Severe MDC1A Congenital Muscular Dystrophy Due to aSplicing Mutation in the LAMA2 Gene Resulting in Exon
Skipping and Significant Decrease of mRNA Level
OLFA SIALA,1 NACIM LOUHICHI,1 CHAHNEZ TRIKI,2 MADELEINE MORINIÈRE,3 AHMED REBAI,4
PASCALE RICHARD,5,6 PASCALE GUICHENEY,5,7 FAOUZI BAKLOUTI,3 and FAIZA FAKHFAKH1
ABSTRACT
Congenital muscular dystrophies (CMDs) are a clinically and genetically heterogeneous group of neuromus-cular disorders, with autosomal recessive inheritance. We report a patient with severe congenital musculardystrophy and total deficiency in the laminin �2 chain. Genetic analyses showed a linkage to the MDC1A lo-cus for the patient’s family, and DNA sequencing revealed in the propositus of a new homozygous mutationin the donor splice site of intron 58 of the LAMA2 gene. RT-PCR experiments performed on total RNA froma patient’s muscle biopsy showed a complete skipping of exon 58 in LAMA2 cDNA and a significant decreasein the LAMA2 mRNA level. This exon skipping altered the open reading frame of the mutant transcript andgenerated a premature termination codon (PTC) within exon 59, which potentially elicits the nonsense mRNAto degradation by NMD (nonsense-mediated mRNA decay). However, the residual exon 58-lacking mRNAcould potentially be translated, and the resulting truncated �2 chain would lack its LG4 and LG5 domainsthat are involved in binding with �-dystroglycan. These results demonstrate the utility of mRNA analysis tounderstand the mutation primary impact and the disease phenotype in the patients.
199
INTRODUCTION
LAMININ2 IS THE PREDOMINANT ISOFORM of laminins presentin the extracellular matrix of skeletal muscle. It is a het-
erotrimeric protein composed of two light chains (�1 and �1)and a heavy �2 chain (merosin), which are assembled into asupramolecular structure (Timpl and Brown 1994; Sewry et al.1995) Like all the �-chains, the C-terminal end of the �2 chainis constituted of five internal homologous repeat domains(LG1–LG5) forming the “G” globular region (Hohenster et al. 1999), and containing the interaction sites with the dystro-phin-associated glycoproteins, essentially the �-dystroglycan(Guicheney et al. 1997). Also, the LG4 and LG5 domains areparticularly important to establish the binding between laminin�2 and �-dystroglycan (Timpl et al. 2000; Wizemann et al.
2003). Laminin �2 is encoded by the LAMA2 gene on the 6q-22 chromosome, and comprises 65 exons encompassing 633 kbof human DNA (Vainzof et al. 2005). Mutations within theLAMA2 gene generate primary deficiency in laminin �2 andcause merosin-deficient congenital muscular dystrophy orMDC1A. This form is characterized by profound hypotonia atbirth or in the first few months of life, delayed motor mile-stones, and instability to reach independent ambulation (Guich-eney et al. 1998). In several cases, brain MRI shows abnormalT2 signals of the periventricular and subcortical white matterdespite normal mental functions (Tezak et al. 2003). Mutationsin the LAMA2 gene have been identified in laminin �2 defi-cient CMD cases without a “hot spot” region. However, it ap-pears that most of the mutations clustered within exons 1 to 31of the LAMA2 gene lead to a loss of laminin �2 protein, whereas
1Laboratoire de Génétique Moléculaire Humaine, Faculté de Médecine de Sfax, 3029 Sfax, Tunisia.2Service de Neurologie, C H U Habib Bourguiba, 3029 Sfax, Tunisia.3Equipe épissage alternatif et différenciation cellulaire, Centre de Génétique moléculaire et cellulaire, CNRS UMR 5534, Université Lyon 1,
69622 Villeurbanne Cedex, France.4Centre de Biotechnologie de Sfax, Tunisia; B P. “K” 3038 Sfax, Tunisia.5INSERM U582, Institut de Myologie, groupe hôspitalier Salpêtrière, Paris 75651, France.6AP-HP, Groupe Hospitalier Pitié Salpêtrière, Service de Biochimie Métabolique, Paris, F-75013.7Université Pierre et Marie Curie, Paris, France.The first two authors contributed equally to this work.
