SASD: the Synthetic Alternative Splicing Databasefor identifying novel isoform from proteomicsFan Zhang1,2, Renee Drabier1*
From Tenth Annual MCBIOS Conference. Discovery in a sea of dataColumbia, MO, USA. 5-6 April 2013
Abstract
Background: Alternative splicing is an important and widespread mechanism for generating protein diversity andregulating protein expression. High-throughput identification and analysis of alternative splicing in the protein levelhas more advantages than in the mRNA level. The combination of alternative splicing database and tandem massspectrometry provides a powerful technique for identification, analysis and characterization of potential novelalternative splicing protein isoforms from proteomics.Therefore, based on the peptidomic database of human protein isoforms for proteomics experiments, our objectiveis to design a new alternative splicing database to 1) provide more coverage of genes, transcripts and alternativesplicing, 2) exclusively focus on the alternative splicing, and 3) perform context-specific alternative splicing analysis.
Results: We used a three-step pipeline to create a synthetic alternative splicing database (SASD) to identify novelalternative splicing isoforms and interpret them at the context of pathway, disease, drug and organ specificity orcustom gene set with maximum coverage and exclusive focus on alternative splicing. First, we extractedinformation on gene structures of all genes in the Ensembl Genes 71 database and incorporated the IntegratedPathway Analysis Database. Then, we compiled artificial splicing transcripts. Lastly, we translated the artificialtranscripts into alternative splicing peptides.The SASD is a comprehensive database containing 56,630 genes (Ensembl gene IDs), 95,260 transcripts (Ensembltranscript IDs), and 11,919,779 Alternative Splicing peptides, and also covering about 1,956 pathways, 6,704diseases, 5,615 drugs, and 52 organs. The database has a web-based user interface that allows users to search,display and download a single gene/transcript/protein, custom gene set, pathway, disease, drug, organ relatedalternative splicing. Moreover, the quality of the database was validated with comparison to other knowndatabases and two case studies: 1) in liver cancer and 2) in breast cancer.
Conclusions: The SASD provides the scientific community with an efficient means to identify, analyze, andcharacterize novel Exon Skipping and Intron Retention protein isoforms from mass spectrometry and interpretthem at the context of pathway, disease, drug and organ specificity or custom gene set with maximum coverageand exclusive focus on alternative splicing.
BackgroundAlternative splicing is a widespread mechanism for gener-ating protein diversity and regulating protein expressionwith multiple splice isoforms. It was thought that at least40-60% of human genes underwent alternative splicing to
encode two or more splice isoforms [1]. Recent advancesin high-throughput technologies have facilitated studies ofgenome-wide alternative splicing. These studies estimatethat the prevalent post-transcriptional gene regulationmechanism affects greater than 95% of roughly 61,000human genes and multiple regulatory processes, includingchromatin modification and signal transduction [2].Furthermore, there are evidences for alternatively splicingevents that are often differentially regulated across tissue
* Correspondence: [email protected] of Academic and Institutional Resources and Technology,University of North Texas Health Science Center, Fort Worth, USAFull list of author information is available at the end of the article
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
types and developmental stages, as well as among indivi-duals and populations, suggesting that individual isoformsmay serve specific spatial or temporal roles [3-5].Alternative splicing is known to be involved in the reg-
ulation of normal physiological functions as well aspathologies. The alternative splicing isoform represents anew class of diagnostic biomarkers. Not only alternativesplicing is thought to increase protein diversity of gen-omes, but also it has been found that splicing variantshave been associated with numerous disease develop-ment and cancer cell growth. For example, David et al.found that aberrant expression of the splicing factorsPTB, hnRNPA1 and hnRNPA2, regulated by the c-Myconcogene, was responsible for the PKM1 to PKM2 switchin cancer [6]. This work helped us understand the alter-native splicing’s role in the cancer cell growth. Eswaranet al. systematically revealed splicing signatures of thethree most common types of breast tumors using RNAsequencing: TNBC, non-TNBC and HER2-positive breastcancer and discovered subtype specific differentiallyspliced genes and splice isoforms not previously recog-nized in human transcriptome. They validated the pre-sence of novel hybrid isoforms of critical molecules likeCDK4, LARP1, ADD3, and PHLPP2 and found that exonskip and intron retention are predominant splice eventsin breast cancer [7]. Yae et al. found that epithelial spli-cing regulatory protein 1 regulates the expression of aCD44 variant isoform (CD44v), and knockdown ofepithelial splicing regulatory protein 1 in CD44v+ cellsresults in an isoform switch from CD44v to CD44 stan-dard (CD44s), leading to reduced cell surface expressionof xCT and suppression of lung colonization. They sug-gested that the epithelial splicing regulatory protein1-CD44v-xCT axis was thus a potential therapeutic targetfor the prevention of metastasis [8].Recent methodological advances, including EST
sequencing, exon array, exon-exon junction array, andnext-generation sequencing of all mRNA transcripts,have made it possible to perform high-throughput alter-native splicing analysis [7]. However, high-throughputidentification and analysis of alternative splicing in theprotein level has several advantages. For example, mRNAabundance in a cell often correlates poorly with theamount of protein synthesized, and proteins rather thanmRNA transcripts are the actual major effector mole-cules in the cell.The combination of alternative splicing database and tan-
dem mass spectrometry provides a powerful technique foridentification, analysis and characterization of potentialnovel alternative splicing protein isoforms from proteomics.In recent years, liquid chromatography tandem mass spec-trometry (LC-MS/MS) has emerged as an innovative analy-tical technology applicable to a wide number of analysesincluding high-throughput identification of proteins [9].
