ORIGINAL CONTRIBUTION Semi-batch control over functional group distributions in thermoresponsive microgels Paniz Sheikholeslami & Christopher M. Ewaschuk & Syed Usman Ahmed & Benjamin A. Greenlay & Todd Hoare Received: 28 November 2011 / Revised: 7 March 2012 / Accepted: 20 March 2012 / Published online: 3 April 2012 # Springer-Verlag 2012 Abstract Thermosensitive poly(N-isopropylacrylamide-co- methacrylic acid) (poly(NIPAM-co-MAA)) microgels were prepared via semi-batch free radical copolymerization in which the functional monomer (methacrylic acid) was con- tinuously fed into the reaction vessel at various speeds. Microgels with the same bulk MAA contents (and thus the same overall compositions) but different radial functional group distributions were produced, with batch copolymer- izations resulting in core-localized functional groups, fast- feed semi-batch copolymerizations resulting in near- uniform functional group distributions, and slow-feed semi-batch copolymerizations resulting in shell-localized functional groups. Functional group distributions in the microgels were probed using titration analysis, electropho- resis, and transmission electron microscopy. The induced functional group distributions have particularly significant impacts on the pH-induced swelling and cationic drug bind- ing behavior of the microgels; slower monomer feeds result in increased pH-induced swelling but lower drug binding. This work suggests that continuous semi-batch feed regimes can be used to synthesize thermoresponsive microgels with well-defined internal morphologies if an understanding of the relative copolymerization kinetics of each comonomer relative to NIPAM is achieved. Keywords Microgels . Semi-batch polymerization . Copolymerization kinetics . Poly(N-isopropylacrylamide) . Functional group distributions Introduction Microgels have recently attracted wide interest in the liter- ature owing to their attractive mix of hydrogel and nano- particle properties [1]. As hydrogels, microgels have a well- defined three-dimensional structure and can shrink and swell according to changes in the solvent conditions; as nanoparticles, microgels can be flocculated at high salt concentrations, can be assembled into two- or three- dimensional nanostructures, and respond quickly to changes in their environment. “Smart” microgels that exhibit well- defined phase transitions as a function of pH [2–5], temper- ature [6, 7], the concentration of a particular chemical [8, 9], or other physical stimuli [10] have attracted particular atten- tion due to their range of potential applications in drug delivery, sensors, optics, purification technologies, and oth- er industries [11–16]. For microgels exhibiting such phase transition behavior, both the chemical composition and the physical morphology present locally throughout the micro- gel play significant roles in determining the types of phys- ical [17–20] and biological [9, 14, 21] responses achieved, making control over microgel morphology a key area of research. Local compositional distributions in microgels are typi- cally controlled by performing multi-stage copolymeriza- tions to create core–shell particles with distinct local compositions. Often, cores and shells have been designed with distinct smart properties that either offer local enhance- ment or frustration in swelling responses as a function of an Electronic supplementary material The online version of this article (doi:10.1007/s00396-012-2642-x) contains supplementary material, which is available to authorized users. P. Sheikholeslami : C. M. Ewaschuk : S. U. Ahmed : B. A. Greenlay : T. Hoare (*) Department of Chemical Engineering, McMaster University, 1280 Main St. W., Hamilton, ON L8S 4L7, Canada e-mail: [email protected]Colloid Polym Sci (2012) 290:1181–1192 DOI 10.1007/s00396-012-2642-x
Transcript
ORIGINAL CONTRIBUTION
Semi-batch control over functional group distributionsin thermoresponsive microgels
Paniz Sheikholeslami & Christopher M. Ewaschuk &
Syed Usman Ahmed & Benjamin A. Greenlay &
Todd Hoare
Received: 28 November 2011 /Revised: 7 March 2012 /Accepted: 20 March 2012 /Published online: 3 April 2012# Springer-Verlag 2012
Abstract Thermosensitive poly(N-isopropylacrylamide-co-methacrylic acid) (poly(NIPAM-co-MAA)) microgels wereprepared via semi-batch free radical copolymerization inwhich the functional monomer (methacrylic acid) was con-tinuously fed into the reaction vessel at various speeds.Microgels with the same bulk MAA contents (and thus thesame overall compositions) but different radial functionalgroup distributions were produced, with batch copolymer-izations resulting in core-localized functional groups, fast-feed semi-batch copolymerizations resulting in near-uniform functional group distributions, and slow-feedsemi-batch copolymerizations resulting in shell-localizedfunctional groups. Functional group distributions in themicrogels were probed using titration analysis, electropho-resis, and transmission electron microscopy. The inducedfunctional group distributions have particularly significantimpacts on the pH-induced swelling and cationic drug bind-ing behavior of the microgels; slower monomer feeds resultin increased pH-induced swelling but lower drug binding.This work suggests that continuous semi-batch feed regimescan be used to synthesize thermoresponsive microgels withwell-defined internal morphologies if an understanding ofthe relative copolymerization kinetics of each comonomerrelative to NIPAM is achieved.
Microgels have recently attracted wide interest in the liter-ature owing to their attractive mix of hydrogel and nano-particle properties [1]. As hydrogels, microgels have a well-defined three-dimensional structure and can shrink andswell according to changes in the solvent conditions; asnanoparticles, microgels can be flocculated at high saltconcentrations, can be assembled into two- or three-dimensional nanostructures, and respond quickly to changesin their environment. “Smart” microgels that exhibit well-defined phase transitions as a function of pH [2–5], temper-ature [6, 7], the concentration of a particular chemical [8, 9],or other physical stimuli [10] have attracted particular atten-tion due to their range of potential applications in drugdelivery, sensors, optics, purification technologies, and oth-er industries [11–16]. For microgels exhibiting such phasetransition behavior, both the chemical composition and thephysical morphology present locally throughout the micro-gel play significant roles in determining the types of phys-ical [17–20] and biological [9, 14, 21] responses achieved,making control over microgel morphology a key area ofresearch.
Local compositional distributions in microgels are typi-cally controlled by performing multi-stage copolymeriza-tions to create core–shell particles with distinct localcompositions. Often, cores and shells have been designedwith distinct smart properties that either offer local enhance-ment or frustration in swelling responses as a function of an
Electronic supplementary material The online version of this article(doi:10.1007/s00396-012-2642-x) contains supplementary material,which is available to authorized users.
P. Sheikholeslami : C. M. Ewaschuk : S. U. Ahmed :B. A. Greenlay : T. Hoare (*)Department of Chemical Engineering, McMaster University,1280 Main St. W.,Hamilton, ON L8S 4L7, Canadae-mail: [email protected]
applied stimulus. Several core–shell approaches have appliedthe use to synthesize microgels with locally distributed pH-sensitive ionizable functional groups [22–25], locally specificpKa ranges for the ionization of a particular functional group[26], local thermal [27–30] or salt [31]-induced phase transi-tion temperatures, or multiple types of chemical [4] or tempo-ral [32] smart responses in the core and shell, facilitating theobservation of two-step phase transitions with distinct swell-ing properties depending on where the smart functionalgroups are located. Multiple shells have also been addedthrough sequential polymerization processes to provide addi-tional control over local swelling [33]. However, it should benoted that local functional group and crosslinker distributionsare observed within a given core or shell of a multi-stagemicrogel due to differences in the copolymerization kineticsof the constituent comonomers, complicating control overlocal functional group distributions. Alternately, surface graft-ing can be used to induce compositional control independentof the microgel synthesis process, with graft-to processes usedto add poly(2-vinylpyridine) [34], chitosan [35], polyvinyl-amine [36], poly(N-isopropylacrylamide) (PNIPAM) [37], orpoly(ethylene glycol) [38, 39] shells to thermoresponsivemicrogels and graft-from processes used to synthesize poly(N,N-dimethylaminoethyl methacrylate)-shell microgels [40].Microfluidics approaches have also been applied to generatelocally structured microgel particles; for example, polyacryl-amide (core)/PNIPAM (shell) microgels have been synthe-sized using microfluidic droplet generation and mixingapproaches [41].
