong evidence that STAT3 signaling is critical to PEL cell biology. STAT3 signaling
was shown to induce the pro-survival protein survivin, which was found to be an important
factor in STAT3-mediated pro-survival effects in PEL cells [152]. However, it is noteworthy
that the JAK-targeting STAT signaling inhibitor curcumin, while demonstrated in an
independent study to induce cell death with concomitant suppression of survivin in most
of the PEL cell lines tested, did not lead to a significant loss of survivin expression in
BCBL-1 cells (used in the present study and that of Aoki et al. [152]) [271]. Thus, survivin
Page 78
64
may not be the only STAT3-regulated factor critical to PEL cell viability. Nonetheless, our
data reveal that gp130 and ER-localized vIL-6 activity through the signal transducer can
contribute to high constitutive levels of active pSTAT3, a factor that has been linked to cell
growth and survival in PEL and other neoplasias as well as under normal physiological
conditions [264,279,280], and to promotion of cell proliferation and viability. It is
noteworthy that gp130-dependent constitutively high levels of active STAT3 have also
been detected in endothelial cells latently infected with HHV-8, although via (paracrine)
mechanisms independent of vIL-6 [281]. Furthermore, de novo infection of blood
endothelial cells leads to STAT3 induction and gp130-dependent, but vIL-6-independent,
lymphatic endothelial cell reprogramming [18]. Thus, STAT3 signaling, in part mediated
via gp130, may be generally important for maintenance of HHV-8 latency and viability of
latently infected cells and may also contribute to virus-associated neoplasia.
In conclusion, the data presented here document for the first time the significance
of gp130-mediated signaling for proliferation and survival of PEL cells, the importance of
gp130 as a contributor to constitutively high levels of functionally significant STAT and/or
ERK signaling in at least some PEL cell lines, and the contribution of ER-localized vIL-
6/gp130 interactions for PEL cell growth and viability. These data broaden our
understanding of the role and mechanisms of action of latently expressed vIL-6 in PEL
cell biology and provide further support for the idea that drug targeting of the viral cytokine
and its signal transducer, within the ER compartment, may represent a useful therapeutic
strategy.
Page 79
65
FIGURES
Figure 3.1. Effects of gp130 depletion on PEL cell growth and viability. (A) Triplicate
cultures of BCBL-1, JSC-1, BC-1, and BC-3 PEL cells and HHV-8-negative BJAB B-cells
were transduced either singly (BCBL-1, JSC-1, and BJAB) or dually (BC-1 and BC-3) with
gp130-directed shRNA(s) by lentiviral infection, and cells were counted for three days
following normalization of cell numbers (see Chapter 2 and Methods above). Error bars
represent divergence from mean values determined for the triplicate cultures. Western
blotting of cell lysates derived from pooled cultures at the end of the growth experiment
(on day 3) was carried out to verify gp130 depletion (shown below the associated growth
curves). For BC-3 cells, complete cytostatic effects of gp130 shRNA transduction resulted
in insufficient cell numbers to process for immunoblotting. Levels of gp130 in BJAB cell
lysates were below the limit of detection; the growth data show lack of general cytostatic
effects of lentiviral-mediated gp130 shRNA transduction. (B) Samples of gp130 and NS
shRNA-transduced BCBL-1 and JSC-1 cultures harvested on day 2 from the growth curve
experiments shown in panel A were analyzed for proliferation. Rates of proliferation were
determined by detection of BrdU incorporation using Alexa Fluor 488-conjugated BrdU-
specific antibody for immunofluorescence visualization of S-phase cells and
counterstaining with DAPI to detect all nuclei (see Chapter 2 and Methods above).
Relative proliferation rates are shown as the percentage of cells in S-phase (BrdU-
positive) for the respective cultures. Data were derived from two random fields (>100
cells/field) for each slide-spotted culture sample. Examples of BrdU and DAPI co-staining
(sections of a single field, BCBL-1 cells) are shown below the quantified data. (C) Rates
of apoptosis in these same PEL cultures were determined by annexin V-Cy3 binding. DAPI
staining was again used to visualize nuclei, enabling calculation of the percentage of cells
undergoing apoptosis. Examples of annexin V-Cy3 and DAPI co-staining (of JSC-1 cells)
are shown below the graphs. Cells were harvested on day 3 for the apoptosis assays.
Page 81
67
Figure 3.2. Contribution of gp130 signaling to levels of active STATs and ERKs in PEL
cells. Phosphotyrosine-specific antibodies were utilized in immunoblotting to detect and
determine the levels of phosphorylated (active) gp130-targeted STAT3 (panel A), STAT1
(panel B), and ERKs 1 and 2 (panel C) (pSTAT3, pSTAT1, and pERK) present in lysates
of PEL cells depleted of gp130 relative to levels of these proteins in NS shRNA-transduced
controls. Antibodies detecting total (phosphorylated and unphosphorylated) STAT3 and
ERK1/2 and β-actin were used to verify equivalent protein loading on SDS-PAGE gels and
transfer to blotting membranes. For each set of lysates (used for single or multiple
phospho-protein detection), effective depletion of endogenous gp130 in the lentiviral-
shRNA infected PEL cultures was verified by immunoblotting for gp130. Monitoring of
growth of NS-shRNA versus gp130-shRNA cultures for BCBL-1, JSC-1, and BC-1 cells
over three days (as in Figure 3.1) confirmed the growth-inhibitory effects of gp130
depletion (data not shown); cells were harvested on the third day for western analyses.
For BC-3 cultures, all cells were harvested 48 hrs after lentiviral infection (“day 0”) to
ensure sufficient material for immunoblotting.
Page 83
69
Figure 3.3. Contributions of gp130-activated STATs to PEL cell growth. (A) Appropriate
lentiviral-shRNA vectors (see Methods above) were used in combination to transduce
BCBL-1 and JSC-1 PEL cells to deplete STAT3. Growth curves were derived from
triplicate cultures transduced separately with the NS and STAT3-targeting lentiviral-
shRNA vectors and monitored in parallel (see Chapter 2). Error bars represent deviations
from mean cell density values at each 24-hr timepoint over the three-day experiment.
Pooled cultures were harvested at the end of the experiment (day 3) for western analysis
of cell lysates to check for depletion of STAT3. (B) Proliferation of STAT3-depleted versus
NS shRNA-transduced BCBL-1 and JSC-1 cells were examined by BrdU incorporation
assay, as outlined in Figure 3.1, applied to cells harvested on day 3 (5 days after shRNA
transduction). (C) Rates of apoptosis in these same culture samples were determined by
annexin V-Cy3 binding assays and co-staining of cell nuclei with DAPI (see above, Figure
3.1 legend, Chapter 2). (D) STAT1 depletion experiments were performed to examine the
potential contributions of STAT1 to BCBL-1 and JSC-1 cell growth. STAT1 depletion was
mediated by transduction of PEL cultures with a lentiviral vector encoding a STAT1
mRNA-targeting shRNA (see Methods above), and NS shRNA-encoding lentivirus was
used as a control as before. While substantial depletion of STAT1 was apparent for each
cell line (immunoblots, right), growth of neither was altered significantly as a result.
Chemiluminescence signals from western blots were digitally captured, quantified using
imager-associated software, and normalized to β-actin signal. Levels of pSTAT1 in JSC-
1 cultures was 1.9-fold reduced relative to STAT1 levels in the corresponding NS shRNA-
transduced controls; no pSTAT1-specific signal was detected in BCBL-1 cell lysates from
STAT1 shRNA-transduced cultures.
Page 85
71
Figure 3.4. Analysis of culture growth, cell proliferation, and apoptosis rates as a function
of ERK depletion. (A) Potential contributions of ERK to the growth of BCBL-1 and JSC-1
cultures were examined by monitoring growth of cells transduced by lentiviral vectors
encoding either ERK-targeting or NS (control) shRNAs. Cell growth assays were
conducted as described in Chapter 2 and the Methods above. Error bars indicate
deviations from average values derived from triplicate cultures. Effective ERK depletion
was verified by immunoblotting of lysates from BCBL-1 and JSC-1 cells pooled from the
respective triplicate cultures and harvested at the end of the experiment. β-actin was used
as a control for equivalent protein loading and membrane transfer. (B) Relative
proliferation rates of BCBL-1 and JSC-1 cultures transduced with NS or ERK1/2-directed
shRNAs were determined by BrdU incorporation assay (see Chapter 2 and Methods
above). Cultures were harvested 4 days after shRNA transduction and 2 days post-
normalization prior to growth monitoring (panel A). The proportions of BrdU-positive, S-
phase cells are expressed as a percentage of the total cell population (DAPI+) for BCBL-
1 and JSC-1 cultures (dark and light bars, respectively). Data, expressed as averages,
were derived from multiple random microscopic fields (>100 cells/field) for each. (C)
Apoptosis in the same cultures (harvested 5 days post-transduction and on day 3 of
counting) was identified by annexin V-Cy3 staining; data are expressed as a percentage
of the total DAPI+ population. Average values from multiple random microscopic fields are
shown. (D) Growth experiments equivalent to those shown in panel A were undertaken to
determine the effects of ERK depletion in two additional PEL cell lines, BC-1 and BC-3, in
addition to HHV-8-negative BJAB cells, previously shown to be refractory to ERK kinase
(MEK) inhibition [272] and used here as a control for potential non-specific cytotoxic
effects of ERK shRNA transduction. The PEL cell lines, but not BJAB cultures, were
growth-inhibited by ERK shRNA transduction relative to NS shRNA-expressing controls.
Effective ERK depletion in each of the three cell lines was verified by immunoblotting for
Page 86
72
ERKs 1 and 2; β-actin antibody was used to check for equal protein loading and
membrane transfer. Blots for each of the cell lines are shown below the respective growth
curves.
Page 88
74
Figure 3.5. ER-localized vIL-6 signal transduction via gp130 supports PEL cell growth. (A)
Previously described vIL-6 variant W167G, which cannot form tetrameric (vIL-62:gp1302,
ER-localized) complexes [159,166], was tested in transfected HEK293T cells to verify its
predicted inability to mediate signal transduction when restricted to the ER compartment.
KDEL-tagged, ER-directed forms of vIL-6 and vIL-6.W167G were expressed together with
Fc-tagged gp130 from appropriate expression plasmids. After 24 hrs, cell lysates were
analyzed by immunoblotting for pSTAT3, an indicator of gp130 signaling, and protein A-
agarose-precipitated gp130-Fc was probed with phosphotyrosine-specific antibody for
detection of activated gp130. Only wild-type vIL-6 (vIL-6.K) was able to signal via gp130
in the ER compartment, inducing gp130 and STAT3 phosphorylation, in contrast to vIL-
6.W167G-KDEL (W167G.K) and empty vector (vec). (B) Testing of VKORC1v2 binding by
vIL-6.W167G was undertaken by co-immunoprecipitation assay, which was used previously
for the detection of the vIL-6/VKORC1v2 interaction [173]. CBD-tagged VKORC1v2 was
precipitated with chitin beads from lysates of transfected cells, and precipitated material
was analyzed by immunoblotting for the presence of vIL-6.W167G, wild-type vIL-6 (positive
control), or KDEL motif-tagged hIL-6 (hIL-6.K, negative control), expressed in separately
transfected cultures. Both vIL-6 proteins were precipitated and therefore competent for
interacting with VKORC1v2; binding of VKORC1v2 by vIL-6.W167G verified that the “site
III” mutation affected only gp130 binding. (C) Experiments utilizing shRNA-mediated vIL-
6 depletion and complementation with KDEL-tagged, ER-restricted vIL-6 (vIL-6.K) and vIL-
6.W167G (W167G.K) were undertaken to determine whether vIL-6 signaling through ER-
localized gp130 was important for PEL cell growth. BCBL-1 and JSC-1 cells were first
transduced with lentiviral vectors expressing either vIL-6.K or vIL-6.W167G.K (designed to
be refractory to vIL-6 shRNA) and then transduced with lentiviral vectors expressing non-
silencing (NS) or vIL-6-directed shRNAs. Triplicate cultures were monitored for growth as
outlined in Chapter 2 and in the Methods above; error bars indicate standard deviations
Page 89
75
from mean values of cell density. Below the plots, ethidium bromide-stained agarose gels
are shown containing RT-PCR-amplified vIL-6 mRNA sequences extracted from lysates
of PEL cell samples derived from the growth assay cultures. The data provide evidence
of appropriate and equivalent expression of each of the vIL-6 constructions. RT-PCR
products amplified with PCR primer pairs for vIL-6.K (v) and β-actin (a), used as a
normalization control, are indicated. Omission of reverse transcriptase (-RT) provided a
control for RNA-derived vIL-6.K PCR products (i.e., lack of DNA contamination).
Page 91
77
Figure 3.6. Inhibitory targeting of vIL-6/gp130 signaling diminishes PEL cell proliferation
and survival. (A) Functional testing of KDEL motif-fused (ER-directed and retained) and
Flag epitope-tagged soluble gp130 (sgp130.K) as an inhibitor of ER-localized vIL-6
signaling in transfected HEK293T cells. Cultures were co-transfected with expression
vectors for sgp130.K (or empty vector control, vec) and either vIL-6, vIL-6-KDEL (vIL-6.K),
or hIL-6. Cell lysates were harvested 48 hrs post-transfection for preparation of cell
lysates. These were analyzed by western blotting for detection of pSTAT3, an indicator of
gp130 signaling, and β-actin (protein loading control). Both vIL-6 (mainly ER-localized but
also secreted) and KDEL-tagged vIL-6 (completely ER-retained [115]) signaling were
inhibited by sgp130.K, whereas signaling by hIL-6 (efficiently secreted) was unaffected.
The data verified the ER-restricted, vIL-6/gp130-inhibitory activity of sgp130.K. (B)
Confirmation of intracellular localization of KDEL motif-fused and Flag-tagged sgp130 and
comparison to (surface-expressed) full-length gp130 via Flag- and gp130-based
immunofluorescence staining of transfected cells. Alexa Fluor 594-conjugated secondary
antibody was used for detection of (red) fluorescence signal. Each protein was expressed
together with KDEL-tagged GFP (GFP.K, used previously as an ER marker [115]) in
vector-co-transfected HEK293T cells. As expected, Flag-specific staining for
sgp130.Flag.KDEL [sgp130(Flag).K] was evident intracellularly and was coincident with
ER-localized GFP signal, whereas gp130 staining was localized mainly to the cell surface.
Stacked (“total”) and representative individual Z-sections (“slice”) are shown for two
examples (1 and 2) taken from each of the transfected cultures. (C) BCBL-1 and JSC-1
cells were transduced with equivalent titers of “empty” (vec) or sgp130-KDEL (sgp130.K)
expressing lentiviral vectors, and growth of cell density-normalized cultures was monitored
after a two-day recovery period as described in Chapter 2 and in the Methods above. Error
bars represent standard deviations from derived average values. (D) Relative proliferation
rates of PEL cells at “day 2” (from growth assays) were determined by BrdU incorporation
Page 92
78
assays (see Methods and Chapter 2). (E) In a separate experiment, transduction of
sgp130.K into BCBL-1 and JSC-1 cultures led to induction of apoptosis, as determined by
annexin V-Cy3 staining. Cells were harvested and analyzed 3 days post-transduction with
test (sgp130.K) or control (vec) lentiviral vectors.
Page 94
80
CHAPTER 4: ROLE OF GP130 IN VIL-6-MEDIATED VIRUS REPLICATION
Page 95
81
Summary
HHV-8 vIL-6 has been implicated in the pathogenesis of HHV-8-associated
disease and plays a role in maintaining PEL cell growth and viability in addition to
contributing to elevated levels of phosphorylated STAT3 via its interaction with gp130.
Furthermore, STAT3 and ERK1/2 are required for optimal PEL cell growth and survival.
Additionally, vIL-6 interactions with VKORC1v2 contribute to the pro-growth and survival
activities of vIL-6. While the previous study (Chapter 3) investigated the role of vIL-6 in
PEL cell latency, the effects of vIL-6/gp130 signaling on virus replication are unknown.
Thus, the goal of this study was to determine the role of vIL-6 and gp130 in HHV-8
productive replication in both PEL and endothelial cells.
To assess the effects of vIL-6 and gp130 signaling on HHV-8 replication, vIL-6,
gp130, and gp130-induced STAT3 were depleted in both PEL and endothelial cultures.
Furthermore, depletion-complementation experiments were utilized to determine the
relevance of the ER-localized vIL-6/gp130 interaction as it relates to virus replication.
Finally, ER-localized gp130 signaling mutants were employed to evaluate the specific
contributions of vIL-6/gp130-mediated STAT and ERK signaling to virus replication.
