UNIVERSITÁ DEGLI STUDI ROMA TRE Facoltà di Scienze Matematiche, Fisiche e Naturali Dipartimento di Biologia Scuola Dottorale in Biologia Sezione di Biologia Applicata alla Salute dell’Uomo (BASU) - XXII MECHANISMS UNDERLYING THE PROTECTIVE EFFECTS OF ESTROGEN IN DIFFERENT CELLULAR CONTEXTS MECCANISMI ALLA BASE DEGLI EFFETTI PROTETTIVI DEGLI ORMONI ESTROGENI IN DIFFERENTI CONTESTI CELLULARI Ph.D. Student: Dr. Paola Galluzzo Supervisor: Prof. Maria Marino
Transcript
UNIVERSITÁ DEGLI STUDI ROMA TRE
Facoltà di Scienze Matematiche, Fisiche e Naturali Dipartimento di Biologia
Scuola Dottorale in Biologia Sezione di Biologia Applicata alla Salute
dell’Uomo (BASU) - XXII
MECHANISMS UNDERLYING THE PROTECTIVE EFFECTS OF ESTROGEN IN DIFFERENT CELLULAR
CONTEXTS
MECCANISMI ALLA BASE DEGLI EFFETTI PROTETTIVI DEGLI ORMONI ESTROGENI IN
DIFFERENTI CONTESTI CELLULARI
Ph.D. Student: Dr. Paola Galluzzo Supervisor: Prof. Maria Marino
To my family for its understanding, endless patience and encouragement
when it was most required
To Alessandro always beside me, here and across the world
With all my heart
INDEX SUMMARY ........................................................................................... I RIASSUNTO ............................................................................... IV 1. BACKGROUND .............................................................................. 1 1.1 OVERVIEW ON ESTROGEN ..................................................................... 1 1.2 ESTROGEN RECEPTORS .......................................................................... 4 1.3 ER ACTION MECHANISMS ...................................................................... 5
1.3.1 Nuclear signals (NS) of estrogens 1.3.2 Membrane initiated signals (MIS) of estrogens
1.4 ER LOCALIZATION .............................................................................. 10 1.5 CELL FUNCTIONS REGULATED BY MIS ............................................... 11 1.6 ERΒ RAPID ACTIONS ........................................................................... 12 1.7 ER TISSUTAL DISTRIBUTION ................................................................ 14 1.8 SELECTIVE ESTROGEN MODULATORS (SERM) ..................................... 14 2. AIMS ................................................................................................ 16 3. ERα-MEDIATED SIGNAL TRANSDUCTION PATHWAYS
IN CANCER CELL PROLIFERATION ................................17 3.1 INTRODUCTION .................................................................................. 17 3.2 RESULTS ............................................................................................ 19
3.2.1 Effect of E2 and naringenin on cell proliferation 3.2.2 Mechanisms underlying the ERα activities modulation by naringenin
Nar decreases ERα palmitoylation Nar rapidly impairs ERα-caveolin-1 association Nar rapidly stimulates p38/MAPK Nar prevents the association of ERα with signaling protein involved in proliferation Nar rapidly stimulates p38/MAPK
3.3 DISCUSSION ....................................................................................... 31 4. ERβ-MEDIATED SIGNAL TRANSDUCTION PATHWAYS
IN CANCER CELL PROLIFERATION ................................34 4.1 INTRODUCTION .................................................................................. 34 4.2 RESULTS ............................................................................................ 37
4.2.1 ERβ is a palmitoylable protein
4.2.2 ERβ palmitoylation is negatively modulated by E2 4.2.3 Role of ERβ palmitoylation
ERβ palmitoylation is necessary for receptor-protein association and plasma membrane localization ERβ palmitoylation is necessary for E2-induced signaling and pro-apoptotic effects ERβ palmitoylation is necessary for DLD-1 cell growth decrease ERβ palmitoylation is important for E2-inducecd ERβ up regulation ERβ palmitoylation is not necessary for E2-induced transcriptional activity
4.3. DISCUSSION ...................................................................................... 56 5. SIGNAL TRANSDUCTION PATHWAYS ACTIVATED BY
5.2.1 ER in skeletal muscle cells 5.2.2 E2 effects on skeletal muscle cell proliferation 5.2.3 E2 effects skeletal muscle differentiation markers 5.2.4 Mechanisms underlying the E2 effects in L6 myoblasts 5.2.5 ERα and ERβ involvement in E2-induced L6 myoblast
differentiation. 5.2.6 E2 effects on C2C12 myoblasts 5.2.7 ERα and ERβ involvement in E2 effect on ROS production in L6
myoblasts. 5.3 DISCUSSION ....................................................................................... 86 6. CONCLUSION ............................................................................... 92 7. REFERENCES ............................................................................... 95 ACKNOWLEDGEMENTS .......................................................118 APPENDIX ...................... Material and Methods available on CD-ROM
I
SUMMARY Estrogens, in particular 17β-estradiol (E2) the most potent estrogen in
humans, play key roles in development and maintenance of normal sexual and reproductive functions. In addition, in both men and women, they exert a vast range of biological effects in the cardiovascular, musculoskeletal, immune, and central nervous systems (Gustafsson, 2003; Deroo and Korach, 2006, Heldring et al., 2007) and they are now generally thought to play protective effects against several degenerative diseases (e.g., cardiovascular and neurodegenerative pathologies). This expanded view of estrogen action reflects the findings of a large number of clinical and epidemiological studies which gathered on the effects of exogenous estrogen administration on the post-menopausal women health (Deroo and Korach, 2006). The biological actions of E2 are mediated by two estrogen receptor isoforms (ERα and ERβ) which belong to the nuclear receptor super-family (NR3A1 and NR3A2, respectively) (Ascenzi et al., 2006). The mechanisms of action of ERs are complex and involve signal pathways starting from plasma membrane that lead to protein kinase activation (membrane-initiated steroid signals, MISS), and direct or indirect transcription of target genes (nuclear mechanism, NS). These pathways seem to synergize each other to determine the overall effects of E2 in target tissues. However, if the nuclear-initiated signals of E2 have been studied for long time, the membrane-started signals of ER need more investigation to be fully understood. The specific role of rapid signals of each ER isoform in estrogen regulating cell functions is still unclear and several controversies related to their physiological relevance are still unsolved.
The overall aim of this thesis is to investigate the molecular mechanisms underlying some E2-induced protective effects evaluating the role played by each ERα- and ERβ-mediated mechanism. For this purpose we choose different experimental models: ER-devoid human cervix epitheloid carcinoma cells (HeLa) transiently transfected with the expression vector of ERα or ERβ; human hepatocellular carcinoma cell line (HepG2) which contains endogenous ERα; human colon cancer cells (DLD-1) that express only ERβ and rat skeletal myoblasts (L6) where both estrogen receptor isoforms (ERα and ERβ) are expressed.
At the present, most of finding point to the concept that ERα membrane starting pathways is mainly involved in E2-induced cell proliferation that may contribute to malignant tumor growth. However, we recently demonstrated that ERα shows anti-proliferative activities in presence of different ligands (e.g., naringenin, Nar) as well (Totta et al., 2004; Virgili et al., 2004) and the mechanisms underlying are here investigated. Present data show that ERα plasma membrane localization and
II
the ERα-caveolin-1 interaction are impaired through the modulation of ERα post-transductional modification (i.e., palmitoylation) by the nutritional compound naringenin. As a consequence, the ERα association with adaptors and/or signaling proteins [e.g., c-Src and the modulator of non genomic action (MNAR)] is prevented such as the ERα ability to activate ERK and AKT phosphorylation important for cell proliferation. On the other hand, a pro-apoptotic cascade [caspase-3 activation and and poly(ADP)-ribose polymersase (PARP) cleavage] involving the p38 activation is induced by the ERα:Nar complex. The ERα-mediated transcriptional activity of an estrogen responsive element (ERE)-containing promoter is not affected by Nar (Galluzzo et al., 2008) which decouples ERα rapid pathways from its direct transcriptional activity. Beside the knowledge of the mechanisms underlying the (anti)estrogenicity of dietary compounds such as flavonoids these data bring to light the new important role played by ERα signals. Due to the allosteric properties of this molecule, different ligands induce ER to assume different conformation responsible for specific protein-protein interaction which, in turn, drive cell to different fate.
At present, inadequate information is available on the role played by ERβ, even if its role as dominant negative of ERα proliferative activities has been reported (Paruthiyil et al., 2004; Strom et al., 2004). Besides this role, these results show that ERβ is also able to mediate specific signal transduction pathways underlying the protective effects of estrogen against cancer cell proliferation. Present results demonstrate that in DLD-1 colon cancer cells in which only the ERβ1 isoform is expressed, like ERα, ERβ undergoes the palmitoylation which allows to a small ERβ pool to localize at the plasma membrane and associate with caveolin-1 and the p38. Upon E2 stimulation, ERβ undergoes de-palmitoylation increasing receptor association to caveolin-1 and p38. The ERβ-caveolin-1 and ERβ-p38 physical association are responsible for the ERβ level increase at the plasma membrane impairing the association of signaling proteins important for ERα-mediated cell survival and proliferation (i.e., Src, ERK and AKT) (Galluzzo et al, 2007). On the other hand, the ERβ-mediated E2-induced p38 activation deeply impact on DLD-1 colon cancer cells. P38 activation is fundamental for the downstream pro-apoptotic cascade which involves the caspase-3 activation and PARP cleavage, but also for both for the rapid increase of ERβ mRNA translation and for the slow ERβ (ESR2) gene transcription (Caiazza et al, 2007). The final consequence is an increased level of ERβ in DLD-1 cells which, in the presence of E2, further increase the hormone protective effect against tumor growth.
Overall these data allow to conclude that E2 in presence of ERα or ERβ activates very different rapid signals important for the final cellular
III
fate. In particular, ERα:E2 is able to activate signal cascades addressing to cell proliferation, even if ERα is also able to exert protective effects against cancer cells proliferation in dependence on the bound ligand (Galluzzo et al., 2008). On the other hand, beside its role as negative modulator of ERα activities previously reported (Matthews and Gustafsson, 2003; Paruthiyil et al, 2004; Strom et al, 2004), ERβ is able to activate specific rapid signals started from plasma membrane which trigger a pro-apoptotic cascade. The presence of both ER isoforms, for instance in skeletal muscle cells, lead us to investigate the presence and the impact of ERα- and ERβ-dependent pathways on the final effects of estrogen in this cellular context.
Present results demonstrate that in actively proliferative rat myoblasts (L6), E2 does not increase cell proliferation or apoptosis, whereas E2 stimulates the cell differentiation increasing the expression of myogenesis markers [i.e., myogenin, myosin heavy chain (MHC), and the glucose transporter type 4 (Glut-4) translocation]. In particular, the rapid extra-nuclear signals (i.e. AKT activation) and the presence of ERα on plasma membrane are necessary and sufficient for the first step of differentiation process (Glut-4 translocation). Although ERα is the isoform less expressed in L6 cells, its contribute has been demonstrated to be significant. In addition, the use of ER specific agonists and antagonist (i.e., PPT and THC) demonstrate that in absence of ERβ activities the E2 effect is more evident, suggesting that ERβ opposes the ERα functions. On the other hand, ERβ is the receptor isoform mainly expressed and even if its contribute seems to be negligible to E2-induced differentiation, ERβ exerts important functions to protect the skeletal muscle cell from H2O2-induced oxidative stress that could occur during muscle activity (Galluzzo et al., 2009a). These data lead us to conclude that the E2 effects in cells co-expressing ERα and ERβ could not only depend on the protein expression level of ERs, but also on the balance between the signals originated by each isoform.
All together reported data argue that both ERα and ERβ activities mediate the estrogen protective effect depending on cellular context and that a dominant protective ER player in the intricate interplay among ERα and ERβ dependent signaling does not exist. Furthermore, the complexity of the mechanism of ER action suggests a more finely tuned control exerted by E2-induced rapid signals on cellular molecular events. In particular, the extra-nuclear signals induced by E2 occur before the appearance of nuclear effects and the cell context in which the genomic events occur will be different depending on which signal pathway is activated. Thus, the integration between these molecular events is required to obtain the complete cellular response.
IV
RIASSUNTO Gli estrogeni, ed in particolare il 17β-estradiolo (E2) il più potente
estrogeno presente nell’uomo, svolgono un ruolo fondamentale nello sviluppo e nel mantenimento delle normali funzioni sessuali e riproduttive, ma la loro azione si esplica, sia nell’uomo che nella donna, anche in sistemi non riproduttivi come, ad esempio, l’apparato cardiovascolare, l’apparato muscolo scheletrico, il sistema immunitario (Gustafsson, 2003; Deroo and Korach, 2006, Heldring et al., 2007). Inoltre più di recente è stato attribuito agli estrogeni un importante ruolo protettivo contro alcune patologie degenerative (e.g., cardiovascolari e neurodegenerative). Questa più ampia conoscenza sulle funzioni degli estrogeni deriva da studi epidemiologi basati sugli effetti sulla salute di donne in post-menopausa sottoposte a terapie estrogeniche (Deroo e Korach, 2006).
Le azioni biologiche esercitate da E2 sono mediate da due isoforme del recettore degli estrogeni (ERα ed ERβ) appartenenti alla superfamiglia dei recettori nucleari (NR3A1 e NR3A2, rispettivamente) (Ascenzi et al., 2006). Il meccanismo d’azione di ERα e ERβ è complesso e coinvolge vie di trasduzione del segnale che partono dalla membrana plasmatica (membrane-initiated steroid signals, MISS) e portano all’attivazione di protein-chinasi, e la trascrizione diretta ed indiretta di geni target (nuclear mechanism, NS). Questi meccanismi sembrano sinergizzare l’un l’altro per determinare l’effetto complessivo di E2 nei tessuti bersaglio, tuttavia, se i meccanismi genomici sono ormai ben noti, i segnali che partono dalla membrana plasmatica richiedono ulteriori studi per essere chiariti. Il ruolo specifico di ERα ed ERβ nel regolare le funzioni cellulari dipendenti da E2 non è completamente noto e le numerose controversie sull’importanza fisiologica ricoperta dai segnali rapid di E2 non sono ancora risolte.
Scopo di questa tesi è proprio quello di investigare i meccanismi molecolari alla base di alcuni effetti protettivi esercitati dagli estrogeni valutando il coinvolgimento dei meccanismi specifici mediati da ciascuna isoforma di ER. Per questo studio differenti linee cellulari sono state scelte come modello sperimentale: cellule umane di carcinoma alla cervice uterina (HeLa) rese responsive ad E2 in seguito a transfezione transiente con i vettori di espressione per ERα o ERβ; cellule di epatocarcinoma umano che contengono endogenamente solo il recettore ERα (HepG2); cellule umane di adenocarcinoma al colon che esprimono solo l’isoforma β di ER e mioblasti scheletrici di ratto (L6) dove sono co-espressi ERα de ERβ.
Al presente, la maggior parte dei dati a nostra disposizione riporta che i segnali rapidi mediati da ERα sono prevalentemente coinvolti negli effetti proliferativi di E2 che possono contribuire a trasformazione e crescita tumorale. Tuttavia, dati ottenuti di recente nel nostro laboratorio, hanno
V
dimostrato che ERα svolge anche attività antiproliferative in presenza di differenti ligandi (e.g., naringenina, Nar) (Totta et al., 2004; Virgili et al., 2004) ed i meccanismi alla base sono oggetto di studio di questa tesi.
I dati qui presentati dimostrano che la localizzazione di ERα e la sua associazione con la caveolina-1 sono modificate dalla molecola di origine nutrizionale Nar attraverso la modulazione della modificazione post-traduzionale di ERα (i.e., palmitoilazione). Di conseguenza, l’associazione di ERα con proteine adattatrici e di seganle [e.g. c-Src e la proteina modulatrice delle attività non genomiche di ER (MNAR)], è prevenuta così come la fosforilazione e attivazione di ERK e AKT importanti per la proliferazione cellulare. D’altra parte il complesso ERα:Nar induce una cascata pro-apoptotica che coinvolge l’attivazione della p38/MAPK, l’attivazione della caspase-3 ed il taglio proteolitico poli(ADP)-ribosio polimerasi (PARP). L’attività trascrizionale mediatada ERα di gene contententi nel promotore l’elemento di risposta agli estrogeni (ERE) non è influenzata dall naringenina (Galluzzo et al., 2008) che disaccoppia, così, le attività rapide di ERα dalla sua capacità trascrizionale diretta. Oltre alla comprensione del meccanismo alla base dell anti(estrogenicità) di composti nutrizionali come i flavonoid, questi dati portano alla luce un nuovo importante ruolo esercitato dai segnali rapidi di ERα. Date le proprietà allosteriche di questa proteina, diversi ligandi possono assumere differenti conformazioni responsabili delle specifiche interazioni protein-proteina che, a loro volta, determinano diversi destini cellulari.
Al presente, le informazioni relative al ruolo rivestito da ERβ non sono sufficienti, anche se gli è stato attribuito un ruolo come regolatore negativo delle attività proliferative di ERα (Couse and Korach, 1999; Weihua et al., 2003; Paruthiyil et al., 2004; Strom et al., 2004). I risultati di questa tesi dimostrano che accanto a questo ruolo, ERβ attiva rapidamente vie specifiche di segnale coinvolte negli effetti protettivi di E2 contro la proliferazione cellulare. In particolare, in cellule di cancro al colon (DLD-1), così come ERα, anche ERβ viene modificato post-traduzionalmente mediante palmitoilazione che consente ad una piccola frazione di ERβ di localizzarsi a livello della membrana plasmatica, associarsi alla caveolina-1 e alla p38/MAPK. La stimolazione con E2 induce la de-palmitoilazione di ERβ e l’aumento della sua associazione con la caveolina-1 e la p38. Queste interazioni protein-proteina sono responsabili dell’aumento di ERβ al livello della membrana plasmatica e prevengono l’associazione (e.g., Src e MNAR) e l’attivazione di proteine di segnale (e.g., ERK e AKT) importanti per la proliferazione mediata da ERα (Galluzzo et al, 2007). D’altra parte, l’attivazione della p38 dipendente da E2 influenza fortemente il destino cellulare delle DLD-1 inducendo una cascata pro-apoptotica (attivazione
VI
della caspase-3 e taglio proteolitico della PARP) ed essendo responsabile contemporaneamente del rapido incremento della traduzione dell’mRNA di ERβ e della lenta trascrizione del gene ESR2 di ERβ (Caiazza et al, 2007). Il complessivo incremento dei livelli proteici di ERβ amplifica ulteriormente l’effetto protettivo dell’ormone contro la crescita tumorale.
L’insieme di questi dati ci permette di concludere che E2 attiva vie di segnale diverse in presenza di ERα o ERβ che portano a proliferazione o ad apoptosi, rispettivamente. Anche se ERα è anche in grado di indurre una cascata apoptica in presenza di differenti ligandi (e.g., Nar).
La contemporanea espressione di entrambe le isoforme di ER per esempio in cellule muscolari scheletriche ci ha spinto a studiare l’impatto delle vie rapide mediate da ERα ed ERβ sull’effetto finale di E2 in questo contesto cellulare. I dati ottenuti dimostrano che in cellule muscolari in attiva proliferazione (L6) E2 non induce un incremento della proliferazione né apoptosi, mentre stimola il differenziamento agendo sui marker del differenziamento [miogenina, catena pesante della miosina (MHC), e trasportatore del glucosio di tipo 4 (Glut-4)]. In particolare le attvitità rapide di ERα (i.e., attivazione AKT) sono necessarie e sufficienti per le prime fasi del differenziamento (traslocazione sulla membrana del Glut-4). Anche se meno rappresentato, l’isoforma α di ER contribuisce in modo significativo. Inoltre, l’eliminazione delle attività di ERβ, mediante l’uso di agonisti e antagonisti selettivi di ER (i.e., PPT and THC) rende l’effetto di E2 più evidente, suggerendo ancora il suo ruolo di regolatore negativo di ERα. Tuttavia, anche se ERβ, l’isoforma maggiormante espressa, non contribuisce all’incremento dei marker del differenziamento, esercita un ruolo fondamentale nella protezione delle cellule da stress ossidativo indotto da H2O2 (Galluzzo et al., 2009a) che protebbe verificarsi durante l’attività muscolare. Questi dati ci permettono di concludere che l’effetto finale di E2 in cellule che co-esprimono ERα e ERβ dipendono non solo dai livelli proteici ma dal bilancio dei segnali rapidi attivati da ogni isoforma.
L’insieme di questi dati ci permette di sostenere che entrambi ERα e ERβ mediano gli effetti protettivi di E2 in dipendenza dal contesto cellulare e che non esiste un attore pricipale nelle intricate azioni tra ERα e ERβ. Inoltre, i segnali extra-nucleari indotti da E2 avvengono prima della comparsa degli effetti nucleari ed il contesto cellulare nel quale gli eventi genomici avvengono saranno diversi in dipendenza dalle vie rapide attivate. Di conseguenza, per ottenere una completa risposta cellulare è richiesta l’integrazione di tutti gli eventi molecolari.
1
1. BACKGROUND 1.1 OVERVIEW ON ESTROGENS
Estrogens, like other steroid hormones, are naturally occurring cyclopentanophenanthrene compounds whose synthesis begins with cholesterol. Estrogens play key roles in development and maintenance of normal sexual and reproductive function in both men and women (Heldring et al., 2007). Although, their levels are significantly higher in women of reproductive age. They are mainly produced by the ovary and in part by the adrenal cortex, and three estrogens occur naturally in the female (Ruggiero et al., 2002; Ascenzi et al., 2006). In premenopausal women, 17β-estradiol (E2), produced by the ovary granulosa, is the estrogen produced in the largest quantity and is the most potent as it has the highest affinity for its receptors. In pre-menopausal women, circulating estradiol levels fluctuate from 40 to 200-400 pg/mL during the menstrual cycle (Ruggiero et al., 2002). After menopause, estradiol levels drop to less than 20 pg/mL. The second estrogen is estrone (E1), a less potent metabolite of estradiol. Estrone is produced from androstenedione, the immediate precursor of estrone, in the liver and adipose tissue. In post-menopausal women, the ovary ceases to produce estradiol while the adrenal gland continues to produce androstenedione. As the result, the level of estrone remains unchanged while the plasma level of estradiol falls significantly. The third endogenous estrogen is estriol (E3), also a metabolite of estradiol. Estriol is the principal estrogen produced by the placenta during pregnancy, and is found in smaller quantities than estradiol and estrone in non-pregnant women (Ruggiero et al., 2002; Ascenzi et al., 2006; Chen et al., 2008).
In the target tissues such as reproductive organs, estrogens exhibit important biological functions. Among these, the ovarian cycle, the development of both sexual primary and secondary characters are dependent on estrogen (Ikeda and Inoue, 2004). Besides regulation of physiological functions in reproductive tissues, estrogens exert a vast range of biological effects and influence many other physiological processes in mammals including cardiovascular health, bone integrity, cognition, and behaviour (Gustafsson, 2003; Heldring et al., 2007).
