Use of inverse gas chromatography to account for the pervaporationperformance in monitoring the oxidation of primary alcohols
Saad Moulay ∗, Hassiba Benguergoura, Taıeb Aouak
Facult´ e des Sciences de l’Ing´ enieur, D´ epartement de Chimie Industrielle, Universit´ e de Blida, Route de Soumˆ aa, B. P. 270, Blida 09000, Algeria
Received 9 May 2006; received in revised form 29 July 2006; accepted 8 September 2006
Available online 2 October 2006
Abstract
The oxidation of n-propanol and n-butanol to their corresponding aldehydes was monitored by the pervaporation technique. Mass transferphenomenon that occurs in the pervaporation process was confirmed by the results of inverse gas chromatography. Polydimethylsiloxane (PDMS),
a hydrophobic polymer widely employed as a membranein pervaporation technique, was evaluated as a stationary phase in this study. The retention
times of the different molecules probes (n-propanol, n-butanol, propionaldehyde, and butyraldehyde), molecules involved as reactants and products
in the oxidation reaction, gave an insight into the extent of the interactions between each of these molecules and the stationary phase. The infinite
dilution conditions allowed to measure the infinite dilution activity coefficient, γ ∞, and the specific retention volume, V 0g , and to estimate the
Flory-Huggins parameter interactions, χ∞12. The magnitudes of these parameters threw some light on the permselectivity of the membranes in the
S. Moulay et al. / J. Chromatogr. A 1135 (2006) 78–84 79
Fig. 1. Set-up of the online pervaporation for monitoring the oxidation reaction
of n-alcohols.
it has been applied to determine the solubility parameters of
polymers and oligomers [10,11], the diffusion coefficients in
crosslinked polymers [12], the polymer–polymer interaction
parameters [13], and the characterization of conducting poly-
mers [14,15]. Recently, we used the inverse gas chromatography
(ICG) technique to validate the pervaporation results of the
breakdown of the microemulsions using PDMS as membrane[16].
2. Experimental
2.1. Chemicals
n-Propanol and n-butanol in 99.5% purity were purchased
from Merck and Panreac Quimica, respectively, and were dis-
tilled in the presence of CaCl2 before use. Potassium dichro-
mate was provided by Fluka. Butyraldehyde, propionalde-
hyde, n-butylbutyrate, and n-propylpropionate in 99% purity,
employed as standards for chromatography analysis, were
purchased from Acros organics. Rhodorsil RTV-2 (a two-component silicon elastomer that crosslinks at room temper-
ature on mixing with a curing agent; RTV means “room
temperature vulcanizing”) and its curing agent, were sup-
plied by Rhodia (ex-Rhone-Poulenc); Rhodorsil RTV-2 con-
sisted of ,-dihydroxypolydimethylsiloxane and the curing
agent consisted of 10% of a catalyst based on dibutyltin lau-
rate, [CH3(CH2)10CO2]2Sn[(CH2)3CH3]. The crosslinking of
PDMS occurred via a condensation mechanism.
2.2. Pervaporation technique
2.2.1. ApparatusThe pervaporation apparatus usedin this workwas previously
described [8]. It comprised the following parts: stainless steel
pervaporation cell (capacity of 125 cm3); pervaporation pyrex-
made receiving setfittedwith vaportraps; primary vacuum pump
(10−1 mmHg), Trivac type.
2.2.2. Membrane preparation
The PDMS membranes were prepared as follows: an amount
necessary for the confection of a membrane with a thickness
of about 125m and an area of about 25 cm2, was mixed with
2% by weight of the crosslinking agent (curing agent). After
a good homogenization at room temperature, this mixture was
spread evenly over a clean teflon-plate surface by means of a
hand coater. A transparent film was obtained after drying for
24 h at room temperature followed by an annealing at tem-
perature of 50 ◦C for an additional 8 h. The thickness of the
membrane was estimated to be 125±1m with a micrometer
(Micro-Italiana).
For every oxidation reaction experiment, a newly made mem-
brane with identical characteristics was employed.
2.2.3. Oxidation reaction on-line with pervaporation
An amount of alcohol was charged into the pervaporation
cell (Fig. 1) and heat was supplied to the system. As soon as
the working temperature was reached, a magnetic stirring was
started. Then, the required quantity of the oxidizing solution
(K2Cr2O7 + H2SO4) was added dropwise within a period of
20 min. The pervaporation vacuum (0.5 Torr) was set at the first
drop of the oxidizing solution. The vapor permeate coming out
from downstream of the cell will condense in the provided cold
traps (traps were immersed in liquid nitrogen), and the liquid
pervaporate was then obtained. The reaction time was arbitrar-ily fixed at 90 min.