LC-MS/MS proteomics has been used to identify candidatemolecular biomarkers in diverse range of samples, includ-ing cells, tissues, serum/plasma, and other types of bodyfluids. Due to the inherent high variability of both clinicalsamples and MS/MS instruments, it is still challenging toquantify minute changes of proteins that exist in traceamount in response to changes in disease states of biologi-cal samples. Identifying alternative splicing isoform relevantto disease can improve both sensitivity and specificity ofcandidate disease biomarkers because many proteins couldgenerate abundant alternative splicing isoforms in a disease,some of them may be exclusively regulated in a given dis-ease condition, and therefore their identification process isoften sufficient to distinguish between disease samples andcontrols [10].However, without a proper alternative splicing data-
base, tandem mass spectrometry could not discriminateagainst novel alternative splicing peptides [10,11].Searching traditional protein sequence databases whichare commonly used by peptide/protein search enginesuch as 1) IPI [12], 2) NCBI nr (ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz), and 3) UniProt [13] biases theresults towards well-understood protein isoformsbecause they contains a rather small set of splicing pep-tides and not enough for the identification of alternativesplicing isoform from mass spectrometry data.There are also currently several alternative splicing data-
bases, for example, ASTD [14], EID [15,16], Fast DB [17],and ECgene [18]. They are not suitable for being directlyapplied to novel alternative splicing isoform identificationwithout proper modifications made in format and content,because either their coverages are all relatively small inpossible combination of alternative splicing junctions suchas intron-exon, exon-intron, or non-neighboring exon, orsingle intron, or their storage formats make the databasesdifficult to use for mass spectrometry analysis and alterna-tive splicing analysis.Therefore, there is an urgent need to build an alternative
splicing database which can be used by tandem mass spec-trometry to identify the novel alternative splicing isoform.In 2010, we developed the PEPtidomics Protein IsoformDatabase (PEPPI [10], http://bio.informatics.iupui.edu/peppi), a database of computationally-synthesized humanpeptides that can identify protein isoforms derived fromeither alternatively spliced mRNA transcripts or SNP var-iations. We collected genome, pre-mRNA alternative spli-cing and SNP information from Ensembl and synthesizedin silico isoform transcripts that cover all exons and theo-retically possible junctions of exons and introns, as well asall their variations derived from known SNPs.Based on the PEPPI [10], our objective is to design a
new alternative splicing database to 1) provide morecoverage of genes, transcripts and alternative splicing, 2)exclusively focus on the alternative splicing (we will
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
build another database excusive to SNP isoform), and 3)perform context-specific alternative splicing analysis.More coverage means more sensitivity in identifyingnovel alternative splicing isoforms. Exclusive focus onalternative splicing can increase the specificity of theidentification of alternative splicing. Context specificityanalysis can improve our understanding of alternativesplicing’s roles in the context.Splicing events often lead to enormous differences
among isoforms in their sequences and structures and inthe interactions, pathway networks, diseases, drugs, andorgans formed. An enormous body of evidence hasdemonstrated the roles of alternative splicing in determin-ing tissue-specific and species-specific differentiation pat-terns [2]. However, of interest is not only how it canrespond to various signaling pathways, disease treatmentsand drug actions that target the splicing machinery butalso what are the differences in pathways, diseases anddrugs between different isoforms are generally overlooked.Therefore, it is crucial to the advance of basic and medicalresearch that alternative splicing isoforms are interpretedand analyzed on a basis of context: pathway, disease, drugand organ because alternative splicing isoforms occur in aparticular pathway, disease, drug action, or organ and weneed to know about not only the isoforms themselves, butalso their context regarding where they develop and stage.We created the Synthetic Alternative Splicing Data-
base (SASD) for users to detect specific alternative spli-cing isoforms and interpret their context at the pathway,disease, drug and organ level with maximum coverageand exclusive focus on alternative splicing. First, weextracted information on gene structures of all genes inthe Ensembl Genes 71 database [19] and incorporatedthe IPAD database [20]. Then, we compiled artificialsplicing transcripts. Lastly, we translated the artificialtranscripts into alternative splicing peptides.In addition, we built a web interface for users to
browse 1) by genes/proteins, 2) by context (customgene/protein set, signaling and metabolic pathway, dis-ease, drug, and organ specificity).In the end, we presented two case studies: 1) in liver
cancer and 2) in breast cancer to demonstrate that theSASD can enable users to 1) identify novel alternativesplicing isoform, and 2) analyze, characterize, andunderstand the impact of alternative splicing on genesinvolved in drug, disease, pathway, function, and organ-specificity.The SASD, located at http://bioinfo.hsc.unt.edu/sasd is a
comprehensive database containing 56,630 genes(Ensembl gene IDs), 95,260 transcripts (Ensembl transcriptIDs), and 11,919,779 Alternative Splicing peptides(1,200,494 EXON_NM; 1,005,388 E_E_NM; 1,005,368E_I_AS; 1,005,344 I_E_AS; 6,709,352 E_E_AS; and 993,833INTRON_AS), and also covering about 1,956 pathways,
6,704 diseases, 5,615 drugs, and 52 organs incorporatedfrom the IPAD [20].It is the first comprehensive database that can be used
for novel alternative splicing identification on the con-text of pathway, disease, drug and organ specificity orcustom gene set. The maximum coverage and exclusivefocus on alternative splicing provide enough sensitivityand specificity in identifying novel alternative splicingisoforms. The context specificity analysis enables us toimprove our understanding of alternative splicing’s rolesin the context (custom gene set, pathway, disease, drugand organ specificity).The SASD provides the scientific community with an
efficient means to identify, analyze, and characterizenovel Exon Skipping and Intron Retention protein iso-forms from mass spectrometry data. We believe that itwill be useful in annotating genome structures usingrapidly accumulating proteomics data and will assistscientific research on signal transduction pathways regu-lating pre-mRNA, clinical therapy, disease prevention,and drug development.
