HAL Id: tel-01558924 https://tel.archives-ouvertes.fr/tel-01558924 Submitted on 10 Jul 2017 HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci- entific research documents, whether they are pub- lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers. L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés. Altérations physiologiques et récupération à long terme dans un modéle murin de séparation associée à une restriction du temps d’accés à l’alimentation : un outil pour l’étude des conséquences de l’anorexie mentale Sara Zgheib To cite this version: Sara Zgheib. Altérations physiologiques et récupération à long terme dans un modéle murin de séparation associée à une restriction du temps d’accés à l’alimentation : un outil pour l’étude des conséquences de l’anorexie mentale. Physiologie [q-bio.TO]. Université du Littoral Côte d’Opale, 2014. Français. NNT : 2014DUNK0428. tel-01558924
Transcript
HAL Id: tel-01558924https://tel.archives-ouvertes.fr/tel-01558924
Submitted on 10 Jul 2017
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
Altérations physiologiques et récupération à long termedans un modéle murin de séparation associée à une
restriction du temps d’accés à l’alimentation : un outilpour l’étude des conséquences de l’anorexie mentale
Sara Zgheib
To cite this version:Sara Zgheib. Altérations physiologiques et récupération à long terme dans un modéle murin deséparation associée à une restriction du temps d’accés à l’alimentation : un outil pour l’étude desconséquences de l’anorexie mentale. Physiologie [q-bio.TO]. Université du Littoral Côte d’Opale,2014. Français. �NNT : 2014DUNK0428�. �tel-01558924�
I-POURQUOI S’ INTÉRESSER À L’ANOREXIE MENTALE ? ............................................................................................. 5
I-A Données épidémiologiques ............................................................................................................................ 5
I-B Principales causes ......................................................................................................................................... 6 II- CONSÉQUENCES DE L’ANOREXIE MENTALE .......................................................................................................... 9 II-A COMPORTEMENT ALIMENTAIRE ......................................................................................................................... 9
II-B Régulation centrale de la prise alimentaire ............................................................................................... 11
II-C Déséquilibre de la balance énergétique et survie ...................................................................................... 12
II-D La masse grasse et les tissus adipeux ........................................................................................................ 19
II-E La masse maigre et les tissus musculaires ................................................................................................. 19
II-F Les altérations osseuses ............................................................................................................................. 20 II-F-1 Structure Osseuse .................................................................................................................................................. 20 II-F-2 Régulations de la masse osseuse ........................................................................................................................... 22
II-F-2.1 Balance des marqueurs de formation et de résorption osseuse...................................................................... 22
II-F-2.2 Les mécanismes de régulation de la masse osseuse ...................................................................................... 25
II-F-2.2.1 Perturbations des facteurs endocriniens impliqués dans la régulation de la masse osseuse .................. 25 La leptine ......................................................................................................................................................... 28 L’adiponectine ................................................................................................................................................. 33 L’aménorrhée et les hormones sexuelles ......................................................................................................... 35
L’axe GH-IGF-1 .............................................................................................................................................. 36 Le cortisol ........................................................................................................................................................ 41 L’insuline ......................................................................................................................................................... 42 La ghréline ....................................................................................................................................................... 43
II-F-2.2.2 L’adiposité médullaire .......................................................................................................................... 44 Quelle est la fonction de l’adipocyte médullaire .............................................................................................. 44
L’adiposité médullaire et la masse osseuse ...................................................................................................... 46
L’adiposité médullaire dans l’anorexie mentale .............................................................................................. 50
II-G- Autres altérations ..................................................................................................................................... 51 II-G-1 Conséquences digestives ...................................................................................................................................... 51 II-G-2 Système nerveux central ....................................................................................................................................... 52 II-G-3 Système immunitaire ............................................................................................................................................ 52
III-COMMENT ÉTUDIER LES CONSÉQUENCES PHYSIOLOGIQUES DE L’AM ? ............................................................. 52 III-A Études cliniques ........................................................................................................................................ 52 III B- Les modèles animaux ............................................................................................................................... 53
III-B-1 Modèles d’induction environementale ................................................................................................................ 54 III-B-1.1 Modèles de dépression ................................................................................................................................ 54 III-B-1.2 Modèles de restriction alimentaire quantitative ........................................................................................... 54
III-B-1.3 Modèle d’activité / restriction ..................................................................................................................... 55 III-B-1.4 Modèles de restriction du temps d’accès à la nourriture ............................................................................. 56
III-B-1.5 Modèle de Séparation / restriction............................................................................................................... 56 III-B-2 Les modèles murins génétiques ........................................................................................................................... 57
TRAVAIL DE THESE ............................................................................................................................................. 60
I- AXES DE RECHERCHE DU PMOI........................................................................................................................... 60 II-SUJET ET OBJECTIF DU PROJET DE THÈSE ............................................................................................................. 61
II-A Définition du cahier des charges ............................................................................................................... 61
II-B Choix du modèle animal ............................................................................................................................. 62
II-B-1 Choix du protocole d’induction ............................................................................................................................ 62 II-B-2 Animaux et conditions d’élevage ......................................................................................................................... 63 II-B-3 Protocoles étudiés ................................................................................................................................................. 64
II-B-3-1 Essais à court terme...................................................................................................................................... 64 II-B-3-2 Essais à long terme ....................................................................................................................................... 66
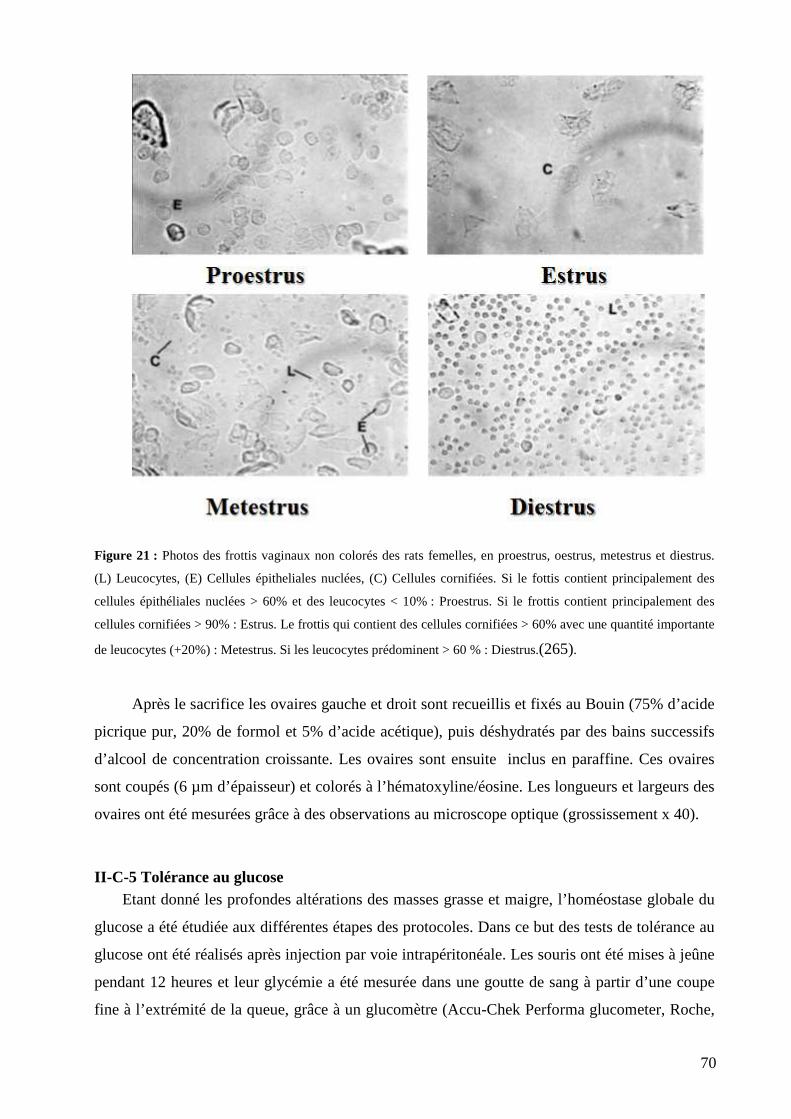
II-C Choix des paramètres étudiés et des méthodes utilisées pour évaluer ces altérations .............................. 67 II-C-1 Prise alimentaire ................................................................................................................................................... 67 II-C-2 Poids corporel ....................................................................................................................................................... 67 II-C-3 Composition corporelle ........................................................................................................................................ 67
III-A Choix du protocole à partir d’essais à court terme .................................................................................. 77
III-A-1 Suivi du poids corporel et de la prise alimentaire à court terme .......................................................................... 78
III-A-2 Composition corporelle ....................................................................................................................................... 79 III-A-3 Conclusion de l’étude à court terme .................................................................................................................... 80
III-B Protocoles SBA et REC à long terme ........................................................................................................ 81
II-B-1 Poids corporel et prise alimentaire ........................................................................................................................ 81 II-B-2 Composition corporelle ........................................................................................................................................ 82 II-B-3 Etude de la microarchitecture osseuse .................................................................................................................. 84 II-B-4 Pesées tissulaires .................................................................................................................................................. 85 II-B-5 Fonctions reproductrices : la taille des ovaires et le cycle œstral .......................................................................... 88
II-B-6 Etude des perturbations endocriniennes ................................................................................................................ 92 II-B-6.1 Leptinémie et adiponectinémie ..................................................................................................................... 92 II-B-6.2 Hormone de croissance GH et Insulin-like growth factor-1 IGF-I ............................................................... 94
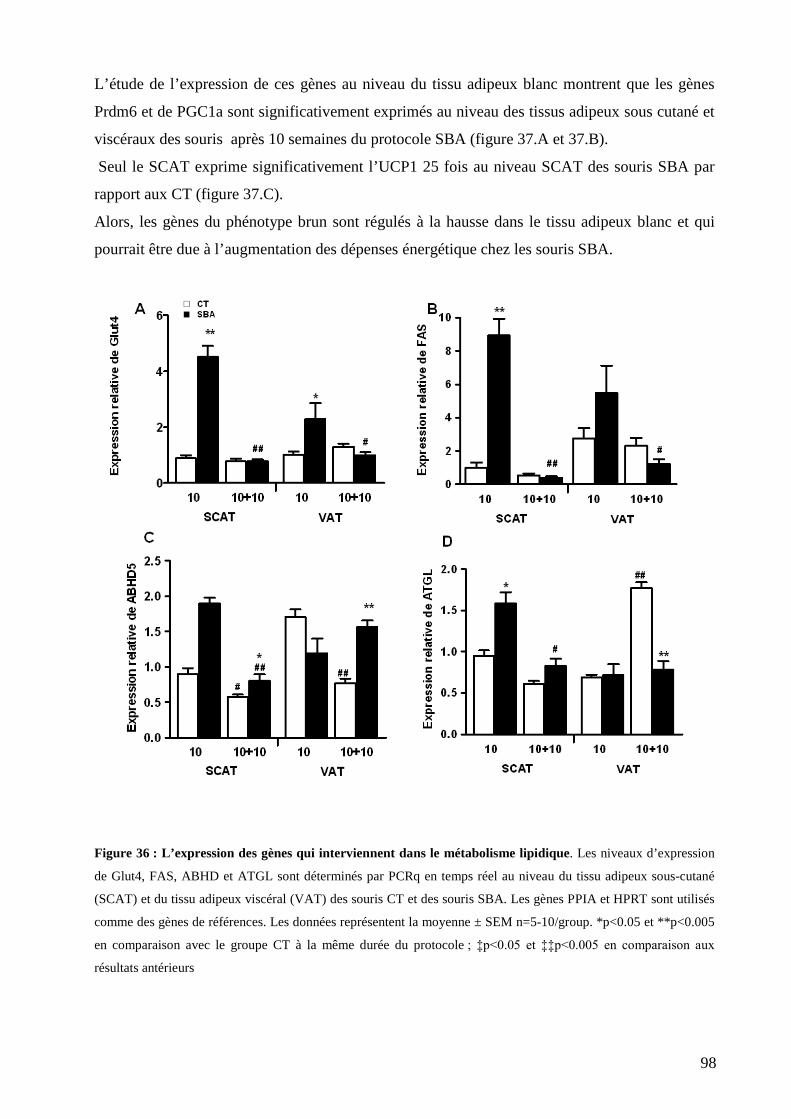
II-B-7 Tolérance au glucose ............................................................................................................................................ 95 II-B-8 Niveaux d’expression des gènes impliqués dans l’oxydation des acides gras et la lipogenèse dans les tissus adipeux blancs au cours du protocole SBA et les niveaux d’expression des gènes spécifiques du tissu brun ................. 97 II-B-9 Étude de l’expression du récepteur bêta 3 adrénergique au niveau du VAT, SCAT et BAT ................................ 99 II-B-10 Etude histologique de l’adiposité médullaire .................................................................................................... 100
IV-A ANALYSE INTÉGRÉE DU MODÈLE SBA ......................................................................................................... 102 IV-B COMPARAISON DU MODÈLE SBA AVEC LES AUTRES MODÈLES .................................................................... 108 IV-C COMPARAISON DES ALTÉRATIONS OBSERVÉES AVEC CELLES DÉCRITES CHEZ LES PATIENTES ...................... 112 IV-D PERSPECTIVES D’ÉTUDE DU MODÈLE SBA ................................................................................................... 113
I-Pourquoi s’intéresser à l’anorexie mentale ? En regard des grands programmes de recherche dédiés aux pathologies induites par les
mauvaises habitudes alimentaires et un mode de vie sédentaire, les études consacrées aux
troubles mentaux font figure de parent pauvre. Au sein de ces troubles mentaux, les troubles du
comportement alimentaire (TCA) sont considérés comme des pathologies de faible prévalence,
alors même qu’ils sont globalement moins recherchés que les autres troubles mentaux. Pourtant
dans le monde occidental, les TCA prennent insidieusement les proportions d’une épidémie, et
comme nous le verrons dans la partie consacrée aux données épidémiologiques et dans celle
consacrée aux conséquences, ils constituent un vrai problème de santé publique.
Parmi ces TCA, l’anorexie mentale (AM) est une affection psychiatrique débutant souvent
à l’adolescence, dont l’expression clinique est somatique (amaigrissement et aménorrhée) et
alimentaire (restriction alimentaire). Elle touche essentiellement les adolescentes et les jeunes
femmes. La forte réduction de la prise alimentaire des patientes anorexiques conduit en quelques
mois à un amaigrissement intense. Le National Collaborating Centre for Mental Health (UK) a
défini que la perte du poids chez les patients représentait 15 à 30% de leur poids initial, et que
leur indice de masse corporelle (IMC) était inférieur à 17.5 kg/m² (1). Dans ces conditions, la
survie des patientes nécessite des adaptations métaboliques importantes, dont certaines
pourraient ensuite constituer des freins au long et incertain processus de guérison.
Dans un premier temps et afin de mieux cerner cette pathologie dans son ensemble nous
allons présenter les connaissances disponibles concernant les causes et l’épidémiologie. Cela
nous permettra ensuite de présenter ses multiples conséquences physiologiques tout en ayant une
image du contexte dans lequel elles se produisent. Dans cette partie introductive nous traiterons
enfin des différentes approches développées pour étudier les mécanismes et conséquences de
l’AM.
I-A Données épidémiologiques Peu de données sont disponibles pour estimer la prévalence des troubles de l'alimentation
dans la population mondiale. Mais on sait bien que ces troubles seraient en augmentation dans
les pays économiquement développés depuis une vingtaine d’années.
Aux États-Unis, l’anorexie est la troisième maladie chronique après l’obésité et l’asthme
chez les adolescentes, avec une prévalence de 0,48 % dans la tranche des 15-19 ans. La
prédominance féminine est nette : 8 à 9 cas sur 10 (2).
6
En France, 0,5 % à 1 % des adolescentes et des jeunes femmes souffrent d’AM soit 30 000
à 40 000 au total (3). le sex-ratio est de 9 filles pour 1 garçon (1). Parmi les troubles
psychiatriques, les TCA entrainent un grand nombre de décès, 15 % des anorexiques finissent
par décéder subitement dont 50% des cas de décès sont dus à un sucide et un certains nombre de
malades ne semblent jamais guérir, d’où la gravité de cette maladie (4).
L’incidence la plus élevée a été trouvée entre l’âge de 10 à 19 ans (3). Mais le
déclenchement de la pathologie a lieu principalement à deux périodes critiques : 13-14 ans et 16-
17 ans. Les psychiatres ont noté que ces périodes correspondaient aux moments où la
dépendance vis-à-vis de la famille est la plus importante (4).
I-B Principales causes Bien qu’ils soient encore relativement mal connus, il a été établi que les facteurs favorisant
l’apparition du comportement anorexique appartenaient à différentes catégories. Classiquement
les spécialistes distinguent les facteurs psychosociaux et les facteurs génétiques (5). Ces facteurs
constituent en fait un terrain favorable et sont à distinguer des évènements déclencheurs qui
entraînent très souvent une modification temporaire du comportement alimentaire.
1- Facteurs de risque psychosociaux
Les troubles alimentaires sont qualifiés de troubles liés à la culture et à l’échelle sociale.
Les premières études réalisées montraient des disparités dans leurs développements en fonction
des différentes ethnies présentes aux États-Unis. Ces troubles étaient décrits dans les sociétés où
la nourriture était abondante et où la minceur était convoitée. Les cultures plus pauvres dans
lesquelles les rondeurs étaient valorisées semblaient relativement épargnées par ces pathologies.
Toutefois, les résultats des recherches sur l’influence de la culture sont discordants. Des études
récentes et de grandes ampleurs montrent que la prévalence des troubles alimentaires est la
même, quelle que soit l’origine ethnique (6).
Néanmoins, l’image de la minceur et la pression sociale à être mince sont entretenues et
amplifiées depuis une trentaine d’année par, les magazines féminins le cinéma et les médias qui
jouent un rôle non négligable en véhiculant une image faussée de la réalité. Les femmes
exposées à des images de femmes avec des corps très minces ressentent des émotions négatives
telles que la tristesse la honte et bien sûr une insatisfaction de leur propre image. Une étude
menée aux États-Unis montre qu’environ 60% des filles et 30% des garçons désirent abaisser
leur poid. Cette insatisfaction corporelle est désormais considérée comme le principal facteur de
développement des troubles alimentaires (7).
7
Les histoires familiale et personnelle semblent également jouer un rôle. Les données de
la littérature sur l'environnement psychosocial de l'adolescente anorexique suggèrent que
l'altération des relations intrafamiliales et de la communication, la maltraitance, les troubles
affectifs et alimentaires parentaux, les pressions parentales inappropriées, les séparations et les
adversités chroniques, seraient impliqués(8). On peut mentionner l'obésité prémorbide, le trouble
obsessionnel-compulsif précédant les troubles dépressifs, les troubles de la personnalité
borderline, et des antécédents d’abus sexuel. Les activités qui favorisent la minceur, comme la
danse classique, le fitness et le sport sont aussi considérées comme des facteurs de susceptibilité
(9).
Enfin, mêlant les facteurs sociaux et héréditaires, certaines études de jumeaux montrent
que ces personnes présentent un lien étroit des troubles du comportement alimentaire (4).
Cependant, d’autres études ne retrouvent pas de corrélation. Parmi les facteurs du même ordre,
les antécédents familiaux de troubles de l'humeur chez un parent au premier degré peuvent
également s’avérer être un facteur de risque (10).
Il est logique de s’interroger sur ce qui fait qu’une personne très mince ou maigre continue
à chercher à perdre du poids. L’explication principale est liée à un trouble psychiatrique qui
entraîne une distorsion de l’image corporelle, autrement dit, les patientes bien que très maigres
se perçoivent malgré cela comme des personnes en surpoids. Ce paradoxe est classiquement
représenté par l’image d’une personne se regardant dans un miroir (figure 1). Une des approches
psychiatriques est notamment consacrée à faire travailler les patientes sur la perception qu’elles
ont de leur corps et de celui des autres, car cette distorsion semble constituer un frein important à
la correction du comportement alimentaire.
8
Figure1: Illustration de la distorsion de l’image corporelle
Source : http://www.medicalorama.com/
2- Facteurs génétiques:
L’évolution technologique considérable de ces dernières années en matière d’analyse
génétique a permis de développer des études sur les facteurs génétiques qui sont susceptibles de
participer au développemnt de l’AM. Ces études font notamment partie des Genome-Wide
Association studies (GWAS) dont l’approche est très prometteuse, mais elles peinent encore à
dégager de résultats significatifs et reproductibles pour l’AM (11). Ces études sont fondées sur
l’hypothèse qu’une partie au moins des personnes qui développent une même pathologie
peuvent partager une ou plusieurs mutations génétiques qui les prédisposent à cette maladie.
Plusieurs études sur des fratries, ont permis d’estimer que 33% à 84% des cas d’AM ont
une origine ou une composante héréditaire (12,13)(14,15)(16).
Parmi ces gènes suspectés, on trouve l’Agouti-related protein (AgRP) qui est un facteur
orexigène qui antagonise le melanocortin-4- receptor (MC4-r). Le gène de l’AgRP a été
séquencé chez 100 patientes anorexiques. Il a été trouvé une fréquence assez élevée d’une
mutation entraînant un remplacement de l’Alanine 67 par une thréonine. Ce résultat a permis de
montrer que cette mutation était significativement associée à l’AM. Ce qui va entrainer une
inhibition altérée de la MC4-r, une diminution de la prise alimentaire et augmentation du risque
de développer l’anorexie (17).
Le facteur neurotrophique dérivé du cerveau (BDNF) est une protéine qui favorise la
croissance, la différenciation et la survie des neurones et des synapses du système nerveux
central et périphérique (18,19). Le BDNF est exprimé dans les régions du cerveau responsables
9
des fonctions cognitives et exécutives supérieures, telles que l'hippocampe, le noyau arqué qui
joue un rôle crucial dans les formes de plasticité synaptique (20,21). Plusieurs études ont montré
que le gène BDNF intervient dans la régulation du comportement alimentaire (22,23). Les études
sur des patientes anorexiques ont montré que le taux sérique de BDNF est moins élevé que celui
de femmes saines, ce taux bas de BDNF en anorexie peut jouer un rôle important dans les
symptômes dépressifs qui accompagnent les troubles du comportement alimentaire de cette
maladie (22,24). Le BDNF jouerait donc un rôle important dans la physiopathologie des troubles
d’aimentation, mais les mécanismes impliqués sont très mal étudiés. Etant donné que plusieurs
études cliniques et précliniques ont montré une implication du BDNF dans les troubles du
comportement alimentaire. Il a été montre que les différences dans le gène BDNF pouvaient
être à l’origine de la grande hétérogénéité comportementale. L’existence d’un polymorphisme
sur un seul nucléotide (SNP) à l’origine d’un changement d’acide aminé (une leucine est
remplacée par une méthionine) en position 32 dans la séquence du prodomaine du BDNF, bien
que ce SNP ne modifie par l’expression basale de BDNF dans le cerveau, est associé au
phénotype « anxieux », et à la perturbation du comportement alimentaire et pourrait participer à
la baisse de l’IMC notamment en anorexie (25).
3- Phénomènes déclencheurs de l’anorexie mentale :
Les facteurs ou évènements susceptibles de déclencher un comportement alimentaire de
type anorexique peuvent être d’origine traumatique, comme des accidents ou des interventions
chirurgicales touchant la face et imposant une alimentation réduite et sous forme liquide pendant
plusieurs semaines. L’AM peut aussi démarrer par des régimes initialement envisagés pour du
court terme. Dans tous les cas, comme le souligne le psychiatre J. Vignau (Service
d’addictologie, CHRU Lille) « les patientes arrivent à un stade où elles perdent le contrôle de
leur comportement alimentaire ».
II- Conséquences de l’anorexie mentale
II-A Comportement alimentaire L’anorexie mentale et la boulimie sont deux troubles du comportement alimentaire. Et s’il
peut paraître logique de distinguer ces deux troubles, les cas rencontrés en clinique font douter
de la pertinence de cette démarche (26). La majorité des anorexiques évoluent vers une
boulimie (27), mais une minorité de patientes boulimiques évoluent vers une anorexie mentale
(28). Cependant l’étude relativement récente de Birmingham et ses collègues (2009) conclue, sur
la base des critères utilisés, à l’existence de deux pathologies différentes.
10
Au sein même de l’AM il est nécessaire de distinguer deux sous-types basés sur des
comportements alimentaires différents. En effet, 40 à 50% des patientes anorexiques
appartiennent au sous-type « mixte » car elles présentent un comportement boulimique suivi de
vomissements volontaires (29).
Figure 2: les deux sous-types de l’AM
Les patientes relevant de ce comportement anorexique mixte présentent des caractéristiques
différentes des patientes anorexiques restrictives, notamment vis-à-vis du poids corporel, de
l’indice de masse corporelle, des perturbations endocriniennes (30,31). Le sous-type restrictif
comprend une restriction sévère de la quantité et de la teneur énergétique des aliments, sans
phase de boulimie/vomissements (Figure 2).
Nous orienterons l’étude bibliographique et le développement d’un modèle animal vers ce
type restrictif qui est lié à des cas généralement à la fois plus sévères et moins hétérogènes que le
type mixte. Dans le processus qui nous a conduit à développer un modèle animal mimant les
conséquences de l’AM restrictive, il était nécessaire de définir plus précisément à quoi
correspondait la restriction alimentaire des patientes. Afin de rationaliser les études sur le sujet,
une échelle a été proposée par Wilson et al en 1989 le « Eating Behavior Rating Scale » qui est
une échelle d’évaluation du comportement alimentaire. Dans ce type d’évaluation les patientes
anorexiques restrictives et boulimiques ensemble ont un score global plus élevé comparé aux
patients boulimiques à poids normal. Les patientes restrictives ont montré plus de dégoûts
11
alimentaires que les autres groupes et leur comportement à table a été estimé comme plus
ritualisé (32).
Comme présenté dans le paragraphe concernant les phénomènes déclencheurs, à l’origine
du comportement anorexique il ne s’agit nullement d’une perte d’appétit, mais souvent d’un
régime pour perdre quelques kilos ou d’une cause accidentelle ou chirurgicale imposant une
modification importante mais temporaire de l’alimentation. La conduite alimentaire anorexique
au début est modérée et devient méthodique, obsessionnelle et radicale. Ce qui se manisfeste par
le dégoût pour certains types d’aliments, la préférence pour une alimentation de basse valeur
calorique. Ce comportement conduit les patientes à éviter le plus souvent les repas en société et
même en famille pour éviter de se jsutifier (32,33).
À ces comportements s’ajoutent des idées reçues comme par exemple la nécessité de
prendre certains aliments selon un ordre rigidement respecté ou d’une façon particulière afin
d’influencer la digestion et la vitesse de la prise du poids (34).
Le retour à une alimentation plus abondante et plus riche est un des premiers objectifs du
traitement, afin de restaurer progressivement le poids corporel. Cette reprise doit également être
continue et conditionne la poursuite de la croissance des jeunes patientes (35–37) et la correction
de nombreux paramètres biologiques altérés. Mais la multiplicité des causes, et comme nous le
verrons par la suite des conséquences, contribue à rendre la prise en charge difficile. De fait, les
patientes bénéficient généralement de soins psychologiques et somatiques grâce à des prises en
charge pluridisciplinaires. Le recours à la prescription des psychotropes est peu développé car il
n’a pas démontré d’efficacité pour diminuer les préoccupations pondérales (38).
La durée de l’hospitalisation dépend de plusieurs facteurs. Ceux-ci comprennent bien
entendu le temps nécessaire pour atteindre le poids souhaité avec une stabilisation d’au moins
deux semaines mais aussi la qualité des repas et des relations avec les parents, la fratrie, et les
relations sociales. Cependant l’AM est une maladie considérée comme chronique et le milieu
psychiatrique étudie les similitudes de cette pathologie avec les comportements addictifs (39).
Les patientes présentent des risques élevés de récidive pendant une longue période d’où les
efforts des cliniciens pour maintenir un suivi spécialisé à long terme (40) malgré la « fuite » des
patientes.
II-B Régulation centrale de la prise alimentaire Le noyau arqué de l’hypothalamus joue un rôle primordial dans la régulation du
comportement alimentaire Il contient deux types de neurones différents : les premiers entraînant
un effet orexigène, les seconds un effet anorexigène (Sam et al. 2012). Les neurones orexigènes
sont les neurones à neuropeptide Y (NPY) et à « agouti related protein » (AgRP) (Sam et al.
2012). Ils ont une action anorexigène en allant inhiber les neurones POMC/CART grâce à
12
l’antagonisme de l’AgRP sur le récepteur à la mélanocortine de type 4 (MCR-4) présent sur les
neurones à POMC (41). L’expression de ces neurones est augmentée lors du jeûne mais inhibée
par la leptine (42)(.
Les neurones à proopiomélanocortine (POMC) ont un effet anorexigène. L’activation des
neurones POMC est notamment effectuée par la leptine. Ils ont un effet anorexigène en
produisant deux molécules différentes : CART et α-MSH. Premièrement, ils réduisent l’apport
alimentaire en produisant du CART, qui active lui-même la famille des récepteurs à la
mélanocortine (MC3-R et MC4-R)(43). L’activation de ces neurones POMC entraîne la
production, dans un second temps, d’ α -MSH qui réduit la prise alimentaire en agissant
principalement sur MCR-4 (41). De nombreuses études menées chez l’animal prouvent que si
l’on élimine ne serait-ce qu’un composant de cette voie, l’animal développe une forte obésité
(43) . Ils activent directement les récepteurs à la mélanocortine MC3-R et MC4-R. Les neurones
NPY/AgRP exercent un tonus inhibiteur sur les neurones POMC (43) .
II-C Déséquilibre de la balance énergétique et survie Afin de mieux appréhender le contexte dans lequel se produisent les différentes altérations
décrites chez les patientes, il serait utile de définir l’état physiologique dans lequel elles se
trouvent. Les adaptations au déficit énergétique se font par la réduction des dépenses
énergétiques, une diminution de la température corporelle et du métabolisme, un retard de la
croissance, et la mobilisation des stocks de graisse dans l’organisme. On peut distinguer
schématiquement trois différentes phases de l’adaptation à la privation alimentaire
prolongée (44) .
La phase I :
Elle commence quelques heures après le dernier repas ingéré et digéré. Le corps entre ainsi
dans l’état post-absorptif. La phase I est une phase d’approvisionnement régulier de glucose
entre les repas. La glycémie est d’environ 5 g (100 mg/dl dans 5 L de sang), il y a aussi 80 g de
glycogène dans le foie et 350 g dans les muscles, qui peuvent se convertir en glucose par
phosphorylation. Les réserves totales de glucose et de glycogène sont donc de l’ordre de 480 g
qui seront épuisés dans les 24 heures (45). Des acides gras sont libérés par les tissus adipeux, ce
qui permet par exemple aux muscles squelettiques d’économiser leur utilisation globale de
glucose.
13
La phase II :
Une fois les réserves de glycogène complètement épuisées, la phase II doit commencer
pour assurer une néoglucogenèse et donc un approvisionnement en glucose notamment pour le
cerveau. Elle peut s’étaler sur plusieurs semaines selon les dépenses énergétiques et les réserves
lipidiques disponibles. Chez l’homme les acides aminés issus de la protéolyse musculaire
représentent la source initiale pour la néoglucogenèse. Mais cette contribution chute rapidement
grâce à la libération d’un autre substrat de la néoglucogenèse par les tissus adipeux, le glycérol.
Car cette phase est caractérisée par une mobilisation importante des réserves de graisse. La
libération de glycérol et d’acides gras libres permet la néoglucogenèse dans le foie et les reins
(46), ainsi que la bêta oxydation et la formation d’acétyl-CoA qui peuvent être métabolisés en
corps cétoniques pour alimenter le cerveau (44).
PPAR alpha et PPAR gamma favorisent la synthèse des graisses et la sensibilité à l’insuline
(47). Si la privation de nourriture est prolongée, les réserves de graisses utilisables sont
complètement épuisées, et le cerveau ne peut plus être alimenté par les corps cétoniques, la
phase III commence.
La phase III :
L’organisme attaque le tissu musculaire, les acides aminés se convertissent en glucose
dans le foie afin de maintenir le bon fonctionnement du cerveau, ce processus connu sous le nom
de « gaspillage de protéines », conduit très rapidement à la mort (44,48).
Voici un schéma qui récapitule les 3 phases d’adaptation au jeûne.
14
Table 1: Metabolic states occurring during food restriction PHASE I: FASTING post-absorptive phase (hours) ↑ Glycogen depletion (from liver stores) ↑ Fatty acid release (from adipose tissues) PHASE II: From FASTING to STARVATION (several weeks) ⇒ End of glycogen stores ↑ Gluconeogenesis * from adipose tissues (rapid) ⇓ Oxydation of fatty acids ⇓ Ketone bodies * from proteolysis of muscle proteins (slow) PHASE III: From STARVATION to DEATH * Dramatic depletion of adipose stores
* Degradation of muscular mass (for gluconeogenesis)
Tableau 1 : Les 3 phases d’adaptation au jeûne d’après Méquinion M et al, 2013
Ce découpage est bien entendu schématique et théorique. Afin de situer l’état des
patientes, l’étape suivante consiste à comparer les paramètres métaboliques utilisés pour
déterminer ces phases, à ceux des patientes. Dans cette démarche nous nous heurtons à plusieurs
difficultés. D’une part, les données disponibles sont limitées chez les patientes. D’autre part, les
groupes de patientes sont relativement hétérogènes en sévérité en fonction des différentes études
fournissant quelques données. Ceci est clairement visible quand on compare les IMC moyens
des patientes de différentes études, comme présenté dans le tableau 2.
15
Premier auteur et
année de publication
IMC (kg/m2)
AM vs contrôles
% IMC par rapport
aux contrôles
Nombre d’effectif
AM – contrôles
Stoving 2003 12.9 vs 20.9 62 6 – 6
Pannaccuilli 2003 16.4 vs 20.7 79 11 – 26
Delporte 2003 14.3 vs 22.4 64 26 – 24
Misra 2003 16.4 vs 21.6 76 21 – 21
Shimizu 2004 13.9 vs 17.7 78 12 – 12
Tagami 2004 14 vs 20.3 69 31 – 11
Mayer 2005 15.95 vs 20.65 77 29 -
Haas 2005 15.2 vs 22.3 57 57-49
Misra 2007 16.6 vs 22.3 74 34 – 34
Ohwada 2007 13.3 vs 22.1 60 26 – 7
Legroux 2007 15.48 vs 20.5 75 113 – 21
Misra 2008 18.4 vs 20.5 74 10 – 10
Ehrlich 2009 15.3 vs 20.8 73 36 – 44
Germain 2010 15.2 vs 20.9 73 22 – 9
Estour 2010 14.6 vs 21.4 68 210 – 42
Lawson 2011 18.2 vs 22.3 81 16 – 12
Faje 2012 17.2 vs 21.1 81 22 – 25
Kavalkova 2012 15.58 vs 21.8 71 18 – 16
Komisiki 2013 13.8 vs 20.6 67 30 – 25
Tableau 2: Dans la littérature les résultats varient largement. Ceci peut s’expliquer par des effectifs souvent
faibles et des IMC très differents ce qui va jouer un impact important sur les conclusions apportées. Voici un extrait
de la littérature de certaines études avec des conditions d’inclusion très variées (IMC entre 13 et 18 kg/m2) avec un
nombre d’individu qui varie entre 6 et 210.