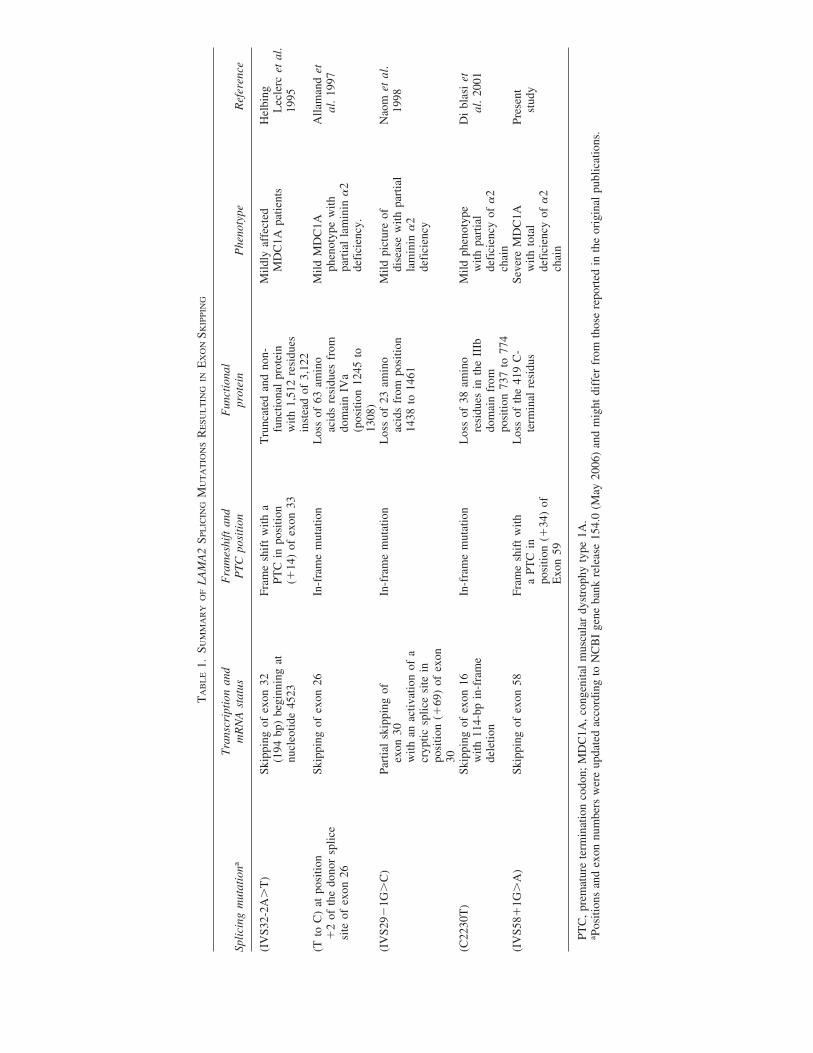
those clustered within exons 58 to 65 produce partial laminin�2 deficiency with a less severe clinical picture (Girgenrath etal. 2004). Among LAMA2 mutations, only few have been de-scribed to affect splicing. These mutations can generate exonskipping (Helbing-Leclerc et al. 1995; Di Blasi et al. 2001), adecrease in spliceosome–mRNA binding (Naom et al. 2000),or/and an activation of cryptic splice site(s) (Naom et al. 1998).
In the present study, we report a new homozygous (G�A)transition in the first nucleotide of the donor splice site of in-tron 58. RT-PCR and semiquantitative RT-PCR analyses re-vealed total skipping of exon 58 with a significant decrease ofthe LAMA2 mRNA level. Functional findings are in agreementwith the total deficiency in laminin �2 revealed by immuno-histochemistry.
PATIENTS AND METHODS
Patient
The patient is a 4-year-old son born after an uneventful preg-nancy and term delivered from consanguineous Tunisian par-ents. He presented at birth with generalized hypotonia andweakness with joint contractures. He had delayed motor mile-stones, and at the age of 2 years, he was not able to stand upand had axial and peripheral hypotonia with joint contracturesand without calf hypertrophy. The deep tendon reflex was abol-ished, and the foot exhibited equinovarus deformity. He hadnormal head circumference and no cognitive impairment. Thelevel of serum CK was high (950 UI/l) and the MRI showedabnormal signal of the periventricular white matter. Blood sam-ples were collected from all family members after informedconsent. Biopsies from the deltoid muscle were obtained afterthe consent of both the propositus’ family and control individ-ual, and then frozen at �80°C, awaiting investigation.
METHODS
Immunohistochemistry
Muscle samples were analyzed by conventional immuno-histochemical methods (Jones et al. 1998), using unfixedfrozen cryostat sections (5 �m). Expression of laminin a2,�1, and �1, dystrophin, �-sarcoglycan (DAG50), and �-dy-stroglycan, was examined using monoclonal antibodies di-rected against the 80-kDa C-terminal fragment of humanlaminin �2 (mAb 1922 Chemicon, Temacula, CA), the N-ter-minal 300-kDa fragment of human merosin (NCL-merosinNovocastra, Newcastle upon Tyne, UK), human �1, and �1chains (mAb 1928 and mAb 1914, Chemicon), dystrophin(NCL Dys2 Novocastra,), �-dystroglycan (VIA4-1-05298,Euromedex, Souffelweyershein, France), and �-sarcoglycan(NCL 50-DAG, Novocastra). All primary antibodies were ap-plied for 1 h on muscular sections, washed in phosphate-buffered saline (PBS 1X, pH 7.4) and then muscle proteinswere revealed with an appropriate fluorescein isothiocyanate(FITC) conjugated secondary antibody (antimouse or antiratimmunoglobulins, Dako, Carpenteria, CA). All dilutions andwashes were made in PBS.