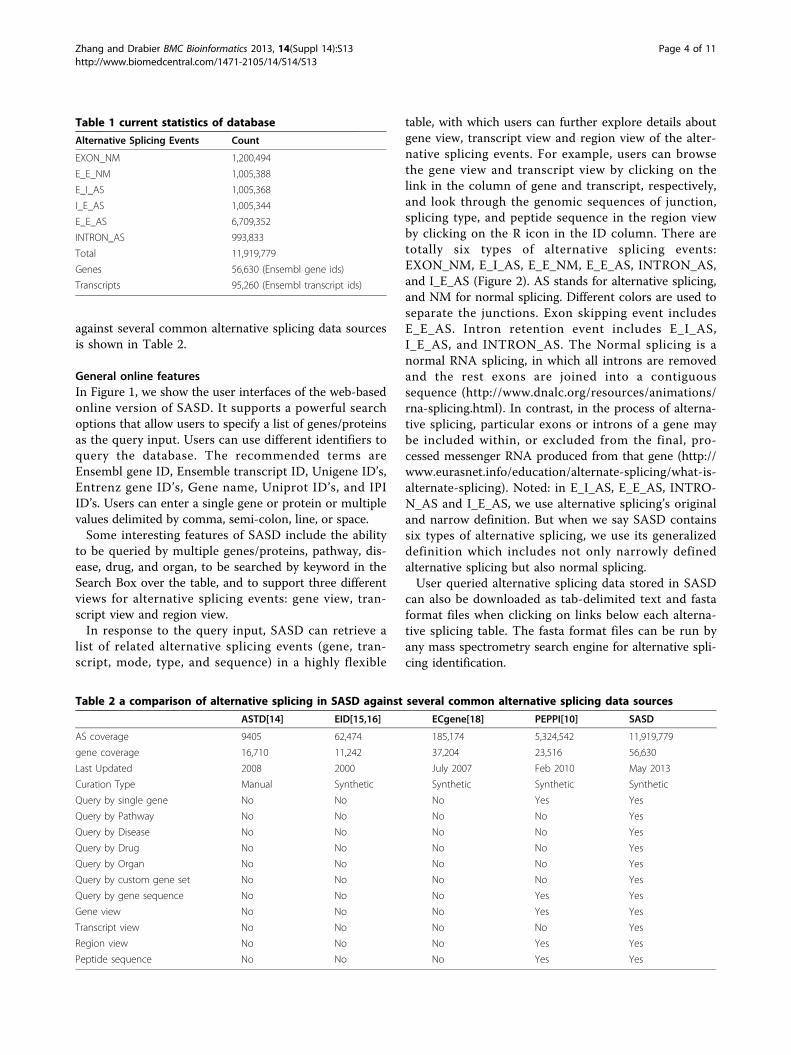
ResultsDatabase content statisticsThe synthetic set of alternative splicing events (ASevents) is derived from Ensembl gene annotation [19].The Ensembl gene set includes both automatic and man-ual annotation, with all transcripts based on experimentalevidence. Alternatively splice from transcripts of anygiven Ensembl gene are computationally synthesized andautomatically annotated to provide a comprehensive listof six types of elementary alternative splicing events.These data can be searched on the website by gene, pro-tein, transcript, peptide sequence, disease, organ, drug,and pathway. The AS events are available for the Homosapiens. In order to reflect specific isoform in the contextof pathway, disease, drug, and organ, the Integrated Path-way Analysis Database (IPAD) [20] is also incorporated.The IPAD [20] is the first comprehensive database forenrichment and inter-association analysis between path-way, disease, drug and organ. It was developed by inte-grating pathway, disease, drug, and organ specificitydatabases including BioCarta[21], KEGG[22], NCI-Nat-ure curated[23], Reactome[24], CTD[25], PharmGKB[26], DrugBank[27], and Homer[28].As of the current release (May 2013), SASD contains
56,630 genes (Ensembl gene IDs), 95,260 transcripts(Ensembl transcript IDs), and 11,919,779 AlternativeSplicing peptides (1,200,494 EXON_NM; 1,005,388E_E_NM; 1,005,368 E_I_AS; 1,005,344 I_E_AS;6,709,352 E_E_AS; and 993,833 INTRON_AS) (Table 1),and also covers about 1,956 pathways, 6,704 diseases,5,615 drugs, and 52 organs incorporated from the IPAD[20]. A comparison of alternative splicing in SASD
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
against several common alternative splicing data sourcesis shown in Table 2.
General online featuresIn Figure 1, we show the user interfaces of the web-basedonline version of SASD. It supports a powerful searchoptions that allow users to specify a list of genes/proteinsas the query input. Users can use different identifiers toquery the database. The recommended terms areEnsembl gene ID, Ensemble transcript ID, Unigene ID’s,Entrenz gene ID’s, Gene name, Uniprot ID’s, and IPIID’s. Users can enter a single gene or protein or multiplevalues delimited by comma, semi-colon, line, or space.Some interesting features of SASD include the ability
to be queried by multiple genes/proteins, pathway, dis-ease, drug, and organ, to be searched by keyword in theSearch Box over the table, and to support three differentviews for alternative splicing events: gene view, tran-script view and region view.In response to the query input, SASD can retrieve a
list of related alternative splicing events (gene, tran-script, mode, type, and sequence) in a highly flexible
table, with which users can further explore details aboutgene view, transcript view and region view of the alter-native splicing events. For example, users can browsethe gene view and transcript view by clicking on thelink in the column of gene and transcript, respectively,and look through the genomic sequences of junction,splicing type, and peptide sequence in the region viewby clicking on the R icon in the ID column. There aretotally six types of alternative splicing events:EXON_NM, E_I_AS, E_E_NM, E_E_AS, INTRON_AS,and I_E_AS (Figure 2). AS stands for alternative splicing,and NM for normal splicing. Different colors are used toseparate the junctions. Exon skipping event includesE_E_AS. Intron retention event includes E_I_AS,I_E_AS, and INTRON_AS. The Normal splicing is anormal RNA splicing, in which all introns are removedand the rest exons are joined into a contiguoussequence (http://www.dnalc.org/resources/animations/rna-splicing.html). In contrast, in the process of alterna-tive splicing, particular exons or introns of a gene maybe included within, or excluded from the final, pro-cessed messenger RNA produced from that gene (http://www.eurasnet.info/education/alternate-splicing/what-is-alternate-splicing). Noted: in E_I_AS, E_E_AS, INTRO-N_AS and I_E_AS, we use alternative splicing’s originaland narrow definition. But when we say SASD containssix types of alternative splicing, we use its generalizeddefinition which includes not only narrowly definedalternative splicing but also normal splicing.User queried alternative splicing data stored in SASD
can also be downloaded as tab-delimited text and fastaformat files when clicking on links below each alterna-tive splicing table. The fasta format files can be run byany mass spectrometry search engine for alternative spli-cing identification.