Enfin, la dernière difficulté réside dans le fait que les trois phases décrites correspondent à
des cas de jeûne et non de restriction sévères des apports caloriques. Malgré tout cela lorsqu’on
essaie de resituer l’état des patientes par rapport aux différnets stades de jeûne on arrive au
constat suivant :
• Réserves en glycogène dans le foie : Il y a très peu de données disponibles (49),
mais qui indiquent que l’organisme en anorexie se protègerait des hypoglycémies
profondes, potentiellement létales, en stockant du glycogène dans le foie. Ce qui
distinguerait donc le métabolisme de l’AM de celui du jeûne.
16
• Lipolyse dans les tissus adipeux : la réduction drastique de la masse grasse permet
de conclure à la mobilisation massive des réserves lipidiques chez les patientes, de
même que les taux plasmatiques élevés d’acides gras libres (50).
• Néoglucogenèse à partir de la bêta -oxydation des acides gras / production de corps
cétoniques : Dans les cas d’AM sévères, les taux plasmatiques d’acides gras libres,
et de corps cétoniques (acétocaétate, �-Hydroxybutyrate) sont élevés. (50)
• Protéolyse musculaire : la teneur en protéines dépend de l’IMC. En effet, Hass et
al. (2009) ont montré dans leur étude que le groupe de patientes ayant une quantité
faible de protéines corporelles ont un IMC moyen de 15,3, alors que celles ayant un
taux proche de celui des contrôles ont un IMC moyen de 17,3. Cela pourrait être
évalué dans le plasma, puisque Halmi et al, ont montré que les concentrations
plasmatiques et érythrocytaires en acides aminés étaient également altérées chez les
personnes atteintes d’AM sévère (51).
• Réserves adipeuses épuisées et protéolyse musculaire : en tenant compte des
données citées ci-dessus, il semble que la combinaison de l’épuisement des
réserves lipidiques mobilisables et de la protéolyse musculaire soit effectivement
observée dans les cas sévères d’AM.
Ce qui caractérise notamment les patientes anorexiques c’est la raréfaction des dépôts
adipeux et une fonte musculaire importante. Ceci les place plutôt dans la phase III, mais avec
des apports caloriques qui permettent la survie et dans la plupart des cas des taux plasmatiques
d’acides gras libres supérieurs à la normale. Le statut métabolique des patientes anorexiques
semble donc combiner les caractéristiques de différentes phases du jeûne, y compris la phase III.
Cependant, les effets de l’activité physique importante, retrouvée chez une partie non
négligeable des patientes (52), ne sont pas encore clairement établis et donc pas pris en compte.
Les études des adaptations physiologiques chez les patientes anorexiques ont abouti à des
résultats contradictoires, mais les méthodes utilisées pour estimer la composition corporelle et
notamment la masse grasse, n’étaient souvent ni optimales, ni comparables. La disparité des
résultats peut aussi être expliquée par les différences de degré de gravité de la restriction
calorique chez les patientes anorexiques (53). Aujourd’hui, il est admis que les modifications du
métabolisme énergétique des patientes aboutissent à une diminution des dépenses énergétiques
(53–56) .
En effet, l’étude récente de Kosmiski et al, a été menée sur des patientes anorexiques (IMC
moyen 13,8) et des personnes en bonne santé et minces (IMC moyen 20,6). Les personnes
anorexiques présentaient une masse grasse inférieure de 68% à celle des contrôles, une masse de
muscles squelettiques inférieure de 21% et la masse restante (masse maigre non musculaire et
17
masse minérale) inférieure de 25%. Cette étude a été basée sur l’hypothèse que la diminution du
métabolisme cellulaire pouvait être évaluée à travers la mesure de la dépense énergétique au
repos corrigée par le poids et la composition corporelle. Les auteurs ont comparé la dépense
énergétique au repos mesurée à celle calculée à partir de la composition corporelle obtenue par
dual-energy X-ray absorptiomery. Ils ont montré que la dépense mesurée était inférieure de 20%
à celle attendue. Cette étude conclue que les patientes anorexiques dépensent 536 kcal/j de
moins que les personnes saines. Lorsque ces données sont corrigées par la composition
corporelle, il apparaît que les patientes dépensent environ 150 à 226 kcal/j de moins que ce que
permettait de prédire leur composition corporelle. Les auteurs ont considéré cette économie
comme étant physiologiquement significative en regard de la prise de poids qu’entraîne chez un
adulte un excès de 10kcal/j (53).
Pour compléter cette réflexion, il est utile de s’intéresser à une expérience de référence sur
la réduction de l’alimentation, la Minnesota Starvation Experiment (57). Cette étude de 1950, a
été menée chez 32 jeunes hommes soumis à 24 semaines de restriction alimentaire de 50%, puis
12 semaines de réalimentation contrôlée et enfin pour certains 12 semaines supplémentaires
d’alimentation à volonté (figure 3.A). La restriction a porté principalement sur les matières
grasses (-80%), les hydrates de carbones étant dimunués de 40% et les protéines d’environ 45%.
Ce régime reproduit en partie les choix alimentaires effectués habituellement par les patientes
anorexiques. La restriction a entraîné en moyenne une perte de poids de 24%. Cette perte de
poids, rapide pendant les premières semaines, tend vers zéro après environ 120 jours de
protocole (figure 3.B). La courbe expérimentale de masse grasse, bien que plus écrasée, présente
le même aspect que celle du poids corporel (figure 3.C). La reprise par réalimentation contrôlée
est beaucoup plus rapide que la perte. Le passage à une alimentation ad libitum, visiblement plus
riche en graisse, accélère encore cette récupération de poids et de masse grasse, jusqu’à placer
les individus à un poids corporel et une masse grasse supérieurs à ceux d’avant l’expérience.
18
Figure 3: Prise alimentaire (A) et données expérimentales de poids corporel (B ■) et de masse grasse (C □)
et courbes obtenues par simulation, pendant les phases basales (B), de restriction de 50% de la prise alimentaire (SS
= semistarvation), de réalimentation contrôlée (CR) et enfin de réalimentation à volonté (ALR = ad libitum
refeeding) pendant l’expérience Minnesota. Keys A, 1950; d’après Hall KD, 2006. En A, CI: carbohydrate intake,
FI : fat intake, PI : protein intake.
Il est logique de s’interroger sur la nature des économies énergétiques. La réduction de la
thermogenèse fait partie des hypothèses avancées pour expliquer cette économie, mais les
résultats à ce sujet restent controversés, notamment à cause des méthodes imprécises utilisées
pour déterminer la composition corporelle des patientes et à cause des différences d’IMC entre
les études.
19
Une fois l’existence et l’importance de cette économie énergétique admises, il est
intéressant de rechercher les régulations qui sont impliquées dans cette économie. Kosmiski et
al, rappellent que l’hypothyroïdisme est associé à une réduction significative des dépenses
énergétiques au repos. Toujours selon ces auteurs, le système nerveux sympathique (SNS)
pourrait aussi être impliqué, mais il n’y a pas d’étude reliant l’activité du SNS et la dépense
énergétique chez les patientes anorexiques.
II-D La masse grasse et les tissus adipeux Chez les femmes adultes atteintes d’AM, la masse grasse diminue fortement par rapport aux
sujets sains, la masse adipeuse est préférentiellement perdue au niveau des membres. Alors que
la reprise de poids se traduira par une récupération significative de la masse grasse au niveau du
tronc et du tissu adipeux viscéral (58,59), ce qui laisse penser que la distribution de la masse
grasse n’est plus la même (60). Plusieurs groupes ont étudié la composition corporelle et la
répartition de la graisse chez les adultes anorexiques, mais très peu d’études ont été faites chez
les adolescentes anorexiques. La physiologie des adolescentes est différente de celles des adultes
et donc les résultats obtenus chez l’adulte ne peuvent pas être extrapolés aux adolescentes. Tout
comme les adultes, les adolescentes anorexiques perdent significativement leur masse grasse et
masse maigre par rapport aux sujets contrôles et la perte de la masse grasse reste plus importante
par rapport à la perte de la masse maigre. Contrairement aux adultes, le pourcentage de la masse
grasse périphérique chez les adolescentes varie peu. C’est la masse adipeuse au niveau du tronc
qui baisse significativement par rapport aux individus sains (61–63). Des études ont montré
qu’une hypercortisolémie est observée chez les adultes anorexiques (64–66). Par contre, les
études chez les adolescentes anorexiques n’ont montré aucune différence du taux de cortisol
plasmatique par rapport à celui des sujets sains (67,68).
Après la récupération, les adolescents anorexiques montrent une augmentation de la masse
grasse et de la masse maigre. Plusieurs études ont conclu que la récupération du poids corporel
résulte principalement de l’augmentation de la masse grasse plutôt que de l’augmentation de la
masse maigre (59,64,69). Cependant, malgré une augmentation importante de l’IMC des
adolescentes, cet indice reste plus faible que celui des sujets sains après 12 mois de récupération.
II-E La masse maigre et les tissus musculaires Concernant la masse maigre en anorexie mentale, il existe dans la littérature des résultats
contradictoires. Par exemple l’étude de Hass et al. (2009) sur un groupe de patientes d’IMC
moyen de 16,7 kg/m2 montre que la masse maigre reste intacte (70), ce résultat semble
20
correspondre à des cas de sévérité modérée si on la compare aux valeurs des différentes études
(voir tableau 2 dans II-B). D’autres travaux montrent que la masse maigre baisse fortement chez
les anorexiques (36,38). Sur une population anorexique d’IMC moyen de 16,4 (21,6 pour les
contrôles) Misra et al, (2003) ont évalué la masse maigre du corps entier à 34,6kg chez les
anorexiques contre 38 kg pour les individus sains. Cette différence ne semble pas très
importante, mais elle englobe l’ensemble de la masse maigre et non pas spécifiquement la masse
musculaire. Cette baisse de la masse maigre en anorexie peut être influencée par
l’hypercortisolemie, car il existe une corrélation négative entre le taux de cortisol et la masse
maigre en anorexie mentale. Les patientes qui présentent des taux trop élevés de cortisol ont une
masse musculaire très basse (74).
II-F Les altérations osseuses
II-F-1 Structure Osseuse Les patientes anorexiques présentent fréquemment une densité minérale osseuse inférieure
à celle de la population de référence. Les patientes peuvent présenter des caractéristiques
permettant de les considérer ostéopéniques ou ostéoporotiques. Une définition densitométrique
de l’ostéopénie et de l’ostéoporose a été proposée par l’Organisation Mondiale de la Santé. Cette
définition est basée sur la comparaison de la densité minérale osseuse (DMO) d’une personne
avec celle de la population de référence, c’est-à-dire d’une population de jeunes adultes pour
laquelle ont été calculée une DMO moyenne et une déviation standard (SD). Cette mesure est
faite de préférence au niveau du rachis lombaire et de l’extrémité proximale du fémur. Lorsque
la DMO individuelle mesurée est inférieure de 1 à 2,5 SD à la moyenne de la population, le sujet
est considéré ostéopénique, c’est-à-dire de masse osseuse faible. En dessous de la moyenne
moins 2,5 SD, la personne est ostéoporotique (75). (Figure 4).
21
Figure 4: Représentation des statuts osseux en function du T-score.
Cette réduction de la densité minérale osseuse (DMO) correspond à une diminution de la
quantité de tissu osseux par unité de volume et est associée à des modifications de la
microarchitecture osseuse. Ces deux phénomènes conduisent à une augmentation de la fragilité
du squelette et à un risque fracturaire accru (76). Il est admis que le risque fracturaire est
multiplié par 2 autant de fois qu’il y a de SD en dessous de la valeur de référence.
L’évolution du status osseux chez les patientes anorexiques doit prendre en compte l’âge.
En effet, si on observe la cinétique d’acquisition de la masse osseuse du corps entier au cours de
la vie, il est communément admis qu’environ 35 % de la masse minérale est acquise pendant les
trois premières années de la vie. Entre 4 ans et l’âge de la puberté, 20 % de la masse osseuse est
obtenue. Enfin, pendant l’adolescence 45 % de la masse osseuse est acquise (77). De plus Del
Rio et al. ont montré qu’il existait une différence de cinétique d’acquisition de la masse osseuse
entre le squelette axial et appendiculaire. En effet, l’acquisition de la masse osseuse axiale est
plus tardive (13 % seulement entre 0 et 3 ans chez les filles) (77).
Ces données doivent nous conduire à considérer séparemment les cas des patientes selon
qu’elles ont développé la maladie avant/pendant l’acquisition de l’essentiel de leur masse
osseuse ou après.
Plus de 90% des femmes adultes anorexiques sont ostéopéniques et 20 à 50% sont
ostéoporotiques lorsqu’on considère un ou plusieurs sites osseux. Une fracture osseuse se
produit chez 44% de patientes (64,78,79). Tous les sites osseux ont une DMO diminuée en AM
(colonne vertébrale, les hanches, col du fémur et le corps considéré dans sa totalité) (76) et les
fractures ont tendance à se produire au niveau des sites habituels des fractures osptéoporotiques :
les vertèbres, le radius et la hanche (80,81). Concernant l’architecture osseuse, les résultats sont
pour certains contradictoires Milos et al. ont montré que chez les patientes adultes, il y avait une
22
diminution du volume et du nombre des travées osseuses (82). Chez les adolescentes et les
adultes anorexiques les mesures en microCT montrent une réduction de l’épaisseur et de la
surface corticale et une augmentation de la surface trabéculaire (82–84). On distingue chez les
adolescentes une porosité corticale élevée (84). Chez les adultes le nombre et l’épaisseur des
travées sont réduits avec une augmentation de la distance qui les sépare (82,83).
En général, l’anorexie mentale survient à l’âge de l’adolescence, de ce fait les patientes
anorexiques perdent de la masse osseuse à l’âge auquel elles devraient acquérir presque la moitié
de leur masse osseuse d’adulte. C’est sans doute pour cela que l’AM à cet âge n’entraine pas
seulement une augmentation de risque fracturaire immédiat, mais aussi des séquelles au niveau
osseux qui sont en grande partie irréversibles (85).
II-F-2 Régulations de la masse osseuse
II-F-2.1 Balance des marqueurs de formation et de résorption osseuse Différents marqueurs de remodelage osseux sont utilisés pour évaluer les activités de formation
et de résorption osseuses. Les marqueurs de formation osseuse fréquemment utilisés sont
l’ostéocalcine, les extrémités c- et n- terminales du propeptide du collagène de type I, la
phosphatase alcaline non spécifique et la phosphatase alcaline osseuse. Les marqueurs de
résorption osseux sont la deoxypyridinoline, les peptides c-terminaux et n-terminaux du
collagène de type I (CTX et NTX respectivement), N-terminal peptides of type 1 collagen
(NTX) et le télopeptide c-terminal du collagène de type I (ICTP).
Lorsqu’on observe les données de la littérature relatives aux marqueurs de formation et de
résorption osseuse chez les patientes anorexiques, il apparait que les concentrations plasmatiques
en marqueurs de formation sont en général inférieures chez les sujets anorexiques et que les
résultats sont moins tranchés pour les marqueurs de résorption (Tableau 3, Méquinion et al,
1 All but two studies found increased GH levels 2 Most of the studies found decreased estrogen levels 3 Only three studies found no significant differences when compared to control group, and one found a decrease, while all the other found increased adiponectin levels
4 All but two studies found decreased T3 and/or T4 levels 5 Most of the studies found increased cortisol levels
27
Comme cela a été souligné précédemment pour d’autres paramètres, l’hétérogénéité
individuelle se traduit par une variation importante des perturbations observées dans les
différentes études. Une étude en 2010, a évalué l’hétérogénéité des perturbations hormonales en
fonction de l’IMC(90). La Figure 5 montre que quand l’IMC diminue, le nombre de
perturbations hormonales augmente. À un IMC<13 le nombre des hormones perturbées est
supérieur à six chez 80% des patientes. Chez les patientes ayant un IMC entre 13 et 16.5, le
pourcentage des personnes qui présentent plus de six hormones dont le niveau est altéré n’est
plus que de 40 %. Enfin si l’IMC est relativement élevé (entre 16.5 et 18) le pourcentage des
anorexiques présentant des perturbations importantes est inférieur à 10.
Figure 5: Les distributions des patients anorexiques et les perturbations hormonales en fonction de l’IMC. Plus de 6 hormones perturbées (barre hachurée), entre 3 et 6 hormones perturbées (barre avec petits points) et moins de 3 hormones perturbées (barre grise), d’après Estour et al, 2010.
Cette même étude de 2010 met donc en évidence la relation forte entre l’IMC est les
perturbations hormonales chez les patientes anorexiques. Mais cette même étude montre aussi
l’hétérogénéité des marqueurs et hormones altétérés pour des patientes de même IMC (Fig 6,
chevauchement des lignes continues et des pointillés). Ceci suggère une adaptation individuelle
variable face à la dénutrition. Ces résultats remettent en question la définition de la normalité et
de l’utilisation de l’IMC simple comme critère de gravité et surtout questionnent sur une
composante génétique de cette adaptation individuelle à la dénutrition (90).
28
Figure 6: Hormones et marqueurs osseux altérés (ligne continue) ou non (pointillés) chez les patientes anorexiques
en fonction de leur IMC D’après Estour et al, 2010 (90) .
La leptine La leptine (du grec leptos qui veut dire mince) est la première hormone dont la description
s’impose dans le cadre de nos travaux, à la fois parce que sa concentration dans le sang est
physiologiquement reliée à la masse grasse présente, et donc cette concentration chute
systématiquement chez les patients anorexiques (71,91), et parce qu’elle est un régulateur
majeur de la formation osseuse.
La leptine a été découverte par Zhang en 1994 par clonage positionnel dans un modèle de souris
obèse portant une mutation homozygote du gène correspondant qui entraîne l’absence de son
expression. La protéine non glycosylée de 146 acides animés est produite par les adipocytes, et
est sécrétée dans la circulation sanguine sans qu’elle ne subisse de modifications post-
traductionnelles.
Sa découverte constitue l’une des avancées majeures dans la compréhension de la
régulation de la prise alimentaire. Le niveau circulant de leptine renseigne le cerveau sur le
niveau des réserves adipeuses et permet ainsi à l’organisme d’adapter sa prise alimentaire et son
métabolisme énergétique (92). Depuis sa découverte l’expression de son messager a également
été mise en évidence dans le placenta (93) le muscle squelettique (94), mais les tissus adipeux
restent la source ultra-majoritaire de leptine.
29
De façon surprenante, lorsqu’on considère les plus de 20000 articles qui ont été consacrés à la
leptine, la régulation de son expression dans les tissus adipeux n’a fait l’objet que de peu
d’études.
On peut ajouter le fait que la déméthylation d’îlots CpG spécifiques a été démontrée
pendant la différenciation adipocytaire, au moment de l’apparition de l’expression de la leptine
(95). Ce qui permet de supposer que ce phénomène contribue également à la régulation de
l’expression de la leptine.
Pour agir via le cerveau, la leptine doit traverser la barrière hémato-encéphalique (96).
Cela est possible grâce à sa fixation à des récepteurs transmembranaires de la leptine, présents
dans les microvaisseaux cérébraux. Seule une forme soluble de ces récepteurs inhibe le
transport. La barrière hémato-encéphalique est une régulateur majeur du taux de leptine dans le
cerveau, comme le montre l’augmentation moindre de leptine dans le liquide cérébrospinal des
individus obèses (x1,3) par rapport à l’augmentation de la leptine dans le sérum de ces mêmes
personnes (x3) (97,98).
Une fois dans le cerveau la leptine peut aller agir sur le noyau arqué, les noyaux ventro et
dorsomédian ou encore sur le noyau paraventriculaire (99) ainsi que de manière indirecte sur
l’aire hypothalamique latérale (100), qui tous expriment la forme active du récepteur à la leptine.
Elle agit au niveau de l’hypothalamus en entraînant une réduction de la prise alimentaire
(effet anorexigène), et en augmentant les dépenses énergétiques. Ces deux conséquences
contribuent à la baisse du poids corporel (101). Pour réduire la prise alimentaire, la leptine agit
au niveau du noyau arqué pour stimuler les peptides anorexigeniques l’α-melanocyte-
stimulating-hormone (α-MSH) et cocaine-and amphetamine-related transcript (CART) et inhiber
les peptides orexigéniques Neuropeptide Y (NPY) et Agouti –related protein (AgRP).
De nombreuses études ont montré que la leptine régule la formation osseuse via deux voies
alternatives. La voie directe stimule la formation osseuse. Cette voie a notamment été démontrée
en évaluant les effets osseux d’administration périphérique de leptine (102) et soutenue par de
nombreuses études in vitro. La voie indirecte qui passe par le système nerveux central, contrôle
négativement la masse osseuse. Cela a notamment été démontré par l’injection intra-
cérébroventriculaire de leptine (103). Les mécanismes impliqués dans ces deux voies sont à
présent relativement bien connus, grâce à des études in vivo et in vitro.
La voie indirecte est la première décrite par plusieurs études du groupe de Karsenty (104–
106) Le mécanisme d’action détaillé de la leptine au niveau du système nerveux central est
schématisé figure 7.
30
Figure 7 : Mécanismes impliqués dans les effets indirects de la leptine sur la masse osseuse. D’après Motyl &
Rosen, 2012.
La majorité des études montre que la leptine secrétée par le tissu adipeux agit au niveau du
tronc cérébral en se fixant sur les récepteurs ObR pour inhiber la production de la sérotonine (5-
HT) dans les neurones contenant de la sérotonine. Ces neurones ont des terminaisons dans
l’hypothalamus ventromédian (VMH) qui permettent notamment la sécrétion de la sérotonine
dans le VMH. La leptine inhibe cette sécrétion qui diminue normalement la stimulation
sympathique sérotonine dépendante de l’os Lorsque la leptine bloque la production de
sérotonine, l’inhibition de la voie sympathique est levée (107). Le système nerveux sympathique
(SNS) libère alors au niveau osseux de la norépinéphrine (NE) qui se fixe sur les récepteurs β2-
adrénergiques exprimés notamment par les ostéoblastes. L’activation de ces récepteurs inhibe la
formation osseuse et stimule la résorption via la production ostéoblastique de RANK Ligand
(RANKL) (108).
Concernant les effets directs de la leptine, les études in vitro ont donné des résultats
contradictoires, comme le présentent Motyl et Rosen dans leur revue de 2012 (107). En effet,
une étude utilisant des doses supraphysiologiques de leptine (0,6 à 2,4 µg/ml) ont montré que la
leptine entraînait une augmentation de la différenciation ostéoblastique et bloquaient la
différenciation adipocytaire (109). Une autre étude toujours en conditions supraphysiologiques
(100 ng/ml) a montré un effet positif de la leptine sur la prolifération d’ostéoblastes humains en
culture primaire et un effet également positif sur la minéralisation de cultures d’ostéoblastes
mâtures (110). Cette étude a aussi mis en évidence la diminution de l’expression de marqueurs
proapoptotiques. L’effet prolifératif de la leptine a aussi était montré dans une étude utilisant des
doses physiologiques descendant jusqu’à 1,6 ng/ml, sur des ostéoblastes de calvaria de rat (111).
serotonin
31
Cependant, d’autres études menées en utilisant de faibles concentrations de sérum et donc
en contrôlant plus précisément la concentration totale de leptine, n’ont pas reproduit les effets
directs décrits précédemment (112,113). Par contre, une d’elles a montré un effet proapoptotique
des faibles doses de leptine sur les les cellules stromales de la moelle osseuse humaine (112).
L’ensemble de ces résultats a conduit Motyl et Rosen à proposer que les effets de la leptine
seraient différents en fonction de sa concentration (figure 8).
Figure 8 : Effets dose dépendants de la leptine (modifié d’après Motyl & Rosen, 2012) A : In vitro, dans des
conditions normales, la leptine stimulerait la prolifération des ostéoblastes et la suppression des adipocytes, et elle
diminuerait aussi l’action des ostéoclastes en stimulant l’expression d’OPG. La leptine dans ces conditions aurait
également un effet négatif direct dur les ostéoclastes. B : Dans des conditions de concentration basse, la leptine
n’aurait pas d’effet sur la différenciation ostéoblastique, mais favoriserait l’apoptose des cellules stromales de la
moelle osseuse.
Depuis cette revue, une étude focalisée sur les effets périphériques de la leptine a été
publiée par Turner et al. (2013). Cette étude in vivo a démontré par différentes approches la
réalité des effets directs et positifs de la leptine sur la physiologie osseuse (114).
La principale question, qui n’est pas encore tranchée, est de savoir s’il y a une voie
prépondérante en fonction de contextes particuliers comme l’anorexie, la restriction calorique,
l’obésité, l’hypoleptinémie ou l’hyperleptinémie. Des éléments de réponse peuvent être trouvés
dans quelques études in vivo. En 2002, Takeda et al. ont publié des travaux montrant que la
surexpression de leptine dans le micro-environnement osseux par la génération de souris
transgéniques n’affectait pas la masse osseuse (115). Ces résultats tendraient à montrer que chez
ces souris nourries normlement le mécanisme préponderant d’action de la leptine sur la masse
osseuse serait central. Mais l’étude de Turner et al, a aboutie à des résultats qui sont
complétements opposés et donc à la conclusion que l’action principale de la leptine était au
niveau périphérique. En effet les auteurs ont montré que le traitement sous-cutané de souris
ob/ob par de la leptine entraîne une augmentation de la formation osseuse. Ils ont aussi
notamment montré que chez des souris wild-type, la greffe de moelle osseuse provenant de
32
souris déficientes pour le récepteur de la leptine (db/db) entraîne une formation osseuse du
niveau de celle déterminée chez les souris db/db, malgré un taux normal de leptine circulante
(114). Il semble difficile de concilier les résultats de ces deux études effectuées sur des souris
élevées dans des conditions standard.
Il est intéressant de noter que les sujets obèses, malgré leur hyperleptinémie, ont une masse
osseuse qui n’est pas basse. Ces personnes sont caracterisées par un état de résistance à l’effet de
la leptine, dont la cause est à ce jour mal connue. Ces personnes peuvent par conséquent être
assimilées à des patients atteints d’un déficit fonctionnel en leptine (116), ce qui expliquerait
leur masse osseuse préservée.
La question reste entière pour les personnes anorexiques ou les modèles animaux de
restriction calorique. Dans ces deux cas l’hypoleptinémie avérée est associée à une faible masse
osseuse. Ce qui permet de poser différentes hypothèses :
- Soit la faible masse osseuse est due notamment au faible taux de leptine périphérique qui
réduit la stimulation directe de la formation osseuse, sans que celle-ci soit suffisamment
déréprimée par l’augmentation de sérotonine centrale et la diminution du tonus sympathique qui
en découle.
- Soit la restriction alimentaire entraîne une augmentation de la perméabilité de la barrière
hématoencéphalique et donc paradoxalement une augmentation de la concentration en leptine
dans le cerveau, et une activation du tonus sympathique. Il y a des premiers arguments
bibliographiques pour soutenir l’hypothèse d’une augmentation de la perméabilité de la barrière
dans un modèle animal de jeûne à très court terme (117).
- soit à l’opposé de la résistance à la leptine décrite chez les obèses, la restriction entraîne
une augmentation de la sensibilité cérébrale à la leptine qui se traduit finalement par une
augmentation du tonus sympathique au niveau osseux.
Ces trois hypothèses ne sont bien sûr pas mutuellement exclusives.
Enfin, le récepteur soluble de la leptine S OB-R est la principale protéine piegeant la
leptine. Sous cette forme de complexe la leptine ne peut franchir la barrière hémato-
encéphalique et empêche la fixation sur le récepteur. Le ratio de la leptine sérique sur le taux de
S OB-R donne le free leptin index (FLI). Le FLI semble refléter plus précisément l’action de la
leptine (118). Misra et al., ont montré que le FLI est très bas chez les patientes AM, c’est à dire
qu’on a une hypoleptinémie associée à une concentration élevée de SOB-R. Ce taux élevé de
SOB-R limite peut être l’action des molécules de leptine (119).
33
Après la récupération de la masse corporelle la FLI augmente significativement avec une
augmentation du taux de leptine associée à une diminution de la concentration de SOB - R.
(119). Le taux bas de la leptine chez les patientes AN n’est pas corrigé après une courte
réalimentation, les anorexiques corrigent progressivement le taux de leptine au bout des 5
premiers mois de récupération (120). Haas et al, ont dosé le taux de la leptine chez des patientes
anorexiques, ils ont remarqué que les patients qui ont un taux élevé de leptine entre le 43ème et
le 84ème jours de récupération, récuppérent lentement leur poids corporel (91). Une autre étude
dans le même sens que l’étude précédente, a proposé que l’augmentation du taux de la leptine
après récupération pourrait représenter une contre régulation et prédisposer le patient à une
rechute du poids. (121). La récupération de la masse corporelle chez les femmes anorexiques est
associée à une augmentation du taux de leptine. Et les adolescentes anorexiques qui ont préservé
leurs règles présentent un taux de leptine plus élevé que les adolescentes anorexiques
aménorrhéiques (122).
L’adiponectine L’adiponectine (AdpN) est une adipocytokine sécrétée par le tissu adipeux. Cette cytokine
est connue sous plusieurs noms : adipocyte complement-related protein of 30kDa (Acrp30)
(123) AdipoQ (124) gelatin binding protein of 28kDa (GBP28) (125) et adipose most abundant
gene transcript1 (apM1) (126). L’adiponectine humaine est constituée de 244 acides aminés
avec une homologie structurale élevée avec le collagène VIII, collagène X et C1q (126).
L’Adiponectine est produite par le gène apM1 qui est exprimé principalement par le tissu
adipeux blanc (126). L’adiponectine est une protéine très présente dans le sérum, de l’ordre de
1000 fois plus que la leptine (127). L’adiponectine circule sous trois formes : trimérique (low
molecular weight, LMW), hexamérique (medium molecular weight, MMW) et multimérique de
12 à 18 sous unités (high molecular weight, HMW), cette dernière forme semble la plus active
(128–130). Les récepteurs de l’adiponectine sont les AdpR1 et AdpR2 (131). AdipR1 est surtout
exprimé au niveau des cellules musculaires squelettiques, tandis que l’AdpR2 est exprimé au
niveau du foie (131). Cet adipokine joue un rôle très important dans l’homéostasie énergétique et
la sensibilité à l’insuline.
L’adiponectine affecte le métabolisme osseux (132–134). La régulation de l’adiponectine
par l’ostéocalcine et l’expression de l’adiponectine et de ses récepteurs au niveau de l’os sont
deux raisons de penser que l’os pourrait être un tissu cible de l’adiponectine. Certaines études
ont indiqué que l’adiponectine affecte la masse osseuse, par contre elles ne fournissent pas les
mécanismes moléculaires ou cellulaires de cette action (135,136). Mais récemment il a été
montré par l’équipe de Karsenty que l’adiponectine régule la masse osseuse via la voie centrale
et la voie périphérique (137) . Ils ont effectué leur étude chez des souris C57Bl/6 nourries ad
34
libitum. Il en ressort que chez ces souris l’adiponectine agit directement sur les osteoblastes pour
inhiber leur prolifération et favoriser leur apoptose, et par conséquent entraîner une diminution
de la masse osseuse. Cette action directe est masquée par l’action de l’adiponectine via le
système nerveux central, plus précisément au niveau du locus coeruleus pour atténuer le tonus
sympathique, ce qui va entrainer une augmentation de la masse osseuse et une diminution des
dépenses énergetiques (Figure 9). Cette étude révèle que l’adiponectine a la capacité de réguler
la même fonction de deux manières complètement opposées selon l’endroit où cette hormone
agit. La leptine et l’adiponectine exercent des effets complétement différents au niveau de la
masse osseuse via deux voies sympathiques distinctes.
Figure 9 : Représentation schématique des différentes fonctions exercées par l’adiponectine chez des souris
nourries ad libitum avec une nourriture standard d’après Kajimura et al. 2013.
Chez les patientes anorexiques, le taux d’adiponectine sérique est négativement
corrélée à l’IMC et au pourcentage de masse grasse corporelle chez les patientes anorexiques
(138). Cette hyperadiponectinémie est observée au dessous d’un certain seuil d’IMC <
13.8kg/m2 (30,120,138–142). Dans ce contexte de déficit énergétique, il a été suggéré que le
taux élevé d’ adiponectine pourrait jouer un rôle dans le maintien de l’homéostasie énergétique
(143). Cependant, les résultats concernant l’AdpN ne sont pas encore tranchés en AM. En effet,
si la grande majorité des études montrent qu’on a une hyperadiponectinémie en AM, il existe
autres études qui révelent que le taux d’adiponectine reste intacte chez les patientes anorexiques
35
par rapport aux sujets contrôles (119,144). Une seule étude de Tagami et al, montre une
diminution de l’adiponectine sérique dosée par ELISA (11µg/ml vs 18.3µg/ml chez les
controles) chez 13 femmes anorexiques de type restrictif, agées de 25 ans dont l’IMC moyen est
égal à 13.8 kg/m2 (145). Il est donc difficile de tirer des conculsions car les études sont très
hétérogènes avec des critères d’inclusion différents et un nombre de sujets relativement bas (pas
plus de 40 sujets/étude).
Plusieurs études ont montré qu’il n’y a pas de rapport significatif entre la concentration de
l’adiponectine et la sensibilité à l’insuline (30,139,140). Par contre, beaucoup d’autres études ont
montré qu’il existe une corrélation positive entre l’augmentation du taux d’adiponectine et la
sensibilité à l’insuline chez les AN (30,138,140). Ces résultats contradictoires sont peut être dus
aux différentes techniques utilisées pour évaluer la sensibilité à l’insuline. Cela peut être aussi
dû aux différences d’IMC des patientes, de sévérité de la restriction et de la durée de la
récupération chez les patientes AN.
Modan-Moses et al. ont constaté que le taux d’adiponectine augmente significativement
pendant le premier mois de récupération, puis se normalise (120).
L’aménorrhée et les hormones sexuelles L’aménorrhée, se définie par l’absence d’au moins trois cycles menstruels consécutifs,
était le 4ème critère de diagnostique de l’anorexie selon la quatrième édition du Manuel
diagnostique et statistique des troubles mentaux (DSM-IV). Cette mise en veille des fonctions
reproductrices en période d’insuffisance d’apports alimentaires est justifiée physiologiquement
par le fait que s’il y a une insuffisance alimentaire. Le fait de limiter les fonctions reproductrices
permet d’économiser de l’énergie.
Le déficit en estradiol associé à l’aménorrhée est considéré comme un facteur étiologique
majeur de la perte osseuse en anorexie. Plus la durée de l’aménorrhée est longue plus la perte
osseuse est importante chez les patientes AN (47).
Plusieurs études se sont intéressées au rôle de la leptine dans la reproduction.
L’hypogonadisme associé à l’hypoleptinémie est retrouvé chez les patientes anorexiques (146),
(147). Des études ont montré que la diminution du taux de la leptine reflète un déficit
énergétique et inhibe la fonction de reproduction chez l’animal (148). Conformément à cette
hypothèse, l’injection de la leptine empêche la réduction du taux d’estradiol chez les souris
femelles en restriction calorique pendant 48h (149). Les femmes en aménorrhée souffrant
d’anorexie mentale ont en moyenne des taux de leptine trop faibles (150). L’hypoleptinemie
donc peut engendrer des réponses adaptatives à la restriction alimentaire notamment en
supprimant les fonctions gonadiques (151,152).