Depletion and depletion-complementation experiments revealed that ER-localized
vIL-6 activity via gp130 and gp130-activated STAT signaling, but not ERK activation, were
critical for vIL-6 pro-replication activity. Additionally, vIL-6 and gp130 depletion did not
affect de novo infection or the establishment of latency. These data significantly extend
current understanding of vIL-6 function and associated mechanisms in HHV-8 biology.
Page 96
82
HUMAN HERPESVIRUS 8 VIRAL INTERLEUKIN-6 SIGNALING THROUGH GP130
PROMOTES VIRUS REPLICATION IN PRIMARY EFFUSION LYMPHOMA AND
ENDOTHELIAL CELLS
This work has been published, and sections are reproduced here (with modifications) with permission from the American Society for Microbiology (Copyright © American Society for Microbiology, [Journal of Virology, 88, 2014, 12167-12172, doi: 10.1128/JVI.01751-14]).
Cousins E, Gao Y, Sandford G, Nicholas J (2014) Human herpesvirus 8 viral interleukin-6 signaling through gp130 promotes virus replication in primary effusion lymphoma and endothelial cells. Journal of Virology 88(20):12167-12172. INTRODUCTION
The previous chapter described the role of vIL-6 in PEL cell growth and viability
and determined that the ER-localized vIL-6/gp130 interaction is critical for these pro-
growth and survival activities of the viral cytokine. Additionally, the elevated levels of
phosphorylated STAT3 commonly observed in PEL could be attributed, in part, to vIL-
6/gp130 signaling. This chapter aims to expand on these findings and to characterize the
role of vIL-6 and its gp130-mediated downstream signaling components in the reactivation
and replication of HHV-8 in both PEL and endothelial cells.
As has been stated previously, HHV-8 is associated with PEL in addition to MCD
and KS [256-258,282]. HHV-8-encoded vIL-6 promotes cell growth and survival,
inflammation, and angiogenesis and is implicated in pathogenesis and promotes disease
progression [115,144,147,169,259]. However, its role in normal virus biology remains
unclear. vIL-6 and hIL-6 share ~25% sequence identity, fold into similar three-dimensional
structures, and initiate signaling through similar complexes [157-159,165,260,261]. While
both hIL-6 and vIL-6 form hexameric signaling complexes comprised of two gp80 receptor
subunits, two gp130 signal transducers, and two IL-6 molecules (gp802-gp1302-IL-62), vIL-
6 is unique in that it can also signal from the ER via tetrameric complexes (gp1302-vIL-62)
[159,165,170]. vIL-6 can be expressed during PEL cell latency in addition to being
Page 97
83
maximally expressed during the lytic stage of the viral life cycle [115,116]. Furthermore,
latently infected PEL cell viability is maintained in part via vIL-6/gp130 signaling from the
ER and also through its interaction with VKORC1v2 [169,173]. The vIL-6/VKORC1v2
interaction has also been shown to contribute to productive replication [175].
Upon vIL-6-induced gp130 dimerization, phosphorylation of specific gp130
tyrosine residues initiates either STAT1/3 (Y767, Y804, Y905, and Y915) or MAPK (Y759)
signaling [156,161-163]. Activated STAT3 and ERK1/2 have been shown to promote
growth and survival of PEL cells, and vIL-6/gp130 signaling contributes [40,152,169].
However, the role of vIL-6 in HHV-8 lytic replication is unclear. One study utilized a vIL-6-
knockout recombinant virus to assess the role of the viral cytokine in virus replication in
HHV-8 negative BJAB cells, but no effect was observed [283]. A second study found that
vIL-6 depletion in BCBL-1 and JSC-1 PEL cells resulted in less HHV-8 virus production
following chemically-induced reactivation in culture. No other studies have been
conducted to assess whether vIL-6 plays a role in HHV-8 productive replication; thus, we
sought to determine the effects of vIL-6/gp130 signaling on HHV-8 lytic replication in both
PEL and endothelial cells.
METHODS
Cell culture, transfections, and lentivirus production
HEK293T, Vero-r219, TRExBCBL1-RTA, JSC-1, and TIME-TRE/RTA cells were
maintained as described in Chapter 2. For all transfections, HEK293T cells were
transfected using standard calcium-phosphate-DNA co-precipitation methods as detailed
in Chapter 2. Cells were harvested 24 to 48 hrs post-transfection, and lysates were
prepared as outlined previously (Chapter 2). Lysates were analyzed immediately via
western blot analysis or stored at -20°C for later use.
Page 98
84
To produce lentivirus, HEK293T cells were transfected with pYNC352-encoded
shRNAs (specific for vIL-6, gp130, NS, or STAT3) or pDUET001-encoded plasmids
(empty vector, wild-type [WT] gp130-Flag, gp130.STAT-Ys, gp130.ERK-Y, and
gp130.ΔYs) or pYNC419-encoded plasmids (mvIL-6.KDEL, mvIL-6-W167G-KDEL, or
luciferase) with Pax2 gag/pol and VSV/G expression vectors as described previously and
in Chapter 2 [173]. To harvest lentivirus, medium was collected 2 days post-transfection,
filtered, and centrifuged as detailed in Chapter 2. Viral pellets were resuspended in RPMI
1640 medium and stored at -80°C until needed. Prior to experimental use, lentiviruses
were titered on HEK293T cells to ensure equivalent titers as described previously and in
Chapter 2 [169].
HHV-8 r219 virus was produced as described in Chapter 2 for the infection of naïve
TIME-TRE/RTA cells. Harvested r219 virus was titered on naïve TIME-TRE/RTA cells via
virus-encoded GFP expression prior to infection of TIME-TRE/RTA cells (Chapter 2). After
allowing cultures to recover for two days, r219-infected TIME-TRE/RTA cells were
transduced with lentiviral vectors to assess HHV-8 reactivation (Chapter 2).
Plasmids and Primers
The pYNC352 shRNA-encoded lentiviral vectors (NS, vIL-6, gp130, and STAT3)
have been described [115,169]. The mvIL-6.KDEL, mvIL-6.W167G.KDEL, and luciferase
lentiviral pYNC419 vectors have been reported [115,169]. pSG5-derived vIL-6 and hIL-6
and the pDUET001 vector have also been reported [166,168,270]. To generate the
pDUET001-encoded gp130 WT and signaling mutants that are resistant to depletion by
gp130-specific shRNA, wild-type gp130, gp130.STAT-Ys (Y759F-mutated), gp130.ERK-
Y (Y759 only, all other Ys mutated to F), and gp130.ΔYs (pan Y-to-F-mutated) (described
in [168]) were PCR-amplified from the pEF-BOS vector using gp130-BamHI-FWD (5’
TTGGATCCATGTTGACGTTGCAGACTTGG 3’) and gp130-1205res-REV (5’
Page 99
85
AGATGGTCTGTCCTCGTACGTAATTCCGCTTGCTTCTTCACTCCAGT 3’) primers to
amplify the 5’ end of the gene. To add a C-terminal FLAG epitope and to amplify the 3’
end of WT gp130 and gp130.STAT-Ys, gp130-1205res-FWD (5’
GAAGAAGCAAGCGGAATTACGTACGAGGACAGACCATCTAAAGCACC 3’) and
gp130-WT-Flag-REV (5’ TTCTCGAGTCACTTGTCATCGTCGTCCTTGTAATCCTGAGG
CATGTAGCCGCCTTG 3’) primers were used for PCR amplification. Similarly, gp130-
1205res-FWD and gp130-deltaY-Flag-REV (5’ TTCTCGAGTCACTTGTCATCGTCGTCC
TTGTAATCCTGAGGCATGAAGCCGCCTTGCCGTACAGT 3’) primers were used to
amplify the 3’ end of gp130.ERK-Y and gp130.ΔYs and to add a FLAG epitope to the C-
terminus. The 5’ and 3’ amplicons for each gp130 gene (WT or signaling mutant) were
then used as templates to amplify across the entire length of gp130-Flag; the resulting
PCR products were resistant to the gp130-specific shRNA and contained C-terminal Flag
epitope tags. Finally, the gp130-shRNA-resistant and Flag epitope-tagged amplicons were
ligated into pDUET001 between the BamHI and XhoI restriction sites, replacing the GFP
ORF. All of the constructions were verified by sequencing.
RT-PCR primers were designed to amplify gp130-Flag sequences in transduced
TRExBCBL1-RTA cells in order to verify the expression of wild-type and gp130 signaling
mutant plasmids. The following primers were used: gp130-Flag-FWD (5’
TAGATGGCGGTGATGGTATTT 3’) and gp130-Flag-REV (5’
CTTGTCATCGTCGTCCTTGTA 3’). Previously validated glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) primers were used to amplify GAPDH as a positive control:
GAPDH-FWD (5’ TTGCCATCAATGACCCCTTCA 3’) and GAPDH-REV (5’
CGCCCCACTTGATTTTGGA 3’); these primers were designed at the RRC Core
Genomics Facility at the University of Illinois at Chicago.
Page 100
86
Antibodies and Western blotting
The following commercially available antibodies were used: β-actin (Sigma, St.
Louis, MO; catalog number A2228), pSTAT3 (Cell Signaling Technology, Beverly, MA;
catalog number 9131), STAT3 (Santa Cruz Biotechnology, Santa Cruz, CA; catalog
number sc-482), pERK (Santa Cruz Biotechnology; catalog number sc-7383), LANA
(Advanced Biotechnologies, Columbia, MD; catalog number 13-210-100), and gp130
(Santa Cruz Biotechnology; catalog number sc-655). Rabbit anti-serum to vIL-6 was
generated previously in our laboratory [166]. Cy3-goat anti-rat IgG fluorescent antibody
(Life Technologies, Carlsbad, CA; catalog number A10522) was used to detect LANA by
immunofluorescence assay.
Western blotting was conducted by separating heat-denatured and reduced lysate
samples via SDS-PAGE and transferring samples to nitrocellulose membranes using
standard protocols described in Chapter 2. Following antibody incubation, immuno-
targeted proteins were visualized via standard chemiluminescence assay.
Quantification of HHV-8 titers following reactivation
For HHV-8 reactivation experiments in TRExBCBL1-RTA and JSC-1 PEL cells,
cells were transduced with pYNC352-encoded shRNAs as described previously [169] and
in Chapter 2, allowed to recover for 2 days, normalized for cell number, and then induced
with Dox (TREx-BCBL1/RTA cells; 1 μg/μl) or TPA (20 nM) and NaB (0.5 μM) for JSC-1
cells. NaB was removed from JSC-1 cultures after 12 hrs of incubation. Transduced PEL
cells were maintained in Dox or TPA for 4 days, and HHV-8 virus was harvested from
culture media as detailed in Chapter 2. Cells were counted at the time of virus harvest to
ensure no drastic differences in cytotoxicity between cultures. Aliquots of harvested virus
were added to naïve TIME-TRE/RTA cells plated at ~90% confluency in 12-well plates,
Page 101
87
centrifuged, and incubated for an additional hr as outlined in Chapter 2. One day post-
infection, cells were trypsinized and added to chamber slides for LANA immunostaining.
For reactivation of HHV-8 in TIME-TRE/RTA endothelial cells, cells were seeded
at ~90% confluency in 12-well plates and then infected with HHV-8 r219 virus (described
above and in Chapter 2). After recovering for 48 hrs, TIME-TRE/RTA GFP fluorescence
was assessed to verify a high viral titer (~90% of cells infected). Cells were then infected
with pYNC352-encoded shRNA-expressing lentiviruses as described above and in
Chapter 2. Two days post-shRNA lentiviral infection, cells were induced with Dox (1 μg/μl).
Medium was removed from infected and induced cells every 48 hrs for 6 days, and fresh
medium containing Dox was added to cell cultures. Harvested media containing virus were
stored at -80°C. Reactivated TIME-TRE/RTA cells were also visualized daily for RFP
fluorescence to assess rates of reactivation. Medium collected from each sample was
pooled and centrifuged at 16,000 rpm for 2 hr at 4°C; viral pellets were resuspended in
EMB-2 without serum or supplements. Naïve TIME-TRE/RTA cells were incubated with
an aliquot of collected virus and then transferred to chamber slides for immunostaining as
described above.
For depletion-complementation experiments in TRExBCBL1-RTA cells, cells were
first transduced with pYNC419-expressing mvIL-6.KDEL, mvIL-6.W167G.KDEL, or
luciferase to assess vIL-6 effects on virus replication or were transduced with pDUET001-
expressing empty vector, WT gp130-Flag, gp130.STAT-Ys, gp130.ERK-Y, or gp130.ΔYs
to determine the contribution of STAT and ERK signaling to virus replication. Cells were
allowed to recover for 2 days prior to being transduced with pYNC352-encoded shRNAs
as described in Chapter 2. Dually-transduced cells were induced with Dox; medium was
collected, and virus was harvested as outlined in Chapter 2. Naïve TIME-TRE/RTA cells
were infected with aliquots of harvested virus as described above, and cells were
transferred to chamber slides prior to immunostaining.
Page 102
88
Chamber slides containing infected TIME-TRE/RTA cells were stained for LANA
expression to quantify HHV-8 infection, as outlined in Chapter 2. Chamber slides were
washed, fixed in ice cold methanol, and incubated with anti-LANA antibody (1:100 dilution
in PBS blocking buffer [Thermo Fisher Scientific, Waltham, MA]) as outlined in Chapter 2.
After thoroughly washing, slides were incubated with Cy3-goat anti-rat secondary antibody
(1:200 dilution in PBS blocking buffer), washed, and then stained with DAPI to detect
nuclei. The percentage of LANA+ cells in the total DAPI+ cell population was determined
from 6 random fields (>50 cells/field) in each duplicate culture.
De Novo Infection
TIME-TRE/RTA cells were transduced with lentivirus-encoding shRNAs specific
for NS, gp130, or vIL-6. Two days post-transduction, cells were infected with a
subsaturating dose of HHV-8 r219 virus. One day post-HHV-8 r219 infection (and 3 days
post-lentiviral transduction), infected and transduced TIME-TRE/RTA cells were washed,
fixed, and stained for LANA and DAPI expression as described in Chapter 2. Multiple (>6)
fields were visualized for each duplicate culture, and the percentage of LANA+ cells in the
total DAPI+ cell population was calculated as outlined above.
Annexin Staining
TIME-TRE/RTA cells were plated on chamber slides at a confluency of ~80%. The
following day, TIME-TRE/RTA cells were transduced with NS-specific or STAT3-specific
lentivirus-encoded shRNAs. One day post-transduction, cells were fixed and stained for
annexin V-Cy3 as described in Chapter 2. Multiple fields were visualized, and mean values
were reported.
Page 103
89
RESULTS
gp130 and vIL-6 promote HHV-8 replication in PEL and endothelial cells. The
gp130 signal transducer was depleted in TRExBCBL1-RTA and JSC-1 PEL cells
[100,111] and in TIME-TRE/RTA endothelial cells [203] latently infected with HHV-8 r219
[254] using previously established techniques [115,169]. Following depletion, lytic
replication was induced with Dox or TPA and NaB. HHV-8 was collected from PEL and
endothelial culture media, and infectious viral titers were determined as undertaken
previously [175]. Viable (trypan blue-excluding) TRExBCBL1-RTA cell densities at the
time of media harvest were not negatively affected by gp130 transduction (1.31x106 and
1.41x106 cells per ml for NS shRNA-transduced duplicate cultures versus 1.75x106 and
1.83x106 cells per ml for gp130 shRNA-transduced cells). Depletion of gp130 led to
decreased HHV-8 titers in PEL (~3-fold) and endothelial cultures (>8-fold) (Figure 4.1A,
B, C). Similarly, vIL-6 depletion in endothelial cells reduced viral titers by ~3-fold (Figure
4.1D). TIME-TRE/RTA cells were visibly unaffected by gp130 or vIL-6 depletion, and rates
of apoptosis (% annexin V-Cy3+ cells) were equivalent in gp130- and vIL-6-depleted
cultures relative to NS shRNA-transduced controls (data not shown). Rates of reactivation,
measured by ORF59-antigen immunofluorescence (PEL cells, data not shown) and RFP
imaging (Figure 4.1C,D, bottom panels), were equivalent in depleted and control cultures.
Combined, these data identify the importance of gp130 for productive replication in PEL
and endothelial cells and the involvement of vIL-6 in support of productive replication in
endothelial cells in addition to BCBL-1 and JSC-1 cells, reported previously [175]. In
contrast to reactivated productive replication, neither gp130 nor vIL-6 depletion had any
substantial effect on latency titers (measured 3 days post-infection) following de novo
infection of endothelial cells (Figure 4.1E), a process known to involve a burst of
expression of a subset of lytic genes, including vIL-6, prior to rapid latency establishment
[284,285].