Hoare and Pelton have reported extensively on an alternateway to control local compositions in microgels by controllingthe copolymerization kinetics of the constituent monomers[42, 43]. In the case of thermosensitive PNIPAM-basedmicro-gels, monomers that react faster than NIPAM (e.g., metha-crylic acid) become localized in the microgel core, monomerswith similar propagation rates to NIPAM (e.g., acrylic acid)are distributed throughout the microgel, and monomers withsignificantly slower propagation rates than NIPAM (e.g.,vinylacetic acid or fumaric acid) are concentrated in the micro-gel shell [42]. In this way, microgels with well-defined localcompositional and morphological features can be prepared ina single step simply by selecting appropriate monomer com-binations, resulting in microgels with controlled swellingresponses [17, 44], ionization behaviors [19], and applicationperformances [21, 45]. However, the types of microgel mor-phologies that can be generated by batch kinetic control arelimited by whether or not an appropriate monomer combina-tion with the required copolymerization kinetics can beidentified.
The use of semi-batch processes, coupled with an under-standing of the copolymerization kinetics of the functionalmonomers used, has the potential to reduce the time re-quired to make structured microgel particles while reducing
the probability of secondary particle formation, an inherentrisk with the use of multi-step polymerization processes.Few reports in the literature have described the use ofsemi-batch techniques, commonly employed in emulsionpolymerizations, for regulating local compositions withinmicrogels. Particular interest has been placed on usingsemi-batch strategies to create uniformly crosslinked micro-gels. For example, Acciaro et al. recently reported the use ofdual monomer-crosslinker semi-batch feeds to produce PNI-PAM microgels with a uniform crosslinking density thatexhibited significantly higher degrees of swelling andtransparency than conventional microgels containing a het-erogeneous crosslinker distribution [46]. Zha et al. alsodemonstrated that the semi-continuous feed of NIPAM,acrylic acid, and crosslinker into an ammonium persulfate(initiator) solution resulted in the formation of significantlysmaller microgels and different internal morphologies thanwould be achieved with conventional batch processes; how-ever, the colloidal stability of the resulting particles wassignificantly lower than that of conventional batch particles[47]. Semi-batch approaches have also been applied to syn-thesize cationic microgels, as many of the cationic mono-mers typically used have lower reactivities and are thusdifficult to incorporate quantitatively into batch microgels.For example, Forcada et al. used a semi-batch approach tosynthesize vinylcaprolactam-based microgels [48], whileSun et al. used a semi-batch feed strategy to synthesize N,N-dimethylaminoethyl methacrylate-co-ethyl acrylate microgels[49]. In both these approaches, semi-batch approaches areused primarily to increase the degree of functional monomerincorporation into the microgel as opposed to induce a partic-ular morphology.
In this paper, we report the synthesis of microgels with thesame composition but different functional group distributionsby controlling the rate at which methacrylic acid (MAA) isadded during the polymerization reaction. Methacrylic acidwas selected given its preference for being incorporated overNIPAM during microgel copolymerization (rMAA02.8,rNIPAM00.2) [50]; as such, by controlling the MAA concen-tration throughout the polymerization process, we anticipatebeing able to control the radial distribution ofMAAwithin themicrogel and thus the swelling behavior and application per-formance of the microgels.
Experimental
Materials N-Isopropylacrylamide (NIPAM, 99%), metha-crylic acid (MAA, 99%), ammonium persulfate (APS,99%), N,N′-methylenebisacrylamide (MBA, 99%), diso-dium phosphate (99%), dihydrogen sodium phosphate(99%), sodium citrate (99%), and dibucaine hydrochloride(99%) were obtained from Sigma-Aldrich (Oakville, ON,
1182 Colloid Polym Sci (2012) 290:1181–1192
Canada). Sodium dodecyl sulfate (SDS, 99.9%) wasobtained from Bioshop Canada (Burlington, ON, Canada).Sodium hydroxide and hydrochloric acid solutions wereobtained by diluting Acculute standards. All water usedwas of Milli-Q grade.
Microgel synthesis Microgels were prepared via a mixedprecipitation-emulsion process in which the functionalmonomer (methacrylic acid) was fed into the reactor atdifferent rates. Each microgel was synthesized using 1.4 gof NIPAM, 0.1 g MBA crosslinker, 0.05 g SDS surfactant,0.068 g methacrylic acid (functional monomer), and 0.1 gAPS initiator. In each case, the NIPAM, MBA, and SDSfractions were dissolved in 150 mL of water inside a 500-mL round-bottom flask and heated to 70 °C under 200 rpmmixing and a nitrogen purge. For the batch microgel, themethacrylic acid was also dissolved in this initial solution,and an additional 5 mL of water was added; for all semi-batch microgels, methacrylic acid was dissolved in 5 mL ofwater, loaded into a 5-mL syringe, and mounted on a KDScientific Model 780210 syringe pump. The syringe wasprimed so that methacrylic acid was added into the reactionvessel immediately upon starting the syringe pump. After30 min of stabilization, APS initiator, dissolved in 5 mL ofwater, was added in a single shot to initiate polymerization.For semi-batch microgels, the syringe pump was started at thesame time to add methacrylic acid into the polymerizationreaction. The flow rate of the syringe pump was changed toalter the availability of methacrylic acid at various points inthe polymerization, as indicated in Table 1.
Recipes are presented both in terms of the flow rate of themethacrylic acid solution into the polymerization reactor (rep-resented in the microgel codes), as well as the total timerequired to add a full 5 mL syringe of methacrylic acidsolution to the reaction vessel. The smaller the number inthe microgel code, the longer the time required to add the fullaliquot of MAA into the reactor. Microgels were purified
through six cycles of dialysis (Spectrum Labs, 12–14 kDaMWCO) against distilled deionized water to remove residualmonomer, surfactant, and the uncrosslinked linear polymerfractions, after which the microgel was lyophilized for storage.All microgels shown in Table 1 were colloidally stable, withno macroscopic aggregate observed either visually or via lightscattering analysis in any of the listed samples.
Titration analysis Potentiometric and conductometric titra-tions were performed using a Burivar-I2 automatic buret(ManTech associates) running PC-Titrate software (version2.0.0.79). Samples of 50 mg of each microgel were suspendedin 50 mL of 10−3 M NaCl. Titrations were conducted using0.1 M HCl and 0.1 M NaOH Acculute standards. For bothtotal –COOH measurements and pKa calculations, the pH ofthe suspension was lowered to pH∼3 (zero microgel ioniza-tion), and 0.1 MNaOHwas added to achieve a titration rate of10 min/unit pH over the course of the titration. Effective pKa
values were calculated using the Henderson–Hasselbachequation as a function of the degree of ionization.
Microgel characterization Particle sizes were measured viadynamic light scattering using a Melles Griot He–Ne laserwith a 632.8 nm light source as the incident light source and aBrookhaven Instruments Corporation (BIC) detector modelBI-APD operating at 90°. Five replicate measurements werecollected (minimum 107 counts/sample), with means andstandard deviations of these sizes reported. Electrophoreticmobility of microgels was measured using a ZetaPlus zetapotential analyzer operating in PALS mode (BIC). A total often samples, 15 cycles/sample, were conducted for eachmicrogel, with means and standard errors of those mobilitiesreported. In both size and mobility measurements, dilutemicrogel suspensions in 10 mM citric acid buffer (pH 4),phosphate buffer (pH 7), and carbonate buffer (pH 10) wereused for analysis. For assessment of the impact of drug (dibu-caine hydrochloride) binding on microgel swelling, 1 mg/mLof microgel was suspended in 10 mM phosphate buffer (pH0
6.5) in the presence of dibucaine hydrochloride at fixed ratiosof moles dibucaine per mole –COOH group in the samples(0.5x, 1x, 2x, 3x, and 5x). Light scattering analysis was thenconducted as described above to track the changes in microgelswelling due to drug–microgel interactions.