Estrogens play important roles in bone homeostasis, being involved in modeling of bone during adolescence. They initiate pubertal growth and later limit longitudinal bone growth by inducing closure of the epiphysial growth plate. Even in adult life, sex steroids appear to influence the remodeling of bone. E2, in particular, is crucial for the maintenance of bone mass in females (Migliaccio et al., 1996) as is evident from the rapid loss of trabecular bone and development of osteoporosis that occurs after
2
ovariectomy or at menopause (Turner, 1990, 1999). Several factors, known to be important in regulating differentiation and function of osteoblasts and osteoclasts, are regulated by E2. In osteoblasts, E2 stimulates synthesis and secretion of the anabolic growth factor IGF-I and inhibits that of the cytokines, IL-1, tumor necrosis factor (TNF), and IL-6 (Roodman, 1996) that are involved in bone resorption (Nilsson et al., 2001).
Estrogen appears to be protective in the cardiovascular system, in part due to favourable modulation of serum lipid profiles (Ettinger et al., 1996; Farish et al., 1996). The 30% of these effects are due to a reduction of total cholesterol by increasing the LDL receptor (LDLr) expression in the liver (Di Croce et al., 1996; Marino et al., 2001a) and inhibiting the LDLr oxidation at vascular level (Zhu et al., 2002). Recent evidence indicates that the 70% of estrogen effect is also due to its direct action on the vasculature, as demonstrated by gender differences in smooth muscle contractility, with a greater response to noradrenaline or phenylephrine in aortas isolated from male than female rats (Stallone et al., 1991; Kanashiro et al., 2001). E2 rapidly induces the activity of endothelial nitric oxide synthase (eNOS), which produces the vasodilator nitric oxide (NO) (Kim and Bender, 2005). Furthermore, estrogens are able to prevent the proliferation and the migration of vascular smooth muscular cells (VSMC) both directly (Razandi et al., 2000) and indirectly blocking the mitogenic action of growth factors (Mendelsohn and Karas, 1999; Incerpi et al., 2003). Prior to menopause, women have a lower incidence of coronary heart disease compared with age-matched men (Van der Schouw et al., 1996; Barrett-Connor, 1997; Phillips et al., 1997). Moreover, estrogen replacement therapy reduces mortality due to coronary heart disease in post-menopausal women (Stampfer et al., 1991; Ettinger et al., 1996). Deprivation of estrogens by ovariectomy results in enhanced vascular contraction in aortas of female rats (Kanashiro et al., 2001). In ovariectomized rats, chronic estrogen replacement suppresses endothelium-dependent contractions mediated by the cyclo-oxygenase/prostaglandin H synthase pathway (Davidge et al., 1998). Studies in humans also demonstrate a beneficial vascular action of estrogen, with endothelium-dependent vasodilatation being enhanced by the administration of 17β-estradiol at physiological levels (Herrington et al., 1994; Gilligan et al., 1994a,b; New et al., 1997; Leung et al. 2007).
In central nervous system, estrogen has been found to contribute to promote synapse formation and plasticity and to limit neuronal damage and death, besides its capability to protect from neurodegenerative process. Furthermore, the 17β-estradiol plays an important role as antioxidant reducing the oxygen reactive species (ROS) by increasing the protein level
3
of tioredoxin, an important protein that protect from lipid peroxidation after oxidative stress (McCullough and Hurn, 2003; Lee et al., 2003; Hammes and Levin, 2007).
Postmenopausal women are in an estrogen-deprived state and are at risk for stroke and other neurodegenerative diseases (Garcia-Segura et al., 2001). Epidemiological evidence suggests that postmenopausal estrogen therapy (ET) reduces the risk or delays the onset of Alzheimer disease (Henderson et al., 1994; Paganini-Hill et al., 1994). Estrogen loss from natural or surgical menopause has been associated with a decline in cognitive function (Sherwin, 1997, 1998) that is reversed by ET. ET has been shown to affect cognitive function during brain aging as well (Resnick et al., 1998; Maki et al., 2001; Resnick and Maki, 2001; Simpkins and Singh, 2008).
Given this widespread role for estrogen in human physiology, it is not surprising that estrogen is also implicated in the development or progression of numerous diseases, which include but are not limited to osteoporosis, neurodegenerative diseases, cardiovascular disease, insulin resistance, lupus erythematosus, endometriosis, obesity and various types of cancer (breast, ovarian, colorectal, prostate, and endometrial). Indeed, in addition to proliferative effects on normal cells, estrogen is considered as a stimulant for the initiation and promotion of tumors in these organs (Ikeda and Inoue, 2004). This expanded view of estrogen action reflects the findings of a large number of clinical and epidemiological studies which gathered on the effects of exogenous estrogen administration on the post-menopausal women health. Based on data from both clinical and animal studies, estrogen is implicated in the development of breast cancer stimulating proliferation of mammary cells by increasing cell division and DNA synthesis (Deroo and Korach, 2006). Risk factors associated with breast cancer reflect cumulative exposure of the breast epithelium to E2 (Henderson and Feigelson, 2000; McEwan, 2004; Ascenzi et al., 2006). There is also evidence that estrogens, together with gonadotropins, contribute to the etiology of ovarian cancer in humans (Chu et al., 2000; Deroo and Korach, 2006). Whereas, clinical studies indicate that the incidence of colon cancer is lower in women than in men (Jemal et al., 2004), and data from the Women’s Health Initiative indicate a significantly reduced incidence of colon cancer in postmenopausal women receiving combined ‘Hormone Replacement Therapy’(HRT; estrogen plus progestin) (Rossouw et al., 2002; Ascenzi et al., 2006).
4
1.2 ESTROGEN RECEPTORS The biological actions of estrogens are traditionally mediated by
binding to one of two specific estrogen receptors (ERs), ERα or ERβ, that are encoded by different genes located on different chromosomes (locus 6q25.1 and locus 14q23-24.1, respectively) (Gosden et al., 1986; Enmark et al., 1997; Luisi et al., 2006; Zhou et al., 2006). ERα and ERβ, like all the members of the nuclear receptor super-family, are modular proteins sharing common regions, named A/B, C, D, and E/F, as well as a high sequence homology (Fig. 1.1). These regions participate in the formation of independent but interacting functional domains. The N-terminal domain (A/B region) is involved in both inter-molecular and intra-molecular interactions as well as in the activation of gene transcription. The DNA binding domain (DBD, C region) allows ER to dimerize and to bind to the specific ERE sequence on DNA through its two “zinc finger” structures. The hinge domain (D region) has a role in receptor dimerization and in binding to chaperone heat-shock proteins (Hsp). The ligand binding domain (LBD, E/F region, C-terminal) comprises the E2-binding domain and works, synergistically with the N-terminal domain in the regulation of gene transcription (Mosselman et al., 1996; Nilsson et al., 2001; Claessens and Gewirth, 2004; Kumar et al., 2004).
Figure 1.1: A schematic structural comparison of human ERα and ERβ functional domains. Receptor domains are illustrated with different colored boxes, and the approximate size of each domain is indicated. The A/B domain contains the ligand-independent transcriptional-activation function AF-1, the C domain represents the DNA-binding-domain (DBD), the D domain corresponds to the hinge region, and the E domain contains the hormone-binding domain (LBD) and the hormone-dependent transcriptional-activation function AF-2. The number inside each box of ERβ refers to the percentage of amino acid identity.
5
ERs contain two regions called activation functions (AFs) important for ligand-dependent transcriptional activity (Fig. 1.1) (Mosselman et al., 1996; Nilsson et al., 2001; Claessens and Gewirth, 2004; Kumar et al., 2004). AF-1 and AF-2 regions of ERs, interacting with a number of trancription co-activators, can activate transcription independently but in most cases, they synergize with one another in a promoter- and cell-context specific manner (McEwan, 2004). AF-1 could be activated even in a ligand-independent manner, depending on the phosphorylation status of ER. In particular, the Ser118 residue in the AF-1 region of ERα, as well as residues Ser106 and Ser124 in the AF-1 region of ERβ, are the phosphorylation sites essential for the ligand-independent activation of ERs through the Ras-mitogen activated protein kinase (MAPK) signaling cascade (see Ortì et al., 1992; Lannigan, 2003).
Recent progress in studies on genomic and cDNA sequences has accelerated the identification of gene splice variants in the NR super-family. Numerous mRNA splice variants exist for both ERs and the best-characterized splice variants are ERα46 and ERβcx, which are frequently co-expressed with their wild-type counterparts. The exact function and potential role of these and other ERs splice variants in physiology and human disease remain to be elucidated (see Herynk and Fuqua, 2004; Marino et al., 2006a).
1.3 ER ACTION MECHANISMS
The mechanisms of ERα and ERβ action are complex pathways that involve two distinct types of signaling which lead to protein kinase activation (rapid membrane-initaited mechanism) and direct or indirect transcription of target genes (nuclear mechanism) (Fig. 1.2). All these pathways synergize each other to determine the overall effects of E2.
1.3.1 Nuclear signals (NS) of estrogens
In the “classical” mechanism of action, estrogens diffuse into the cell membrane and bind to ERs causing ERs to dissociate from heat shock proteins, to dimerize and to traslocate into the nucleus. The nuclear ERα- or ERβ-E2 complex directly binds DNA through the ERE (estrogen responsive element) sequences or indirectly through protein-protein interactions with activator protein-1 (AP-1) or stimulating protein (Sp-1), resulting in recruitment of coregulatory proteins (coactivators or corepressors) to the promoter, increased or decreased mRNA levels, protein synthesis, and physiological responses (Ascenzi et al, 2006; Deroo and Korach, 2006). A large subset of coregulatory proteins (e.g., steroid receptor coactivator-1, 2, and 3) helps the hormone-receptor complex to recruit histone
6
acetyltransferases and methyltransferases which, in turn, possess chromatin-remodeling ability and tether activated receptors to the basal transcriptional machinery (Smith and O’Malley, 2004). Both ERα and ERβ are capable of regulating gene transcription through this classical mechanism involving ERE. ERβ seems to be a weaker transactivator (Cowley and Parker, 1999). AF-1 activity of ERβ is weak compared with that of ERα on ERE, whereas their AF-2 activities are similar (Cowley and Parker, 1999). In turn, when both AF-1 and AF-2 functions are active in a particular cell and/or on a particular promoter, the activity of ERα greatly exceeds that of ERβ, whereas ERα and ERβ activities are similar when only AF-2 is required (McInerney et al, 1998; Cowley and Parker, 1999; Ascenzi et al, 2006). It has been postulated that differences in the ERα and ERβ activities are due to differences in the ability of the receptors to interact with coregulatory proteins, because of the low amino acid identity in A/B domain of ERs (Figure 1.1) (Smith and O’Malley, 2004; Ascenzi et al, 2006).
Only a fraction of the known mammalian EREs reflects the consensus palindromic element ERE (GGTCAnnnTGACC), initially described based on the ERE in the Xenopus laevis vitellogenin A2 promoter (Klein-Hitpass et al., 1986; Ponglikitmongkol et al., 1990). For instance, for the 38 estrogen-responsive genes reviewed by Klinge (Klinge, 2001), most of the functional EREs located within the promoters or 30-untranslated regions are not the traditional consensus sequence. Thus, many target genes contain response elements that bear little similarity to consensus EREs and affects the affinity that a given receptor isoform has for binding DNA (Loven et al., 2001).
Another category of gene promoters, lacking any ERE-like sequences, requires a second DNA-binding transcription factor (e.g., Sp-1 and AP-1) to mediate ER association with the DNA (O’Lone et al, 2004). Although ERα and ERβ have similar effects on ERE-mediated gene transcription, the receptors show opposite effects on promoters containing AP-1 (Paech et al, 1997). E2 activates AP-1-mediated gene transcription when bound to ERα but inhibits promoter activity when bound to ERβ (Paech et al, 1997). The converse is true for anti-estrogens, such as tamoxifen, raloxifene, and ICI 164384, which are AP-1 transcriptional suppressors via ERα and activators via ERβ (Paech et al, 1997; Weyant, et al, 2001; Loven et al., 2001). Similar to AP-1, E2 binding to ERα induces transcriptional activation when associated with Sp-1 in GC-rich regions. However, E2 binding to ERβ does not result in the formation of a transcriptionally active complex at a promoter containing Sp-1 elements (Saville et al, 2000). As an example ERα and ERβ, in the presence of E2, oppose each other’s function in the regulation of the cyclin D1 promoter (Liu et al, 2002). There is considerable
7
evidence that cyclin D1, important for the progression of cells through the G1 phase of the cell cycle, is a well defined target for ERα-E2 action in mammary carcinoma cells (Altucci et al, 1996; Foster and Wimalasena, 1996; Prall et al, 1997), although no detectable “perfect” or ERE-like sequence in the cyclin D1 gene promoter has been reported (Herber et al, 1994). Deletion of AP-1 and Sp-1 responsive element motifs in the cyclin D1 gene promoter resulted in attenuation of promoter responsiveness to E2 (Marino et al, 2002, 2003). Unlike ERα, E2-bound ERβ represses cyclin D1 expression (Acconcia et al, 2005a) and blocks ERα-E2-mediated induction when both receptor isoforms are present (Matthew and Gustafsson, 2003). Consequently, these differences in transcriptional activity between the ERα and ERβ may account for the major differences in their tissue specific biological actions.
Figure 1.2: Schematic model illustrating the action mechanisms of E2. In the first panel (a) is depicted the classical interaction of the activated receptor with ERE on DNA. In panels b and c are representations of the indirect effects of ERs on transcription interactions. This occurs through protein-protein interactions with the Sp1 (b), AP-1 (c). The panel d represents the E2-non-genomic mechanism. AP-1, activating factor-1; Sp-1, stimulating factor-1.
8
1.3.2 Membrane initiated signals (MIS) of estrogens The nuclear initiated signals of steroid hormones occurs after a time-
lag of at least 2 hours after E2 stimulation and explains some of hormone functions in physiological and pathological situations (Farach-Carson and Davis, 2003; Marino et al., 2005). This picture was challenged when a physiological dose of E2 was reported to increase the uterine cAMP level in ovariectomized rats within 15 seconds (Szego and Davis, 1998), an effect too rapid to be accounted for genomic action(s). This event was not abrogated by transcriptional inhibitors and was termed “rapid or non-genomic”. Actually the term “non-genomic” is not adequate when referring to rapid changes that may also initiate new gene transcription (Farach-Carson and Davis, 2003; Kampa and Castanas, 2006). Various signaling pathways are activated upon E2 binding to ERs. These rapid events may be classified into four main signaling cascade: phospholipase C (PLC)/protein kinase C (PKCs) (Morley et al., 1992; Marino et al., 1998, 2001a, 2001b; Picotto et al., 1999; Perret et al., 2001; Incerpi et al., 2003), Ras/Raf/MAPK (Marino et al., 2002; Watter et al., 1997; Russel et al., 2000; Dos Santos et al., 2002; Migliaccio et al., 2002; Tanaka et al., 2003; Klinge et al., 2005; Woo et al., 2005), phosphatidyl inositol 3 kinase (PI3K)/AKT (Castoria et al., 1999, 2001; Simoncini et al., 2000; Marino et al., 2003; Björnström and Sjöberg , 2005; Levin, 2005; Acconcia et al., 2005a; Marino et al., 2005; Chambliss et al., 2005), and cAMP/protein kinase A (PKA) (Gu and Moss, 1996; Farhat et al., 1996; Picotto et al., 1996; Chen et al., 1998; Malyala et al., 2005). These pathways present numerous interactions with several other pathways. The ERα:E2 complex interacts with the IGF-1 receptor, leading to IGF-1 receptor activation and hence to MAPK signaling pathway activation (Kahlert et al., 2000). In addition, the ERαE2 complex activates the EGF receptor by a mechanism that involves activation of guanine nucleotide exchange proteins (G-proteins), Src, and matrix metalloproteinases, leading to an increase in extracellular regulated kinases (ERK) and PI3K/AKT activities (Dos Santos et al., 2002; Driggers et al., 2002; Improta-Brears et al., 1999; Razandi et al., 2003; Zhang et al., 2004; Kupzig et al., 2005). In endothelial cells the Src/PI3K/AKT pathway mediates rapid E2-dependent activation of eNOS and the release of nitric oxide. AKT and PKC could also modulate the MAPK pathway through Raf phosphorylation (Chambliss et al., 2000, 2005; Marino et al., 2005; Kim and Bender, 2005). It is important to note that activation of signaling pathways by E2 is cell type-specific. Indeed, the effect of E2 on PKC activity has been observed in the preoptic area of female rat brain slices, but not in the hypothalamus or cortex (Ansonoff and Etgen, 1998). The activation of G-protein/Src/PI3K/MAPK pathway by E2 was evident in late,
9
but not early, differentiated rat pre-adipocytes (Dos Santos et al., 2002). The differential requirement of Src/PI3K or intracellular calcium for MAPK activation is also observed in diverse cell types (Dos Santos et al., 2002; Björnström and Sjöberg, 2005; Kupzig et al., 2005). Different PKC isoforms are rapidly activated by E2 in HepG2 and MCF7 cells (Marino et al., 2001b). As a whole, these studies indicate that the rapid actions of E2 depend on a number of conditions such as the set of signal transduction molecules and downstream targets present in the target cell, thus the responses are likely to be diverse. All these results point to the concept that ERαis the primary endogenous mediator of rapid E2 actions. Figure 1.3: Schematic model illustrating the relationship between the mechanisms of action of E2. Palmitoylation (PA) allows the estrogen receptor (ER) localization at the plasma membrane. E2 binding induces ER association to signaling proteins, and triggers the activation of signaling cascades. The kinase activations phosphorylate ER, modulate transcriptional coactivators recruitment, and enhance AP-1 and Sp-1 activation. After dimerization ERs directly interact with ERE on DNA. ERs-DNA indirect association occurs through protein-protein interactions with the Sp-1 and AP-1 transcription factors.
10
1.4 ER LOCALIZATION The rapidity by which E2 induces rapid signals as well as the
localization of signaling complex outside the nucleus raises the requirement of a plasma membrane ER. Debate continues over whether structural changes target nuclear ERs in separate pools localizing them to the membrane (Chambliss, et al., 2000; Acconcia and Kumar, 2005; Marino et al., 2005; Kampa and Castanas, 2006), or whether membrane ER represents a novel receptor (Ahola et al., 2002; Filardo et al., 2002; Ropero et al., 2002; Toran-Allerand et al., 2002; Thomas et al., 2005; Vivacqua et al., 2006). Besides these data, much evidence favors the idea that the membrane-localized ER is the same protein as the nuclear-localized receptor (Pappas et al., 1995; Norfleet et al., 1999; Razandi et al., 1999; Marino et al., 2002, 2003) and that ERα and ERβ must be considered a population of protein(s) which localization in the cell is able to dynamically change, shuttling from membrane to cytosol and to the nucleus, depending on ligand binding (Razandi et al., 1999; Dan et al., 2003; Marino et al., 2005; Leclercq et al., 2006). Current evidence indicates that a small population of ERα and ERβ localize at the plasma membrane exists within caveolar rafts. It is at the plasma membrane that E2-liganded ER associates with the scaffolding protein caveolin-1 and a variety of signal transduction cascade activation occurs [e.g., PLC, PKC, ERK, PI3K, and nitric oxide synthase (NOS)]. ERs do not contain a trans-membrane domain (Björnström and Sjöberg, 2005; Ascenzi et al., 2006), thus the ability of ERα and ERβ to associate with the plasma membrane could be due to its association with membrane proteins and/or by post-translational addition of lipids to ERα (Acconcia et al., 2005b; Levin, 2005).
Fatty acids and isoprenoids are two of the most common lipid moieties found on post-translational modified proteins bound to membranes. No consensus sequences for N-acylation (i.e., miristoylation) or S-prenylation have been found in ERα and ERβ (Acconcia et al., 2003). On the contrary, S-acylation (i.e., palmitoylation) does not require any consensus sequence, but just reactive Cys residues (Bijlmakers and Marsh, 2003). Cys residues present in the ERαand ERβLBD could undergo S-acylation. In particular, the amino acid sequence encompassing the Cys447 residue of ERαand Cys399 of ERβis highly homologous to that surrounding the S-palmitoylated Cys132 residue of human caveolin-1 (Acconcia et al., 2003). Based on this observation we demonstrated that ERαundergoes S-palmitoylation which represents the major determinant for its residence at the plasma membrane, for its association with caveolin-1 (Acconcia et al., 2003; 2005b; Marino et al., 2006b) and ERα-mediated activation of signal transduction pathways important for cell proliferation
11
(Acconcia et al., 2004; Acconcia et al., 2005b). The Cys 399 of ERβ is also palmitoylated as demonstrated in this thesis.
A physiological E2 concentration reduces by 50% the amount of palmitate incorporated in ERα within 60 min without any change in the protein level, while E2 induces a 90% reduction in the ability of ERα to form a complex with caveolin-1 (Acconcia et al., 2005b). E2-induced reversible S-palmitoylation of ERα could account for the coexistence of both membrane-bound and soluble isoforms of ERα (Marino and Ascenzi, 2006). S-palmitoylation is necessary for E2-induced rapid events. In fact, neither the ERα Cys447Ala mutant nor palmitoyl-acyl-transferase inhibition supports E2-induced ERK and PI3K/AKT pathway activation in human cancer cells (Acconcia et al., 2005b). These results have been recently confirmed by others authors (Pietras et al., 2005; Pedram et al., 2007).