Linkage analyses and LAMA2 gene sequencing
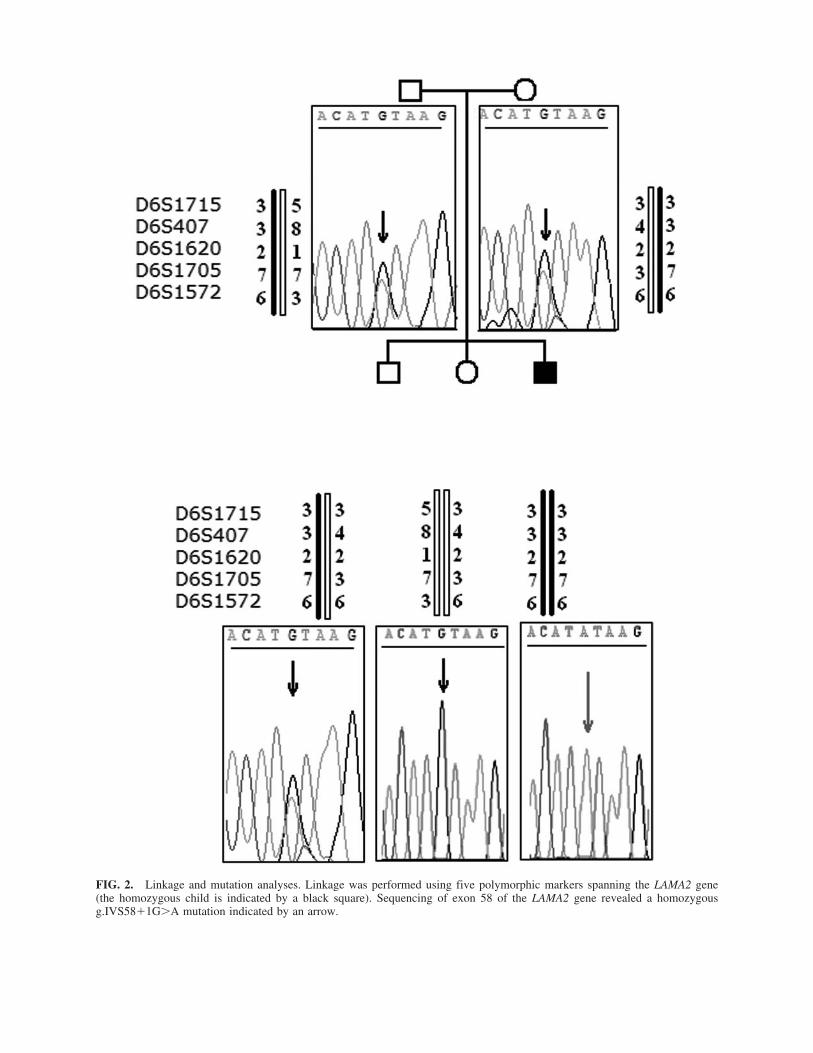
Genetic linkage was performed on genomic DNA extracted fromblood leucocytes using standard phenol/chloroform procedure. Fivemicrosatellite markers spanning the LAMA2 locus (D6S1715,D6S407 upstream of the gene, and D6S1620, D6S1705, andD6S1572 downstream of the gene) were analyzed. Mutation anal-ysis was performed by PCR amplification of each of the 65 en-coding exons of the LAMA2 gene and intron–exon boundaries aspreviously published (Guicheney et al. 1998), then sequenced us-ing an ABI Prism Big Dye Terminator Cycle Sequencing Kit (Ap-plied Biosystems, Bedford, MA), and loaded on an “ABI 3100Avant” automated sequencer. The g.IVS58�1G�A mutation foundin the affected boy induces the loss of NspI restriction site (NewEngland Biolabs, Beverly, MA), and was used to confirm the mu-tation in relatives and in 100 unrelated Tunisian healthy controls.
RNA extraction and RT-PCR
Total RNA was extracted from muscle biopsies using “Ex-tract-all” solution from Eurobio (Lille, France) according to themanufacturer’s recommendations. RT-PCR experiments werecarried out as previously described (Moriniere et al. 2000). Thesequence in LAMA2 cDNA spanning exons 56–59 was ampli-fied using the following primers: 5�GAAAACGAAGGCA-GACTGGAC 3� (forward primer 23 pb downstream of exon55) and 5�GGAAGTAAGAACCGAAGCTG 3� (reverse primer35 pb upstream of exon 60). The PCR reaction was performedfor 35 rounds, each consisting of 30 sec at 95°C, 30 sec at 58°C,and 30 sec at 72°C followed by a final elongation step of 10min at 72°C. The expected PCR products of 666 bp were elec-trophoresis on a 2% agarose gel in TAE buffer (40 mM Trisacetate, 1 mM EDTA, pH 7.7), stained with 0.5 �g/ml ethid-ium bromide and then visualized under UV light.
Cloning and sequencing of RT-PCR products
PCR products were gel-purified with the Qiaquick PCR pu-rification Kit (Qiagen, Chatsworth, CA) then cloned into “PSTBlue-1” vector (perfectly Blunt cloning Kit 70-191-3, Novagen,Madison, WI) according to the manufacturer’s instructions. Re-combinant plasmid DNAs were isolated using a SV Wizardplasmid miniprep Kit, Promega (Madison, WI), and sequencedusing the ABI Prism 3100-Avant automated DNA sequencer.