Page 104
90
ER-localized vIL-6/gp130 signaling contributes to productive replication.
Dox-inducible TRExBCBL1-RTA PEL cells were used to test the ability of gp130 signaling-
competent and ER-retained vIL-6 to rescue the vIL-6-depletion phenotype. As before,
lentiviral vectors encoding either control (NS) or vIL-6 mRNA-directed shRNAs were used
for transduction. Depletion of vIL-6 led to reduced virus production (luciferase control),
and this phenotype could be rescued with lentiviral vector-transduced vIL-6.KDEL (ER-
retained [115]) but not vIL-6.W167G.KDEL (ER-retained and gp130-dimerization
incompetent in the ER [166,169]) (Figure 4.2). These data identify the involvement of ER-
localized vIL-6/gp130 signaling in support of HHV-8 productive replication.
gp130-mediated STAT signaling, but not ERK signaling, supports HHV-8
replication. To determine the significance of gp130-activated STAT versus ERK signaling
in HHV-8 replication, we employed signaling-altered gp130 variants in gp130 depletion-
rescue experiments. Four C-tail tyrosines of gp130 initiate STAT1/3 signaling, and one
induces MAPK signaling [156,161-163]. Lentiviral vector-cloned gp130 ORFs for
complementation encoded wild-type gp130 (WT), gp130.STAT-Ys (Y759F-mutated,
ERK1/2 signaling-null), gp130.ERK-Y (Y759 only, STAT1/3 signaling-null), and
gp130.∆Ys (inactive). Functional testing in transfected HEK293T cells verified that in the
presence of vIL-6, introduced WT and gp130.STAT-Ys induced STAT3 phosphorylation
above levels supported by endogenous gp130, and gp130.ERK-Y exclusively induced
pERK levels (Figure 4.3A). Unexpectedly, WT gp130 did not support detectable ERK
signaling in response to vIL-6, but the same result was seen for hIL-6 in the presence of
overexpressed gp80 (data not shown). In TRExBCBL1-RTA-based depletion-
complementation experiments, WT and gp130.STAT-Ys, but not gp130.ERK-Y or
negative controls gp130.ΔY and empty vector, fully rescued replication (Figure 4.3B),
indicating that STAT signaling alone is sufficient for vIL-6/gp130-mediated support of
HHV-8 replication.
Page 105
91
Contribution of STAT3 to HHV-8 replication. As previous reports have
demonstrated the importance of STAT3 for latent PEL cell proliferation and viability
[152,169], we examined its role in productive replication. STAT3 depletion led to reduced
HHV-8 titers induced from both PEL and endothelial cultures, although effects in
TPA/NaB-induced JSC-1 cultures were modest despite robust STAT3 depletion (Figure
4.4). The latter may reflect different PEL cell-specific thresholds of STAT3 functionality or
different dependencies on STAT3 for support of HHV-8 replication. It is important to note
that viable cell densities at the end of the experiment were equivalent between NS and
STAT3-directed shRNA-transduced cultures (Figure 4.4 panels A and B, bottom),
indicating that pro-proliferative and anti-apoptotic activities of STAT3 operating in latently
infected PEL cells [152,169] do not account directly for diminished replication resulting
from STAT3 depletion in reactivated cultures. Also, STAT3 depletion in TIME-TRE/RTA
cells had no detectable effect on cell viability (Figure 4.4D). Our data show that STAT3
supports HHV-8 replication, and together with the vIL-6 and gp130 depletion-rescue data
(Figures 4.2 and 4.3) indicate that vIL-6/gp130 signaling via STAT3 contributes
significantly to the pro-replication activities of vIL-6 and gp130.
DISCUSSION
The likely role of vIL-6 in pathogenesis has been described in numerous reports
[115,144,147,169,258,259]. However, investigation of the role of vIL-6 in HHV-8
replication is restricted to one study of vIL-6-null virus replication in (HHV-8-negative)
BJAB cells, which noted no phenotype [283], and our own preliminary vIL-6 depletion
experiments in PEL cells [175]. Previously, vIL-6 was shown to promote HHV-8 replication
in BCBL-1 and JSC-1 PEL cells, but its role in endothelial cells and additional PEL cells
was not evaluated. The present study significantly extends these previous reports,
demonstrating that vIL-6 promotes HHV-8 productive replication in endothelial cells in
Page 106
92
addition to PEL cells, that gp130 contributes significantly to HHV-8 replication in these cell
types, that vIL-6 activity via gp130 can be mediated largely or exclusively through ER-
localized signaling via tetrameric complexes, and that STAT3, but not gp130-activated
MAPK signaling, is likely critical for vIL-6/gp130-enhanced replication.
While it possible that gp130-activated STAT1 signaling could also contribute, lack
of detectable effects of STAT1 depletion on PEL cell growth [169] coupled with the well-
established role of STAT1 in promotion of anti-viral interferon and apoptotic signaling [286]
indicate that this is unlikely. It is noteworthy that previous studies have reported the
importance of STAT3 signaling for replication of viruses, including VZV and hepatitis C
virus [287,288]. For VZV, STAT3-activated survivin was found to be involved in pro-
replication activity, reflecting the pro-survival activities of STAT3 via survivin reported in
PEL cells [152]. However, HCMV blocks STAT3 phosphorylation, though requires it for
optimal replication, and mouse cytomegalovirus induces phosphorylation of STAT3 but
not STAT3-responsive cellular genes, suggesting novel, virus-redirected activities of the
transcription factor [289,290]. Our findings of vIL-6/gp130 and STAT3 involvement in HHV-
8 productive replication could facilitate the development of therapeutic interventions for
the treatment of HHV-8-associated diseases, in which productive replication contributes
significantly.
Page 107
93
FIGURES
Figure 4.1. Involvement of gp130 and vIL-6 in promotion of HHV-8 replication. (A) HHV-8
titers following gp130 depletion in TRExBCBL1-RTA cells. The gp130 signal transducer
was depleted in TRExBCBL1-RTA cells using lentiviral-encoded shRNAs (NS, control or
gp130-specific) described previously [169]. Cell numbers were normalized, and cultures
were reactivated with Dox. Aliquots of harvested virus were applied to naïve TIME-
TRE/RTA cells to determine infectious virus titers by staining for LANA, as described
previously [175]. Multiple random fields were assessed to quantify the number of LANA-
positive cells in the total DAPI-positive population. (B) Importance of gp130 in HHV-8
replication in JSC-1 cells. JSC-1 cells were depleted of gp130 (as described in 4.1A) and
induced with NaB and TPA. Released virus was pelleted from culture media and quantified
as before. (C) and (D) Influence of gp130 and vIL-6 on HHV-8 replication in endothelial
cells. TIME-TRE/RTA cells were first infected with HHV-8 r219 virus [254], then
transduced with shRNA-encoding lentiviral vectors (NS, control; gp130-specific; or vIL-6-
specific), and finally induced with Dox. Lentiviral vector-encoded GFP, expressed above
levels arising from latent r219 HHV-8 genomes, was used to monitor lentiviral infection,
achieved in >90% of cells. HHV-8 reactivation was monitored by visualizing RTA-induced
RFP expression from r219 viral genomes via fluorescence after Dox treatment (examples
shown). Media containing virus were collected for six days and pooled prior to virus
concentration and titration. (E) NS, gp130, and vIL-6 shRNA-transduced TIME-TRE/RTA
cells were infected with HHV-8 r219 two days post-lentiviral transduction to determine
possible effects of gp130 and vIL-6 depletion on de novo infection and establishment of
latency, as determined by LANA staining three days post-HHV-8 r219 infection. For all
panels, data were derived by counting of LANA+ cells in multiple fields to obtain the
percentage of LANA+/DAPI+ cells. Average values from duplicate cultures are shown; error
bars represent deviations of individual values from the means.
Page 109
95
Figure 4.2. Importance of ER-localized vIL-6/gp130 interactions for HHV-8 replication.
TRExBCBL1-RTA cells were transduced with lentiviral expression vectors encoding KDEL
motif-tagged (ER-retained) and shRNA-resistant forms of vIL-6 (vIL-6.K) or vIL-6.W167G
(W167G.K) [115,169] or with lentivirus encoding luciferase (negative control). Two days
post-transduction, cells were transduced with control (NS) shRNA-encoding lentivirus or
lentivirus expressing vIL-6-specific shRNA. Dually-transduced cells were then normalized
for cell density and induced the following day with Dox. Virus was collected and titered as
before (see Figure 4.1 legend). RNA was harvested from a subset of cells using TRIzol,
and cDNA was amplified for vIL-6.KDEL [169] and GAPDH (normalization and positive
control) mRNA to verify expression of the introduced vIL-6 genes (v, vIL-6 RT-PCR; g,
GAPDH RT-PCR; -RT, no reverse transcriptase). Viable cell densities at the time of Dox
treatment (day 0, normalized) and at virus harvest (day 4) are shown (bottom panel). v,
vIL-6-KDEL; v167.K, vIL-6.W167G-KDEL; luc, luciferase.
Page 111
97
Figure 4.3. Contributions of gp130-mediated STAT and ERK signaling to HHV-8
productive replication. (A) Functional assessment of gp130 signaling-tyrosine variants.
HEK293T cells were co-transfected with empty vector (-) or vIL-6 plasmid (+) together with
empty lentiviral vector or lentiviral vectors expressing wild-type gp130, gp130.STAT-Ys
(Y759F-mutated), gp130.ERK-Y (Y759 only, other C-tail tyrosines mutated to F), or
gp130.∆Ys (pan Y-to-F-mutated). Two days post-transduction, cells were lysed for
western blot analysis to detect pSTAT3 and pERK as indicators of gp130-activated STAT
and MAPK pathways. (B) Role of STAT and ERK signaling in vIL-6/gp130-promotion of
HHV-8 replication. TRExBCBL1-RTA PEL cells were transduced with empty lentiviral
vector (pDUET001 [270]) or vectors expressing Flag-tagged wild-type gp130,
gp130.STAT-Ys, gp130.ERK-Y, or gp130.∆Ys. Two days post-transduction, cells were
transduced with either NS (control) or a gp130-specific shRNA-encoding lentiviral vector.
Following Dox-induced HHV-8 reactivation for four days, virus was collected and titered
as described previously. To verify appropriate expression of wild-type gp130 and the
gp130 signaling mutants, a subset of cells was lysed in TRIzol for preparation of RNA
samples. RNA was reverse transcribed and amplified using 3’ Flag- and 5’ gp130-directed
primers to assess expression of transduced gp130-Flag mRNAs. GAPDH-specific primers
were used for normalization and to provide a positive control. Flag, gp130-Flag RT-PCR;
g, GADPH RT-PCR; -RT, no reverse transcriptase.
Page 113
99
Figure 4.4. Role of STAT3 in HHV-8 replication in PEL and endothelial cells. (A)
TRExBCBL1-RTA cells were transduced with control (NS) and STAT3-specific shRNA-
encoding lentiviral vectors. After two days, lytic reactivation was induced with Dox for four
days, and virus was then collected and concentrated from culture media for titration.
Experimental procedures and data collection and calculations were as outlined in the
legend to Figure 4.1. Subsets of transduced cells were harvested at the time of induction
and lysed for western blot analysis to verify STAT3 depletion. Densities of viable (trypan
blue-excluding) cells were normalized at the time of lytic induction (day 0) and determined
at the end of the experiment (day 4) (bottom panel). (B) The same analyses were
undertaken for JSC-1 cultures, induced with TPA/NaB and harvested after four days for
virus titration. A subset of cells was lysed for western blotting at the time of induction to
verify STAT3 depletion. Viable cell densities at the start and end of the experiment are
shown (bottom). (C) Dox-induced TIME-TRE/RTA cultures were analyzed in the same
way except that virus was harvested following six days of Dox treatment; parallel cultures
were harvested at the time of induction for western blot analysis. (D) Rates of apoptosis
(% annexin V-Cy3+/DAPI+ cells) in similarly transduced TIME-TRE/RTA cultures were
unaffected by STAT3 depletion.
Page 115
101
CHAPTER 5: DISCUSSION AND PERSPECTIVES
Page 116
102
DISCUSSION OF MAJOR FINDINGS
The goal of this project was to further characterize the function of vIL-6 as it relates
to normal HHV-8 biology and pathogenesis. Many previous studies have sought to
determine the role of vIL-6 in viral pathogenesis and tumorigenesis. While pathogenesis
studies are important, virus-induced tumorigenesis is an unintentional byproduct of HHV-
8 infection and occurs in rare cases. In most immunocompetent individuals, host immune
responses are able to control the virus, and HHV-8-associated malignancies are not
observed. When the immune system is incapable of controlling virus replication and
spread, the resulting increase in infection events provides many more opportunities for the
virus to cause malignant transformation. Thus, determining how vIL-6 facilitates viral
infection under normal physiological conditions is crucial for understanding how the virus
maintains its fitness and expands within the host. The development of therapeutics to
combat viral infection and HHV-8-mediated neoplasia will require a detailed understanding
of virus-host interactions under normal, non-transformative conditions.
Studies presented here indicate that vIL-6 promotes latently infected PEL cell
growth and survival, and they identify an important and therapeutically-targetable
mechanism of PEL cell maintenance. Additionally, these studies demonstrate a role of
vIL-6 signaling in normal virus latency in untransformed B-cells and possibly other cell
types. Thus, disruption of vIL-6 signaling through specific abrogation of its interaction with
gp130 could be a promising means not only to target PEL therapeutically but also to attack
latently infected cells more generally and limit viral load. Furthermore, this project has
identified intracellular signaling by vIL-6 specifically within the ER as leading to high levels
of activated (phosphorylated) STATs 1 and 3 and ERKs 1 and 2. Moreover, STAT3 and
ERK1/2 expression are important for PEL cell growth and survival, and these pathways
could be targeted pharmacologically. These findings are in agreement with a previous
study that determined that STAT3 promotes PEL cell growth and survival via the
Page 117
103
upregulation of survivin though data described in this dissertation project were generated
using an alternative method to assess the contribution of STAT3 to PEL cell proliferation
and viability. This current work (described in Chapter 3) utilized shRNA-encoding lentiviral
vectors to effectively and specifically deplete STAT3 in multiple PEL cell lines in order to
confirm previous findings and to establish that vIL-6/gp130 signaling from the ER
compartment contributes significantly to the increased levels of phosphorylated STAT3.
Furthermore, a similar approach was undertaken to assess the role of ERK1/2 signaling
as it relates to PEL cell growth and survival. For the first time, ERK1/2 was found to play
a major role in the proliferation and viability of PEL cells in culture. This finding, along with
data demonstrating that depletion of ERK1/2 in HHV-8 negative BJAB cells had no effect
on cell growth, suggests that ERK1/2 activity may be specifically required by HHV-8-
infected PEL cells. The effects of STAT3 and ERK1/2 on PEL cell growth were assessed
by measuring rates of both cell proliferation and apoptosis; depletion of both signaling
molecules led to decreased rates of cell growth and increased rates of apoptosis.
Moreover, vIL-6-mediated signaling through gp130 was found to be responsible, at least
in part, for high levels of both phosphorylated STAT3 (in all PEL cell lines tested) and
ERK1/2 (in some PEL cell lines). These data also suggest that the constitutively high levels
of phosphorylated STAT3 observed in PEL cells can be attributed largely to vIL-6/gp130
signaling. Finally, additional experiments were performed to evaluate the importance of
ER-localized vIL-6 signaling; data from these depletion-complementation experiments
indicate that vIL-6/gp130 ER-localized signaling is responsible for promoting PEL cell
growth and survival in culture, as a vIL-6 mutant (W167G, incapable of dimerizing gp130 in
the ER) was unable to rescue PEL cell growth following vIL-6 depletion. Thus, the study
described in Chapter 3 adds to the body of work regarding vIL-6 function and intracellular
signaling in the context of PEL cell proliferation and viability and has implications not only
for PEL therapy but also for normal HHV-8 latency.
Page 118
104
In addition to functioning as a pro-growth and pro-survival viral protein in PEL cells,
we hypothesized that vIL-6 might also play a positive role in productive virus replication
due to its role in prolonging cell survival. Thus, vIL-6 function in HHV-8 lytic replication
was assessed in both PEL and endothelial cells. Using shRNA-encoding lentiviral vectors,
vIL-6 and gp130 were depleted in both cell types. Data from these experiments indicate
that vIL-6 and gp130 are required for efficient HHV-8 replication. Prior to these
experiments, little was known regarding the function of vIL-6 in HHV-8 productive
replication. One study utilized a vIL-6 knock-out recombinant virus to assess replication in
HHV-8 negative BJAB cells, and in this setting, vIL-6 appeared to have no effect on virus
replication [283]. In a second study, the function of vIL-6 in HHV-8 replication was
evaluated in two PEL cell lines (BCBL-1 and JSC-1), and decreased viral titers were
observed following vIL-6 depletion via vIL-6-specific lentivirus-encoded shRNA [175].