Because ERα has no intrinsic kinase domains the localization of ERs at the plasma membrane facilitate the association between ER and signaling proteins allowing the activation of rapid events. Src, Shc, proline-, glutamic acid-, leucine- rich protein /modulator of non-genomic activity of estrogen receptor (PELP1/MNAR), the p85α subunit of PI3K, receptor tyrosine kinases (i.e., EGF and IGF-1 receptors), as well as G-protein isoforms (i.e., Gαs and Gαq) have all been reported to serve as components of large complexes of interacting proteins. Through the mediation of these molecules, E2 activates the MAPK and PI3K/AKT pathways (Acconcia and Kumar, 2005; Kennedy et al., 2005; Levin, 2005; Song et al., 2005; Greger et al., 2006). Protein-protein complex formation occurs only 5 to 15 min after E2 stimulation (Greger et al., 2006), thus, the conformational changes of the ER LBD domain, which follows E2 entry into the cell, seems to be important in allowing the ER:E2 complex to detach from the membrane and allocate with growth factor receptors or adapter proteins to activate downstream signals
1.5 CELL FUNCTIONS REGULATED BY MIS
The rapid activities of ERs are widely accepted and disagreement on the involvement of nuclear receptors is quite settled. However, other controversies in this field are still present and related to whether or not all of these rapid effects are of physiological relevance (Warner and Gustafsson, 2006). The main difficulties are linked to the experimental models used. In fact, the study of signaling pathways can be done mainly on isolated, often immortalized, cells and it is very complicated to obtain similar information on a whole organism in which the use of signaling inhibitors could have many side effects other than to inhibit just one kinase. Nevertheless, the physiological significance of rapid membrane-starting pathways has been
12
clarified at least for some E2 targets. In the nervous system, E2 affects neural functions (e.g., cognition, behavior, stress responses, and reproduction) in part by inducing such rapid responses (Farach-Carson and Davis, 2003). In the skeleton, ERα, present in caveolae of bone-forming osteoblasts, transmits survival signals through activation of the Src/Shc/ERK pathway and prolongs the life span of osteoblasts (Kousteni et al., 2003). At the same time, E2 delivers a pro-apoptotic signal to bone-resorbing osteoclasts, shortening their life span (Kousteni et al., 2003). Although these studies have been done mainly in cell-culture systems, their results suggest that ER rapid signaling actions have also a role in vivo. In the liver, rapid E2- induced signals (i.e., PLC/PKC) are deeply linked to the expression of the LDL receptor and to a decreased level of serum LDL-cholesterol (Marino et al., 2001a). Finally, vascular protection by E2 in ischemia/reperfusion injury in vivo requires E2-induced activation of endothelial NOS, as mediated by the PI3K/AKT pathway (Chambliss et al., 2000; Simoncini et al., 2000). The mechanism(s) by which E2 exerts proliferative effects is assumed to be exclusively mediated by rapid membrane-starting actions (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003). E2 treatment of mammary-derived MCF-7 cells triggers the association of ERα with Src and p85α leading to DNA synthesis (Castoria et al., 2001). In HepG2 cells multiple and parallel membrane starting pathways are rapidly activated by the ERα-E2 complex (Marino et al., 1998, 2002, 2003) and the blockade of PLC/PKC, ERK, and PI3K/AKT pathways completely prevents the E2-induced DNA synthesis (Marino et al., 2002, 2003). ERK/MAPK and PI3K/AKT pathways, rapidly activated by the ERα-E2 complex, also have a critical role in E2 action as a survival agent. In fact, these pathways enhance the expression of the anti-apoptotic protein Bcl-2, block the activation of the p38/MAPK, reduce the pro-apoptotic caspase-3 activation, and promote G1-to-S phase transition via the enhancement of the cyclin D1 expression (Marino et al., 2002, 2003; Acconcia et al., 2005a).
1.6 ERβ RAPID ACTIONS
Less information is available on the role played by the ERβ:E2 complex to activate rapid non-genomic mechanisms and its contribution in E2 proliferative effects.
ERβ appears to act as a dominant regulator in E2 signaling, and when co-expressed with ERα it causes a concentration-dependent reduction of ERα-mediated transcriptional activation (Matthews and Gustafsson, 2003) and the repression of ERα-mediated effects including cell proliferation. Consistent with this notion, E2 increases cell proliferation and causes tumor
13
formation in MCF-7 cells expressing only ERα (Matthews and Gustafsson, 2003). On the other hand, ERβ inhibits the E2-induced proliferation of transfected MCF-7 cells and prevents tumor formation in a mouse xenograft model in response to E2 (Paruthiyil et al., 2004). This effect is linked to the ERβ repressive effect on ERα-induced gene transcription of cell cycle components (e.g., c-myc, cyclin D1, and cyclin E) which are associated with proliferation. These findings suggested a possible role for ERβ as tumor suppressor in breast cancer (Matthews and Gustafsson, 2003; Paruthiyil et al, 2004; Strom et al, 2004). Furthermore these studies support a functional antagonism between ERα and ERβ with respect to the E2-induced cell proliferation, suggesting without clarify the signal transduction pathways involved.
However, the ability of the ERβ:E2 complex to activate rapid non-genomic mechanisms has been reported, even if these data are limited and conflicting. A sub-population of ERβ transfected into Chinese Hamster ovary cells is capable of stimulating IP3 production, ERK/MAPK activation, and c-JNK phosphorylation (Razandi et al, 1999). Geraldes and coworkers reported that E2 reduces ERK activity through ERβ stimulation in porcine smooth muscle cells (Geraldes et al, 2003). Recently, ERβ has been reported to rapidly induce a persistent membrane-initiated activation of p38/MAPK without any interference on survival proliferative pathways, thus impairing the activation of cell cycle components (i.e., cyclin D1 expression) (Acconcia et al., 2005a). Although the scarce information does not allow a complete discussion on the contribution of ERβ in E2-induced rapid signals, these data indicate that also ERβ could originate cell-specific signal transduction cascade. Also for ERβ, the rapidity by which these cellular cascades are activated raises the need for a receptor localized at the plasma membrane. Although a subpopulation of ERβ localized within caveolar rafts, responsible for rapid endothelial nitric oxide synthase stimulation by E2 has been reported in the plasma membrane of endothelial cells (Chambliss et al. 2000), the mechanism allowing ERβ localization at the plasma is completely unknown.
As a whole, these evidence demonstrate the mechanism by which E2 exerts proliferative properties is to be exclusively mediated by ERα-induced rapid membrane-starting actions (Marino et al., 1998; Castoria et al., 1999; Lobenhofer et al., 2000; Marino et al., 2001b, 2002, 2003; Castoria et al., 2001; Acconcia et al., 2005a), whereas E2 induces cell death through ERβ non-genomic signaling (Acconcia et al., 2005a). All these results point to the concept that the ability of ER:E2 membrane starting pathways to signal through multiple cascades, undoubtedly has an impact on all aspects of cellular function, contributing to E2-induced cell proliferation and survival,
14
all essential features of cell physiology as well as of tumor biology (Levin, 2005). Thus the membrane-initiated signals of ERα and ERβ are both required to obtain the complete cellular response to E2.
1.7 ER TISSUTAL DISTRIBUTION
Both ERs are widely distributed throughout the body, displaying distinct or overlapping expression patterns in a variety of tissues (Couse and Korach, 1999; Pettersson and Gustafsson, 2001). In particular, ERα mRNA is highly expressed in epididymis, testis, ovary, kidney, and adrenal. Moderate amounts of ERα are also present in the prostate gland, bladder, liver, and thymus. The highest amounts of ERβ mRNA were detected in the prostate gland, brain, ovary, gastrointestinal tract and bladder, hematopoietic and central nervous systems. ERα and β are, however, coexpressed in a number of tissues including the mammary gland, epididymis, thyroid, adrenal, bone, and certain regions of the brain. Although both ER subtypes may be expressed in the same tissue, they might not be expressed in the same cell type. In the rat ovary, ERβ is the predominant ER in the granulosa cells, whereas ERα is largely present in the thecal and interstitial cells (Hiroi et al., 1999; Sar and Welsch, 1999; Nilsson et al., 2001). The uterus and pituitary gland are special in that ERβ is expressed during development and ERα when the tissue matures (Brandenberger et al., 1997; Nishihara et al., 2000). Uterus, bladder, lung, and testis show intermediate levels of ERβ, whereas low but detectable levels of ERα are observed in the pituitary gland, thymus, various brain sections, and spinal cord (Deroo and Korach, 2006). Nonetheless, ERα and ERβ proteins have been simultaneously detected in many cell types including neurons and thymocytes (Greco et al., 2001; Mor et al., 2001), and these as well as other cell types that coexpress both ER subtypes are targets for potential interplay between the two receptors. As previously reported, when coexpressed with ERα, ERβ appears to act as a dominant regulator of estrogen signaling causing a concentration dependent reduction in ERα-mediated transcriptional and rapid activities (Pettersson et al., 2000; Liu et al., 2002; Matthews and Gustafsson, 2003). Form these data raises the hypothesis that the final effect of E2 of cell physiology could dependent on the subtle balance of ERα and ERβ expression level, even if other underlying mechanisms can not be excluded.
1.8 SELECTIVE ESTROGEN MODULATORS (SERM)
The central role of the ER signaling network in cancer, cardiovascular disease, osteoporosis, and neurological disease and an increasingly detailed understanding to the underlying cell biology have made ER an attractive
15
target for pharmacological intervention. Selective estrogen receptor modulators (SERMs) are ER ligands that can have varying agonist or antagonist activities given the cell context (Burger, 2000; Osborne et al., 2000). They have been developed and characterized to obtain more favorable tissue and receptor subtype selective effects (Veeneman, 2005; Heldring et al., 2007). These ligands that display tissue-selective pharmacology: as anti-estrogens (or antagonists) they oppose the action of estrogens in certain tissues, while mimicking the action of endogenous estrogens (agonists) in others. For instance, Tamoxifen is the prototypical SERM that, because of its agonist activity in the liver, reduces serum total cholesterol and LDL levels (Williams et al., 1997). Unfortunately, its strong agonist activity in the endometrium leads to endometrial hyperplasia and low-grade cancers.
Non steroidal plant-derived flavonoids present in the human diet show high degree of structural resemblance to certain selective estrogen modulators (SERMs) (Brzezinski and Debi, 1999). In fact, the flavonoids compete with E2 in binding to ERα and induce the classical genomic activity of ER through the transcription of ERE-containing genes (Kuiper et al., 1998). These observations raise the hypothesis that flavonoids may act as an estrogen ‘mimetic’ in different tissues and organs. However, flavonoids may have protective roles in several E2-related diseases (i.e., osteoporosis and cardiovascular diseases in post-menopausal women) (Lissin and Cooke, 2000; Ewies, 2002; Wuttke et al., 2003). Furthermore, epidemiological and in vitro studies indicate that this family of dietary components displays divergent effects from E2 in affecting cancer growth (Middleton et al., 2000; Wuttke et al., 2003). In particular, the ability of flavonoids (e.g., naringenin and quercetin) to hamper cell proliferation via their binding to ERα and to interfere with its rapid signaling has been showed (Totta et al., 2004; Virgili et al., 2004; Galluzzo et al., 2009b). From this evidence, these dietary compounds represent a very selective tool, which might be of great relevance for fully understanding the ER rapid signal transduction pathways and their impact on E2-dependent cell functions.
16
2. AIMS The nuclear-initiated signals of E2 have been studied for long time.
However, in addition to its well known role as a regulator of gene transcription sustained by the classical nuclear ERs (Nilsson et al., 2001; Acevedo and Kraus, 2004), E2 also shows rapid extra-nuclear actions (Levin, 2005; Marino et al., 2005; Ascenzi et al., 2006). At present it has been assumed that the rapid membrane initiated signals after E2-bound to ERα are mainly involved in E2-dependent proliferative effects (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003; Lobenhofer et al., 2000; Fernando and Wimalasena, 2004). For instance, as previously reported, non-nuclear recruitment of MAPK signaling cascades by ERα has been demonstrated in cardiomyocytes (Nuedling et al., 1999) breast cancer (Castoria et al., 1999) and bone (Endoh et al., 1997; Jessop et al., 2001) leading to responses that include cell growth, cell cycle progression, and survival. Furthermore, recent data point to ERα palmitoylation as the mechanism responsible for the receptor localization to the cell membrane and the regulation of the E2-induced cell proliferation (Acconcia et al., 2005b). From these data, it is reasonable to assume that the stimulatory effect of estrogen on cell proliferation also contribute to malignant tumor growth in these tissue through ERα. At present if ERα and the downstream rapid signals are only involved in mediating these adverse E2 effects is still unclear.
On the other hand, less information are available on membrane-started action of ERβ and its physiological role. Contradictory evidence on the ability of ERβ to activate or inactivate Src and p38 kinases has also been reported (Castoria et al. 2001, Kousteni et al. 2001, Geraldes et al. 2003, Mori-Abe et al. 2003; Acconcia et al., 2005a). At present, the contribution of ERβ on E2-induced cell proliferation is due only to its action as dominant negative of ERα activities (Couse and Korach, 1999; Weihua et al., 2003; Paruthiyil e al., 2004; Strom et al., 2004). Thus, a full understanding of the existence of ERβ specific extranuclear signals and their impact on the final E2 effect remains to be determined.
Aim of present thesis is to investigate the presence and the impact of membrane-initiated signals mediated by ERα and ERβ on E2-dependent cell functions. In particular the role played by each ER rapid signal in protective effects of E2 versus proliferation and their integration when both ERs are co-expressed in the same cellular context will be studied.
.
17
3. ERα-MEDIATED SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELL PROLIFERATION
3.1 INTRODUCTION
The mechanism by wich E2 exerts proliferative properties has been assumed to be exclusively mediated by ERα-dependent membrane-initiated signals (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003; Lobenhofer et al., 2000; Fernando and Wimalasena, 2004) pointing to ERα as the isoform mainly involved in E2 adverse effects responsible for tumor growth. On the other hand, data from animal model and clinical studies support the involvement of ERα in the protective role for estrogens, for instance on cardiovascular system (Mendelsohn, 2000; Mendelsohn and Karas, 2005; Kim and Bender, 2005; Deroo and Korach, 2006). Indeed, the estrogens are able to directly prevent the proliferation and the migration of vascular smooth muscular cells (VSMC) (Razandi et al., 2000). In humans, both ERα and ERβ are expressed in vascular endothelial cells and vascular smooth muscle cells (Mendelsohn, 2000; Incerpi et al., 2003; Mendelsohn and Karas, 2005). Although it is possible that ERβ inhibits the vascular smooth muscle cell proliferation acting as dominant negative of ERα activities (Watanabe et al., 2003; Kim and Levin, 2006), we recently demonstrated that also ERα may activate rapid signals devoted to the block of cancer cell proliferation These signals started upon exogenous ligands bind to ERα (Totta et al., 2004; Virgili et al., 2004).
Naringenin (5,7,4’-trihydroxyflavanone, Nar; chemical structure Fig. 3.1), especially abundant in citrus fruits and in tomatoes, is reported to have anti-proliferative effects in different cancer cell lines (e.g., colon, breast, and uterus cancer cell lines) (So et al., 1996; Kawaii et al., 1999; Birt et al., 2001; Manthey et al., 2002; Frydoonfar et al., 2003; Harmon and Patel, 2004, Totta et al., 2004; Virgili et al., 2004). Among several mechanisms proposed for Nar induced anti-proliferative effects (i.e., antioxidant activities and kinase and glucose uptake inhibition) (Harmon and Patel, 2004; Moon et al., 2006), the ability of Nar to hamper cell proliferation by binding to ERs is particularly intriguing. Thus the study of the ER mediated anti-cancer action of nutritional flavonoids, could help to clarify the involvement of ER activities in regulating cell proliferation.
We previously showed that Nar concentrations, physiologically achieved in the plasma (1-10 µM) after the consumption of meals rich in Nar, enhanced ERβ-mediated signals important for cancer cell apoptosis and impaired ERα-mediated signaling important for E2-induced cancer cell proliferation (Totta et al., 2004; Virgili et al., 2004). In the presence of ERα, Nar prevented the activation of ERK/MAPK and PI3K/AKT signaling
18
without impairing the transcription of an ERE-containing gene construct. Moreover, Nar activated the rapid phosphorylation of p38/MAPK which, in turn, induced a pro-apoptotic cascade (e.g., caspase-3 activation) (Totta et al., 2004; Galluzzo and Marino, 2006). These data raised the possibility that Nar antagonistic effects on E2-related cancers were dependent on flavonoid ability to modulate ERα association to the plasma membrane and the protein-protein interaction important for ERα-mediate proliferative effects of E2.
We hypothesize that Nar binding to ERα could selectively modulate the receptor post-translational lipid modification palmitoylation. Here this possibility has been investigated comparing the effect of E2 and Nar on ERα palmitoylation and on ERα association to either membrane (i.e., caveolin-1) or adaptors (i.e., MNAR) or signaling proteins (i.e., c-Src). Finally, the influence of Nar-induced regulation of ERα palmitoylation on signaling cascade activation has been evaluated.
This study was conducted on human cervix epitheloid carcinoma cells (HeLa), devoid of any ERs and rendered E2-sensitive by transient transfection with a human ERα expression vector. This model allows analyzing the flavonoids effects on ERα activities without any interference of ERβ or ER splice variants. Furthermore, HeLa cell transfected with empty vector were used as control. Figure 3.1: Chemical structures of 17β-estradiol and naringenin.
19
3.2 RESULTS
3.2.1 Effect of E2 and naringenin on cell proliferation Our first target was to confirm the effects of naringenin in comparison
with E2, on well known cell functions modulated by ERα:E2 complex such as promotion of cell growth. In a different way from E2 (Fig. 3.2a), the treatment of ERα-transfected HeLa cells with naringenin significantly decreased cell number (Fig. 3.2b). These effects of naringenin, in line with results reported for other flavonoids in several cell lines (Kuiper et al., 1998), were time and dose-dependent within the range utilized (0.01 µM-100 µM) (data not shown). However, none of flavonoid concentration utilized, as well as E2, influenced the growth of Hela cells transfected with the empty plasmid (Fig. 3.2a and 3.2b). The date confirms that Nar impairs cancer cell growth through an ERα-dependent mechanism. Figure 3.2: Effect of E2 and Nar on cell growth. Time course analysis of cell growth in empty plasmid or ERα transfected-HeLa cells in the absence (Vehicle) or in the presence of E2 (10 nM) (a) or Nar (10 µM) (b). The data are the mean values ± S.D. of three independent experiments carried out in duplicate. *P<0.001, compared with un-stimulated cell values (Vehicle), was determined using Student’s t-test.
a
b
20
3.2.2 Mechanisms underlying the ERα activities modulation by naringenin Nar decreases ERα palmitoylation
As previously demonstrated, ERα is a palmitoylated protein (Acconcia et al., 2004; Marino et al., 2006). In fact, [3H]-palmitate incorporated into ERα-transfected HeLa cells for up to 240 min reached a steady-state regimen after 10 min, the half-time of ERα palmitoylation was about 1.5 min (Fig. 3.3a). This is consistent with a rapid turnover of fatty acid on ERα supporting the idea that ERα-palmitoylation is a dynamic event involving cycles of acylation and deacylation (Linder and Deschenes, 2006). As previously reported (Acconcia et al., 2005b), 10 nM E2 stimulation induced the decrease of [3H]-palmitate incorporation with a half-time of about 30 min. Two hundred forty min after E2 stimulation 25 ± 0.5 % of ERα was still palmitoylated (Fig. 3.3b). A similar kinetic behavior was obtained pre-treating cells with the palmitoyl acyl transferase (PAT) inhibitor 2-Bromo palmitate (2-Br) 30 min before E2 stimulation (Fig. 3.3b). The Nar concentration (i.e., 1 to 10 µM) rapidly decreased the amount of [3H]-palmitate incorporated in HeLa cells transfected with wild type ERα; the same result was obtained at higher Nar concentrations (i.e., 100 µM), whereas a lower Nar concentration (i.e., 0.01 and 0.1 µM) was ineffective (data not shown). The half-time of Nar-induced ERα-de-palmitoylation (Nar concentration = 10 μM) in cells containing transfected ERα (HeLa) (Fig. 3b) was about 8 min. In cells containing endogenous ERα (HepG2), the half-time of Nar-induced ERα-de-palmitoylation was about 1 min (Fig. 3.3c). Two hundred forty min after Nar stimulation 6.0 ± 0.5 % and 8.1 ± 1.2 % of ERα was still palmitoylated in HeLa and HepG2 cells, respectively (Fig. 3.3b and 3.3c). No change in the ERα protein level accompanied the E2- and Nar-induced decrease of [3H]-palmitate incorporation into ERα in both cell lines (Fig. 3.4).
21
Figure 3.3: Effect of Nar on ERα palmitoylation. (a) Time course of [3H]-palmitate incorporation in ERα transfected HeLa cells. Data are the mean of six independent experiments ± S.D. (b) Effect of ligands on [3H]-palmitated ERα in transfected HeLa cells. After [3H]-palmitate incorporation (120 min), cells were stimulated with either 10 nM E2 or 10 µM Nar or vehicle or 10 µM PAT inhibitor 2-Br (added 30 min before E2 administration). At different times, ERα was immuno-precipitated and radioactivity determined. Data are the mean of five independent experiments ± S.D. (c) Effect of Nar on [3H]-palmitated ERα in HepG2 cells. After [3H]-palmitate incorporation (120 min), cells were stimulated with 10 µM Nar. At different times, ERα was immunoprecipitated and radioactivity determined. Data are the mean of five independent experiments ± S.D.
a b
c
22
Figure 3.4: Effect of Nar on ERα protein level. Western blot (a) and densitometric analysis (a’) of immuno-precipitated ERα in transfected HeLa cells and in HepG2 cells. Data, normalized by comparison with β-actin expression (data not shown), are the mean of five independent experiments ± S.D.
a
a’
23
Nar rapidly impairs ER-caveolin-1 association At steady state ERα palmitoylation is necessary for receptor
association to the scaffold protein caveolin-1 (Acconcia et al 2005b). Thus, we would assess if ligand induced ERα de-palmitoylation could affect the association between receptor and this membrane protein. As expected (Chambliss et al., 2000), ERα was constitutively associated with caveolin-1 and this association was still present 30 min after E2 stimulation. ERα-caveolin-1 association decreased, by half, 60 min after hormone treatment and it was barely detectable 240 min after stimulation (Fig. 3.5a). On the other hand, 10 min after Nar treatment the residual ERα-caveolin-1 complex was only ~ 10% (Fig. 3.6b and 3.6c). This tendency corresponded to the time of E2-induced and Nar-induced ERα de-palmitoylation suggesting that receptor de-palmitoylation and ERα-caveolin-1 dissociation were parallel events driven by ligand binding to ERα (compare Fig. 3.3b with Figs. 3.5a and 3.5b).
Nar prevents the association of ERα with signaling protein involved in proliferation
ERα, like other steroid hormone receptors, does not possess kinase activity, thus its ability to mediate E2-induced proliferation depends on protein-protein complex formation which, in turn, triggers the activation of proliferative signals (Cheskis, 2004: Ascenzi et al., 2006; Song et al., 2006). We and other hypothesised that the E2-dependent ERα de-palmitoylation and ERα-caveolin-1 dissociation could be a prerequisite for ERα docking to the partner proteins (Marino et al., 2005; Pedram et al., 2007). In order to verify this hypothesis, the Nar ability to drive ERα association to the non receptor tyrosine kinase c-Src and to the adaptor protein MNAR has been evaluated and compared with the effect of E2. In the presence of E2, there was a rapid (10 min, data not shown) increase in ERα association to c-Src that was still detectable 60 min after hormone stimulation (Fig. 3.6a). Nar stimulation of ERα transfected HeLa cells prevented such increase. Moreover, prolonged stimulation with Nar induced the dissociation of the ERα-c-Src complex. A similar result was obtained by immunoprecipitation experiments with the adaptor protein MNAR (Fig. 3.6b). The E2-induced ERα-c-Src association was paralleled by an increase of c-Src activation (Fig. 3.6c). On the other hand, Nar stimulation was unable to induce c-Src activation. These data suggest that the very fast Nar-induced ERα de-palmitoylation and ERα-caveolin-1 dissociation impede ERα re-localization by preventing ERα binding to other molecular partners. To verify this hypothesis, HeLa cells were transiently transfected with the un-palmitoylable Cys447Ala ERα point mutant (Acconcia et al., 2004) and
24
stimulated with E2. E2 did not increase ERα-c-Src (Fig. 3.7a) or ERα-MNAR (Fig. 3.7b) association in the presence of the un-palmitoylable ERα mutant, thus strongly confirming that the slow E2-induced ERα de-palmitoylation is required for molecular complex formation (i.e., ERα-c-Src and ERα-MNAR). Accordingly, Nar stimulation does not allow the activation of down-stream kinases involved in E2-induced cell proliferation (Marino et al., 2002, 2003). In fact, no activation of ERK and AKT phosphorylation was observed after Nar stimulation (Fig. 3.8) suggesting an antagonistic role of Nar on this ERα activity.