36
Le traitement des patientes anorexiques par la leptine est en discussion mais les données
relatives à la leptine amènent des arguments pour et des arguments contre cette stratégie. Welt et
al, ont administré de la leptine à des patientes présentant une aménorrhée hypothalamique, ce qui
entraîne des cycles ovulatoire chez trois femmes parmi huit femmes étudiées (151) à poids
normal (IMC de 18.8 à 24.4kg/m2). Hebebrand et al, pensent que la leptine pourrait corriger
l’hyperactivité, la dépression et les fonctions de reproduction après récupération du poids
corporel. Parmi les effets secondaires de ce traitement : la perte de l’appétit et la perte de poids.
Cependant les auteurs estiment que ces deux préoccupations comme négligeable, car les
patientes sont sous surveillance (153).
L’axe GH-IGF-1 L’axe GH/IGF-1 est quasiment systématiquement altéré chez les patientes anorexiques
(Tableau 5 paragraphe II-E-2.2.1). Ces deux hormones participent à la régulation de la
physiologie osseuse et sont plus particulièrement essentielles pour la formation osseuse. Stoving
et al., ont reporté des perturbations de sécrétion de la GH chez les patientes anorexiques (154).
GH et IGF-1 régulent mutuellement leur sécrétion. En effet, dans le cadre d‘un fonctionnement
physiologique normal, la GH est sécrétée par l’hypophyse antérieure (anterior pituitary) et va
stimuler la production et la sécrétion d’IGF-1 au niveau de différents tissus. L’IGF-1 circulant et
celui produit par l’hypohyse vont à leur tour réguler négativement la sécrétion de GH en
stimulant les neurones inhibiteurs de la production de GH (SRIF neurons) et en bloquant l’action
des neurones stimulant la sécrétion de GH (GHRH neurons) au niveau de l’hypothalamus.
L’IGF-1 a également un effet inhibiteur direct sur l’hypohyse. Cette régulation complexe qui fait
également intervenir d’autres facteurs (comme la Ghréline mentionnée dans la figure 10) aboutie
à une pulsatilité des taux de GH circulante (figure 10) (155).
La GH (somatotrophine, somatotropine ou somatropine) est une protéine de 191 a.a
sécrétée par l’hypophyse. Elle stimule principalement la croissance osseuse et a un effet
anabolisant et lipolytique. Chez l’adulte, le pic principal de libération de GH se produit durant
les deux premières heures de sommeil, lors du sommeil profond. Physiologiquement, deux
hormones sécrétées par l’hypothalamus contrôlent la sécrétion de GH. La somatoréline ou
GHRH, peptide de 44 acides aminés, stimule la sécrétion de GH en se liant à un RCPG couplé à
une protéine G activatrices (Gs). La somatostatine (14 acides aminés) inhibe la sécrétion de GH,
en se liant à des RCPG couplé à la protéine G inhibitrice (Gi). La GH agit comme une hormone
trophique pour stimuler la sécrétion d’IGF principalement dans le foie (156).
IGF-I est une hormone avec une activité métabolique. L’IGF-I a une fonction systémique
et une fonction locale (157). L’IGF-I circulante est essentiellement synthétisée par le foie et sa
37
synthèse est GH dépendant. IGF-I est un régulateur important de l’homéostasie osseuse et
stimule la croissance des os longs d’une manière autocrine et endocrine (158,159) . La sécrétion
de l’IGF-I comme la sécrétion de l’insuline est stimulée par la prise alimentaire et inhibée par le
jeûne (159). GH régule la sécrétion d’IGF-I systémique et de l’IGF-I local au niveau osseux. La
production locale d’IGF-I est aussi contrôlée par la PTH, l’oestradiol, la testosterone mais le rôle
de l’IGF-I local est très peu étudié (160).
38
Figure 10 : Régulation réciproque de GH et d’IGF-1 en contexte physiologique. D’après Okada et Kopchick 2001.
39
Chez les patientes anorexiques, comme le montre le tableau 5 (paragraphe II-E-2.2.1) les
taux d’IGF-1 sont classiquement bas alors que ceux de GH sont élevés. Concernant la GH, les
adolescentes anorexiques présentent une augmentation de la sécrétion basale (20 fois par rapport
aux controles) et de l’amplitude des pics (4 fois par rapport aux individus sains), une diminution
du taux d’IGF-I circulant est aussi présente chez ces personnes. Le mécanisme classiquement
admis pour la perturbation de cet axe GH/IGF-1est que le taux d’IGF-1 est diminué par la sous-
alimentation, ce qui diminue le contrôle négatif central de la production de GH par l’IGF-1. Le
taux de GH circulant augmente donc, sans que cela entraîne une augmentation du taux d’IGF-1
limité par la dénutrition. (161,162). Afin de déterminer s’il y avait bien une forme de résistance à
la GH, Fazeli et al. ont réalisé un traitment supraphysiologique de GH (rhGH) chez des patientes
anorexiques, pendant 12 semaines. Ce traitment n’a pas entraîné de différences significatives au
niveau de l’IGF-I, du glucose, de l’insuline, ni des acides gras libres. On peut noter cependant
que chez les patientes traitées avec GH, la masse grasse totale diminue fortement. Il semble donc
bien que les personnes anorexiques développent une résistance hépatique à la GH en termes de
sécrétion d’IGF-1 (163).
Les mécanismes associés à la résistance à la GH en restriction alimentaire ont été étudiés
chez la souris et sont synthétisés dans la figure 11. Il a ainsi été montré qu’une restriction
alimentaire sévère entraînait une diminution de l’expression de récepteurs de la GH au niveau
des hépatocytes. Mais cette diminution ne suffisait pas à justifier la forte résistance à la GH
(164). D’autres études ont montré une implication du Fibroblast Growth Factor (FGF) 21 dans
la résistance à la GH liée à la restriction. Le FGF21 fait partie de la sous-famille des FGF qui ne
posède pas de domaine de liaison à l’héparine, ce qui lui permet de circuler et d’avoir une action
endocrine à partir du foie et des adipocytes qui le produisent. Durant le jeûne l’augmentation du
taux circulant de FGF21 induit la néoglucogenèse, l’oxydation des acides gras et la production
de corps cétoniques (165). Enfin, la restriction alimentaire entraîne une augmentation de la
concentration en acides gras libres circulants, ce qui induit une activation de PPARα qui lui
même stimule la production de FGF21 (166).
40
Figure 11 : Présentation synthétique des mécanismes de la résistance à la GH induite par la restriction alimentaire.
Ce mécanisme décrit chez des souris en restriction est-il représentatif de celui qui aboutie à
la résistance à la GH chez les patientes ? On peut difficilement avoir accès aux mêmes types
d’échantillons chez les patientes anorexiques pour répondre définitivement à cette question.
Mais deux études à ce jour ont permis d’effectuer des dosages de FGF21 circulant chez ces
patientes. En 2008, Dostalova et al. ont mesuré chez des femmes anorexiques des taux de FGF21
inférieurs de moitié à ceux d’individus contrôles (161). Alors qu’en 2010, Fazeli et al. ont
effectué ces mesures chez des patientes adolescentes et n’ont pas trouvé de différences avec les
adolescentes contrôles (167). Ils ont cependant trouvé une différence après correction par le
pourcentage de masse grasse corporelle. Le FGF21 étant produit principalement par le foie et les
tissus adipeux, ils ont supposé que la production hépatique de FGF21 était augmentée. Malgré
cela, ces deux études laissent supposer qu’il pourrait y avoir des différences entre les patientes
adultes et les patientes adolescentes. Et il est évident que cela appelle d’autres études pour
clarifier la situation.
Il a été montré chez la souris que l’IGF-1 circulant et l’IGF-1 produit dans l’os jouaient
chacun un rôle important dans la formation osseuse (168,169). Si on suppose qu’il peut en être
de même chez l’humain, il est légitime de se demander si la résistance à la GH et éventuellement
les mécanismes associés comme la faible production IGF-1 sont également présents en intra-
41
osseux chez les patientes anorexiques. Kubicky et al. (2012) ont montré grâce à des souris
FGF21 -/-, que la perte de poids corporel et la diminution de la croissance tibiale induites par la
restriction alimentaire étaient dépendantes de l’expression de FGF21. Ils ont également montré
chez des souris wild-type que la restriction alimentaire entraînait une augmentation de
l’expression (transcrits et protéines) de FGF21 et une diminution d’IGF-1 au niveau de la plaque
de croissance tibiale (164). Ces deux résultats plaident en faveur d’une forte similitude entre les
perturbations de l’axe GH/IGF-1 dans le foie et dans l’os, au moins chez la souris.
Le cortisol L’axe hypothalamo-hypophyso-surrénalien est connu pour être stimulé chez les patientes
anorexiques (122,170–172). Cette stimulation entraîne une augmentation de la sécrétion de
cortisol par les glandes corticosurrénales. Le cortisol (ou hydro-cortisone) est une hormone
corticostéroïdienne sécrétée par le cortex de la glande surrénale à partir du cholestérol, sous la
dépendance de l'adrenocroticotropin hormone (ACTH) hypophysaire. Ses fonctions principales
sont l’augmentation de la glycémie par le biais de la néoglucogenèse, l'inhibition de certaines
réponses du système immunitaire et la régulation du métabolisme des graisses, protéines et
glucides. À ce titre il est inversement corrélé à l’IMC et à la masse grasse des patientes
anorexiques (171).
Les femmes anorexiques ont un taux de cortisol plus élevé que les femmes saines
(172,173). Le cortisol étant associé à l’anxiété et à des symptômes dépressifs, ce taux élevé est
considéré impliqué dans les troubles de l’humeur observés. Il participerait aussi à la perte
osseuse chez ces patientes, mais Grinspoon et al, ont rapporté que l’hypercorticisme était
présent chez seulement 22 % des patientes souffrant d’AM avec une perte osseuse sévère (67).
L’hypercorticisme ne serait donc qu’un facteur parmi d’autres favorisant la faible DMO (174).
Cependant, sur la base d’autres études le taux élevé de cortisol en AM est considéré comme un
indice de faible DMO (172). Enfin, de nombreux patients présentent des altérations de la
dynamique de sécrétion du cortisol, sans pour autant présenter les signes d’un syndrome de
Cushing. Ces patients présentant donc un syndrome de Cushing subclinique ont néanmoins une
DMO réduite (175). Ce qui montre l’importance de l’hypercorticisme même modéré, pour la
physiologie osseuse.
42
L’insuline L’insuline est une hormone peptidique synthétisée sous forme de pro-hormone et activée
avant sa sécrétion par les cellules β-pancréatiques. Le glucose est un stimulus important de la
sécrétion d’insuline. L’insuline se combine à un récepteur membranaire sur les cellules cibles.
Les cibles essentielles de l’insuline sont le foie, le tissu adipeux et les muscles squelettiques.
L’insuline agit sur la glycémie selon quatre modes : elle augmente l’entrée du glucose dans la
plupart des cellules insulinosensibles, elle stimule l’utilisation et le stockage du glucose dans la
cellule, elle active la glycolyse la glycogenèse et la lipogenèse. L’insuline stimule l’utilisation
des acides aminés (156).
Mais l’insuline possède d’autres fonctions qui ne sont pas encore toutes élucidées.
D’autres cellules que celles des tissus précités expriment des récepteurs de l’insuline. C’est le
cas de l’ostéoblaste (176). Karsenty et al, ont identifié la voie de signalisation de l’insuline au
niveau de l’ostéoblaste. L’insuline se fixe sur son récepteur InsR ce qui, inhibe la FoxO1 et
entraîne une diminution de l’OPG. Ceci entraîne la résorption osseuse et l’expression de la
Tcigr1 au niveau des ostéoclastes. La Tcigr1 est responsable de l’acidification de la matrice
extracellulaire osseuse. Une fois le milieu acidifié, l’osteocalcine est décarboxylée (OCN
active). Cette OCN active stimule la prolifération des cellules bêta-pancréatiques, ainsi que leur
sécrétion d’insuline. Elle augmente la sensibilité à l’insuline des tissus cibles (177).
L’insuline aurait également un effet anabolisant sur le squelette (178). Cet effet
anabolisant est suggéré par différentes études. Les enfants brûlés recoivent un traitement
hyperinsulinémique qui entraîne une augmentation de la masse maigre mais aussi de la masse
osseuse enregistrée à la fin des 6 semaines à 3 mois d’hospitalisation (179). Les mécanismes
cellulaires impliqués font l’objet de nombreuses études reprises dans une revue récente (180) et
synthétisées dans une figure de cette revue (figure 12). Cette figure met en évidence les effets
positifs directs de l’insuline sur la prolifération et la différenciation ostéoblastiques.
43
Figure 12 : Signalisation et effets directs de l’insuline sur les ostéoblastes.D’après Pramojanee et al, 2014.
L’insuline chez les patientes anorexiques, présente des taux inférieurs à la normale selon
la plupart des études (30,139,142). Mais il est évident que chez ces patientes la sensibilité à
l’insuline est également perturbée. Les études concluent généralement à une augmentation
importante de la sensibilité à l’insuline, mais à l’aide d’indices indirects. C’est le cas notamment
de Misra et al. qui ont évalué la sensibilité à l’insuline chez des adolescentes anorexiques à
travers deux méthodes de calcul. D’une part, le ratio insuline/glucose était de 1,54 µU/mmol
chez les adolescentes anorexiques contre 3,06 µU/mmol chez les adolescentes saines et d’autre
part le HOMA-IR (homeostase model assessment of insulin resistance) est égal à la
concentration en glucose (mM) multipliée par la concentration en insuline (mU/ml) divisées par
22,5. Ce dernier calcul a donné des valeurs de HOMA-IR de 1,36 pour les patientes anorexiques
contre 3,08 pour les sujets sains (119). Ces valeurs confirment l’hypothèse d’une grande
sensibilité à l’insuline chez les patientes.
La ghréline La ghréline est une hormone naturelle isolée de l'estomac de certains mammifères, dont
l'homme, par l'équipe japonaise de Kojima en 1999. La ghreline est l’unique ligand connu pour
se fixer sur le récepteur Growth hormone secretagogue receptor 1a (GHS-R1a) qui est couplé à
la protéine G, les deux variantes de ce récepteur sont la GHS-R1a (fonctionnel) et GHS-R1b
(non fonctionnel). Le GHS-R1a est exprimé au niveau central (noyau arqué, l’hypothalamus), et
au niveau périphérique (l’estomac, l’intestin, le cœur, la thyroïde, les gonades, les muscles
44
squelettiques, les tissus adipeux et le foie) pour lequel on ne connait pas encore le rôle exact. La
ghréline qui est une hormone orexigène agit via l’hypothalamus, pour activer les neurones
orexigéniques AgRP/NPY et inhiber les neurones anorexigéniques (POMC /CART) (181).
Cependant, elle agit aussi directement au niveau hypophysaire sur la sécrétion de GH (182).
La ghréline a pour fonction principale la stimulation de l'appétit. Sécrétée par des cellules
spécifiques de l'estomac peu avant le repas, elle déclenche la sensation de faim, provoquant la
prise de nourriture. La ghréline semble réguler le métabolisme lipidique et glucidique. Elle peut
maintenir le niveau du glucose dans le sang. L’injection de la GH stimule la prise alimentaire
chez l’animal et chez l’homme (183).
En plus, la restriction calorique prolongée entraine une élévation de la concentration de la
ghreline sérique. Et elle participe à la stabilisation de la glycémie au cours de la restriction
calorique (184,185).
Il existe une corrélation négative entre l’indice de masse corporelle et les concentrations
plasmatiques de ghréline à jeûne : l’amaigrissement chez les sujets obèses s’accompagne d’une
augmentation des niveaux de ghréline (186) tandis que l’inverse est observé avec la prise de
poids en cas d’anorexie mentale (187). À noter que l’AM se caractérise par un taux élevé de
ghréline.
L’effet de la ghréline sur la masse osseuse n’est pas encore bien identifié. Une étude in vitro a
montré que la ghréline stimule la prolifération ostéoblastique chez les cellules de Calvaria de
rat(188) .
II-F-2.2.2 L’adiposité médullaire
Quelle est la fonction de l’adipocyte médullaire ? Les fonctions de l’adiposité médullaire (AdpM) font encore l’objet de nombreuses
hypothèses et discussions. Plusieurs éléments suggèrent un rôle de l’AdpM sur le
microenvironnement local. On pense que l’AdpM intervient dans la thermogenèse (111).
D’autres estiment que l’AdpM peut fournir de l’énergie pour la formation osseuse. Enfin sur le
plan sécrétoire, il a récemment été suggéré que l’adiponectine circulante proviendrait notamment
de l’AdpM ce qui expliquerait le « paradoxe de l’adiponectine « c'est-à-dire la corrélation
inverse observée entre la quantité de masse grasse et le taux circulant d’adiponectine (189). Ainsi
les adipocytes médullaires pourraient modifier le phénotype des ostéoblastes (190), et favorisent
la différenciation ostéoclastique (191).
L’adiposité médullaire au cours de la vie
45
La présence de dépôts adipeux et d’adipocytes dans la moelle osseuse est connue de longue date.
Son volume est estimé à 7% de la masse grasse totale (192), ce qui correspond à entre 1,5 et 3kg
chez l’adulte d’âge et d’IMC moyens. Cette masse représente un stock équivalent à environ
23 000 calories (189). La quantité et la distribution de ces dépôts adipeux ne sont pas constantes
au cours de la vie. En effet, une conversion de la moelle rouge (hématopoiétique) en moelle
jaune (adipeuse) se produit avec l’âge. Une étude a montré que de 20 à 65 ans le volume de tissu
adipeux médullaire augmentait en passant de 15 % à 65 % (193). Cette évolution est différente
dans les os longs et les os plats. La conversion moelle rouge / moelle jaune dans les os longs a
été illustrée par Blebea et al. (Figure 15) (194). Dans ces os, toute la moelle est hématopoiétique
à la naissance. La conversion commence classiquement au niveau des phalanges distales et
évolue de façon centripète. Cette évolution s’arrête vers l’âge de 25 ans. La moelle
hématopoiétique est alors principalement localisée dans le squelette axial, le sternum, les côtes,
les fémurs et humérus proximaux.
Figure 13 : Illustration de la conversion fémorale moelle rouge / moelle jaune avec l’âge (années). Bone marrow:
normal and pathologic aspects in MRI. D’après Turki MW (195)
Pour illustrer plus précisément, ce qui se produit dans les os longs, on peut observer
l’adiposité médullaire fémorale. Cette évolution a été schématisée notamment par Turki et al.
(2012) (figure 13). Il apparaît que la quasi-totalité de la cavité médullaire est occupée par les
dépôts adipeux dès le début de l’âge adulte. Mais cette représentation masque en réalité des
évolutions très variables avec l’âge, en fonction des personnes comme le montrent les clichés de
la figure 14 effectués sur des hémi-humérus humains (photographies par de G. Wavreille,
PMOI).
46
Figure 14 : Clichés de deux hémi-humérus d’hommes d’environ 80 ans. L’humérus de gauche présente une
proportion élevée de moelle rouge par rapport a ce qui est décrit classiquement et illustré par le cliché de droite.
(Clichés de G. Wavreille, PMOI- Laboratoire d’Anatomie de Lille).
L’adiposité médullaire et la masse osseuse Comme cela a été présenté dans la partie précédente, dans les os longs la conversion en
moelle jaune se produit relativement tôt au cours de la vie, à un âge auquel la masse minérale
n’est pas encore affectée. L’augmentation de l’adiposité médullaire débute chez l’homme avant
la troisième décennie et donc avant la diminution de la masse osseuse (196). Cela laisse supposer
que les adipocytes présents pendant cette période n’ont pas d’impact négatif sur la physiologie
osseuse. À la naissance, les cavités osseuses sont principalement remplies de moelle rouge
hématopoitiques. Puis se produit au cours de l’enfance une « conversion » de la moelle rouge qui
est progressivement remplacée par de la moelle jaune, adipeuse (figure 15). Cette conversion
médullaire débute dans les phalanges peu après la naissance et suit une évolution centripète
jusqu’au squelette axial (197). D’importantes variations individuelles sont retrouvées mais il
existe globalement une corrélation positive entre l’adiposité médullaire et l’âge (192,193).
47
Figure 15 : Représentation schématique de la distribution de moelle rouge et de moelle jaune dans les os longs
humains, de la naissance à l’âge adulte. D’après Blebea et al (194)
L’implication potentielle des adipocytes dans la perte osseuse pourrait donc s’expliquer soit
par l’apparition de nouveaux adipocytes ayant des caractéristiques différentes leur permettant
d’influencer négativement la balance formation / résorption, soit par l’acquisition de ces
nouvelles propriétés par les adipocytes pré-existants.
La perte osseuse, liée au vieillissement, s’accompagne d’une augmentation de l’adiposité
médullaire (198) (figure 16). Le vieillissement augmente l’expression de PPARγ dans la moelle
(199) ce qui favorise la différenciation adipocytaire. Chez l’animal, l’ovariectomie augmente
l’adiposité médullaire (200–202), ce qui est réversible par l’administration d’estradiol (203). Ces
résultats expérimentaux ne sont pas cependant toujours dans le même sens, certaines études ont
montré que l’interaction entre adipogenèse et ostéogenèse ne se résume pas à une simple
régulation inverse (204,205).
Chez l’humain, une augmentation de l’adiposité médullaire, en taille et en nombre, est
constatée lors de l’ostéoporose (206). Également on a remarqué que l’augmentation de l’Ad Med
qui accompagne la perte osseuse après la ménopause est corrigée par l’administration
d’oestrogènes (207).
48
Les corrélations avec la densité minérale osseuse montrent que l’adiposité médullaire est
plus importante chez les sujets ostéoporotiques que chez les ostéopéniques, et qu’elle est plus
élevée chez les sujets avec fractures vertébrales (193). L’adiposité médullaire est fortement
corrélée de manière inverse à la DMO (192).
Figure 16 : adiposité médullaire humaine de la crête iliaque. (A) 18 ans et (B) 80ans. D’après Pouneh K et al,
2013 (198).
Afin de déterminer le rôle des adipocytes médullaires et d’établir leurs éventuelles
implications dans la régulation de la masse osseuse, il est nécessaire d’étudier leurs activités.
Tout d’abord, il a été établi que les cellules souches mésenchymateuses présentes dans la moelle
osseuse sont à l’origine de nombreux types cellulaires (figure 17), dont les ostéoblastes et les
adipocytes médullaires. Ceci suggère notamment une compétition entre ostéogenèse et
adipogenèse (208). Par ailleurs, il est unanimement reconnu que les adipocytes des dépôts
adipeux extraosseux ont des fonctions métaboliques et endocriniennes majeures et que ces
fonctions varient en fonction de la localisation des dépôts. Néanmoins, les adipocytes
médullaires ont été longtemps considérés comme de simples cellules de comblement entre les
cellules hématopiétiques et le tissu minéralisé. Même si de nombreuses études à ce sujet sont en
cours, il semble que les adipocytes médullaires présentent non seulement des spécificités par
rapport aux adipocytes des autres sites, mais aussi des fonctions sécrétoires qui pourraient
influencer les populations cellulaires voisines.
49
Figure 17 : Schéma représentant la pluripotentialité des cellules souches mésenchymateuses. D’après Harada et A
Rodan 2003 (209).
Parmi les spécificités des adipocytes médullaires, il peut être noté que sauf lorsqu’ils
occupent tout un volume médullaire, les adipocytes sont dispersés au sein du tissu
hématopoïétique. Morphologiquement, le diamètre de ces adipocytes est de l’ordre de 50 µm
chez l’humain. Ils sont donc plus petits que les adipocytes viscéraux (210). Fonctionnellement,
des travaux récents ont comparé l’expression génique d’adipocytes médullaires à celle
d’adipocytes périgonadiques chez la souris, et à celle d’adipocytes sous-cutanés chez l’humain.
Chez la souris les adipocytes médullaires ont un moindre niveau d’expression des gènes
spécifiques de l’adipocyte, et un niveau d’expression plus élevé des gènes inflammatoires, pro-
apoptotique et des cytokines comme la TNF-α , IL-6 et TGF β1 (211). Chez l’humain, les
adipocytes médullaires expriment de manière plus marquée des gènes importants pour la
pluuripotence et pour la reprogrammation cellulaire. Ils expriment des gènes de cellules souches
embryonnaires tels que Oct4, KLf4, c-myc, Gata4, Tbx1 et Sox17 suggérant une plasticité
cellulaire marquée (212).
50
L’adiposité médullaire dans l’anorexie mentale L’anorexie mentale représente un cas particulier en raison du caractère surprenant de
l’augmentation de l’AdpM chez des femmes très maigres (213,214) et indique que le stockage
de graisse ne semble donc pas obéir aux mêmes lois dans la moelle osseuse et dans les autres
dépôts graisseux. La spectroscopie (« 1 H MR spectroscopy ») permet de quantifier les
composants de la moelle osseuse (215) en séparant la graisse et l’eau et donc une mesure de la
fraction chimique de la graisse, la quantité de la graisse médullaire est estimée à 73 % chez les
patientes anorexiques (216) (figure 18). Dans cette pathologie, l’augmentation de l’AdpM
mesurée par IRM/contrôle, est inversement corréle au DMO et s’accompagne d’une diminution
de la formation osseuse (214). Cette augmentation de l’AdpM est fortement suspectée de
participer à la perte osseuse au cours de l’anorexie mentale (B Cortet, et al. Relationships
between bone and fat in anorexia nervosa. In: Seventh Meeting on Bone Quality 2012: Bone-Fat
Figure 18 : Quantification de l’adiposité médullaire en 1 H MR spectroscopie. (A) chez les patientes anorexiques
(B) chez les femmes saines D’après Singhal 2014 (88).
Le facteur préadipocytaire 1 (PREF1) appartient à la famille des facteurs « epidermal growth
factor-like family » inhibe la différenciation des CSM en ostéoblastes. Le taux élevé de PREF-1
en anorexie est inversement corrélé à la DMO et positivement corrélé à l’Ad Med (Fazeli 2010a,
2012).
Il existe d’autres hypothèses qui expliqueraient cette augmentation d’AdpM en AM :
- les adipocytes de la moelle secrétent des cytokines ostéoclastogénique comme l’IL-6
responsable de l’inhibition de l’activité ostéoblastique
- cette augmentation de l’AdpM est peut être associée à une augmentation de la taille de
ces adipocytes qui vont compresser les capillaires sanguins intraosseux, perturbant ainsi
l’irrigation correcte du tissu osseux.
51
Il n’existe pas de relation entre la durée de la maladie et l’AdpM chez les anorexiques (214,217)
ce qui suggère que le taux élevé de l’adiposité médullaire en anorexie mentale est fortement lié à
la perte de la masse corporelle et non pas à la chronicité de la maladie (213)
II-G- Autres altérations
II-G-1 Conséquences digestives Les troubles fonctionnels digestifs sont pratiquement constants au cours de l’anorexie mentale,
et constituent une gêne majeure pour les patients et un frein à la réalimentation (218). Une
perméabilité intestinale élevée due à l’inflammation de la paroi intestinale, va entrainer la
translocation des bactéries et des protéines alimentaires dans la circulation générale. Une flore
intestinale de mauvaise qualité induit une altération de la muqueuse colique (219). L’existence
d’un ralentissement de la vidange gastrique au cours de l’anorexie mentale est bien établie mais
les mécanismes impliqués dans ce trouble ne sont pas encore élucidés (220–222). En effet, le
degré de retard de la vidange gastrique est corrélé avec le degré de dénutrition et la vidange
gastrique tend vers une normalisation lors de la correction de la dénutrition (220,221,223).
Hypokaliémie, hypomagnesemie et hypocalcémie sont très fréquents chez les patientes (224).
Les patientes anorexiques sont les plus exposées au risque de perforation gastrique car une
structure déformée de la paroi gastrique peut entrainer une nécrose et par la suite une perforation
du tractus gastro- intestinal (225–227). L’augmentation des concentrations d’amylase sérique
résultent des vomissements fréquents en AM. Les vomissements chroniques ont des effets
profonds sur divers organes. Près de 20% des patientes souffrent d’ulcères (224,228).
La constipation est très fréquente chez les patientes anorexiques (environ 40%) parfois
extrêmement sévère (229). La constipation est souvent vécue de façon très négative par les
patientes (218). La physiopathologie des troubles au cours de l’anorexie mentale reste mal
connue (230). Le stress dû à cette maladie peut favoriser la survenue d’anomalies de la barrière
intestinale et de l’immunité ce qui conduit à une diminution de la fonction de la barrière
intestinale et une réponse inflammatoire locale (218).
L’inflammation locale pourrait à son tour avoir des conséquences sur la production
gastrointestinale impliquée dans la régulation du comportement alimentaire. Ainsi
l’inflammation intestinale est accompagnée d’une augmentation de la libération de CCK, qui
peut à la fois ralentir la vidange gastrique et inhiber la prise alimentaire au niveau
hypothalamique (231). L’intestin pourrait ainsi être une des sources de cytokines
proinflammatoire circulants en excès au cours de l’anorexie mentale (232).
52
II-G-2 Système nerveux central Parmi les facteurs biologiques perturbés en anorexie mentale on trouve les neuropeptides
qui régulent la prise alimentaire. Les résultats des études effectués à ce sujet sont très
hétérogènes pour la plupart des facteurs analysés. Cette hétérogénéité peut être expliquée par des
caractéristiques cliniques différentes d’une étude à une autre (sévérité et durée de la maladie).
Concernant les hormones anorexigènes le taux de la leptine, diminue, tandis que il y’a d’autres
hormones qui sont surexprimées comme PYY 3-36. Les résultats sont contradictoires concernant
les facteurs anorexigéniques cholecystokinie (CKK) et le glucagon-like peptide 1 (GLP-1) ce qui
rend difficile l’interprétation de ces variations dans la pathologie humaine et nécessite des études
plus approfondies. L’expression des facteurs orexigéniques comme les facteurs anorexigènes
n’est pas encore bien définie (89).
II-G-3 Système immunitaire Un certain nombre d’études cliniques et animales montrent que les cytokines pro-
inflammatoires peuvent jouer un rôle dans les troubles alimentaires comme l’anorexie mentale.
Corcos et al, ont effectué des dosages sériques des cytokines pro-inflammatoires, chez des
patientes anorexiques, (IL-1, IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ et TGF-β) les résultats des
montrent que seulement le taux d’IL-2 est bas en anorexie tandis que l’expression des autres
cytokines reste intacte. Cette baisse sérique de l’IL-2 qui est principalement exprimée au niveau
de la muqueuse intestinale est peut être dûe au déficit nutritionel ou/et à l’altération ou l’atrophie
de la mqueuse intestinale observée en anorexie (233).
Une autre étude, celle de Brambilla et al, a montré que les taux sériques de la TNFα, l’IL1 β et
l’IL-6 et leurs récepteurs solubles sIL1β-R, sIL-6-R, sTNF-α RI et RII restent intacts chez les
anorexiques (234).
En conclusion, le rôle du système immunitaire en AM n’est pas encore bien clair.
III-Comment étudier les conséquences physiologiques de l’AM ?
III-A Études cliniques Les études cliniques sur cette maladie sont souvent disparates, il est difficile de suivre les
patientes à long terme, les effectifs sont souvent très faibles, les populations sont très
hétérogènes avec des critères d’inclusion et d’exclusion qui varient d’une étude à une autre
notamment en terme d’IMC. Ces études cliniques sont aussi hétérogènes (anorexie restrictive
boulimique), et que certaines patientes sont atteintes avant la puberté et d'autres bien après. La
53
durée de la maladie est également un facteur très variable selon les études et les patientes ne sont
pas toutes en aménorrhée. Enfin, l’impossibilité d’accéder aux tissus pour étudier en profondeur
cette maladie s’ajoute aux éléments précedement cités pour conclure que l’utilisation de modèles
animaux adaptés est indispensable.
En effet, les études cliniques réalisées sur l’anorexie mentale ont des critères d’inclusion et
d’exclusion des sujets très variables notamment au niveau de l’index de masse corporelle. Dans
la plupart des études les patientes considérées anorexiques ont des IMC compris entre 15 et 16
kg/m2 (31,71,91,167,235,236). Et autres études avec des IMC qui varient entre 13 et 14 kg/m2
(53,90). Tous les composants corporels sont sévèrement touchés en AM. La masse adipeuse
comme la totalité de la masse musculaire est significativement réduite en anorexie mentale.
Chez les sujets sains le pourcentage de la masse grasse varie entre 26% et 32 % par contre chez
les patientes anorexiques il varie entre 9% et 16% (71,91).
Ainsi la privation de nourriture en anorexie mentale est très souvent associée à une hyperactivité
et ceci concerne un certain pourcentage des anorexiques. Elle concerne 20 % des cas d’anorexie
mentale chez les femmes et 8 % chez les hommes (237). Dans d’autres études l’hyperactivité
concerne 30-80% des anorexiques (238–240) Kohl et al, ont comparé des patientes souffrant
d'anorexie mentale en hyperactivité et patientes sans hyperactivité. Les patientes hyperactives
sont plus insatisfaites de leur image corporelle. De nombreux facteurs peuvent favoriser
l'émergence et le maintien de l’hyperactivité, en particulier les exigences sociales et culturelles,
de l'environnement sportif, les influences familiales. L’exercice excessif peut être considéré
comme un symptôme de l'anorexie mentale. Il peut également favoriser le développement de
troubles de l’alimentation (52).
III B- Les modèles animaux Le développement d’un modèle animal idéal reproduisant tous les aspects de l’AM n’est pas
envisageable. D’une part, parce que l’origine de cette maladie est psychiatrique et d’autre part,
parce que les interactions complexes entre les facteurs génétiques, environnementaux, les
facteurs sociaux et culturels ne sont pas reproductibles chez les animaux. Des modèles ont donc
été développés pour mimer et étudier plus spécifiquement certains aspects de cette pahologie.
54
III-B-1 Modèles d’induction environementale
III-B-1.1 Modèles de dépression Le stress et la dépression sont des paramètres retrouvés chez de nombreuses patientes
anorexiques. Ce sont aussi des facteurs pouvant entraîner de multiples altérations
comportementales, nerveuses et physiologiques. Ils sont donc à la base de différents modèles
animaux visant à reproduire certaines de ces altérations. Le modèle considéré comme le plus
réaliste entrainant un comportement de type dépressif est celui développé par Willner et al.
(241). C’est un modèle de stress chronique modéré (chronic mild stress – CMS) qui est basé sur
l’application quotidienne de séries aléatoires de différents stimuli (Tableau 6). Il est considéré
comme un modèle de dépression car il entraîne une anhédonie d’après la perte de préférence
pour la nourriture sucrée, l’anhédonie étant elle-même considérée comme un symptôme majeur
de la dépression. Ce modèle entraîne une diminution de la prise alimentaire, une prise de poids
corporel ralentie (95% de celle des contrôles après 8 semaines de protocole chez le rat). De
façon intéressante, une étude chez la souris a montré qu’il entraînait aussi une diminution de la
densité du volume osseux trabéculaire mesuré en fémur distal et en vertèbre lombaire, de plus de
20 et 10 % respectivement (242). Ce protocole présente cependant deux inconvénients. En effet,
il ne reproduit pas la sévérité des altérations de prise alimentaire, ni de perte de poids. Et d’un
point de vue pratique, l’application de ce type de protocole nécessite beaucoup de manipulations
et de temps de travail quotidiens sur une période longue. Néanmoins, ce modèle suggère que le
stress chronique serait un facteur important pour mimer chez l’animal certains aspects de l’AM.