Table 1 current statistics of database
Alternative Splicing Events Count
EXON_NM 1,200,494
E_E_NM 1,005,388
E_I_AS 1,005,368
I_E_AS 1,005,344
E_E_AS 6,709,352
INTRON_AS 993,833
Total 11,919,779
Genes 56,630 (Ensembl gene ids)
Transcripts 95,260 (Ensembl transcript ids)
Table 2 a comparison of alternative splicing in SASD against several common alternative splicing data sources
ASTD[14] EID[15,16] ECgene[18] PEPPI[10] SASD
AS coverage 9405 62,474 185,174 5,324,542 11,919,779
gene coverage 16,710 11,242 37,204 23,516 56,630
Last Updated 2008 2000 July 2007 Feb 2010 May 2013
Curation Type Manual Synthetic Synthetic Synthetic Synthetic
Query by single gene No No No Yes Yes
Query by Pathway No No No No Yes
Query by Disease No No No No Yes
Query by Drug No No No No Yes
Query by Organ No No No No Yes
Query by custom gene set No No No No Yes
Query by gene sequence No No No Yes Yes
Gene view No No No Yes Yes
Transcript view No No No No Yes
Region view No No No Yes Yes
Peptide sequence No No No Yes Yes
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
Figure 1 Web interface structure. a) Query by genes or proteins. For example, Ensembl Gene ID, Ensembl Transcript ID, UniGene IDs, Entrez geneIDs, gene names, UniProt IDs, UniProt Accessions or IPI IDs are all supported. To enter multiple values, delimit them by comma, semi-colon, line orspace. b) search result. In the alternative splicing analysis table, it shows Query ID, SASD ID, Ensembl Gene ID, Ensembl Transcript ID, Mode, and Type.For each alternative splicing event, users can further browse its region view, gene view and transcript view by clicking on the links at the right cornersof Column ID, Column Gene, and Column Transcript. c) gene view d) transcript view e) region view. f) molecule inter-association. It shows molecule,Gene Symbol, Pathway ID (Disease ID, Drug ID, Organ ID), and Pathway Name(Disease Name, Drug Name, Organ Name).
Figure 2 Six types of as events. The thick boxes represent exons and the thin boxes stand for introns. The orange lines stand for combinationof exon-exon or exon-intron.
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
Page 5 of 11
Case studiesWe presented two case studies: 1)in liver cancer and 2)in breast cancer, to demonstrate that the SASD canenable users: 1) to identify novel alternative splicing iso-form, and 2) to analyze, characterize, and understandthe impact of alternative splicing on genes involved indrug, disease, pathway, function, and organ-specificity.Case study 1: identification of novel alternative splicingisoformsThe Human Liver Proteome Project (HLPP) is a large-scale international collaborative initiative focusing onthe proteomic analysis of the human liver. It aims togenerate a comprehensive protein atlas of the humanliver, uncover the proteomic basis of liver development,physiology and pathology and develop liver-specificdiagnostics and therapeutics. We downloaded eighthuman fetal liver cytoplasm proteome data sets betweenweeks 16 and 24 of gestation available through thehuman liver proteome project web site (http://hlpic.hupo.org.cn). The human fetal liver between weeks 16and 24 of gestation is a major site of fetal hematopoiesisand is at the critical turning point between immigrationand emigration of the hematopoietic system. Proteinprofiling of its unique characteristics can help research-ers understand the immigration and emigration processand improve conventional liver therapy [29].OMSSA [30] is an efficient search engine for identify-
ing MS/MS peptide spectra by searching libraries ofknown protein sequences. OMSSA scores significant hitswith a probability score developed using classical hypoth-esis testing, the same statistical method used in BLAST.OMSSA searches protein libraries formatted for BLAST.In order to use OMSSA to identify alternative splicingisoforms, we first used the program formatdb to createour own alternative splicing sequence library from thefasta format file. Then we set OMSSA search parametersand run OMSSA to search the alternative splicingsequence library against the DTA files we merged fromthe eight raw liver proteome data. Totally, we identified17 Novel alternative splicing isoforms which were undis-covered previously by the PEPPI [10] when we choseonly peptides with at least two hits of samples as truepeptides, of which 5 are left intron retention events, 2right intron retention events, 6 single intron retentionevents, and 4 exon skipping events (Table 3). Bold text isthe left part of the junction and italic text is the rightpart. Splicing site is marked by ^ or (). ‘()’ means the spli-cing site is shared by the left region and right region. Forexample, the first peptide LISQIVSSIT(A)SLR is a syn-thetic product of the ENST00000473885 in geneENSG00000243910 when its third intron is retained andcombined together with its forth exon. The alanine is theshared splicing site between the intron and the exon.Although it can be mapped to ENSP00000449325,
ENSP00000396212, ENSP00000412646, and ENSP00000443475, there is no hit when mapping it to ENSG00000243910’s currently existed proteins. Thus it can beviewed as a novel alternative splicing isoform ofENSG00000243910. OMSSA search engine also providesa very good spectrum display and peak labeling andmatching. For example, the matched MS/MS spectrumof the first peptide is shown in Figure 3.Pathway enrichment analysis of the alternative splicing