Nar rapidly stimulates p38/MAPK
The results reported here indicate, for the first time, the mechanism(s) underling the different effects of the ERα:Nar and ERα:E2 complexes on cell proliferation, but they do not explain the Nar-mediated anti-proliferative effect(s). Nar rapidly induces the ERα-dependent activation of the p38 member of the MAPK family which, in turn, activates a pro-apoptotic cascade (Totta et al., 2004). Here, we verified the impact of Nar-induced rapid ERα de-palmitoylation on this signal pathway. Figure 3.9a, a’, and a” confirm that both Nar and E2 stimulation induced a rapid (0.25 h) p38 phosphorylation. In addition, Nar, but not E2, induced the persistent (24 h) activation of this kinase. The Nar-induced p38 activation was ERα-dependent being prevented by cell pre-treatment with the pure anti-estrogen ICI (Fig. 3.9b). On the contrary, cells pre-treatment with the PAT inhibitor 2-Br (Fig. 3.9b) did not affect Nar-induced p38 activation (Fig. 9b). A similar effect was observed by stimulating HeLa cells transiently transfected with the un-palmitoylable Cys447Ala ERα point mutant with E2 or Nar (Fig. 3.9b). This palmitoylation independent mechanism allows the Nar-induced block of cell cancer growth (Fig. 3.10a). Moreover, Nar hampered the ERα transfected HeLa cell proliferation even in the presence of E2, confirming the antagonistic role played by this flavonoid on E2-induced proliferation (Fig. 3.10b).
25
Figure 3.5: Effect of E2 and Nar on ERα association to caveolin-1 (cav). ERα transfected Hela cells were stimulated with 10 nM E2 (a and a’) or with 10 µM Nar (b, b’, c, and c’). Then cells were lysated and subjected to caveolin-1 immuno-precipitation (a, a’, b, and b’) or to ERα immuno-precipitation (c and c’) followed by Western blot with anti-caveolin-1 or with anti-ERα antibodies. Typical Western blots are shown in panels a, b, and c. Densitometric analyses of four different experiments are shown in panels a’, b’, and c’; data are the mean ± S.D. *P<0.001, calculated with Student’s t test, compared with respective unstimulated values (0 min).
a a’
b b’
c c’
26
Figure 3.6: Effect of E2 and Nar on ERα association to c-Src and MNAR. ERα transfected Hela cells were stimulated with either vehicle (60 min) or with 10 nM E2 (60 min) or with 10 µM Nar (1, 10, and 60 min). ERα was immuno-precipitated with anti-ERα antibody followed by Western blot with anti-c-Src (a) or with anti-MNAR (b) and anti-ERα antibodies (a and b). In panels c and c’ the cells were lysed after stimulation and Western blot was performed. Typical Western blots are shown in panels a, b, and c. Densitometric analyses of four different experiments are shown in panels a’, b’, and c’; data are the mean ± S.D. *P<0.001, calculated with Student’s t test, compared with vehicle values.
c
b
a a’
b’
c’
27
Figure 3.7: Effect of E2 on the association of the Cys447Ala ERα mutant to c-Src and MNAR. HeLa cells were transfected with the un-palmitoylable Cys447Ala ERα mutant and were stimulated with either vehicle (60 min) or with 10 nM E2 (1, 10, and 60 min). ERα was immuno-precipitated with anti-ERα antibody followed by Western blot with anti-c-Src (a) or with anti-MNAR (b) and anti-ERα antibodies (a and b). Typical Western blots are shown in panels a and b. Densitometric analyses of four different experiments are shown in panels a’ and b’; data are the mean ± S.D.
a a’
b b’
28
Figure 3.8: Effect of E2 and Nar on ERK and AKT activation. ERα transfected HeLa cells were stimulated with either vehicle (60 min) or with 10 nM E2 (15 min) or with 10 µM Nar (1, 10, and 60 min). Cells were lysed and Western blot was performed. The amount of protein levels were normalized by comparison with β-actin expression. Typical Western blots are shown in panels a and b. Densitometric analyses of four different experiments are shown in panels a’ and b’; data are the mean ± S.D. P<0.001, calculated with Student’s t test, was compared with vehicle (*) or with E2-stimulated (°) values.
a a’
b b’
29
Figure 3.9: Effect of E2 and Nar on p38 activation. (a, a’, and a”) ERα transfected HeLa cells were un-stimulated (0) or stimulated with either 10 nM E2 (0.25 to 24 h) or with 10 µM Nar (0.25 to 24 h) or for 1 h with different Nar concentrations (0.01 to 100 µM). Cells were lysed and Western blot was performed. The amount of protein levels were normalized by comparison with β-actin expression. Typical Western blots are shown. (b and b’) HeLa cells were transfected with ERα wild type (HE0) or with the un-palmitoylable Cys447Ala ERα mutant (Cys447Ala). Cells were stimulated with either vehicle (15 min) or 10 nM E2 (15 min) or 10 µM Nar (15 min) or 10 µM PAT inhibitor 2-Br (added 30 min before Nar administration) or 1 µM ER inhibitor ICI (added 30 min before Nar administration). Typical Western blots are shown in panel b. Densitometric analysis of four different experiments is shown in panels b’; data are the mean ± S.D. P<0.001, calculated with Student’s t test, compared with respective vehicle values (*) or with the Nar-stimulated values (°).
a’ a’’
b’
a
b
30
Figure 3.10: Effet of E2 and Nar on cell proliferation. (a) ERα transfected HeLa cells were grown for 30 h in the presence of either vehicle or E2 (10 nM) or 10 µM Nar and/or 5 µM p38 inhibitor, SB (SB, added 30 min before E2 or Nar administration) and then counted; data are the mean ± S.D. of five independent experiments carried out in duplicate. P<0.001, calculated with Student’s t test, compared with the control values (*) or with Nar-stimulated values (°). (b) ERα transfected HeLa cells were grown for 30 h in the presence of either vehicle or E2 (10.0 nM) and different concentration of Nar (0.01 to 100 µM) and then counted; data are the mean ± S.D. of four independent experiments carried out in duplicate. P<0.001, calculated with Student’s t test, compared with the control values (*) or with E2- stimulated values (°).
b
a
31
3.3 DISCUSSION The data reported here strongly indicate that Nar binding to ERα
induces fast receptor de-palmitoylation, the major determinant of ERα localization at the plasma membrane. In turn, the ERα association with membrane (i.e., caveolin-1) and signaling (i.e., c-Src) proteins is prevented probably due to a different conformational change.
We speculated that the E2-dependent ERα de-palmitoylation, decreasing receptor-caveolin-1 association, could allow ERα redistribution and its association with adaptors and/or signaling proteins (e.g., MNAR, c-Src, tyrosine kinase receptors), which in turn contribute to rapid signaling cascades (e.g., ERK/MAPK and PI3K/AKT) (Totta et al., 2004; Marino and Galluzzo 2007). Present data corroborate the validity of the hypothesis. Actually, E2 stimulation of HeLa cells transfected with the un-palmitoylable ERα mutant does not increase ERα association with MNAR or c-Src (compare Figs. 3.6 and 3.7), impairing ERα ability to activate ERK and AKT phosphorylation (Acconcia et al., 2005b). Correspondingly, Nar rapidly modifies ERα palmitoylation status, and modulates ERα membrane-started activities impairing ERα association with adaptors and/or signaling proteins (i.e. MNAR and c-Src). This prevents the ERα-dependent activation of mitogenic signaling cascades (e.g., ERK/MAPK and PI3K/AKT). Notably, no decrease of constitutive ERK and AKT phosphorylation was observed, suggesting that the Nar concentration used here was unable to inhibit enzyme activities. The data reported here specify the molecular bases of Nar-E2 antagonism and explain the mechanism of Nar induced apoptosis in cancer cells (Totta et al., 2004). In fact, both E2 and Nar rapidly stimulate (15 min), via ERα, the activation of the pro-apoptotic kinase p38/MAPK (Totta et al., 2004 and present data). The ligand-induced p38 activation results independent from ERα palmitoylation being activated even in HeLa cells transfected with the ERα un-palmitoylable mutant, but only Nar induces the persistent activation of such a kinase. Apoptosis signal regulating kinase 1 (ASK1) is one of the upstream activators of p38. In quiescent cells, ASK1 is a cytosolic kinase in which activity is tightly regulated. Raf-1, ERK activator, and AKT suppress ASK1 death-promoting activity (Kim et al., 2001; Yuan et al., 2003; Du et al., 2004; Mabuchi et al., 2004). Moreover, E2 induces ASK1 phosphorylation at Ser83 via ERα-AKT cascade (Mabuchi et al., 2004). Thus, the ability of the ERα-E2 complex to activate rapidly ERK and AKT avoiding the persistent p38 activation guarantees both cell proliferation and cell survival. On the other hand, by inducing the rapid ERα de-palmitoylation, Nar allows ERα association to signal proteins and activation of ERK and AKT. Therefore, the persistent activation of p38, pivotal for
32
pro-apoptotic cascade initiation, occurs. Nar in part impairs the ERα genomic mechanisms. In fact, Nar stimulates the activity of estrogen responsive element luciferase reporter gene construct in the presence of ERα (Kuiper te al., 1998; Totta et al., 2004; Virgili et al., 2004), even if to a lesser extent than E2. Conversely, this flavanone prevents the indirect ERα-mediated transcriptional activity of cyclin D1 promoter (Virgili et al., 2004), which occurs through receptor association with other transcription factors (e.g., activator protein-1) and requires ERα-mediated non-genomic mechanisms (Marino et al., 2002, 2003). Moreover, Nar influences cancer cell proliferation (Fig. 3.9) by acting as a selective antagonist of ERα-mediated non-genomic activities. This implies Nar to work as an E2 antagonist on specific ERα-mediated pathways. In addition, Nar and other nutritional flavonoids (i.e., quercetin and daidzein) act as E2 mimetic, thus mediating the anti-proliferative and protective effects mediated by ERβ against cancer (Totta et al., 2004).
Flavonoids have been studied for more than 50 yr, and now it is definite that they exert a wide range of biochemical and pharmacological effects. Among others, the most investigated effects refer to their cancer preventive activities, which have been predominantly associated with their antioxidant proprieties (Amic et al., 2007), and their inhibitory effects on many enzyme activities such as those of drug metabolism (Moon et al., 2006), aromatase (Lacey et al., 2005), signal kinases (Harmon et al., 2004), and cyclooxygenase (Laughton et al., 1991). However, much of these studies have been conducted in vitro using pharmacological doses of flavonoids (e.g., 100 µm) with little regard to the bioavailability and metabolism of the compounds studied. In addition, the cellular mechanisms involved in the flavonoid anticancer activities are still largely unknown. Although flavonoids have estrogenic activity, bind weakly to ERs, and initiate E2-dependent transcription (Kuiper et al., 1998), the impact of these mechanisms on cancer protective effects is largely confused. The mechanisms through which phytoestrogens may stimulate or inhibit growth of ER-positive cancer cells are controversial, probably due to the different growth-stimulatory properties that E2 mediates in the presence of ERα or ERβ. Here we demonstrate that mechanism underlying the Nar anti-cancer effect is due to its capability to modulate rapid membrane initiated signal of ERα.
In conclusion, this molecule shifts the bioactivity of ERα from its major involvement in promoting cell proliferation by the kinase cascade activation toward anti-proliferative functions by modulating ERα localization and ERα specific signaling pathways starting from plasma membrane. Beside the knowledge of the mechanisms underlying the
33
(anti)estrogenicity of dietary compounds such as flavonoids, overall these data bring to light the new important role played by ERα signals. Due to the allosteric properties of this molecule, different ligands induce ER to assume different conformation responsible for specific protein-protein interaction which, in turn, drive cell to different fate (Fig. 3.11).
All together these findings point to a new perspective in E2 effect suggesting that the signal transduction pathways activated are pivotal in modulating cell function more than ERα/ERβ protein level balance. Figure 3.11: Schematic representation of ERα activities. Protein-protein interactions and E2 final effects after modulation of ERα palmitoylation by E2 and Nar.
34
4. ERβ-MEDIATED SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELL PROLIFERATION
4.1 INTRODUCTION
Recently, various studies have shown decreased expression of ERβ mRNA and protein (or an increased ERα/ERβ mRNA ratio) in cancer vs normal tissues in many tumors, including breast, ovary, colon, and prostate (Foley et al. 2000; Campbell-Thompson et al. 2001: Roger et al. 2001: Bardin et al. 2004). Moreover, ERβ gene is localized on chromosome 14q (see Ascenzi et al., 2006 and references therein), the loss of which has been detected in breast (Roger et al. 2001), ovarian, prostate (Horvath et al. 2001), and colon cancers (Young et al. 1993; Bandera et al. 1997; Loveday et al. 2000; Kasahara et al. 2002; Konstantinopoulos et al. 2003; Wada-Hiraike et al. 2006a, 2006b). These overall findings suggest a potential tumor-suppressive function for ERβ (Iwao et al. 2000) and support a functional antagonism between ERα and ERβ with respect to the E2-induced cell proliferation, but do not clarify either the putative role of ERβ in E2-induced apoptosis or the signal transduction pathways involved. However, recent reports indicate that ERβ seems to participate in the E2-inducing blockages of cell growth and proliferation increasing apoptosis in several cell types (Horvath et al. 2001; Konstantinopoulos et al. 2003; Song and Santen 2003; Bardin et al. 2004; Koehler et al. 2005).
We recently demonstrated that E2-induced rapid signal transduction pathways in ERβ-transfected HeLa cells appear to play a major role in mediating anti-proliferative properties of this steroid hormone. The action of E2 in these cells results from binding to ERβ which, in turn, acutely promotes the rapid and persistent phosphorylation of the p38/MAPK thus triggering downstream activation of a pro-apoptotic cascade (Acconcia et al. 2005a). The rapidity by which these cellular cascades are activated raises the need for a receptor localized at the plasma membrane. Although a subpopulation of ERβ localized within caveolar rafts, responsible for rapid endothelial nitric oxide synthase stimulation by E2, has been reported in the plasma membrane of endothelial cells (Chambliss et al. 2000), the mechanism allowing ERβ localization at the plasma membrane and its putative involvement in the anti-proliferative effect mediated by ERβ:E2 complex is completely unknown. We previously demonstrated that the Cys447 residue present in the ERα ligand binding domain (LBD) is palmitoylable and this lipid modification is necessary for the induction of the non-genomic ERK/MAPK signal transduction pathway, which is relevant to E2-induced cell proliferation (Acconcia et al. 2005b). Although, the homology between ERα and ERβ LBDs is only 59% (Ascenzi et al.
35
2006), the amino acid sequence encompassing the palmitoylated Cys447 and Cys132 residues of ERα and caveolin-1, respectively, is highly homologous to that surrounding the Cys399 residue of ERβ (Acconcia et al., 2003). These findings prompted us to investigate on the involvement of ERβ mediated membrane started signals in the protective E2-dependent pro-apoptotic cascade in colon cancer cells.
Estrogen, and their receptors are involved in the development of many types of malignant tumors, but the role of estrogen in digestive cancers (colorectal, esophageal, and liver) appears to be different from the other categories such as breast and ovarian cancers. Epidemiological studies have ascertained that colorectal cancer (CRC) is the second to fourth most common fatal malignancy in industrialized countries (Potter, 1999; Slattery et al., 2001) and is a common malignancy in both sexes (DeCosse et al, 1993). However, several sex-related differences in incidence, molecular characteristics and response to chemotherapy have been reported. It has therefore been suggested that exposure to E2 and/or estrogenic compounds may underlie these differences. CRC is more common in men than women, the difference being more striking amongst premenopausal women and age-matched men (DeCosse et al., 1993; Wong et al., 2005). In the early 1970s, a transient decrease in colon cancer incidence occurred among women aged 35-44 years old, but not among men (McMichael and Potter, 1980). This observation correlated with a peak in fertility and the use of high dose oral contraceptives during the preceding decade. The authors concluded that either high fertility or exposure to exogenous steroid hormones protected women from colon cancer. Based on meta-analysis of 18 epidemiological studies, the use of hormone replacement therapy by postmenopausal women was associated with a 20% decrease in colon cancer risk (Bhat et al., 2003; Guo et al., 2004). Other studies also demonstrated that women with a history of current or past hormone replacement therapy had a significantly decreased risk of colon cancer and showed that there are gender differences regarding cancer location and type within the colorectum (Cho et al., 2007). These findings have led many investigators to search for the ER isoform involved in E2 protection against colorectal cancer. Since ERα is reported to be minimally expressed in normal colon mucosa and colon cancer cells (Waliszewski et al., 1997; Campbell-Thompson et al., 2001), the effects of E2 on colon cancer susceptibility could be mediated by ERβ (Bardin et al., 2004). The presence of ERβ and its splicing isoforms (ERβ1, ERβ2 and ERβ5) have been demonstrated in normal colorectal mucosa and at much higher levels than ERα (Foley et al., 2000; Campbell-Thompson et al., 2001). But, only the full-length ERβ protein, equivalent to the ERβ1 isoform, can activate ERE in reporter assays (Peng et al., 2003; Wong et al.,
36
2005). On the other hand, the presence of different ERβ-isoforms could differentially modulate E2 by their capability to form homo-dimeri and hetero-dimeri. For instance, ERβ2 (ERβcx), which is unable to bind ligands or coactivators and has no transcriptional activity in reporter assays, shows preferential hetero-dimerization with ERα rather than with ERβ. ERβ2 inhibits ERα DNA binding and has a dominant-negative effect on ligand-dependent ERβ reporter gene activity (Ogawa et al., 1998). These data suggest that the ERβ isoforms could differentially modulate E2 action (Ascenzi et al., 2006) and make elucidating the physiological role of this receptor more difficult. Thus, the main difficulty in studying ERs action mechanism in cancer cells derives from the expression of these ERs splice variants. To bypass this problem we chose human colon adenocarcinoma (DLD-1) cells as an experimental model, which contain only one ERβ isoform corresponding to 54 kDa protein (Fig. 4.1a and 4.1b). In this model the ability of ERβ:E2 complex to activate rapid signals devoted to inhibition of the colon cancer cell growth has been studied. Figure 1: Level of ERs in DLD-1 cells (a) Western blot analysis of ERα and ERβ levels were performed in DLD-1. 54 (kDa) of human ERβ recombinant protein (5 ng, Invitrogen) was used as control. (b) Identification of ERα and ERβ mRNA by RT-PCR using specific sense/antisense primers. Amplification products were separated by electrophoresis on 2% agarose and identified by ethidium bromide staining. ERβ=108 bp; ERα=248 bp.
a
b
37
4.2 RESULTS
4.2.1 ERβ is a palmitoylable protein As previously performed on ERα (Acconcia et al., 2004, 2005b), we
first verified the occurrence of ERβ palmitoylation in DLD-1 cells. Cells were incubated with [3H]-palmitate at 37 °C for different periods (from 0 to 240 min) and the amount of radioactivity in both the immunoprecipitate and the supernatant was determined (Fig. 4.2a). ERβ was not detected in the supernatant fractions (Fig. 4.2a). [3H]-palmitate incorporation in immunoprecipitated ERβ was complete within 120 min and remained unchanged over 240 min (Fig. 4.2b). As a positive control, the radioactivity present in the palmitoylated caveolin-1 (Resh, 1999), immunoprecipitated from DLD-1 cells, was measured (Cav-1) (Fig. 4.2c). A significant decrease in ERβ and caveolin-1 palmitoylation occurred in DLD-1 cells pre-treated with the PAT inhibitor, 2-bromopalmitate (2-Br) (Fig. 4.2c). These findings indicate that ERβ, like caveolin-1, undergoes PAT-dependent palmitoylation.
The time course for [3H]-palmitate incorporation in ERβ was different to that reported for ERα (Acconcia et al. 2004). Since, the PATs are a heterogeneous group of enzymes, which differ depending on the cell type (Smotrys and Linder, 2004), we compared the kinetics of [3H]-palmitate incorporation in the same cell line. The ERs-devoid HeLa cells were transiently transfected with either ERα- or ERβ-encoding vectors and then incubated with [3H]-palmitate for 4 h at 37 °C. After ERα or ERβ immunoprecipitation, the radioactivity present in the supernatant was determined. ERα palmitoylation was very rapid being complete within 10 min and remaining constant over 240 min (Fig. 4.2d). Kinetics of [3H]-palmitate incorporation in ERβ was slow in both transfected HeLa and DLD-1 cells (Fig. 4.2, compare panels b and d). This suggests that ERβ is a worse substrate than ERα for PAT.