Supernatant analysis To assay the characteristics of thelinear polymer waste fraction produced during microgelsynthesis, raw microgels were ultracentrifuged (Beckman-Coulter Optima L-80 XP Ultracentrifuge, 50,000 rpm, 3×1 h cycles) at both the reaction pH (∼3.5) and at higher pHvalues (pH 9, achieved by addition of 0.1 M NaOH). Underthese latter conditions, MAA residues in the linear polymerswould become ionized and thus easier to remove from themicrogel given that NIPAM-MAA hydrogen bonding can
Table 1 Recipes and methacrylic acid solution addition rates for semi-batch microgels tested
Microgel code Addition rate(mL/min)
Total time ofaddition (min)
SB-2.5 2.5 2
SB-1.9 1.9 2.6
SB-1.5 1.5 3.3
SB-1.1 1.1 4.4
SB-0.75 0.75 6.7
SB-0.37 0.37 13.5
SB-0.18 0.18 27.7
SB-0.03 0.03 180.3
SB-0.01 0.01 500.0
Colloid Polym Sci (2012) 290:1181–1192 1183
occur in this system. The microgel and supernatant fractionswere collected and weighed to calculate the mass percentageof total monomer added that is ultimately present in themicrogels. Supernatants were also titrated (using the sameapproach described above) to probe supernatant compositionand analyzed via gel permeation chromatography (GPC,Waters, DMF solvent, 0.5 mL/min flow rate) to confirmeffective removal via dialysis. GPC results were standardizedbased on a poly(N-isopropylacrylamide) standard (polymersource, Mv010,250, Mw/Mn01.12).
Transmission electron microscopy (TEM) TEM imageswere obtained using a JEOL 1200EX TEMSCAN micro-scope operating at 80 kV. Microgels were suspended in1 mL of a 1-mM uranyl acetate solution at a concentrationof 0.05 wt.% for 2 h to selectively stain –COO− groups inthe microgel with cationic uranium species, enabling differ-entiation of functionalized and non-functionalized regionswithin the microgels. Stained microgels were subsequentlycentrifuged and redispersed in 0.05 wt.% suspensions in wa-ter. A total of 5 μL of microgel suspension was dropped on aFormvar-coated copper TEM grid and dried overnight.Images were analyzed using Image-J software. Pixel densitywas measured for 20 particles per microgel, taking four cross-sections per particle to account for any possible anisotropy;reported pixel density graphs represent the averages of these80 independent pixel density distributions.
Drug uptake measurements The binding of dibucaine hydro-chloride to microgels prepared under different semi-batchflow regimes was assayed by suspending (in 10-mM pH 6.5phosphate buffer) (a) a 5-mg/mL microgel in a 1-mg/mLdibucaine hydrochloride solution and (b) a 1-mg/mLmicrogelin a 5-mg/mL dibucaine hydrochloride solution. These con-centrations were selected to assay the effect of changing themicrogel:drug ratio on the drug binding observed. The sam-ples were placed on a shaker and mixed for 24 h; at whichpoint, the microgels were removed from suspension via ul-tracentrifugation (Sorvall RC M720, 73,000 rpm, 1 h, 25 °C).The supernatant solution was recovered and assayed usingUV/Vis spectrophotometry using a wavelength of 239 nm tocalculate the residual dibucaine concentration based on acalibration curve prepared in the same buffer (R200.99); thedifference between the original and supernatant concentra-tions is the amount of drug bound to the microgel.
Results
Degree of functional group incorporation Conductometrictitration was performed to investigate the effect of the semi-batch feed protocols used on the total incorporation of the semi-batch fed monomer (methacrylic acid) into the microgels.
Figure 1 shows the percentage of MAA incorporation into themicrogels (relative to the total amount of MAA added to thereaction vessel) as a function of the time over which MAAwasadded to the reaction.
Quantitative incorporation of MAA was observed formicrogels prepared using a batch strategy or using fast-to-moderate speed semi-batch feed strategies (addition timesless than 30 min). Slower feed strategies resulted in signif-icantly lower incorporations of MAA, despite the fact thatample non-degraded persulfate initiator should be present inthe reaction vessel even at these longer times (t1/2∼7 h or420 min for ammonium persulfate at the 70 °C reactiontemperature) [51]. Indeed, experiments in which bothMAA and fresh persulfate initiator were simultaneouslyfed into the reaction vessel did not result in significantincreases in MAA incorporation into the microgels; MAAincorporations for SB-0.01 recipes were 24±6% when ini-tiator (1 mg/mL) was added together with MAA and 21±6% when MAA was fed without added initiator.
The turbidity of the reactor contents does not significant-ly change after the first 30–40 min of reaction time, suggest-ing that the NIPAM fraction present has largely polymerizedinto microgels by this time; furthermore, light scattering didnot indicate the presence of a secondary particle fraction inany of the semi-batch microgels reported. Together, theseobservations suggest the formation of a larger fraction of“waste” linear polymer (i.e., polymer not incorporated intothe microgels) with a high MAA content in cases whereMAA is added very slowly into the reactor. In this case, asMAA is added into the reactor and subsequently polymer-izes in solution at later times, there is no driving force for the
Fig. 1 Conductometric titration of total carboxylic acid monomercontent of batch and semi-batch microgels (expressed as the molepercent of MAA in the resulting microgel) as a function of the timeover which MAA was added into the reaction
1184 Colloid Polym Sci (2012) 290:1181–1192
growing chains to precipitate on to the microgel particles(i.e., all NIPAM is consumed) or to chemically attach to themicrogel particles (i.e., all crosslinker is consumed), result-ing in lower MAA incorporation into the microgels and ahigher fraction of MAA-rich linear polymer waste (which isremoved from the suspension via dialysis prior to sampleanalysis). GPC results confirm that the molecular weight ofthe supernatant polymers is similar to the molecular weightcut-off of the membrane used for these experiments (12–14 kDa, Supplementary Information, Fig. S3) such that thewaste polymer fraction should be removed from the micro-gel using the protocol described.
The hypothesis that a large fraction of MAA-rich linearpolymer formation is formed in slow-feed semi-batchmicrogels is further supported based on gravimetric analysisfollowing isolation of the microgel via ultracentrifugationand subsequent titration of the recovered microgel and su-pernatant fractions, as shown in Table 2. Note that experi-ments were conducted both at the synthesis pH (pH∼3.5)and a higher pH value (pH 9) since methacrylic acid resi-dues are known to hydrogen bond with NIPAM residueswhen protonated; as such, MAA-rich linear polymer frac-tions may remain in the microgel fraction when centrifugedat pH 3.5 but would be expected to be removed into thesupernatant when centrifuged at pH 9.