For a matter of convenience, the oxidation reactions of n-
propanol and n-butanol are herein referred to as P and B, respec-
tively.
2.2.4. Characterization
The liquid permeate was analyzed by gas chromatography
using Shimadzu-14B with the following parts: Thermal conduc-
tivity detector (TCD, katharometer) as a detection technique, N2
as a carrier gas, Carbowax 20M as a stationary phase, column
(packed with Chromosorb W, 80 mesh, as a stationary phase
solid support, 2 m in length, 3.17 mm in diameter); injection-and detector-ports temperature, 180 ◦C; column temperature,
130 ◦C.
The swelling degrees of the membrane with n-propanol, n-
butanol, propionaldehyde, and butyraldehyde, were determined
by Eq. (1):
S (%) =
w−w0
w0
× 100 (1)
wherew0 andw are the sample weight before and after swelling.
Before weightingand afterthe swellingoperation,the membrane
was rapidly wiped off in a cold area to prevent theloss of swelling
probe. The total mass flux J and the molar flux J M were providedby Eqs. (2) and (3), respectively:
J (g/h m2) =W
tA(2)
J M (moles/h m2) =N
tA(3)
where W is the weight of the condensate (g), N the sum of the
number of moles of formed aldehyde and the number of moles of
alcohols in the permeate, t the trapping time (h) and A the surface
area of the membrane (m2); the surface area of the membrane
was 24.62 cm2
.
7/29/2019 sdarticle 7
http://slidepdf.com/reader/full/sdarticle-7 3/7
80 S. Moulay et al. / J. Chromatogr. A 1135 (2006) 78–84
2.3. Inverse gas chromatography technique
2.3.1. Equipment preparation
Gas chromatography used in this part of work was the
one cited above, Shimadzu-14B fitted with a TCD detector
(katharometer); N2 was as a carrier gas flowing at 5 mL/min
and a soap-film meter was employed to measure the gas flow
rates. For binary systems, the molecule probes were injected
individually with a 10L Hamilton syringe. Also, to assess
the effect of one component on the activity of the other, that
is the aldehyde in the presence of its corresponding alcohol, the
mixtures of two probes (propionaldehyde and n-propanol, and
butyraldehyde and n-butanol) were injected. These two mixtures
are herein designated M PP and M BB, respectively. The injection-
and detector-port temperatures were set at 160 ◦C. The column
was prepared as follows:
1. Chromosorb W was employed as a stationary phase solid
support (80 mesh, specific area of 1 m2 /g). Prior to use, it
was deactivatedwith an acidic washingfollowed by treatmentwith dimethyldichlorosilane (DMCS).
2. Impregnation of the solid support: Approximately 0.2 g of
polymer (PDMS employed was thenon-crosslinked one) was
dissolved in 20 mL of ethyl acetate, and the solution was
placed into a 250 mL round-bottomed flask. After addition
of 2 g of the solid support, the slurry was well swirled before
evaporation of the solvent to dryness by means of a rota-
evaporator. Afterwards, the impregnated support was further
dried in vacuo in a drying-oven at a temperature of 40 ◦C for
48 h. The dried solid was then sieved before the following
step.
3. Column packing: A 1.50 m× 3.17 mm stainlesssteel columnwas thoroughly filled with the impregnated solid support.
To ensure a homogeneous packing, the filling operation was
secured with an electric vibrator.
4. Column conditioning: After the packing process, the column
was subjected to heat at a temperature of 200 ◦C for 48h
under a gas carrier flow rate of 3 mL/min.
2.3.2. Inverse gas chromatography parameters
The specific retention volume V 0g , the normalized volume of
the carrier gas necessary to elute the molecule probe (solute) out
of the column, was calculated using Eqs. (4) and (5):
V 0g =jF (t R − t 0)
W s
273.15
T c(4)
j =3
2
(P in/P ou)2 − 1
(P in/P ou)3 − 1(5)
where t R is the measured retention time at maximum peak of
the solute, t 0 the retention time of methane, j the James-Martin
pressure drop correction, F the saturated carrier gas flow rate, T cthe column temperature, W s the weight of the stationary phase,
Pin and Pou the pressures at the inlet and outlet, respectively. W swas taken 0.2 g, assuming that all the coated packing ends up in
the column.
Theinfinite dilution activity coefficient of the molecule probe
(γ ∞1 ), was estimated from the solute specific retention volume
using the following equation [17], Eq. (6):
ln(γ ∞1 ) = ln
273.15R
M 1P 01V
0g
−P 01 (B11 − V 1)
RT (6)
where R is the universal gas constant, M 1 the molar mass of thesolute, P 01 the saturated pressure of solute at temperature T , V 1the molar volume of the solute, B11 the second virial coefficient
of the probe, its values at the different temperatures were taken
from [18].