Tableau 6 : Exemple de séries de stress appliquées chez le rat dans le modèle de stress chronique modéré d’après
Wiborg (243).
III-B-1.2 Modèles de restriction alimentaire quantitative Le protocole murin de restriction alimentaire est un modèle avantageux dans l’étude des
mécanismes central et périphérique et des effets de la perturbation de la prise alimentaire chez
les humains, et tester les potentiels approches thérapeutiques. Toutefois, le développement et
l'utilisation de modèles animaux de troubles psychiatriques sont intrinsèquement difficiles en
raison de la nature complexe de ces maladies comme par exemple la privation volontaire de
55
nourriture en AM. Donc le modèle murin de restriction calorique permet de donner un aperçu
sur la façon dont la maladie peut évoluer vers l’épuisement, et de reproduire les caractéristiques
squelettiques de la maladie humaine comme la diminution de la densité de la masse minérale,
baisse du volume trabéculaire, diminution de la formation osseuse et augmentation de la
résorption osseuse. Les effets de la restriction calorique sur la composition corporelle et les
hormones sont similaires à ceux observés en maladie humaine.
Une des principales critiques faites aux différents modèles de restriction alimentaire est que
la restriction est imposée par l’expérimentateur. Ce n’est en fait un problème majeur que si on
souhaite étudier les raisons et les mécanismes de l’autoprivation telle qu’elle se produit chez les
patientes. Par contre ce type de modèle mime de nombreuses perturbations neuroendocriniennes
décrites dans la pathologie humaine (244–246).
En effet, les protocoles courants de restriction alimentaire comportent une suppression de
30 à 40% de la prise alimentaire des groupes d’animaux contrôles (ad libitum). Ce qui signifie
une alimentation avec 60 à 70% de ce que mangent les contrôles. Or, les contrôles nourris ad
libitum se suralimentent classiquement d’environ 30% par rapport à leurs besoins. Cette
suralimentation entraîne une prise de poids notable avec le temps (247). Ce qui revient à dire que
les souris en restriction consomment finalement à peine moins que leurs besoins et que les
études de ce type comparent ces animaux à des contrôles en surpoids. Ces protocoles de
restriction sont en général appliqués sans supplémentation minérale ni vitaminique ce qui
conduit à cumuler les effets du déficit d’apport énergétique avec ceux des carences induites
proportionnellement à la restriction (248).
Les protocoles sévères sont beaucoup moins courants. Les restrictions sont alors de 50 à
70% de ce que mangent les contrôles (249,250). Ces protocoles sont nécessairement plus courts
pour maintenir les animaux en vie et incluent également des carences importantes. Cette courte
durée entraîne des limites à ces études car certaines perturbations, y compris en dehors de celles
touchant le tissu osseux, nécessitent plus de quelques semaines pour se développer (251).
III-B-1.3 Modèle d’activité / restriction Selon les études, 30 à 80 % des patientes anorexiques utilisent l’activité physique pour
accentuer la perte de poids, mais cette activité physique peut aussi leur apporter une sensation de
bien être par des mécanismes bien connus impliquant notamment la libération d’endorphines.
Elles peuvent de ce fait développer, comme cela est décrit chez des sportifs, une relation de type
addictif avec l’activité physique. Un modèle dit « d’autoprivation » a été développé chez le rat
par Routtenberg and Kuznesof en 1967 (252). Ce modèle comporte une activité physique
volontaire qui peut être importante. En effet, le modèle consiste à placer un rat dans une cage
équipée d’une roue en libre accès, avec de l’eau à volonté. Le rat a accès à l’alimentation à
56
raison d’une heure par jour seulement. Ce modèle, ensuite désigné comme modèle d’anorexie
basée sur l’activité (activity-based anorexia - ABA) entraîne une perte rapide et importante du
poids corporel et de la prise alimentaire, une hyperactivité, une hypothermie, une suppression
des estrus et une diminution de l’activité de l’axe hypothalamo-hypohyso-ovarien.
Dans ce modèle les rats ABA mangent moins que les contrôles et se privent de manger jusqu’à
mourir. La sévérité de ce modèle ABA est fortement corrélée à la souche animale (plus ou
moins résistant), au sexe de l’animal (les femelles sont plus vulnérables) et à l’âge (les jeunes
sont plus sensibles à l’ABA) (239–241).
III-B-1.4 Modèles de restriction du temps d’accès à la nourriture Les souris sont logées dans des cages classiques avec un accès limité à la nourriture (TR).
Dans ce modèle on n’a pas de régime de restriction calorique qui est parfois pauvre en minéraux
entrainant une malnutrition. Ce modèle TR régule l’horloge biologique et empêche l’induction
de l’accumulation de la graisse, l’hyperinsulinemie, et entraine une diminution des cytokines
pro-inflammatoire et améliore le métabolisme glucidique (253).
III-B-1.5 Modèle de Séparation / restriction Les souris sont logées dans une cage divisée en six compartiments. Les souris se voient
mais sans contact physique. Elles sont transférées dans des cages classiques pour se nourrir. En
outre, ce modèle est facile à réaliser sans équipement compliqué ou intervention.
L’équipe de Van Leeuwen en 1997 était le premier à décrire le modèle de séparation (254). Ce
modèle consiste à séparer les souris les une des autres par des cloisons en plexiglas. Cette
séparation va entraîner une perte importante du poids. Avant cette étude, les modèles animaux
utilisés pour induire une perte de poids sont basés sur l’hyperactivité et sur autres sources de
stress, ou sur la restriction alimentaire. Les modèles basés sur le stress utilisent les pincements
de la queue, la nage en eau froide, ces stimuli peuvent physiquement nuire aux animaux. Le
stress chronique peut être un modèle plus représentatif des troubles alimentaires humains d’un
stress temporaire. Ce modèle permet de surmonter certaines des limites présentes dans d’autres
modèles de stress d’induction de la perte de poids. Il est facile à réaliser, en obtenant une perte
rapide de poids et un stress chronique (254–256). C’est un modèle intéressant dans l’étude des
troubles alimentaires humaines.
57
Tableau 7 : Synthèse des caractéristiques décrites dans les différents modèles environementaux
III-B-2 Les modèles murins génétiques Dans la littérature, de nombreux modèles de souris déficientes génétiques pour un ou
plusieurs gènes impliqués dans la régulation de l'alimentation équilibre comportement/
rendement / énergie ont été développés (256). Ces modèles génétiques fournissent des données
mécanistiques essentiels relatifs à une voie spécifique, mais ne reflètent pas complètement les
symptômes observés dans la maladie humaine (257). Au cours des dernières années, il y a eu
une explosion de l’information en ce qui concerne la régulation génétique de la prise alimentaire
et la balance énergétique. Chez la souris les mutations génétiques généralement effectuées sont
des mutations donnant un phénotype d’obésité. En revanche, il y a peu de modèles animaux
génétiques d’anorexie mentale. Cela peut être dû à plusieurs facteurs. Une possibilité est qu’un
phénotype anorexigène pourrait conduire à la mort de faim ou de malnutrition.
III-B-2.1 Lou/C Rats : Modèle animal de restriction alimentaire volontaire Les rats Lou/C issus de la souche Wistar présentent un intérêt car c’est un modèle de
résistance à l’obésité. Sous un régime alimentaire normal le rat Lou/c ingère moins de calories
par jour que le rat Wistar. Alors ce modèle peut être proposé comme un modèle de restriction
calorique spontonnée. En outre, lorsque ces rats sont soumis à une auto-sélection, ils
sélectionnent spontanément une forte propotion de matières grasses (70% de l’apport calorique
quotidien) sans aucune modification de l’apport calorique. Le rat Wistar, devient généralement
obèse avec l’âge et développe rapidement l’obésité et la résistance à l’insuline lorsque il est
soumis à une alimentation riche en graisse (258).
58
III-B-2.2 Anx/anx La seule mutation spontanée de la souris qui provoque l’anorexie mentale et qui a été
largement étudiée est la mutation récessive anx/anx. Les souris anx/anx sont caractérisées par un
manque d’appétit. Les données expérimentales ne révèlent aucun problème anatomique mais
montrent que les souris ne parviennent pas à réguler la quantité de nourriture consommée. Les
souris sont caractérisées par un poids corporel réduit, des tremblements corporels, une
hyperactivité et une démarche non coordonnée. Ce phénotype apparaît au cinquième ou huitième
jour après la naissance, les animaux meurent à l’âge de cinq semaines en fonction du fond
génétique. Les souris anx/anx présentent une réduction des niveaux sérique de leptine à partir du
huitième jour postnatal, et expriment moins la leptine dans le tissu adipeux. Les neuropeptides
régulés par la leptine sont également étaient étudiés. Les études imunohistochimiques au niveau
du noyau arqué montrent une accumulation importante des neuropeptides Y et AGRP à
l’intérieur de la cellule que dans les extensions dendritique, une diminution des taux de pro-
opiomelanocortin (POMC), des récepteurs neuropeptidiques Y1R, Y5R et la cocaine and
amphetamine –related transcript (CART) (259,260).
III-B-3 Les modèles pharmacologiques
Modèle LPS (lipopolysaccaride): Les deux principales cytokines pro-inflammatoires impliquées dans la manifestation du
comportement de certaines maladies comme l’anorexie mentale est l’IL-1β et la TNF-α.
L’administration systémique de LPS induit l’expression de l’IL-1 bêta et autres cytokines pro-
inflammatoires dans le cerveau.
Les études pharmacologiques ont démontré que l’administration systémique ou centrale de l’IL-
1β ou TNF-α à des rats et des souris induient des signes comportementaux dose-temps
dépendant. En général, les animaux injectés avec de l’IL-1 β ou TNF-α s’isolent dans un coin de
leur cage, le dos vouté et ne montrent pas d’intérêt dans leur environement physique et social.
Plus précisement, ils montrent une diminution de l’activité motrice, un retrait social, une
réduction de la prise alimentaire et une sensibilité accrue à la douleur (261).
59
IV-Conclusions L’anorexie mentale est très mal étudiée, les études cliniques sont rares, le suivi des malades
est difficile, les cliniciens ne sont pas encore arrivés à des prises en charges efficaces notamment
par manque de connaissance des mécanismes qui interviennent dans cette maladie.
L’impossibilité d’accéder aux tissus chez les patientes rend indispensable l’utilisation des
modèles animaux. La restriction calorique chronique et sévère est fréquement utilisée dans les
études. Le développement d’un modèle animal approprié à l’anorexie mentale est très difficile
car cette maladie est complexe elle implique des interactions entre les facteurs génétiques,
environnementaux, facteurs sociaux et culturels. Aucun animal ne décide volontier de cesser de
manger. Alors au lieu d’un modèle basé sur la cause, les chercheurs se sont tournés vers des
modèles animaux qui miment certains aspects physiopathologiques de la maladie humaine
comme la perte du poids, la restriction alimentaire, l’augmentation de l’activité, la perturbation
des fonctions neurœndocriniennes, l’aménnohrée, la perte osseuse, et les perturbations des
facteurs sanguins. Très peu de modèles animaux ont réussi à englober tous les aspects
somatiques de la maladie.
Dans notre étude on a choisi un modèle murin qui associe la restriction calorique à un stress
chronique Separation Based Anorexia (SBA) pour induire les aspects physiopathologiques
observés en anorexie mentale, ces aspects physiopathologiques ont été étudiés à differents temps
à 2 semaines, 5 semaines et 10 semaines du protocole SBA. Et on a complété cette étude par une
phase de récupération (Rec) pour voir si les paramètres perturbés se corrigeaint et pour
comprendre les raisons ou les mécanismes qui empechent la récupération de certains facteurs.
60
TRAVAIL DE THESE
I- Axes de recherche du PMOI L’hypothèse sur laquelle travaille l’ensemble du PMOI est que la modification des
relations entre tissu osseux et adipocytes médullaires participe à la pathogénie de l’ostéoporose
et de l’ostéonécrose.
En s’appuyant sur les compétences cliniques des membres du laboratoire, trois pathologies
sont plus spécifiquement étudiées : l’ostéoporose de l’anorexie mentale, l’ostéonécrose de la tête
fémorale, l’ostéonécrose des maxillaires.
Le PMOI utilise différentes approches pour développer cette thématique. En effet, des
études basées sur les prélèvements et observations cliniques permettent d’étayer les hypothèses
proposées et de déterminer si certains résultats obtenus avec d’autres approches sont pertinents
dans la pathologie humaine étudiée.
Des modèles animaux permettent de compléter les résultats obtenus chez l’humain. Des
modèles murins d’ostéoporose sont plus particulièrement étudiés. Le modèle de souris
ovariectomisée permet de disposer d’un modèle dont les conséquences en termes de composition
corporelle et d’altération osseuse sont déjà bien décrites. Il présente l’avantage d’induire une
perte de masse osseuse liée à une augmentation de la formation osseuse associée et à une
augmentation plus importante de la résorption (79,262). Les mécanismes impliqués dans
l’augmentation de la résorption incluent une augmentation des taux de cytokines inflammatoires
circulantes consécutive à une chute des taux d’estrogènes, à l’image de ce qui est décrit chez les
femmes post ménopausiques. Ce modèle induit également une augmentation de l’adiposité
médullaire. L’autre modèle développé au laboratoire vise à reproduire les conséquences
physiologiques de l’AM chez la souris. La définition et l’étude de ce modèle font l’objet du
présent projet de thèse.
Le PMOI développe également des modèles d’études des interactions adipocytes /
ostéoblastes et d’étude des régulations des leurs voies de différenciation. Ces modèles incluant la
mise en co-culture d’adipocytes et d’ostéoblastes préalablement différenciés à partir de cellules
souches mésenchymateuses ainsi que la co-différenciation de cellules stromales en culture
primaire permettent de mettre en évidence des interactions (190) et de rechercher les facteurs
potentiellement impliqués.
61
II-Sujet et objectif du projet de thèse Le projet de ma thèse a été conçu sur la base de premiers essais de mise au point d’un
modèle reproduisant une partie des altérations physiologiques décrites chez les patients
anorexiques et en tenant compte des données scientifiques récentes qui ont modifié la
compréhension de la régulation de la balance énergétique.
Il en ressort la nécessité d’étudier de manière approfondie les liens étroits entre os, cerveau et
tissu adipeux. Le cerveau participe à la régulation du comportement alimentaire, de la masse
adipeuse et de la masse osseuse, notamment par l’intermédiaire du système nerveux
sympathique. Le tissu adipeux sécrète des facteurs protéiques et lipidiques qui influencent le
contrôle de la balance énergétique et de la masse osseuse par le cerveau. L’os sécrète des
facteurs protéiques qui participent à la régulation du métabolisme énergétique. Les
caractéristiques de ces trois acteurs sont perturbées chez les patients anorexiques. Comme cela a
été décrit dans la partie introductive, les modèles murins couramment étudiés sont basés sur une
restriction alimentaire modérée ou une restriction sévère mais sur du court terme. De plus ils
sont pour la plupart appliqués à des rats ou des souris mâles. Le PMOI souhaitait développer un
modèle plus fidèle à la pathologie humaine.
Les objectifs de ma thèse étaient donc de :
1. Développer un modèle murin qui mime de nombreuses perturbations
physiopathologiques observées chez les patientes anorexiques,
2. Évaluer ce modèle par rapport aux autres modèles animaux existant,
3. Évaluer l’intérêt de ce modèle pour des études de récupération,
4. Étudier la perte et la récupération osseuse et les mécanismes potentiellement impliqués
dans ces deux processus pour le modèle étudié.
II-A Définition du cahier des charges Le cahier des charges du modèle est établi sur la base des altérations observées en
clinique :
- Perte d’au moins 20 % du poids initial comme pour la maladie humaine (même s’il ne
peut y avoir de comparaison directe car les compositions corporelles et les physiologies
humaines et murines.
- Altérations des fonctions de reproduction, car l’aménorrhée quasiment systématique chez
les patientes anorexiques est liée à des perturbations hormonales impactant le
métabolisme et la masse osseuse.
- Baisse de la masse osseuse par rapport aux contrôles.
62
- Perturbations de la sécrétion de certaines hormones comme la leptine, l’adiponectine, la
GH, l’IGF-1, dont les niveaux sont très fréquemment altérés chez les patientes, ce qui
participe aux adaptations métaboliques et à la régulation de la masse osseuse.
- Possibilité de maintenir la phase « anorexique » pendant une durée longue, de l’ordre de
10 semaines afin de laisser se développer de potentielles altérations tardives.
A ces critères s’est ajouté la possibilité de replacer les animaux en conditions standards
pendant 10 semaines, après 10 semaines de protocole inducteur, afin d’étudier les capacités de
récupération. Cela suppose donc qu’après le premier protocole les animaux soient dans un état
physique permettant de les garder 10 semaines de plus. L’étude de cette phase de récupération
est destinée à déterminer les capacités de récupération et identifier d’éventuelles altérations
persistantes qui pourraient influencer le processus de récupération.
II-B Choix du modèle animal
II-B-1 Choix du protocole d’induction On a choisi de tester le modèle Separation Based Anorexia (SBA) qui associe le stress
chronique et le contrôle de l’alimentation. Ces deux facteurs étant décrits chez les patientes, ce
modèle mime ces deux aspects, on réduit la durée d’accès à la nourriture et on les sépare les unes
des autres pour créer le stress chez eux. Les souris utilisées pour ce protocole sont des souris
femelles C57Bl/6 elles sont décrites comme sensibles aux stress. Ces souris au début du
protocole sont âgées de 8 semaines, elles sont donc relativement jeunes et en fin de croissance
rapide.
Une première phase d’essais sur deux semaines était destinée à choisir les conditions et tester les
réponses de souris. Par la suite, le protocole SBA s’étale sur 10 semaines, afin d’étudier les
différentes phases de la pathologie (la phase de perte rapide de poids, la phase d’adaptation) car
en clinique on n’a pas accès à ces deux phases. La phase SBA est suivie de la phase de
récupération (REC) pour mieux comprendre les mécanismes impliqués dans ce processus.
La restriction alimentaire en phase SBA est progressive allant de 6h jusqu’à 2h/jour sur une
période de 10 semaines, ce qui permet aux souris de s’adapter progressivement et réduit
considérablement le nombre de souris devait être exclues de l’étude à cause de leur épuisement
extrême. Ce protocole SBA est suivi d’une phase de récupération (REC) de 10 semaines en
condition standard pour voir s’il y’a des altérations durables et si elles pourraient constituer un
frein à la récupération complète.
63
II-B-2 Animaux et conditions d’élevage Les travaux décrivant partiellement le modèle de séparation associée à la restriction du
temps d’accès à l’alimentation, avaient été effectués sur une lignée de souris peu répandue la
lignée SABRA (254). Cette lignée semble posséder des caractéristiques différentes de celles
classiquement étudiées et utilisées notamment pour des travaux sur la restriction calorique, le
métabolisme et l’évolution de la masse osseuse. Afin de pouvoir par la suite comparer nos
données à celles de la littérature, nous avons choisi d’adapter le modèle à des souris femelles
C57Bl6/J.
Les animaux sont réceptionnés à l’âge de 7 semaines est acclimatés pendant une semaine
en conditions classiques d’élevage, à 6 souris par cage. La température de l’animalerie est
maintenue à 22-23°C, avec un cycle jour/ nuit de 12 :12h. L’eau et la nourriture sont disponibles
à volonté.
La nourriture choisie est une nourriture standard qui contient tout les éléments nécessaires
à la reproduction, à la lactation et à la croissance des animaux (Special Diets Services, St
Gratien, France). Elle contient 22 % de proteines brutes, 5% de matière grasses brutes et 50.5%
de glucides (Composition détaillée dans le tableau 7). Cet aliment présente une teneur
relativement basse en lipides par rapport à d’autres types d’aliments. Cela permet de limiter la
prise de poids importante qui est souvent observée chez les groupes contrôles au cours des
protocoles longs. L’objectif était de comparer nos groupes expérimentaux à un groupe contrôle
« sain » et non en surpoids (247).
64
Tableau 8 : Analyse nutritionnelle de la nourriture donnée à nos souris
Les différents protocoles utilisés dans cette étude ont bénéficié d’une autorisation du
Comité d’Ethique en Expérimentation Animale du Nord-Pas-de-Calais (autorisation CEEA
022012). Au cours des différents protocoles et pour des raisons éthiques, les souris qui avaient
perdu 30% ou plus de leur poids initial pendant plusieurs jours d’affilé, et ce malgré une durée
d’alimentation augmentée, étaient écartées du protocole. Pour les mêmes raisons, des souris
montrant des signes d’affaiblissement important (prostration, absence de réaction à la
manipulation, absence de prise de nourriture) étaient également écartées de l’étude et sacrifiées.
II-B-3 Protocoles étudiés
II-B-3-1 Essais à court terme Dans un premier temps, les effets des quatre protocoles différents ont été étudiés, pendant
2 semaines. Les quatre protocoles sont présentés dans le tableau 8.
Analyses nutritionnelle (% moyen par kg/ brut)
Protéines brutes 22.5 %
Matières grasses brutes 5.0 %
Cellulose brute 4.5 %
Cendres brutes 6.5 %
Humidité 11.0 %
Glucides 50.5 %
Calcium 0.90 %
Phosphore 0.70%
Magnésium 0.20%
Sodium 0.20%
Potassium 0.90
Energie métabolisme 2 952.8 kcal/kg brut
65
Groupe d’animaux Description Durée du protocole
Contrôle (CT) 6 souris maintenues en
condition classique, eau et
nourriture à volonté
2 semaines
Separation (SEP) 6 souris separées avec eau et
nourriture à volonté
2 semaines
Time restriction (TR) 6 souris maintenues en cage
collective avec de l’eau à
volonté, mais la nourriture
n’est pas disponible que
pendant 6h à 2h de temps par
jour
2 semaines
Separation Based Anorexia
(SBA)
6 souris placées dans une cage
divisée en 6 compartiments
avec une restriction
progressive du temps d’accès
à l’alimentation de 6h jusqu’à
2h par jour (figure 19)
2 semaines
Tableau 9 : Description detaillée des 4 groupes d’animaux
66
Figure 19 : Les souris dites SBA sont soumises à la séparation et à la restriction du temps d’accès à l’alimentation
afin d’empêcher une éventuelle sur-alimentation. Pour ces souris, l’alimentation a lieu en cage collective, comme
dans la publication décrivant le premier modèle associant séparation et restriction du temps d’accès à l’alimentation
(254).
II-B-3-2 Essais à long terme Le protocole SBA entraînant d’importantes modifications au cours des 2 semaines, a été
ensuite appliqué sur une période de 10 semaines. Cette durée a été choisie pour répondre à 2
objectifs. D’une part, nous souhaitions étudier les adaptations précoces mais aussi celles qui se
produisent plus tardivement, une fois que les réserves sont épuisées. Une étude basée sur une
restriction quantitative de la nourriture (-40%) chez la souris a montré que certaines
modifications nécessitent de nombreuses semaines avant de se produire (251).
D’autre part, nous souhaitions étudier les altérations osseuses induites, et il est bien connu que
dans les modèles de restriction alimentaire la masse osseuse n’est pas atteinte aussi rapidement
que la masse grasse ou la masse maigre. Toujours en regard des caractéristiques osseuses, nous
avons également pris comme repère les durées couramment utilisées pour évaluer la perte
osseuse après ovariectomie chez la souris, c’est-à-dire après 4 semaines et 12 semaines de
l’opération (263,264).
Ce protocole SBA est suivi d’une phase de Rec de 10 semaines en conditions standard. Les
souris CT sont alors logées dans des cages classiques, 6 souris/cage, avec de l’eau et de la
nourriture à volonté.
67
II-C Choix des paramètres étudiés et des méthodes utilisées pour évaluer ces altérations Les paramètres étudiés sont en priorité ceux qui sont altérés dans l’anorexie mentale et
dont on souhaite reproduire les altérations : le poids, les masses grasse, maigre et minérale, les
fonctions reproductrices, la leptinémie, le taux de GH, d’IGF-1, et de cortisol. À ces paramètres
s’ajoutent des paramètres que nous avons sélectionnés afin d’approfondir la caractérisation du
modèle et afin d’initier l’étude des mécanismes potentiellement impliqués : la microarchitecture
osseuse, l’adiposité médullaire, l’adiponectinémie, la tolérance au glucose, l’expression génique
des marqueurs du métabolisme énergétique et lipidique dans les tissus adipeux.
II-C-1 Prise alimentaire La prise alimentaire est suivie en pesant la nourriture avant et après la période d’alimentation
des souris en restriction du temps d’accès à l’alimentation. La prise alimentaire des souris
nourries ad libitum est évaluée en pesant quotidiennement la nourriture restante avant le début
du cycle nocturne. Les souris CT, TR et SBA s’alimentant en cage collective de 6 souris, la prise
alimentaire est mesurée pour la cage puis la valeur divisée par le nombre d’individus est
exprimée en g d’aliment/souris/jour. Afin de mettre en évidence l’évolution de la prise
alimentaire et les différences pouvant apparaître entre les groupes, la prise alimentaire est
exprimée sous forme cumulée tout au long des protocoles. Ainsi la valeur d’une journée donnée
se rajoute à la somme des valeurs déterminées les jours précédents.
II-C-2 Poids corporel Le poids corporel est déterminé avant chaque phase de prise alimentaire, c’est-à-dire que les
animaux sont pesés individuellement avant le début du cycle nocturne.
II-C-3 Composition corporelle Les mesures sont effectuées par absorptiométrie DEXA en utilisant le densitomètre
(PIXImus) entre 9h et 11h du matin. Les souris sont injectées d’un myorelaxant (Kétamine®)
puis d’un anesthésiant (Xylazine®)
La souris est couchée sur le ventre et les captures sont effectuées sur corps entier sans la
tête. Les calculs de masses grasse, maigre et minérale sont ensuite effectués sur le corps entier.
La masse minérale est également calculée sur des régions d’intérêt encadrant le fémur droit ou
les vertèbres lombaires (figure 20.A, B et C).
68
A
B
C
Figure 20 : Captures d’écran du logiciel d’analyse Piximus. A- Les résultats des mesures densitométriques du corps entier. B- Mesure de la BMC du fémur. C- Mesure de la BMC des vertèbres L1-L5.
69
II-C-4 Les fonctions reproductrices Les altérations des fonctions reproductrices représentent des données importantes dans
l’étude du modèle, non seulement parce qu’elles sont quasi-systématiques chez les patientes,
mais aussi parce qu’elles supposent une diminution des taux d’hormones sexuelles, dont les
estrogènes qui sont impliqués dans la régulation de la masse osseuse.
Deux paramètres ont été suivis pour évaluer la perturbation de ces fonctions. Le cycle
œstral des souris a été suivi tout au long des protocoles SBA et Rec. Lors des sacrifices les
ovaires ont été récupérés et leur taille déterminée histologiquement.
Pour le suivi du cycle œstral, des frottis vaginaux ont été réalisés avant l’accès à la
nourriture. Pour cela, la pointe de la pipette remplie de solution saline est introduite de 5 mm
dans le vagin. Le contenu est aspiré/refoulé environ 5 fois. La suspension cellulaire ainsi
obtenue est placée sur une lame de verre. Les cellules sont ensuite observées, sans coloration, en
microscopie optique Axio Skop (Zeiss, Allemagne) équipé d’une caméra Digital Interface
(Sony, Japon) avec un grossissement final de 100x. La nature et les proportions des cellules
présentes permettent de déterminer le stade du cycle œstral dans lequel se trouve la souris,
comme l’illustre la figure 21.
70
Figure 21 : Photos des frottis vaginaux non colorés des rats femelles, en proestrus, oestrus, metestrus et diestrus.
(L) Leucocytes, (E) Cellules épitheliales nuclées, (C) Cellules cornifiées. Si le fottis contient principalement des
cellules épithéliales nuclées > 60% et des leucocytes < 10% : Proestrus. Si le frottis contient principalement des
cellules cornifiées > 90% : Estrus. Le frottis qui contient des cellules cornifiées > 60% avec une quantité importante
de leucocytes (+20%) : Metestrus. Si les leucocytes prédominent > 60 % : Diestrus.(265).
Après le sacrifice les ovaires gauche et droit sont recueillis et fixés au Bouin (75% d’acide
picrique pur, 20% de formol et 5% d’acide acétique), puis déshydratés par des bains successifs
d’alcool de concentration croissante. Les ovaires sont ensuite inclus en paraffine. Ces ovaires
sont coupés (6 µm d’épaisseur) et colorés à l’hématoxyline/éosine. Les longueurs et largeurs des
ovaires ont été mesurées grâce à des observations au microscope optique (grossissement x 40).
II-C-5 Tolérance au glucose Etant donné les profondes altérations des masses grasse et maigre, l’homéostase globale du
glucose a été étudiée aux différentes étapes des protocoles. Dans ce but des tests de tolérance au
glucose ont été réalisés après injection par voie intrapéritonéale. Les souris ont été mises à jeûne
pendant 12 heures et leur glycémie a été mesurée dans une goutte de sang à partir d’une coupe
fine à l’extrémité de la queue, grâce à un glucomètre (Accu-Chek Performa glucometer, Roche,
71
Switzerland). Au temps zéro, les souris ont été injectées avec une solution de glucose (1g/kg de
poids corporel), et le taux de glucose a ensuite été mesuré aux temps 5, 10, 15, 30, 45, 70, et 90
minutes post-injection. Les quantités de glucose injectées sont proportionnelles aux poids
corporels et donc différentes entre les 2 groupes. Afin de vérifier que les différences de courbes
de glycémie n’étaient pas liées à cette différence de quantités de glucose injecté, nous avons
également réalisé un test de tolérance au glucose annexe, en injectant au groupe CT la même
quantité de glucose que celle injectée aux souris SBA.
II-C-6 Sacrifice À différents temps dans le protocole (2 semaines, 10 semaines, 12 semaines et 20
semaines), les souris ont été sacrifiées. Afin de récupérer et étudier différents tissus et récupérer
le sang. Les souris ont été mises à jeun pendant 6 heures avant d’être anesthésiées par le
pentobarbital. Le sang a été recueilli par ponction cardiaque, immédiatement centrifugé (3500
tours par minute, pendant 10 min, 4°C) et surgelé dans de l’azote liquide avant stockage (-80°C),
puis dosages.
La glycémie a été mesurée. Les triceps, le foie, l’hypothalamus, ainsi que des tissus adipeux
sous-cutanés (SCAT) (inguinal et glutéal), périutérins (VAT) (tissu adipeux viscéral) et le tissu
adipeux brun interscapulaire, ont été collectés, pesés et congelés en azote liquide (Figure 22).
Les tibias, les fémurs et les vertèbres lombaires ont également été disséqués, débarrassés des
tissus mous périphériques puis préparés pour l’inclusion en résine, l’analyse
microtomographique et enfin la coupe histologique.
72
Figure 22 : Croquis des dépôts de tissu adipeux chez la souris mâle. Les tissus colorés en brun correspondent au
tissu adipeux brun (BAT), en brun clair pour « brite » et le blanc pour les tissus adipeux blancs (WAT). Les dépôts
graisseux prélevés au moment du sacrifice sont entourés. iBAT : BAT interscapulaire, glWAT : Tissu adipeux blanc
sous cutané glutéal, iWAT : Tissu adipeux blanc sous cutané inguinal, eWAT : tissu adipeux épididimique, dans
notre modèle on a utilisé des souris femelles, ce tissu adipeux est donc du tissu adipeux périutérin.
II-C-7 Microarchitecture osseuse Les tibias, les vertèbres lombaires et les fémurs sont récupérés après 2 et 10 semaines du
protocole SBA et après 2 et 10 semaines du protocole Rec, ils sont fixés dans du formol à 4 %
et transférés dans de l’éthanol à 70% après 24h. Et les os sont déshydratés dans des bains
d’acétone sous agitation (3 bains) puis incuber pendant 1 heure dans du cyclohexane sous
agitation puis 2h d’incubation dans du MMA, une fois ces deux heures sont écoulées on ajoute
10 µl d’initiateur pour 10 ml de MMA et on les incube pour une nuit à -20º C. Ensuite les os
sont inclus dans une résine de méthylméthacrylate. La résine en excès est poncée afin de limiter
ses interférences sur les paramètres osseux qui sont évalués au microtomographe. L’étude de la
microarchitecture osseuse est effectuée en 3 phases : D’abord, l’acquisition est réalisée par
microtomographe SkyScan 1172, l’appareil effectue 4 scans pour couvrir la longueur d’un tibia
à une résolution de 5 µm. Puis la reconstrution de ces 4 scans est faite par le programme de
reconstitution NRecon, et enfin l’analyse est effectuée par le progromme d’analyse CTAn
(Tableau 9).
73
Acquisition
Nombre de Scans : 4
Résolution : 5.02µm
Voltage : 59 Kv
Courant : 100 µA
Degré de rotation : 0.400°
Temps d’exposition : 240 ms
Profondeur : 16 bits
Reconstitution
Fusion et réalignement des 4 sous scans
Postalignement = 1.00
Temps de reconstitution par tranche : 0.54 ms
Fenêtre d’intensité entre 0 et 0.101333
Analyse
Point de départ : fin de la spongieuse primaire
Sélection de la Région d’intérêt (ROI)
Puis Binarisation
Tableau 10 : Paramètres d’acquisition et de reconstitution utilisés pour l’analyse microtomographique des tibias
et vertèbres des souris.
74
II-C-8 Histologie osseuse Après avoir été scannés au microtomographe, les blocs en méthylméthacrylate sont coupés
en longitudinal au microtome (Leica) avec une épaisseur de 7 µm. Les coupes sont faites selon
des plans parallèles à ceux passant par les deux épiphyses (figure 23). Ces coupes sont colorées
avec de l’hématoxyline et observées au microscope au grossissement 100, et le comptage des
adipocytes médullaires a été effectué en tibia proximal, dans une zone de 1mm au-dessous de la
plaque de croissance en utilisant le logiciel Image J. Illustration par photo PMOI avec les 4
secteurs.
Figure 23 : Coupe histologique d’un tibia proximal colorée par l’hématoxyline, ou on voit les quatres secteurs de
comptage d’adipocytes effectué dans une zone de 1mm au dessous de la plaque de croissance.
II-C-9 Dosages plasmatiques Pour le dosage des facteurs circulants on a utilisé différentes techniques. On a utilisé le kit
Milliplex pour le dosage de la leptine (Millipore, Billerica, USA) et des plaques de la
technologie Luminex (Luminex Corporation, Austin, USA). Le taux d’IGF-I est dosé en utilisant
une plaque pré- coatée (kit ELISA Quantikine, R&D Systems Inc., Minneapolis, USA). La GH
est dosée par ELISA Sandwich sur sang entier. Un volume de 4µl de sang est prélevé de la
queue de la souris et homogénisé dans 116µl de 1X PBS-T Buffer (0.05%). L’anticorps de
capture utilisé est le monkey anti-rGH-IC-14 (AFP411S) et le rabbit anti-r GH (AFP5672099)
comme anticorps de détection, avec une dilution finale de 1/40.000.