Semiquantitative RT-PCR assay
RT-PCR was performed with a coamplification of theLAMA2 minigene transcript using primers listed previously, andthe HPRT minigene to provide a semiquantitative internal con-trol for RNA template quantity and PCR reaction efficiency.The HPRT gene encodes the “Hypoxanthine phosphoribosyltransferase,” a constitutively expressed enzyme (Johnson et al.2004). The sequences of HPRT primers used were: forward,5�ATGAGAGCTATTGTAATGACCAGTC3�, reverse, 5�CT-GGCGATGTCAATAGGACTC 3�, leading to a 430-bp PCRproduct. The cycle’s number is chosen in order to fall into theexponentially increasing phase of detectable products, thus en-abling quantification. For a significant result, at least triplicateessays for each sample have been performed (Deguillien et al.2001). The RT step was performed as previously described ina final volume of 30 �l corresponding to a master RT reaction.For duplex PCR, the PCR reagents were then added as described
SIALA ET AL.200
previously, and the resulting master mix was dispensed in iceinto three time-course tubes. The PCR coamplification was car-ried out for 24, 26, and 28 cycles. Each time point contained25 pmol of each LAMA2 primer and 10 pmol of each HPRTprimer. When the desired number of cycles was completed, thecorresponding time-point PCR was stopped by chilling the tubeon ice. The PCR products were resolved on 5% polyacrylamide,gel stained with SYBR Green I (Amersham, Arlington Heights,IL), and quantified on a FluoroImager 595 system (MolecularDynamics, Sunnyvale, CA). The LAMA2 mRNA level was mea-sured as the ratio of LAMA2 to HPRT in each sample by usingImageQuant Mac software (vesion 1.2).
Statistical and bioinformatic analysis
Multiple pair-wise comparisons using the Student–New-man–Keuls test were performed to analyze the differences inmRNA expression assayed by semiquantitative RT-PCR. Allvalues were expressed as mean � standard deviation (�SD),and statistical significance was assumed at p � 0.05. The ef-fect of the g.IVS58�1G�A mutation was first assessed usingthe consensus score calculation method to quantify the influ-ence of the mutation on the formation of splicing loops (Carmelet al. 2004) and corresponding software available on-line athttp://ast.bioinfo.tau.ac.il/splicesite.
RESULTS
Complete absence of laminin �2 chain associated withabnormal distribution of dystrophin
Immunofluorescent microscopy observations of the patientmuscle sections showed a marked variation in the fibers diam-
eter, with a marked increase in fibrous connective tissue at theexpense of muscular fibers. These experiments showed a totaldeficiency in the laminin �2 chain using either of the two an-tibodies: (anti-80) (Fig. 1a and b) or (anti-300) (Fig. 1g and h).Expression of laminin �1 and �1 also showed no epitope im-munostaining around the muscular fibers (Fig. 1c and d, i, andj, respectively). �-Sarcoglycan (Fig. 1k and l) and �-dystro-glycan (not shown) were normally expressed in the patient.However, dystrophin was partially reduced. Indeed, the anti-body directed against the C-terminal domain of dystrophinshowed uneven immunostaining at the sarcolemma (Fig. 1e and f).
A splicing mutation triggers exon skipping
Genetic linkage study was performed using five markersaround the LAMA2 locus. These experiments revealed a link-age to the LAMA2 gene based on homozygosity in the affectedchild (Fig. 2). All the 65 coding exons of the LAMA2 gene andtheir intron–exon junctions were then PCR-amplified and se-quenced. DNA sequencing revealed in the patient a homozy-gous g.IVS58�1G�A mutation at the first position of the donorsplicing site of intron 58. The mutation was also found in theparents and one healthy carrier brother at the heterozygous state,and absent in the unaffected sister (Fig. 2). The IVS58�1G�Amutation abolishes a NspI restriction site at the exon–intronjunction. However, NspI digestion consistently showed the ab-sence of the mutation in 200 control chromosomes.
The splicing mutation triggers exon 58 skipping
The g.IVS58�1G�A substitution was first assessed usingthe consensus score calculation method to quantify the influ-ence of the mutation on the formation of splicing loops. The
LAMA2 EXON SKIPPING IN MDC1A FORM 201
FIG. 1. Immunohistochemistry. Immunostaining of skeletal muscle biopsies from the control and the patient, with antibodiesagainst the 80-kDa fragment (a and b; �2/80) and the 300-kDa fragment of laminin alpha2 (g and h; �2/300), laminin beta1 (cand d), laminin gamma1 (i and j), to dystrophin (e and f) and to �-sarcoglycan (k and l). Note the total deficiency of laminin al-pha2, beta1, and gamma1 in the patient compared to the control. Scale bar: 100 �m.