variants identified suggested that the involvement ofthese proteins especially in apoptosis. For example, thetight junction protein 2 (ENSG00000119139, TJP2) hasbeen proposed to be a tumor suppressor gene. TJP2protein and/or mRNA expression is either lost ordecreased in pancreatic, prostate, breast and lung adeno-carcinomas, in testicular in situ carcinoma and in lungsquamous carcinoma. Exon 21 in the alternative splicingLL^AKTQNK (non-neighboring combination of exon 15and exon 21) we identified has been involved in 3 of 5isoforms previously identified.Case study 2: identification of novel alternative splicingbiomarkersOne of advantage of using SASD for alterative splicinganalysis is that it supports query by pathway, disease,drug, organ, or user input gene set, which can be usedfor context specific alternative splicing analysis at thelevel of pathway, disease, drug, organ or any user speci-fied gene set. In this case, we demonstrate how to use thenew function of SASD to identify, analyze and character-ize cancer-specific alternative splicing isoforms deriveddirectly from cancer-specific genes. The ‘specific’ heremeans: 1) the genes from which the alternative splicingpeptides are synthesized are linked to breast cancer inprevious findings; and 2) the alternative splicing markersidentified are significantly differentially expressedbetween breast cancer samples and normal samples.Breast cancer is the fifth most common cause of cancer
death (after lung cancer, stomach cancer, liver cancer andcolon cancer). Among women worldwide, breast cancer isthe most common cause of cancer death. In 2012, an esti-mated 226, 870 new cases of invasive breast cancer wereexpected to be diagnosed in women in the U.S., along with63,300 new cases of non-invasive breast cancer. Tradi-tional methods such as mammogram or biopsy can oftendetect a tumor only after a malignancy is advanced andmay have metastasized to other organs. There is an urgentneed for developing new methods for earlier detection ofbreast cancer. Cancer-specific splice isoforms has beenobserved in a few cases. Cancer-specific alterations insplice site selection can affect genes controlling cellularproliferation, cellular invasion, angiogenesis, apoptosis andmultidrug resistance. Therefore, it is essential to developcancer-specific alternative splicing isoform to enable bio-marker discoveries for early detection of breast cancer.
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
We first downloaded 15 breast cancer related genesfrom the Cancer Gene Census[31]: AKT1, BAP1,BRCA2, CCND1, CDH1, EP300, ERBB2, ETV6, GATA3,MAP2K4, NTRK3, PBRM1, PIK3CA, RB1, and TP53.Then we created the 15 genes’ alternative splicingsequence library from the fasta format file which wedownloaded from the database SASD after a query bythe 15 genes in the SASD. Lastly, we used the 15 breastcancer related genes’ alternative splicing sequencelibrary to run OMSSA search against 40 normal plasmaand 40 breast cancer plasma. The plasma protein pro-files of 40 samples from women diagnosed with breastcancer and 40 samples from healthy volunteer womenas control were collected by the Hoosier Oncology
Group (HOG) (Indianapolis, IN, USA). Most of patientsinvolved were diagnosed with a stage II or III or earlierbreast cancer.The following options for OMSSA were used when
identifying MS/MS peptide spectra: -e Trypsin (selectingtrypsin to use for theoretical protein digestion), -y 1(allowing maximum 1 missed cleavage), -hl 10 (Maximum10 peptide hitlist length per spectrum), -he 0.1 (EValuecutoff 0.1), -x human (Homo sapiens to search), -te 2.0(Mass tolerance 2.0Da), -tem monoisotopic (Mass searchtype monoisotopic), -zl 1 (Charge handle: low bound 1),-zh 3 (Charge handle: upper bound 3), -zt 3 (Minimumcharge to start using multiple charged products 3), -to0.8 (Mass tolerance 0.8 Da), -tom monoisotopic (mass
Table 3 17 novel peptide isoforms identified in human fetal liver project
Figure 3 Spectrum of LISQIVSSIT(A)SLR. The blue lines represent b-ions. And the red lines represent y-ions.
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
Page 7 of 11
search type monoisotopic), and -zoh 2 (Maximum 2charge state allowed for product ions).With the one-sided Wilcoxon signed-rank test [32], 8
alternative splicing markers (Table 4) were found differ-entially present (pvalue < 0.05) at cancer state, out ofwhich there are five exon skipping, two single intronretention, and one left intron retention. All the 8 alterna-tive splicing markers are not identified by the traditionalalternative splicing database including the IPI database[12], the NCBI-nr database, and the UniProt knowledgebase [33], and the PEPPI [10].Pathway analysis identified the cancer pathways
including Pancreatic cancer, Pathways in cancer, Pros-tate cancer, Bladder cancer, Endometrial cancer, Non-small cell lung cancer, which are linked with the eightalternative splicing isoforms. The cancer-specific differ-entially expressed variants offer novel biomarker candi-dates that may function in breast cancer progressionand metastasis. For example, the BRCA2 gene belongsto a class of genes known as tumor suppressor genesand is the most well-known gene linked to breast cancerrisk. Bonnet et al. detected 20 variants of BRCA1 orBRCA2 that happened on exons 3, 16, 17, 18 or 25from 17 index cases selected from families undergoingoncogenic consultations [34]. We identified a novelalternative splicing variant of BRCA2 QTPKHISESL-GAEVDPDMSWSSSLATPPTLSSTVLI(G)LLHSSVK onthe exon7 and exon11 as a non-neighboring exon-exonevent.This case study shows that compared to traditional
alternative splicing database, the SASD can be moreuseful in identification of novel alternative splicing mar-kers specific to some pathways, diseases, drugs or organspecification.