As indicated in Table 2, significantly more linear polymerby-product is formed when the addition rate of MAA isslowed (SB-0.03 versus SB-1.9); however, that additionalby-product amount can only be recovered when the microgelis ionized (pH 9). In contrast, no difference in supernatantrecovery was observed based on purification pH for the fast-feed microgel SB-1.9. This observation suggests that thelinear polymer product formed during SB-0.03 synthesis hasa high MAA content, while the linear polymer fraction in SB-1.9 production has a significantly lower MAA content, anobservation confirmed by the titration analysis. Table 2 indi-cates that SB-0.03 supernatants had significantly higher–COOH (and thus MAA residue) contents than SB-1.9 super-natants; this was particularly true in analyzing the supernatantsrecovered when microgels were centrifuged at pH 9, suggest-ing that very high MAA content chains are recovered into thissupernatant upon basic dialysis. Thus, semi-batch addition of
MAA produces microgels with the same overall functionalgroup content at faster addition rates (<30 min) but reducedMAA contents if the addition period extends beyond theeffective polymerization period of the available NIPAM.
The effective pKa of the carboxylic acid groups in thevarious semi-batch microgels as a function of the degree ofionization, measured via potentiometric titration, is shownin Fig. 2. The effective pKa of the functional groups changesas a function of the degree of ionization due to the poly-electrolyte effect, in which the ionization of one functionalgroup in a polymer changes the electronic environmentaround an adjacent ionizable functional group; the higherthe observed increase in effective pKa over the full ioniza-tion range, the more clustered the functional groups are inthe microgel (i.e., the higher effect the ionization of onefunctional group has on an adjacent functional group) [19].All semi-batch microgels start with a similar pKa at lowdegrees of ionization (5.0<pKa<5.2) slightly higher thanthe pKa of methacrylic acid (4.7), while the batch microgelhas a significantly higher pKa (∼5.5) even at low degrees ofionization. As ionization proceeds, microgels prepared us-ing slow semi-batch feeds show large increases in effectivepKa over a broad ionization range, while microgels preparedwith fast to intermediate speed semi-batch feeds exhibitsmaller increases in effective pKa that are manifested onlyat relatively high degrees of ionization (>0.8). Note thatwhile SB-1.9 and SB-0.03 follow similar pKa trends atlower degrees of ionization, SB-0.03 has only ∼40% ofthe total number of –COOH groups of SB-1.9; as a result,fewer functional groups are ionized (and thus a lower mag-nitude polyelectrolyte effect is observed, regardless of thedistribution of the functional groups) at higher degrees ofionization in SB-0.03. This difference in total –COOH con-tent leads to the similar pKa profiles observed at lowerdegrees of ionization, while the higher functional groupclustering in SB-0.03 results in the significantly sharperincrease observed in pKa over a narrow ionization range athigher degrees of ionization. Together, these results suggestthat functional groups are clustered together in the slow feedsemi-batch microgels, while functional groups are moreuniformly distributed in microgels prepared with moderateto fast feed semi-batch feeds.
Table 2 Percentage of microgel recovery and –COOH content (normalized by mass) of supernatant and microgel phases recovered following threesuccessive ultracentrifugation cycles
This observation is echoed by calculation of the excessGibbs free energy of ionization of the microgels (Supplemen-tary Information, Fig. S1), in which slow-feed semi-batchmicrogels exhibit higher excess ionization energies. Thehigher inherent pKa of the same functional monomer residuesin the batch microgel suggests that –COOH groups are clus-tered together in the batch microgel but exist in a differentchemical environment than the clustered functional groups inthe slow-feed semi-batch microgels. This observation wouldbe consistent with the presence of functional groups in a morehighly crosslinked microgel core [15, 39], as the lower localdegree of swelling inhibits functional group ionization. Hy-drogen bonding between MAA and NIPAM and/or two MAAresidues locally may also impact the observed pKa curves;however, such effects are likely to magnify any observedfunctional group distribution effects in that the local MAA:NIPAM ratio would drive any observed differences in thetitration curves due to hydrogen bonding.
Electrophoretic mobility To assay the distribution of theMAA monomer residues within the microgel, electrophoreticmobility data were collected at pH 4 (MAA residues proton-ated), pH 7 (most MAA residues ionized), and pH 10 (allMAA residues ionized). Results are shown in Fig. 3 as afunction of the total addition time of MAA into the reactor.
At pH 4 (no effective MAA ionization), all microgels testedhad a similar, small negative mobility value derived primarilyfrom the anionic persulfate initiator groups present on themicrogels. The similarity of this value across all batch andsemi-batch compositions tested suggests similar fundamentalparticle structures in each case, in contrast to other semi-batchstrategies previously reported [47]. Titration results suggest thatthe microgels are mostly ionized at pH 7 (0.75<DI<0.88),
while all microgels tested are fully ionized at pH 10 (seeSupplementary Information, Fig. S2). For microgels with thesame total carboxylic acid content (addition times <30 min),absolute electrophoretic mobilities increase as the rate at whichMAA is added to the reaction is decreased. While electropho-retic mobility is not a direct function of surface charge densityin ion-penetratable microgels in which electroosmosis may alsooccur through the microgel in the presence of an electric field[52], this result suggests that MAA residues are successivelyincorporated closer toward the surface of the microgel as theMAA feed rate into the reaction vessel is reduced. As the totalincorporation of MAA into the microgels is reduced at longerfeed times (from quantitative incorporation at ∼30min additiontime to ∼40% incorporation at 180 min addition time and∼20% incorporation at 500 min addition time, Fig. 1), theabsolute electrophoretic mobility decreases due to the presenceof fewer charged residues in the microgels. However, it shouldbe emphasized that the electrophoretic mobilities of SB-0.03 atpH 7 and pH 10 are ∼40% higher than those of the batch-synthesized microgel even though the slow addition of semi-batch microgels contain only ∼40% of the total number offunctional groups; furthermore, SB-0.01 has a significantlyhigher absolute mobility at pH 7 and pH 10 than SB-0.03(p00.01) despite containing only half the total MAA residues.Together, these observations suggest improved localization ofthe fewer number of functional groups at the microgel surface;that is, a successively higher fraction of the available MAAresidues is located at or near the microgel surface as the speedof the MAA semi-batch feed is slowed.
Transmission electron microscopy To confirm the changingradial functional group distributions of the microgels as the
Fig. 2 Effective pKa of carboxylic acid residues from MAA mono-mers as a function of the degree of ionization of the –COOH groups inthe microgel
Fig. 3 Electrophoretic mobilities of batch and semi-batch microgels asa function of pH
1186 Colloid Polym Sci (2012) 290:1181–1192
feed rate of MAA into the reactor was varied, anionic chargedgroups were stained with uranyl acetate so they appear darkerin TEM images. TEM images for representative microgelsprepared via batch and semi-batch feed strategies over the fullrange of feed rates explored are shown in Fig. 4. Pixel densitywas measured across a series of particles to translate the imagedata into a radial functional group distribution plot for micro-gels prepared with differentMAA feed regimes; the results areshown in Fig. 5.
The TEM images (Fig. 4) and image analysis (Fig. 5)confirm the general trend of functional group distributionssuggested by the electrophoretic mobility results: the slowerthe MAA is added to the reactor, the more surface-localizedthe –COOH distribution becomes within the microgels. Theobservation of decreased functional group content in the veryslow feed semi-batch microgels SB-0.03 and SB-0.01 is alsoreflected in the TEM images (Fig. 4), as lower contrast isobserved between the background and the stained particles asthe total number of –COO− available to stain decreases at theslowest feed rates. The images also support the pKa datapresented in Fig. 2. Semi-batch microgels with clustered func-tional groups in the images exhibit larger changes in effectivefunctional group pKa over a broader ionization range. Micro-gels with functional groups clustered in the shell (SB-0.03 andSB-0.01) exhibited the largest and most continuous changes inpKa over the full ionization range, while functional groups aremore generally distributed in faster and intermediate feedsemi-batch microgels (SB-1.9, SB-0.75, and SB-0.18) thatshow smaller pKa jumps only at higher degrees of ionization.In contrast, the batch microgel contains functional groupsalmost exclusively in the microgel core, consistent with thehigher inherent functional group pKa observed via titration.