75
II-C-10 Expression génique des tissus adipeux Les ARNs sont extraits des tissus adipeux sous cutanés (SCAT) et périutériens (VAT) à
l’aide d’Extract-All (Eurobio, Les Ulis, France). 4 µg d’ADN sont traités à la DNase I (Roche
Diagnostics, Penzberg, Allemagne). La synthèse des molécules d’ADN complémentaires a été
réalisée avec la transcriptase inverse RT ThermoScientific Maxima First strand cDNA synthesis.
La PCR en temps réel est faite en utilisant le Nano LightCycler et le FastStart Essential DNA
Green Master. Les amorces sont designées en utilisant le logiciel Oligo 6 et le logiciel crée par
Roche (tableau 10). Le test d’efficacité est réalisé sur ces amorces, l’amorce est considérée
efficace si l’efficacité se trouve entre 1.85 et 2. Les deux gènes de référence utilisés sont le
cyclophilin A (PPIA) et l’Hypoxanthine-Guanine Phosphoribosyl Transferase (HPRT).
76
Gène Efficacité Le choix
HPRT 1.97 Gène de référence
expression stable
dans les tissus et les
protocoles utilisés
PPIA 2.01 Gène de référence
expression stable
dans les tissus et les
protocoles utilisés
Leptine 1.96 Suivi de l’évolution
de la masse grasse
Glut-4 2.03 Impliqué dans la
synthèse lipidique
FAS 1.98 Impliqué dans la
synthèse lipidique
ATGL 1.90 Impliqué dans la
lipolyse
PRDM16 2.07 Impliqué dans la
régulation
transcriptionnelle
des adipocytes bruns
PGC1 alpha 2.06 Impliqué dans la
régulation
transcriptionnelle
des adipocytes bruns
ACOX 1 1.88 Co facteur de ATGL
ABDH5 1.82 Co facteur de ATGL
UCP-1 et b3AdR
(récepteur bêta 3
adrénergique)
1.96-.1.98 Gène impliqué dans
la thermogenèse
Tableau 11 : liste des gènes étudiés.
77
II-C-11 Tests statistiques Les données sont présentées sous forme de moyenne ± SEM et ont été générées en utilisant
GraphPad (GraphPad Software Inc., Etats-Unis). Le Mann-Whitney U Test est utilisé pour la
comparaison des différences entre 2 groupes et/ou entre deux temps. Le two-away ANOVA est
utilisé pour tester deux droites de régression représentant des populations indépendantes, suivi
du test de Bonferroni post-hoc pour comparer les différences entre deux points correspondant au
même temps. Les différences sont considérées comme significatives à p< 0.05.
III-Résultats
III-A Choix du protocole à partir d’essais à court terme Afin de vérifier l’intérêt de la combinaison de la séparation et de la restriction du temps
d’accès à l’alimentation, des essais à court terme ont été réalisés (pendant deux semaines).
Quatre conditions différentes ont été testées sur 4 groupes de 6 souris :
- Un groupe CT, les souris sont hébergées dans des cages classiques avec nourriture à
volonté,
Un groupe TR : les souris sont placées dans des cages classiques et elles sont soumises à
une restriction du temps d’accès à l’alimentation,
- Un groupe SEP : les souris ont un accès libre à la nourriture mais elles sont séparées par
des cloisons de plexiglas,
- Un groupe SBA : Les souris sont séparées par des cloisons en plexiglas et soumises à une
restriction du temps d’accès à l’alimentation.
Pour cette étude à court terme, seuls les poids corporels, les prises alimentaires et les
compositions corporelles ont été déterminées, afin de vérifier l’efficacité du protocole SBA sur
ces premiers paramètres basiques et déterminer la progressivité optimale de réduction du temps
d’alimentation. Après des essais préliminaires, l’étude telle qu’elle est présentée a été répétée
une fois. Les résultats présentés sont ceux d’une des expériences et sont représentatifs de ce qui
a été obtenu pour chacune.
78
III-A-1 Suivi du poids corporel et de la prise alimentaire à court terme Les mesures du poids corporel et de la prise alimentaire ont été effectuées pour les 4 groupes
tout au long du protocole. Les résultats montrent une diminution significative du poids corporel
pour les groupes SBA et FR par rapport au groupe CT (figure 24). Pour le groupe SBA, cette
diminution représente plus de 25 % du poids initial. Elle est de 12 % seulement pour le groupe
TR. Les valeurs du groupe SBA sont significativement différentes de celles du groupe CT dès le
jour 1 de l’expérience (p < 0.001). À la fin des 2 semaines, le poids des souris du groupe SBA
est inférieur de 37 % à celui des souris du groupe CT. Les poids des souris du groupe SBA sont
significativement différents de ceux des souris TR à partir du 6ème jour (p < 0.05). Les valeurs du
groupe TR sont significativement inférieures à celles du groupe CT dès le 1er jour (p<0.05).
Le suivi de la prise alimentaire n’a pas fait l’objet d’une analyse statistique car dans 3
protocoles sur 4 les souris sont nourries en groupe et il n’y a donc qu’une valeur moyenne par
jour pour chaque groupe. Néanmoins, il apparait clairement que les souris SEP ont tendance à se
nourrir plus que les autres groupes, alors que les souris TR et SBA ont des prises alimentaires
proches. A partir du 10ème jour, ces 2 groupes se nourrissent moins que les souris CT. Il en
résulte qu’à la fin du protocole les souris SBA ou TR ont mangé, selon les expériences, 10 à 20
% de nourriture en moins.
79
Figure 24 : Suivi du poids corporel et de la prise alimentaire tout au long du protocole.
A-La prise de la masse corporelle est faite au début de la phase noir. La comparaison est faite par 2- way ANOVA
suivit du test de Bonferroni. La moyenne des poids corporels du groupe SBA est significativement basse par rapport
a la moyenne des poids corporels du groupe Ctrl du J1 Jusqu’à la fin du protocole SBA (** P < 0.001). La moyenne
des poids corporels du groupe SBA est significativement basse par rapport à la moyenne des poids corporels du
groupe TR du J6 jusqu’à la fin du protocole (‡ P < 0.05). La moyenne des valeurs du groupe TR est
significativement plus basse que les valeurs du groupe Ctrl du J1 jusqu’à la fin du protocole. **P<0.001 CT vs
SBA ; ‡ P<0.05 SBA vs TR ; *P<0.05 CT vs TR
B-L’apport de nourriture a été calculé pour chaque groupe en faisant la somme de la consommation alimentaire
moyenne par souris du premier jusqu’au dernier jour du protocole. Pas possible de faire les tests statistiques car le
nombre d’effectif est faible.
III-A-2 Composition corporelle La composition corporelle a été évaluée par absorptiométrie des rayons X (Dual Energy X
ray Absorptiométrie) appliquée au corps entier en excluant la tête (figure 25). La perte de poids
importante relevée pour le groupe SBA correspond principalement à une diminution de la masse
grasse et de la masse maigre. La diminution de la masse grasse même si elle est quantitativement
moins importante que celle de la masse maigre représente 34% de la masse grasse initiale. Alors
que la masse maigre ne diminue que de 10%.
La masse grasse du groupe SBA présente une fonte significative au bout de 14 jours par
rapport au J0 du même groupe et par rapport au groupe CT à J14. Par contre, pour tous les autres
groupes (CT, SEP, FR) cette masse grasse reste stable tout au long du protocole.
La masse maigre du groupe SBA baisse significativement à J14 par rapport à celle du J0 et
par rapport à celle des CT à J14.
Le contenu minéral osseux a une tendance d’augmenter pour tous les groupes, mais cette
augmentation n’est significative que pour les CT et SEP, autrement dit toutes les souris nourries
à volonté.
80
Figure 25 : La masse maigre, la masse grasse et la masse minérale osseuse sont mesurées pour chaque animal
à j0 et à j14. A, B, C-Mesures densitométrique effectuées sur les souris des quatres groupes *P<0.05 et **P<0.005
comparé au même groupe à j0 ; ‡P<0.05 et ‡‡P<0.005 comparé au groupe CT au même temps du protocole.
III-A-3 Conclusion de l’étude à court terme Les résultats obtenus lors de l’étude à court terme permettent plusieurs conclusions.
Concernant le poids corporel, l’étude montre clairement que la combinaison de la séparation et
de la restriction du temps d’accès à l’alimentation est nécessaire pour obtenir une perte de poids
à la fois rapide et supérieure à 20% du poids initial, et une perte de 34% de la masse grasse et de
10% de la masse maigre chez les souris du groupe SBA par rapport aux CT. Par contre, on
n’observe pas de différence significative entre les 4 groupes à 2 semaines. C’est donc cette
combinaison qui sera retenue pour les essais à long terme afin d’observer les altérations osseuses
qui généralement surviennent plus tard. Cette perte de poids est obtenue alors même que les
souris SBA ont une prise alimentaire relativement proche de celle des CT. Ce qui est un premier
résultat laissant supposer que les souris SBA pourraient avoir des besoins énergétiques accrus.
mas
se m
aigr
e (
g)
0
5
10
15
20
CT SBA SEP TR
B
*
**
‡ ‡ ‡ ‡
CT SBA SEP TR
mas
se g
rass
e (
g)
0.0
0.5
1.0
1.5
2.0
2.5
A
** ‡
□ Jour 0
■ Jour 14
Mas
se m
iné
rale
(g)
0.0
0.1
0.2
0.3
0.4
CT SBA SEP TR
C
* **
81
Les souris soumises uniquement à la restriction du temps d’accès à l’alimentation (TR)
présentent une perte de poids significative par rapport aux souris CT, mais cette perte reste
modérée et significativement moindre que celle des souris SBA. Par contre leur alimentation
semble très proche de ces souris SBA. Cela semble indiquer que la séparation est bien impliquée
dans une partie de la perte de poids observée chez les SBA.
Les souris uniquement soumises à la séparation (SEP) ont une courbe de poids proche de
celle des souris CT, alors que leur prise alimentaire semble nettement supérieure à celle de ces
souris. Ce résultat confirme l’implication de la séparation dans la perte de poids des SBA.
III-B Protocoles SBA et REC à long terme Les effets du protocole SBA ont été déterminés principalement après 2 et 10 semaines, afin
d’identifier et évaluer les altérations induites tardivement. Pour une partie des groupes, ce
protocole SBA a été suivi d’une phase de récupération (REC) équivalente (10 semaines) afin de
déterminer dans quelle mesure les différentes altérations observées dans le protocole SBA
pouvaient être corrigées.
II-B-1 Poids corporel et prise alimentaire Le poids corporel et la prise alimentaire ont été suivis tout au long du protocole SBA puis
du protocole REC (figure 26).
Les souris SBA présentent une perte rapide et importante de poids (≈ 25 %) comme lors du
protocole à court terme. Là encore, cette perte de poids est associée à une prise alimentaire
cumulée qui n’est inférieure que de 10% à celle des souris CT. Ce qui laisse supposer que le
protocole SBA induit une augmentation des dépenses ou des besoins énergétiques. Les souris
SBA, après la perte de poids des 2 premières semaines, sont maintenues dans le même protocole
qui avait entraîné cette perte pendant 8 semaines supplémentaires durant lesquelles leur poids est
stabilisé. Cela laisse supposer que le métabolisme des souris SBA s’est adapté afin d’assurer
leurs survies et stabiliser leurs masses corporelles. Pendant ces mêmes 10 semaines les souris CT
prennent régulièrement du poids.
Après 10 semaines de protocole SBA, la courbe de poids et la courbe de prise alimentaire
cumulée des souris remises en conditions standard (REC) rattrapent celles des souris CT en
moins d’une semaine. Au cours de cette première semaine de protocole REC les souris
précédemment soumises au protocole SBA vont manger jusqu’à plus de 8 g par jour et par
souris, alors que l’alimentation quotidienne des CT pendant cette période est de l’ordre de 3 à
3,4 g. Cette brève phase hyperphagique prend fin rapidement avec la normalisation du poids
82
corporel. Il semble que pendant ces 10 semaines les souris CT prennent toujours du poids, mais
moins rapidement que pendant les 10 premières semaines.
Figure 26 : Suivi du poids corporel et de la prise alimentaire pendant 10 semaines du protocole SBA suivi de
10 semaines de Rec. A : les pesées sont effectuées avant le début de la phase noire. B : La prise alimentaire
cumulée pour chaque souris, exprimée en gramme est suivie du début de la phase SBA jusqu’à la fin de la phase de
Rec. Dans A : le test statistique effectué est le 2-way Anova suivi du test de Bonferroni post-hoc. Le poids corporel
des souris du groupe SBA est significativement différent de celui des souris CT, *P<0.0001.
II-B-2 Composition corporelle Les mesures effectuées par absorptiométrie DEXA (PIXImus) nous ont permis d’étudier la
composition corporelle à différents temps du protocole SBA et du protocole REC. Chacune des
masses grasse, maigre et minérale osseuse présente une évolution qui lui est propre. Nous allons
commencer l’analyse des résultats obtenus, par le protocole SBA.
La masse grasse diminue rapidement de 40% puis se stabilise jusqu’à la fin du protocole
SBA (figure 27.A). Il semble donc que sa diminution atteigne une limite au-delà de laquelle
l’organisme utilise d’autres formes de réserves. De 2 à 10 semaines de protocole les souris CT
prennent régulièrement de la masse grasse.
La masse maigre diminue de 15% pendant les 2 semaines du protocole SBA puis semble
tendre à diminuer progressivement jusqu’à la fin des 10 semaines (figure 27.B). Néanmoins
seule la valeur relevée après 2 semaines est significativement différente de la précédente (jour 0
dans ce cas). La tendance à la diminution observée jusqu’à la fin du protocole SBA laisse
supposer que l’organisme de ces souris utilise progressivement ses protéines musculaires. Les
souris CT au contraire ont tendance à gagner lentement de la masse maigre.
83
Les mesures de masse minérale osseuse du corps entier (sans la tête) ont montré que cette
masse est identique chez les souris SBA après 2 et 5 semaines de protocole, ce qui n’est pas le
cas pour les souris CT dont la masse minérale augmente encore significativement entre 2 et 5
semaines (figure 27.C). Ceci semble montrer qu’il y a chez les souris SBA un arrêt de
l’acquisition de masse osseuse du corps entier, à partir de la 2ème semaine de protocole.
Afin d’affiner les résultats obtenus pour la masse minérale osseuse nous avons déterminé
si celle-ci évolue différemment entre le squelette axial et le squelette appendiculaire la masse
minérale osseuse a également été déterminée pour le fémur (figure 27.E) et pour les vertèbres
lombaires L1 à L5 (figure 27.D). Et lorsqu’on mesure spécifiquement le contenu minéral osseux
du fémur gauche, afin d’évaluer les altérations du squelette appendiculaire, l’augmentation entre
les semaines 0 et 2 reste significative chez les souris SBA et chez les CT. À la 2ème semaine du
protocole SBA les deux groupes présentent un contenu minéral comparable. La différence entre
les deux groupes apparaît à la 5ème semaine, comme pour le corps entier. Contrairement à la
stabilisation observée sur le corps entier, la masse osseuse fémorale diminue significativement
chez les souris SBA entre 5 et 10 semaines de protocole. La masse osseuse des vertèbres
lombaires L1 à L5 des souris SBA présente une évolution proche de celle du fémur. La
principale différence est qu’il n’apparaît pas de diminution significative, seulement une
stagnation à partir de la 5ème semaine.
La restauration de cette masse osseuse est complète à 10 semaines de protocole REC. Les
résultats obtenus lors du protocole REC montrent que les 2 premières semaines permettent la
récupération complète des masses grasse et maigre. La récupération de la masse minérale
nécessite plus de 2 semaines, puisqu’après cette période les souris ont une masse minérale qui
tend à être inférieure à celle des souris ayant subi seulement les 10 semaines de protocole SBA.
Par contre, après 10 semaines de protocole REC, les souris ont une masse minérale corporelle
totalement normalisée.
Il est à noter que pour les trois types de masse osseuse la récupération est complète à 10
semaines de protocole REC, alors qu’après 2 semaines cette masse osseuse n’a pas encore
commencé à augmenter. Ce qui laisse supposer l’existence d’un temps de latence précédant une
reprise rapide.
84
Figure 27 : Suivi de la composition corporelle à differents temps du protocole SBA et à 2 et 10 semaines
après Rec. A-E. : Les suivis de la masse maigre, La masse grasse et le contenu de la masse minérale (BMC) du
corps entier, la BMC du fémur, la BMC des lombaires Sont fait après 2, 5 et 10 semaines du protocole SBA et après
2 et 10 semaines de récupération, *P<0.05, **P<0.05 en comparaison avec le groupe CT. ‡P<0.05, ‡‡P<0.005 en
comparaison avec les souris du même groupe au temps précédent.
II-B-3 Etude de la microarchitecture osseuse Le laboratoire a reçu le microtomographe (Skyscan 1172) en début d’année 2014, ce qui n’a
pas permis d’achever les analyses de la microarchitecture à differents temps du protocole SBA et
du protocole de Rec. À l’heure actuelle la microarchitecture a été analysée à 10 semaines du
protocole SBA et après 10 semaines de REC. Les altérations de l’os trabéculaire du tibia
proximal prélevé chez les souris SBA ont été analysées par microCT et montrent une forte
réduction (-25%) de la densité minérale (BV/TV) (figure 28.A) et une réduction de 30% de
l’épaisseur des travées (Tb.Th) environ par rapport aux souris CT (figure 28.B). Par contre, le
nombre des travées reste identique (figure 28.C). La densité minérale corticale mesurée au
milieu de la diaphyse est réduite de 15% (Figure 28.D). Les 10 semaines de protocole REC
permettent une récupération complète des caractéristiques osseuses. Ces résultats indiquent donc
que la récupération osseuse serait à la fois quantitative et architecturale. Il serait donc intéressant
85
de déterminer quels sont les mécanismes ou facteurs qui permettent cette récupération. Dans un
second temps, il faudra ensuite déterminer pourquoi ces éléments ne fonctionnent chez les
patientes en rémission.
Figure 28 : Etude de la microarchitecture osseuse à 10 semaines du protocole SBA et à 10 semaines de REC.
A- Densité volumique osseuse trabéculaire (Trab BV/TV) à 10 semaines du protocole SBA et à 10 semaines de la
phase Rec. B- Epaisseur des travées trabéculaires (Trab Tb.Th).C- Nombre des travées trabéculaires (Trab Tb.N) D-
Densité volumique osseuse corticale (Cort BV/TV). A, B, C et D les analyses sont effectués à 10 semaines du
protocole SBA et à semaines de la phase REC. *P<0.05 et**P< 0.005 en comparaison au groupe CT ; ‡P<0.05 et
‡‡P<0.005 en comparaison aux valeurs antérieures du même groupe.
II-B-4 Pesées tissulaires Les résultats obtenus par absorptiométrie ne permettent pas de distinguer strictement la
masse musculaire du reste de masse maigre, ni les différents tissus adipeux entre eux. Afin de
confirmer et préciser les données obtenues par absorptiométrie, des pesées de tissus adipeux et
musculaires ont été effectuées lors des différents sacrifices.
Pour représenter le tissu adipeux viscéral (VAT), les masses adipeuses périutérines ont été
prélevées. De la même manière, les masses adipeuses glutéales et inguinales ont été collectées et
86
regroupées pour étudier le tissu adipeux sous-cutané (SCAT). Enfin, le tissu adipeux brun (BAT)
a été récupéré au niveau interscapulaire.
Dès 2 semaines du protocole SBA, le VAT disparait pratiquement. Parmi tous ceux qui ont été
pesés, c’est le tissu qui connait la plus forte et la plus rapide diminution de poids (figure 29.A).
Ceci est logiquement lié à son importante activité métabolique notamment par rapport au SCAT.
Le SCAT des souris SBA est rapidement diminué de 75% par rapport à celui des souris CT et
remonte à 60% après 10 semaines de protocole SBA (figure 29.B). Le tissu adipeux brun des
souris SBA augmente après deux semaines du protocole SBA et se stabilisent jusqu’à la fin du
protocole SBA (figure 29.C).
D’après les pesées des triceps surae, une fonte musculaire de l’ordre de 35% survient dans les 2
premières semaines de protocole SBA. Dans les 8 semaines suivantes ce poids musculaire
diminue très modérément mais significativement (figure 29.D).
D’une part, les pesées tissulaires representent une vue d’ensemble de la composition chez les
souris SBA par rapport aux CT. D’autre part, les mesures densitométriques par Piximus, qui un
outil d’analyse quantitative, donnent plus de précision sur l’évolution quantitative de chaque
masse tissulaire (masse osseuse, masse grasse, masse maigre), le piximus nous offre aussi la
possibilité de mesurer une région d’intérêt (par exemple, mesurer la DMO du fémur).
87
Figure 29 : Pesées de la masse adipeuse viscérale, sous cutanée, du triceps et du tissu adipeux brun. Les
pesées sont effectuées au moment du sacrifice à différents temps après 2, 10 semaines SBA et après 2 et 10
semaines de récupération, A : pour l’estimation de la masse viscérale on a utilisé le tissu adipeux périutérien, B :
pour estimer la masse de SCAT on a pesé la masse adipeuse prélevé autour des pattes. C : Pesée du tissu adipeux
brun (BAT). D : le triceps est utilisé pour déterminer l’évolution de la masse maigre. Chez A, B, C et D *P<0.05
et**P< 0.005 en comparaison au groupe CT ; ‡P<0.05 et ‡‡P<0.005 en comparaison aux valeurs antérieures du
même groupe.
La mesure de ces différentes masses tissulaires au cours du protocole de récupération met en
évidence une même cinétique de normalisation pour le SCAT et le VAT. En effet, dès la
deuxième semaine de protocole REC les poids de ces deux tissus ont rattrapé et même
significativement dépassé ceux des souris CT. Après 10 semaines, les souris en protocole de
REC et les souris CT ont strictement les mêmes poids de SCAT et les mêmes poids de VAT.
La masse musculaire des souris en protocole REC est progressivement restaurée pour
rejoindre celle des souris CT après 10 semaines.
Le protocole REC semble induire une augmentation modérée du poids du foie par rapport
à celui des souris CT.
88
II-B-5 Fonctions reproductrices : la taille des ovaires et le cycle œstral Dès 2 semaines de protocole SBA, les études histologiques montrent une atrophie
importante des ovaires. La longueur et la largeur des ovaires SBA sont alors inférieures à celles
des ovaires CT de 30% en moyenne. Après 10 semaines de protocole SBA, l’atrophie s’est
amplifiée et atteint 40% pour la longueur et pour la largeur (figure 30).
Au-delà de l’atrophie constatée, l’étude fonctionnelle révèle également des perturbations
du cycle oestrale (figure 31). En effet, les œstrus caractérisés par la présence des cellules
cornifiées (90%) (figure 32.A) tendent à disparaître dès la première semaine de protocole (figure
32.B). Ce qui révèle une absence de phase féconde pour ces souris.
Les perturbations morphologiques et fonctionnelles observées chez les souris SBA
permettent de supposer que des perturbations hormonales associées se produisent également.
Deux semaines de protocole REC permettent aux souris SBA de retrouver une taille
ovarienne comparable à celle des CT. Au cours de ces 2 premières semaines, les souris SBA
présentent à nouveau un ou plusieurs œstrus, même si comme montré en (figure 32.C), certaines
souris ne retrouvent pas immédiatement un cycle classique pendant cette période.
Figure 30 : Taille des ovaires. A- Longueur des ovaires en µm B-Largeur des ovaires en µm mesurées chez les
souris après 2 et 10 semaines du protocole SBA et de 2 et 10 semaines du protocole de Rec *P<0.05 et **P<0.005
comparant au groupe CT au même temps du protocole. ‡P<0.05 en comparant aux valeurs antérieurs du même
groupe.
0
500
1000
1500
2000
2500
Lo
ng
ue
ur
de
s o
vair
es
(µm
) □ CT
■ SBA
2 10 10+2 10+10
Durée du protocole (semaines)
A
**
‡
*
‡ ‡ ‡
** *
B
□ CT
■ SBA
2 10 10+2 10+10
Durée du protocole (semaines)
*
‡
**
0
500
1000
1500
2000
Lar
geur
de
s ov
aire
s (µ
m)
89
Figure 31 : Altération du cycle oestral. Le cycle oestral a été déterminé en fonction de la population des cellules
issues de lavages vaginaux quotidiens des souris CT et des souris SBA du jour 0 jusqu’à J70 du protocole et suivi de
20 jours de récupération A- Suivi d’une souris CT. B, C Suivi de deux souris SBA.
90
Figure 32.A : Suivi du cycle oestral d’une souris SBA par des lavages vaginaux à J1, J2, J8 et J10 du protocole
SBA.
J 1
J 10
J 2
J 8
91
Figure 32.B : Aspect des frottis vaginaux après deux semaines du protocole SBA, chez la même souris à J12, J30
et J45 ces images montrent qu’il n’y a plus de cycle normal chez les souris SBA et que la plupart des lavages
vaginaux ne permettent de récupérer de cellules.
J 12 J 30
J 45
92
Figure 32.C : Aspect des frottis vaginaux après le retour aux conditions standard, le cycle oestral est reprit
progressivement dès la première semaine du retour aux conditions standard (J5, J6, J9 et J11).
II-B-6 Etude des perturbations endocriniennes
II-B-6.1 Leptinémie et adiponectinémie La leptine est une hormone clé impliquée notamment dans la régulation de la prise
alimentaire, du métabolisme de l’énergie et de la masse osseuse. Elle contrôle aussi la sécrétion
thyroïdienne et récemment il a été montré que la leptine joue un rôle essentiel dans la régulation
des hormones sexuelles. Comme le taux de cette hormone informe le cerveau du niveau des
réserves lipidiques, c’est logiquement que l’hypoleptinémie est systématiquement décrite chez
les patients anorexiques.
Chez les souris en protocole SBA, les taux plasmatiques de leptine sont très bas et
relativement homogènes par rapport à ceux des CT (figure 33.A) après 2 et 10 semaines. Après 2
J 5 Rec
J 11 Rec
J 6 Rec
J 9 Rec
93
et 10 semaines de protocole REC, les souris qui ont pourtant récupéré la totalité de leur masse
adipeuse restent hypoleptinémiques. En effet, non seulement leur leptinémie reste inférieure à
celle des souris CT, mais en plus elle n’augmente pratiquement pas.
Ce résultat surprenant a été confirmé par l’étude des niveaux d’expression du gène de la
leptine dans les tissus adipeux viscéraux et sous-cutanés après 10 semaines de protocole SBA et
après 10 semaines de REC (figure 33.B). Après 10 semaines de REC, le niveau d’ARNm de la
leptine dans le SCAT est bien normalisé, mais ce tissu adipeux est une source secondaire de
leptine pour l’organisme. Au contraire dans le VAT, principal tissu sécréteur de leptine, le
niveau d’ARNm de la leptine n’atteint que 50% environ de celui des souris CT.
L’adiponectine a également été analysée car les études chez les patients anorexiques ont
montré des résultats contradictoires même si la majorité conclue à une augmentation. Des études
récentes ont montré des effets opposés à ceux de la leptine. Dans notre modèle, l’adiponectine
diminue significativement après 2 semaines de protocole SBA puis tend à se normaliser à 10
semaines. Au cours du protocole REC, les souris SBA et CT ont des taux d’adiponectine
comparables. L’étude des niveaux d’ARNm de l’adiponectine indique une diminution de 50% à
10 semaines dans le VAT, le niveau dans le SCAT étant identique à celui des CT.
94
Figure 33 :Dosage et étude de l’expression des gènes de la leptine et d’adiponectine A : Les taux de leptine et
d’adiponectine sont dosés chez les souris du groupe CT et les souris du groupe SBA à 2 et 10 semaines du
protocole SBA et après 2 et 10 semaines de récupération.B : Les niveaux d’expression de la leptine et de
l’adiponectine dans le SCAT et le VAT vs HPRT et PPIA (les 2 gènes de références).Les données représentent la
moyenne ±SEM ; n=6-10/group. *p<0.05 et **p<0.005 en comparaison avec le groupe CT au même temps du
protocole ; ‡p<0.05 et ‡‡p<0.005 en comparaison aux valeurs antérieures du même groupe.
II-B-6.2 Hormone de croissance GH et Insulin-like growth factor-1 IGF-I Le taux sérique d’IGF-1 dont on connaît l’activité endocrine et paracrine sur le cartilage, le
développement et le remodelage osseux, est effondré chez les patientes anorexiques, du fait de la
dénutrition. Cette chute s’accompagne d’une augmentation de GH plasmatique en absence de
rétrocontrôle négatif par l’IGF-1. Une résistance à la GH contribue également à la diminution de
l’expression du gène de l’IGF-1 au niveau hépatique (266).
Comme la montre la figure 34 ces résultats sont également obtenus chez nos souris SBA. En
effet, après 2 et 10 semaines du protocole SBA le taux de GH est 10 fois supérieur chez les
souris SBA (P<0.005 vs CT). Ce taux très élevé de GH est associé à une diminution significative
95
du taux d’IGF-I de l’ordre de 50%, chez les SBA par rapport aux CT (P<0.05 et P<0.005 après 2
et 10 semaines, respectivement).
Les taux de GH et d’IGF-1 des souris en protocole REC dépassent significativement ceux
des CT après 2 semaines (P<0.005), puis se normalisent après 10 semaines.
Figure 34: Les taux de GH et d’IGF-I sont dosés chez les souris du groupe CT et les souris du groupe SBA à 2 et
10 semaines du protocole SBA et après 2 et 10 semaines de récupération. Les données représentent la moyenne
±SEM ; n=6-10/group. *p<0.05 et **p<0.005 en comparaison avec le groupe CT au même temps du protocole ;
‡p<0.05 et ‡‡p<0.005 en comparaison aux valeurs antérieurs du même groupe.
II-B-7 Tolérance au glucose Dans le contexte du protocole SBA, des données concernant les capacités de capture du
glucose par l’organisme pouvaient participer à la description et la compréhension des
adaptations métaboliques de ces animaux. Des tests de tolérance au glucose ont donc été réalisés
après injection par voie intrapéritonéale, aux différents temps des protocoles SBA et REC. Pour
ne pas induire un choc glycémique trop important chez les souris SBA, la dose de glucose
injectée était de 1mg/kg de poids corporel et non de 2mg/kg comme plus classiquement utilisé.
À la suite des premiers essais, les protocoles courants ont également été modifiés en effectuant
des mesures très tôt après l’injection, afin de ne pas ignorer d’éventuels pics de glycémie trop
brefs pour persister au-delà de 15 minutes.
Après deux semaines du protocole les souris SBA et les souris CT à jeûne affichent des
glycémies basales très proches (figure 35.A). Ce qui laisse supposer que les souris SBA ont
adapté leur métabolisme glucidique pour compenser à la fois la période courte d’accès à
l’alimentation et la période longue sans alimentation disponible. Après injection du glucose le
taux de glycémie n’augmente pratiquement pas chez les souris SBA alors que la dose injectée
aux souris CT entraîne un doublement de la glycémie au temps 15 minutes (P<0.0001). Ces
résultats suggèrent une forte capacité de correction de la glycémie chez les souris SBA après 2
semaines de protocole.
GH
(ng
/ml)
0
5
10
15
20
25
2 10 10+2 10+10
** **
* ‡ ‡ ‡ IG
F-1
(ng/
ml)
0
100
200
300
400
* **
** ‡
‡ ‡
□ CT
■ SBA
Durée du protocole (semaines)
2 10 10+2 10+10
Durée du protocole (semaines)
96
Après 10 semaines de protocole SBA, cette capacité de correction de la glycémie apparaît
moins efficace que lors de la deuxième semaine du protocole SBA mais elle reste supérieure à
celle du groupe CT (P<0.0001) (figure 35.B). Il faut noter qu’à cette période la glycémie basale
des souris SBA est inférieure à celle des souris CT, ce qui pourrait perturber le test. Néanmoins,
une différence significative persiste lorsqu’on corrige les données de chaque groupe par leur
glycémie basale (données non présentées). Les différences entre les mesures à 2 et 10 semaines
de protocole permettent de supposer que l’allongement de la durée du protocole SBA a entraîné
de nouvelles altérations ou adaptations métaboliques.
Le protocole de récupération semble corriger la tolérance au glucose élevée des souris
SBA. En effet, après 2 semaines de récupération les souris CT et SBA ont le même profil
glycémique (figure 35.C). Il faut noter une différence significative qui réapparaît entre les 2
groupes après 10 semaines de protocole REC. Cependant cette différence reste faible et donc sa
significativité biologique discutable.
Figure 35 : Test de tolérance au glucose chez les souris dans des conditions standard (CT) et chez les souris
du groupe SBA à 2 et à 10 semaines du protocole SBA suivi de 2 et de 10 semaines de récupération. A-Mesure
de la glycémie des souris à 2 semaines du protocole SBA. B-Mesure de la glycémie à 10 semaines du protocole
SBA. C-Mesure de la glycémie après 2 semaines de REC. D-Mesure de la glycémie après 10 semaines de Rec. les
données représentent la moyenne ± SEM ; n=6-10/groupe. *p<0.05 et **p<0.0001 Two-way ANOVA et
Bonferroni post-test.
О CT ■ SBA
97
II-B-8 Niveaux d’expression des gènes impliqués dans l’oxydation des acides gras et la lipogenèse dans les tissus adipeux blancs au cours du protocole SBA et les niveaux d’expression des gènes spécifiques du tissu brun
Dans notre étude, on s’est intéressé au métabolisme énergétique au niveau du tissu
adipeux. Afin de déterminer les niveaux d’oxydation et les taux de synthèses de la masse grasse
dans notre modèle SBA.
Dans notre modèle les tissus adipeuxe VAT et SCAT sont prélevés après 10 semaines du
protocole SBA, et les niveaux relatifs d’ARNm de différents gènes d’intérêt et de deux gènes de
références ont été déterminés par la technique PCR en temps réel, des gènes impliqués notament
dans la synthèse de triglycérides (Glut4, FAS) et des gènes impliqués dans la lipolyse comme la
triglycéride lipase acyl (ATGL) et son cofacteur ABDH5/CGI-58 ont ainsi été éudiés.
Les résultats montrent qu’ après 10 semaines du protocole SBA on a une augmentation
significative de l’expression de Glut-4 chez les SBA par rapport aux CT (figure 36.A) (le niveau
d’expression de Glut-4 est 4 fois plus élevé chez les SBA par rapport aux CT) et une
augmentation prononcée du taux d’expression de FAS chez les SBA par rapport aux Ctrls
(figure 36.B) (le niveau d’expression de FAS est 8 fois plus élevé chez les SBA par rapport aux
CT). Nos résultats sont comparables aux résultats d’autres études de restriction calorique chez
les souris qui ont montré une augmentation de la lipogenèse au niveau du tissu adipeux qui est le
site principal de synthèse lipidique.
La lipogenèse est plus prononcée dans le SCAT que dans la VAT c’est ce qu’on a trouvé dans
notre modèle (267).
Chez les souris en restriction calorique on a une élévation spectaculaire de l’oxydation d’acides
gras (267). Les gènes impliqués dans la lipolyse tels que l’Acyl triglycéride lipase (ATGL) et
son cofacteur ABDH5/CGI-58 : l’expression significative de ces gènes a été seulement retrouvé
dans le modèle SBA au niveau du SCAT (figure 36.Cet D).
Le peroxisomal-coenzyme A oxydase (ACOX1) est une enzyme qui intervient dans la bêta
oxydation des acides gras, son taux d’expression est significativement élevé. Et ce taux
d’expression est plus prononcé au niveau du SCAT qu’au niveau VAT.