38
Figure 4.2: ERβ palmitoylation. (a) Western blot analysis of immunoprecipitated ERβ in the pellet (P) or the supernatant (S). 54kDa of human ERβ recombinant protein (5 ng, Invitrogen) was used as control. (b) Time course of [3H]-palmitate incorporation in immunoprecipitated ERβ in DLD-1 cells. Data are the means of six independent experiments ± S.D. (c) [3H]-palmitate incorporation (120 min) in immunoprecipitated ERβ or caveolin-1 (Cav-1) in the presence or absence of PAT inhibitor (2-Br, 10 µM). (d) Time course of [3H]-palmitate incorporation in HeLa cells transfected with ERα or ERβ expression vectors. ERs were immunoprecipitated and radioactivity counted. Data are the means of four independent experiments ± S.D. of duplicate analyses.
a b
c
d
39
4.2.2 ERβ palmitoylation is negatively modulated by E2 To assess the ability of E2 to modulate ERβ palmitoylation, DLD-1
cells were incubated with [3H]-palmitate for 4 h in the presence of different E2 concentrations. Physiological E2 concentration (1-10 nM) decreased the amount of [3H]-palmitate incorporated in ERβ by more than half; a higher E2 concentration (i.e., 100 nM) was more efficient to decrease palmitate incorporation in ERβ, whereas hormone lower concentration (i.e., 0.1 nM; Fig. 4.3a) was ineffective. The time course of E2 (10 nM) stimulation in DLD-1 cells showed that, after 60 min, E2 reduced the [3H]-palmitate incorporation in ERβ by 38% (Fig. 4.3b) with an increase in the protein level (Fig. 4.3c). The increase in ERβ levels were also detected 120 and 240 min after E2 stimulation (Fig. 3c), which is in good accordance with data reported in the literature (Chiang et al. 2000; Matthews and Gustafsson, 2003 and literature therein). The kinetics of E2-induced de-palmitoylation is very similar for both ERs, as demonstrated in transfected HeLa cells (Fig. 4.4). Figure 4.3: E2 effect on ERβ palmitoylation. (a) Dose-response curve of E2. [3H]-palmitate was incorporated in DLD-1 cells for 120 min, then different E2 concentrations were administrated for further 120 min and ERβ was immunoprecipitated. Data are the means of five independent experiments ± S.D. (b) Time course of E2 stimulation. [3H]-palmitate was incorporated in DLD-1 cells for 120 min, then cells were stimulated with 10 nM E2 at different time and ERβ was immunoprecipitated. Data are the means of five independent experiments ± S.D. (c) Typical Western blot of immunoprecipitated ERβ from DLD-1 cells stimulated with 10 nM E2 at different time.
a b
c
40
Figure 4.4: E2 effect on ERα and ERβ palmitoylation. HeLa cells were transfected with human ERα or human ERβ expression vectors. [3H]-palmitate was incorporated in HeLa cells for 120 min, then cells were stimulated with 10 nM E2 at different time and ERβ or ERα were immunoprecipitated. Data are the means of four independent experiments ± S.D. 4.2.3 Role of ERβ palmitoylation ERβ palmitoylation is necessary for receptor-protein association and plasma membrane localization
The ability of ERβ to bind the scaffolding plasma membrane protein caveolin-1 was examined by coimmunoprecipitation. The association of ERβ with caveolin-1 was present even in the absence of E2 (Fig. 4.5a and 4.5b). This association increased 60 min after E2 stimulation (Fig. 4.5a and 4.5b). Notably, E2-induced ERβ-caveolin-1 association was completely prevented by pre-treatment with the PAT inhibitor, 2-Br (Fig. 4.5c). No variation in the level of caveolin-1 was present after E2 stimulation (Fig. 4.5b). As a whole, these results suggest that ERβ palmitoylation is necessary for receptor localization at the plasma membrane. Immunofluorescence and cell fractionation results were consistent with these results (Fig. 4.6). Under basal conditions, ERβ was expressed in both membrane and cytoplasm (Fig. 4,6a, left panel and Fig. 4.6b), while upon E2 stimulation, the receptor was mainly present at the cell periphery corresponding to the plasma membrane (see the arrows in Fig. 4.6a, central panel and Fig. 4.6b). Pre-treatment with the PAT inhibitor abrogated the ability of E2 to re-localizate ERβ to the plasma membrane compartment (Fig. 4.6a, right panel and Fig. 4.6b).
41
Figure 4.5. Role of ERβ palmitoylation in ERβ association to caveolin-1. DLD-1 cells were stimulated with 10 nM E2 for different times then were subjected to ERβ immunoprecipitation (a and a’) or caveolin-1 immunoprecipitation (b and b’) followed by Western blot with anti-caveolin-1 or with anti-ERβ antibodies. (c and c’). DLD-1 cells were pre-treated for 30 min with 10 µM PAT inhibitor 2-Br then stimulated with 10 nM E2 for different times and subjected to ERβ immunoprecipitation or caveolin-1 immunoprecipitation followed by Western blot with anti-caveolin-1 or with anti-ERβ antibodies. (a, b, and c) Typical Western blot; (a’, b’, and c’) densitometric analysis of four different experiments. Data are the mean ± S.D.
a a’
b b’
c c’
42
Figure 4.6: ERβ plasma membrane localization in DLD-1 cells. (a) Immuno-fluorescence analysis of un-stimulated (a left panel), 10 nM E2 stimulated for 120 min (a central panel), and pre-treated for 30 min with 10 µM PAT inhibitor 2-Br then stimulated with 10 nM E2 for 120 min (a right panel) DLD-1 cells. White arrows indicate the membrane localization of ERβ. (b) Cell fractionation assay of un-stimulated (1, vehicle), 10 nM E2 stimulated for 120 min (2, E2), and pre-treated for 30 min with 10 µM PAT inhibitor 2-Br then stimulated with 10 nM E2 for 120 min (3, E+2Br) DLD-1 cells. Typical Western blot (upper panel) and densitometric analysis (bottom panel) of four different experiments. Data are the mean ± S.D.
a
b
43
ERβ palmitoylation is necessary for E2-induced signaling and pro-apoptotic effects
We next determined whether ERβ palmitoylation could have an impact on the rapid non genomic ERβ activities. After E2 binding, ERα is able to associate to adaptor and/or signaling proteins, which in turn are responsible for signaling cascade activation important for cell proliferation (Greger et al. 2006). This prompted us to evaluate the ability of ERβ to interact with some of these signaling proteins, both in the absence and in the presence of 10 nM E2 for 15 min. No association between ERβ and Src or MNAR was observed before or after E2 stimulation of DLD-1 cells (Fig. 4.7a and 4.7b, respectively). This result was followed by the inability of ERβ to induce ERK or AKT phosphorylation (Fig. 4.7c and 4.7c, respectively). On the other hand, E2 stimulation induced a rapid (15 min) and persistent (24h) increase of p38/MAPK phosphorylation in DLD-1 cells (Fig. 4.8a). This effect was mimicked by the E2 membrane-impermeable E2-BSA and prevented by pretreatment with ICI 182,780 or with the p38/MAPK inhibitor SB 203,580 (Fig. 4.8b). Figure 4.7: ERβ association to Src and MNAR and activation of ERK/MAPK and PI3K/AKT pathways in DLD-1 cells. Cells were grown in the absence (0) or stimulated for 15 min with 10 nM E2, ERβ was immunoprecipitated with anti-ERβ antibody followed by Western blot with anti-Src (c) or anti-MNAR (d) and anti-ERβ antibodies. Western blot analysis of ERK (C) and AKT (D) phosphorylation were performed on un-stimulated (0) and stimulated DLD-1cells for 10–30 min with E2 (10 nM). Data represent a typical Western blot of three different experiments.
c d
a b
44
Figure 4.8: Effect of E2 on p38/MAPK activation. Time course analysis of p38/MAPK phosphorylation was performed on untreated (0) and E2-treated (10 nM). Analysis of p38/MAPK phosphorylation was performed on (a and a’) untreated (0; Vehicle) and E2-treated (10 nM) DLD-1 cells at the indicated times or (b and b’) cells treated for 15 min with E2 (10 nM) or E2-BSA (10 nM) or ICI (1 µM) or 30 min p38/MAPK inhibitor, SB (5 µM). The amount of protein levels were normalized by comparison with actin expression. (a) and (b) show the typical blot of three independent experiments; (a’) and (b’) show the data obtained by densitometric analysis, mean values ± SD. P<0.001, calculated with Student’s t-test, compared with respective un-stimulated (0, Vehicle) values (*) or with E2-stimulated values (°).
a
b
a’
b’
45
We next investigated whether a physical association existed between p38 and ERβ by immunoprecipitation. Under basal conditions a complex formed by the un-phosphorylated form of p38 and ERβ has been detected (Fig. 4.9a and 4.9a’). After 10 min of E2 treatment, ERβ-p38 association as well as p38 phosphorylation significantly increased (Fig. 4.9a and 4.9a’). However, when the cells were pre-treated with the PAT inhibitor, 2-Br, the E2-induced activation of this signaling kinase was completely blocked even though the basal p38 levels were unaffected (Fig. 4.9b and 4.9b’) Furthermore, E2 induced the cleavage of the caspase-3 proform (32-kDa band), resulting in the production of the active subunit of the protease (17-kDa band; Fig. 4.10). To confirm that the appearance of the 17-kDa band was associated with an increase in caspase-3 activity, we analysed one of the known substrates of caspase-3, namely PARP. This 116-kDa, DNA repair enzyme, is cleaved by active caspase-3, so produces the inactive 85-kDa fragment. The E2 treatment of DLD-1 cells resulted in the conversion of PARP into the inactive 85-kDa fragment (Fig. 4.10). In contrast, neither caspase-3 nor PARP was affected by E2 after the pre-treatment of DLD-1 cells with the PAT inhibitor 2-Br or with the p38 inhibitor SB 203,580. Notably, no changes in the pro-apoptotic cascade were detected after treatment with both inhibitors, when used alone (Fig. 4.10)
Thus, palmitoylated ERβ mediates the E2-induced p38 and caspase-3 activation as well as PARP cleavage. These findings demonstrate the critical role played by palmitoylation in ERβ-mediated anti proliferative E2-induced effects, strongly indicating that a membrane-localized ERβ is required for the E2-dependent p38/MAPK activation and for the E2 protective effect against cancer cell proliferation.
46
Figure 4.9: Role of ERβ palmitoylation on p38/MAPK activation. DLD-1 cells were stimulated with 10 nM E2 for different times then were subjected to ERβ or p38 immunoprecipitation followed by Western blot with anti-p38 or anti-p38 phosphorylated (p38-P) or anti-ERβ antibodies. (a and a’) DLD-1 cells were pre-treated for 30 min with 10 µM PAT inhibitor 2-Br then stimulated with 10 nM E2 for different times and subjected to ERβ or p38 immunoprecipitation followed by Western blot with anti-p38 or anti-p38 phosphorylated (p38-P) or anti-ERβ antibodies. (b and b’) Typical Western blot (a and b); densitometric analysis (a’ and b’) of four different experiments. Data are the mean ± S.D.
a a’
b b’
47
Figure 4.10: Role of ERβ palmitoylation on pro-apoptotic cascade activation. Western blot analysis of caspase-3 (a) and PARP (b) activation were performed on un-stimulated or 24 h E2-treated (10 nM) DLD-11 cells. When indicated p38/MAPK cascade inhibitor SB (5 µM) or PAT inhibitor 2-Br (10 µM) were added 30 min before E2 administration. The amount of protein levels were normalized by comparison with actin expression. (a and b) Typical Western blot; (a’ and b’) densitometric analysis of five different experiments. Data are the mean ± S.D.
a a’
b b’
48
ERβ palmitoylation is necessary for DLD-1 cell growth decrease Finally we also evaluated the E2 effect on DLD-1 cell growth. E2
stimulation decreased DLD-1 cell number. This effect required ERβ, since it was completely prevented by ICI 182,780 (Fig. 4.11a) and it was confirmed by flow cytometry assay of the DLD-1 cell cycle distribution after 30 h of E2 treatment. The typical plot of DLD-1 cell population is illustrated in Fig. 4.11b (Vehicle). The first peak indicates the cell number in the G1 phase of the cell cycle (45.4 ± 5.0%), followed by the S phase (10.3 ± 3.2%), and by the peak of the G2/M phase (44.3 ± 2.8%). Thirty hours after E2 stimulation, a peak in the sub-G1 region appeared (7.5 ± 1.0%) (Fig. 4.11b), indicating the presence of DNA fragmentation. Cell pre-treatment with ICI 182,780 prevented the E2-induced sub-G1 peak appearance (Fig. 4.11b). Stimulation of DLD-1 cells, with the E2 cell membrane impermeable E2-BSA, a well-known agent able to discriminate between non-genomic versus genomic effects of ER(s) (Marino et al. 2003), affected DLD-1 cell growth, as did E2 (Fig. 4.11b). This indicates the pivotal role of plasma membrane-starting signals in E2-induced anti-proliferative effects. Thus we next evaluated whether ERβ palmitoylation and ERβ non genomic activities were necessary for the E2-induced DLD-1 cell growth decrease. As a matter of fact, DLD-1 cells pre-treated with either the ERβ palmitoylation inhibitor or ERβ non-genomic activities inhibitor (i.e., 2-Br and SB 203,580, Fig. 4.11c) completely abrogated the E2 effect on cell growth.
49
Figure 4.11: E2 effect on DLD-1 cell growth and role of ERβ palmitoylation on DLD-1 cell growth (a) DLD-1 cells were grown in the presence of E2 (10 nM) and/or ICI (1 µM), and counted at the indicated times. The data are means ± S.D. of five independent experiments carried out in duplicate. P<0.001, calculated with Student’s t-test compared with respective unstimulated values (Vehicle) (*). (b) Flow cytometric analysis of DLD-1 cells after 30 h of E2 or E2-BSA treatment in the presence or the absence of ICI (1 µM) compared with unstimulated cells (Vehicle). The plots indicate cell cycle distribution present in sub-G1, G1, S, and G2/M phases respectively.(c) Cells were treated with E2 (10 nM) or 2-Br (10 µM) or SB (5 µM) for 30 h. Cells were then harvested and counted in a hemocytometer with Trypan Blue solution. Data are mean values ± SD of four different experiments. P<0.001, calculated with Student’s t test compared with vehicle- (*) or with E2-stimulated values (#).
a
b
c
50
ERβ palmitoylation is important for E2-inducecd ERβ up regulation The time course of DLD-1 stimulation by E2 (10 nM) showed that
ERβ protein levels increased from 1 to 24 h (Fig. 4.12a and 4.12a’). The E2-induced ERβ increase was ER-dependent since it was prevented by a pre-treatment with the pure antiestrogen ICI (Fig. 4.12a and 4.12a’). The dose-dependent effect showed that, after 2 h of stimulation, 1 nM E2 induced ERβ up-regulation with a peak at 10 nM. No further increase was detectable at higher E2 concentration (data not shown). To verify that the E2-induced ERβ up-regulation was independent of a block in protein degradation, DLD-1 cells were stimulated with 10 nM E2 in the presence or absence of the proteasome inhibitor MG-132 (15 µM). E2 or MG-132 stimulation induced a similar increase of ERβ levels, while the co-treatment was additive (Fig. 4.12b and 4.12b’). This data indicates that the E2 effect was not mediated by a block of proteasomal protein degradation. We next evaluated the mechanisms underlying this E2 effect assessing the involvement of E2-induced genomic mechanisms. After pre-treating DLD-1 cells with actinomycin (Act), a well-known inhibitor of transcription, the increased ERβ protein level was still observed 2 and 4 h after E2 stimulation, but was completely prevented 24 h after hormone stimulation (Fig. 4.13a and 4.13a’). RNA expression analysis by qRT-PCR confirmed that the E2-induced ERβ gene transcription occurs only 24 h after stimulation. In fact, E2 increased the ERβ mRNA levels only after long incubation time, whereas at shorter stimulation time (i.e., 2 and 4 h) a decrease of ERβ mRNA levels was detected (Fig. 4.13b). This suggests that the rapid E2-induced ERβ up-regulation could be dependent on the rapid induction of ERβ mRNA translation. Indeed, DLD-1 cells pre-treatment with cycloheximide (Cyc), a well-known translation inhibitor, completely prevented the rapid E2-induced ERβ protein up-regulation (Fig. 4.14a and 4.14a’). Thus, we verified the putative role played by E2-induced rapid signal transduction mechanisms. The cell pre-treatment with 2-Br and SB demonstrated that a membrane-localized ERβ as well the rapid E2-induced p38/MAPK activation are required for the rapid E2-induced ERβ up-regulation. Indeed, just 2 h of pre-treatment with either the palmitoylation or the p38/MAPK inhibitor was sufficient to influence the E2-induced ERβ up-regulation because receptor levels were maintained at control values (Fig. 4.15a). Interestingly, these inhibitors impaired the E2-induced ERβ up-regulation even after 24 h of stimulation (Fig. 4.15b). This result was further confirmed by the decrease in the E2-induced ERβ mRNA level occurring 24 h after palmitoylation and p38/ MAPK inhibitor pre-treatment (Fig. 4.16).
51
Figure 4.12: ERβ levels upon E2 treatment of DLD-1 cells. Western blot analysis of ERβ was performed on cells treated with vehicle, E2 (10 nM) or E2 (10 nM) + ICI (1 µM) in DLD-1 cells at the indicated times. (a). Densitometric analysis of four different experiments. Data are mean values ± S.D. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with E2-stimulated values (°) (a’). (b) Western blot analysis of ERβ was performed on cells treated for 4 h with vehicle, E2 (10 nM) or E2 (10 nM) +MG-132 (15 µM) in DLD-1 cells. β-Actin expression (a, b) was used for protein level normalization (b). Densitometric analysis of four different experiments. Data are mean values ± S.D. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with E2- or MG-132-stimulated values (°) (b’).
a
a’
b b’
52
Figure 4.13: Mechanisms involved in E2-induced ERβ expression. (a) Western blot analysis of ERβ was performed on cells treated with vehicle, E2 (10 nM) or E2 (10 nM) + actinomycin (Act, 1 µg/ml) in DLD-1 cells at the time indicated. β-Actin expression was used for protein level normalization (a). Densitometric analysis of four different experiments. Data are mean values ± S.D. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with 4 h Act + E2-stimulated values (°) (a’). (b) qRT-PCR analysis was performed on total RNA extracted from DLD-1 cells treated with E2 (10 nM) at the time indicated. ERβ mRNA levels are expressed as % change versus vehicle stimulated samples. Data are mean values ± SD of four different experiments. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with 2 h E2-stimulated values (#) or with 4 h E2-stimulated values (°).
a
a’
b
53
Figure 4.14: Mechanisms involved in E2-induced ERβ expression. (a) Western blot analysis of ERβ was performed on cells treated with vehicle, E2 (10 nM) or E2 (10 nM) + cycloheximide (Cyc, 10 µg/ml) in DLD-1 cells at the indicated times. β-Actin expression was used for protein level normalization (a). Densitometric analysis of four different experiments. Data are mean values ± S.D. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with E2-stimulated values (°) (a’).
a
a’
54
Figure 4.15: Involvement of E2-induced p38/MAPK activation in ERβ levels. (a) Time course analysis of ERβ level expression was performed on cells treated with vehicle, E2 (10 nM), E2 (10 nM) + 2-Br (10 µM) or E2 (10 nM) + SB (5 µM) at the time indicated. β-Actin expression was used for protein level normalization. Representative Western blot of three different experiments (b) qRT-PCR analysis was performed on total RNA extracted from DLD-1 cells treated with E2 (10 nM), E2 (10 nM) + 2-Br (10 µM) or E2 (10 nM) + SB (5 μM) for 24 h. ERβ mRNA levels are expressed as % change versus DLD-1 cells treated with vehicle. Data are mean values ± S.D. of four different experiments. P<0.001, calculated with Student’s t test, compared with vehicle stimulated values (*) or with E2- stimulated values (°)
a
b
55
ERβ palmitoylation is not necessary for E2-induced transcriptional activity
We previously demonstrated that ERα palmitoylation is required for E2-induced gene transcription (Acconcia et al. 2005b). Since, we wanted to compare the ERα and ERβ transcriptional activity in the same cellular context, the ERs devoid HeLa cells were used as experimental model. HeLa cells were co-transfected with the ERE containing pC3 promoter and ERβ or ERα expression vectors. As expected, in HeLa cells transfected with ERα, the E2 treatment induced a three- and a two-fold increase of pC3 and pD1 promoter activities, respectively (Fig. 4.16). On the other hand, in ERβ-transfected HeLa cells E2 induced a twofold increase only on the pC3 promoter activity (Fig. 4.16). Notably, the pre-treatment of ERα or ERβ transfected HeLa cells with the PAT inhibitor 2-Br reduced the E2-inducible pC3 promoter activity by 30% without affecting the basal pC3 promoter activity (Fig. 4.16). As expected, the ERα palmitoylation has a powerful effect on the E2-induced cyclin D1 promoter activity, which is totally impaired by 2-Br treatment, whereas ERβ was unable to mediate cyclin D1 promoter activity, both in the presence or absence of 2-Br.
Figure 4.16: Role of ERs palmitoylation on the E2-induced genomic activity in HeLa cells. Cells were co-transfected with human ERβ or human ERα expression vectors together with pC3-luciferase (pC3) or pD1-luciferase (pD1) constructs and pre-treated 30 min with PAT inhibitor 2-Br (10 µM) before E2 administration (10 nM for 6 h). The data are the mean values ± S.D. of five different experiments. P<0.001 was calculated with Bonferroni’s test: a, significantly different from control value (open bar); b, significantly different from E2 stimulated samples; and c, significantly different from ERα transfected HeLa cells.
56
4.3 DISCUSSION As previously reported ERα and ERβ show significant differences in
genomic mechanism of action (see Background), but much more significant differences are reported between ERβ and ERa actions with respect to their ability to activate rapid E2-induced signals. The activation of ERK/MAPK, PI3K/AKT, and PKC, rapidly generated after E2 binding to ERα in different cell lines, are all defined as necessary and sufficient for E2-induced G1 to S phase progression, to increase survival pathways (e.g., Bcl-2), and to regulate the transcription of AP-1- and Sp1-dependent genes important for cell cycle modulation (e.g., cyclin D1). Limited, conflicting data are reported for ERβ mediated rapid signals (Razandi et al., 1999, Geraldes et al., 2003; Castoria et al., 2001, Kousteni et al., 2001, Mori-Abe et al., 2003). We recently reported the ERβ:E2 complex ability to activate p38/MAPK in the ER-devoid HeLa cells transiently transfected with the ERβ expression vector (Acconcia et al. 2005a). The discrepancies reported in these studies could be due to the different cellular models utilized, in which ERβ is over-expressed, or both ERα and ERβ are co-expressed, or different ERβ splicing forms could even be present. This further enhances the complexity in the spectrum of potential cellular responses to E2. Here, using colon cancer cells which contain only one ERβ isoform, we demonstrated that ERβ is localized to the plasma membrane and originate rapid signal transduction cascades important for anti-proliferative effects of E2 (Fig. 4.11). In fact, we prove that ERβ undergoes PAT-dependent palmitoylation even if this isoform is a poor substrate for PAT as compared to ERα (Fig. 4.3). In addition, the localization to the membrane is dependent on ERβ palmitoylation since the PAT inhibitor impairs ERβ localization at the membrane and its interaction with caveolin-1. Similar to that reported for ERα is the time- and concentration-dependent negative regulation of ERβ palmitoylation exerted by E2 (Fig. 4.4). Palmitoylation function must be considered more than a simple membrane association of otherwise soluble proteins. In fact, the palmitoylation status of several proteins has also been linked to their activation and their movement within membrane subdomains (Robinson et al., 1995, Smotrys and Linder, 2004). Thus, palmitate addition is a dynamic modification that is continually turning over on cellular proteins. ERβ and ERα do not contain a trans-membrane domain (Zhang et al., 2004) or consensus sequences for miristoylation or prenylation (Acconcia et al., 2003), thus their ability to associate with scaffolding or/and signaling proteins at the plasma membrane seems principally due to palmitoylation (Acconcia et al., 2005b; Levin, 2005). In the resting state, ERβ is localized mainly in the cytosol and nucleus of DLD-1 cells and only a little amount of receptor is tethered with caveolin-1 (Fig. 4.5). After E2
57
stimulation, ERβ undergoes de-palmitoylation, which increases receptor–caveolin-1 association (Fig. 4.5) and, thus, its presence at the plasma membrane (Fig. 4.6). E2 stimulation of ERα-containing cells decreases receptor palmitoylation with a kinetics similar to that reported for ERβ (Fig. 4.3). The E2-dependent de-palmitoylation decreases ERα-caveolin-1 association allowing ERα association with adaptors and/or signaling proteins (e.g., MNAR, Src, tyrosine kinase receptors), which in turn give rise to rapid signaling cascades (e.g., MAPK and PI3K; Levin, 2005, Marino et al., 2005, Song et al., 2005, Leclercq et al., 2006). This does not occur in the presence of ERβ. It has been reported that intact A/B domain and tyrosine 537 in E domain of ERα are both required for receptor interaction with Src in the MNAR–ERα–Src complex and the in vitro association between ERβ and MNAR has been reported (Barletta et al., 2004; Greger et al., 2006). Although ERβ possess a tyrosine residue at 488, which could be subjected to phosphorylation, the ERα and ERβ A/B domain differ in both length and amino acid sequence, exhibiting a low amino acid identity (Ascenzi et al., 2006). Moreover, present data indicate that no association between ERβ and MNAR and Src is present in DLD-1 cells before and after E2 stimulation (Fig. 4.7). On the other hand, E2 increases ERβ level (Chiang et al., 2000, Matthews and Gustafsson, 2003 and present data) and its association with caveolin-1 (Fig. 4.5). As a whole, these data raise the intriguing possibility that the short A/B domain of ERβ could facilitate the E2-induced association between ERβ and caveolin-1, impairing its association with MNAR and Src. As a consequence, ERK and AKT activation does not occur. On the other hand, ERK/MAPK as well as PI3K/AKT cascades cooperate in ERα-E2 induced cell proliferation and cell survival, enhancing the expression of the antiapoptotic protein (Bcl-2) and promoting the G1/S transition via the enhancement of cyclin D1 expression (Marino et al., 2002, 2003). In addition, the ERα:E2 complex rapidly increased p38/MAPK phosphorylation but the contemporary increase of Bcl-2 levels, mediated by ERK/MAPK and PI3K/AKT pathways, impairs the prolonged p38 activation and the downstream effects of this kinase (Acconcia et al. 2005a). On the contrary, the rapid increase of p38 phosphorylation induced by the ERβ:E2 complex is not modulated by ERK/MAPK and PI3K/AKT pathways, thus a more prolonged p38 phosphorylation occurs (Acconcia et al. 2005a; Fig. 4.8). The ERβ palmitoylation is important for E2-induced cell functions. In fact, ERβ-p38 association and E2-induced prolonged activation of this kinase is prevented by the PAT inhibitor 2-Br (Fig. 4.9). Moreover, ERβ palmitoylation is necessary for the p38-dependent activation of downstream pro-apoptotic
58
cascade, which involves the caspase-3 activation and PARP cleavage (Fig. 4.10).