The Flory Huggins interaction parameter χ∞12 is related to the
specific retention volume according to the following relation
[19], Eq. (7), where d 1 and d 2 are the densities of the solute and
the stationary phase, respectively, and V 2 is the molar volume of
the stationary phase. By assuming ln d 1 / d 2 =0 and V 1 /V 2→0
[19], Eq. (7) can be simplified to Eq. (8).
χ∞12 = ln 273.15R
M 1P 01V
0g
−P 0
1(B
11− V
1)
RT − ln
d 1d 2−
1−V
1V 2
(7)
χ∞12 = ln γ ∞1 − 1 (8)
3. Results and discussion
3.1. Sorption/pervaporation
The swelling capacity of the PDMS membrane with the reac-
tants, n-propanol and n-butanol, and their corresponding prod-
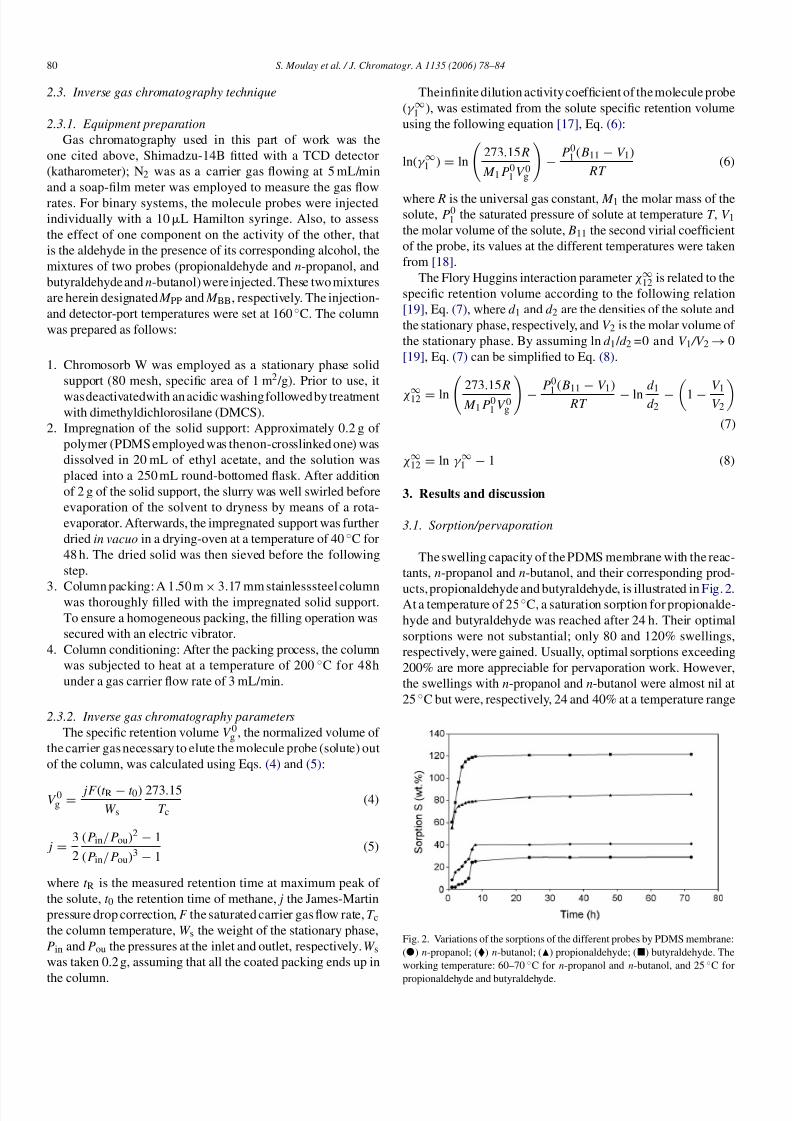
ucts, propionaldehyde and butyraldehyde, is illustrated in Fig. 2.At a temperature of 25 ◦C, a saturation sorption for propionalde-
hyde and butyraldehyde was reached after 24 h. Their optimal
sorptions were not substantial; only 80 and 120% swellings,
respectively, were gained. Usually, optimal sorptions exceeding
200% are more appreciable for pervaporation work. However,
the swellings with n-propanol and n-butanol were almost nil at
25 ◦C but were, respectively, 24 and 40% at a temperature range
Fig. 2. Variations of the sorptions of the different probes by PDMS membrane:
() n-propanol; () n-butanol; () propionaldehyde; () butyraldehyde. The
working temperature: 60–70 ◦C for n-propanol and n-butanol, and 25 ◦C for
propionaldehyde and butyraldehyde.