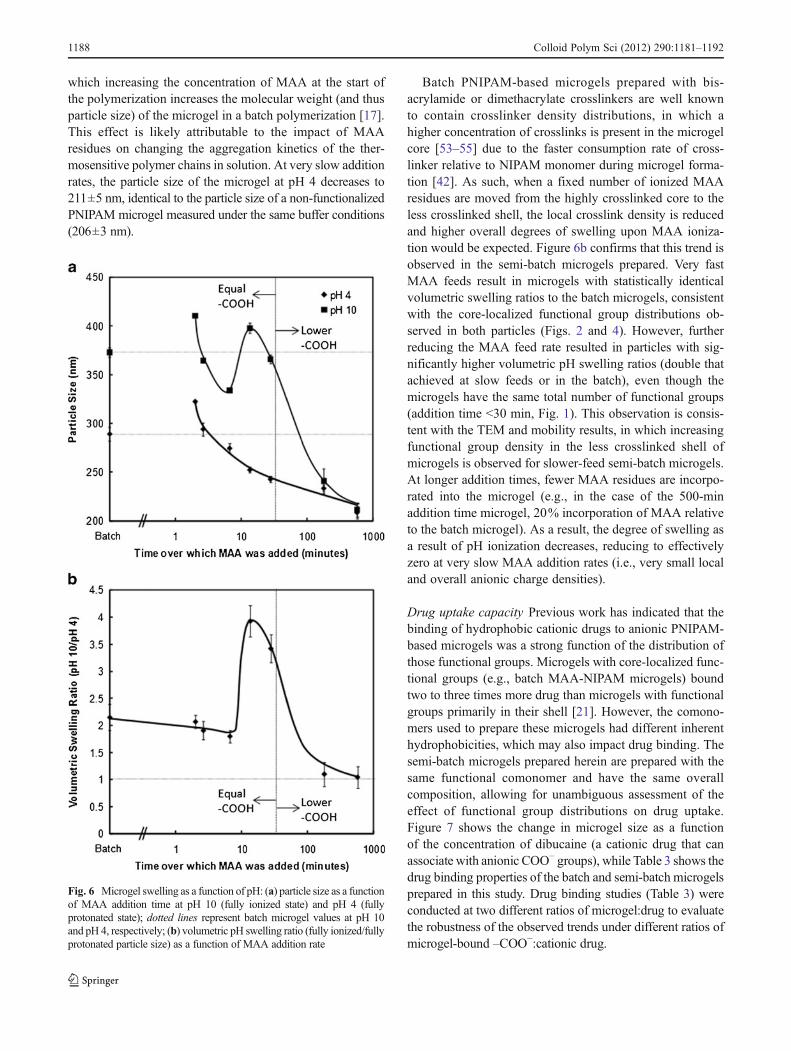
Particle size and swelling control Figure 6 shows the pH-induced swelling properties of the batch and semi-batchmicrogels, with Fig. 6a illustrating the raw particle sizes inthe fully protonated state (pH 4) and the fully ionized state(pH 10), and Fig. 6b illustrating the volumetric swellingratio observed for each of the microgels upon ionization.
Figure 6a indicates that the particle size of the microgel inthe protonated state (pH 4) decreases as the time over whichMAA was added is increased under semi-batch feed con-ditions. This is consistent with previous observations, by
Fig. 4 TEM images ofmicrogels prepared using batchand semi-batch protocols withvarious MAA feed rates; uranylacetate staining; ×150,000magnification
Fig. 5 Relative pixel density (normalized versus the background in-tensity of the TEM images) as a function of relative radius (r/ro) inTEM images shown in Fig. 4
Colloid Polym Sci (2012) 290:1181–1192 1187
which increasing the concentration of MAA at the start ofthe polymerization increases the molecular weight (and thusparticle size) of the microgel in a batch polymerization [17].This effect is likely attributable to the impact of MAAresidues on changing the aggregation kinetics of the ther-mosensitive polymer chains in solution. At very slow additionrates, the particle size of the microgel at pH 4 decreases to211±5 nm, identical to the particle size of a non-functionalizedPNIPAM microgel measured under the same buffer conditions(206±3 nm).
Batch PNIPAM-based microgels prepared with bis-acrylamide or dimethacrylate crosslinkers are well knownto contain crosslinker density distributions, in which ahigher concentration of crosslinks is present in the microgelcore [53–55] due to the faster consumption rate of cross-linker relative to NIPAM monomer during microgel forma-tion [42]. As such, when a fixed number of ionized MAAresidues are moved from the highly crosslinked core to theless crosslinked shell, the local crosslink density is reducedand higher overall degrees of swelling upon MAA ioniza-tion would be expected. Figure 6b confirms that this trend isobserved in the semi-batch microgels prepared. Very fastMAA feeds result in microgels with statistically identicalvolumetric swelling ratios to the batch microgels, consistentwith the core-localized functional group distributions ob-served in both particles (Figs. 2 and 4). However, furtherreducing the MAA feed rate resulted in particles with sig-nificantly higher volumetric pH swelling ratios (double thatachieved at slow feeds or in the batch), even though themicrogels have the same total number of functional groups(addition time <30 min, Fig. 1). This observation is consis-tent with the TEM and mobility results, in which increasingfunctional group density in the less crosslinked shell ofmicrogels is observed for slower-feed semi-batch microgels.At longer addition times, fewer MAA residues are incorpo-rated into the microgel (e.g., in the case of the 500-minaddition time microgel, 20% incorporation of MAA relativeto the batch microgel). As a result, the degree of swelling asa result of pH ionization decreases, reducing to effectivelyzero at very slow MAA addition rates (i.e., very small localand overall anionic charge densities).