Moreover, here, we reported for the first time that rapid and prolonged E2-induced p38/MAPK activation is also important for the up-regulation of ERβ levels in DLD-1 colon cancer cells. The E2-induced p38/MAPK activation is crucial to rapidly increase the level of ERβ even in the absence of ERβ gene transcription as demonstrated by actinomycin effect and mRNA levels. In addition, p38/MAPK activation is also required for the initiation of transcription. The mRNAs encoding hormone receptors are commonly regulated by their own hormones to create auto-regulatory loops of feedback. Moreover, different hormones, including steroid hormones, regulate concentrations of various gene products primarily by altering mRNA translation and stability (Ing, 2005 and literature cited therein). Our data demonstrate that E2-induced p38/MAPK is fundamental both for the rapid increase of ERβ mRNA translation and for the slow ERβ gene transcription. The final consequence is an increased level of ERβ which, in the presence of E2, will further increase the hormone protective effect against tumor growth. Indeed, in the presence of the p38/MAPK inhibitor the ERβ levels remain similar to the control and the E2-induced DLD-1 cell number reduction is completely prevented. These results reinforce the interpretation of a role for ERβ levels as a negative regulator of colon tumor growth.
To evaluate the impact of palmitoylation on E2-transcriptional effects, we compared ERβ and ERα in a cellular context, which contained the same co-activators. As expected, ERβ is a weaker transactivator than ERα. The palmitoylation of ERβ scarcely influences ERβ genomic activities (i.e., ERE-containing gene promoter transcription). Similar results were obtained in ERα-containing cells, indicating that the rapid palmitoylation-dependent signal transduction pathways are important for the complete transcriptional activity of ERs. This could be due to the ability of such cytosolic signals to modulate the co-activator recruitment and the chromatin activation status (Smith and O’Malley, 2004). ERβ and ERα have opposite effects in mediating the E2-induced transcription of cyclin D1 (i.e., non-ERE-containing gene promoter). As expected, ERβ is unable to induce cyclin D1 promoter transcription, whereas a critical requirement of ERα palmitoylation for cyclin D1 promoter transcription is present.
In conclusion, present findings (schematically summarized in Fig. 4.17) indicate that palmitoylation localizes a little quantity of ERβ at the plasma membrane, thus directing several E2 effects, which allow the protective effect of this hormone in colon cancer. These data, showing the molecular mechanism, which rapidly follows E2 entry in ERβ-containing
59
cells, further sustain the tumor suppressor function played by this receptor isoform. Although ERα is palmitoylated, the outcome effects in cell physiology are opposite to that reported for ERβ. Thus, the expression of each ER isoform and the rapid signal cascade that results activated in the cells could account for the different E2-dependent modulation of cell proliferation reported. Figure 4.17: Model representing the mechanism underlying the ERβ-mediated E2 protective effect against tumor growth
60
5. SIGNAL TRANSDUCTION PATHWAYS ACTIVATED BY ERα AND ERβ COEXPRESSION
5.1 INTRODUCTION
The results previously reported show that E2 in presence of ERα or ERβ activates very different rapid signals important for the final cellular fate. In particular, ERα:E2 is able to activate signal cascades addressing to the proliferation, while ERβ:E2 mediates the protective effects of E2 activating a pro-apoptotic cascade. Because of the ER isoforms are often co-expressed in the E2 target tissues, the major purpose of this study is to evaluate the impact of rapid signals in a tissue where both ERα and ERβ are expressed.
As previously reported, while estrogen’s effects on the reproductive system, bone, and cardiovascular system are quite well established, less is known about the physiological role of E2 on other so call non canonical target tissues such as skeletal muscle. However, several gender-related differences in skeletal muscle between sexes and in women treated with hormone replacement therapy (HRT) and prevention in the menopause-related decline in muscle performance have been reported (Phillips et al., 1993, Heikkinen et al., 1997, Sirola and Rikkonenn, 2005). The depletion of ovarian hormones due to ovariectomy induces an anabolic environment characterized by increased circulating growth factor levels and a positive energy balance (Fisher et al., 1998). Variations in skeletal muscle strength have been described during the human menstrual cycle and muscle mass and strength diminish during the postmenopausal years leading to sarcopenia (Dionne et al., 2000; Lemoine et al., 2003; Wiik et al., 2003), even if the studies are still controversial. In addition, female muscles are more fatigue-resistant and recover faster than male muscles and estrogens increase skeletal muscle force production (Glenmark et al., 2004). All together these data indicate that estrogens can regulate skeletal muscle mass. Congruent with this observation, the two estrogen receptors (i.e., ERα and ERβ) have earlier been reported to be expressed at mRNA level in human skeletal muscle in both sexes (Lemoine et al., 2003; Wiik et al., 2003). At the protein level, only ERβ was identified in human skeletal muscle (Wiik et al., 2003). Very recently, it has been established that, besides mouse and pig, even human skeletal muscle contains both functional estrogen receptor isoforms (Wiik et al., 2009). Thus the skeletal muscle is a E2 target tissue, even if, the mechanism(s) behind the sex-related differences in skeletal muscle as well as the physiological role of estrogen and its receptors in this tissue remains to be clarified.
61
Regulation of skeletal muscle formation (myogenesis) is an essential step for normal development as well as for the maintenance, repair, and regeneration of skeletal muscle (Lluìs et al., 2006; Wilson and Rotwein, 2006). Myogenesis is a dynamic process in which mononucleated undifferentiated myoblasts first proliferate, then withdraw from the cell cycle, and finally differentiate and fuse to form the multinucleated mature muscle fibers in the animal. This process is controlled by myogenic regulatory factors (MRFs), which in concert with other general or muscle specific proteins activate the differentiation program by inducing transcription of regulatory and structural muscle specific genes (Berkes and Tapscott, 2005).
Several families of peptide growth factors influence muscle growth, metabolism, and repair. Myostatin, a member of the transforming growth factor-β family, is a dominant inhibitor of muscle growth (Solomon and Bouloux, 2006). In contrast to myostatin, the insulin and insulin-like growth factors, IGF-I and IGF-II, are the only extracellular growth factors known to promote terminal differentiation of myoblasts enhancing muscle growth (Sheffield-Moore and Urban, 2004; Lluìs et al., 2006). Other hormones such as sex steroid hormones (i.e., testosterone and E2), growth hormone, and glucocorticoids strongly influence the metabolic flexibility of muscle and its substrate storage capacity, being important regulators of skeletal muscle protein remodelling process (Solomon and Bouloux, 2006; Lluìs et al., 2006). Among several hormones, the effect of estrogens on skeletal muscle functions and the mechanism underlying and the ER involvement have received minimal mechanistic scientific investigation.
To evaluate the effects and action mechanisms of E2 in skeletal muscle, rat myoblast L6 cells are chosen as experimental model. L6 are actively proliferating cells if maintained in high serum concentration (10% Fetal Bovine Serum, FBS), while differentiate forming the myotubes when the serum concentration is below 2%.
62
5.2 RESULTS 5.2.1 ER in skeletal muscle cells
Both ER isoforms in rat L6 myoblast are present as confirmed by Western blot analysis (Fig. 5.1a). A single band at 66 kDa or 57 kDa corresponding to ERα or ERβ, respectively, was evident. ERβ represents the main receptor isoform in these cells. Indeed, the ERα/ERβ ratio (Fig. 5.1b) was 0.4 ± 0.02 as determined by comparing the density of tubulin-normalized ER isoform bands with the density of a known amount of ERα or ERβ recombinant protein (5 ng). Figure 5.1: ER isoform levels in L6 cells. Level of estrogen receptors (ERs) in L6 cells and recombinant proteins (a) with the relative densitometric analyses (b).
a
b
63
5.2.2 E2 effects on skeletal muscle cell proliferation The impact of E2 on proliferation-apoptosis balance was assessed in
L6 myoblast. E2 stimulation (10 nM, 24 h) did not affect L6 cell number (Fig. 5.2a) nor caspase-3 activation or the cleavage of caspase-3 substrate, PARP (Fig. 5.2b). Figure 5.2c shows the distribution of cell population in different cell cycle phases in vehicle and E2-stimulated cells: G1 phase (50.0 ± 5.0% and 54 ± 4.8%, respectively) followed by S phase (12.3 ± 3.2% and 13.1 ± 2.4%, respectively), and by the peak of G2/M phase (34.8 ± 2.8% and 32.9 ± 3.1%, respectively). These data further confirmed that E2 did not influence skeletal muscle cell proliferation or apoptosis.
On the other hand, in actively proliferating L6 myoblasts (i.e., grown in 10% serum), E2 rapidly increased (15 min) the Glut-4 translocation to cell membrane (Fig. 5.3a and 5.3a’); successively, the increase of myogenin (Fig. 5.3b and 5.3b’) and MHC levels (Fig. 5.3c and 5.3c’) was observed at 6 and 24 h, respectively, after E2 stimulation. E2 effects on skeletal muscle differentiation markers were dose-dependent with a maximum at 1 and 10 nM (Fig. 5.4). E2 effects was not seen at the higher concentrations tested. Seven days after E2 treatment (10 nM) the alignment and fusion of L6 myoblasts into multinucleated myotubes were visible in E2-treated cells compared to the vehicle-treated cells (Fig. 5.4). In addition, E2 increased the differentiation (0.11) and fusion (3.0) indexes over the control (0.08 and 0.03, respectively) (Fig. 5.5). The E2 effects on skeletal muscle differentiation markers has been also verified in L6 myoblasts growing for 0 or 7 days in low serum percentage (differentiation medium). The E2 effects on earlier differentiation markers (Glut-4 translocation and Myo levels) were similar to that reported in L6 growing in the proliferation medium (Fig. 5.6), whereas the E2 had further effect on the increased levels of MHC over time when cells were cultured in low serum medium (Fig. 5.6).
IGF-I is one of the few extra-cellular growth factors known to promote myoblast differentiation; thus, we compared the effect of E2 and IGF-I in L6 myoblasts growing in proliferation medium. Both hormones influenced the three muscle differentiation markers considered, even if the E2 efficacy was ~ 40 % less than IGF-I (Fig. 5.7). In addition, E2 effects were completely prevented by the ER inhibitor, ICI 182,780, whereas the cell pre-treatment with the specific IGF-I receptor inhibitor, PPP, did not impair E2 properties in L6 cells (Fig. 5.7) even if it completely prevented IGF-I effects (data not shown). These results demonstrate that E2 possesses the ability to modulate the levels of muscle differentiation markers as IGF-I does in a ER-dependent manner.
64
Figure 5.2: E2 effect on L6 cell proliferation. L6 cells were grown in proliferating medium with 10% serum in presence of vehicle or E2 (10 nM) and counted at the indicated time (a). The data are the mean values ± S.D. of 5 independent experiments carried out in duplicate. *P<0.001, calculated with ANOVA followed by Turkey-Kramer post test, comparing the samples at different time of stimulation to the samples at time 0. Western blot analyses of caspase-3 activation and PARP cleavage were performed on un-stimulated (vehicle) and 24 h E2-treated (10 nM) L6 cells (b). The amount of proteins was normalized by comparison with tubulin level. Flow cytometric analysis of L6 cells treated for 24 h with vehicle or E2 (10 nM). The plots indicate cell cycle distribution present in G1, S, and G2/M phases, respectively (c).
a
b
c
65
Figure 5.3: E2 effect on skeletal muscle differentiation markers in L6 cells. Time-course analysis of E2 treatment (10 nM) on Glut-4 plasma membrane translocation (a) and on myogenin (Myo, b) and MHC (c) levels, and related densitometric analyses (a’, b’, and c’, respectively) in L6 cells grown in proliferating medium containing 10% serum. Data are the mean of 3 different experiments ± S.D. *P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test with respect to the vehicle-treated samples. E2 dose-dependent (0.1 to 1000 nM) effects on plasma membrane Glut-4 translocation (30 min of stimulation) and on myogenin (Myo) and MHC levels (24 hrs of stimulation) (d). The amount of protein was normalized by comparison with cav-1 (a) or tubulin levels (b, c). The data are typical Western blots of 3 independent experiments.
c c’
b b’
a a’
66
Figure 5.4: E2 effect on skeletal muscle differentiation markers in L6 cells. E2 dose-dependent (0.1 to 1000 nM) effects on plasma membrane Glut-4 translocation (30 min of stimulation) and on myogenin (Myo) and MHC levels (24 hrs of stimulation). The amount of protein was normalized by comparison with cav-1 (upper panel) or tubulin (middle and lower panel) levels. The data are typical Western blots of 3 independent experiments.
67
Figure 5.5: E2 effect on skeletal muscle differentiation markers in L6 cells. Morphological analysis of L6 cells maintained in growing medium were performed in fixed and permeabilized L6 myoblasts treated for 0 day or 7 days with vehicle or E2. The analysis of myotube formation was performed by incubating cells with anti-MHC antibody and with anti-mouse Alexa488-conjugated antibody (Invitrogen, Carlsbad, CA). Cells were counterstained with DAPI dye. The images, representative of 3 independent experiments (20× magnification), represent anti-MHC immunostaining (left panels) and DAPI staining (right panels).
68
Figure 5.6: E2 Effect in differentiation-induced L6 cells. Time-course analysis of E2 treatment (10 nM) on Glut-4 plasma membrane translocation (a) and on myogenin (Myo, b) and MHC (c) levels in L6 cells grown in differentiation medium containing 2% serum for 0 day or 7 days. Related densitometric analysis are shown in a’, b’ and c’ respectively. Data are the mean of 3 different experiments ± S.D. P>0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing stimulated samples to samples at time 0 of stimulation (empty bar) (*) and comparing samples in differentiation medium at 7 days with respect to 0 days (°).
c
b
a a’
b’
c’
69
Figure 5.7: Cross-talk between E2 and IGF-I receptors. Western blot analyses of Glut-4 plasma membrane translocation (a) and myogenin (Myo, b) and MHC (c) levels in L6 cell maintained in growing medium for 30 min (a) or 24 h (b, c) in the presence of either vehicle or E2 (10 nM) or IGF-I (100 ng/ml). When indicated, the cells were pre-treated with the ER inhibitor, ICI (1 µM), or the IGF-I receptor inhibitor, PPP (100 µM). The amount of proteins was normalized by comparison with cav-1 (a) or tubulin (b, c) levels. Data are representative Western blots of 5 independent experiments, related densitometric analysis are shown in a’, b’, and c’, respectively. Data are the mean ± S.D. P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing samples to vehicle- (*) or to E2- (°) treated samples.
a a’
b b’
c
c’
70
5.2.4 Mechanisms underlying the E2 effects in L6 myoblasts The contribute of both nuclear and extra-nuclear action mechanisms
on E2-induced modification of differentiation markers was thus evaluated. The E2-dependent increase of myogenin (Fig. 5.8a and 5.8a’) and MHC (Fig. 5.8b and 5.8b’) levels was prevented by cell pre-treatment with the translation inhibitor, cycloheximide, and by the transcription inhibitor, actinomycin, thus suggesting that the nuclear mechanism of ER action was involved in E2-induced L6 differentiation. Moreover, to evaluate the impact of the rapid extra-nuclear ER-mediated signaling in E2 effects L6 cells were pre-treated with the PAT inhibitor 2-Br , that prevents the membrane starting signals of both receptor isoforms (Acconcia et al., 2005b; Galluzzo et al., 2007). Thirty min of 2-Br pre-treatment completely prevented the E2 effect on myogenin and MHC protein levels (Fig. 5.9). As expected, the E2 effect on Glut-4 translocation was only prevented by 2-Br, whereas transcription and translation inhibitors did not impair this rapid effect (data not shown). As a whole these data strongly suggest that both E2-dependent rapid signals and nuclear action are required for in E2-induced L6 differentiation.
71
Figure 5.8: Mechanisms underlying E2 effects. Western blot analyses of myogenin (Myo, a) and MHC (b) levels in L6 cells maintained in growing medium for 24 h in the presence or absence of E2 (10 nM), the transcription inhibitor actinomycin (Ac, 1 µg/ml), the translation inhibitor cycloheximide (Cx, 10 µg/ml), and the palmitoylation inhibitor (2-Br, 10 µM). The amount of proteins was normalized by comparison with tubulin levels. Data are representative Western blots of 3 independent experiments. Relative densitometric analyses are shown in Panels a’ and b’. Data are mean values ± S.D. P<0.001, was calculated with ANOVA followed by Turkey-Kramer post test comparing samples to vehicle- (*) or to E2- (°) treated samples.
a a’
b b’
72
Figure 5.9: Mechanisms underlying E2 effects. Western blot analyses of myogenin (Myo, a) and MHC (b) levels in L6 cells maintained in growing medium for 24 h in the presence or absence of E2 (10 nM), 2-Br (10 µM). The amount of proteins was normalized by comparison with tubulin levels. Data are representative Western blots of 3 independent experiments. Relative densitometric analyses are shown in a’ and b’. Data are mean values ± S.D. P<0.001, was calculated with ANOVA followed by Turkey-Kramer post test comparing samples to vehicle- (*) or to E2- (°) treated samples.
a a’
b b’
73
These results prompted us to identify which membrane-starting cascade was involved in E2 effects. As previously reported, upon E2 binding to ERs various signaling pathways are activated (i.e., AKT, ERK, and p38) (Björnström and Sjöberg, 2005; Ascenzi et al., 2006), thus we focused our study on the involment of these kinases in E2-induced myoblasts differentiation. E2 stimulation (10 nM) induced the rapid (15 min) phosphorylation of AKT and p38 without any effect on ERK activation (Fig. 5.7a, 5.7b, and 5.7c, respectively). A significant increase of ERK phosphorylation was evident only 60 min after E2 stimulation. E2-induced kinase activation was dependent by membrane localized ER being prevented by ER inhibitor ICI (Fig. 5.10) and by palmitoylation inhibitor 2-Br (Fig. 5.11) pre-treatment.
Figure 5.10: Rapid signal transduction pathways activated by E2. Time-course analyses of phosphorylated and un-phosphorylated AKT (a), p38 (b) and ERK (c) in L6 cell maintained in growing medium and stimulated for 0, 15, 30, or 60 min with E2 (10 nM) and/or the ER inhibitor, ICI (1 µM). Data are representative Western blots of 3 independent experiments.
a
b
c
74
Figure 5.11: Rapid signal transduction pathways activated by E2. Western Blot analysis of phosphorylated and un-phosphorylated AKT (a), p38 (b) and ERK (c) in L6 cell maintained in growing medium and stimulated for 60 min with E2 (10 nM) and/or the palmitoylation inhibitor, 2-Br (10 µM). Data are representative Western blots of 3 independent experiments. The amount of proteins was normalized by comparison with tubulin levels.
The impact of these rapid signals on E2-dependent effects in L6
differentiation was next determined by using a pharmacologic approach. The E2-induced rapid activation of AKT was necessary for Glut-4 translocation to membrane (Fig. 5.12a and 5.12a’) and for the increase of myogenin level (Fig. 5.12b), but it was dispensable for E2-induced MHC level increase (Fig. 5.12c). On the other hand, the p38 activation was involved in both myogenin and MHC protein level increase. However, this kinase did not affect the rapid E2-induced Glut-4 translocation (Fig. 5.12a). Remarkably, PD-dependent inhibition of ERK did not impact any E2 tested effects on L6 cells (Fig. 5.12). Finally, the influence of these E2-dependent rapid signal pathways on ER nuclear activities has been evaluated by analysing the effect of the kinase inhibitors on myogenin and MHC mRNA levels. In good accordance with the results obtained by Western blot, the pre-treatment with AKT inhibitor prevented the E2-dependent accumulation of myogenin mRNA without any effect on MHC mRNA, whereas p38 inhibitor blocked the E2-induced myogenin and MHC mRNA increase (Fig. 5.13).
a b
c
75
Figure 5.12: Impact of E2-dependent rapid signal inhibitors on differentiation marker levels. Western blot analyses of Glut-4 plasma membrane translocation (a) and myogenin (Myo, b) and MHC (c) levels in L6 cells maintained in growing medium for 30 min (a) or 24 hrs (b and c) in the presence of vehicle or E2 (10 nM) and/or the AKT inhibitor (1 µM), or the p38 inhibitor, SB (5 µM), or ERK inhibitor PD (10 µM). The amount of proteins was normalized by comparison with cav-1 (a) or tubulin (b and c) levels. Data are representative Western blots of 4 independent experiments. Related densitometric analyses are shown in a’, b’, and c’, respectively. Data are the mean ± S.D. P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing samples with respect to vehicle- (*) or E2- (°) treated samples.
a
b
c
a’
c’
b’
76
Figure 5.13: Impact of E2-dependent rapid signal inhibitors on differentiation marker levels. qRT-PCR analyses were performed on total RNA extracted from L6 cells treated for 6 h with E2 (10 nM) and/or the p38 inhibitor, SB (5 µM), or the AKT inhibitor (1 µM) or the ERK inhibitor, PD (10 µM). Myogenin (Myo) and MHC mRNA levels are expressed as % change versus vehicle stimulated samples. Data are mean values ± S.D. of 4 different experiments. P<0.001, calculated with ANOVA followed by Turkey-Kramer post test, comparing samples to vehicle- (*) or E2- (°) stimulated samples.