These studies also identified vIL-6-targeted VKORC1v2 and associated suppression of
pro-apoptotic CatD were involved in promoting HHV-8 replication. The findings described
in Chapter 4 are novel in that they demonstrate that vIL-6 promotes virus replication in
both endothelial cells and in an additional PEL cell line (TREx-BCBL1/RTA). Additionally,
the contributions of gp130 to HHV-8 productive replication were evaluated using similar
methods, and gp130 depletion led to decreased viral titers in both cell types. Furthermore,
vIL-6 depletion-complementation experiments were conducted using both wild-type and
mutant vIL-6 (W167G) to determine the contribution of vIL-6/gp130 intracellular signaling
on productive replication in PEL cells. This experiment demonstrated that vIL-6/gp130 ER-
localized signaling is required for effective virus replication, as the ER-localized vIL-6
mutant (which was unaffected with respect to VKORC1v2 interaction) was incapable of
rescuing HHV-8 replication following vIL-6 depletion. As expected, ER-directed wild-type
vIL-6 was capable of rescuing replicative titers following lentivirus-mediated shRNA
depletion of vIL-6. STAT and MAPK signaling are activated by vIL-6/gp130 signaling, and
Page 119
105
these downstream signaling molecules were evaluated to determine their contributions to
HHV-8 productive replication. A set of gp130 signaling mutants were over-expressed
following gp130 depletion in a depletion-complementation assay, and data from this
experiment indicate that vIL-6/gp130-mediated STAT3 signaling is required for optimal
HHV-8 replication and that MAPK signaling is not required for HHV-8 replication. Thus,
vIL-6, gp130, and STAT3 have been identified for the first time as positive regulators of
HHV-8 replication.
The studies described here and in other reports suggest that STAT signaling is
critically important for virus replication and host cell proliferation and survival. STATs are
known to be upregulated following viral infection and the activation of type 1 interferons.
While STATs 1 and 2 facilitate and promote the host IFN response, STAT3 negatively
regulates type 1 interferons [291]. Depletion and/or knockout of STAT3 in cell lines leads
to an enhanced type 1 IFN response, and the addition of STAT3 following depletion
dampens the type 1 IFN response [291]. Additionally, STAT3 knockout mice are
embryonic lethal; conversely, STAT1 knockout mice are viable but are unresponsive to
IFNα and IFNƔ, and STAT2 knockout mice also display greater susceptibility to viral
infections [292-294]. Therefore, the activation of STAT3 following virus infection is
beneficial to the virus because STAT3 attenuates the host antiviral response by
suppressing the pro-type 1 IFN response promoted by STATs 1 and 2.
Many viruses, including HHV-8, modulate STAT3 activity to promote virus
replication and to evade host immune responses. For example, VZV induces STAT3
phosphorylation and upregulates survivin both in cell culture and in virus-infected SCID
mice; inhibition of STAT3 and/or survivin expression limits VZV replication and spread in
cell-based models [287]. VZV also inhibits STAT1, which ultimately attenuates the host
type 1 IFN response [287]. HCMV inhibits STAT3 phosphorylation and sequesters the
transcription factor (in its unphosphorylated form) in the nucleus; consequently, IL-6-
Page 120
106
mediated gene expression is inhibited [289]. However, STAT3 activity is necessary for
maximal HCMV replication, and inhibition of STAT3 by various chemical inhibitors resulted
in drastically reduced viral titers [289]. Murine CMV (MCMV) inhibits the transcription of
STAT3-responsive genes (including suppressor of cytokine signaling 3 [SOC3], a negative
regulator of STAT3) while simultaneously upregulating STAT3 phosphorylation [290].
HSV1 latency is maintained by STAT3 expression, and the inactivation of STAT3 via
inhibitors or a dominant negative STAT3 induces HSV1 productive replication [295]. In
addition to herpesviruses, the rabies virus P protein interacts with STAT3, inhibiting the
accumulation of the transcription factor in the nucleus [296]. Furthermore, targeting of
STAT3 by the rabies virus abrogates gp130-mediated signaling [296]. The mumps virus,
a member of the paramyxovirus family, also interacts with STAT3 via its V protein; this
interaction leads to the proteasomal degradation of the transcription factor and the
inhibition of IL-6-induced signaling [297]. HIV-1-encoded Nef activates STAT3 by inducing
the expression of soluble factors, including Mip-1α, in monocyte-derived macrophages
[298]. Hepatitis C virus (HCV) also modulates STAT3 activity via interactions with the HCV
core protein [288]. Addition of constitutively active STAT3 leads to increased HCV
replication, and the inhibition of STAT3 via siRNA or chemicals (AG490, STA-21, and S31-
201) results in lower HCV RNA levels [288]. Thus, the targeting and modulation of STAT3
activity is conserved by many viruses across multiple families, suggesting that STAT3
function is crucial to virus biology. Whether STAT3 acts to enhance or inhibit virus
replication and whether a particular virus induces or suppresses STAT3 activation
presumably is dependent on the particular biological requirements of the viruses, their
specific abilities to modify STAT3 activity, and the cell types infected.
STAT3 not only plays a role in regulating virus infection, but it is also involved in
oncogenesis. STAT3 interacts with RelA, a nuclear factor ĸB (NFĸB) family member,
sequestering it within the nucleus; ultimately, this interaction leads to constitutive NFĸB
Page 121
107
signaling in tumor cells [299]. Furthermore, STAT3 and NFĸB are linked via IL-6. NFĸB
regulates IL-6 expression at the transcriptional level, and IL-6 signaling promotes STAT3
phosphorylation and activation [300]. Thus, NFkB signaling leads to elevated levels of IL-
6, and in turn, high levels of phosphorylated STAT3; this positive feedback loop between
NFkB and STAT3 results in increased inflammation via the upregulation of pro-
inflammatory cytokines [301]. Inflammation is known to contribute to tumorigenesis, and
several cancers have been linked to inflammation caused by virus infection. Virus-induced
inflammatory cancers include HHV-8-associated KS, liver cancer (associated with
hepatitis B and/or C virus), cervical cancer (linked to human papillomavirus [HPV]), and T
cell leukemias and lymphomas (associated with human T lymphotrophic virus [HTLV-1])
[255,302-304]. Viral and cellular cytokines, VEGF, host immune cell extravasation, and
repetitive DNA damage promote inflammation. Therefore, STAT3 activation can lead to
inflammation and a microenvironment that is hospitable for tumor development and
growth.
In immunocompetent individuals, HHV-8 infection does not generally result in
oncogenesis. However, virus infection promotes key pathways, such as angiogenesis and
inflammation, which are often involved in tumor development. HHV-8-encoded vIL-6
increases the expression of VEGF in cell culture and mouse models [147]. VEGF
promotes angiogenesis and causes increased permeability of existing vasculature, which
enhances the infiltration of immune cells into the lesion [305]. HHV-8-encoded vIL-6 also
inhibits neutrophil recruitment during acute inflammation in a murine peritoneal
inflammation model [148]. Data from this mouse model indicate that vIL-6 promotes
STAT3 phosphorylation, CCL-2 production, and inhibition of IL-1β-mediated release of
CXCL8 (IL-8) [148]. CCL-2 recruits monocytes and basophils to the lesion and directs the
immune response away from the anti-viral Th1 response and toward the antibody-
mediated Th2 response [148,306]. HHV-8-encoded viral chemokines (vCCL-1, vCCL-2,
Page 122
108
and vCCL-3) also promote Th2 polarization, which weakens the host anti-viral immune
response and promotes maintenance of the virus (Table 1.1). IL-8 is a chemotactic factor
for neutrophils and promotes angiogenesis. HHV-8-encoded vFLIP induces IL-8
production that counteracts CCL-2-mediated suppression of IL-8 release, and vFLIP-
regulated induction of IL-8 is dependent on functional NFĸB [226,307]. Thus, STAT3 is a
pleiotropic factor that promotes the growth and viability of virus-infected cells, enhances
inflammation and IL-8 expression via interactions with NFkB, and drives Th2 polarization.
HHV-8 vIL-6 induces STAT3 phosphorylation; therefore, the viral cytokine mediates pro-
inflammatory, pro-angiogenic, and pro-survival signaling in virally infected cells.
IMPLICATIONS FOR CLINICAL RESEARCH
Treatment of viral infections and virus-associated neoplasias has been difficult due
to the fact that some viruses, such as herpesviruses and HIV, have both latent and lytic
phases. Traditional antiviral drugs were designed to target virus-encoded proteins such
as TK, DNA polymerase, reverse transcriptase, and protease. Unfortunately, these drugs
require that the virus be actively replicating. Because the virus is not productively
replicating during latency, many proteins are not expressed and cannot be effectively
targeted during this phase of the viral life cycle. A minority of viral proteins are expressed
during latency. These proteins interact with host proteins to promote latency and genome
maintenance. Therapeutic targeting of intracellular interactions between viral and host
proteins during viral latency may provide a means to target the latent virus population.
Data presented in this body of work suggest means of novel therapeutic
interventions for the treatment of HHV-8-associated disease. Specifically, the
establishment that interactions between vIL-6 and gp130 are critical for viral latency and
viral replication supports the targeting of this protein-protein interaction in future drug
Page 123
109
design efforts. By preventing this interaction with small molecules or peptides, critical viral
processes might be inhibited. Many experimental approaches were undertaken to
evaluate the vIL-6/gp130 interaction. Specific findings are stated below.
1) Expression of both gp130 and vIL-6 in HHV-8-positive PEL cells is required for cell
growth and survival in culture.
2) Gp130 contributes to elevated levels of active (phosphorylated) STAT1 and STAT3
in PEL cells.
3) Gp130 contributes to levels of active (phosphorylated) ERK1/2 in a subset of PEL
cell lines.
4) STAT3 and ERK1/2 signaling are required for PEL cell proliferation and survival,
but STAT1 is not.
5) ER-localized vIL-6/gp130-mediated signaling promotes PEL cell growth and
survival.
6) Optimal HHV-8 replication requires expression of vIL-6, gp130, and STAT3 in both
PEL and endothelial cells; these proteins do not play a substantial role in de novo
infection (of endothelial cells) or the establishment of latency.
7) The ER-localized vIL-6/gp130 interaction promotes HHV-8 productive replication.
8) Gp130-mediated STAT3 signaling promotes virus replication.
Taken together, these data suggest that the ER-localized vIL-6/gp130 interaction is critical
for multiple viral processes, including growth of latently-infected cells, cell viability, and
productive viral replication. Therapeutic interventions targeting the vIL-6/gp130 interaction
or downstream signaling could theoretically inhibit the growth of latently infected cells
while also limiting the production of virus in lytically infected cells.
Page 124
110
FUTURE DIRECTIONS
While these studies have shown that vIL-6-mediated signaling through gp130
helps maintain viral latency and is required for optimal lytic replication, many questions
remain. Data indicate that STAT3 and ERK1/2 participate in vIL-6 signaling downstream
of gp130, but the effects of STAT3 and ERK1/2 signaling on cellular gene expression in
virally-infected cells are not known. Future studies should investigate virus-induced
transcriptional changes downstream of STAT3 and ERK1/2 signaling. Results from these
studies may provide additional insight into the ways in which the virus harnesses host
signaling cascades to enhance its own fitness.
Due to the importance of vIL-6/gp130 interactions within the ER, screens should
be conducted to identify small molecule or peptide inhibitors of this interaction within the
ER compartment. Should an in vitro screen identify robust inhibitors of this interaction,
potential inhibitors should be utilized in functional assays to assess their effects on the
growth, survival, and replication potential of virally infected cells. Validation of potential
inhibitors in cell-based assays is the first step in the lengthy process of developing FDA-
approved therapeutics for the treatment of human disease.
Page 125
111
FIGURES
Figure 5.1. ER-localized vIL-6 and gp130 signaling in PEL cell growth and survival. vIL-6
interacts with both gp130 and VKORC1v2 within the ER. A previous study indicated that
the vIL-6/VKORC1v2 interaction was important for the growth and viability of PEL cells in
culture [173]. The study outlined in Chapter 3 demonstrated that vIL-6 signaling through
gp130 in the ER was critical for the proliferation and survival of PEL cells. Depletion of
gp130 led to reduced cell growth and increased apoptosis in all four PEL cell lines tested
(BCBL-1, JSC-1, BC-1, and BC-3). Furthermore, gp130 was found to contribute to levels
of phosphorylated STATs 1 and 3 (in all PEL cell lines tested) and phosphorylated ERKs
1 and 2 in a subset of PEL cells (BCBL-1 and BC-1). STAT3 had been previously shown
to promote PEL cell growth/viability in culture via the upregulation of survivin [152], but the
role of ERK1/2 in the growth and viability of PEL cells had not been established previously.
Data described in Chapter 3 indicate that ERK1/2 expression is critical for PEL cell
proliferation and survival in all four PEL cell lines tested. The asterisks (*) denote that
STAT3 was required for PEL cell growth and survival in the two cell lines tested (BCBL-1
and JSC-1). To establish that ER-localized vIL-6/gp130 signaling is responsible for vIL-6-
mediated pro-growth and pro-survival functions, two approaches were utilized. First, vIL-
6 depletion-complementation experiments were conducted, and ER-directed and mutated
vIL-6 (W167G mutation, incapable of dimerizing gp130 in the ER though capable of binding
to VKORC1v2) was unable to rescue the growth of vIL-6-depleted PEL cells; ER-directed
and wild-type vIL-6 was able to rescue the vIL-6-depletion phenotype. Secondly,
introduction of an ER-directed soluble gp130 molecule (sgp130.K) into PEL cells inhibited
the vIL-6/gp130 interaction in the ER, leading to decreased PEL cell growth and increased
apoptosis. In summary, ER-localized vIL-6/gp130 signaling promotes cell growth and
survival via the activation of STAT3 and ERK1/2.
Page 127
113
Figure 5.2. Role of vIL-6 and gp130 in HHV-8 replication in PEL and endothelial cells. vIL-
6 and gp130 are required for maximal HHV-8 productive replication in both PEL and
endothelial cells. Depletion of vIL-6 or gp130 results in lower HHV-8 titers following
reactivation. Additionally, STAT3, which is activated by vIL-6/gp130 signaling, promotes
HHV-8 replication; depletion of STAT3 leads to decreased viral titers in both PEL and
endothelial cells. To assess the role of ERK signaling in HHV-8 replication, a series of
gp130 mutants were employed as described in Chapter 4. The gp130.ERK.Y mutant was
capable of initiating ERK signaling but not STAT3 signaling, and gp130.STAT.Ys could
activate STAT3 (and STAT1) signaling but not ERK signaling. In a depletion-
complementation experiment, gp130.STAT.Ys, but not gp130.ERK.Y, was capable of
rescuing HHV-8 titers following depletion of endogenous gp130. These results indicate
that vIL-6/gp130-mediated STAT signaling, but not ERK signaling, is necessary for
maximal HHV-8 replication. Finally, a vIL-6 depletion and complementation experiment
was conducted to determine the contribution of ER-localized vIL-6/gp130 signaling to
productive replication. ER-directed wild-type vIL-6 was capable of rescuing HHV-8 titers
following vIL-6 depletion, but ER-directed and mutated vIL-6 (W167G; incapable of
dimerizing gp130 in the ER) was unable to rescue the vIL-6-depletion phenotype. In
summary, ER-localized vIL-6/gp130-mediated STAT3, but not ERK1/2, signaling is
required for maximal HHV-8 replicative titers.
Page 129
115
REFERENCES
1. Friedman-Kien AE (1981) Disseminated Kaposi's sarcoma syndrome in young
homosexual men. J Am Acad Dermatol 5: 468-471.
2. Haverkos HW, Drotman DP (1985) Prevalence of Kaposi's sarcoma among patients
with AIDS. N Engl J Med 312: 1518.
3. Safai B, Johnson KG, Myskowski PL, Koziner B, Yang SY, et al. (1985) The natural
history of Kaposi's sarcoma in the acquired immunodeficiency syndrome. Ann
Intern Med 103: 744-750.
4. Slavin G, Cameron HM, Singh H (1969) Kaposi's sarcoma in mainland Tanzania: a
report of 117 cases. Br J Cancer 23: 349-357.
5. Taylor JF, Smith PG, Bull D, Pike MC (1972) Kaposi's sarcoma in Uganda: geographic
and ethnic distribution. Br J Cancer 26: 483-497.