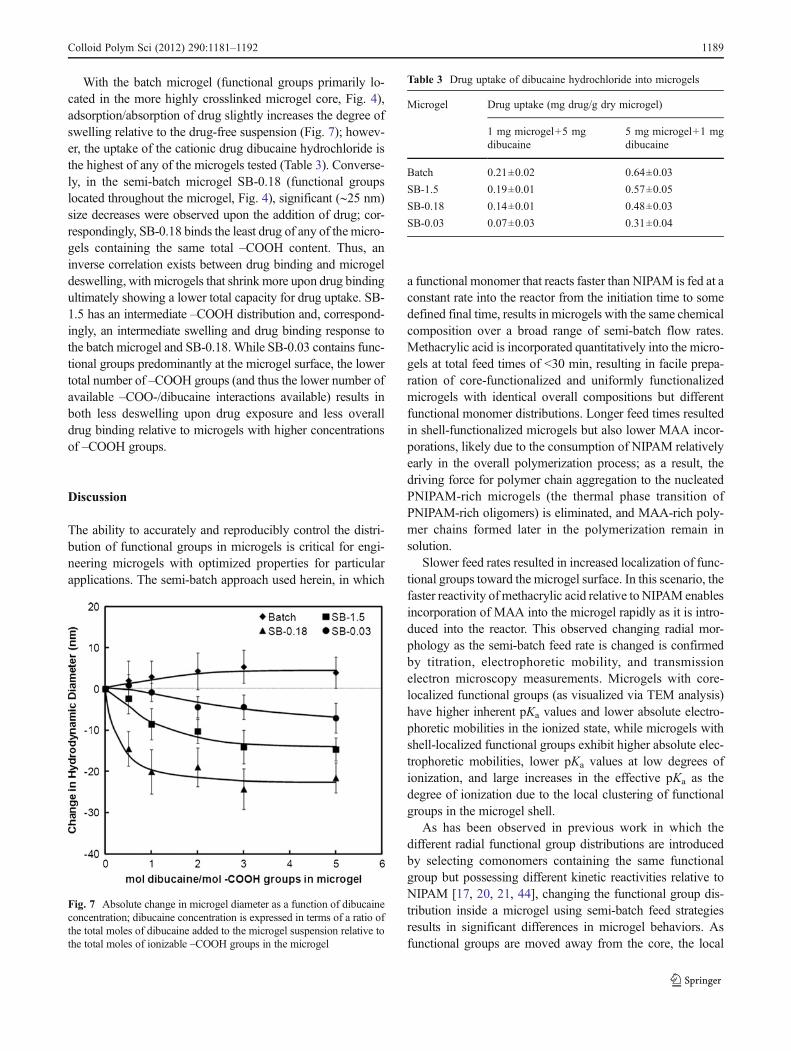
Drug uptake capacity Previous work has indicated that thebinding of hydrophobic cationic drugs to anionic PNIPAM-based microgels was a strong function of the distribution ofthose functional groups. Microgels with core-localized func-tional groups (e.g., batch MAA-NIPAM microgels) boundtwo to three times more drug than microgels with functionalgroups primarily in their shell [21]. However, the comono-mers used to prepare these microgels had different inherenthydrophobicities, which may also impact drug binding. Thesemi-batch microgels prepared herein are prepared with thesame functional comonomer and have the same overallcomposition, allowing for unambiguous assessment of theeffect of functional group distributions on drug uptake.Figure 7 shows the change in microgel size as a functionof the concentration of dibucaine (a cationic drug that canassociate with anionic COO− groups), while Table 3 shows thedrug binding properties of the batch and semi-batch microgelsprepared in this study. Drug binding studies (Table 3) wereconducted at two different ratios of microgel:drug to evaluatethe robustness of the observed trends under different ratios ofmicrogel-bound –COO−:cationic drug.
Fig. 6 Microgel swelling as a function of pH: (a) particle size as a functionof MAA addition time at pH 10 (fully ionized state) and pH 4 (fullyprotonated state); dotted lines represent batch microgel values at pH 10and pH 4, respectively; (b) volumetric pH swelling ratio (fully ionized/fullyprotonated particle size) as a function of MAA addition rate
1188 Colloid Polym Sci (2012) 290:1181–1192
With the batch microgel (functional groups primarily lo-cated in the more highly crosslinked microgel core, Fig. 4),adsorption/absorption of drug slightly increases the degree ofswelling relative to the drug-free suspension (Fig. 7); howev-er, the uptake of the cationic drug dibucaine hydrochloride isthe highest of any of the microgels tested (Table 3). Converse-ly, in the semi-batch microgel SB-0.18 (functional groupslocated throughout the microgel, Fig. 4), significant (∼25 nm)size decreases were observed upon the addition of drug; cor-respondingly, SB-0.18 binds the least drug of any of the micro-gels containing the same total –COOH content. Thus, aninverse correlation exists between drug binding and microgeldeswelling, with microgels that shrink more upon drug bindingultimately showing a lower total capacity for drug uptake. SB-1.5 has an intermediate –COOH distribution and, correspond-ingly, an intermediate swelling and drug binding response tothe batch microgel and SB-0.18.While SB-0.03 contains func-tional groups predominantly at the microgel surface, the lowertotal number of –COOH groups (and thus the lower number ofavailable –COO-/dibucaine interactions available) results inboth less deswelling upon drug exposure and less overalldrug binding relative to microgels with higher concentrationsof –COOH groups.
Discussion
The ability to accurately and reproducibly control the distri-bution of functional groups in microgels is critical for engi-neering microgels with optimized properties for particularapplications. The semi-batch approach used herein, in which
a functional monomer that reacts faster than NIPAM is fed at aconstant rate into the reactor from the initiation time to somedefined final time, results in microgels with the same chemicalcomposition over a broad range of semi-batch flow rates.Methacrylic acid is incorporated quantitatively into the micro-gels at total feed times of <30 min, resulting in facile prepa-ration of core-functionalized and uniformly functionalizedmicrogels with identical overall compositions but differentfunctional monomer distributions. Longer feed times resultedin shell-functionalized microgels but also lower MAA incor-porations, likely due to the consumption of NIPAM relativelyearly in the overall polymerization process; as a result, thedriving force for polymer chain aggregation to the nucleatedPNIPAM-rich microgels (the thermal phase transition ofPNIPAM-rich oligomers) is eliminated, and MAA-rich poly-mer chains formed later in the polymerization remain insolution.
Slower feed rates resulted in increased localization of func-tional groups toward the microgel surface. In this scenario, thefaster reactivity of methacrylic acid relative to NIPAM enablesincorporation of MAA into the microgel rapidly as it is intro-duced into the reactor. This observed changing radial mor-phology as the semi-batch feed rate is changed is confirmedby titration, electrophoretic mobility, and transmissionelectron microscopy measurements. Microgels with core-localized functional groups (as visualized via TEM analysis)have higher inherent pKa values and lower absolute electro-phoretic mobilities in the ionized state, while microgels withshell-localized functional groups exhibit higher absolute elec-trophoretic mobilities, lower pKa values at low degrees ofionization, and large increases in the effective pKa as thedegree of ionization due to the local clustering of functionalgroups in the microgel shell.
As has been observed in previous work in which thedifferent radial functional group distributions are introducedby selecting comonomers containing the same functionalgroup but possessing different kinetic reactivities relative toNIPAM [17, 20, 21, 44], changing the functional group dis-tribution inside a microgel using semi-batch feed strategiesresults in significant differences in microgel behaviors. Asfunctional groups are moved away from the core, the local
Fig. 7 Absolute change in microgel diameter as a function of dibucaineconcentration; dibucaine concentration is expressed in terms of a ratio ofthe total moles of dibucaine added to the microgel suspension relative tothe total moles of ionizable –COOH groups in the microgel
Table 3 Drug uptake of dibucaine hydrochloride into microgels
Microgel Drug uptake (mg drug/g dry microgel)
1 mg microgel+5 mgdibucaine
5 mg microgel+1 mgdibucaine
Batch 0.21±0.02 0.64±0.03
SB-1.5 0.19±0.01 0.57±0.05
SB-0.18 0.14±0.01 0.48±0.03
SB-0.03 0.07±0.03 0.31±0.04
Colloid Polym Sci (2012) 290:1181–1192 1189
crosslink density surrounding the ionizable functional groupsdecreases and the microgels swell more (relative to their fullyprotonated state) as they are ionized. Microgels with function-al groups localized primarily in the core bind more cationicdrug (and deswell less in the presence of cationic drug) thanmicrogels with functional groups distributed throughout themicrogel or localized primarily in the shell. This latter obser-vation is likely attributable to the formation of a collapsed skinlayer at the periphery of the microgels when binding sites arelocalized toward the microgel surface that inhibits further druguptake into the microgel bulk. Both these trends are identicalto those observed using different functional comonomers inprevious work [17, 21]. As a result, semi-batch strategies canreproduce the general morphologies achievable by batch co-polymerization as well as facilitate the design of functionalgroup distributions that are not readily accessible based on theabsence of a functional comonomer with appropriate relativepropagation kinetics to NIPAM.