77
5.2.5 ERα and ERβ involvement in E2-induced L6 myoblast differentiation
The contribution of each ER isoform in activating the previously identified E2-dependent effects has been evaluated by using three different experimental approaches. First, we tested both ERα and ERβ selective agonists, PPT (4,4',4''-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol) and DPN (2,3-bis(4-Hydroxyphenyl)-propionitrile), respectively. PPT mimicked the E2-induced rapid AKT phosphorylation (Fig. 5.14), whereas the ERβ selective agonist DPN was unable to induce the activation of this kinase (Fig. 5.14). Conversely, the cell stimulation with either PPT or DPN induced p38 phosphorylation to the same extent as E2, thus indicating that both ERs are involved in p38 activation (Fig. 5.14). These data suggest that only in the presence of ERα, E2 can trigger the required signals for L6 differentiation marker expression. Notably, ERα-selective agonist PPT induced the Glut-4 membrane translocation and myogenin protein level to a greater extent than E2 (Fig. 5.15). To further confirm the role played by ERα in E2-induced differentiation we reduced ERα isoform with specific siRNAs. ERα siRNA oligo-nucleotides led to a significant reduction in the levels of the corresponding protein without affecting ERβ levels (Fig. 5.16a). As expected, E2 was unable to induce AKT phosphorylation, whereas the E2-dependent activation of p38 was still present, in ERα-silenced cells (Fig. 5.16b). Intriguingly, E2 was not able any more to induce the Glut-4 translocation and the increase of myogenin and MHC level in ERα-depleted cells (Fig. 5.16c), thus confirming the pivotal role of ERα signaling in the E2-induced L6 differentiation. To evaluate the possibility that ERα know down affected the cell system and not only E2 signal transduction pathways, ERα-silenced myoblasts were treated with IGF-I. As shown in Figure 5.17a, this growth factor was still able to increase AKT and p38 activation as well as myogenin and MHC levels even in the absence of ERα. Finally, to evaluate the role played by ERβ in E2-induced L6 differentiation, cells were pre-treated with the selective ERβ antagonist, THC (1 µM, 30 min). E2-induced p38 activation is reduced by THC pre-treatment, while an significant increase in E2-induced AKT phosphorylation and Myogenin and MHC levels was present (Fig. 5.17b and 5.17c, respectively).
78
Figure 5.14: Impact of ERα and ERβ on muscle differentiation markers. Western blot analyses (a) and relative densitometric analyses (a’) of phosphorylated and un-phosphorylated AKT and p38 in L6 cells maintained in growing medium for 30 min in the presence of E2 (10 nM) or the ERα agonist, PPT (10 nM) or the ERβ agonist, DPN (10 nM). Data are representative Western blot of 4 independent experiments. Data are the mean ± S.D. P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing the samples to the vehicle- (*) or to E2- (°) treated samples.
a
a’
79
Figure 5.15: Impact of ERα and ERβ on muscle differentiation markers. Western blot analyses of Glut-4 (a) plasma membrane translocation, myogenin (Myo) (b) and MHC (c) levels in L6 cells treated with E2 (10 nM) or the ERα agonist, PPT (10 nM). The amount of proteins was normalized by comparison with caveolin-1 (a) or tubulin (b and c) levels. Data are representative Western blot of 4 independent experiments. Related densitometric analyses are shown in a’, b’, c’. Data are the mean ± S.D. P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing the samples to the vehicle- (*) or to E2- (°) treated samples. The amount of proteins was normalized by comparison with caveolin-1 or tubulin levels.
a a’
b b’
c c’
80
Figure 5.16: Impact of ERα and ERβ on muscle differentiation markers. Western blot analyses of ERα and ERβ protein levels (a), phosphorylated and un-phosphorylated AKT and p38 (b), Glut-4 plasma membrane translocation, myogenin (Myo) and MHC levels (c) in L6 cells un-transfected or transfected with ERα oligonucleotides in the absence or in the presence of E2 (10 nM). The amount of proteins was normalized by comparison with tubulin or caveolin-1 levels. Typical blots of 3 independent experiments.
a
b c
81
Figure 5.17: Impact of ERα and ERβ on muscle differentiation markers. Western blot analyses (a) of phosphorylated and un-phosphorylated AKT and p38, myogenin (Myo) and MHC levels, and ERα level in L6 cells un-transfected or transfected with ERα oligonucleotides in the absence or in the presence of IGF-I (100 ng/ml). The amount of proteins was normalized by comparison with tubulin levels. Typical blots of 3 independent experiments. Western blot analyses (b) of phosphorylated and un-phosphorylated AKT and p38 in L6 cells maintained in growing medium for 30 min in the presence of E2 (10 nM) or the ERβ antagonist, THC (1 µM). Western blot analyses (c) of myogenin (Myo) and MHC levels in L6 cells treated with E2 (10 nM) or with the ERβ antagonist, THC (1 µM). The amount of proteins was normalized by comparison with tubulin levels. Data are representative Western blot of 4 independent experiments.
a b
c
82
5.2.6 E2 effects on C2C12 myoblasts To establish that the E2 effect on L6 cells was not linked to this
particular cellular context, some experiments were also performed in C2C12 mouse myoblasts. Figure 5.14 confirmed that E2 rapidly increased (15 min) the Glut-4 translocation to cell membrane (Fig. 5.18a upper panel) which was followed (24 h) by the increase of myogenin (Fig. 5.18a, middle panel) and MHC levels (Fig. 5.18a, lower panel) in C2C12 cells. Even in these cells the E2 efficacy was ~40 % less than IGF-I and E2 effects were completely prevented by the ER inhibitor, ICI 182,780 (Fig. 5.18a). Western blot analysis confirmed that both ER isoforms were present in mouse C2C12 myoblasts and ERβ represented the main receptor isoform also in these cells (Fig. 5.18b). Finally, the C2C12 cells stimulation with the ERα agonist, PPT, strongly increased the rapid AKT phosphorylation more than E2 (Fig. 5.18c), whereas the ERβ selective agonist, DPN confirmed its failure to induce the activation of this kinase (Fig. 5.18c) sustaining the pivotal role of ERα also in E2-induced C2C12 differentiation.
83
Figure 5.18: E2 effect on skeletal muscle differentiation markers in C2C12 cells. Western blot analyses of Glut-4 plasma membrane translocation and myogenin (Myo) and MHC levels in C2C12 cells maintained in growing medium for 30 min or 24 h in the presence of either vehicle or E2 (10 nM) or IGF-I (100 ng/ml) or with the ER inhibitor, ICI (1 µM) (a). The amount of proteins was normalized by comparison with caveolin-1 or tubulin levels. Data are representative Western blots of 3 independent experiments. In b the level of estrogen receptors in C2C12 cells and recombinant proteins is reported. Western blot analyses (c) of phosphorylated and un-phosphorylated AKT in C2C12 cells maintained in growing medium for 30 min in the presence of E2 (10 nM) or the ERα agonist, PPT (10 nM) or the ERβ agonist, DPN (10 nM). The data are typical Western blots of 3 independent experiments.
a
b c
84
5.2.7 ERα and ERβ involvement in E2 effect on ROS production in L6 myoblasts.
All together these results point to a role of ERα signals in E2-induced myoblasts differentiation. We than would assess if also ERβ signals may play a role in E2 protective effects.
Recent studies support the evidence for estrogens as strong antioxidant and important factor in maintaining membrane stability and protection from damaged muscle in female animals (Tiidus, 2005). Therefore, the E2 effect on H2O2-induced ROS production and the contribution of each ER isoform in mediating this E2 effect has been evaluated.
In order to determine the pro-oxidant/antioxidant effect of E2, L6 cells were exposed to H2O2 (200 µmol/l) for 15 minutes, and then ROS generation (Fig. 5.19a, -FeSO4) and the state of oxidation of cellular component (i.e., lipid peroxidation) (Fig. 5.19a, +FeSO4) were measured evaluating the changing in DCF (2',7'-Dichlorofluorescein) fluorescence. The stimulation with E2 (10nM) for 24h before H2O2 addition caused a marked decrease in ROS generation preventing the H2O2 effect. The contribution of each ER isoforms in the E2 antioxidant effect has been evaluated by the use of ERα and ERβ selective agonists, PPT and DPN, respectively, and ERβ selective antagonists THC. The results show that DPN completely mimicked the E2 effect, whereas ERβ selective antagonist, THC, and the ERα selective agonist, PPT, were unable to prevent the ROS production (Fig. 5.19b). These data suggest that in L6 myoblasts ERβ is the only ER isoform involved in the E2 protective effect against H2O2-induced ROS production dangerous for cells.
85
Figure 5.19: E2 effect on H2O2-induced ROS production in L6 myoblasts. (a) L6 cells were exposed to H2O2 (200 µmol/l) after 24h of stimulation with E2 (10 nM) and changes in DCF fluorescence were measured in presence or not of FeSO4. Panels represent original outputs (arbitrary units) of the registrations captured by the spectrofluorimeter during 15 minutes substance administration. (b) L6 cells were pre-treated, as indicated, with vehicle, E2 (10 nM) or PPT (10 nM) or DPN (10 nM), THC (1 µM) before exposition to H2O2 (200 µmol/l) for 15 minutes. Data, expressed as % of variation between H2O2-stimulated fluorescence versus basal fluorescence for each stimulation, are the mean ± S.D. of 3 independent experiments carried out in duplicate. P<0.001 was calculated with ANOVA followed by Turkey-Kramer post test comparing the samples to the vehicle- (*) or to E2- (°) treated samples.
a
b
86
5.3 DISCUSSION 17-estradiol (E2) is much more than a female reproductive
hormone. Skeletal muscle has recently been included in non-reproductive estrogen-responsive tissues due to the presence of both ER isoforms (Dahlberg et al., 1982; Lemoine et al.; 2003, Wiik et al., 2009) and to the relevant epidemiological and in vivo studies (Fisher et al., 1998; Fulco et al., 1999; Dionne et al., 2000; Lemoine et al., 2003; Wiik et al., 2003; Glenmark et al., 2004; McCormick et al., 2004;) which in concert sustain the possible impact of E2 on muscle physiology of the E2-induced different signals
The present study was aimed to determine the contribute and the impact of ERα:E2- and ERβ:E2-mediated rapid signals on the E2 final effect in non cancerous cells when both receptor isoforms are present. In particular, a more physiological experimental model such as actively proliferating L6 rat myoblasts has been used. The L6 muscle cell line derived from one-day-old rat muscle retains many skeletal muscle properties and is usually used as a model to study skeletal muscle differentiation. In other studies, the authors used L6 cells myoblasts growing in 10% serum, the proliferating medium, then, to induce quiescence and promote differentiation, the percentage of serum was lowered to 2%. After E2 treatment, we detected a dose- and time-dependent increase of well known skeletal muscle differentiation markers (i.e., Glut-4 membrane translocation, the transcription factor myogenin and the contractile protein MHC expression) followed by changes in cell morphology. Notably, the hormone dose response displays a bell-shaped curve, as is already known for other hormones that typically interact with plasma membrane receptors (i.e., insulin, atrial natriuretic factor). The lack of effect at higher concentrations could be considered the expression of a receptor down-regulation phenomenon, by which the cells protect themselves against high hormone levels. Although to minor extent, E2 activates similar pathways as IGF-I, a hypertrophic factor for muscle cells (Jacquemin et al., 2004; Harridge et al., 2007). In fact, both hormones rapidly increase the AKT-dependent translocation of Glut-4 glucose transporter. Glut-4 translocation to the plasma membrane precedes other muscle specific protein increase (i.e., myogenin and MHC) which are necessary for the appearance of the morphological muscle phenotype (Lluìs et al., 2006). This E2-induced effect, further confirmed in C2C12 cells, indicates, for the first time, that the hormone promotes the differentiation of actively proliferating myoblasts.
Under normal circumstances, mammalian adult skeletal muscle is a stable tissue with very little turnover of nuclei. However, upon injury,
87
skeletal muscle has the remarkable ability to initiate a rapid and extensive repair process preventing the loss of muscle mass (Chargé and Rudnicki, 2004). Skeletal muscle repair is a highly synchronized process involving the activation of myogenic cells to proliferate, differentiate, and fuse leading to new myofiber formation (Chargé and Rudnicki, 2004). Several trophic factors have been involved in maintaining a balance between growth and differentiation of myogenic cells to restore normal muscle architecture after injury (Sheffield-Moore and Urban, 2004). Among several muscle trophic factors sex steroid hormones have been reported to play a role in maintaining muscle mass and strength in humans. Testosterone supplementation of healthy, hypogonadal men results in muscle hypertrophy (Sheffield-Moore and Urban, 2004) and there are reports that estrogen replacement therapy in postmenopausal women enhances both muscle mass and strength (Sipila et al., 2001; Jacobsen et al., 2007). Moreover, sarcopenia, the age-dependent muscle mass decline, is more extensive in women, who lose an additional 15% of muscle mass around the time of the menopause (Jacobsen et al., 2007). Our results, in addition to strength a role of E2 in muscle physiology, strongly indicate that E2 should be included in the list of skeletal muscle trophic factors.
We, thus, turn our attention to the mechanism(s) at the root of E2 effects. In non-reproductive rat tissues, Ciana and co-workers (Ciana et al., 2003) found that the maximal activation of ER transcriptional activities was detected during diestrus when low levels of serum estrogen were present and they suggested that an hormone-independent mechanism may trigger this effect. Moreover, the administration of growth factors such as IGF-I and epidermal growth factor, even in the absence of E2, induces the activation of MAPK-signal transduction cascade which, in turn, increases the transcriptional activity of ERs (Bunone et al., 1996; Cenni and Picard, 1999; Tremblay et al., 1999; Ciana et al., 2003). In addition, accumulating evidence suggests that E2 is able to enhance the expression and the activation status of IGF-I receptor (Mendez et al., 2006); thus, a cross-talk between IGF-I and E2 and their receptors could be at the root of E2-induced L6 differentiation. To verify the existence of a putative cross-talk between ER and IGF-I receptors the cells were pre-treated with the IGF-I receptor inhibitor. The inability of IGF-I receptor inhibitor to impair E2-induced effects on differentiation markers levels led to exclude a contribution of IGF-I signals in E2 effects, at least in the first phases of myoblast differentiation. However, it has been reported that muscle contractions are paralleled by IGF-I levels increase (Mendez et al., 2006) and this may be a pre-requisite for an hormone-independent ER activation during physical exercise (Wiik et al., 2009). Present results indicate that E2-induced L6 cell
88
differentiation requires the presence of both rapid membrane starting and transcriptional ER activities.
Ligand binding causes the activation of ERs which culminate in their direct binding to specific estrogen response element (ERE), in the indirect binding via other transcriptional factors (e.g., AP-1), and in E2-induced gene transcription (O’Lone et al., 2004; Piccone et al., 2005; Ascenzi et al., 2006). Interestingly, the rat mhc-IIx gene promoter region [rat genome database (RGD):735061] has two half-site ERE motifs, making it possible that E2 regulates directly the MHC expression. Indeed, the use of the transcription inhibitor actimomycin impairs the E2 effect outlining the role played by transcription signals in modulating the differentiation markers expression. On the other hand, ERα and ER must be considered a population of proteins which localization in the cell dynamically change, shuttling from membrane to cytosol and to the nucleus, depending on ligand binding (Dan et al., 2003; Ascenzi et al., 2006; Leclercq et al., 2006). Present data indicate that the pre-treatment with the palmitoylation inhibitor, which impedes ER localization at membranes (Acconcia et al., 2005b; Galluzzo et al., 2007), prevents the E2 effect on myogenin and MHC protein levels. These data suggest that membrane pools of ERs are present in L6 cells and able to initiate rapid signals important for E2-dependent L6 differentiation. As reported in other cellular contexts (Castoria et al., 2001; Dos Santos et al., 2002; Marino et al., 2003; Glenmark et al., 2004; Ascenzi et al., 2006), E2 rapidly induced the activation of AKT and p38/MAPK in myoblasts cells.
AKT has been linked to muscle development, regeneration, and hypertrophy (Lawlor et al., 2000; Rommel et al, 2001; Lai et al., 2004; Sandri et al., 2004). In addition, independent studies have unambiguously demonstrated that the p38/MAPK signaling pathway is crucial for the transcriptional control of skeletal muscle differentiation and for the fusion of myoblasts into myotubes (see Lluìs et al., 2006). Although several studies suggest that the two pathways are parallel (see Keren et al., 2006), they may affect different downstream targets or may converge on shared targets that require input from both signaling pathways. This could occur in L6 cells after E2 stimulation.
E2 stimulation in myoblasts induced the rapid AKT phosphorylation which is necessary for the rapid (15 min) translocation of Glut-4 at membranes. This seems to be the first event necessary for the E2 promotion of differentiation process in L6 cells. E2-induced AKT phosphorylation is also required for the subsequent (6-24 h) expression of myogenin confirming the role played by this kinase in muscle differentiation (Vandromme et al., 2001). In addition, E2-induced p38 phosphorylation is
89
mainly required for the expression of both myogenin and MHC. On the other hand, ERK1/2 did not seem to play a role in E2-induced expression of muscle differentiation markers. In fact, even if the hormone activates ERK1/2 phosphorylation in L6 cells the pre-treatment with specific ERK1/2 inhibitor did not impair E2-induced differentiation marker levels. The role played by ERK/MAPK on muscle cell differentiation is controversial (Bennett and Tonks, 1997; Gredinger et al., 1998). It has been reported that ERK activation could inhibit myogenic transcription in myoblasts, but it could contribute to the activation of myogenic transcription and regulates post-mitotic responses in myotubes (Wu et al., 2000). Thus, we cannot exclude that E2-induced ERK1/2 activation is necessary in the myotubes, the ultimate steps of differentiation. In line with this, it has been reported that E2 activates ERK1/2 in the C2C12 skeletal muscle cell maintained in serum-free medium (differentiation condition) (Ronda et al., 2007); however, the role of this activation in myoblast differentiation remains vague.
As reported above, both ERα (low amount) and ERβ (high amount) are present in myoblast cells. In a very elegant paper Glenmark and coworkers (Glenmark et al., 2004) showed that muscles of male mice had shorter contraction and relaxation times than those of female mice, and ER deficiency (ER-/-) had no effect on this. Moreover, fatigue and recovery of female muscles were not affected by ER deficiency (Glenmark et al., 2004). In mouse gastrocnemius muscle, in which ERα and ER are co-expressed, Glut-4 expression on the cell membrane was not affected by loss of ER but was extremely reduced in ERα-/- mice (Barros et al., 2006). All together these in vivo experiments suggest that ERα could act as the principal mediator of E2-induced effect in skeletal muscle. Our data, obtained by using selective agonists and ERα silencing procedure, demonstrate for the first time, that the rapid ERα-dependent AKT activation is pivotal for E2-induced differentiation marker expression. In fact, both in the cells stimulated with the selective ER agonist (DPN) and in ERα silenced ones stimulated with E2, the AKT activation and the differentiation markers were impaired even in the presence of ER-mediated p38 phosphorylation. Intriguingly, the cell stimulation with ERα selective agonist (PPT) induced a significant increase in Glut-4 translocation at cell membranes and in myogenin levels. Similar results have been obtained with the E2 stimulation of ERα silenced cells, which present a lower Glut-4 translocation at cell membranes and myogenin levels respect to the control cells. This datum was further confirmed by cell pre-treatment with ER antagonist (THC), which induced an increase in ERα:E2-induced AKT
90
phosphorylation and Myogenin and MHC levels. Taken together, these results indicate that ER may elicit a suppressive role in E2-induced skeletal differentiation. The lack of ER signals in ovariectomized ERα-/- mice determined a reduction of body and fat pad weights while improving insulin and glucose metabolism (Jones et al., 2000). Furthermore, administration of the selective ER ligand, DPN, to knock-out mice for aromatase resulted in a substantial reduction in expression of Glut-4 (Barros et al., 2006). These data and our results strengthen the idea that ER opposes the effect of ERα on Glut-4 translocation in skeletal muscle.
The ERβ high content in L6 cells and the possibility that it could exert specific function besides its role as negative regulator of ERα activities, lead us to investigate the possible protective effects estrogen against insults as oxidative stress. In muscle cells, reactive oxygen species (ROS) are continually generated. It is believed that these molecules have a well-established role as physiological modulators of skeletal muscle functions, including development, metabolism and contractile functions. Moreover, studies in the past two decades suggest that, during strenuous muscle activity, in some pathological conditions, or in aging, the generation of ROS in the skeletal muscle cells may be elevated to a level that overwhelms the antioxidant defense systems (Ji, 1995, 2001) and can contribute to the development of muscle fatigue, inflammation, and degeneration (Meydani et al.,, 1992) leading to many muscle diseases. When L6C5 rat myoblasts were exposed to moderate or high intensity H2O2-induced oxidative stress (50–300 µM for 1–6 h), cell death induction became evident and the activation of the apoptotic pathway could be evaluated very early (2 h) after treatment. H2O2 cytotoxicity in rat myoblasts seemed correlated to the induction of apoptosis, since cell loss evaluated by MTS assay was related to the percentage of Hoechst positive nuclei and to the degree of caspase-3 activation (Caporossi et al., 2003). The apoptosis of myoblasts is a physiological process during myogenesis and regeneration (Miller and Stockdale, 1986), but inappropriate myoblasts apoptosis may contribute to the pathological degeneration seen in various muscular dystrophies and spinal muscular atrophies (reviewed in Adams et al., 2001). Thus, it seems very important to define the factors that in skeletal muscle cells could determine a protection from oxidative damage (Caporossi et al., 2003).
Estrogen has been shown to protect skeletal muscle from damage and to exert antioxidant activities (Persky et al., 2000). A decrease in E2 level can increase free radicals, thereby potentially causing adverse effect in a variety of tissues in post-menopausal women (Persky et al., 2000). Similar to the negative effects that the estrogen deprivation has on brain, bone and cardiovascular system, estrogen deprivation could increase vulnerability of
91
skeletal muscle to damage. The increased vulnerability may lead potentially to muscle wasting and decreased strength and can partially account for the increased incidence of fall in elderly woman and generally decline in the quality life (Persky et al., 1999).