DiscussionIn this paper, we have demonstrated that SASD can beused to identify novel alternative splicing isoforms onthe context of pathway, disease, drug and organ specifi-city or custom gene set. Its maximum coverage andexclusive focus on alternative splicing provide enoughsensitivity and specificity in identifying novel alternative
splicing isoforms. The context specificity analysisenables us to improve our understanding of alternativesplicing’s roles in the context (custom gene set, pathway,disease, drug and organ specificity). In Case Study 1, weillustrated the SASD’s ability to identify novel alternativesplicing isoform. In Case Study 2, we demonstrated howto use the new function of SASD to identify cancer-spe-cific markers for distinguishing breast cancer from nor-mal samples.Alternative splicing is a widespread mechanism for
generating protein diversity and regulating proteinexpression. Five basic types of alternative splicing eventsare generally recognized: 1) exon skipping, 2) intronretention, 3) mutually exclusive exons, 4) alternativedonor site, and 5) alternative acceptor site. In exon skip-ping, an exon may be spliced out of the primary tran-script or retained. This is the most common mode inmammalian pre-mRNAs. Intron retention is an eventwhere a sequence is spliced out as an intron or remainsin the mature mRNA transcript. Mutually exclusiveexons event happens when one of two exons is retainedin mRNAs after splicing, but not both. Alternative donorsite is an event where an alternative 5’ splice junction(donor site) is used, changing the 3’ boundary of theupstream exon. And alternative acceptor site is an eventwhere an alternative 3’ splice junction (acceptor site) isused, changing the 5’ boundary of the downstream exon.The SASD does not contain the last three types of
events. But actually, they all can be derived indirectlyfrom the two basic types: exon skipping and intron reten-tion which are included in the SASD in the form ofE_E_AS, and E_I_AS, INTRON_AS, and I_E_AS, respec-tively. For example, if two modes such as E1E3 and E2E4happen at the same time, it is actually a mutually exclu-sive exons event. Alternative 5’ donor site can bedetected by the type E_I_AS such as E1i1 in the SASDwhere donor site i1 is remained. Alternative 3’ acceptorsite can be detected by the type I_E_AS such as i4E5 inthe SASD where acceptor site i4 is remained.In addition, the SASD contains EXON_NM, E_E_NM,
and INTRON_AS. The two normal splicing types:EXON_NM and E_E_NM are included as a contrast to
Table 4 8 cancer-specific peptide markers identified in breast cancer
Peptide sequence gene transcript mode type pvalue h c
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
Page 8 of 11
the alternative splicing events. The INTRON_AS isactually a complement of intron retention.Some artificially synthetized peptides in the database
may not exist biologically. This will reduce computa-tional efficiency but won’t affect the usage of the data-base, as long as the following assumptions are met: ifsome artificially synthetized peptides in the databasedon’t actually exist biologically, they will less likely ornever match with proteome experimental spectra.The SASD is similar to a modified peptide database
for Post Translational Modification (PTM) identificationfrom MS/MS. The first approach to PTM identificationproposed by Yates et al. [35], enumerating all possiblemodifications for each peptide from the database stillworks very well with small database. Enumerating allpossible mutations and modifications in the databasemakes the database prohibitively large and is computa-tionally expensive, so that using this kind of database tosearch for modifications remains limited to smallerdatabases.One advantage of using SASD is context-based alter-
native splicing identification. Users can build a relativelysmall database based on the context (pathway, disease,disease, drug, and other user input gene set). This fea-ture enables users not only to identify context-specificalternative splicing, but also with significantly-improvedcomputational efficiency.Fortunately, all search engines provide their own scores
as thresholds such as expectation value which is thenumber of matches with equal or better scores that areexpected to occur by chance alone. In order to increasethe true discovery rate for these artificially synthetizedpeptides that don’t exit biologically and that is less likelyor never to be identified by proteome experiments, whenusing SASD, we recommend to use 1) the p-value (Mas-cot) or evalue (OMSSA) as thresholds provided by var-ious search engines and 2) cross-validation experiments.That is, if a synthetic AS peptide is identified with signifi-cant score and from more than n samples (for example,n = 2), we say the synthetic AS peptide is identified andthe synthetic AS peptide exists biologically. More strin-gent threshold or more experiment validations fromother labs or by other methods such as PCR are definitelyneeded for further validation because SASD is an in-silicodatabase after all.
MethodsDatabase pipelineThe overall pipeline of SASD comprises three steps: 1)extracting information on gene structures of all genes inthe human genome and incorporating the IPAD data-base [20], 2)compiling artificial splicing transcripts, and3) translating the artificial transcripts into alternativesplicing peptides.