It should be emphasized that the semi-batch feed strategydiscussed (i.e., continuous monomer feed starting at thesame time as initiator injection) was one of several protocolsinvestigated over the course of the study but was found to bethe most effective method of controlling local functionalgroup distributions without significantly changing the un-derlying size or morphology of the microgel. Shot additionstrategies, by which MAA was added over a short period(<2 min) following a prescribed delay after polymerizationwas initiated, resulted in microgels with generally poorercolloidal stability at all pH values, with visible aggregatesforming in the reactor as the MAAwas added in some cases.As an example, if MAA is added to the reaction vessel over2.5 min but only after a 20-min delay following the initia-tion of the polymerization, quantitative incorporation ofMAAwas achieved (106±6%), and functional groups werelocalized primarily in the microgel shell (mobility0(−2.59±0.09)×10−8 m2/Vs at pH 10), but the colloidal stability waspoor both in the protonated (particle size01,930±430 nm atpH 4) and ionized (particle size0562±63 nm at pH 10)states. In contrast, all microgels produced via the continuousaddition approach maintained high colloidal stability andredispersibility, even following lyophilization. Longer delaytimes resulted in lower functional monomer incorporationsat corresponding times (i.e., shot time versus total additiontime) in all cases, similar to the trend observed in Fig. 1 asthe monomer feed rate is decreased, with a concurrent lossof pH-sensitive properties. As an example, if the delay timewas increased to 30 min following polymerization initiation(maintaining the short 2.5 min addition time following thedelay), the percentage of MAA incorporation (33±5%), theabsolute electrophoretic mobility (mobility0(−1.22±0.06)×10−8 m2/Vs at pH 10), and the pH-induced swelling ratio(particle size0218±4 nm at pH 4 and 222±8 nm at pH 10)all significantly decreased. Slower addition of MAA into the
reactor following a delay resulted in larger reductions in thetotal percentage of MAA incorporation relative to thoseobserved in Fig. 1 at equivalent MAA feed rates, likelyowing to the high consumption of NIPAM prior to thepresence of any MAA in the reactor. Simultaneous feedingof MAA together with an additional initiator shot resulted inno significant change in MAA incorporation or MAA dis-tribution relative to microgels prepared using MAA-onlyfeeds at the same flow rate, likely due to the large excessof initiator present in the original polymerization solution.Simultaneous feeding of NIPAM and MAA resulted insecondary nucleation, smaller microgels, and lower colloi-dal stability, similar to the observations of Zha et al. [47].Thus, the semi-batch MAA feed method reported in thispaper appears to be the most robust method by which micro-gels with the same chemical composition and similar parti-cle morphologies but different functional group distributionscan be synthesized.
While only functional monomer distribution control wasinvestigated in this work, we anticipate that the observationsof this study could be directly translated to controlling theradial distributions of crosslinker or other desired comono-mers within microgels. In any case, knowledge of the rela-tive reactivity ratios and propagation rate constants of eachmonomer would be required such that the faster reactingmonomers/crosslinkers are fed into a reactor containing theslower reacting monomer(s) at a desired rate, controlling thetemporal concentration of the faster reacting monomersthroughout the polymerization process. Ideally, the como-nomers chosen should react faster than NIPAM under thereaction conditions, avoiding the risk of secondary nucle-ation observed in other semi-batch strategies in whichNIPAM is also fed into the reactor as a function of time.Non-constant feed rate regimes may also be used to main-tain the ratio between NIPAM and the target co-monomerconstant throughout the reaction if uniform distributions offunctional monomer or crosslinker were particularly impor-tant for a given application.
Conclusions
1. Functionalized thermosensitive microgels with the sameoverall composition but different radial functional groupdistributions can be synthesized using semi-batch feedstrategies.
2. Functional groups can be localized in the core (batch orfast feed semi-batch microgels), the shell (slow feedsemi-batch microgels), or distributed uniformlythroughout the microgel (intermediate feed semi-batchmicrogels) according to the rate at which the functionalmonomer is added to the reactor relative to the rate ofNIPAM polymerization.
1190 Colloid Polym Sci (2012) 290:1181–1192
3. Core-functionalized microgels swell less upon ioniza-tion but bind higher fractions of oppositely chargeddrugs than shell-functionalized microgels, with uni-formly functionalized microgels exhibiting intermediatebehavior.
4. Simultaneous shot addition of initiator and initiation ofthe functional monomer feed into the reactor results inmicrogels with the most similar morphologies and high-est colloidal stabilities relative to other single shot orshot-feed strategies evaluated.
We anticipate that semi-batch feed strategies should resultin control of the radial distribution of any comonomer orcrosslinker inside microgels if the faster reacting comonomer(s) are added to the slower reacting comonomer(s) (ideallyincluding all the NIPAM to be used in the polymerization tominimize secondary particle nucleation) at a rate relative to theconsumption rate of NIPAM and the comonomer(s) under thefree radical polymerization conditions used.
Acknowledgments The Natural Sciences and Engineering ResearchCouncil of Canada (NSERC) is acknowledged for funding. KevinDeFrance is acknowledged for his assistance in synthesizing thesemi-batch microgels.
2. Snowden MJ, Chowdhry BZ, Vincent B, Morris GE (1996) Col-loidal copolymer microgels of N-isopropylacrylamide and acrylicacid: pH, ionic strength and temperature effects. J Chem Soc-Faraday Trans 92(24):5013–5016
3. Tan BH, Ravi P, Tam KC (2006) Synthesis and characterization ofnovel pH-responsive polyampholyte microgels. Macromol RapidCommun 27:522–528
4. Liu R, Milani AH, Saunders JM, Freemont TJ, Saunders BR(2011) Tuning the swelling and mechanical properties of pH-responsive doubly crosslinked microgels using particle composition.Soft Matter 7:9297–9306
5. Tan BH, Tam KC, Lam YC, Tan CB (2005) Microstructure andrheological properties of pH-responsive core-shell particles. Polymer46:10066–10076
6. Pelton RH, Chibante P (1986) Preparation of aqueous lattices withN-isopropylacrylamide. Colloids Surf 20(3):247–256
7. Lu Y, Ballauff M (2011) Thermosensitive core-shell microgels:from colloidal model systems to nanoreactors. Prog Polym Sci36:767–792
8. Lapeyre V, Gosse I, Chevreux S, Ravaine V (2006) Monodis-persed glucose-responsive microgels operating at physiologicalsalinity. Biomacromolecules 7(12):3356–3363
9. Hoare T, Pelton R (2008) Charge-switching, amphoteric glucose-responsive microgels with physiological swelling activity. Bioma-cromolecules 9(2):733–740
10. Zhao Y, He J, Yan B, Tremblay L (2011) Both core- and shell-cross-linked nanogels: photoinduced size change, intraparticleLCST, and interparticle UCST thermal behaviors. Langmuir 27(1):436–444