6. Geddes M, Franceschi S, Balzi D, Arniani S, Gafa L, et al. (1995) Birthplace and classic
Kaposi's sarcoma in Italy. Associazione Italiana Registri Tumori. J Natl Cancer Inst
87: 1015-1017.
7. Franceschi S, Geddes M (1995) Epidemiology of classic Kaposi's sarcoma, with special
reference to mediterranean population. Tumori 81: 308-314.
8. Penn I (1979) Kaposi's sarcoma in organ transplant recipients: report of 20 cases.
Transplantation 27: 8-11.
9. Kaposi (1872) Idiopathisches multiples Pigmentsarkom der Haut. Archiv für
Dermatologie und Syphilis 4: 265-273.
10. DiGiovanna JJ, Safai B (1981) Kaposi's sarcoma. Retrospective study of 90 cases
with particular emphasis on the familial occurrence, ethnic background and
prevalence of other diseases. Am J Med 71: 779-783.
11. Beral V, Peterman TA, Berkelman RL, Jaffe HW (1990) Kaposi's sarcoma among
persons with AIDS: a sexually transmitted infection? The Lancet 335: 123-128.
Page 130
116
12. Gingues S, Gill MJ (2006) The impact of highly active antiretroviral therapy on the
incidence and outcomes of AIDS-defining cancers in Southern Alberta. HIV Med
7: 369-377.
13. Beral V, Newton R (1998) Overview of the epidemiology of immunodeficiency-
associated cancers. J Natl Cancer Inst Monogr: 1-6.
14. Stein ME, Spencer D, Ruff P, Lakier R, MacPhail P, et al. (1994) Endemic African
Kaposi's sarcoma: clinical and therapeutic implications. 10-year experience in the
Johannesburg Hospital (1980-1990). Oncology 51: 63-69.
15. Andreoni M, Goletti D, Pezzotti P, Pozzetto A, Monini P, et al. (2001) Prevalence,
incidence and correlates of HHV-8/KSHV infection and Kaposi's sarcoma in renal
and liver transplant recipients. J Infect 43: 195-199.
16. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, et al. (1999) Distribution of human
herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's
disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A 96: 4546-4551.
17. Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C, et al. (1995) Kaposi's
sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat Med 1:
1274-1278.
18. Morris VA, Punjabi AS, Lagunoff M (2008) Activation of Akt through gp130 receptor
signaling is required for Kaposi's sarcoma-associated herpesvirus-induced
lymphatic reprogramming of endothelial cells. J Virol 82: 8771-8779.
19. Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, et al. (2004) Lymphatic
reprogramming of blood vascular endothelium by Kaposi sarcoma-associated
herpesvirus. Nat Genet 36: 683-685.
20. Hansen A, Henderson S, Lagos D, Nikitenko L, Coulter E, et al. (2010) KSHV-encoded
miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev 24: 195-
205.
Page 131
117
21. Duprez R, Lacoste V, Briere J, Couppie P, Frances C, et al. (2007) Evidence for a
multiclonal origin of multicentric advanced lesions of Kaposi sarcoma. J Natl
Cancer Inst 99: 1086-1094.
22. Judde JG, Lacoste V, Briere J, Kassa-Kelembho E, Clyti E, et al. (2000) Monoclonality
or oligoclonality of human herpesvirus 8 terminal repeat sequences in Kaposi's
sarcoma and other diseases. J Natl Cancer Inst 92: 729-736.
23. Corbeil J, Evans LA, Vasak E, Cooper DA, Penny R (1991) Culture and properties of
cells derived from Kaposi sarcoma. J Immunol 146: 2972-2976.
24. Ensoli B, Nakamura S, Salahuddin SZ, Biberfeld P, Larsson L, et al. (1989) AIDS-
Kaposi's sarcoma-derived cells express cytokines with autocrine and paracrine
growth effects. Science 243: 223-226.
25. Miles SA, Rezai AR, Salazar-Gonzalez JF, Vander Meyden M, Stevens RH, et al.
(1990) AIDS Kaposi sarcoma-derived cells produce and respond to interleukin 6.
Proc Natl Acad Sci U S A 87: 4068-4072.
26. Roth WK, Werner S, Schirren CG, Hofschneider PH (1989) Depletion of PDGF from
serum inhibits growth of AIDS-related and sporadic Kaposi's sarcoma cells in
culture. Oncogene 4: 483-487.
27. Sciacca FL, Sturzl M, Bussolino F, Sironi M, Brandstetter H, et al. (1994) Expression
of adhesion molecules, platelet-activating factor, and chemokines by Kaposi's
sarcoma cells. J Immunol 153: 4816-4825.
28. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM (1995) Kaposi's sarcoma-
associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based
lymphomas. N Engl J Med 332: 1186-1191.
29. Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, et al. (1996) Primary
effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood 88: 645-656.
Page 132
118
30. Carbone A, Cilia AM, Gloghini A, Capello D, Perin T, et al. (2000) Primary effusion
lymphoma cell lines harbouring human herpesvirus type-8. Leuk Lymphoma 36:
447-456.
31. Carbone A, Gloghini A (2008) KSHV/HHV8-associated lymphomas. Br J Haematol
140: 13-24.
32. Cesarman E, Knowles DM (1999) The role of Kaposi's sarcoma-associated
herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Semin Cancer Biol 9:
165-174.
33. Gaidano G, Gloghini A, Gattei V, Rossi MF, Cilia AM, et al. (1997) Association of
Kaposi's sarcoma-associated herpesvirus-positive primary effusion lymphoma
with expression of the CD138/syndecan-1 antigen. Blood 90: 4894-4900.
34. Renne R, Lagunoff M, Zhong W, Ganem D (1996) The size and conformation of
Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected
cells and virions. J Virol 70: 8151-8154.
35. Renne R, Zhong W, Herndier B, McGrath M, Abbey N, et al. (1996) Lytic growth of
Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat
Med 2: 342-346.
36. Carbone A, Gloghini A, Vaccher E, Cerri M, Gaidano G, et al. (2005) Kaposi's
sarcoma-associated herpesvirus/human herpesvirus type 8-positive solid
lymphomas: a tissue-based variant of primary effusion lymphoma. J Mol Diagn 7:
17-27.
37. Boulanger E, Gerard L, Gabarre J, Molina JM, Rapp C, et al. (2005) Prognostic factors
and outcome of human herpesvirus 8-associated primary effusion lymphoma in
patients with AIDS. J Clin Oncol 23: 4372-4380.
Page 133
119
38. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, et al. (1995)
Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric
Castleman's disease. Blood 86: 1276-1280.
39. Radaszkiewicz T, Hansmann ML, Lennert K (1989) Monoclonality and polyclonality of
plasma cells in Castleman's disease of the plasma cell variant. Histopathology 14:
11-24.
40. Yoshizaki K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, et al. (1989) Pathogenic
significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood 74: 1360-
1367.
41. Nishi J, Arimura K, Utsunomiya A, Yonezawa S, Kawakami K, et al. (1999) Expression
of vascular endothelial growth factor in sera and lymph nodes of the plasma cell
type of Castleman's disease. Br J Haematol 104: 482-485.
42. Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, et al. (2005) Humanized
anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease.
Blood 106: 2627-2632.
43. Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, et al. (2000) High levels
of human herpesvirus 8 viral load, human interleukin-6, interleukin 10, and C
reactive protein correlate with exacerbation of multicentric Castleman disease in
HIV-infected patients. Blood 96: 2069-2073.
44. Uldrick TS, Wang V, O'Mahony D, Aleman K, Wyvill KM, et al. (2010) An interleukin-
6-related systemic inflammatory syndrome in patients co-infected with Kaposi
sarcoma-associated herpesvirus and HIV but without Multicentric Castleman
disease. Clin Infect Dis 51: 350-358.
45. Polizzotto MN, Uldrick TS, Hu D, Yarchoan R (2012) Clinical Manifestations of Kaposi
Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-
MCD) and the KSHV Inflammatory Cytokine Syndrome. Front Microbiol 3: 73.
Page 134
120
46. Tamburro KM, Yang D, Poisson J, Fedoriw Y, Roy D, et al. (2012) Vironome of Kaposi
sarcoma associated herpesvirus-inflammatory cytokine syndrome in an AIDS
patient reveals co-infection of human herpesvirus 8 and human herpesvirus 6A.
Virology 433: 220-225.
47. McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA (1995) Molecular phylogeny
and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol
247: 443-458.
48. Pellett P, Roizman B (2007) The Family Herpesviridae: A Brief Introduction. In: Knipe
D, Howley P, editors. Fields Virology, 5th Edition. 5 ed: Lippincott Williams &
Wilkins.
49. Roizman B, Furlong D (1974) The replication of herpesviruses. 229-403.
50. Whitley R (1996) Herpesviruses. In: Baron S, editor. Medical Microbiology. Galveston,
TX: University of Texas Medical Branch at Galveston.
51. Roizman B, Carmichael LE, Deinhardt F, de-The G, Nahmias AJ, et al. (1981)
Herpesviridae. Definition, provisional nomenclature, and taxonomy. The
Herpesvirus Study Group, the International Committee on Taxonomy of Viruses.
Intervirology 16: 201-217.
52. Crough T, Khanna R (2009) Immunobiology of human cytomegalovirus: from bench
to bedside. Clin Microbiol Rev 22: 76-98.
53. Papazian L, Fraisse A, Garbe L, Zandotti C, Thomas P, et al. (1996) Cytomegalovirus.
An unexpected cause of ventilator-associated pneumonia. Anesthesiology 84:
280-287.
54. Halme L, Lempinen M, Arola J, Sarkio S, Hockerstedt K, et al. (2008) High frequency
of gastroduodenal cytomegalovirus infection in liver transplant patients. APMIS
116: 99-106.
Page 135
121
55. Rafailidis PI, Kapaskelis A, Falagas ME (2007) Cytomegalovirus meningitis in an
immunocompetent patient. Med Sci Monit 13: CS107-109.
56. Campbell PT, Li JS, Wall TC, O'Connor CM, Van Trigt P, et al. (1995) Cytomegalovirus
pericarditis: a case series and review of the literature. Am J Med Sci 309: 229-234.
57. Braun DK, Dominguez G, Pellett PE (1997) Human herpesvirus 6. Clin Microbiol Rev
10: 521-567.
58. Black JB, Pellett PE (1999) Human herpesvirus 7. Rev Med Virol 9: 245-262.
59. Irving WL, Chang J, Raymond DR, Dunstan R, Grattan-Smith P, et al. (1990) Roseola
infantum and other syndromes associated with acute HHV6 infection. Arch Dis
Child 65: 1297-1300.
60. Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, et al. (1988) Identification
of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet 1: 1065-
1067.
61. Tanaka K, Kondo T, Torigoe S, Okada S, Mukai T, et al. (1994) Human herpesvirus
7: another causal agent for roseola (exanthem subitum). J Pediatr 125: 1-5.
62. Zahorsky J (1910) Roseola infantilis. Pediatrics 22: 60-64.
63. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, et al. (1994) Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science
266: 1865-1869.
64. Damania B (2004) Oncogenic gamma-herpesviruses: comparison of viral proteins
involved in tumorigenesis. Nat Rev Microbiol 2: 656-668.
65. Mocarski Jr. E (2007) Comparative analysis of herpesvirus-common proteins. In: Arvin
A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B et al., editors. Human
Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge.
Page 136
122
66. Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, et al. (1990) Homology
of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI.
Science 248: 1230-1234.
67. Moore PS, Boshoff C, Weiss RA, Chang Y (1996) Molecular mimicry of human
cytokine and cytokine response pathway genes by KSHV. Science 274: 1739-
1744.
68. Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, et al. (1997) Human herpesvirus 8
encodes a homolog of interleukin-6. J Virol 71: 839-842.
69. Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan X, et al. (1997) Kaposi's
sarcoma-associated human herpesvirus-8 encodes homologues of macrophage
inflammatory protein-1 and interleukin-6. Nat Med 3: 287-292.
70. Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, et al. (1996) Cyclin encoded
by KS herpesvirus. Nature 382: 410.
71. Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, et al. (1997) Kaposi's sarcoma-
associated herpesvirus encodes a functional cyclin. J Virol 71: 1984-1991.
72. McSharry BP, Avdic S, Slobedman B (2012) Human cytomegalovirus encoded
homologs of cytokines, chemokines and their receptors: roles in
immunomodulation. Viruses 4: 2448-2470.
73. Nicholas J (2010) Human herpesvirus 8-encoded cytokines. Future Virology 5: 197-
206.
74. Oudejans JJ, van den Brule AJ, Jiwa NM, de Bruin PC, Ossenkoppele GJ, et al. (1995)
BHRF1, the Epstein-Barr virus (EBV) homologue of the BCL-2 protooncogene, is
transcribed in EBV-associated B-cell lymphomas and in reactive lymphocytes.
Blood 86: 1893-1902.
Page 137
123
75. Cleary ML, Smith SD, Sklar J (1986) Cloning and structural analysis of cDNAs for bcl-
2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18)
translocation. Cell 47: 19-28.
76. Henderson S, Huen D, Rowe M, Dawson C, Johnson G, et al. (1993) Epstein-Barr
virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells
from programmed cell death. Proc Natl Acad Sci U S A 90: 8479-8483.
77. Nicholas J, Ruvolo V, Zong J, Ciufo D, Guo HG, et al. (1997) A single 13-kilobase
divergent locus in the Kaposi sarcoma-associated herpesvirus (human
herpesvirus 8) genome contains nine open reading frames that are homologous to
or related to cellular proteins. J Virol 71: 1963-1974.
78. Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, et al. (1997) Viral FLICE-
inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature
386: 517-521.
79. Kincaid RP, Sullivan CS (2012) Virus-encoded microRNAs: an overview and a look to
the future. PLoS Pathog 8: e1003018.
80. Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, et al. (1995) In vitro
establishment and characterization of two acquired immunodeficiency syndrome-
related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-
associated herpesvirus-like (KSHV) DNA sequences. Blood 86: 2708-2714.
81. Lagunoff M, Ganem D (1997) The structure and coding organization of the genomic
termini of Kaposi's sarcoma-associated herpesvirus. Virology 236: 147-154.
82. Ganem D (2007) Kaposi's Sarcoma-associated Herpesvirus. In: Knipe D, Howley P,
editors. Fields Virology, 5th Edition: Lippincott Williams & Wilkins. pp. 2848-2931.
83. Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, et al. (2005)
Identification of microRNAs of the herpesvirus family. Nat Methods 2: 269-276.
Page 138
124
84. Samols MA, Hu J, Skalsky RL, Renne R (2005) Cloning and identification of a
microRNA cluster within the latency-associated region of Kaposi's sarcoma-
associated herpesvirus. J Virol 79: 9301-9305.
85. McAllister SC, Hansen SG, Messaoudi I, Nikolich-Zugich J, Moses AV (2005)
Increased efficiency of phorbol ester-induced lytic reactivation of Kaposi's
sarcoma-associated herpesvirus during S phase. J Virol 79: 2626-2630.
86. Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, et al. (1997) Selective switch
between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-
Barr virus in dually infected body cavity lymphoma cells. J Virol 71: 314-324.
87. Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, et al. (1999) Induction of human
herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1
cells. J Gen Virol 80: 83-90.
88. Chang J, Renne R, Dittmer D, Ganem D (2000) Inflammatory cytokines and the
reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology
266: 17-25.
89. Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, et al. (2009) Toll-like receptor
signaling controls reactivation of KSHV from latency. Proc Natl Acad Sci U S A
106: 11725-11730.
90. Ye F, Zhou F, Bedolla RG, Jones T, Lei X, et al. (2011) Reactive oxygen species
hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus
reactivation from latency. PLoS Pathog 7: e1002054.
91. Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, et al. (2007) B cell terminal
differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated
herpesvirus. FEBS Lett 581: 3485-3488.
92. Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, et al. (1999) Kinetics of Kaposi's
sarcoma-associated herpesvirus gene expression. J Virol 73: 2232-2242.
Page 139
125
93. Zhu L, Wang R, Sweat A, Goldstein E, Horvat R, et al. (1999) Comparison of human
sera reactivities in immunoblots with recombinant human herpesvirus (HHV)-8
proteins associated with the latent (ORF73) and lytic (ORFs 65, K8.1A, and K8.1B)
replicative cycles and in immunofluorescence assays with HHV-8-infected BCBL-
1 cells. Virology 256: 381-392.
94. Rimessi P, Bonaccorsi A, Sturzl M, Fabris M, Brocca-Cofano E, et al. (2001)
Transcription pattern of human herpesvirus 8 open reading frame K3 in primary
effusion lymphoma and Kaposi's sarcoma. J Virol 75: 7161-7174.