In the first step, we use the BioMart to extract infor-mation on all human genes in the Ensembl [19] fromthe Homo sapiens genes dataset (GRCh37.p10) in theEnsembl Genes 71 database. We then extract informa-tion on each human gene’s position, name, exon/introncoordinates, exon phase, sequences, and annotation. Theinformation is organized in a relational database hostedin a local SQL server 2012 database server.In the second step, we generate artificial splicing tran-
script (AST), which is an exhaustive compilation of twocategories of peptides (the first is the peptides translatedfrom all single exons and introns, the second is the pep-tides that covers all theoretically possible exon/intronjunction regions of all genes in the human genome). Withthese two categories of peptides, both the whole sequenceof the genome and all possibilities of alternative splicingare covered. In addition to single exon(EXON_NM) andsingle intron (INTRON_AS), four types of exon/intronsequence junctions are considered when generating ASTs:intron-exon (I_E_AS, left intron retention junction), exon-intron (E_I_AS, right intron retention junction), neighbor-ing exon-exon (E_E_NM, normal splicing junction) andnon-neighboring exon-exon (E_E_AS, exon skipping junc-tion). For each type, 120 nucleotides both upstream anddownstream of the joined sequence beside the junctionsite are extracted, resulting in a computationally synthe-sized virtual transcript of 240 nucleotides long. Determina-tion of the number 120 is based on the length distributionof fragment obtained from protein digestion in MS/MSexperiments. The boundaries where two components arespliced are known as splicing sites. Totally, there are threetypes of common splicing events: Normal Splicing, ExonSkipping, and Intron Retention in the SASD. The NormalSplicing includes single exon (EXON_NM) and neighbor-ing exon-exon junction (E_E_NM), the Exon Skippingincludes non-neighboring exon-exon junction (E_E_AS),and the Intron Retention includes single intron (INTRO-N_AS), left intron retention junction (I_E_AS), rightintron retention junction (E_I_AS).In the third step, we directly use the phase to translate
the sequence for the exons with the phase information inEnsembl transcript. For the exons without the phase infor-mation in Ensembl transcript, three translations are firstderived, each of which corresponds to a possible openingreading frame (ORF) and generates one peptide. Then, theautomatic translation procedure calculates the length ofthe peptide that map across the splicing site. Lastly, thetranslation which contains the longest peptide is reservedas alternative splicing peptide for SASD.
Online SASD server designThe online version of SASD database is a typical 3-tierweb application [10], with an SQL Server2012 databaseat the backend database service layer as Data Access
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
Page 9 of 11
Tier, Apache/PHP server scripts to the middlewareapplication web server layer as Logic or ApplicationTier, and CSS-driven web pages presented on the brow-ser as Presentation Tier.The result tables derived from the data generation
steps are imported into the SQL Server2012 database(Figure 4). The pathway-gene, disease-gene, drug-gene,organ-gene, protein-gene, gene-gene mapping tablesenable users to query the database with different IDs.
Pathway analysisPathway analysis is performed using the following data-bases: Integrated Pathway Analysis Database (IPAD)(http://bioinfo.hsc.unt.edu/ipad/) [36].
ConclusionWe developed SASD as a complement to the currentlyexisting alternative splicing databases to perform novelalternative splicing identification on a biological con-text such as pathway, disease, drug and organ specifi-city or custom gene set with maximum coverage andexclusive focus on alternative splicing. SASD integratesthe gene structure from Ensembl [19] and the context(pathway, disease, drug and organ specificity) fromIPAD [20].A single gene/transcript/protein, custom gene set,
pathway, disease, drug, organ related alternative splicingcan be searched, displayed, and downloaded from ouronline user interface. The current SASD database canhelp users discover novel alternative splicing from massspectrometry and interpret them at the context of path-way, disease, drug and organ specificity or custom gene
set with maximum coverage and exclusive focus onalternative splicing. We believe that it could help gener-ate novel hypothesis on molecular risk factors and mole-cular mechanisms of complex diseases, leading toidentification of potentially highly specific protein bio-markers. Lastly, our database was demonstrated by com-parison to other known databases and two case studies.
Competing interestsThe authors declare that they have no competing financial interests.
Authors’ contributionsRD conceived the initial work and designed the method for the databaseconstruction. FZ generated the datasets, developed the statistics method,the database backend and the web-based interface, and performed thealternative splicing analyses for the case studies. All authors are involved inthe drafting and revisions of the manuscript.
AcknowledgementsWe thank Hoosier Oncology Group for collecting breast cancer plasmasamples and Drs. Mu Wang and Jake Chen for providing LC/MS/MSproteomics experimental data for this analysis. We also thank MatthewAndrews, Kathryn Taylor, Brian Denton, Woody Hagar, Anthony Tissera, andLynley Dungan for help with database design and web development.
DeclarationsThe publication costs for this article were funded by the bioinformaticsprogram in University of North Texas Health Science Center.This article has been published as part of BMC Bioinformatics Volume 14Supplement 14, 2013: Proceedings of the Tenth Annual MCBIOS Conference.Discovery in a sea of data. The full contents of the supplement are availableonline at http://www.biomedcentral.com/bmcbioinformatics/supplements/14/S14.
Authors’ details1Department of Academic and Institutional Resources and Technology,University of North Texas Health Science Center, Fort Worth, USA.2Department of Forensic and Investigative Genetics, University of NorthTexas Health Science Center, Fort Worth, USA.
Figure 4 Relational metadata model. The datasets were derived by the data generation pipeline.
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13
References1. Modrek B, Lee C: A genomic view of alternative splicing. Nature genetics
2002, 30(1):13-19.2. Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ: Alternative
splicing: a pivotal step between eukaryotic transcription and translation.Nature reviews Molecular cell biology 2013, 14(3):153-165.
3. Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ,Slobodeniuc V, Kutter C, Watt S, Colak R, et al: The evolutionary landscapeof alternative splicing in vertebrate species. Science 2012,338(6114):1587-1593.
4. Yeo G, Holste D, Kreiman G, Burge CB: Variation in alternative splicingacross human tissues. Genome biology 2004, 5(10):R74.
5. Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ:Deciphering the splicing code. Nature 2010, 465(7294):53-59.
6. David CJ, Chen M, Assanah M, Canoll P, Manley JL: HnRNP proteinscontrolled by c-Myc deregulate pyruvate kinase mRNA splicing incancer. Nature 2010, 463(7279):364-368.
7. Eswaran J, Horvath A, Godbole S, Reddy SD, Mudvari P, Ohshiro K,Cyanam D, Nair S, Fuqua SA, Polyak K, et al: RNA sequencing of cancerreveals novel splicing alterations. Scientific reports 2013, 3:1689.
8. Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ,Wada T, Masuko T, Mogushi K, Tanaka H, et al: Alternative splicing of CD44mRNA by ESRP1 enhances lung colonization of metastatic cancer cell.Nature communications 2012, 3:883.
9. Vogeser M, Parhofer KG: Liquid chromatography tandem-massspectrometry (LC-MS/MS)–technique and applications in endocrinology.Experimental and clinical endocrinology & diabetes: official journal, GermanSociety of Endocrinology [and] German Diabetes Association 2007,115(9):559-570.
10. Zhou A, Zhang F, Chen JY: PEPPI: a peptidomic database of humanprotein isoforms for proteomics experiments. BMC bioinformatics 2010,11(Suppl 6):S7.
11. Edwards NJ: Novel peptide identification from tandem mass spectrausing ESTs and sequence database compression. Molecular systemsbiology 2007, 3:102.
12. Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R: TheInternational Protein Index: an integrated database for proteomicsexperiments. Proteomics 2004, 4(7):1985-1988.
13. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res 2009,37(Database):D169-174.
14. Koscielny G, Le Texier V, Gopalakrishnan C, Kumanduri V, Riethoven JJ,Nardone F, Stanley E, Fallsehr C, Hofmann O, Kull M, et al: ASTD: TheAlternative Splicing and Transcript Diversity database. Genomics 2009,93(3):213-220.
15. Shepelev V, Fedorov A: Advances in the Exon-Intron Database (EID).Briefings in bioinformatics 2006, 7(2):178-185.
16. Saxonov S, Daizadeh I, Fedorov A, Gilbert W: EID: the Exon-IntronDatabase-an exhaustive database of protein-coding intron-containinggenes. Nucleic Acids Res 2000, 28(1):185-190.
17. de la Grange P, Dutertre M, Martin N, Auboeuf D: FAST DB: a websiteresource for the study of the expression regulation of human geneproducts. Nucleic Acids Res 2005, 33(13):4276-4284.
18. Lee Y, Lee Y, Kim B, Shin Y, Nam S, Kim P, Kim N, Chung WH, Kim J, Lee S:ECgene: an alternative splicing database update. Nucleic Acids Res 2007,35(Database):D99-103.
19. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D,Clapham P, Coates G, Fairley S, et al: Ensembl 2013. Nucleic Acids Res 2013,41(Database):D48-55.
21. Hanyu H, Inoue Y, Sakurai H, Kanetaka H, Nakamura M, Miyamoto T, Sasai T,Iwamoto T: Voxel-based magnetic resonance imaging study of structuralbrain changes in patients with idiopathic REM sleep behavior disorder.Parkinsonism & related disorders 2012, 18(2):136-139.
22. Victor KG, Rady JM, Cross JV, Templeton DJ: Proteomic Profile ofReversible Protein Oxidation Using PROP, Purification of ReversiblyOxidized Proteins. PloS one 2012, 7(2):e32527.
23. Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH:PID: the Pathway Interaction Database. Nucleic Acids Res 2009,37(Database):D674-679.
24. Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M,Garapati P, Gopinath G, Jassal B, et al: Reactome: a database of reactions,pathways and biological processes. Nucleic Acids Res 2011, 39(Database):D691-697.
25. Davis AP, King BL, Mockus S, Murphy CG, Saraceni-Richards C, Rosenstein M,Wiegers T, Mattingly CJ: The Comparative Toxicogenomics Database:update 2011. Nucleic Acids Res 2011, 39(Database):D1067-1072.
26. McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE: Frompharmacogenomic knowledge acquisition to clinical applications: thePharmGKB as a clinical pharmacogenomic biomarker resource.Biomarkers in medicine 2011, 5(6):795-806.
27. Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C,Neveu V, et al: DrugBank 3.0: a comprehensive resource for ‘omics’research on drugs. Nucleic Acids Res 2011, 39(Database):D1035-1041.
28. Zhang F, Chen JY: HOMER: a human organ-specific molecular electronicrepository. BMC bioinformatics 2011, 12(Suppl 10):S4.
29. Ying W, Jiang Y, Guo L, Hao Y, Zhang Y, Wu S, Zhong F, Wang J, Shi R,Li D, et al: A dataset of human fetal liver proteome identified bysubcellular fractionation and multiple protein separation andidentification technology. Molecular & cellular proteomics: MCP 2006,5(9):1703-1707.
30. Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X,Shi W, Bryant SH: Open mass spectrometry search algorithm. Journal ofproteome research 2004, 3(5):958-964.
31. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N,Stratton MR: A census of human cancer genes. Nature reviews Cancer2004, 4(3):177-183.
33. Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S,Gasteiger E, Huang H, Lopez R, et al: The Universal Protein Resource(UniProt): an expanding universe of protein information. Nucleic Acids Res2006, 34(Database):D187-191.
34. Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P,Chevrier A, Dugast C, Layet V, et al: Screening BRCA1 and BRCA2unclassified variants for splicing mutations using reverse transcriptionPCR on patient RNA and an ex vivo assay based on a splicing reporterminigene. Journal of medical genetics 2008, 45(7):438-446.
35. Yates JR, Eng JK, McCormack AL: Mining genomes: correlating tandemmass spectra of modified and unmodified peptides to sequences innucleotide databases. Analytical chemistry 1995, 67(18):3202-3210.
doi:10.1186/1471-2105-14-S14-S13Cite this article as: Zhang and Drabier: SASD: the Synthetic AlternativeSplicing Database for identifying novel isoform from proteomics. BMCBioinformatics 2013 14(Suppl 14):S13.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Zhang and Drabier BMC Bioinformatics 2013, 14(Suppl 14):S13http://www.biomedcentral.com/1471-2105/14/S14/S13