FIG. 2. Linkage and mutation analyses. Linkage was performed using five polymorphic markers spanning the LAMA2 gene(the homozygous child is indicated by a black square). Sequencing of exon 58 of the LAMA2 gene revealed a homozygousg.IVS58�1G�A mutation indicated by an arrow.
calculated values of the numerical score for the altered sequence(AT/ataagt) were 57.36% compared to 69.13% for the wild-type splice donor site (AT/gtaagt) eliciting an abnormal splic-ing process. In order to establish the effect of this mutation onRNA splicing, we analyzed LAMA2 mRNA obtained from mus-cle biopsies. Using primers within exon 56 and exon 60, RT-PCR revealed an amplified fragment of 666 bp in the control,which corresponds to the expected splicing product. In contrast,RT-PCR experiments displayed a shorter band of 497 bp in thepatient (Fig. 3A). This result suggested a splicing defect, prob-ably affecting exon 58. In order to verify this hypothesis, theRT-PCR products from both the control and the patient weresubcloned into “PST Blue-1” vector and sequenced. As ex-pected, sequence analysis revealed a total lack of exon 58 inthe patient (Fig. 3B), thus confirming that the (G�A) mutationat the highly invariable GU dinucleotide completely abolishesexon 58 5� splicing site selection.
The splicing mutation is associated with decreasedlevel of LAMA2 mRNA
The skipping of the 169 bp of exon 58 leads to a translationframe shift, and the occurrence of a PTC at position �34 ofexon 59. We therefore asked whether these changes would leadto mRNA degradation by NMD. The LAMA2 mRNA level wasestimated by a semiquantitative RT-PCR approach using HPRTmRNA as an internal control (Fig. 4A and B). The steady-statelevel of the LAMA2 transcript was provided as the ratio ofLAMA2 mRNA levels relative to those of HPRT mRNA, andresults are expressed as mean � SD. As shown in Figure 4, thenormalized LAMA2 mRNA level is dramatically low in the patient, compared to the control. The estimated decrease isabout 2.6-fold in the MDC1A affected patient (p-value �0.05)(Fig. 4C).
Predicted effect of g.IVS58�1G�A mutation on thestructure and the function of laminin �2 protein
In order to predict the effect of (IVS58�1G�A) mutationon protein structure and function, we investigated in silico theeffect of exon 58 skipping. We noted first that the skipping ofexon 58 generates a PTC at position 34 within exon 59 of theLAMA2 gene. The truncated protein, if translated, would con-tain only 2703 residues over a total of 3122, and would lack its last 419 residues clustering the LG4 and LG5 domains, re-sulting in the loss of its functional binding region to the �-dystroglycan.
DISCUSSION
In the present study, we perform genetic and functional stud-ies in a patient affected with severe CMD form. Genetic andmolecular analyses showed linkage to the MDC1A locus and anew homozygous splice mutation in the donor splice site of in-tron 58 of the LAMA2 gene g.IVS58�1G�A. RT-PCR analy-sis of LAMA2 mRNA isolated from the patient’s muscle biopsyshowed an abnormally spliced transcript missing exon 58,which causes the occurrence of a PTC at position 34 withinexon 59. The resulting nonsense mRNA is elicited to NMDdegradation according to the PTC position rule previously stated(Nagy and Maquat, 1998). In order to study the consequenceof g.IVS58�1G�A mutation on the LAMA2 mRNA level inthe patient, we have performed semiquantitative RT-PCR as-says using the HPRT gene as control for normalization. Weshowed a 2.6-fold decrease in the LAMA2 mRNA level in thestudied patient compared to the control. It is generally recog-nized that the mRNA expression level is lowered when non-sense or frame-shift mutations occur within the reading frameor when a mutation associated with an RNA splice defect oc-
LAMA2 EXON SKIPPING IN MDC1A FORM 203
FIG. 3. LAMA2 mRNA analysis. (A) Gel electrophoresis of RT-PCR amplification product of the region spanning exons (56–60)of the LAMA2 gene showing: a single band in the control at the expected size of 666 bp (1) and a smaller one of 497 bp in thepatient (2). A control without RNA (3). The size marker is a 100-bp DNA ladder from Fermentas (M). (B) Sequencing of sub-cloned amplified cDNA products in the control and the patient. Note the total skipping of exon 58 in the patient.
A B
curs in the gene (Cooper 1993); the reduction of mutant tran-script quantity may be due to abnormal splice site selection (Di-etz and Kendzior 1994). Surprisingly, a milder phenotype hasbeen described in association with a fivefold decrease inLAMA2 mRNA level and a frame shift mutation at the adjacentexon 57 (Prandini et al. 2004).