95. Lu M, Suen J, Frias C, Pfeiffer R, Tsai MH, et al. (2004) Dissection of the Kaposi's
sarcoma-associated herpesvirus gene expression program by using the viral DNA
replication inhibitor cidofovir. J Virol 78: 13637-13652.
96. Lukac DM, Renne R, Kirshner JR, Ganem D (1998) Reactivation of Kaposi's sarcoma-
associated herpesvirus infection from latency by expression of the ORF 50
transactivator, a homolog of the EBV R protein. Virology 252: 304-312.
97. Lukac DM, Kirshner JR, Ganem D (1999) Transcriptional activation by the product of
open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for
lytic viral reactivation in B cells. J Virol 73: 9348-9361.
98. Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, et al. (1998) A viral gene that activates
lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad
Sci U S A 95: 10866-10871.
99. Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, et al. (2005) A Kaposi's sarcoma-
associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective
for reactivation of latent virus and DNA replication. J Virol 79: 3479-3487.
100. Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, et al. (2003) Global changes
in Kaposi's sarcoma-associated virus gene expression patterns following
expression of a tetracycline-inducible Rta transactivator. J Virol 77: 4205-4220.
Page 140
126
101. Guito J, Lukac DM (2012) KSHV Rta promoter specification and viral reactivation.
Front Microbiol 3: 30.
102. Bu W, Carroll KD, Palmeri D, Lukac DM (2007) Kaposi's sarcoma-associated
herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a
tetramer. J Virol 81: 5788-5806.
103. Lin CL, Li H, Wang Y, Zhu FX, Kudchodkar S, et al. (2003) Kaposi's sarcoma-
associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication:
identification of the ori-Lyt and association of K8 bZip protein with the origin. J Virol
77: 5578-5588.
104. Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, et al. (2001) Origin-independent
assembly of Kaposi's sarcoma-associated herpesvirus DNA replication
compartments in transient cotransfection assays and association with the ORF-K8
protein and cellular PML. J Virol 75: 1487-1506.
105. Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J (2012) Reconstitution of
the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and
formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69
gene products. J Virol 86: 594-598.
106. Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, et al. (2013) Interactions
of the Kaposi's Sarcoma-associated herpesvirus nuclear egress complex: ORF69
is a potent factor for remodeling cellular membranes. J Virol 87: 3915-3929.
107. Krishnan HH, Sharma-Walia N, Zeng L, Gao SJ, Chandran B (2005) Envelope
glycoprotein gB of Kaposi's sarcoma-associated herpesvirus is essential for
egress from infected cells. J Virol 79: 10952-10967.
108. Subramanian R, Sehgal I, D'Auvergne O, Kousoulas KG (2010) Kaposi's sarcoma-
associated herpesvirus glycoproteins B and K8.1 regulate virion egress and
Page 141
127
synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1
cells. J Virol 84: 1704-1714.
109. Mettenleiter TC (2002) Herpesvirus assembly and egress. J Virol 76: 1537-1547.
110. Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, et al. (1997) Kaposi's
sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor
cells. J Virol 71: 715-719.
111. Cannon JS, Ciufo D, Hawkins AL, Griffin CA, Borowitz MJ, et al. (2000) A new primary
effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma
herpesvirus-containing supernatant. J Virol 74: 10187-10193.
112. Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, et al. (1998) A cluster of
latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:
8309-8315.
113. Pearce M, Matsumura S, Wilson AC (2005) Transcripts encoding K12, v-FLIP, v-
cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus
originate from a common promoter. J Virol 79: 14457-14464.
114. Talbot SJ, Weiss RA, Kellam P, Boshoff C (1999) Transcriptional analysis of human
herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion
lymphoma cell line. Virology 257: 84-94.
115. Chen D, Sandford G, Nicholas J (2009) Intracellular signaling mechanisms and
activities of human herpesvirus 8 interleukin-6. J Virol 83: 722-733.
116. Chandriani S, Ganem D (2010) Array-based transcript profiling and limiting-dilution
reverse transcription-PCR analysis identify additional latent genes in Kaposi's
sarcoma-associated herpesvirus. J Virol 84: 5565-5573.
117. Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y (2001) Kaposi's sarcoma-
associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits
p53. J Virol 75: 429-438.
Page 142
128
118. Dittmer DP (2003) Transcription profile of Kaposi's sarcoma-associated herpesvirus
in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays.
Cancer Res 63: 2010-2015.
119. Burysek L, Pitha PM (2001) Latently expressed human herpesvirus 8-encoded
interferon regulatory factor 2 inhibits double-stranded RNA-activated protein
kinase. J Virol 75: 2345-2352.
120. Barbera AJ, Ballestas ME, Kaye KM (2004) The Kaposi's sarcoma-associated
herpesvirus latency-associated nuclear antigen 1 N terminus is essential for
chromosome association, DNA replication, and episome persistence. J Virol 78:
294-301.
121. Cotter MA, 2nd, Robertson ES (1999) The latency-associated nuclear antigen tethers
the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in
body cavity-based lymphoma cells. Virology 264: 254-264.
122. Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, et al. (2006)
The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus
LANA. Science 311: 856-861.
123. Ballestas ME, Kaye KM (2001) Kaposi's sarcoma-associated herpesvirus latency-
associated nuclear antigen 1 mediates episome persistence through cis-acting
terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75: 3250-
3258.
124. Hu J, Garber AC, Renne R (2002) The latency-associated nuclear antigen of Kaposi's
sarcoma-associated herpesvirus supports latent DNA replication in dividing cells.
J Virol 76: 11677-11687.
125. Verma SC, Choudhuri T, Kaul R, Robertson ES (2006) Latency-associated nuclear
antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin
Page 143
129
recognition complexes at the LANA binding sequence within the terminal repeats.
J Virol 80: 2243-2256.
126. Grundhoff A, Ganem D (2003) The latency-associated nuclear antigen of Kaposi's
sarcoma-associated herpesvirus permits replication of terminal repeat-containing
plasmids. J Virol 77: 2779-2783.
127. Lim C, Sohn H, Lee D, Gwack Y, Choe J (2002) Functional dissection of latency-
associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved
in latent DNA replication and transcription of terminal repeats of the viral genome.
J Virol 76: 10320-10331.
128. Shamay M, Krithivas A, Zhang J, Hayward SD (2006) Recruitment of the de novo
DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus
LANA. Proc Natl Acad Sci U S A 103: 14554-14559.
129. An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, et al. (2005) The
latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus
modulates cellular gene expression and protects lymphoid cells from p16 INK4A-
induced cell cycle arrest. J Biol Chem 280: 3862-3874.
130. Friborg J, Jr., Kong W, Hottiger MO, Nabel GJ (1999) p53 inhibition by the LANA
protein of KSHV protects against cell death. Nature 402: 889-894.
131. Radkov SA, Kellam P, Boshoff C (2000) The latent nuclear antigen of Kaposi
sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with
the oncogene Hras transforms primary rat cells. Nat Med 6: 1121-1127.
132. Jeong JH, Orvis J, Kim JW, McMurtrey CP, Renne R, et al. (2004) Regulation and
autoregulation of the promoter for the latency-associated nuclear antigen of
Kaposi's sarcoma-associated herpesvirus. J Biol Chem 279: 16822-16831.
133. Lan K, Kuppers DA, Verma SC, Robertson ES (2004) Kaposi's sarcoma-associated
herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication
Page 144
130
by targeting Rta: a potential mechanism for virus-mediated control of latency. J
Virol 78: 6585-6594.
134. Lan K, Kuppers DA, Robertson ES (2005) Kaposi's sarcoma-associated herpesvirus
reactivation is regulated by interaction of latency-associated nuclear antigen with
recombination signal sequence-binding protein Jkappa, the major downstream
effector of the Notch signaling pathway. J Virol 79: 3468-3478.
135. Fujimuro M, Hayward SD (2003) The latency-associated nuclear antigen of Kaposi's
sarcoma-associated herpesvirus manipulates the activity of glycogen synthase
kinase-3beta. J Virol 77: 8019-8030.
136. Marcu KB, Bossone SA, Patel AJ (1992) myc function and regulation. Annu Rev
Biochem 61: 809-860.
137. Liu J, Martin HJ, Liao G, Hayward SD (2007) The Kaposi's sarcoma-associated
herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81: 10451-10459.
138. Bubman D, Guasparri I, Cesarman E (2007) Deregulation of c-Myc in primary
effusion lymphoma by Kaposi's sarcoma herpesvirus latency-associated nuclear
antigen. Oncogene 26: 4979-4986.
139. Verma SC, Borah S, Robertson ES (2004) Latency-associated nuclear antigen of
Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human
telomerase reverse transcriptase promoter through interaction with transcription
factor Sp1. J Virol 78: 10348-10359.
140. Fakhari FD, Jeong JH, Kanan Y, Dittmer DP (2006) The latency-associated nuclear
antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and
lymphoma. J Clin Invest 116: 735-742.
141. Neipel F, Albrecht JC, Fleckenstein B (1997) Cell-homologous genes in the Kaposi's
sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its
pathogenicity? J Virol 71: 4187-4192.
Page 145
131
142. Heinrich PC, Castell JV, Andus T (1990) Interleukin-6 and the acute phase response.
Biochem J 265: 621-636.
143. Aoki Y, Tosato G, Fonville TW, Pittaluga S (2001) Serum viral interleukin-6 in AIDS-
related multicentric Castleman disease. Blood 97: 2526-2527.
144. Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, et al. (1999) Involvement of
interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi's sarcoma
herpesvirus-associated infected primary effusion lymphoma cells. Blood 94: 2871-
2879.
145. Zhang YJ, Patel D, Nan Y, Fan S (2011) Inhibition of primary effusion lymphoma
engraftment in SCID mice by morpholino oligomers against early lytic genes of
Kaposi's sarcoma-associated herpesvirus. Antivir Ther 16: 657-666.
146. Aoki Y, Tosato G (1999) Role of vascular endothelial growth factor/vascular
permeability factor in the pathogenesis of Kaposi's sarcoma-associated
herpesvirus-infected primary effusion lymphomas. Blood 94: 4247-4254.
147. Aoki Y, Jaffe ES, Chang Y, Jones K, Teruya-Feldstein J, et al. (1999) Angiogenesis
and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded
interleukin-6. Blood 93: 4034-4043.
148. Fielding CA, McLoughlin RM, Colmont CS, Kovaleva M, Harris DA, et al. (2005) Viral
IL-6 blocks neutrophil infiltration during acute inflammation. J Immunol 175: 4024-
4029.
149. Mori Y, Nishimoto N, Ohno M, Inagi R, Dhepakson P, et al. (2000) Human
herpesvirus 8-encoded interleukin-6 homologue (viral IL-6) induces endogenous
human IL-6 secretion. J Med Virol 61: 332-335.
150. Kovaleva M, Bussmeyer I, Rabe B, Grotzinger J, Sudarman E, et al. (2006)
Abrogation of viral interleukin-6 (vIL-6)-induced signaling by intracellular retention
Page 146
132
and neutralization of vIL-6 with an anti-vIL-6 single-chain antibody selected by
phage display. J Virol 80: 8510-8520.
151. Zhang YJ, Bonaparte RS, Patel D, Stein DA, Iversen PL (2008) Blockade of viral
interleukin-6 expression of Kaposi's sarcoma-associated herpesvirus. Mol Cancer
Ther 7: 712-720.
152. Aoki Y, Feldman GM, Tosato G (2003) Inhibition of STAT3 signaling induces
apoptosis and decreases survivin expression in primary effusion lymphoma. Blood
101: 1535-1542.
153. Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, et al. (1996) A central role
for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia
cells. EMBO J 15: 3651-3658.
154. Ambrosini G, Adida C, Altieri DC (1997) A novel anti-apoptosis gene, survivin,
expressed in cancer and lymphoma. Nat Med 3: 917-921.
155. Bartoli M, Gu X, Tsai NT, Venema RC, Brooks SE, et al. (2000) Vascular endothelial
growth factor activates STAT proteins in aortic endothelial cells. J Biol Chem 275:
33189-33192.
156. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998) Interleukin-
6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334 (
Pt 2): 297-314.
157. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, et al. (2003)
Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem
J 374: 1-20.
158. Chow D, He X, Snow AL, Rose-John S, Garcia KC (2001) Structure of an extracellular
gp130 cytokine receptor signaling complex. Science 291: 2150-2155.
Page 147
133
159. Boulanger MJ, Chow DC, Brevnova E, Martick M, Sandford G, et al. (2004) Molecular
mechanisms for viral mimicry of a human cytokine: activation of gp130 by HHV-8
interleukin-6. J Mol Biol 335: 641-654.
160. Kishimoto T, Akira S, Narazaki M, Taga T (1995) Interleukin-6 family of cytokines
and gp130. Blood 86: 1243-1254.
161. Yamanaka Y, Nakajima K, Fukada T, Hibi M, Hirano T (1996) Differentiation and
growth arrest signals are generated through the cytoplasmic region of gp130 that
is essential for Stat3 activation. EMBO J 15: 1557-1565.
162. Schiemann WP, Bartoe JL, Nathanson NM (1997) Box 3-independent signaling
mechanisms are involved in leukemia inhibitory factor receptor alpha- and gp130-
mediated stimulation of mitogen-activated protein kinase. Evidence for
participation of multiple signaling pathways which converge at Ras. J Biol Chem
272: 16631-16636.
163. Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, et al. (1998) Activation of the
protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing
receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase
protein expression. Biochem J 335: 557-565.
164. Aoki Y, Narazaki M, Kishimoto T, Tosato G (2001) Receptor engagement by viral
interleukin-6 encoded by Kaposi sarcoma-associated herpesvirus. Blood 98: 3042-
3049.
165. Chen D, Nicholas J (2006) Structural requirements for gp80 independence of human
herpesvirus 8 interleukin-6 (vIL-6) and evidence for gp80 stabilization of gp130
signaling complexes induced by vIL-6. J Virol 80: 9811-9821.
166. Wan X, Wang H, Nicholas J (1999) Human herpesvirus 8 interleukin-6 (vIL-6) signals
through gp130 but has structural and receptor-binding properties distinct from
those of human IL-6. J Virol 73: 8268-8278.
Page 148
134
167. Hu F, Nicholas J (2006) Signal transduction by human herpesvirus 8 viral interleukin-
6 (vIL-6) is modulated by the nonsignaling gp80 subunit of the IL-6 receptor
complex and is distinct from signaling induced by human IL-6. J Virol 80: 10874-
10878.
168. Li H, Wang H, Nicholas J (2001) Detection of direct binding of human herpesvirus 8-
encoded interleukin-6 (vIL-6) to both gp130 and IL-6 receptor (IL-6R) and
identification of amino acid residues of vIL-6 important for IL-6R-dependent and -
independent signaling. J Virol 75: 3325-3334.
169. Cousins E, Nicholas J (2013) Role of human herpesvirus 8 interleukin-6-activated
gp130 signal transducer in primary effusion lymphoma cell growth and viability. J
Virol 87: 10816-10827.
170. Meads MB, Medveczky PG (2004) Kaposi's sarcoma-associated herpesvirus-
encoded viral interleukin-6 is secreted and modified differently than human
interleukin-6: evidence for a unique autocrine signaling mechanism. J Biol Chem
279: 51793-51803.
171. Chen D, Choi YB, Sandford G, Nicholas J (2009) Determinants of secretion and
intracellular localization of human herpesvirus 8 interleukin-6. J Virol 83: 6874-
6882.
172. Dela Cruz CS, Lee Y, Viswanathan SR, El-Guindy AS, Gerlach J, et al. (2004) N-
linked glycosylation is required for optimal function of Kaposi's sarcoma
herpesvirus-encoded, but not cellular, interleukin 6. J Exp Med 199: 503-514.
173. Chen D, Cousins E, Sandford G, Nicholas J (2012) Human herpesvirus 8 viral
interleukin-6 (vIL-6) interacts with splice variant-2 of vitamin K epoxide reductase
complex subunit 1. J Virol 86: 1577-1588.
Page 149
135
174. Oldenburg J, Bevans CG, Muller CR, Watzka M (2006) Vitamin K epoxide reductase
complex subunit 1 (VKORC1): the key protein of the vitamin K cycle. Antioxid
Redox Signal 8: 347-353.
175. Chen D, Gao Y, Nicholas J (2014) Human herpesvirus 8 interleukin-6 contributes to
primary effusion lymphoma cell viability via suppression of proapoptotic cathepsin
D, a cointeraction partner of vitamin K epoxide reductase complex subunit 1 variant
2. J Virol 88: 1025-1038.
176. Benes P, Vetvicka V, Fusek M (2008) Cathepsin D--many functions of one aspartic
protease. Crit Rev Oncol Hematol 68: 12-28.