Les adipocytes bruns possèdent beaucoup de mitochondries et participent activement aux
dépenses énergétiques sous forme de thermogenèse en exprimant UCP-1. L’UCP-1 est une
protéine de 32 kDa présente à l’intérieur de la membrane mitochondriale interne qui permet la
dissipation du gradient électrochimique de protons sous forme de chaleur (268). La régulation
transcriptionnelle des adipocytes bruns est faite par PPARgamma coactivator-1 alpha (PGC-1α)
et le facteur transcriptionnelle de la lignée des adipocytes bruns Prdm 16 (couplé à C/EBPβ) qui
jouent un rôle très important dans l’adipogenèse du tissu adipeux brun (BAT) (269).
98
L’étude de l’expression de ces gènes au niveau du tissu adipeux blanc montrent que les gènes
Prdm6 et de PGC1a sont significativement exprimés au niveau des tissus adipeux sous cutané et
viscéraux des souris après 10 semaines du protocole SBA (figure 37.A et 37.B).
Seul le SCAT exprime significativement l’UCP1 25 fois au niveau SCAT des souris SBA par
rapport aux CT (figure 37.C).
Alors, les gènes du phénotype brun sont régulés à la hausse dans le tissu adipeux blanc et qui
pourrait être due à l’augmentation des dépenses énergétique chez les souris SBA.
Figure 36 : L’expression des gènes qui interviennent dans le métabolisme lipidique. Les niveaux d’expression
de Glut4, FAS, ABHD et ATGL sont déterminés par PCRq en temps réel au niveau du tissu adipeux sous-cutané
(SCAT) et du tissu adipeux viscéral (VAT) des souris CT et des souris SBA. Les gènes PPIA et HPRT sont utilisés
comme des gènes de références. Les données représentent la moyenne ± SEM n=5-10/group. *p<0.05 et **p<0.005
en comparaison avec le groupe CT à la même durée du protocole ; ‡p<0.05 et ‡‡p<0.005 en comparaison aux
résultats antérieurs
99
Figure 37 : L’expression des gènes du phénotype des adipocytes bruns. L’expression d’UCP1, PGC1a, PRDM6
est déterminée par PCRq en temps réel au niveau du tissu adipeux sous-cutané (SCAT) et du tissu adipeux viscéral
(VAT) des souris CT et des souris SBA. Les gènes PPIA et HPRT sont utilisés comme des gènes de références. Les
données représentent la moyenne ± SEM n=5-10/groupe. *p<0.05 et **p<0.005 en comparaison avec le groupe CT
à la même durée du protocole ; ‡p<0.05 et ‡‡p<0.005 en comparaison aux résultats antérieurs du même groupe.
II-B-9 Étude de l’expression du récepteur bêta 3 adrénergique au niveau du VAT, SCAT et BAT Les récepteurs bêta 3 adrénergiques (b3AdR) sont localisés principalement sur les
adipocytes, l’activation de ces récepteurs stimule la lipolyse (101). Des études ont montré que
l’exposition au froid ou la stimulation pharmacologique du récepteur bêta 3 adrénergique induit
le phénotype brun ou l’aspect « brite » au sein du tissu adipeux blanc.
Dans notre modèle SBA, on a voulu étudier le niveau d’expression du gène b3AdR impliqué
dans l’expression du phénotype brun au sein du tissu blanc « le britening » et au niveau du BAT.
Les résultats montrent que l’expression des récepteurs b3AdR au niveau du VAT et du SCAT
100
diminue significativement après 10 semaines du protocole SBA et se corrige en phase de REC.
(figure 38.A). Par contre, au niveau BAT la diminution de l’expression des récepteurs b3AdR
persiste après REC (figure 38.B).
D’après ces résultats on peut déduire que la restriction calorique dans notre modèle SBA ne peut
pas stimuler l’expression génique des récepteurs b3AdR, un parmi d’autres facteurs,
responsable de l’induction du phénotype brun au sein des tissus SCAT et VAT.
Figure 38 : L’expression des récepteurs β3AdR au niveau des tissus BAT, VAT et SCAT. L’expression de ces
récepteurs au niveau de ces 3 tissus est déterminée par PCRq en temps réel chez les souris SBA et CT à 10 semaines
du protocole SBA et 10 semaines de Rec. Les gènes PPIA et HPRT sont utilisés comme des gènes de références.
Les données représentent la moyenne ± SEM n=5-10/groupe. *p<0.05 et **p<0.005 en comparaison avec le groupe
CT à la même durée du protocole ; ‡p<0.05 et ‡‡p<0.005 en comparaison aux résultats antérieurs.
II-B-10 Etude histologique de l’adiposité médullaire L’adiposité médullaire est définie comme la densité d’adipocytes dans la moelle osseuse.
L’ostéoporose induite par le vieillissement à la période post-ménopausique ou même à l’AM est
associée à une augmentation de cette adiposité médullaire, la même chose a été décrite dans des
modèles de restriction alimentaire. Afin de déterminer si celà était également le cas dans notre
modèle nous avons réalisé une étude histologique. Le comptage des adipocytes médullaire a été
effectué en tibia proximal, dans une zone de 1mm au-dessous de la plaque de croissance. Les
résultats sont exprimés en nombre d’adipocytes par mm2.
D’après ce graphique on peut comprendre que le modèle SBA même si il a réussi à reproduire
les diffèrentes perturbations observées dans les modèles murins de restriction n’a pas pu montrer
une augmentation de l’adiposité médullaire (figure 39).
10 10+10 10 10+100.0
0.5
1.0
1.5
*
**
Exp
ress
ion
rela
tive
dem
b3A
dR
*
SCAT VAT10 10+10
0
1
2
3
*
**
Exp
ress
ion
rela
tive
dem
b3A
dRBAT
‡
A B
101
Figure 39 : nombre d’adipocytes médullaires/mm2/ par secteur à différents temps du protocole A- à 2 semaines SBA ; B-10 semaines SBA ; C- après 2 semaines de REC ; D- Après 10 semaines de REC
102
Discussion Nous avons choisi de présenter la discussion de nos travaux en quatre parties principales. Nous
commencerons par une analyse intégrée du modèle SBA, c’est-à-dire une analyse des résultats
dans leur ensemble et reliés entre eux. Ceci permettra de tirer les premières conclusions sur ce
modèle. Puis, nous comparerons les résultats obtenus avec ceux de la littérature qui concernent
d’autres modèles utilisés pour mimer certains aspects de l’AM. Ceci permettra de situer le
modèle SBA par rapport aux autres. Ensuite, nous comparerons autant que possible les résultats
obtenus avec ce qui est décrit chez les patientes anorexiques, afin de dégager de ces
comparaisons les aspects pathologiques qui sont mimés et étudiables dans le modèle. Pour finir,
nous évoquerons les questions soulevées par les résultats obtenus et les perspectives d’études sur
ce modèle.
IV-A Analyse intégrée du modèle SBA L’anorexie mentale est une maladie humaine peu étudiée. L’étude des facteurs et des
mécanismes qui interviennent dans le développement de cette maladie est limitée chez les
patientes. Pour mieux comprendre l’AM et les mécanismes impliqués dans la physiopathologie
osseuse de cette maladie nous avons choisi de développer un modèle animal qui associe la
restriction du temps d’accès à l’alimentation à un stress chronique. Nous avons appelé ce modèle
Separation-Based Anorexia (SBA) en référence au modèle déjà décrit basé sur l’activité Activity-
Based Anorexia (ABA). Nous avons tout d’abord réalisé des essais préliminaires pour déterminer
le niveau et la vitesse de perte de poids qui étaient supportables pour les souris. Une fois le
protocole validé, une étude à court terme (deux semaines) a montré clairement que la
combinaison de la séparation et la restriction calorique est nécessaire pour obtenir une perte de
poids rapide et importante. Elle a aussi permis de constater que deux semaines de protocole ne
permettaient pas d’induire d’altérations osseuses détectables et significatives. Ce constat était
cependant attendu dans la mesure où les travaux sur les conséquences osseuses de la restriction
alimentaire ou même de l’ovariectomie sont classiquement réalisés sur au moins un mois. Le
modèle SBA est utilisé pour mimer les aspects de dénutrition et de stress chronique observés
chez les patientes anorexiques. Comme tout modèle, il présente certaines limites, en premier lieu,
la restriction alimentaire n’est pas volontaire mais elle est imposée par l’expérimentateur, ce qui
ne pose pas de problème pour reproduire les conséquences physiopatologiques de la maladie. Par
ailleurs la physiologie de la souris présente bien entendu des différences importantes avec
physiologie humaine.
103
Lorsqu’on étudie le protocole SBA à court terme, on constate qu’au bout de deux semaines,
les souris SBA ont perdu 25 % de leur poids initial. C’est le seul groupe qui perd autant de poids,
malgré une prise alimentaire proche des souris en simple restriction du temps d’accès à
l’alimentation (TR). Il est intéressant de noter que les souris uniquement séparées (SEP) se
nourrissent plus que les CT, mais que leur poids corporel a tendance à se maintenir sensiblement
en dessous de celui des CT. Ces constatations appellent plusieurs remarques. D’une part, ils
suggèrent que la séparation seule entraine déjà des perturbations qui aboutissent à une
augmentation des dépenses énergétiques. Cette augmentation pourrait être due à des besoins
accrus de production de chaleur, nécessaires pour assurer le maintien de la température
corporelle d’un animal isolé. D’autre part, le stress chronique induit par la séparation peut aussi
engendrer une augmentation des dépenses énergétiques. On peut également supposer que le
mode d’alimentation imposé induise par lui-même des modifications métaboliques entrainant des
dépenses énergétiques plus élevées (181). Enfin, on ne peut pas exclure la possibilité d’une
moins bonne absorption des nutriments à cause de ce régime. Ces différentes hypothèses ne sont
bien entendues pas exclusives.
Au cours du protocole SBA long, le poids bas obtenu après deux semaines est à peu près
stabilisé par la suite. Néanmoins cette stabilité a été obtenue en modifiant légèrement le temps
d’accès à l’alimentation au cours du protocole. En effet, dans plusieurs essais, nous avons dû
descendre provisoirement en dessous des deux heures quotidiennes d’alimentation afin d’éviter
une légère reprise de poids corporel. Des résultats similaires ont été observés après 35 jours dans
le modèle d’activité associée à une restriction quantitative de la nourriture, développé par M.
Méquinion (travaux soumis à Am J Physiol, juillet 2014) (261). Cela suppose qu’avec le temps
une adaptation nouvelle permet une économie d’énergie suffisante pour se traduire par une
modeste reprise de poids.
La quantité de nourriture ingérée chez les souris du groupe SBA est proche de celle ingérée par
les souris CT nourries ad libitum. La restriction du temps d’accès à la nourriture permet aux
souris de recevoir le même apport énergetique (Kcal) avec une baisse du poids corporel par
rapport aux CT. Une fois encore cela suggère une augmentation importante des dépenses
énergetiques (253).
Cette perte importante du poids est associée à une fonte de la masse grasse, avec notamment une
quasi disparition du tissu adipeux périutérin. Néanmoins, les extractions d’ARN à partir de ce
tissu adipeux permettent d’obtenir à peu près autant d’ARN qu’à partir du même tissu prélevé
chez les souris CT. Cela laisse supposer qu’il y a une réduction drastique des réserves lipidiques
du tissu adipeux périutérin, mais que les adipocytes sont toujours présents et actifs. Cette
104
remarque est à mettre en relation avec la reprise très rapide de poids et notamment de réserves
adipeuses lors de la phase de récupération.
Cette perte de poids en phase SBA s’accompagne aussi d’une diminution de la masse maigre,
c’est-à-dire de tout ce qui n’est ni adipeux, ni minéral. Parmi cette masse maigre on retrouve
donc la masse musculaire qui est effectivement diminuée d’après les pesées effectuées sur le
triceps postérieur. L’amplitude de cette diminution de la masse musculaire (environ 50% après
dix semaines, par rapport au temps zéro) permet de supposer qu’une protéolyse et une
conversion des acides aminés en sucres a eu lieu. Selon ce critère, les souris seraient donc dans
ce qui est décrit comme la phase III d’adaptation au jeûne qui conduit au décès, malgré leur
alimentation.
Chez les souris SBA, la stagnation de la masse minérale osseuse du corps entier après deux
semaines suggère un blocage de l’acquisition, plus qu’une perte, alors que les souris CT
poursuivent une acquisition lente. Cette stagnation peut être due à une diminution de la formatin,
qui s’alignerait alors sur la résorption, inchangée. Elle peut aussi être le résultat d’une baisse
conjointe de la formation et de la résorption, aboutissant à une diminution globale du remodelage
osseux.
Différentes études ont décrit au moins partiellement les adaptations métaboliques induites par la
restriction calorique. Il a ainsi été montré qu’il pouvait y avoir une forme « d’emballement
métabolique » qui correspondait à une augmentation de la lipogenèse et de la lipolyse (267),
accompagnée d’une augmentation de la biogenèse mitochondriale (270,271) aboutissant à une
augmentation des dépenses énergétiques nécessaires à ces processus. Pour initier l’étude des
adaptations métaboliques induites par le protocole SBA, nous avons déterminé les niveaux
d’ARNm des gènes impliqués dans le métabolisme au niveau du tissu adipeux sous cutanée et
viscéral. L’augmentation importante des ARNm de gènes potentiellement reliés à la lipogenèse
dans les adipocytes (Glut4, FAS) et des gènes potentiellement reliés à la lipolyse (ATGL,
ABHD5) suggère que dans le modèle SBA « l’emballement métabolique » est présent et pourriat
participer à l’augmentation des besoins énergétiques. Les gènes liés à la lipolyse présentent des
altérations de moindre amplitude probablement à cause de leur régulation qui est principalement
post-traductionnelle (272).
Comme les souris SBA sont isolées et stressées, nous avons supposé qu’elles pouvaient
présenter une augmentation de leur production de chaleur. Nous avons donc déterminé les
niveaux relatifs d’ARNm de gènes impliqués dans la régulation transcriptionnelle des adipocytes
bruns au niveau des tissus adipeux sous-cutanés, viscéraux et bruns. Nous avons ainsi montré
que les taux d’expression d’UCP-1, PGC1α et Prdm16 étaient quasiment inchangés dans le
tissus adipeux brun, mais qu’ils étaient tous fortement augmentés dans le SCAT des souris SBA.
105
Ces résultats suggèrent l’émergence du tissu adipeux brun au sein du tissu adipeux blanc sous-
cutané et l’augmentation de la thermogenèse. Ce type d’évolution du SCAT a déjà été montré
dans d’autres modèles, mais sans relation avec la restriction alimentaire. Il s’agissait de modèles
d’exposition au froid ou de traitement par des agonistes des récepteurs bêta 3 adrénergiques
(273,274). Il serait intéressant de déterminer les mécanismes impliqués dans le changement de
phénotype adipocytes blancs/adipocytes bruns dans notre modèle. Dans la littérature, plusieurs
hypothèses co-existent quant à l’origine de ces adipocytes. Klaus et al, suggèrent que les
adipocytes bruns apparaissant au sein d’un dépôt de tissu adipeux blanc proviendraient d’un pool
d’adipocytes bruns, déjà présents dans le dépôt graisseux, et qui sous certaines conditions,
proliféreraient de manière plus ou moins importante (275). Une étude in vivo a montré que le
phénotype adipocytes blancs/adipocytes bruns « brite » diminue significativement avec l’âge et
de façon indépendante de la masse corporelle chez des souris mâles C57 BL/6 (276).
L’anorexie mentale est associée à des perturbations endocriniennes majeures. Ces perturbations
participent aux adaptations métaboliques permettant le maintien des fonctions vitales. Parmi ces
perturbations, certaines auraient des effets délétères sur la masse osseuse. Nous avons donc dosé
différentes hormones dans la circulation sanguine, afin de déterminer quelles altérations étaient
présentes dans le modèle SBA.
En relation avec l’étude des tissus adipeux, nous avons dosé deux adipokines, la leptine et
l’adiponectine. La leptine est une hormone principalement secrétée par le tissu adipeux. Le taux
de leptine circulante informe notamment le cerveau de la quantité de réserves adipeuses, ce qui
se traduit par un effet anorexigène initié au niveau de l’hypothalamus. La leptine est aussi
responsable d’une augmentation des dépenses énergétiques. Un taux minimum de leptine
circulante est nécessaire au maintien des fonctions liées à la reproduction. Enfin, la leptine
participe de façon complexe à la régulation de la masse osseuse. La fonte de la masse grasse au
cours du protocole SBA a entrainé une réduction importante du taux de leptine sérique. Dans le
modèle SBA, l’hypoleptinémie importante que nous avons observée, pourrait donc être
impliquée dans les perturbations des fonctions reproductrices et de l’acquisition de masse
osseuse. Concernant la masse osseuse, beaucoup d’études ont montré que la leptine est
impliquée dans la régulation de la formation osseuse via deux voies alternatives, la voie directe,
qui stimulerait la formation osseuse et la voie indirecte qui impacterait négativement sur la
formation osseuse. In vitro, la leptine favorise la différenciation des CSM en ostéoblastes au
détrimant de la voie adipocytaire. Si le débat est encore ouvert dans la littérature concernant la
voie principale de l’action de la leptine sur la physiologie osseuse de souris élevées en
conditions standards (107), il est encore inexistant pour les modèles de restriction alimentaire.
Seul un article récent (117) a montré chez la souris qu’un jeûne de courte durée entraînait une
106
augmentation de la perméabilité de la barrière hémato-encéphalique et donc potentiellement une
entrée facilitée de la leptine dans le cerveau. Si ce phénomène était retrouvé chez les souris
SBA, il faudrait reconsidérer le poids des effets centraux potentiels de la leptine, malgré sa faible
concentration dans la circulation sanguine.
La seconde adipokine que nous avons étudiée est l’adiponectine. Ses mulitples fonctions sur le
métabolisme énergétique et la formation osseuse notamment sont en train d’être décrites.
Généralement la restriction calorique altère l’expression génique des adipocytokines au niveau
du tissu adipeux blanc, particulèrement chez les rongeurs dans lesquels la restriction calorique
stimule l’expression génique de PPAR α et par conséquent entraine une hyperadiponectinemie
(187–191). Dans le modèle SBA le taux d’adiponectine sérique reste plus ou moins stable tout
au long du protocole. Ces résultats ne vont pas dans le même sens que ceux de plusieurs études
qui associent l’hyperadiponectinemie à la restriction calorique. Une explication possible est que
notre modèle est trop sévère en termes de perte de poids et de masse grasse pour permettre
l’expression de cette adipokine. Cela pourrait être vérifié au cours d’une étude ultérieure
comprenant un protocole moins drastique. On peut noter néanmoins que les remarques
concernant la barrière hémato-encéphalique et la leptine peuvent être appliquées à
l’adiponectine, dont il a avait été établi jusqu’à récemment qu’elle ne franchissait pas cette
barrière (137).
L’axe GH/IGF-1, en conditions physiologiques, régule la masse osseuse en stimulant la
formation. Cet axe a été décrit comme étant perturbé dans différents contextes, notamment ceux
de l’anorexie mentale et de la restriction calorique. Les dosages sériques de la GH et de l’IGF-1
effectués chez nos souris SBA montrent une réduction importante du taux d’IGF-1 malgré une
concentration élevée de GH. Ces résultats suggèrent une résistance à la GH au niveau du foie.
(277). L’importance d’IGF-1 pour la formation osseuse permet de penser que sa diminution
importante est en partie impliquée dans les conséquences osseuses observées dans le modèle
SBA. Localement, la GH agit aussi au niveau de l’os en stimulant la production intraosseuse
d’IGF-I. L’IGF-1 osseux est aussi régulé par la PTH, l’oestradiol, la testosterone. L’importance
de l’IGF-1 osseux pour la formation a été démontrée par une série de travaux complémentaires
(168,169,278). Dans notre modèle, la forte perturbation des fonctions de reproduction nous
laisse supposer que les taux d’hormones sexuelles sont réduits, le taux IGF-1 circulant est bas,
mais le taux de GH très élevé. Il serait donc utile pour la compréhension des mécanismes
impactant la physiologie osseuse des souris SBA, de déterminer s’il y a également une forme de
résistance osseuse à la GH.
Le modèle SBA a été développé sur du long terme notamment pour faire apparaître de
potentielles conséquences osseuses. Les mesures densitométriques ont révélé un arrêt
107
d’acquisition de la masse osseuse au niveau du corps entier, du fémur et des vertèbres. Ces
résultats permettent de penser que le protocole SBA impacte négativement à la fois le squelette
axial et le squelette appendiculaire. Les études microarchitecturales par microtomographie ont
montré une forte diminution de la densité et de l’épaisseur des travées en tibia proximal. Elles
ont aussi mis en évidence une diminution de la densité volumique de l’os cortical en diaphyse
tibiale. Il apparait donc que le protocole SBA entraine des répercutions négatives sur la masse et
l’architecture osseuses, donc probablement sur les propriétés mécaniques.
La question principale sur laquelle travaille le laboratoire est la détermination de l’implication
des adipocytes médulaires ou de l’augmentation de l’adiposité médullaire dans la physiologie
osseuse. Après avoir démontré qu’il existait des altérations osseuses significatives, nous avons
cherché à déterminer par une approche histologique, si ces altérations étaient accompagnées
d’une augmentation de la densité des adipocytes médullaires (nombre d’adipocytes / mm2 de
moelle). La première étude que nous avons menée à ce sujet, au niveau du tibia proximal, n’a
pas permis de mettre en évidence de différences significatives entre les deux groupes SBA et
CT. A ce stade de l’étude nous avons supposé que la sévérité du protocole SBA que nous avons
appliqué ne permettait pas de voir une augmentation de l’adiposité médullaire. Nous avons
appuyé cette hypothèse sur ce qui est à notre connaissance la seule étude histologique réalisée
sur des biopsies osseuses de patients anorexiques qui a montré que l’adiposité médullaire chez
les patientes anorexiques n’était coorélée qu’à la perte de poids (213). Afin d’étayer cette
hypothèse, une nouvelle étude est en cours, incluant des protocoles différant par la perte de poids
induite.
En phase de récupération, les souris ayant subi dix semaines de protocole SBA sont ensuite
placées dans des cages classiques avec nourriture à volonté. Les souris récupèrent rapidement du
poids corporel, jusqu’à ce que celui-ci rejoigne celui des souris CT de même âge. Cette
récupération est accompagnée d’une hyperphagie qui dure jusqu’à ce que la courbe
d’alimentation cumulée des souris en récupération se superpose à celle des souris CT. Cette
durée n’est que de l’ordre de quatre à cinq jours. Après deux semaines de récupération, les souris
ont une masse grasse au-dessus de celle des CT et une masse maigre normalisée. La masse
osseuse reste alors basse et sera à son tour normalisée avant les dix semaines de récupération.
L’étude microarchitecturale des tibias de ces souris montre que la normalisation est également
qualitative. Cette récupération osseuse peut en grande partie être expliquée par le fait que la
quasi totalité des perturbations décrites pendant la phase SBA semble corrigée pendant la phase
SBA. Etonnamment, l’hypoleptinémie intimement liée au niveau des réserves adipeuses n’est
pas corrigée même après les dix semaines de protocole de récupération. Ce résultat a été
108
confirmé par des niveaux bas d’expression du gène de la leptine dans les tissus adipeux. Cette
exception laisse penser que d’autres facteurs que nous n’avons pas encore étudiés ont également
été perturbés de façon durable, si ce n’est définitif. Probablement à cause de la complexité du
phénomène, les facteurs participant à la régulation de la production de leptine par les tissus
adipeux sont encore très discutés (279,280). Néanmoins, plusieurs explications à cette
hypoletinémie persistante sont envisageables. D’une part, le tonus adrénergique qui contrôle en
partie les activités des tissus adipeux, et la sensibilité des tissus adipeux, n’ont pas été évalués.
En effet, les niveaux d’ARNm des récepteurs bêta 3 adrénergiques n’ont été évalués qu’à titre
indicatif, car la sensibilité des tissus aux catécholamines est principalement régulée par un jeu
d’internalisation et d’externalisation des récepteurs. D’autre part, des modifications de type
épigénétique ont pu se produire au cours du protocole long et limiter durablement l’expression
du gène de la leptine.
Concernant la masse osseuse, la période de récupération a permis une normalisation des
paramètres densotimétriques et architecturaux mesurés. On peut supposer que la normalisation
de l’axe GH/IGF-I et des fonctions de reproductions (donc des hormones associées), couplée à
une alimentation « normale », représente des conditions favorables pour la formation osseuse.
Pour une réflexion plus complète sur les mécanismes de récupération, il est cependant
intéressant de se demander si l’hypoleptinémie persistante ne joue pas également un rôle. En
effet, elle pourrait contribuer à limiter les dépenses énergétiques pendant la phase de
récupération et donc favoriser la normalisation de différents paramètres. Elle pourrait également
réduire le contrôle négatif de la masse osseuse exercé par la leptine via le système nerveux
central et le système symphatique. Par contre, il faut également noter que cette hypoleptinémie
n’empêche pas la récupération progressive des fonctions de reproduction.
IV-B Comparaison du modèle SBA avec les autres modèles
Le modèle SBA représente un nouveau modèle développé au PMOI à partir de deux articles
relativement anciens et ne décrivant que très partiellement le modèle d’origine. Ce modèle a été
choisi et développé afin de dépasser les limites des modèles existant que sont notamment la
restriction ou la perte de poids modérée et dans d’autres cas, la durée limitée des protocoles. En
effet, le modèle SBA que nous avons validé présente l’avantage d’induire une perte de poids
rapide et importante tout en permettant une longue durée de protocole puis une phase de
récupération.
109
Lorsque les résultats du modèle SBA sont comparés à ceux du modèle de référence dans la
littérature, c’est-à-dire à la restriction alimentaire, on constate de nombreuses similitudes.
En effet, plusieurs études ont démontré l’impact osseux de la restriction calorique. Bien que les
paramètres mesurés ou les conditions dans lesquelles ont été faites les mesures ne soient pas
strictement comparables, il apparait que les altérations osseuses décrites dans la littérature sont
globalement du même ordre et de la même ampleur que celles décrites dans notre étude
(244,245,281). Seule l’équipe de Hamrick décrit une absence d’effet de la restriction sur le
contenu minéral osseux du corps entier associée à un effet négatif sur l’os cortical (fémoral et
vertébral), mais aussi à un effet positif sur l’os trabéculaire vertébral (sans effet sur l’os
trabéculaire fémoral) (282). Ce type de résultat n’a pas été retrouvé dans d’autres études de la
littérature. D’après nos recherches bibliographiques, les capacités de récupération après
restriction longue n’ont fait étudiées que dans les travaux de Tatsumi et al. (245). Cette
récupération n’a été déterminée que pour la densité osseuse volumique en tibia proximal. Il en
ressort que 6 mois de restriction entraînent une diminution de 40% et que cette diminution est
ramenée à 20% après 1 mois d’alimentation à volonté. Ce début d’étude de récupération osseuse,
laissait déjà entrevoir les capacités de rétablissment importantes des souris soumises pendant de
nombreuses semaines à une restriction alimentaire, comme nous l’avons présentement montré
pour de nombreux paramètres.
Les altérations de l’axe GH/IGF-1 ne sont pas sytématiquement évaluées dans les études de
restriction alimentaire à long terme. Dans les travaux de Devlin et al, et de Tatsumi et al, la
diminution du taux d’IGF-1 plasmatique est de l’ordre de 33% (244,245). L’étude de Hamrick et
al. est la seule à présenter une chute de 80% du taux d’IGF-1 par rapport aux animaux contrôles
(282). Nous avons obtenu dans le modèle SBA une diminution de l’ordre de 50%, ce qui semble
correspondre à un niveau intermédiaire par rapport aux autres études. Il est donc difficile de
conclure à une différence ou une similitude entre le modèle SBA et les modèles de restriction
alimentaire à propos du niveau de perturbation de l’axe GH/IGF-1. Par contre, il est évident que
tous ces modèles ont en commun une altération de cet axe. C’est aussi le cas d’études de la
restriction sévère à court terme qui ont mis en évidence une potentielle résistance hépatique à la
GH se traduisant par une augmentation importante du taux de GH est une diminution du taux
d’IGF-1 circulant (283,284). Comme nous l’avons montré dans notre étude, dès deux semaines
de protocole SBA.
Les altérations des fonctions reproductrices par la restriction alimentaire ont été décrites dans de
nombreuses publications. Dès 1978, Zamiri a montré chez la souris que soixante jours
d’alimentation avec 55% de la ration consommée à volonté permettaient de perturber le cycle
110
oestral. Il a également remarqué que des rations moindres (70% de ce qui était consommé à
volonté) n’entraînaient pas les mêmes perturbations (285). Par la suite, Seki et al ont montré
chez la rate qu’une restriction alimentaire de 45% (55% consommés) entraînait dès la septième
semaine un arrêt ou un allongement important du cycle estral chez la plupart des individus (286).
Ce qui n’était pas le cas des régimes à 30 ou 15% de restriction. Enfin, dans une étude complète
chez la rate, Tropp et al. ont conclu que l’interruption du cycle estral était dépendante à la fois de
la vitesse et du pourcentage de perte de poids corporel (246). Les altérations des fonctions
reproductrices observées dans le modèle SBA sont donc communes aux modèles de restriction
alimentaire sévère, malgré une alimentation proche de celle des souris nourries à volonté.
Les adaptations métaboliques que nous avons décrites dans le modèle SBA ont pour la plupart
été déjà décrites de façon éparse dans des modèles de restriction alimentaire. L’originalité du
modèle SBA concernant ces adaptations vient en partie du fait qu’elles se produisent chez des
souris ayant une alimentation proche de celle des souris contrôles. Quelques particularités sont
toutefois soulignées dans la partie ci-dessous.
Le modèle SBA présente aussi des caractéristiques originales ou peu répandues parmi les
autres modèles.
La première est l’évolution du poids corporel. En effet, Devlin et al, ont montré qu’une
restriction alimentaire de 30 % chez des mâles de 3 semaines entraîne un ralentissement de la
prise de poids pendant les trois premières semaines de protocole, puis une stabilisation du poids
pendant les six semaines suivantes (244). Chez des souris mâles agées de trois mois, une
restriction alimentaire équivalant à 40 % et appliquée pendant un an n’entraîne qu’un
ralentissement de la prise de poids, puis une stagnation (245). Il n’y a donc pas de perte de poids
corporel dans ces deux modèles. Chez des rates âgées de douze semaines, une restriction de 40
% entraîne une perte de poids progressive, qui atteint environ 4% du poids initial en deux
semaines, moins de 10 % en quatre semaines et 20 % du poids initial au bout de douze semaines
de protocole (281). Il apparait clairement que dans ces études l’évolution du poids corporel est
différente de celle observée dans le modèle SBA. Hamrick et al. ont montré chez des souris
mâles de 14 semaines, qu’une restriction calorique de 10 semaines dont les 8 dernières semaines
à 40% de restriction entraînait une différence finale de poids de 33% ainsi qu’une diminution
importante des masses grasse et maigre et une chute des taux d’IGF-1, de leptine (-80 % et -90
% respectivement) (282).
Il est intéressant de noter également que dans l’étude de Baeck et al, l’adiposité médullaire est
doublée au niveau du fémur proximal (281) et que dans celle de Devlin et al. elle est multipliée
111
par 8 en fémur distal après 12 semaines (244). Par contre dans le modèle plus sévère de Hamrick
et al, l’adiposité médullaire est nulle aussi bien en site fémoral que vertébral (282) Il est alors
logique de supposer que l’augmentation de l’adiposité médullaire n’est induite par la restriction
alimentaire que lorsque celle-ci entraîne une altération du poids corporel qui reste modérée ou
lente, ce qui au regard de la littérature n’est pas le cas du modèle SBA.
Le modèle SBA est à notre connaissance le seul modèle de restriction dans lequel l’apparition
des caractéristiques de tissu adipeux brun dans le tissu adipeux blanc sous-cutané ait été décrite.
On ne peut cependant pas affirmer que cela ne se produit pas dans d’autres modèles de
restriction, car il semble que cela n’y est pas été étudié. Il n’y a que dans des modèles comme
l’exposition au froid ou le traitement par des agonistes bêta-3-adrénergiques que ce phénomène a
été décrit. Les résultats obtenus dans ces deux types de modèle nous conduisent à nous
demander si le tonus sympathique, et/ou l’exposition au froid simulée par l’isolement des
animaux pourraient être impliqués dans les altérations observées dans notre modèle. Répondre à
cette question nécessiterait d’autres types d’expériences qui n’ont pu être entreprises dans le
cadre de ces travaux de thèse.
La correction rapide de presque tous les paramètres altérés semble ne jamais avoir été décrite
dans la littérature. Comme cela a été discuté précédemment, seule l’étude de Tatsumi et al. avait
déjà permis d’entrevoir l’importante capacité de récupération osseuse des souris après une
longue période de restriction alimentaire (245). Par ailleurs, l’étude de Tropp et al. a montré que
les rates retrouvaient un cycle estral normal à partir du moment où leur poids corporel avait
rejoint celui des animaux nourris à volonté (246).
Enfin, la longue phase de récupération a aussi permis de révéler une hypoleptinémie persistante,
malgré la reconstitution rapide et totale des réserves adipeuses. Ceci n’a jamais été décrit dans la
littérature, mais comme peu d’études ont été effectuées avec des phases longues de restriction
puis de récupération nous ne pouvons pas affirmer que cette hypoleptinémie persistante n’existe
pas aussi dans d’autres modèles.
En conclusion, la comparaison de nos résultats avec les modèles de restriction alimentaire de la
littérature met en évidence de nombreuses similitudes qui permettent de valider le modèle
comme un modèle mimant des altérations associées à des restrictions alimentaires sévères. Cette
comparaison permet aussi de conclure que le modèle SBA présente des particularités, notamment
une perte de poids rapide et importante, un « brunissement » réversible du tissu adipeux sous-
cutané, et une hypoleptinémie persistante. La recherche de particularités éventuelles au niveau
des mécanismes responsables des altérations osseuses reste à développer. Ces particularités
112
pourraient notamment provenir de l’absence d’inflammation qui n’est aujourd’hui qu’une
hypothèse fondée sur l’analogie avec ce qui est décrit pour l’anorexie mentale.
IV-C Comparaison des altérations observées avec celles décrites chez les patientes
Notre modèle SBA semble mimer de nombreux aspects physiopathologiques de la maladie. La
perte importante de la masse corporelle (25%), la fonte de la masse grasse et la baisse de la
masse maigre observées dans le modele SBA sont comparables à celles décrites chez les
patientes anorexiques (86,88,174).
Les études réalisées après rémission montrent que la récupération osseuse chez les patientes est
seulement de 1 à 3% par an (88), autrement dit cette récupération reste dans tous les cas très
partielle. Ces résultats représentent des différences majeures entre d’un côté le modèle SBA et
les modèles de restriction alimentaire, et de l’autre les patientes.
A partir de ce constat on peut proposer une stratégie consistant à rechercher les différences
d’altération qui existent entre le modèle et la pathologie, pour essayer d’expliquer les
diffénrences de capacité de récupération. Ceci nous amène à deux autres différences majeures.