In the LAMA2 gene, all exons are constitutive, except exon31, which is alternatively spliced. The skipping of exon 31leads to an isoform that is unevenly distributed in the musclefiber of basal lamina but preferentially expressed in discreteareas (Pegoraro et al. 2000). Exon skipping has been reportedin several cases of MDC1A. In Table 1, we give a summaryof splicing mutations described up to now in the LAMA2 geneand resulting in total or partial exon skipping. Skipping of exon25 was previously reported in two patients harboring a ho-mozygous mutation in the splice consensus sequence and pre-sented with a mild MDC1A phenotype with partial merosindeficiency. This exon 25 skipping leads to a predicted laminina2 chain lacking 63 amino acid residues in domain IVa (Alla-mand et al. 1997). The skipping of exon 31 following g.IVS31-2A�T mutation was also reported in mildly affected MDC1Apatients (Helbing-Leclerc et al. 1995). In addition, anotherstudy demonstrated that g.IVS28-1G�C and g.IVS37�5G�Ain the LAMA2 gene cause an activation of cryptic splice siteswithin exon 29 following the first mutation (Naom et al. 1998)and within position �10 of intron 37 following the second mu-tation (Tezak et al. 2003). On the other hand, a C to T transi-tion at position 2230 of the LAMA2 gene, outside of the con-sensus splice site sequences, causes a skipping of exon 15 ina mildly affected MDC1A case (Di Blasi et al. 2001). How-ever, in some cases, mutations within the conserved consen-sus sequence do not systematically cause exon skipping
from all mRNA molecules. In two mildly affected girls,g.IVS37�5G � C and a g.IVS63�6T � C mutations in theLAMA2 gene decreased the binding of the spliceosome to theprecursor mRNA 15-fold for the first mutation and 2.6-fold forthe second (Naom et al. 2000). Although mRNA analysis sug-gests a residual amount of LAMA2 exon 58-lacking molecules,immunohistochemical studies using two antibodies against theC-terminal and the N-terminal regions of the laminin �2 chain,showed no detectable level of the laminin �2 protein assem-bled at the myofiber junctions. These results are in agreementwith the MDC1A form often characterized by a total laminin�2 deficiency and the most severe clinical picture (Jones et al.2001; Ralte et al. 2003). Immunohistochemical findings pre-sented here also showed the complete absence of associatedlaminins �1 and �1 chains; this result was often showed in �2laminin-deficient congenital dystrophy eliciting secondaryconsequences of myopathy, denervation, regeneration, and theinability of the mutated �2 chain (merosin) to form polymers.The skipping of exon 58 leads to the loss of the C-terminal419 amino acids of merosin including the fourth and the fifthLG modules of the “G” globular domain of laminin �2 repre-senting the functional binding region of merosin. This regionof laminin �2 is also important to assume a wide range of con-formations relative to the LG1–LG3 region, and it contains twoincompletely coordinated calcium ion sites implicated in the�-dystroglycan binding (Timpl et al. 2000). Indeed, if the aber-rant LAMA2 mRNA is translated, it would generate a truncatedprotein containing the N-terminal 2703 residues; therefore,both mAb1922 anti-80-kDa antibody (epitope betweenresidues 2581 and 2756 of �2 chain) (He et al. 2001) and anti-300 antibody would have been able to detect the truncated pro-tein. Equally possible, the truncated protein would have an
SIALA ET AL.204
0
2
Control
control
LAMA2 levelHPRT Patient
patient
1.8
1.6
1.4
1.2
1
0.8
0.6
0.4
0.2
5 6 7 8 1
497 bp
430 bp
2 3 4
Semi-quantitative RT PCR of LAMA2 mRNA2
(B) (C)
FIG. 4. Semiquantitative RT-PCR analysis of LAMA2 mRNA. Gel electrophoresis of coamplified RT-PCR products of LAMA2and HPRT cDNAs on 5% polyacrylamide gel. The RT-PCR coamplification was interrupted at cycles 24, 26, and 28 in the con-trol (lanes 7, 6, and 5, respectively) (A), and in the patient (lanes 3, 2, and 1) (B). Lanes 4 and 8: (PBR322/ HaeIII) marker. (C)Expression level of LAMA2 gene in the patient (dotted) compared to the control (hatched). Values are expressed as mean � SDand statistical significance was assessed by Student–Newman–Keuls test with (p � 0.05).
TA
BL
E1.
SUM
MA
RY
OF
LA
MA
2SP
LIC
ING
MU
TA
TIO
NS
RE
SU
LT
ING
INE
XO
NSK
IPP
ING
Tra
nscr
ipti
on a
ndF
ram
eshi
ft a
ndF
unct
iona
lSp
lici
ng m
utat
iona
mR
NA
sta
tus
PT
C p
osit
ion
prot
ein
Phe
noty
peR
efer
ence
(IV
S32-
2A�
T)
Skip
ping
of
exon
32
Fram
e sh
ift
with
aT
runc
ated
and
non
-M
ildly
aff
ecte
dH
elbi
ng(1
94 b
p) b
egin
ning
at
PTC
in
posi
tion
func
tiona
l pr
otei
nM
DC
1A p
atie
nts
Lec
lerc
et
al.
nucl
eotid
e 45
23(�
14)
of e
xon
33w
ith 1
,512
res
idue
s19
95in
stea
d of
3,1
22(T
to
C)
at p
ositi
onSk
ippi
ng o
f ex
on 2
6In
-fra
me
mut
atio
nL
oss
of 6
3 am
ino
Mild
MD
C1A
Alla
man
d et
�2
of t
he d
onor
spl
ice
acid
s re
sidu
es f
rom
phen
otyp
e w
ithal
. 19
97si
te o
f ex
on 2
6do
mai
n IV
apa
rtia
l la
min
in �
2(p
ositi
on 1
245
tode
fici
ency
.13
08)
(IV
S29�
1G�
C)
Part
ial
skip
ping
of
In-f
ram
e m
utat
ion
Los
s of
23
amin
oM
ild p
ictu
re o
fN
aom
et
al.
exon
30
acid
s fr
om p
ositi
ondi
seas
e w
ith p
artia
l19
98w
ith a
n ac
tivat
ion
of a
1438
to
1461
lam
inin
�2
cryp
tic s
plic
e si
te i
nde
fici
ency
posi
tion
(�69
) of
exo
n30
(C22
30T
)Sk
ippi
ng o
f ex
on 1
6In
-fra
me
mut
atio
nL
oss
of 3
8 am
ino
Mild
phe
noty
peD
i bl
asi
etw
ith 1
14-b
p in
-fra
me
resi
dues
in
the
IIIb
with
par
tial
al.