177. Prakash O, Tang ZY, Peng X, Coleman R, Gill J, et al. (2002) Tumorigenesis and
aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1
gene. J Natl Cancer Inst 94: 926-935.
178. Lee BS, Lee SH, Feng P, Chang H, Cho NH, et al. (2005) Characterization of the
Kaposi's sarcoma-associated herpesvirus K1 signalosome. J Virol 79: 12173-
12184.
179. Tomlinson CC, Damania B (2004) The K1 protein of Kaposi's sarcoma-associated
herpesvirus activates the Akt signaling pathway. J Virol 78: 1918-1927.
180. Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B (2006) Immortalization
of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated
herpesvirus. Cancer Res 66: 3658-3666.
181. Lee BS, Alvarez X, Ishido S, Lackner AA, Jung JU (2000) Inhibition of intracellular
transport of B cell antigen receptor complexes by Kaposi's sarcoma-associated
herpesvirus K1. J Exp Med 192: 11-21.
182. Prakash O, Swamy OR, Peng X, Tang ZY, Li L, et al. (2005) Activation of Src kinase
Lyn by the Kaposi sarcoma-associated herpesvirus K1 protein: implications for
lymphomagenesis. Blood 105: 3987-3994.
Page 150
136
183. Cousins E, Nicholas J (2014) Molecular biology of human herpesvirus 8: novel
functions and virus-host interactions implicated in viral pathogenesis and
replication. Recent Results Cancer Res 193: 227-268.
184. Coscoy L, Ganem D (2000) Kaposi's sarcoma-associated herpesvirus encodes two
proteins that block cell surface display of MHC class I chains by enhancing their
endocytosis. Proc Natl Acad Sci U S A 97: 8051-8056.
185. Ishido S, Wang C, Lee BS, Cohen GB, Jung JU (2000) Downregulation of major
histocompatibility complex class I molecules by Kaposi's sarcoma-associated
herpesvirus K3 and K5 proteins. J Virol 74: 5300-5309.
186. Ishido S, Choi JK, Lee BS, Wang C, DeMaria M, et al. (2000) Inhibition of natural
killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5
protein. Immunity 13: 365-374.
187. Choi YB, Nicholas J (2008) Autocrine and paracrine promotion of cell survival and
virus replication by human herpesvirus 8 chemokines. J Virol 82: 6501-6513.
188. Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, et al. (1997) Angiogenic and
HIV-inhibitory functions of KSHV-encoded chemokines. Science 278: 290-294.
189. Nakano K, Isegawa Y, Zou P, Tadagaki K, Inagi R, et al. (2003) Kaposi's sarcoma-
associated herpesvirus (KSHV)-encoded vMIP-I and vMIP-II induce signal
transduction and chemotaxis in monocytic cells. Arch Virol 148: 871-890.
190. Chen S, Bacon KB, Li L, Garcia GE, Xia Y, et al. (1998) In vivo inhibition of CC and
CX3C chemokine-induced leukocyte infiltration and attenuation of
glomerulonephritis in Wistar-Kyoto (WKY) rats by vMIP-II. J Exp Med 188: 193-
198.
191. Kledal TN, Rosenkilde MM, Coulin F, Simmons G, Johnsen AH, et al. (1997) A broad-
spectrum chemokine antagonist encoded by Kaposi's sarcoma-associated
herpesvirus. Science 277: 1656-1659.
Page 151
137
192. Luttichau HR, Lewis IC, Gerstoft J, Schwartz TW (2001) The herpesvirus 8-encoded
chemokine vMIP-II, but not the poxvirus-encoded chemokine MC148, inhibits the
CCR10 receptor. Eur J Immunol 31: 1217-1220.
193. Luttichau HR, Johnsen AH, Jurlander J, Rosenkilde MM, Schwartz TW (2007) Kaposi
sarcoma-associated herpes virus targets the lymphotactin receptor with both a
broad spectrum antagonist vCCL2 and a highly selective and potent agonist
vCCL3. J Biol Chem 282: 17794-17805.
194. Stine JT, Wood C, Hill M, Epp A, Raport CJ, et al. (2000) KSHV-encoded CC
chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively
chemoattracts TH2 cells. Blood 95: 1151-1157.
195. Haque M, Chen J, Ueda K, Mori Y, Nakano K, et al. (2000) Identification and analysis
of the K5 gene of Kaposi's sarcoma-associated herpesvirus. J Virol 74: 2867-2875.
196. Dairaghi DJ, Fan RA, McMaster BE, Hanley MR, Schall TJ (1999) HHV8-encoded
vMIP-I selectively engages chemokine receptor CCR8. Agonist and antagonist
profiles of viral chemokines. J Biol Chem 274: 21569-21574.
197. Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C (2002) Characterization of
an anti-apoptotic glycoprotein encoded by Kaposi's sarcoma-associated
herpesvirus which resembles a spliced variant of human survivin. EMBO J 21:
2602-2615.
198. Feng P, Park J, Lee BS, Lee SH, Bram RJ, et al. (2002) Kaposi's sarcoma-associated
herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating
cyclophilin ligand to modulate intracellular calcium concentration and inhibit
apoptosis. J Virol 76: 11491-11504.
199. Feng P, Scott CW, Cho NH, Nakamura H, Chung YH, et al. (2004) Kaposi's sarcoma-
associated herpesvirus K7 protein targets a ubiquitin-like/ubiquitin-associated
Page 152
138
domain-containing protein to promote protein degradation. Mol Cell Biol 24: 3938-
3948.
200. Chandran B, Bloomer C, Chan SR, Zhu L, Goldstein E, et al. (1998) Human
herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by
spliced transcripts. Virology 249: 140-149.
201. Wang FZ, Dahl H, Ljungman P, Linde A (1999) Lymphoproliferative responses to
human herpesvirus-6 variant A and variant B in healthy adults. J Med Virol 57: 134-
139.
202. Cunningham C, Barnard S, Blackbourn DJ, Davison AJ (2003) Transcription mapping
of human herpesvirus 8 genes encoding viral interferon regulatory factors. J Gen
Virol 84: 1471-1483.
203. Choi YB, Sandford G, Nicholas J (2012) Human herpesvirus 8 interferon regulatory
factor-mediated BH3-only protein inhibition via Bid BH3-B mimicry. PLoS Pathog
8: e1002748.
204. Burysek L, Yeow WS, Lubyova B, Kellum M, Schafer SL, et al. (1999) Functional
analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and
its association with cellular interferon regulatory factors and p300. J Virol 73: 7334-
7342.
205. Li M, Damania B, Alvarez X, Ogryzko V, Ozato K, et al. (2000) Inhibition of p300
histone acetyltransferase by viral interferon regulatory factor. Mol Cell Biol 20:
8254-8263.
206. Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, et al. (2001) HHV-8 encoded
vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment
of the CBP/p300 coactivators. Oncogene 20: 800-811.
207. Nakamura H, Li M, Zarycki J, Jung JU (2001) Inhibition of p53 tumor suppressor by
viral interferon regulatory factor. J Virol 75: 7572-7582.
Page 153
139
208. Seo T, Park J, Lee D, Hwang SG, Choe J (2001) Viral interferon regulatory factor 1
of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-
dependent transcription and apoptosis. J Virol 75: 6193-6198.
209. Seo T, Lee D, Shim YS, Angell JE, Chidambaram NV, et al. (2002) Viral interferon
regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus interacts with a cell
death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J
Virol 76: 8797-8807.
210. Seo T, Park J, Choe J (2005) Kaposi's sarcoma-associated herpesvirus viral IFN
regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res
65: 1738-1747.
211. Shin YC, Nakamura H, Liang X, Feng P, Chang H, et al. (2006) Inhibition of the
ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated
herpesvirus interferon regulatory factor 1. J Virol 80: 2257-2266.
212. Lee HR, Toth Z, Shin YC, Lee JS, Chang H, et al. (2009) Kaposi's sarcoma-
associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to
deregulate the p53 tumor suppressor pathway. J Virol 83: 6739-6747.
213. Lee HR, Choi WC, Lee S, Hwang J, Hwang E, et al. (2011) Bilateral inhibition of
HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat Struct
Mol Biol 18: 1336-1344.
214. Joo CH, Shin YC, Gack M, Wu L, Levy D, et al. (2007) Inhibition of interferon
regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's
sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol 81: 8282-8292.
215. Munoz-Fontela C, Marcos-Villar L, Gallego P, Arroyo J, Da Costa M, et al. (2007)
Latent protein LANA2 from Kaposi's sarcoma-associated herpesvirus interacts
with 14-3-3 proteins and inhibits FOXO3a transcription factor. J Virol 81: 1511-
1516.
Page 154
140
216. Shin YC, Joo CH, Gack MU, Lee HR, Jung JU (2008) Kaposi's sarcoma-associated
herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha
to induce vascular endothelial growth factor expression. Cancer Res 68: 1751-
1759.
217. Wies E, Mori Y, Hahn A, Kremmer E, Sturzl M, et al. (2008) The viral interferon-
regulatory factor-3 is required for the survival of KSHV-infected primary effusion
lymphoma cells. Blood 111: 320-327.
218. Burysek L, Yeow WS, Pitha PM (1999) Unique properties of a second human
herpesvirus 8-encoded interferon regulatory factor (vIRF-2). J Hum Virol 2: 19-32.
219. Sadler R, Wu L, Forghani B, Renne R, Zhong W, et al. (1999) A complex translational
program generates multiple novel proteins from the latently expressed kaposin
(K12) locus of Kaposi's sarcoma-associated herpesvirus. J Virol 73: 5722-5730.
220. Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, et al. (2001) Signaling by human
herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol
Cell 7: 833-843.
221. McCormick C, Ganem D (2005) The kaposin B protein of KSHV activates the
p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307: 739-741.
222. McCormick C, Ganem D (2006) Phosphorylation and function of the kaposin B direct
repeats of Kaposi's sarcoma-associated herpesvirus. J Virol 80: 6165-6170.
223. Yoo J, Kang J, Lee HN, Aguilar B, Kafka D, et al. (2010) Kaposin-B enhances the
PROX1 mRNA stability during lymphatic reprogramming of vascular endothelial
cells by Kaposi's sarcoma herpes virus. PLoS Pathog 6: e1001046.
224. Belanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, et al. (2001) Human
herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis
through binding and prevention of procaspase-8 maturation. J Hum Virol 4: 62-73.
Page 155
141
225. Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, et al. (2002) The human herpes virus
8-encoded viral FLICE inhibitory protein physically associates with and persistently
activates the Ikappa B kinase complex. J Biol Chem 277: 13745-13751.
226. Sun Q, Matta H, Lu G, Chaudhary PM (2006) Induction of IL-8 expression by human
herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene 25: 2717-
2726.
227. Foster-Cuevas M, Wright GJ, Puklavec MJ, Brown MH, Barclay AN (2004) Human
herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage
activation through CD200 receptor. J Virol 78: 7667-7676.
228. Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, et al. (2003)
Activation of mitogen-activated protein kinase and NF-kappaB pathways by a
Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol 77:
9346-9358.
229. Lim CS, Seet BT, Ingham RJ, Gish G, Matskova L, et al. (2007) The K15 protein of
Kaposi's sarcoma-associated herpesvirus recruits the endocytic regulator
intersectin 2 through a selective SH3 domain interaction. Biochemistry 46: 9874-
9885.
230. Pietrek M, Brinkmann MM, Glowacka I, Enlund A, Havemeier A, et al. (2010) Role of
the Kaposi's sarcoma-associated herpesvirus K15 SH3 binding site in
inflammatory signaling and B-cell activation. J Virol 84: 8231-8240.
231. Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, et al. (2002) K15 protein of
Kaposi's sarcoma-associated herpesvirus is latently expressed and binds to HAX-
1, a protein with antiapoptotic function. J Virol 76: 802-816.
232. Gottwein E, Cullen BR (2010) A human herpesvirus microRNA inhibits p21
expression and attenuates p21-mediated cell cycle arrest. J Virol 84: 5229-5237.
Page 156
142
233. Lei X, Bai Z, Ye F, Xie J, Kim CG, et al. (2010) Regulation of NF-kappaB inhibitor
IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12: 193-
199.
234. Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, et al. (2011) Kaposi's
sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS
Pathog 7: e1002405.
235. Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, et al. (2007)
Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS
Pathog 3: e65.
236. Catrina Ene AM, Borze I, Guled M, Costache M, Leen G, et al. (2014) MicroRNA
expression profiles in Kaposi's sarcoma. Pathol Oncol Res 20: 153-159.
237. Lu CC, Li Z, Chu CY, Feng J, Feng J, et al. (2010) MicroRNAs encoded by Kaposi's
sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep 11: 784-790.
238. Lu F, Stedman W, Yousef M, Renne R, Lieberman PM (2010) Epigenetic regulation
of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs
that target Rta and the cellular Rbl2-DNMT pathway. J Virol 84: 2697-2706.
239. Ziegelbauer JM, Sullivan CS, Ganem D (2009) Tandem array-based expression
screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet 41:
130-134.
240. Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, et al. (2012)
Kaposi's sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88,
two components of the toll-like receptor/interleukin-1R signaling cascade, to
reduce inflammatory-cytokine expression. J Virol 86: 11663-11674.
241. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O (2009) Diverse herpesvirus
microRNAs target the stress-induced immune ligand MICB to escape recognition
by natural killer cells. Cell Host Microbe 5: 376-385.
Page 157
143
242. Lin X, Liang D, He Z, Deng Q, Robertson ES, et al. (2011) miR-K12-7-5p encoded
by Kaposi's sarcoma-associated herpesvirus stabilizes the latent state by targeting
viral ORF50/RTA. PLoS One 6: e16224.
243. Bellare P, Ganem D (2009) Regulation of KSHV lytic switch protein expression by a
virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic
reactivation. Cell Host Microbe 6: 570-575.
244. Abend JR, Uldrick T, Ziegelbauer JM (2010) Regulation of tumor necrosis factor-like
weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's
sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis
and inflammatory cytokine expression. J Virol 84: 12139-12151.
245. Liu Y, Sun R, Lin X, Liang D, Deng Q, et al. (2012) Kaposi's sarcoma-associated
herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth
factor beta signaling through suppression of SMAD5. J Virol 86: 1372-1381.
246. Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, et al. (2007) A viral
microRNA functions as an orthologue of cellular miR-155. Nature 450: 1096-1099.
247. Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, et al. (2007) Kaposi's
sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 81:
12836-12845.
248. Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, et al. (2010) Upregulation of xCT
by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in
an environment of oxidative stress. PLoS Pathog 6: e1000742.
249. Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, et al. (2005) Kaposi's sarcoma-
associated herpesvirus expresses an array of viral microRNAs in latently infected
cells. Proc Natl Acad Sci U S A 102: 5570-5575.
Page 158
144
250. Lin YT, Kincaid RP, Arasappan D, Dowd SE, Hunicke-Smith SP, et al. (2010) Small
RNA profiling reveals antisense transcription throughout the KSHV genome and
novel small RNAs. RNA 16: 1540-1558.
251. Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, et al. (2000) The
primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence
similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey
rhadinovirus isolate 17577. J Virol 74: 3388-3398.
252. Searles RP, Bergquam EP, Axthelm MK, Wong SW (1999) Sequence and genomic
analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-
associated herpesvirus/human herpesvirus 8. J Virol 73: 3040-3053.
253. Estep RD, Hansen SG, Rogers KS, Axthelm MK, Wong SW (2013) Genomic
characterization of Japanese macaque rhadinovirus, a novel herpesvirus isolated
from a nonhuman primate with a spontaneous inflammatory demyelinating
disease. J Virol 87: 512-523.
254. Vieira J, O'Hearn PM (2004) Use of the red fluorescent protein as a marker of
Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:
225-240.
255. Ensoli B, Sturzl M, Monini P (2001) Reactivation and role of HHV-8 in Kaposi's
sarcoma initiation. Adv Cancer Res 81: 161-200.
256. Schulz TF (2006) The pleiotropic effects of Kaposi's sarcoma herpesvirus. J Pathol
208: 187-198.
257. Uldrick TS, Polizzotto MN, Yarchoan R (2012) Recent advances in Kaposi sarcoma
herpesvirus-associated multicentric Castleman disease. Curr Opin Oncol 24: 495-
505.
258. Nicholas J (2007) Human herpesvirus 8-encoded proteins with potential roles in
virus-associated neoplasia. Frontiers in Bioscience 12: 265-281.