11. Das M, Zhang H, Kumacheva E (2006) Microgels: old materialswith new applications. Ann Rev Mat Res 36:117–142
12. Lu Y, Ballauff M (2007) “Smart” nanoparticles: preparation, char-acterization and applications. Polymer 48(7):1815–1823
13. Lyon LA, Meng ZY, Singh N, Sorrell CD, John AS (2009) Thermor-esponsive microgel-based materials. Chem Soc Rev 38(4):865–874
14. Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K (2008) Thedevelopment of microgels/nanogels for drug delivery applications.Prog Polym Sci 33(4):448–477
15. Saunders BR, Laajam N, Daly E, Teow S, Hu XH, Stepto R (2009)Microgels: from responsive polymer colloids to biomaterials. AdvColloid Interface Sci 147–48:251–262
16. Vinogradov SV (2006) Colloidal microgels in drug delivery appli-cations. Curr Pharm Design 12(36):4703–4712
17. Hoare T, Pelton R (2004) Highly pH and temperature responsivemicrogels functionalized with vinylacetic acid. Macromolecules 37(7):2544–2550
18. Hoare T, Pelton R (2005) Electrophoresis of functionalized micro-gels: morphological insights. Polymer 46(4):1139–1150
19. Hoare T, Pelton R (2006) Titrametric characterization of pH-induced phase transitions in functionalized microgels. Langmuir22(17):7342–7350
20. Hoare T, Pelton R (2007) Calorimetric analysis of thermal phasetransitions in functionalized microgels. J Phys Chem B 111(6):1334–1342
21. Hoare T, Pelton R (2008) Impact of microgel morphology on func-tionalized microgel-drug interactions. Langmuir 24(3):1005–1012
22. Sahiner N, Ozay O, Aktas N (2011) 4-Vinylpyridine-based smartnanoparticles with N-isopropylacrylamide, 2-hydroxyethyl meth-acrylate, acrylic acid, and methacrylic acid for potential biomedicalapplications. Curr Nanosci 7(3):453–462
23. Bradley M, Vincent B (2008) Poly(vinylpyridine) core/poly(N-isopropylacrylamide) shell microgel particles: their characteriza-tion and the uptake and release of an anionic surfactant. Langmuir24(6):2421–2425
24. Liu WJ, Huang YM, Liu HL (2007) Preparation and characteriza-tion of temperature and pH responsive core-shell microgel. ActaChim Sinica 65(2):91–94
25. Richtering W, Kleinen J, Klee A (2010) Influence of architectureon the interaction of negatively charged multisensitive poly(N-isopropylacrylamide)-co-methacrylic acid microgels with oppo-sitely charged polyelectrolyte: absorption vs adsorption. Langmuir26(13):11258–11265
26. Bradley M, Vincent B, Burnett G (2007) Uptake and release ofanionic surfactant into and from cationic core-shell microgel par-ticles. Langmuir 23(18):9237–9241
27. Gan DJ, Lyon LA (2001) Tunable swelling kinetics in core-shellhydrogel nanoparticles. J Am Chem Soc 123(31):7511–7517
28. Gan DJ, Lyon LA (2003) Fluorescence nonradiative energy trans-fer analysis of crosslinker heterogeneity in core-shell hydrogelnanoparticles. Anal Chim Acta 496(1–2):53–63
29. Berndt I, Richtering W (2003) Doubly temperature sensitive core-shell microgels. Macromolecules 36(23):8780–8785
30. Richtering W, Scherzinger C, Lindner P, Keerl M (2010) Conon-solvency of poly(N,N-diethylacrylamide) (PDEAAM) and poly(N-isopropylacrylamide) (PNIPAM) based microgels in water/metha-nol mixtures: copolymer vs core-shell microgel. Macromolecules43(16):6829–6833
31. Hu ZB, Chi CL, Cai T (2009) Oligo(ethylene glycol)-based ther-moresponsive core-shell microgels. Langmuir 25(6):3814–3819
32. Suzuki D, Yoshida R (2010) Self-oscillating core/shell microgels:effect of a crosslinked nanoshell on autonomous oscillation of thecore. Polym J 42(6):501–508
33. Lyon LA, Suzuki D, McGrath JG, Kawaguchi H (2007) Colloidalcrystals of thermosensitive, core/shell hybrid microgels. J PhysChem C 111(15):5667–5672
Colloid Polym Sci (2012) 290:1181–1192 1191
34. Kuckling D, Vo CD, Wohlrab SE (2002) Preparation of nanogelswith temperature-responsive core and pH-responsive arms byphoto-cross-linking. Langmuir 18(11):4263–4269
35. Huang YM, Liu WJ, Liu HL, Hu Y (2007) Composite structure oftemperature sensitive chitosan microgel and anomalous behaviorin alcohol solutions. J Colloid Interface Sci 313(1):117–121
36. Wang PX, Chen Q, Xu K, Zhang WD, Song CL (2009) Prepara-tion and characterization of poly(N-isopropylacrylamide)/polyvi-nylamine core-shell microgels. Colloid Polym Sci 287(11):1339–1346
37. Wu C, Hu TJ, You YZ, Pan CY (2002) The coil-to-globule-to-brush transition of linear thermally sensitive poly(N-isopropylacry-lamide) chains grafted on a spherical microgel. J Phys Chem B 106(26):6659–6662
38. Charleux B, Rieger J, Grazon C, Alaimo D, Jerome C (2009)Pegylated thermally responsive block copolymer micelles andnanogels via in situ RAFT aqueous dispersion polymerization. JPolym Sci Pol Chem 47(9):2373–2390
39. Nagasaki Y, Oishi M (2007) Synthesis, characterization, and bio-medical applications of core-shell-type stimuli-responsive nano-gels—nanogel composed of poly[2-(N,N-diethylamino)ethylmethacrylate] core and PEG tethered chains. React Funct Polym67(11):1311–1329
40. Thurecht KJ, Zheng Y, Turner W, Zong MM, Irvine DJ, HowdleSM (2011) Biodegradable core-shell materials via RAFT and ROP:characterization and comparison of hyperbranched and microgelparticles. Macromolecules 44(6):1347–1354
41. Seiffert S, Thiele J, Abate AR, Weitz DA (2010) Smart microgelcapsules from macromolecular precursors. J Am Chem Soc 132(18):6606–6609
42. Hoare T, McLean D (2006) Kinetic prediction of functional groupdistributions in thermosensitive microgels. J Phys Chem B 110(41):20327–20336
43. Hoare T, McLean D (2006) Multi-component kinetic modeling forcontrolling local compositions in thermosensitive polymers. Mac-romol Theory Simul 15(8):619–632
44. Hoare T, Pelton R (2007) Functionalized microgel swelling: com-paring theory and experiment. J Phys Chem B 111(41):11895–11906
46. Acciaro R, Gilányi T, Varga I (2011) Preparation of monodispersepoly(N-isopropylacrylamide) microgel particles with homogenouscross-link density distribution. Langmuir 27(12):7917–7925.doi:10.1021/la2010387
47. Zha LS, Zhang QS, Ma JH, Liang BR (2009) A novel route toprepare pH- and temperature-sensitive nanogels via a semibatchprocess. J Colloid Interf Sci 330(2):330–336
48. Forcada J, Imaz A, Miranda JI, Ramos J (2008) Evidences of ahydrolysis process in the synthesis of N-vinylcaprolactam-basedmicrogels. Eur Polym J 44(12):4002–4011
49. Sun GX, Zhang MZ, Xu Y, Lu YM, Ni PH (2009) Synthesis andproperties of pH-responsive cationic microgels. Acta Chim Sinica67(14):1685–1690
50. Ponratnam S, Kapur SL (1977) Reactivity ratios of ionizing mono-mers in aqueous-solution—copolymerization of acrylic and meth-acrylic acids with acrylamide. Makromol Chem 178(4):1029–1038
51. Brandrup J, Immergut EH, Grulke EA (eds) (1999) Polymer hand-book, 4th edn. Wiley, New York
52. Ohshima H (1994) Electrophoretic mobility of soft particles. JColloid Interface Sci 163(2):474–483
53. Elmas B, Tuncel M, Senel S, Patir S, Tuncel A (2007) Hydroxylfunctionalized thermosensitive microgels with quadratic crosslink-ing density distribution. J Colloid Interface Sci 313(1):174–183
54. Fernandez-Barbero A, Fernandez-Nieves A, Grillo I, Lopez-Cabarcos E (2002) Structural modifications in the swelling ofinhomogeneous microgels by light and neutron scattering. PhysRev E 66(5):051803
55. Stieger M, RichteringW, Pedersen JS, Lindner P (2004) Small-angleneutron scattering study of structural changes in temperature sensi-tive microgel colloids. J Chem Phys 120(13):6197–6206