D’une part, les souris n’ont jamais perdu leur appétit et leur capacité à s’alimenter voire se
suralimenter. Cette capacité est probablement décisive pour leur permettre de récupérer très
rapidement leur poids corporel, leurs masses grasse et maigre et leur équilibre hormonal et
métabolique. D’autre part, l’hypoleptinémie persistante malgré une masse grasse et de très
nombreux autres paramètres normalisés ne se produit pas chez les patientes, dans la mesure où
les patientes ne récupèrent que très partiellement leur poids corporel, leur masse grasse et donc
leur leptinémie(287–290).
D’après ce qui précéde on peut considérer que le modèle SBA mis au point par le PMOI
représente un modèle murin prometteur pour l’étude des conséquences physiophatologiques de
l’anorexie mentale. L’évolution des nombreuses altérations après une longue période de
restriction a été très peu étudiée chez les rongeurs femelles ce qui renforce l’intérêt et
l’originalité de notre étude. Mais il reste beaucoup des travaux à réaliser afin d’élucider les
mécanismes d’adaptation et de normalisation tout au long des deux phases SBA et REC.
113
IV-D Perspectives d’étude du modèle SBA À court terme les travaux sur le modèle SBA devraient être focalisés sur l’étude approfondie de
la microarchitecture osseuse au niveau du tibia et des lombaires aux différents temps. Pour
aborder la physiologie osseuse de ces souris il faudra ensuite évaluer les concentrations des
marqueurs de formation et de résorption osseuse. L’étude de l’adiposité médullaire en fonction
de la sévérité du protocole a été initiée à la fin du projet de thèse, elle devra donc être poursuivie
afin de clarifier ces relations et afin de pouvoir positionner le modèle SBA dans les principales
hypothèses et la stratégie du laboratoire.
Il a été démontré que l’IGF-1 produit en intra- osseux jouait un rôle important dans la formation
osseuse. Il sera utile pour la compréhension des mécanismes aboutissant au blocage de
l’acquisition de la masse osseuse, de déterminer si la résistance hépatique à la GH qui semble
être présente dans le modèle SBA est également développée au niveau osseux.
L’hypoleptinémie persistante est un aspect qui suscite de nombreuses questions qui elles-mêmes
nécessiteraient plusieurs études pour y répondre. D’une part, il serait utile d’identifier les
altérations durables qui entraînent une sous-production de leptine par les tissus adipeux des
animaux après récupération. D’autre part, étant ses différents rôles et ses relations majeures avec
le système nerveux central, il serait aussi utile de déterminer comment le cervea u intègre cette
hypoleptinémie persistante. En effet, la stabilisation rapide de la prise alimentaire et des réserves
lipidiques pendant la phase de récupération laisse supposer qu’il y a eu une recalibration de la
sensibilité du cerveau à la leptine, ce qui lui permettrait d’interpréter les taux bas de leptine
comme un message signifiant que les réserves adipeuses sont normales.
114
Publication Mes activités de recherche développées au cours de la these m’ont permis de participer à plusieurs publications. A la fin de la thèse, une publication ortait directement sur mon sujet de thèse. Cette publication est présentée ci-dessous, sans les figures qui sont déjà rpésentées dans la partie « résultat » du travail de thèse.
“Long –Term Physiological Alterations and Recovery in a Mouse Model of Separation Associated with time-Restricted Feeding: A Tool to Study Anorexia Nervosa Related Consequences “ Sara Zgheib, Mathieu Méquinion, Stéphanie Lucas, Damien Leterme, Olfa Ghali, Virginie Tolle,
Philippe Zizzari, Nicole Bellefontaine, Isabelle Legroux-Gérot, Pierre Hardouin, Odile Broux, Odile Viltart, Christophe Chauveau
PLOS one Journal, 2014 Aug 4;9(8):e103775. doi:10.1371/journal.pone.0103775.
115
Long-term physiological alterations and recovery in a mouse model of
separation associated with time-restricted feeding: a tool to study anorexia
nervosa related consequences
Sara Zgheib 1,2*, Mathieu Méquinion 1,2,3*, Stéphanie Lucas 1,2, Damien Leterme 1,2, Olfa Ghali
1,2, Virginie Tolle4, Philippe Zizzari4, Nicole Bellefontaine 1,3, Isabelle Legroux-Gérot1,2,5, Pierre
analysis of light-phase restricted feeding reveals metabolic dyssynchrony in mice. Int J
Obes (Lond) 37: 843-852.
39. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA (2012) Time-restricted
feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat
diet. Cell Metab 15: 848-860.
40. Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, et al. (2010) The emergence of
cold-induced brown adipocytes in mouse white fat depots is determined predominantly by
141
white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab 298:
E1244-E1253.
41. Cousin B, Cinti S, Morroni M, S. Raimbault S, Ricquier D, et al. (1992) Occurrence of
brown adipocytes in rat white adipose tissue: molecular and morphological
characterization. J Cell Sci 103: 931-942.
42. Ferguson M, Sohal BH, Forster MJ, Sohal RS (2007) Effect of long-term caloric restriction
on oxygen consumption and body temperature in two different strains of mice. Mech
Ageing Dev 128: 539-545.
43. Veyrat-Durebex C, Montet X, Vinciguerra M, Gjinovci A, Meda P, et al. (2009) The
Lou/C rat: a model of spontaneous food restriction associated with improved insulin
sensitivity and decreased lipid storage in adipose tissue. Am J Physiol Endocrinol Metab
296: E1120-E1132.
44. Rogers NH, Landa A, Park S, Smith RG (2012) Aging leads to a programmed loss of
brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell 11: 1074-1083.
45. Holtkamp K, Hebebrand J, Mika C, Grzella I, Heer M, et al. (2003) The effect of
therapeutically induced weight gain on plasma leptin levels in patients with anorexia
nervosa. J Psychiatr Res 37:165-169.
46. Hebebrand J, Blum WF, Barth N, Coners H, Englaro P, et al. (1997) Leptin levels in
patients with anorexia nervosa are reduced in the acute stage and elevated upon short-term
weight restoration. Mol Psychiatry 2: 330-334.
47. Gendall KA, Kaye WH, Altemus M, McConaha CW, La Via MC (1999) Leptin,
Neuropeptide Y, and peptide YY in long-term recovered eating disorder patients. Biol
Psychiatry 46: 292-299.
48. Holtkamp K, Hebebrand J, Mika C, Heer M, Heussen N, et al. (2004) High serum leptin
levels subsequent to weight gain predict renewed weight loss in patients with anorexia
nervosa. Psychoneuroendocrinol 29: 791-797.
142
Figure 1. Design of the study. Forty mice were submitted to separation and time-restriction
feeding (Separation-Based Anorexia, SBA). After 2 weeks, 10 mice were sacrificed. The others
were kept in SBA conditions. 10 weeks after the beginning of the experiment, 10 mice were
sacrificed and the 20 other mice were placed in standard conditions (Recovery, REC) during 2 or
10 more weeks, before sacrifice. Estrous cycles were followed all along the experiment. Forty
other mice kept in standard housing conditions all along the experiment were studied and
sacrificed according to the pattern used for SBA and REC mice.
Figure 2. Weight, food intake and body composition of mice submitted to a 2-week study.
Measures were performed on mice in standard conditions (CT), separated with food ad libitum
(SEP), submitted to time-restricted feeding (TR) or separated and submitted to food access
restriction (SBA). A: body weights were recorded daily before the eating period (beginning of
the dark phase). B: Cumulative food intake was recorded for each group as the sum of the mean
food intake per mouse from day 1 to day 15. C-E: Fat mass, lean mass and bone mineral content
respectively were evaluated for each animal at day 0 and day 14, before food access. Data
represent mean ± SEM; n= 6/group. In A, differences were tested by a 2-way Anova followed by
a Bonferroni post-hoc test. SBA values are significantly different from CT values from day 1 to
the end (** P < 0.001). SBA values are significantly different from TR values from day 6 to the
end (‡ P < 0.05). TR values are significantly different from CT values from day 1 to the end (*
p<0.05). In C, D, and E, * p<0.05 and ** p<0.005 when compared to day 0 of the same group; ‡
p<0.05 and ‡‡ p<0.005 when compared to CT group at the same duration.
Figure 3. Weight, food intake and body composition of mice submitted to a 10-week SBA
protocol followed by a 10-week recovery protocol (REC). Measures were performed on mice in
standard conditions (CT) or separated and submitted to food access restriction (SBA). A: body
weights were recorded before the eating period (beginning of the dark phase). B: Cumulative
143
food intake was recorded for each group as the sum of the mean food intake per mouse from day
1 to day 140. C-E: Fat mass, lean mass and bone mineral content respectively were evaluated for
each animal at the beginning and after 2, 5 and 10 weeks of SBA protocol or after 10 weeks of
SBA protocol followed by 2 or 10 weeks of housing in standard conditions. Data represent mean
± SEM; n= 6-10/group. In A, differences were tested by a 2-way Anova followed by a
Bonferroni post-hoc test. SBA values are significantly different from CT values from day 1 to
day 70 (* p<0.0001). In B, C, D and E, * p<0.05, ** p<0.005, when compared to corresponding
CT value; ‡ p<0.05, ‡‡ p<0.005 when compared to the previous value of the same group.
Figure 4. Weight evolution of visceral adipose tissue (AT), subcutaneous AT (SCAT) and triceps
surae. Soft tissues from control and SBA mice were weighted after 2 or 10 weeks of protocol or
after 10 weeks of SBA protocol followed by 2 or 10 weeks of housing in standard conditions. A:
perigonadal fat was used to estimate the visceral fat mass evolution. B: SCAT, which gathers
inguinal AT and AT around the leg, was used to estimate the sub-cutaneous fat mass evolution.
C: Triceps surae were weighted to determine the muscle mass evolution. * p<0.05 and **
p<0.005 when compared to corresponding CT group; ‡ p<0.05 and ‡‡ p<0.005 when compared to
the previous value of the same group.
Figure 5. Leptin and adiponectin. A: Plasma concentrations of leptin and adiponectin of mice in
standard conditions, CT(□), or separated and submitted to food access restriction, SBA(■) after 2
and 10 weeks of protocol, followed by 2 and 10 weeks of standard housing conditions. B:
Relative leptin and adiponectin mRNA levels in SCAT and VAT vs HPRT and PPIA
housekeeping genes. Data represent mean ± SEM; n= 6-10/group. * p<0.05 and ** p<0.005
when compared to CT group at the same duration; ‡ p<0.05 and ‡‡ p<0.05 when compared to the
previous value of the same group.
144
Figure 6. GH and IGF-1. Whole blood GH levels and plasma IGF-1 levels were assayed on mice
in standard conditions (CT), or separated and submitted to food access restriction (SBA) after 2
and 10 weeks of protocol, followed by 2 and 10 weeks of standard housing conditions. Data
represent mean ± SEM; n= 6-10/group. * p<0.05 and ** p<0.005 when compared to CT group at
the same duration; ‡ p<0.05 and ‡ ‡ p<0.005 when compared to the previous value of the same
group.
Figure 7. Intraperitoneal glucose tolerance test in mice in standard conditions (CT), or separated
and submitted to time-restricted feeding (SBA) after 2 and 10 weeks of protocol, followed by 2
and 10 weeks of standard housing conditions. Data represent mean ± SEM; n= 6-10/group. *
p<0.05 and ** p<0.0001 significant differences between the two curves using Two-way
ANOVA.
Figure 8. Estrous cycle alteration. Estrous cycle determined according to the observation of the
cell population of vaginal washes was daily followed on mice in standard conditions (CT), or
separated and submitted to food access restriction (SBA) from day 0 to day 70, followed by 20
days of standard housing conditions. D=diestrus, M=metestrus, E=estrus, P=proestrus, 0= no cell
observed. A: A representative example of estrous cycle of CT mice. B: A representative example
of cycles observed in SBA mice, with the onset of long duration diestrus during the recovery
period. C: A representative example of cycles observed in SBA mice, with the onset of estrus
during the recovery period.
Figure 9. Alterations of reproduction. Ovary size of mice in standard conditions (CT), or
separated and submitted to food access restriction (SBA) after 2 and 10 weeks of protocol,
followed by 2 and 10 weeks of standard housing conditions. A: Ovary length measured on ovary
slices. B: Ovary width measured on ovary slices. Data represent mean ± SEM; n= 6/group. *
145
p<0.05 and ** p<0.005 when compared with CT group at the same duration; ‡ p<0.05 when
compared with previous value of the same group.
Figure 10. Expression analysis in adipose tissues of genes involved in lipid metabolism. Relative
mRNA levels of Glut4, FASn, ABHD5 and ATGL were determined by real-time PCR
experiments, in subcutaneous (SCAT) and visceral adipose tissues (VAT) of control □ and SBA
■ mice. PPIA and HPRT were used as housekeeping genes. All results are expressed as fold-
change compared to one SCAT of the control group after 10 weeks. Analyses were done after 10
weeks of SBA protocol and 10 additional weeks of REC protocol. Data represent mean ± SEM;
n= 5-10/group. * p<0.05 and ** p<0.005 when compared to CT group at the same duration; ‡
p<0.05 and ‡ ‡ p<0.005 when compared to the previous value of the same group.
Figure 11. Expression analysis in adipose tissues of genes involved in brown adipocyte
phenotype. Relative mRNA levels of UCP1, PGC1α, PRDM16 and ACOX1 were determined by
real-time PCR experiments, in subcutaneous (SCAT) and visceral adipose tissues (VAT) of
control □ and SBA ■ mice. PPIA and HPRT were used as housekeeping genes. All results are
expressed as fold-change compared to one SCAT of the control group after 10 weeks. Analyses
were done after 10 weeks of SBA protocol and 10 additional weeks of REC protocol. Data
represent mean ± SEM; n= 5-10/group. * p<0.05 and ** p<0.005 when compared to CT group at
the same duration; ‡ p<0.05 and ‡ ‡ p<0.005 when compared to the previous value of the same
group.
146
Références 1. National Collaborating Centre for Mental Health (UK). Eating Disorders: Core Interventions in the Treatment and Management of Anorexia Nervosa, Bulimia Nervosa and Related Eating Disorders [Internet]. Leicester (UK): British Psychological Society (UK); 2004 [cité 19 avr 2014]. Disponible sur: http://www.ncbi.nlm.nih.gov/books/NBK49304/ 2. Corcos M. Approche psychosomatique des conduites addictives alimentaires. Dialogue. mars 2005;(169):97‑109. 3. Simon Y. Epidémiologie et facteurs de risque psychosociaux dans l’anorexie mentale. Nutrition Clinique et Métabolisme. déc 2007;21(4):137‑42. 4. Doucet M. Modalités de prise en charge de l’anorexie mentale restrictivez pure. Universié du droit et de la santé-Lille 2; 2011. 5. Bulik C, Van Furth E, Sullivan P. The genetics of anorexia nervosa. Annual Review of Nutrition. 2007;27:263‑75. 6. Simon Y. Épidémiologie et facteurs de risque psychosociaux dans l’anorexie mentale. Nutr Clin Métabolisme. déc 2007;21(4):137‑42. 7. Stice E, Shaw HE. Role of body dissatisfaction in the onset and maintenance of eating pathology: a synthesis of research findings. J Psychosom Res. nov 2002;53(5):985‑93. 8. Chambaud-Peycher C, Doyen C, M. -C Mouren-Siméoni. Anorexie mentale et situations psychosociales anormales. Annales de psychiatrie. 2000;15(3):200‑13. 9. Kay A Wilhelm, Simon D Clarke. Eating disorders from a primary care perspective. MJA Practice Essentials. :71‑6. 10. Pritts Sarah D, Susman Jeffrey. Diagnosis of Eating Disorders in Primary Care. American Family Physician. 15 janv 2003;67(2). 11. Boraska V, Franklin CS, Floyd J a. B, Thornton LM, Huckins LM, Southam L, et al. A genome-wide association study of anorexia nervosa. Mol Psychiatry. 11 févr 2014; 12. Bulik CM, Thornton LM, Root TL, Pisetsky EM, Lichtenstein P, Pedersen NL. Understanding the relation between anorexia nervosa and bulimia nervosa in a Swedish national twin sample. Biol Psychiatry. 1 janv 2010;67(1):71‑7. 13. Bulik CM, Sullivan PF, Tozzi F, Furberg H, Lichtenstein P, Pedersen NL. Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry. mars 2006;63(3):305‑12. 14. Klump KL, Miller KB, Keel PK, McGue M, Iacono WG. Genetic and environmental influences on anorexia nervosa syndromes in a population-based twin sample. Psychol Med. mai 2001;31(4):737‑40. 15. Wade TD, Bulik CM, Neale M, Kendler KS. Anorexia nervosa and major depression: shared genetic and environmental risk factors. Am J Psychiatry. mars 2000;157(3):469‑71. 16. Kortegaard LS, Hoerder K, Joergensen J, Gillberg C, Kyvik KO. A preliminary population-based twin study of self-reported eating disorder. Psychol Med. févr 2001;31(2):361‑5. 17. Vink T, Hinney A, van Elburg AA, van Goozen SH, Sandkuijl LA, Sinke RJ, et al. Association between an agouti-related protein gene polymorphism and anorexia nervosa. Mol Psychiatry. mai 2001;6(3):325‑8. 18. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677‑736.
147
19. Xiao J, Wong AW, Willingham MM, van den Buuse M, Kilpatrick TJ, Murray SS. Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neurosignals. 2010;18(3):186‑202. 20. Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. oct 2009;42(2):81‑9. 21. Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci. avr 2003;91(4):267‑70. 22. Nakazato M, Hashimoto K, Shimizu E, Kumakiri C, Koizumi H, Okamura N, et al. Decreased levels of serum brain-derived neurotrophic factor in female patients with eating disorders. Biol Psychiatry. 15 août 2003;54(4):485‑90. 23. Radka SF, Holst PA, Fritsche M, Altar CA. Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res. 12 févr 1996;709(1):122‑301. 24. Monteleone P, Tortorella A, Martiadis V, Serritella C, Fuschino A, Maj M. Opposite changes in the serum brain-derived neurotrophic factor in anorexia nervosa and obesity. Psychosom Med. oct 2004;66(5):744‑8. 25. IBARGUEN VARGAS NY. Implication du BDNF dans l’etiopathogenèse et le traitement des troubles anxio-dépressifs Aspects précliniques. [Tours]: UNIVERSITÉ FRANÇOIS - RABELAIS DE TOURS; 2008. 26. Da Costa M, Halmi K. Classifications of anorexia nervosa: question of subtypes. Int J Eat Disord. 1992;11. 27. Tchanturia K, Anderluh MB, Morris RG, Rabe-Hesketh S, Collier DA, Sanchez P, et al. Cognitive flexibility in anorexia nervosa and bulimia nervosa. J Int Neuropsychol Soc JINS. juill 2004;10(4):513‑20. 28. Dancyger IF, Garfinkel PE. The relationship of partial syndrome eating disorders to anorexia nervosa and bulimia nervosa. Psychol Med. sept 1995;25(5):1019‑25. 29. Godart N, Perdereau F, Jeammet P. Données épidémiologiques : Anorexie chez l’adolescent. journal de pédiatrie et de puériculture. Sseptembre 2004;327‑30. 30. Housova J, Anderlova K, Krizová J, Haluzikova D, Kremen J, Kumstyrová T, et al. Serum adiponectin and resistin concentrations in patients with restrictive and binge/purge form of anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. mars 2005;90(3):1366‑70. 31. Legroux-Gérot I, Vignau J, D’Herbomez M, Collier F, Marchandise X, Duquesnoy B, et al. Evaluation of bone loss and its mechanisms in anorexia nervosa. Calcif Tissue Int. sept 2007;81(3):174‑82. 32. Wilson AJ, Touyz SW, O’Connor M, Beumont PJ. Correcting the eating disorder in anorexia nervosa. J Psychiatr Res. 1985;19(2-3):449‑51. 33. Samuel-Lajeunesse B, Silva G, Divas S. , Evaluation of Eating Behaviour and Eating Disorders Abnormalities. European Council of Eating Disorders; 1993; Prague, Czech Republic. 34. Divac S. Prise en Charge Cognitivo-Comportementale de l’Anorexie Mentale. Université René Descartes, Paris V; 1994. 35. Swenne I, Thurfjell B. Clinical onset and diagnosis of eating disorders in premenarcheal girls is preceded by inadequate weight gain and growth retardation. Acta Paediatr Oslo Nor 1992. oct 2003;92(10):1133‑7. 36. Swenne I. Weight requirements for catch-up growth in girls with eating disorders and onset of weight loss before menarche. Int J Eat Disord. déc 2005;38(4):340‑5. 37. Rozé C, Doyen C, Le Heuzey M-F, Armoogum P, Mouren M-C, Léger J. Predictors of late menarche and adult height in children with anorexia nervosa. Clin Endocrinol (Oxf). sept 2007;67(3):462‑7.
148
38. Kaye WH, Nagata T, Weltzin TE, Hsu LK, Sokol MS, McConaha C, et al. Double-blind placebo-controlled administration of fluoxetine in restricting- and restricting-purging-type anorexia nervosa. Biol Psychiatry. 1 avr 2001;49(7):644‑52. 39. Barbarich-Marsteller NC, Foltin RW, Walsh BT. Does anorexia nervosa resemble an addiction? Curr Drug Abuse Rev. sept 2011;4(3):197‑200. 40. Yon L, Doyen C, Asch M, Cook-Darzens S, Mouren M-C. [Treatment of anorexia nervosa in young patients in a special care unit at Robert-Debré Hospital (Paris): guidelines and practical methods]. Arch Pédiatrie Organe Off Sociéte Fr Pédiatrie. nov 2009;16(11):1491‑8. 41. Pénicaud L, Meillon S, Brondel L. Leptin and the central control of feeding behavior. Biochimie. oct 2012;94(10):2069‑74. 42. Aubert G, Burnier M, Dulloo A, Perregaux C, Mazzolai L, Pralong F, et al. Neuroendocrine characterization and anorexigenic effects of telmisartan in diet- and glitazone-induced weight gain. Metabolism. janv 2010;59(1):25‑32. 43. Hernández Vallejo SJ, Alqub M, Luquet S, Cruciani-Guglielmacci C, Delerive P, Lobaccaro J-M, et al. Short-term adaptation of postprandial lipoprotein secretion and intestinal gene expression to a high-fat diet. Am J Physiol Gastrointest Liver Physiol. avr 2009;296(4):G782‑92. 44. McCue MD. Starvation physiology: reviewing the different strategies animals use to survive a common challenge. Comp Biochem Physiol A Mol Integr Physiol. mai 2010;156(1):1‑18. 45. Anghel SI, Wahli W. Fat poetry: a kingdom for PPAR gamma. Cell Res. juin 2007;17(6):486‑511. 46. Reshef L, Olswang Y, Cassuto H, Blum B, Croniger CM, Kalhan SC, et al. Glyceroneogenesis and the triglyceride/fatty acid cycle. J Biol Chem. 15 août 2003;278(33):30413‑6. 47. Guerre-Millo M. Adipose tissue hormones. J Endocrinol Invest. nov 2002;25(10):855‑61. 48. Wang T, Hung CCY, Randall DJ. The comparative physiology of food deprivation: from feast to famine. Annu Rev Physiol. 2006;68:223‑51. 49. Komuta M, Harada M, Ueno T, Uchimura Y, Inada C, Mitsuyama K, et al. Unusual accumulation of glycogen in liver parenchymal cells in a patient with anorexia nervosa. Intern Med Tokyo Jpn. août 1998;37(8):678‑82. 50. Casper RC. Carbohydrate metabolism and its regulatory hormones in anorexia nervosa. Psychiatry Res. 16 avr 1996;62(1):85‑96. 51. Halmi KA, Struss AL, Owen WP, Stegink LD. Plasma and erythrocyte amino acid concentrations in anorexia nervosa. JPEN J Parenter Enteral Nutr. oct 1987;11(5):458‑64. 52. Kohl M, Foulon C, Guelfi J-D. [Hyperactivity and anorexia nervosa: behavioural and biological perspective]. L’Encéphale. oct 2004;30(5):492‑9. 53. Kosmiski L, Schmiege SJ, Mascolo M, Gaudiani J, Mehler PS. Chronic starvation secondary to anorexia nervosa is associated with an adaptive suppression of resting energy expenditure. J Clin Endocrinol Metab. 4 déc 2013;jc20131694. 54. Polito A, Fabbri A, Ferro-Luzzi A, Cuzzolaro M, Censi L, Ciarapica D, et al. Basal metabolic rate in anorexia nervosa: relation to body composition and leptin concentrations. Am J Clin Nutr. juin 2000;71(6):1495‑502. 55. Onur S, Haas V, Bosy-Westphal A, Hauer M, Paul T, Nutzinger D, et al. L-tri-iodothyronine is a major determinant of resting energy expenditure in underweight patients with anorexia nervosa and during weight gain. Eur J Endocrinol Eur Fed Endocr Soc. févr 2005;152(2):179‑84. 56. Russell J, Baur LA, Beumont PJ, Byrnes S, Gross G, Touyz S, et al. Altered energy metabolism in anorexia nervosa. Psychoneuroendocrinology. janv 2001;26(1):51‑63.
149
57. Hall KD. Computational model of in vivo human energy metabolism during semistarvation and refeeding. Am J Physiol Endocrinol Metab. juill 2006;291(1):E23‑37. 58. Iketani T, Kiriike N, Nagata T, Yamagami S. Altered body fat distribution after recovery of weight in patients with anorexia nervosa. Int J Eat Disord. nov 1999;26(3):275‑82. 59. Scalfi L, Polito A, Bianchi L, Marra M, Caldara A, Nicolai E, et al. Body composition changes in patients with anorexia nervosa after complete weight recovery. Eur J Clin Nutr. janv 2002;56(1):15‑20. 60. Franzoni E, Ciccarese F, Di Pietro E, Facchini G, Moscano F, Iero L, et al. Follow-up of bone mineral density and body composition in adolescents with restrictive anorexia nervosa: role of dual-energy X-ray absorptiometry. Eur J Clin Nutr. févr 2014;68(2):247‑52. 61. Kooh SW, Noriega E, Leslie K, Müller C, Harrison JE. Bone mass and soft tissue composition in adolescents with anorexia nervosa. Bone. août 1996;19(2):181‑8. 62. Kerruish KP, O’Connor J, Humphries IRJ, Kohn MR, Clarke SD, Briody JN, et al. Body composition in adolescents with anorexia nervosa. Am J Clin Nutr. janv 2002;75(1):31‑7. 63. Vaisman N, Rossi MF, Goldberg E, Dibden LJ, Wykes LJ, Pencharz PB. Energy expenditure and body composition in patients with anorexia nervosa. J Pediatr. nov 1988;113(5):919‑24. 64. Grinspoon S, Thomas L, Miller K, Pitts S, Herzog D, Klibanski A. Changes in regional fat redistribution and the effects of estrogen during spontaneous weight gain in women with anorexia nervosa. Am J Clin Nutr. mai 2001;73(5):865‑9. 65. Laessle RG, Fischer M, Fichter MM, Pirke KM, Krieg JC. Cortisol levels and vigilance in eating disorder patients. Psychoneuroendocrinology. oct 1992;17(5):475‑84. 66. Herpertz S, Albers N, Wagner R, Pelz B, Köpp W, Mann K, et al. Longitudinal changes of circadian leptin, insulin and cortisol plasma levels and their correlation during refeeding in patients with anorexia nervosa. Eur J Endocrinol Eur Fed Endocr Soc. avr 2000;142(4):373‑9. 67. Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab. déc 1999;84(12):4489‑96. 68. Audí L, Vargas DM, Gussinyé M, Yeste D, Martí G, Carrascosa A. Clinical and biochemical determinants of bone metabolism and bone mass in adolescent female patients with anorexia nervosa. Pediatr Res. avr 2002;51(4):497‑504. 69. Orphanidou CI, McCargar LJ, Birmingham CL, Belzberg AS. Changes in body composition and fat distribution after short-term weight gain in patients with anorexia nervosa. Am J Clin Nutr. avr 1997;65(4):1034‑41. 70. Haas VK, Kohn MR, Clarke SD, Allen JR, Madden S, Müller MJ, et al. Body composition changes in female adolescents with anorexia nervosa. Am J Clin Nutr. avr 2009;89(4):1005‑10. 71. Misra M, Soyka LA, Miller KK, Grinspoon S, Levitsky LL, Klibanski A. Regional body composition in adolescents with anorexia nervosa and changes with weight recovery. Am J Clin Nutr. juin 2003;77(6):1361‑7. 72. Melchior J, Rigaud D, Rozen R, Malon D, Apfelbaum M. Energy expenditure economy induced by decrease in lean body mass in anorexia nervosa. Eur J Clin Nutr. nov 1989;43(11):793‑9. 73. Rigaud D, Hassid J, Meulemans A, Poupard AT, Boulier A. A paradoxical increase in resting energy expenditure in malnourished patients near death: the king penguin syndrome [Internet]. [cité 19 avr 2014]. Disponible sur: http://ajcn.nutrition.org 74. Misra M, Miller KK, Almazan C, Worley M, Herzog DB, Klibanski A. Hormonal Determinants of Regional Body Composition in Adolescent Girls with Anorexia Nervosa and Controls. J Clin Endocrinol Metab. 1 mai 2005;90(5):2580‑7. 75. Roux C. Osteopenia : is it a problem ? lettre du Rhumatologue. oct 2009;(355):16‑20.
150
76. Grinspoon S, Thomas E, Pitts S, Gross E, Mickley D, Miller K, et al. Prevalence and predictive factors for regional osteopenia in women with anorexia nervosa. Ann Intern Med. 2000;133(10):790‑4. 77. Del Rio L, Carrascosa A, Pons F, Gusinyé M, Yeste D, Domenech FM. Bone mineral density of the lumbar spine in white Mediterranean Spanish children and adolescents: changes related to age, sex, and puberty. Pediatr Res. mars 1994;35(3):362‑6. 78. Muñoz MT, Argente J. Anorexia nervosa in female adolescents: endocrine and bone mineral density disturbances. Eur J Endocrinol Eur Fed Endocr Soc. sept 2002;147(3):275‑86. 79. Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab. déc 1999;84(12):4489‑96. 80. Herzog W, Minne H, Deter C, Leidig G, Schellberg D, Wüster C, et al. Outcome of bone mineral density in anorexia nervosa patients 11.7 years after first admission. J Bone Miner Res Off J Am Soc Bone Miner Res. mai 1993;8(5):597‑605. 81. Brotman AW, Stern TA. Osteoporosis and pathologic fractures in anorexia nervosa. Am J Psychiatry. avr 1985;142(4):495‑6. 82. Milos G, Spindler A, Rüegsegger P, Seifert B, Mühlebach S, Uebelhart D, et al. Cortical and trabecular bone density and structure in anorexia nervosa. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. juill 2005;16(7):783‑90. 83. Lawson EA, Miller KK, Bredella MA, Phan C, Misra M, Meenaghan E, et al. Hormone predictors of abnormal bone microarchitecture in women with anorexia nervosa. Bone. févr 2010;46(2):458‑63. 84. Faje AT, Karim L, Taylor A, Lee H, Miller KK, Mendes N, et al. Adolescent girls with anorexia nervosa have impaired cortical and trabecular microarchitecture and lower estimated bone strength at the distal radius. J Clin Endocrinol Metab. mai 2013;98(5):1923‑9. 85. Misra M, Klibanski A. The neuroendocrine basis of anorexia nervosa and its impact on bone metabolism. Neuroendocrinology. 2011;93(2):65‑73. 86. Misra M, Klibanski A. Anorexia nervosa and osteoporosis. Rev Endocr Metab Disord. 2006;7(1-2):91‑9. 87. Soyka LA, Misra M, Frenchman A, Miller KK, Grinspoon S, Schoenfeld DA, et al. Abnormal bone mineral accrual in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab. sept 2002;87(9):4177‑85. 88. Singhal V, Misra M, Klibanski A. Endocrinology of anorexia nervosa in young people: recent insights. Curr Opin Endocrinol Diabetes Obes. févr 2014;21(1):64‑70. 89. Méquinion M, Langlet F, Zgheib S, Dickson S, Dehouck B, Chauveau C, et al. Ghrelin: central and peripheral implications in anorexia nervosa. Front Endocrinol. 2013;4:15. 90. Estour B, Germain N, Diconne E, Frere D, Cottet-Emard J-M, Carrot G, et al. Hormonal profile heterogeneity and short-term physical risk in restrictive anorexia nervosa. J Clin Endocrinol Metab. mai 2010;95(5):2203‑10. 91. Haas V, Onur S, Paul T, Nutzinger DO, Bosy-Westphal A, Hauer M, et al. Leptin and body weight regulation in patients with anorexia nervosa before and during weight recovery. Am J Clin Nutr. avr 2005;81(4):889‑96. 92. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. déc 1995;1(12):1311‑4. 93. Bi S, Gavrilova O, Gong DW, Mason MM, Reitman M. Identification of a placental enhancer for the human leptin gene. J Biol Chem. 28 nov 1997;272(48):30583‑8. 94. Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature. 18 juin 1998;393(6686):684‑8.
151
95. Melzner I, Scott V, Dorsch K, Fischer P, Wabitsch M, Brüderlein S, et al. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem. 22 nov 2002;277(47):45420‑7. 96. Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17(2):305‑11. 97. Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 20 juill 1996;348(9021):159‑61. 98. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. mai 1996;2(5):589‑93. 99. Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A. 20 janv 1998;95(2):741‑6. 100. Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, et al. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. déc 1998;21(6):1375‑85. 101. Landry Y, Gies J-P. Pharmacologie Des cibles vers l’indication thérapeutique. deuxième édition. DUNOD; 2009. 102. Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 25 août 2000;92(1-3):73‑8. 103. Karsenty G. Leptin controls bone formation through a hypothalamic relay. Recent Prog Horm Res. 2001;56:401‑15. 104. Yadav VK, Karsenty G. Leptin-dependent co-regulation of bone and energy metabolism. Aging. nov 2009;1(11):954‑6. 105. Karsenty G, Oury F. The central regulation of bone mass, the first link between bone remodeling and energy metabolism. J Clin Endocrinol Metab. nov 2010;95(11):4795‑801. 106. Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature. 19 janv 2012;481(7381):314‑20. 107. Motyl KJ, Rosen CJ. Understanding leptin-dependent regulation of skeletal homeostasis. Biochimie. oct 2012;94(10):2089‑96. 108. Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 15 mai 2003;423(6937):349‑55. 109. Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. avr 1999;140(4):1630‑8. 110. Gordeladze JO, Drevon CA, Syversen U, Reseland JE. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem. 2002;85(4):825‑36. 111. Krings A, Rahman S, Huang S, Lu Y, Czernik PJ, Lecka-Czernik B. Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone. févr 2012;50(2):546‑52. 112. Kim GS, Hong JS, Kim SW, Koh J-M, An CS, Choi J-Y, et al. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem. 13 juin 2003;278(24):21920‑9. 113. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 21 janv 2000;100(2):197‑207.