2001
dele
tion
dom
ain
from
defi
cien
cy o
f �
2po
sitio
n 73
7 to
774
chai
n(I
VS5
8�1G
�A
)Sk
ippi
ng o
f ex
on 5
8Fr
ame
shif
t w
ithL
oss
of t
he 4
19 C
-Se
vere
MD
C1A
Pres
ent
a PT
C i
nte
rmin
al r
esid
usw
ith t
otal
stud
ypo
sitio
n (�
34)
ofde
fici
ency
of
�2
Exo
n 59
chai
n
PTC
, pr
emat
ure
term
inat
ion
codo
n; M
DC
1A,
cong
enita
l m
uscu
lar
dyst
roph
y ty
pe 1
A.
a Pos
ition
s an
d ex
on n
umbe
rs w
ere
upda
ted
acco
rdin
g to
NC
BI
gene
ban
k re
leas
e 15
4.0
(May
200
6) a
nd m
ight
dif
fer
from
tho
se r
epor
ted
in t
he o
rigi
nal
publ
icat
ions
.
aberrant folding and be targeted for degradation by a “proteinquality control” mechanism requiring molecular chaperonesand cellular proteases (Bross et al. 1999).
We, therefore, conclude that the complete skipping of exon58 leads to complete deficiency of functional Laminin �2,mainly through degradation of the nonsense mRNA, and pos-sibly through translation from mRNA molecules that escape theNMD targeting, of a truncated laminin �2 lacking the C-ter-minal end. Complete absence of a functional laminin �2 mostlikely results in the severe MDC1A phenotype observed in thisfamily. In addition, our findings demonstrate that mRNA anal-ysis and quantification are important to clarify the primary im-pact of genomic mutation on splicing efficiency and expressionlevel. These results, in complementarity with immunohisto-chemical results, allow explaination of the phenotype and theclinical severity of disease.
ACKNOWLEDGMENTS
We thank the patient and his family for their cooperation inthe present study, and Mr. Jamil Jaoua for having proofread thispaper. This work was supported by funds from the Ministèrede Recherche Scientifique et de Technologie (Tunisia), the As-sociation Française contre les Myopathies (AFM), the RegionRhône-Alpes (MIRA), and the Centre National de RechercheScientifique (project CNRS/DGRST project).
REFERENCES
Allamand V, Sunada Y, Salih MA, Straub V, Ozo CO, Al-Turaiki MH,Akbar M, Kolo T, Colognato H, Zhang X, Sorokin LM, YurchencoPD, Tryggvason K, Campbell KP (1997) Mild congenital musculardystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet 6:747–752.
Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L,Gregersen N (1999) Protein misfolding and degradation in geneticdiseases. Hum Mutat 14:186–198.
Carmel I, Tal S, Vig I, Ast G (2004) Comparative analysis detects de-pendencies among the 5_ splice-site positions. RNA 10:828–840.
Cooper DN (1993) Human gene mutations affecting RNA processingand translation. Ann Med 1:11–17.
Deguillien M, Huang S, Moriniere M, Dreumont N, Benz E.J, BakloutiF (2001) Multiple cis elements regulate an alternative splicing eventat 4.1R pre-mRNA during erythroid differentiation. Blood98:3809–3816.
Di Blasi C, He Y, Morandi L, Cornelio F, Guicheney P, Mora M (2001)Mild muscular dystrophy due to a nonsense mutation in the LAMA2gene resulting in exon skipping. Brain 124:698–704.
Dietz HC, Kendzior RJ Jr (1994) Maintenance of an open reading frameas an additional level of scrutiny during splice site selection. NatGenet 8:183–188.
Girgenrath M, Dominov JA, Kostek CA, Miller JB (2004) Inhibitionof apoptosis improves outcome in a model of congenital musculardystrophy. J Clin Invest 114:1635–1639.
Guicheney P, Vignier N, Helbling-Leclerc A, Nissinen M, Zhang X,Cruaud C, Lambert JC, Richelme C, Topaloglu H, Merlini L, BaroisA, Schwartz K, Tome FM, Tryggvason K, Fardeau M (1997) Ge-netics of laminin alpha 2 chain (or merosin) deficient congenital mus-cular dystrophy: from identification of mutations to prenatal diag-nosis. Neuromuscul Disord 7:180–186.
Guicheney P, Vignier N, Zhang X, He Y, Cruaud C, Frey V, Helbling-Leclerc A, Richard P, Estournet B, Merlini L, Topaloglu H, MoraM, Harpey JP, Haenggeli CA, Barois A, Hainque B, Schwartz K,Tome FM, Fardeau M, Tryggvason K (1998) PCR based mutationscreening of the laminin alpha2 chain gene (LAMA2): application toprenatal diagnosis and search for founder effects in congenital mus-cular dystrophy. J Med Genet 35:211–217.