Page 159
145
259. Sakakibara S, Tosato G (2011) Viral interleukin-6: role in Kaposi's sarcoma-
associated herpesvirus: associated malignancies. J Interferon Cytokine Res 31:
791-801.
260. Molden J, Chang Y, You Y, Moore PS, Goldsmith MA (1997) A Kaposi's sarcoma-
associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling
through the shared gp130 receptor subunit. J Biol Chem 272: 19625-19631.
261. Adam N, Rabe B, Suthaus J, Grotzinger J, Rose-John S, et al. (2009) Unraveling
viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways
independently of the interleukin-6 receptor. J Virol 83: 5117-5126.
262. Verzijl D, Pardo L, van Dijk M, Gruijthuijsen YK, Jongejan A, et al. (2006) Helix 8 of
the viral chemokine receptor ORF74 directs chemokine binding. J Biol Chem 281:
35327-35335.
263. Benekli M, Baumann H, Wetzler M (2009) Targeting signal transducer and activator
of transcription signaling pathway in leukemias. J Clin Oncol 27: 4422-4432.
264. Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene
19: 2474-2488.
265. Hodge DR, Hurt EM, Farrar WL (2005) The role of IL-6 and STAT3 in inflammation
and cancer. Eur J Cancer 41: 2502-2512.
266. Ho HH, Ivashkiv LB (2006) Role of STAT3 in type I interferon responses. Negative
regulation of STAT1-dependent inflammatory gene activation. J Biol Chem 281:
14111-14118.
267. Huang F, Tong X, Fu L, Zhang R (2008) Knockdown of STAT3 by shRNA inhibits the
growth of CAOV3 ovarian cancer cell line in vitro and in vivo. Acta Biochim Biophys
Sin (Shanghai) 40: 519-525.
268. Chatterjee M, Stuhmer T, Herrmann P, Bommert K, Dorken B, et al. (2004) Combined
disruption of both the MEK/ERK and the IL-6R/STAT3 pathways is required to
Page 160
146
induce apoptosis of multiple myeloma cells in the presence of bone marrow
stromal cells. Blood 104: 3712-3721.
269. Chang LJ, Zaiss AK (2003) Self-inactivating lentiviral vectors and a sensitive Cre-
loxP reporter system. Methods Mol Med 76: 367-382.
270. Zhou BY, Ye Z, Chen G, Gao ZP, Zhang YA, et al. (2007) Inducible and reversible
transgene expression in human stem cells after efficient and stable gene transfer.
Stem Cells 25: 779-789.
271. Uddin S, Hussain AR, Manogaran PS, Al-Hussein K, Platanias LC, et al. (2005)
Curcumin suppresses growth and induces apoptosis in primary effusion
lymphoma. Oncogene 24: 7022-7030.
272. Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, et al. (2005) c-Jun N-
terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells.
Blood 106: 1382-1391.
273. Cohen A, Brodie C, Sarid R (2006) An essential role of ERK signalling in TPA-induced
reactivation of Kaposi's sarcoma-associated herpesvirus. J Gen Virol 87: 795-802.
274. Ford PW, Bryan BA, Dyson OF, Weidner DA, Chintalgattu V, et al. (2006)
Raf/MEK/ERK signalling triggers reactivation of Kaposi's sarcoma-associated
herpesvirus latency. J Gen Virol 87: 1139-1144.
275. Qin Z, DeFee M, Isaacs JS, Parsons C (2010) Extracellular Hsp90 serves as a co-
factor for MAPK activation and latent viral gene expression during de novo
infection by KSHV. Virology 403: 92-102.
276. Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ (2008) Reactivation of Kaposi's sarcoma-
associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple
mitogen-activated protein kinase pathways. Virology 371: 139-154.
Page 161
147
277. Pan H, Xie J, Ye F, Gao SJ (2006) Modulation of Kaposi's sarcoma-associated
herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-
activated protein kinase pathways during primary infection. J Virol 80: 5371-5382.
278. Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, et al. (2005) ERK1/2
and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human
herpesvirus 8) early during infection of target cells are essential for expression of
viral genes and for establishment of infection. J Virol 79: 10308-10329.
279. Haura EB, Turkson J, Jove R (2005) Mechanisms of disease: Insights into the
emerging role of signal transducers and activators of transcription in cancer. Nat
Clin Pract Oncol 2: 315-324.
280. Hirano T, Ishihara K, Hibi M (2000) Roles of STAT3 in mediating the cell growth,
differentiation and survival signals relayed through the IL-6 family of cytokine
receptors. Oncogene 19: 2548-2556.
281. Punjabi AS, Carroll PA, Chen L, Lagunoff M (2007) Persistent activation of STAT3
by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J
Virol 81: 2449-2458.
282. Ensoli B, Sturzl M (1998) Kaposi's sarcoma: a result of the interplay among
inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth
Factor Rev 9: 63-83.
283. Chen L, Lagunoff M (2007) The KSHV viral interleukin-6 is not essential for latency
or lytic replication in BJAB cells. Virology 359: 425-435.
284. Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, et al. (2004) Concurrent
expression of latent and a limited number of lytic genes with immune modulation
and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early
during infection of primary endothelial and fibroblast cells and subsequent decline
of lytic gene expression. J Virol 78: 3601-3620.
Page 162
148
285. Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, et al. (2013) Biphasic euchromatin-
to-heterochromatin transition on the KSHV genome following de novo infection.
PLoS Pathog 9: e1003813.
286. Najjar I, Fagard R (2010) STAT1 and pathogens, not a friendly relationship. Biochimie
92: 425-444.
287. Sen N, Che X, Rajamani J, Zerboni L, Sung P, et al. (2012) Signal transducer and
activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus
promote replication and skin pathogenesis. Proc Natl Acad Sci U S A 109: 600-
605.
288. McCartney EM, Helbig KJ, Narayana SK, Eyre NS, Aloia AL, et al. (2013) Signal
transducer and activator of transcription 3 is a proviral host factor for hepatitis C
virus. Hepatology 58: 1558-1568.
289. Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C (2013) Human
cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering
STAT3 in the nucleus. J Virol 87: 10763-10776.
290. Trilling M, Le VT, Rashidi-Alavijeh J, Katschinski B, Scheller J, et al. (2014)
"Activated" STAT proteins: a paradoxical consequence of inhibited JAK-STAT
signaling in cytomegalovirus-infected cells. J Immunol 192: 447-458.
291. Wang WB, Levy DE, Lee CK (2011) STAT3 negatively regulates type I IFN-mediated
antiviral response. J Immunol 187: 2578-2585.
292. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, et al. (1996) Targeted
disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in
the JAK-STAT signaling pathway. Cell 84: 431-442.
293. Park C, Li S, Cha E, Schindler C (2000) Immune response in Stat2 knockout mice.
Immunity 13: 795-804.
Page 163
149
294. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, et al. (1997) Targeted
disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl
Acad Sci U S A 94: 3801-3804.
295. Du T, Zhou G, Roizman B (2013) Modulation of reactivation of latent herpes simplex
virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc Natl Acad Sci
U S A 110: E2621-2628.
296. Lieu KG, Brice A, Wiltzer L, Hirst B, Jans DA, et al. (2013) The rabies virus interferon
antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor
signaling. J Virol 87: 8261-8265.
297. Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM (2003) STAT3 ubiquitylation and
degradation by mumps virus suppress cytokine and oncogene signaling. J Virol
77: 6385-6393.
298. Percario Z, Olivetta E, Fiorucci G, Mangino G, Peretti S, et al. (2003) Human
immunodeficiency virus type 1 (HIV-1) Nef activates STAT3 in primary human
monocyte/macrophages through the release of soluble factors: involvement of Nef
domains interacting with the cell endocytotic machinery. J Leukoc Biol 74: 821-
832.
299. Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, et al. (2009) Persistently activated
Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15: 283-
293.
300. Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a
leading role for STAT3. Nat Rev Cancer 9: 798-809.
301. Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation.
Nature 454: 436-444.
Page 164
150
302. Yoshizawa H (2002) Hepatocellular carcinoma associated with hepatitis C virus
infection in Japan: projection to other countries in the foreseeable future. Oncology
62 Suppl 1: 8-17.
303. Boccardo E, Lepique AP, Villa LL (2010) The role of inflammation in HPV
carcinogenesis. Carcinogenesis 31: 1905-1912.
304. Morrison WB (2012) Inflammation and cancer: a comparative view. J Vet Intern Med
26: 18-31.
305. Croll SD, Ransohoff RM, Cai N, Zhang Q, Martin FJ, et al. (2004) VEGF-mediated
inflammation precedes angiogenesis in adult brain. Exp Neurol 187: 388-402.
306. Luther SA, Cyster JG (2001) Chemokines as regulators of T cell differentiation. Nat
Immunol 2: 102-107.
307. Pati S, Cavrois M, Guo HG, Foulke JS, Jr., Kim J, et al. (2001) Activation of NF-
kappaB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a
paracrine model of Kaposi's sarcoma pathogenesis. J Virol 75: 8660-8673.
Page 165
151
CURRICULUM VITAE
Emily M. Cousins EDUCATION 2014 PhD, Cellular and Molecular Medicine
Johns Hopkins University School of Medicine, Baltimore, MD 2008 B.S. Biochemistry
Calhoun Honors College, Clemson University, Clemson, SC RESEARCH EXPERIENCE 2009–2014 Graduate Student, Cellular and Molecular Medicine
Johns Hopkins University School of Medicine, Baltimore, MD Advisor: John Nicholas, PhD
Research: Viral Oncology. Described the role of endoplasmic reticulum-localized HHV-8 viral interleukin-6 (vIL-6) on cell growth, viability, cell signaling, and virus production. Developed an assay to determine infectious viral titers from cells following depletion or overexpression of viral and/or host proteins. Research findings will facilitate the search for small molecule inhibitors of viral-host interactions within the endoplasmic reticulum, advancing therapeutic options for those suffering from HHV-8 infection.
2008 Summer Intern, Pediatric Oncology Education
St. Jude Children’s Research Hospital, Memphis, TN Advisor: Phil Potter, MD Research: Molecular Pharmacology research. Mutagenized human cholinesterase I at key catalytic residues to evaluate substrate specificity. Research findings will help to elucidate the differences in human cholinesterase specificities for chemotherapeutic pro-drugs, impacting future pro-drug design.
2007–2008 Undergraduate Research
Clemson University, Clemson, SC Advisor: Michael Sehorn, PhD Research: DNA repair research. Designed and cloned bacterial DNA repair protein homologs to determine key protein-protein interaction residues. Research findings from the bacterial system have driven experiments utilizing repair proteins in an effort to characterize homologous recombination repair pathways.
Page 166
152
TEACHING EXPERIENCE 2011 Pollard Scholar – Genetics JHU School of Medicine 2007-2008 Student Athlete Tutor Clemson University SELECTED HONORS AND AWARDS 2008 General and Departmental Honors Clemson University 2005-2008 Out of State Tuition Scholarship Clemson University 2005-2008 Presidential Scholar Clemson University 2005-2008 Philip Prince Alumni Presid. Scholar Clemson University 2006 President’s List Clemson University 2005 Dean’s List Clemson University 2007, 2008 Dean’s List Clemson University 2006-2008 Dixon Fellow Clemson University 2005-2008 Alpha Lambda Delta Natl Hon Soc Clemson University PUBLICATIONS Peer-reviewed scientific articles: 1. Chen D, Cousins E, Sandford G, and Nicholas J. 2012. Human herpesvirus 8 viral
interleukin-6 interacts with splice variant 2 of vitamin K epoxide reductase complex subunit 1. J. Virol. 2012; 86(3):1577-1588
2. Cousins E and Nicholas J. 2013. Role of human herpesvirus 8 interleukin-6-activated gp130 signal transducer in primary effusion lymphoma cell growth and viability. J. Virol. 2013; 87(19):10816-10827
3. Cousins E, Gao Y, Sandford G, and Nicholas J. 2014. Human herpesvirus 8 viral
interleukin-6 signaling through gp130 promotes virus replication in primary effusion lymphoma and endothelial cells. J. Virol. 2014; 88(20):12167-12172
Book chapter 1. Cousins E and Nicholas J. 2014. Molecular biology of human herpesvirus 8: Novel
functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 2014; 193:227-68. doi:10.1007/978-3-642-38965-8_13
Abstracts 1. Cousins E and Nicholas J. 2013. Role of human herpesvirus 8 interleukin-6-
activated gp130 signal transducer in primary effusion lymphoma cell growth and viability. Abstract #31. 16th International Workshop on Kaposi’s Sarcoma Herpesvirus (KSHV) and Related Agents. Oral presentation. Puerto Vallarta, Mexico. July 2013
Page 167
153
SERVICE AND LEADERSHIP 2011–2014 Contract editor American Journal Exp., Durham, NC 2010–2014 Mentor Incentive Mentoring Program, Baltimore, MD PROFESSIONAL SOCIETY MEMBERSHIPS 2011– American Association for the Advancement of Science (AAAS) FUNDING 2012–2014 Ruth Kirschstein Fellowship F31-CA171933, NIH NCI 2008 Calhoun Honors College Research Grant, Clemson University

































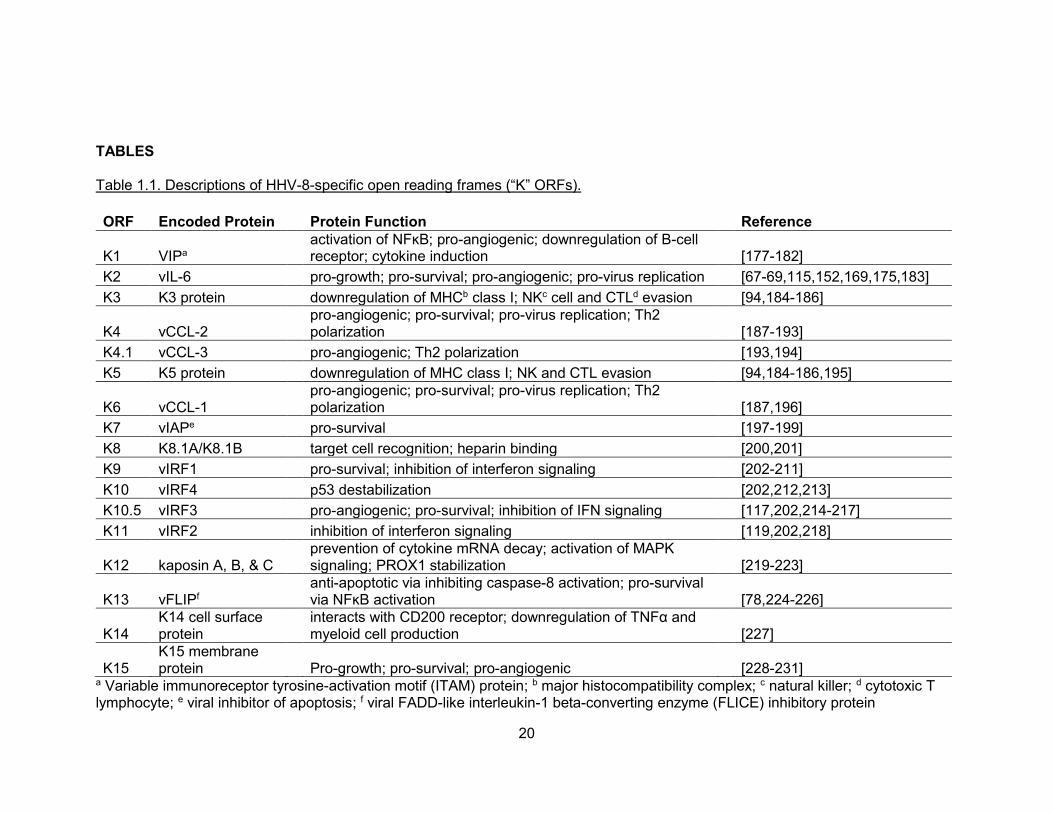









































































































































![BMC Biotechnology BioMed Central...BAC technology created new possibilities for the targeted mutagenesis of the herpesvirus genome (for recent reviews, see [19,20]). Since the viral](https://static.documents.pub/doc/80x56/61076e3eec9cb5684264f3f7/bmc-biotechnology-biomed-central-bac-technology-created-new-possibilities-for.jpg)