7/29/2019 sdarticle 7
http://slidepdf.com/reader/full/sdarticle-7 4/7
S. Moulay et al. / J. Chromatogr. A 1135 (2006) 78–84 81
Fig. 3. Variations of the total mass flux J for systems P and B vs. temperature.
Reaction time, 90 min.
of 60–70 ◦C after 30 h. The course of the oxidation reaction
of n-propanol and n-butanol to their aldehydes was monitored
by the on-line pervaporation to avoid the over-oxidation which
may afford carboxylic acids, hemiacetals, and the corresponding
esters. In fact, the analysis of the permeate by chromatographyrevealed the presence of aldehyde as the exclusive product and
the non-reacted alcohol under the actual oxidation reaction con-
ditions. The total mass and molar fluxes J and J M as a function
of temperature are shown in Figs. 3 and 4. It can be noticed that
the total fluxes were found to increase with the reaction tem-
perature and more significantly beyond a temperature of 70 ◦C.
Raising thetemperature of thefeed would speed up theoxidation
reaction leading to more substantial formation of the aldehyde
and, consequently, the flux rises. In the case of the system B,
the fluxes were higher than those for the system P which is in
a good agreement with the fact that to the better sorption cor-
responds the higher flux [20]. Fig. 5 shows the evolution of the
aldehyde concentration in the pervaporate as a function of thereaction temperature. In both cases, B and P, the aldehyde con-
tent increased at temperatures lower than 65 ◦C, but dropped
beyond the latter temperature. The ester started to form at tem-
peratures higher than 65 ◦C. Overall, the butyraldehyde fraction
was nearly always higher than that of the propionaldehyde.
Again, this can be explained by the difference in the sorption
capacities of these aldehydes by PDMS membrane.
Fig. 4. Variations of thetotalmolar flux J M for systems P and B vs. temperature.
Reaction time, 90 min.
Fig. 5. Variations of the concentration of aldehyde in the permeate (wt.%) vs.
temperature: () propionaldehyde; () butyraldehyde. Reaction time, 90 min.
3.2. IGC results
The inverse gas chromatography technique was used as analternative to validate our results in the oxidation reaction of pri-
mary alcohols to their corresponding aldehydes using the perva-
poration technique. Although the pervaporation and the inverse
gas chromatographytechniquesare quitedifferent in theirproper
functions, they share one feature which is the involvement of the
sorption-diffusion-desorption phenomenon. On the one hand,
the probe in IGC isin a gaseous state, but it isin a liquid phase in
pervaporation (in the feed). On the other hand, its concentration
in theformer techniqueis lowerthan that in thelatter one. Yet,the
performances of both techniques are linked to the interactions
between the probe and the polymeric material (as membrane in
pervaporation and as stationary phase in IGC). Thus, an IGCstudy would give a first insight into the magnitude of the inter-
action of the probe with membrane in pervaporation technique
and, consequently, into the permselectivity performance.
The methodology of this present study was first to assess
the interactions within a binary system consisting of a
solute (molecule probe) and the solvent (polymeric stationary
phase), and, second, to consider those within M PP /PDMS and
M BB /PDMS systems. The working temperatures were between
40and100 ◦C. The differentresults aregatheredin Tables1and2
in which the results for only the lowest and highest temperatures
are given.
The profile of the plot ln V g = f (1/ T ) is claimed to bea straight
line with a positive slope for temperatures higher than the glasstransition temperature, T g, of the polymer used as a sorbent.
In our case, the working temperatures were higher than the T gof PDMS (−123 ◦C) and the results indicate that the expected
straight lines were indeed observed for all systems studied.
Figs. 6–9 illustrate the plots ln V g = f (1/ T ) for the different sor-
bates. With the exceptionof propionaldehyde,straight lineswere
found for all sorbates; the curves of propionaldehyde in the sys-
tems showed a deviation starting at 60 ◦C as can be seen in
Fig. 7. A general observation is that the retention volume of
every sorbate remained unaffected by the presence of the other
one beyond temperatures higher than 80–90 ◦C; however, the
effect of the presence of other component gradually manifested
7/29/2019 sdarticle 7
http://slidepdf.com/reader/full/sdarticle-7 5/7
82 S. Moulay et al. / J. Chromatogr. A 1135 (2006) 78–84
Table 1
Thermodynamic and chromatographic results for the sorbates: n-propanol and propionaldehyde