152
114. Turner RT, Kalra SP, Wong CP, Philbrick KA, Lindenmaier LB, Boghossian S, et al. Peripheral leptin regulates bone formation. J Bone Miner Res Off J Am Soc Bone Miner Res. janv 2013;28(1):22‑34. 115. Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 1 nov 2002;111(3):305‑17. 116. Elefteriou F, Karsenty G. [Bone mass regulation by leptin: a hypothalamic control of bone formation]. Pathol Biol (Paris). avr 2004;52(3):148‑53. 117. Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab. 2 avr 2013;17(4):607‑17. 118. Yannakoulia M, Yiannakouris N, Blüher S, Matalas A-L, Klimis-Zacas D, Mantzoros CS. Body fat mass and macronutrient intake in relation to circulating soluble leptin receptor, free leptin index, adiponectin, and resistin concentrations in healthy humans. J Clin Endocrinol Metab. avr 2003;88(4):1730‑6. 119. Misra M, Miller KK, Almazan C, Ramaswamy K, Aggarwal A, Herzog DB, et al. Hormonal and body composition predictors of soluble leptin receptor, leptin, and free leptin index in adolescent girls with anorexia nervosa and controls and relation to insulin sensitivity. J Clin Endocrinol Metab. juill 2004;89(7):3486‑95. 120. Modan-Moses D, Stein D, Pariente C, Yaroslavsky A, Ram A, Faigin M, et al. Modulation of adiponectin and leptin during refeeding of female anorexia nervosa patients. J Clin Endocrinol Metab. mai 2007;92(5):1843‑7. 121. Hebebrand J, Blum WF, Barth N, Coners H, Englaro P, Juul A, et al. Leptin levels in patients with anorexia nervosa are reduced in the acute stage and elevated upon short-term weight restoration. Mol Psychiatry. juill 1997;2(4):330‑4. 122. Misra M, Prabhakaran R, Miller KK, Tsai P, Lin A, Lee N, et al. Role of cortisol in menstrual recovery in adolescent girls with anorexia nervosa. Pediatr Res. avr 2006;59(4 Pt 1):598‑603. 123. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 10 nov 1995;270(45):26746‑9. 124. Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 3 mai 1996;271(18):10697‑703. 125. Hamada T, Tashiro K, Tada H, Inazawa J, Shirozu M, Shibahara K, et al. Isolation and characterization of a novel secretory protein, stromal cell-derived factor-2 (SDF-2) using the signal sequence trap method. Gene. 17 oct 1996;176(1-2):211‑4. 126. Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun. 16 avr 1996;221(2):286‑9. 127. Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care. août 2003;26(8):2442‑50. 128. Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. juin 2006;55(6):1537‑45. 129. Tsao T-S, Tomas E, Murrey HE, Hug C, Lee DH, Ruderman NB, et al. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J Biol Chem. 12 déc 2003;278(50):50810‑7. 130. Hara K, Horikoshi M, Yamauchi T, Yago H, Miyazaki O, Ebinuma H, et al. Measurement of the high-molecular weight form of adiponectin in plasma is useful for the prediction of insulin resistance and metabolic syndrome. Diabetes Care. juin 2006;29(6):1357‑62.
153
131. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 12 juin 2003;423(6941):762‑9. 132. Denzel MS, Scimia M-C, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. déc 2010;120(12):4342‑52. 133. Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. mai 2008;118(5):1645‑56. 134. Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. févr 2007;117(2):375‑86. 135. Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, et al. Adiponectin increases bone mass by suppressing osteoclast and activating osteoblast. Biochem Biophys Res Commun. 3 juin 2005;331(2):520‑6. 136. Shinoda Y, Yamaguchi M, Ogata N, Akune T, Kubota N, Yamauchi T, et al. Regulation of bone formation by adiponectin through autocrine/paracrine and endocrine pathways. J Cell Biochem. 1 sept 2006;99(1):196‑208. 137. Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, et al. Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab. 4 juin 2013;17(6):901‑15. 138. Dostálová I, Smitka K, Papezová H, Kvasnicková H, Nedvídková J. Increased insulin sensitivity in patients with anorexia nervosa: the role of adipocytokines. Physiol Res Acad Sci Bohemoslov. 2007;56(5):587‑94. 139. Karczewska-Kupczewska M, Straczkowski M, Adamska A, Nikołajuk A, Otziomek E, Górska M, et al. Insulin sensitivity, metabolic flexibility, and serum adiponectin concentration in women with anorexia nervosa. Metabolism. avr 2010;59(4):473‑7. 140. Delporte ML, Brichard SM, Hermans MP, Beguin C, Lambert M. Hyperadiponectinaemia in anorexia nervosa. Clin Endocrinol (Oxf). janv 2003;58(1):22‑9. 141. Pannacciulli N, Vettor R, Milan G, Granzotto M, Catucci A, Federspil G, et al. Anorexia nervosa is characterized by increased adiponectin plasma levels and reduced nonoxidative glucose metabolism. J Clin Endocrinol Metab. avr 2003;88(4):1748‑52. 142. Bosy-Westphal A, Brabant G, Haas V, Onur S, Paul T, Nutzinger D, et al. Determinants of plasma adiponectin levels in patients with anorexia nervosa examined before and after weight gain. Eur J Nutr. sept 2005;44(6):355‑9. 143. Saito K, Arata S, Hosono T, Sano Y, Takahashi K, Choi-Miura N-H, et al. Adiponectin plays an important role in efficient energy usage under energy shortage. Biochim Biophys Acta. juill 2006;1761(7):709‑16. 144. Iwahashi H, Funahashi T, Kurokawa N, Sayama K, Fukuda E, Okita K, et al. Plasma adiponectin levels in women with anorexia nervosa. Horm Metab Res Horm Stoffwechselforschung Horm Métabolisme. sept 2003;35(9):537‑40. 145. Tagami T, Satoh N, Usui T, Yamada K, Shimatsu A, Kuzuya H. Adiponectin in anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. avr 2004;89(4):1833‑7. 146. Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. oct 2002;110(8):1093‑103. 147. Kowalska I, Karczewska-Kupczewska M, Strączkowski M. Adipocytokines, gut hormones and growth factors in anorexia nervosa. Clin Chim Acta Int J Clin Chem. 18 sept 2011;412(19-20):1702‑11. 148. Miller KK. Endocrine dysregulation in anorexia nervosa update. J Clin Endocrinol Metab. oct 2011;96(10):2939‑49.
154
149. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 18 juill 1996;382(6588):250‑2. 150. Grinspoon S, Gulick T, Askari H, Landt M, Lee K, Anderson E, et al. Serum leptin levels in women with anorexia nervosa. J Clin Endocrinol Metab. nov 1996;81(11):3861‑3. 151. Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2 sept 2004;351(10):987‑97. 152. Brichard SM, Delporte ML, Lambert M. Adipocytokines in anorexia nervosa: a review focusing on leptin and adiponectin. Horm Metab Res Horm Stoffwechselforschung Horm Métabolisme. juin 2003;35(6):337‑42. 153. Hebebrand J, Albayrak Ö. Leptin treatment of patients with anorexia nervosa? The urgent need for initiation of clinical studies. Eur Child Adolesc Psychiatry. févr 2012;21(2):63‑6. 154. Støving RK, Veldhuis JD, Flyvbjerg A, Vinten J, Hangaard J, Koldkjaer OG, et al. Jointly amplified basal and pulsatile growth hormone (GH) secretion and increased process irregularity in women with anorexia nervosa: indirect evidence for disruption of feedback regulation within the GH-insulin-like growth factor I axis. J Clin Endocrinol Metab. juin 1999;84(6):2056‑63. 155. Okada S, Kopchick JJ. Biological effects of growth hormone and its antagonist. Trends Mol Med. mars 2001;7(3):126‑32. 156. Dee Unglaub S. Physiologie Animale Une approche intégrée. PEARSON Education Inc. 157. Melmed S. Insulin-like growth factor I--a prototypic peripheral-paracrine hormone? Endocrinology. sept 1999;140(9):3879‑80. 158. Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. août 2008;29(5):535‑59. 159. Ohlsson C, Mohan S, Sjögren K, Tivesten A, Isgaard J, Isaksson O, et al. The role of liver-derived insulin-like growth factor-I. Endocr Rev. août 2009;30(5):494‑535. 160. Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, et al. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology. mars 2003;144(3):929‑36. 161. Dostálová I, Kaválková P, Haluzíková D, Lacinová Z, Mráz M, Papezová H, et al. Plasma concentrations of fibroblast growth factors 19 and 21 in patients with anorexia nervosa. J Clin Endocrinol Metab. sept 2008;93(9):3627‑32. 162. Støving RK, Chen J-W, Glintborg D, Brixen K, Flyvbjerg A, Hørder K, et al. Bioactive insulin-like growth factor (IGF) I and IGF-binding protein-1 in anorexia nervosa. J Clin Endocrinol Metab. juin 2007;92(6):2323‑9. 163. Fazeli PK, Lawson EA, Prabhakaran R, Miller KK, Donoho DA, Clemmons DR, et al. Effects of recombinant human growth hormone in anorexia nervosa: a randomized, placebo-controlled study. J Clin Endocrinol Metab. nov 2010;95(11):4889‑97. 164. Kubicky RA, Wu S, Kharitonenkov A, De Luca F. Role of fibroblast growth factor 21 (FGF21) in undernutrition-related attenuation of growth in mice. Endocrinology. mai 2012;153(5):2287‑95. 165. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. juin 2007;5(6):415‑25. 166. Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. juill 2008;8(1):77‑83. 167. Fazeli PK, Misra M, Goldstein M, Miller KK, Klibanski A. Fibroblast growth factor-21 may mediate growth hormone resistance in anorexia nervosa. J Clin Endocrinol Metab. janv 2010;95(1):369‑74.
155
168. Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 22 juin 1999;96(13):7324‑9. 169. Elis S, Courtland H-W, Wu Y, Rosen CJ, Sun H, Jepsen KJ, et al. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res Off J Am Soc Bone Miner Res. juin 2010;25(6):1257‑66. 170. Biller BM, Federoff HJ, Koenig JI, Klibanski A. Abnormal cortisol secretion and responses to corticotropin-releasing hormone in women with hypothalamic amenorrhea. J Clin Endocrinol Metab. févr 1990;70(2):311‑7. 171. Misra M, Miller KK, Almazan C, Ramaswamy K, Lapcharoensap W, Worley M, et al. Alterations in cortisol secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab. oct 2004;89(10):4972‑80. 172. Lawson EA, Donoho D, Miller KK, Misra M, Meenaghan E, Lydecker J, et al. Hypercortisolemia is associated with severity of bone loss and depression in hypothalamic amenorrhea and anorexia nervosa. J Clin Endocrinol Metab. déc 2009;94(12):4710‑6. 173. Lawson EA, Misra M, Meenaghan E, Rosenblum L, Donoho DA, Herzog D, et al. Adrenal glucocorticoid and androgen precursor dissociation in anorexia nervosa. J Clin Endocrinol Metab. avr 2009;94(4):1367‑71. 174. Legroux-Gerot I, Vignau J, Collier F, Cortet B. Bone loss associated with anorexia nervosa. Jt Bone Spine Rev Rhum. déc 2005;72(6):489‑95. 175. Chiodini I, Torlontano M, Carnevale V, Trischitta V, Scillitani A. Skeletal involvement in adult patients with endogenous hypercortisolism. J Endocrinol Invest. mars 2008;31(3):267‑76. 176. Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 23 juill 2010;142(2):296‑308. 177. Lacombe J, Karsenty G, Ferron M. In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol Metab. 2013;2(4):498‑504. 178. Klein GL. Insulin and bone: Recent developments. World J Diabetes. 15 févr 2014;5(1):14‑6. 179. Thomas SJ, Morimoto K, Herndon DN, Ferrando AA, Wolfe RR, Klein GL, et al. The effect of prolonged euglycemic hyperinsulinemia on lean body mass after severe burn. Surgery. août 2002;132(2):341‑7. 180. Pramojanee SN, Phimphilai M, Chattipakorn N, Chattipakorn SC. Possible roles of insulin signaling in osteoblasts. Endocr Res. 28 mars 2014; 181. Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, et al. A role for ghrelin in the central regulation of feeding. Nature. 11 janv 2001;409(6817):194‑8. 182. Tolle V, Zizzari P, Tomasetto C, Rio MC, Epelbaum J, Bluet-Pajot MT. In vivo and in vitro effects of ghrelin/motilin-related peptide on growth hormone secretion in the rat. Neuroendocrinology. janv 2001;73(1):54‑61. 183. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 9 déc 1999;402(6762):656‑60. 184. Li RL, Sherbet DP, Elsbernd BL, Goldstein JL, Brown MS, Zhao T-J. Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. J Biol Chem. 25 mai 2012;287(22):17942‑50. 185. Kang K, Zmuda E, Sleeman MW. Physiological role of ghrelin as revealed by the ghrelin and GOAT knockout mice. Peptides. nov 2011;32(11):2236‑41. 186. Hansen TK, Dall R, Hosoda H, Kojima M, Kangawa K, Christiansen JS, et al. Weight loss increases circulating levels of ghrelin in human obesity. Clin Endocrinol (Oxf). févr 2002;56(2):203‑6.
156
187. Tolle V, Kadem M, Bluet-Pajot M-T, Frere D, Foulon C, Bossu C, et al. Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. J Clin Endocrinol Metab. janv 2003;88(1):109‑16. 188. Maccarinelli G, Sibilia V, Torsello A, Raimondo F, Pitto M, Giustina A, et al. Ghrelin regulates proliferation and differentiation of osteoblastic cells. J Endocrinol. janv 2005;184(1):249‑56. 189. Scheller E, Cawthron W, Learman B, Mori H, Simon B, Parlee S. the metabolic nature of marrow fat:insulin signaling, CREB phosphorylation and the adiponectin paradox. 2013. 190. Clabaut A, Delplace S, Chauveau C, Hardouin P, Broux O. Human osteoblasts derived from mesenchymal stem cells express adipogenic markers upon coculture with bone marrow adipocytes. Differ Res Biol Divers. juill 2010;80(1):40‑5. 191. Goto H, Hozumi A, Osaki M, Fukushima T, Sakamoto K, Yonekura A, et al. Primary human bone marrow adipocytes support TNF-α-induced osteoclast differentiation and function through RANKL expression. Cytokine. déc 2011;56(3):662‑8. 192. Shen W, Chen J, Punyanitya M, Shapses S, Heshka S, Heymsfield SB. MRI-measured bone marrow adipose tissue is inversely related to DXA-measured bone mineral in Caucasian women. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. mai 2007;18(5):641‑7. 193. Wehrli FW, Hopkins JA, Hwang SN, Song HK, Snyder PJ, Haddad JG. Cross-sectional study of osteopenia with quantitative MR imaging and bone densitometry. Radiology. nov 2000;217(2):527‑38. 194. Blebea JS, Houseni M, Torigian DA, Fan C, Mavi A, Zhuge Y, et al. Structural and functional imaging of normal bone marrow and evaluation of its age-related changes. Semin Nucl Med. mai 2007;37(3):185‑94. 195. Maalej A, Guermazi Y, Fakhfakh K, Akid F, Haddar S, Fourati H. 2012. 196. Di Iorgi N, Mittelman SD, Gilsanz V. Differential effect of marrow adiposity and visceral and subcutaneous fat on cardiovascular risk in young, healthy adults. Int J Obes 2005. déc 2008;32(12):1854‑60. 197. Vande Berg BC, Malghem J, Lecouvet FE, Maldague B. Magnetic resonance imaging of normal bone marrow. Eur Radiol. 1998;8(8):1327‑34. 198. Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, et al. Marrow fat and bone--new perspectives. J Clin Endocrinol Metab. mars 2013;98(3):935‑45. 199. Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. déc 2004;3(6):379‑89. 200. Elbaz A, Rivas D, Duque G. Effect of estrogens on bone marrow adipogenesis and Sirt1 in aging C57BL/6J mice. Biogerontology. déc 2009;10(6):747‑55. 201. Martin RB, Chow BD, Lucas PA. Bone marrow fat content in relation to bone remodeling and serum chemistry in intact and ovariectomized dogs. Calcif Tissue Int. mars 1990;46(3):189‑94. 202. Martin RB, Zissimos SL. Relationships between marrow fat and bone turnover in ovariectomized and intact rats. Bone. 1991;12(2):123‑31. 203. Somjen D, Katzburg S, Kohen F, Gayer B, Posner GH, Yoles I, et al. The effects of native and synthetic estrogenic compounds as well as vitamin D less-calcemic analogs on adipocytes content in rat bone marrow. J Endocrinol Invest. févr 2011;34(2):106‑10. 204. Iwaniec UT, Turner RT. Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone. mars 2013;53(1):145‑53.
157
205. Tornvig L, Mosekilde LI, Justesen J, Falk E, Kassem M. Troglitazone treatment increases bone marrow adipose tissue volume but does not affect trabecular bone volume in mice. Calcif Tissue Int. juill 2001;69(1):46‑50. 206. Meunier P, Aaron J, Edouard C, Vignon G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop. oct 1971;80:147‑54. 207. Syed FA, Oursler MJ, Hefferanm TE, Peterson JM, Riggs BL, Khosla S. Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. sept 2008;19(9):1323‑30. 208. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 2 avr 1999;284(5411):143‑7. 209. Harada S, A.Rodan G. Control of osteoblast function and regulation of bone mass. 210. Veilleux A, Caron-Jobin M, Noël S, Laberge PY, Tchernof A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes. mai 2011;60(5):1504‑11. 211. Liu L-F, Shen W-J, Ueno M, Patel S, Kraemer FB. Characterization of age-related gene expression profiling in bone marrow and epididymal adipocytes. BMC Genomics. 2011;12:212. 212. Poloni A, Maurizi G, Serrani F, Mancini S, Zingaretti MC, Frontini A, et al. Molecular and functional characterization of human bone marrow adipocytes. Exp Hematol. juin 2013;41(6):558‑66.e2. 213. Abella E, Feliu E, Granada I, Millá F, Oriol A, Ribera JM, et al. Bone marrow changes in anorexia nervosa are correlated with the amount of weight loss and not with other clinical findings. Am J Clin Pathol. oct 2002;118(4):582‑8. 214. Bredella MA, Fazeli PK, Miller KK, Misra M, Torriani M, Thomas BJ, et al. Increased bone marrow fat in anorexia nervosa. J Clin Endocrinol Metab. juin 2009;94(6):2129‑36. 215. Griffith JF, Yeung DKW, Antonio GE, Lee FKH, Hong AWL, Wong SYS, et al. Vertebral bone mineral density, marrow perfusion, and fat content in healthy men and men with osteoporosis: dynamic contrast-enhanced MR imaging and MR spectroscopy. Radiology. sept 2005;236(3):945‑51. 216. Hardouin P, Pansini V, Cortet B. Bone marrow fat. Jt Bone Spine Rev Rhum. juill 2014;81(4):313‑9. 217. Ecklund K, Vajapeyam S, Feldman HA, Buzney CD, Mulkern RV, Kleinman PK, et al. Bone marrow changes in adolescent girls with anorexia nervosa. J Bone Miner Res Off J Am Soc Bone Miner Res. févr 2010;25(2):298‑304. 218. Déchelotte P, Grigioni S, Fetissov S. Conséquences digestives de l’anorexie mentale. Nutr Clin Métabolisme. déc 2007;21(4):166‑71. 219. Roulet M, Cheseaux M, Coti P. Conséquences de la dénutrition chez l’enfant et l’adolescent. Mortalité, morbidité, conséquences médicoéconomiques. Nutr Clin Métabolisme. déc 2005;19(4):207‑13. 220. Rigaud D, Bedig G, Merrouche M, Vulpillat M, Bonfils S, Apfelbaum M. Delayed gastric emptying in anorexia nervosa is improved by completion of a renutrition program. Dig Dis Sci. août 1988;33(8):919‑25. 221. Szmukler GI, Young GP, Lichtenstein M, Andrews JT. A serial study of gastric emptying in anorexia nervosa and bulimia. Aust N Z J Med. juin 1990;20(3):220‑5. 222. Kamal N, Chami T, Andersen A, Rosell FA, Schuster MM, Whitehead WE. Delayed gastrointestinal transit times in anorexia nervosa and bulimia nervosa. Gastroenterology. nov 1991;101(5):1320‑4.
158
223. Buchman AL, Ament ME, Weiner M, Kodner A, Mayer EA. Reversal of megaduodenum and duodenal dysmotility associated with improvement in nutritional status in primary anorexia nervosa. Dig Dis Sci. févr 1994;39(2):433‑40. 224. Mitchell J., Pyle R., Lentz R. Electrolyte and other physiological abnormalities in patients with bulimia. 1983; 225. Abdu R., Garritano D, Culver O. Acute gastric necrosis in anorexia nervosa and bulimia. Two case reports. Archives of Surgery. 1987; 226. Stacher G, Kiss S, Bergmann H, Hobart C. Oesophageal and gastric motility disorders in patients categorised as having primary anorexia nervosa. Gut. 1986; 227. Ravelli A., Helps B., Milla P. Normal gastric antral myoelectrical activity in early onset anorexia nervosa. 1993; 228. Hall R., Beresford T. Medical complications of anorexia and bulimia. Psychiatric Medicine. 1989; 229. Chiarioni G, Bassotti G, Monsignori A, Menegotti M, Salandini L, Di Matteo G, et al. Anorectal dysfunction in constipated women with anorexia nervosa. Mayo Clin Proc. oct 2000;75(10):1015‑9. 230. Zipfel S, Sammet I, Rapps N, Herzog W, Herpertz S, Martens U. Gastrointestinal disturbances in eating disorders: clinical and neurobiological aspects. Auton Neurosci Basic Clin. 30 oct 2006;129(1-2):99‑106. 231. McDermott JR, Leslie FC, D’Amato M, Thompson DG, Grencis RK, McLaughlin JT. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut. avr 2006;55(4):492‑7. 232. Marcos A, Nova E, Montero A. Changes in the immune system are conditioned by nutrition. Eur J Clin Nutr. sept 2003;57 Suppl 1:S66‑9. 233. Corcos M, Guilbaud O, Chaouat G, Cayol V, Speranza M, Chambry J, et al. Cytokines and anorexia nervosa. Psychosom Med. juin 2001;63(3):502‑4. 234. Brambilla F, Monti D, Franceschi C. Plasma concentrations of interleukin-1-beta, interleukin-6 and tumor necrosis factor-alpha, and of their soluble receptors and receptor antagonist in anorexia nervosa. Psychiatry Res. 20 sept 2001;103(2-3):107‑14. 235. Ehrlich S, Burghardt R, Schneider N, Broecker-Preuss M, Weiss D, Merle JV, et al. The role of leptin and cortisol in hyperactivity in patients with acute and weight-recovered anorexia nervosa. Prog Neuropsychopharmacol Biol Psychiatry. 15 juin 2009;33(4):658‑62. 236. Mayer L, Walsh BT, Pierson RN Jr, Heymsfield SB, Gallagher D, Wang J, et al. Body fat redistribution after weight gain in women with anorexia nervosa. Am J Clin Nutr. juin 2005;81(6):1286‑91. 237. Purper-Ouakil D, Michel G, Baup N, Mouren-Siméoni M-C. Aspects psychopathologiques de l’exercice physique intensif chez l’enfant et l’adolescent : mise au point à partir d’une situation clinique. Ann Méd-Psychol Rev Psychiatr. oct 2002;160(8):543‑9. 238. Duclos M, Ouerdani A, Mormède P, Konsman JP. Food restriction-induced hyperactivity: addiction or adaptation to famine? Psychoneuroendocrinology. juin 2013;38(6):884‑97. 239. Klein DA, Mayer LES, Schebendach JE, Walsh BT. Physical activity and cortisol in anorexia nervosa. Psychoneuroendocrinology. juin 2007;32(5):539‑47. 240. Davis C, Kennedy SH, Ravelski E, Dionne M. The role of physical activity in the development and maintenance of eating disorders. Psychol Med. nov 1994;24(4):957‑67. 241. Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci Biobehav Rev. 1992;16(4):525‑34. 242. Yirmiya R, Goshen I, Bajayo A, Kreisel T, Feldman S, Tam J, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci U S A. 7 nov 2006;103(45):16876‑81.
159
243. Wiborg O. Chronic mild stress for modeling anhedonia. Cell Tissue Res. oct 2013;354(1):155‑69. 244. Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, et al. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res Off J Am Soc Bone Miner Res. sept 2010;25(9):2078‑88. 245. Tatsumi S, Ito M, Asaba Y, Tsutsumi K, Ikeda K. Life-long caloric restriction reveals biphasic and dimorphic effects on bone metabolism in rodents. Endocrinology. févr 2008;149(2):634‑41. 246. Tropp J, Markus EJ. Effects of mild food deprivation on the estrous cycle of rats. Physiol Behav. juill 2001;73(4):553‑9. 247. Austad SN. Does caloric restriction in the laboratory simply prevent overfeeding and return house mice to their natural level of food intake? Sci Aging Knowl Environ SAGE KE. 7 nov 2001;2001(6):pe3. 248. Cerqueira FM, Kowaltowski AJ. Commonly adopted caloric restriction protocols often involve malnutrition. Ageing Res Rev. oct 2010;9(4):424‑30. 249. Gairdner SE, Amara CE. Serum leptin is not correlated with body fat in severe food restriction. Appl Physiol Nutr Metab Physiol Appliquée Nutr Métabolisme. déc 2012;37(6):1063‑71. 250. Heresi G, Chandra RK. Effects of severe calorie restriction on thymic factor activity and lymphocyte stimulation response in rats. J Nutr. sept 1980;110(9):1888‑93. 251. Dhahbi JM, Kim H-J, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc Natl Acad Sci U S A. 13 avr 2004;101(15):5524‑9. 252. Routtenberg A, Kuznesof AW. Self-starvation of rats living in activity wheels on a restricted feeding schedule. J Comp Physiol Psychol. déc 1967;64(3):414‑21. 253. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 6 juin 2012;15(6):848‑60. 254. Van Leeuwen SD, Bonne OB, Avraham Y, Berry EM. Separation as a new animal model for self-induced weight loss. Physiol Behav. juill 1997;62(1):77‑81. 255. Hao S, Avraham Y, Bonne O, Berry EM. Separation-induced body weight loss, impairment in alternation behavior, and autonomic tone: effects of tyrosine. Pharmacol Biochem Behav. févr 2001;68(2):273‑81. 256. Kim SF. Animal models of eating disorders. Neuroscience. 1 juin 2012;211:2‑12. 257. Willner P. The validity of animal models of depression. Psychopharmacology (Berl). 1984;83(1):1‑16. 258. Veyrat-Durebex C, Montet X, Vinciguerra M, Gjinovci A, Meda P, Foti M, et al. The Lou/C rat: a model of spontaneous food restriction associated with improved insulin sensitivity and decreased lipid storage in adipose tissue. Am J Physiol Endocrinol Metab. mai 2009;296(5):E1120‑32. 259. Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A. 8 déc 1998;95(25):15043‑8. 260. Maltais LJ, Lane PW, Beamer WG. Anorexia, a recessive mutation causing starvation in preweanling mice. J Hered. déc 1984;75(6):468‑72. 261. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. janv 2008;9(1):46‑56.
160
262. Grinspoon S, Miller K, Coyle C, Krempin J, Armstrong C, Pitts S, et al. Severity of osteopenia in estrogen-deficient women with anorexia nervosa and hypothalamic amenorrhea. J Clin Endocrinol Metab. juin 1999;84(6):2049‑55. 263. Bouxsein ML, Myers KS, Shultz KL, Donahue LR, Rosen CJ, Beamer WG. Ovariectomy-induced bone loss varies among inbred strains of mice. J Bone Miner Res Off J Am Soc Bone Miner Res. juill 2005;20(7):1085‑92. 264. Iwaniec UT, Yuan D, Power RA, Wronski TJ. Strain-dependent variations in the response of cancellous bone to ovariectomy in mice. J Bone Miner Res Off J Am Soc Bone Miner Res. juill 2006;21(7):1068‑74. 265. Marcondes FK, Bianchi FJ, Tanno AP. Determination of the estrous cycle phases of rats: some helpful considerations. Braz J Biol Rev Brasleira Biol. nov 2002;62(4A):609‑14. 266. Brihaye Abadie I, de Tournemire R, Alvin P. Anorexie mentale : conséquences sur la croissance et la minéralisation osseuse. Arch Pédiatrie. sept 2003;10(9):836‑40. 267. Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, Hellerstein MK. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am J Physiol Endocrinol Metab. janv 2010;298(1):E108‑16. 268. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. janv 2004;84(1):277‑359. 269. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. janv 2011;121(1):96‑105. 270. Cerqueira FM, da Cunha FM, Caldeira da Silva CC, Chausse B, Romano RL, Garcia CCM, et al. Long-term intermittent feeding, but not caloric restriction, leads to redox imbalance, insulin receptor nitration, and glucose intolerance. Free Radic Biol Med. 1 oct 2011;51(7):1454‑60. 271. Park S, Komatsu T, Hayashi H, Yamaza H, Chiba T, Higami Y, et al. Calorie restriction initiated at a young age activates the Akt/PKC zeta/lambda-Glut4 pathway in rat white adipose tissue in an insulin-independent manner. Age Dordr Neth. déc 2008;30(4):293‑302. 272. Higami Y, Pugh TD, Page GP, Allison DB, Prolla TA, Weindruch R. Adipose tissue energy metabolism: altered gene expression profile of mice subjected to long-term caloric restriction. FASEB J Off Publ Fed Am Soc Exp Biol. févr 2004;18(2):415‑7. 273. Okita N, Hayashida Y, Kojima Y, Fukushima M, Yuguchi K, Mikami K, et al. Differential responses of white adipose tissue and brown adipose tissue to caloric restriction in rats. Mech Ageing Dev. mai 2012;133(5):255‑66. 274. Bray MS, Ratcliffe WF, Grenett MH, Brewer RA, Gamble KL, Young ME. Quantitative analysis of light-phase restricted feeding reveals metabolic dyssynchrony in mice. Int J Obes 2005. juin 2013;37(6):843‑52. 275. Klaus S, Ely M, Encke D, Heldmaier G. Functional assessment of white and brown adipocyte development and energy metabolism in cell culture. Dissociation of terminal differentiation and thermogenesis in brown adipocytes. J Cell Sci. oct 1995;108 ( Pt 10):3171‑80. 276. Rogers NH, Landa A, Park S, Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. déc 2012;11(6):1074‑83. 277. Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 15 nov 2002;277(46):44005‑12. 278. Liu JL, Yakar S, LeRoith D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N. avr 2000;223(4):344‑51.
161
279. Lee M-J, Fried SK. Integration of hormonal and nutrient signals that regulate leptin synthesis and secretion. Am J Physiol Endocrinol Metab. juin 2009;296(6):E1230‑8. 280. Wrann CD, Rosen ED. New insights into adipocyte-specific leptin gene expression. Adipocyte. 1 juill 2012;1(3):168‑72. 281. Baek K, Bloomfield SA. Blocking β-adrenergic signaling attenuates reductions in circulating leptin, cancellous bone mass, and marrow adiposity seen with dietary energy restriction. J Appl Physiol Bethesda Md 1985. 1 déc 2012;113(11):1792‑801. 282. Hamrick MW, Ding K-H, Ponnala S, Ferrari SL, Isales CM. Caloric restriction decreases cortical bone mass but spares trabecular bone in the mouse skeleton: implications for the regulation of bone mass by body weight. J Bone Miner Res Off J Am Soc Bone Miner Res. juin 2008;23(6):870‑8. 283. Gahete MD, Córdoba-Chacón J, Luque RM, Kineman RD. The rise in growth hormone during starvation does not serve to maintain glucose levels or lean mass but is required for appropriate adipose tissue response in female mice. Endocrinology. janv 2013;154(1):263‑9. 284. Zhao T-J, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A. 20 avr 2010;107(16):7467‑72. 285. Zamiri MJ. Effects of reduced food intake on reproduction in mice. Aust J Biol Sci. déc 1978;31(6):629‑39. 286. Seki M, Yamaguchi K, Marumo H, Imai K. Effects of food restriction on reproductive and toxicological parameters in rats--in search of suitable feeding regimen in long-term tests. J Toxicol Sci. déc 1997;22(5):427‑37. 287. Nakai Y, Hamagaki S, Kato S, Seino Y, Takagi R, Kurimoto F. Role of leptin in women with eating disorders. Int J Eat Disord. juill 1999;26(1):29‑35. 288. Casanueva FF, Dieguez C, Popovic V, Peino R, Considine RV, Caro JF. Serum immunoreactive leptin concentrations in patients with anorexia nervosa before and after partial weight recovery. Biochem Mol Med. avr 1997;60(2):116‑20. 289. Nakai Y, Hamagaki S, Takagi R, Taniguchi A, Kurimoto F. Plasma concentrations of tumor necrosis factor-alpha (TNF-alpha) and soluble TNF receptors in patients with bulimia nervosa. Clin Endocrinol (Oxf). sept 2000;53(3):383‑8. 290. Frühbeck G, Jebb SA, Prentice AM. Leptin: physiology and pathophysiology. Clin Physiol Oxf Engl. sept 1998;18(5):399‑419.
162
Résumé
L’anorexie mentale (AM) est un trouble du comportement alimentaire qui se caractérise par une
recherche obsessionnelle de minceur, une forte réduction de la prise alimentaire et une distorsion de
l’image de soi. Elle est associée à de multiples perturbations endocriniennes et métaboliques, et à une
altération de la masse et de la microarchitecture osseuses. Les facteurs et les mécanismes qui
interviennent dans cette maladie sont très mal connus ce qui limite les options thérapeutiques. Il est donc
nécessaire de développer un modèle animal qui reproduise les perturbations physiologiques observées en
AM et permette d’étudier les facteurs associés à l’altération osseuse.
Dans ce but nous avons développé un modèle murin avec une restriction du temps d’accès à
l’alimentation associée à un stress induit par la séparation (Separation-based anorexia, SBA). Cette
phase SBA de 10 semaines est suivie d’une phase de récupération en conditions standards (REC) de 10
semaines. Chez des souris femelles C57Bl/6 en fin de croissance rapide, la phase SBA induit une perte
rapide et importante du poids corporel. L’analyse de la composition corporelle par DEXA révèle une
diminution rapide de près de 40% de la masse grasse ainsi qu’une baisse progressive de la masse maigre
et un arrêt de l’acquisition de la masse osseuse. Au niveau des tibias, la densité minérale corticale et la
microarchitecture trabéculaire sont altérées. L’observation des frottis vaginaux et la mesure des ovaires
révèlent une perturbation importante des fonctions reproductrices. Les tests de tolérance au glucose ont
montré que les souris SBA ont une capacité très élevée à corriger la glycémie. Ces animaux sont
fortement hypoleptinémiques, et l’axe GH-IGF-1 est très perturbé. L’étude de l’expression génique de
différents tissus adipeux a montré une augmentation du niveau des marqueurs de lipogenèse et de
lipolyse, ainsi qu’une forte induction du phénotype « adipocyte brun » dans le tissu adipeux sous-cutané.
Après deux semaines de REC, les souris SBA retrouvent très rapidement leur poids corporel, leurs masses
maigre et grasse. La masse minérale toujours basse à ce stade est corrigée après 10 semaines de REC,
ainsi que la microarchitecture osseuse (étude préliminaire). Tous les autres paramètres étudiés sont
normalisés, sauf l’hypoleptinémie qui étonnement persiste même après 10 semaines de protocole REC et
malgré la normalisation de la masse adipeuse.
D’après ces résultats, on peut conclure que le modèle SBA reproduit de nombreuses perturbations
physiologiques observées en AM. La phase de REC révèle que ces souris ont une importante capacité de
récupération. L’hypoleptinémie persistante pourrait favoriser la récupération.
L’identification des mécanismes impliqués pourrait fournir des pistes thérapeutiques afin de favoriser la
reconstitution du capital osseux des patientes anorexiques.