He Y, Jones KJ, Vignier N, Morgan G, Chevallay M, Barois A, Es-tournet-Mathiaud B, Hori H, Mizuta T, Tome FM, North KN, Guich-eney P (2001) Congenital muscular dystrophy with primary partiallaminin alpha2 chain deficiency: molecular study. Neurology57:1319–1322.
Helbing-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weis-senbach J, Tome F.M, Schwartz K, Fardeu M, Tryggvason K, Guich-eney P (1995) Mutations in the laminin �2 chain gene (LAMA2) causemerosin-deficient congenital muscular dystrophy. Nat Genet11:216–218.
Hohenster E, Tisi D, Talts JF, Timpl R (1999) The crystal structureof Laminin G-like module reveals the molecular basis of �-Dys-troglycan binding to laminins, perlecan, and agrin. Mol Cell4:783–792.
Johnson CM, Young S, Sellins K.S, Frank G.R (2004) Selection ofHPRT primers as controls for determination of mRNA expression indogs by RT-PCR. Vet Immunol Immunopathol 99:47–51.
Jones K.J, Kim SS, North KN (1998) Abnormalities of Dystrophin, theSarcoglycan and Laminine alpha 2 in the muscular dystrophies. JMed Genet 35:379–386.
Jones K.J, Morgan G, Johnston H, Tobias V, Ouvrier R, Wilkinson I,North K. N (2001) The expanding phenotype of laminin�2 (merosin)deficiency abnormalities: case series and review. J Med Genet38:649–657.
Moriniere M, Ribeiro L, Dalla Venezia N, Deguillien M, Maillet P,Cynober T, Delhommeau F, Almeida H, Tamagnini G, Delaunay J,Baklouti F (2000) Elliptocytosis in patients with C-terminal domainmutations of protein 4.1 correlates with encoded messenger RNAlevels rather than with alterations in primary protein structure. Blood95:1834–1841.
Nagy E, Maquat LE (1998). A rule for termination-codon positionwithin intron-containing genes: when nonsense affects RNA abun-dance. Trends Biochem Sci 6:198–199.
Naom I, D’Alessandro M, Sewry C.A, Jardine P , Philpot J, ManzurA.Y, Dubowitz V, Muntoni F (1998) Laminin alpha 2-chain genemutations in two siblings presenting with limb-girdle muscular dys-trophy. Neuromuscul Disord 8:495–501.
Naom I, D’Alessandro M, Sewry C. A, Jardine P, Ferlini A, Moss T,Dubowitz V, Muntoni F (2000) Mutations in the laminin �2-chain genein two children with early-onset muscular dystrophy. Brain 123:31–41.
Pegoraro E, Fanin M, Trevisan CP, Angelini C, Hoffman EP (2000) Anovel laminin alpha2 isoform in severe laminin alpha2 deficient con-genital muscular dystrophy. Neurology 55:1128–1134.
Prandini P, Berardinelli A, Fanin M, Morello F, Zardini E, PichiecchioA, Uggetti C, Lanzi G, Angelini C, Pegoraro E (2004) LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dys-trophy. Neurology 63:1118–1121.
Ralte AM, Sharma MC, Gulati S, Das M, Sarkar C (2003) Merosinnegative congenital muscular dystrophy: a short report. Neurol India51:417–441.
Sewry CA, Chevallay M, Tome FM (1995) Expression of laminin sub-units in human fetal skeletal muscle. Histochem J 27:497–504.
Tezak Z, Prandini P, Boscono M, Monin A, Devaney J, Morino M,Fanin M, Trevizan CP, Park J, Tyson W, Finkel R, Garcia C, Angelini C, Hoffman EP, Pegarono E (2003) Clinical and molecularstudy in congenital muscular dystrophy with partial laminin alpha 2(LAMA2) deficiency. Hum Mutat 21:103–111.
Timpl R, Brown JC (1994) The laminins. Matrix Biol 14:275–281.
SIALA ET AL.206
Timpl R, Tisi D, Talts JF, Andac Z, Sasaki T, Hohenester E (2000)Structure and function of laminin LG modules. Matrix Biol4:309–317.
Vainzof M., Richard P, Herrmann R, Jimenez-Mallebrera C, Talim B,Yamamoto L.U, Ledeuil C, Mein R, Abbs S, Brockington M, RomeroN.B, Zatz M, Topaloglu H, Voit T, Sewry CA, Muntoni F, Guich-eney P, Tome F.M (2005) Prenatal diagnosis in laminin a2 chain(merosin)-deficient congenital muscular dystrophy: a collective ex-perience of five international centers. Neuromuscul Disord15:588–594.
Wizemann H, Garbe J, Friedrich M, Timpl R, Sasaki T, Hohenester E(2003) Distinct requirements for Heparin and �-Dystroglycan bind-
ing revealed by structure-based mutagenesis of the laminin (2 LG4-LG5 domain pair. J Mol Biol 332:635–642.
Address reprint requests to:Faiza Fakhfakh
Laboratoire de Génétique Moléculaire HumaineFaculté de Médecine de Sfax