Although mounting evidence suggests antioxidant effects contribute to the protective effects of estrogens, at present the mechanism underlying are still not clarified. Our data, obtained by using ER selective agonists, demonstrate for the first time, that the ERβ activities are necessary and sufficient for preventing the H2O2-induced ROS production in skeletal muscle cells. In fact, both in the cells stimulated with the selective ERα agonist (PPT) and ERβ selective antagonist (THC) H2O2-induced ROS production was not impaired. Whereas, ERβ selective agonist (DPN) completely mimicked the E2 effect on ROS production. Therefore, beside its role as negative regulator of ERα activities, the involvement of ERβ specific activities in the regulation of important cellular function is here reported, even in a cellular context where both ERs are expressed.
All together these data (summarized in Fig. 5.20) indicate that E2, like other extra-cellular growth factors, modulates specific cell signals affecting the skeletal muscle development. Moreover, they provide the basis of gender-related physiological differences in skeletal muscle recovery after damage and define new molecular targets for the therapeutic treatment of skeletal muscle degenerative diseases. Figure 5.20: Schematic representation of E2 action on skeletal muscle cells.
92
6. CONCLUSION
At the present, rapid activities of ERs as well the recognition of ER signaling complexes outside the nucleus in the estrogen target tissue are widely accepted, but a full understanding of the nature of the mechanisms underlying these actions and their physiological relevance are still far from being accomplished. The rapidity by which E2 induces rapid signals raise the requirement of a plasma membrane ER, although the debate continues over whether structural changes target nuclear ERs in separate pools localizing them to the membrane (Simoncini et al., 2000; Chambliss and Shaul, 2002; Acconcia and Kumar, 2005; Marino et al., 2005; Kampa and Castanas, 2006), or whether membrane ER represent a novel receptor. Some cell types that do not express either ERα or ERβ could still exhibit rapid responses to E2, but at present a membrane receptor for estrogen unrelated to ER has neither been isolated nor characterized. However, a novel ER, ER-X, functionally distinct from ERα and ERβ, has been reported in caveolar-like microdomains (Toran-Allerand et al., 2002). Furthermore, a membrane progesterone receptor (Falkenstein et al., 1999), γ-adrenergic receptor (Ropero et al., 2002) and a membrane glucocorticoid-binding protein homologous to k-opioid receptors (Evans et al., 2000) have been suggested as membrane estrogen-binding proteins. In addition, the ability of E2 to activate G-proteins through an orphan G-protein-coupled receptor-30 (GPR30) has been reported (Ahola et al., 2002; Filardo et al., 2002; Vivacqua et al., 2006). GPR30 shows a low E2 binding capacity and supports a modest generation of cAMP (Thomas et al., 2005), but if this heptahelical G-protein coupled receptors contributes to the overall signaling by these hormones at the plasma membrane is unclear.
Besides these data, much evidence favors the idea that the membrane-localized ER is the same protein as the nuclear-localized receptor. This idea is based on the immuno-histochemistry of the endogenous membrane ER which uses a panel of antibodies directed against multiple epitopes of nuclear ER (Pappas et al., 1995), loss of endogenous ER protein detection at the membrane in cells transfected with an anti-sense oligonucleotide to nuclear ERα (Norfleet et al., 1999), and the co-detection of membrane and nuclear ER after nuclear ER cDNA expression in ER null cells (Razandi et al., 1999; Marino et al., 2002, 2003). More recent data indicate that cells from the DERKO mice fail to show endogenous membrane or nuclear ERα or ERβ, by Western blot, E2 binding, and rapid signaling (Levin, 2005). Current evidence indicates that a small population of ERα and ERβ localized in the plasma membrane exist within caveolar rafts. It is at the plasma membrane that E2-liganded ER associates with the scaffolding
93
protein caveolin-1 leading to the activation of rapid membrane started signals (Acconcia et al., 2005b; Levin, 2005; Galluzzo et al., 2007). Even if the mechanisms of translocation of ER to the plasma membrane are still under investigation, recently, a highly conserved nine amino acid motif in the ligand binding domains of ER was identified which is critically relevant to ERs palmitoylation and membrane localization (Acconcia et al., 2005b; Marino and Ascenzi, 2006; Pedram et al., 2007).
The recent finding that ER also resides at plasma membrane have opened a new spectrum on E2 rapid signaling and raised several new concerns in the field of estrogen biology, which has focused its attention on unveiling many unknown mechanistic actions of estrogen in cellular physiology. Although the characterization of the nature of ER membrane and the rapid signals starting from there have been approached, further efforts are still needed to finally identify these proteins and the mechanism underlying the membrane started signals.
However, the debate has been now partially redirected away from the ER membrane nature and membrane localization and towards the physiological significance of the rapid estrogen effects. The main difficulties are linked to the experimental models used. In fact, most of the studies of signaling pathways can be done in vitro with cell culture systems, and it is very complicated to obtain similar information on a whole organism in which the use of signaling inhibitors could have many side effects other than to inhibit just one kinase. However, as previously reported, the physiological significance of rapid membrane-starting pathways has been clarified at least for some E2 targets such as the nervous (Farach-Carson and Davis, 2003), skeleton (Kousteni et al., 2003) and vascular system (Simoncini et al., 2000, 2002). Although these studies have been done mainly in cell-culture systems, their results suggest that ER rapid signaling actions have also a role in vivo. However, much remains to be done for a full understanding of the contribute of rapid activities of both ER isoforms to the final E2 effect.
The overall aim of this thesis was to investigate the presence and the impact of membrane started signals mediated by ERα and ERβ on the E2-dependent cell functions. All together present data demonstrate that one important characteristic of both ERα and ERβ is their capability to activate specific membrane-started pathways that are involved in the final impact of E2 on cell functions. In particular, ERα:E2 complex is able to activate signal cascades addressing to the cell proliferation (Galluzzo et al., 2008), while ERβ:E2 complex induces specific rapid signals started from plasma membrane which trigger a pro-apoptotic cascade (Galluzzo et al., 2007). However, the allosteric properties of ER and the individuation of specific
94
ligand that may selectively modulate nuclear and membrane rapid signals has created exciting opportunities for further study the physiological involvement of ER rapid actions. Therefore, this precious tool allowed us to demonstrate that impairing the ERα membrane localization and the protein-protein interaction important for E2-dependent functions, ER rapid activities can be modulated and are responsible for a completely different final cell fate (Galluzzo et al., 2008; Galluzzo et al., 2009b).
However, ERα and ERβ have some overlapping tissue distribution and display high relative tissue-specific expression and numerous clinical. Furthermore, in vitro studies suggest that the ratio ERα/ERβ expression is a critical point in several E2-dependent effects. On the other hand, present data demonstrate that the complessive effect of E2 could be different from cell proliferation or apoptosis when both ERα and ERβ are expressed in the same cellular context. E2 effects depend not only on the relative expression of ER isoform, but also on the balance between the signals originated by each isoform (Galluzzo et al., 2009a).
Overall, reported data argue that both ERα and ERβ activities are involved in mediating the estrogen protective effect depending on cellular context and that a dominant protective ER player in the intricate interplay among ERα and ERβ dependent signaling does not exist. Furthermore, the complexity of the mechanism of ER action suggests a more finely tuned control exerted by E2-induced rapid signals on cellular molecular events. In particular, the extra-nuclear signals induced by E2 occur before the appearance of nuclear effects and the cell context in which the genomic events occur will be different depending on which signal pathway is activated. Thus, the integration between these molecular events is required to obtain the complete cellular response.
95
7. REFERENCES Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A,
Visca P, Marino M. (2005b) Palmitoylation-dependent estrogen receptor α membrane localization: regulation by 17β-estradiol. Mol Biol Cell 16: 231-237.
Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. (2004) S-Palmitoylation modulates human estrogen receptor-α functions. Biochem Biophys Res Commun 316: 878-883.
Acconcia F, Bocedi A, Ascenzi P, Marino M. (2003) Does palmitoylation target estrogen receptors to plasma membrane caveolae? IUBMB Life 55: 33-35.
Acconcia F, Kumar R. (2005) Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 238: 1-14.
Acconcia F, Totta P, Ogawa S, Cardillo I, Inoue S, Leone S, Trentalance A, Muramatsu M, Marino M. (2005a) Survival versus apoptotic 17β-estradiol effect: role of ERα and ERβ activated non-genomic signaling. J Cell Physiol 203: 193-201.
Acevedo ML, Kraus WL. (2004) Transcriptional activation by nuclear receptors. In: McEwan IJ, editor. Essays in biochemistry: the nuclear receptor superfamily. Oxford: Portland Press Ltd. p. 73-88.
Adams V, Gielen S, Hambrecht R, Schuler G. (2001) Apoptosis in skeletal muscle. Front Biosci 6: d1–d11.
Ahola TM., Manninen T, Alkio N, Ylikomi T. (2002) G proteincoupled receptor 30 is critical for a progestin-induced growth inhibition in MCF-7 breast cancer cells. Endocrinology 143: 3376-3384.
Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, Beato M, Sica V, Bresciani F, Weisz A. (1996) 17β-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene 12: 2315-2324.
Amic´ D, Davidovic´-Amic´ D, Beslo D, Rastija V, Lucic´ B, Trinajstic´ N. (2007) SAR and QSAR of the antioxidant activity of flavonoids. Curr Med Chem 14: 827-845
Ansonoff MA, Etgen AM. (1998) Estradiol elevates protein kinase C catalytic activity in the preoptic area of female rats. Endocrinology 139: 3050-3056.
Ascenzi P, Bocedi A, Marino M. (2006) Structure-function relationship of estrogen receptor α and β: impact on human health. Mol Aspects Med 27: 299-402.
Bandera CA, Takahashi H, Behbakht K, Liu PC, LiVolsi VA, Benjamin I,
96
Morgan MA, King SA, Rubin SC, Boyd J. (1997) Deletion mapping of two potential chromosome 14 tumor suppressor gene loci in ovarian carcinoma. Cancer Res 57: 513-515.
Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. (2004) Loss of ERβ expression as a common step in estrogen dependent tumor progression. Endocr-Relat Cancer 11: 537-551.
Barletta F, Wong CW, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. (2004) Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Molecular endocrinology 18: 1096-1108.
Barrett-Connor E. (1997) Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation 95: 252-264.
Barros RP, Machado UF, Warner M, Gustafsson J-Å. (2006) Muscle GLUT4 regulation by estrogen receptors ERβ and ERα. Proc Natl Acad Sci USA 103: 1605-1608
Bennett AM, Tonks NK. (1997) Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278: 1288-1291
Berkes CA, Tapscott SJ. (2005) MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16: 585-95.
Bhat HK, Calaf G, Hei TK, Loya T, Vadgama JV. (2003) Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc Natl Acad Sci USA 100: 3913-3918.
Bijlmakers MJ, Marsh M. (2003) The on-off story of protein palmitoylation. Trends Cell Biol 13: 32-42.
Birt DF, Hendrich S, Wang W. (2001) Dietary agents in cancer prevention: flavonoids and isoflavonoids. Pharmacol Therap 90: 157-177.
Björnström L, Sjöberg M. (2005) Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endo 19: 833-842.
Brandenberger AW, Tee MK, Lee JY, Chao V, Jaffe RB. (1997) Tissue distribution of estrogen receptors alpha (ER-α) and beta (ER-β) mRNA in the midgestational human fetus. J Clin Endocrinol Metab 82: 3509-3512.
Brzezinski A, Debi A. (1999) Phytoestrogens: the ‘‘natural’’ selective estrogen receptor modulators? Eur J Obstet Gynecol Reprod Biol 85: 47-51.
Bulzomi P, Bolli A, Galluzzo P, Leone S, Acconcia F, Marino M. (2009) Naringenin and 17β-estradiol coadministration prevents hormone-induced human cancer cell growth IUBMB Life in press
97
Bunone G, Briand PA, Miksicek RJ, Picard D. (1996) Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 15: 2174-2183
Caiazza F, Galluzzo P, Lorenzetti S, Marino M. (2007) 17β-estradiol induces ERβ up-regulation via p38/MAPK activation in colon cancer cells. Biochem Biophys Res Commun 359: 102-107.
Campbell-Thompson M, Lynch IJ, Bhardwaj B. (2001) Expression of estrogen receptor (ER) subtypes and ERβ isoforms in colon cancer. Cancer Res 61: 632-640.
Caporossi D, Ciafrè SA, Pittaluga M, Savini I, Farace MG. (2003) Cellular responses to H2O2 and bleomycin-induced oxidative stress in L6C5 rat myoblasts. Free Radic Biol Med 35: 1355-64.
Castoria G, Barone MV, Di Domenico M, Bilancio A, Ametrano D, Migliaccio A, Auricchio F. (1999) Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J 18: 2500-2510.
Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. (2001) PI3-kinase in concert with Src promotes the S phase entry of oestradiol-stimulated MCF-7 cells. EMBO J 20: 6050-6059.
Cenni B, Picard D. (1999) Ligand-independent activation of steroid receptors: new roles for old players. Trends Endocrinol Metab 10: 41-46.
Chambliss KL, Shaul PW. (2002). Rapid activation of endothelial NO synthase by estrogen: evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids 67: 413-419.
Chambliss KL, Simon L, Yuhanna I., Mineo C, Shaul PW. (2005) Dissecting the basis of nongenomic activation of eNOS by estradiol: role of ERα domains with known nuclear functions. Mol Endocrinol 19: 277-289.
Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, Mendelsohn ME, Anderson RG, Shaul PW (2000) Estrogen receptor α and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res 87: E44-E52.
Chargé SBP, Rudnicki MA. (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84: 209-238.
Chen GG, Zeng Q, Tse GM. (2008) Estrogen and its receptors in cancer. Med Res Rev. 28: 954-974.
Chen ZJ, Yu L, Chang CH. (1998) Stimulation of membrane-bound
98
guanylate cyclase activity by 17β-estradiol. Biochem Biophys Res Commun 252: 639-642.
Cheskis BJ. (2004) Regulation of cell signalling cascades by steroid hormones. J Cell Biochem 93: 20-27.
Chiang C-H, Cheng KW, Igarashi S, Nathwani PS, Leung PCK. (2000) Hormonal regulation of estrogen receptor α and β gene expression in human granulosa-luteal cells in vitro. The Journal of Clinical Endocrinology and Metabolism 85: 3828-3839.
Cho NL, Javid SH, Carothers AM, Redston M, Bertagnolli MM. (2007) Estrogen receptors α and β are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+mice. Cancer Res 67: 2366-2372.
Ciana P, Raviscioni M, Mussi P, Vegeto E, Que I, Parker MG, Lowik C, Maggi A. (2003) In vivo imaging of transcriptionally active estrogen receptors. Nat Med 9: 82-86.
Claessens F, Gewirth DT. (2004) DNA recognition by nuclear receptors. In: McEwan, IJ. (Ed.), Essay in Biochemistry: The Nuclear Receptor Superfamily. Portland Press, London, pp. 59-72.
Couse JF, Korach KS. (1999) Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 20: 358-417.
Cowley SM, Parker MG. (1999) A comparison of transcriptional activation by ERα and ERβ. J Steroid Biochem Mol Biol 69: 165-175.
Dahlberg E. (1982) Characterization of the cytosolic estrogen receptor in rat skeletal muscle. Biochim Biophys Acta 717: 65-75.
Dan P, Cheung JC, Scriven DR, Moore ED. (2003) Epitope dependent localization of estrogen receptor-α, but not -β, in en face arterial endothelium. Am J Physiol 284: H1295-H1306.
Davidge ST, Zhang Y. (1998) Estrogen replacement suppresses a prostaglandin H synthase-dependent vasoconstrictor in rat mesenteric arteries. Circ Res 83: 388-395.
DeCosse JJ, Ngoi SS, Jacobson JS, Cennerazzo WJ (1993) Gender and colorectal cancer. Eur J Cancer Prev 2: 105-115.
Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. (2006) 116: 561-570.
Di Croce L, Bruscalupi G, Trentalance A. (1996) Independent behavior of rat liver LDL receptor and HMGCoA reductase under estrogen treatment. Biochem Biophys Res Commun 224: 345-350.
Dionne IJ, Kinaman KA, Poehlman ET. (2000) Sarcopenia and muscle
99
function during menopause and hormone-replacement therapy. J Nutr Health Aging 4: 156-161.
Dos Santos EG, Dieudonne MN, Pecquery R, Le Moal V, Giudicelli Y, Lacasa D. (2002) Rapid nongenomic E2 effects on p42/p44 MAPK, activator protein-1, and cAMP response element binding protein in rat white adipocytes. Endocrinology 143: 930-940.
Driggers PH, Segars JH. (2002) Estrogen action and cytoplasmic signaling pathways: Part II. The role of growth factors and phosphory lation in estrogen signaling. Trends Endocrinol Metab 13: 422-427.
Du J, Cai SH, Shi Z, Nagase F. (2004) Binding activity of H-Ras is necessary for in vivo inhibition of ASK1 activity. Cell Res 14: 148-154.
Endoh H, Sasaki H, Maruyama K, Takeyama K, Waga I, Shimizu T, Kato S, Kawashima H. (1997) Rapid activation of MAP kinase by estrogen in the bone cell line. Biochem Biophys Res Commun 235: 99-102.
Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjold M, Gustafsson J-Å. (1997) Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab 82: 4258-4265.
Ettinger B, Friedman GD, Bush T, Quesenberry Jr CP. (1996) Reduced mortality associated with long-term postmenopausal estrogen therapy. Obstet Gynecol 87: 6-12.
Ewies AA. (2002) Phytoestrogens in the management of the menopause: up-to-date. Obstet Gynecol Surv 57: 306-313.
Falkenstein E, Heck M, Gerdes D, Grube D, Christ M, Weigel M, Buddhikot M, Meizel S, Wehling M. (1999) Specific progesterone binding to a membrane protein and related nongenomic effects on Ca2+-fluxes in sperm. Endocrinology 140: 5999-6002.
Farach-Carson MC, Davis PJ. (2003) Steroid hormone interactions with target cells: cross talk between membrane and nuclear pathways. J Pharmacol Exper Therap 30: 839-845.
Farhat MY, Abi-Younes S, Dingaan B, Vargas R, Ramwell PW. (1996) Estradiol increases cyclic adenosine monophosphate in rat pulmonary vascular smooth muscle cells by a nongenomic mechanism. J Pharmacol Exp Ther 276: 652-657.
Farish E, Spowart K, Barnes JF, Fletcher CD, Calder A, Brown A, Hart DM. (1996) Effects of postmenopausal hormone replacement therapy on lipoproteins including lipoprotein(a) and LDL subfractions. Atherosclerosis 126: 77-84.
Fernando RI, Wimalasena J. (2004) Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras dependent nongenomic pathways requiring signaling through ERK and Akt. Mol Biol Cell 15:
100
3266-3284. Filardo EJ, Quinn JA, Frackelton AR. Jr, Bland KI. (2002) Estrogen action
via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16: 70-84.
Fisher JS, Hasser EM, Brown M. (1998) Effects of ovariectomy and hindlimb unloading on skeletal muscle. J Appl Physiol 85: 1316-1321.
Foley EF, Jazaeri AA, Shupnik MA, Jazaeri O, Rice LW. (2000) Selective loss of estrogen receptor β in malignant human colon. Cancer Res 60: 245-248.
Foster JS, Wimalasena J. (1996) Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol Endocrinol 10: 488-498.
Frydoonfar HR, McGrath DR, Spigelman AD. (2003) The variable effect on proliferation of a colon cancer cell line by the citrus fruit flavonoid Naringenin. Colorectal Dis. 5: 149-152.
Fulco CS, Rock PB, Muza SR, Lammi E, Cymerman A, Butterfield G, Moore LG, Braun B, Lewis SF. (1999) Slower fatigue and faster recovery of the adductor pollicis muscle in women matched for strength with men. Acta Physiol Scand 167: 233-239.
Galluzzo P, Ascenzi P, Bulzomi P, Marino M. (2008) The nutritional flavanone naringenin triggers antiestrogenic effect by regulating estrogen receptor α palmitoylation. Endocrinology 149: 2567-2575
Galluzzo P, Caiazza F, Moreno S, Marino M. (2007) Role of ERβ pamitoylation in the inhibition of human colon cancer cell proliferation Endocr Relat Cancer 14: 153-167.
Galluzzo P, Marino M. (2006) Nutritional flavonoid impact on nuclear and extranuclear estrogen receptor activities. Genes Nutr 1: 161-176.
Galluzzo P, Martini C, Bulzomi P, Leone S, Bolli A, Pallottini V, Marino M (2009b) Quercetin induced apoptotic cascade in cancer cells: antioxidant versus estrogen receptor α-dependent mechanisms. Mol Nutr Food Res 53: 699-708.
Galluzzo P, Rastelli C, Bulzomi P, Acconcia F, Pallottini V, Marino M. (2009a) 17β-estradiol regulates the first steps of skeletal muscle cell differentiation via ERα-mediated signals. Am J Physiol Cell Physiol in press
Garcia-Segura LM, Azcoitia I, DonCarlos LL. (2001) Neuroprotection by estradiol. Prog Neurobiol 63: 29-60.
Geraldes P, Sirois MG, Tanguay JF. (2003) Specific contribution of estrogen receptors on mitogen-activated protein kinase pathways and vascular cell activation. Circ Res 93: 399-405.
Gilligan DM, Quyyumi AA, Cannon RO. (1994a) Effects of physiological levels of estrogen on coronary vasomotor function in postmenopausal women. Circulation. 89: 2545-2551.
Glenmark B, Nilsson M, Gao H, Gustafsson J-Å, Dahlman-Wright K, Westerblad H. (2004) Difference in skeletal muscle function in males vs. females: role of estrogen receptor-β. Am J Physiol Endocrinol Metab 287: E1125-E1131.
Gosden JR, Middleton PG, Rout D. (1986) Localization of the human oestrogen receptor gene to chromosome 6q24–q27 by in situ hybridization. Cytogenet Cell Genet 43: 218-220.
Greco B, Allegretto EA, Tetel MJ, Blaustein JD. (2001) Coexpression of ERβ with ERα and progestin receptor proteins in the female rat forebrain: Effects of estradiol treatment. Endocrinology 142: 5172-5181.
Gredinger E, Gerber AN, Tamir Y, Tapscott SJ, Bengal E. (1998) Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J Biol Chem 273: 10436-10444.
Greger JG, Guo Y, Henderson R, Ross JF, Cheskis BJ. (2006) Characterization of MNAR expression. Steroids 71: 317-322.
Gu Q, Moss RL. (1996) 17β-estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci 16: 3620-3629.
Guo JY, Li X, Browning JD Jr, Rottinghaus GE, Lubahn DB, Constantinou A, Bennink M, MacDonald RS. (2004) Dietary soy isoflavones and estrone protect ovariectomized ERαKO and wild-type mice from carcinogen-induced colon cancer. J Nutr 134: 179-182.
Gustafsson J-Å. (2003) What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol Sci 24: 479-485.
Harmon AW, Patel YM. (2004) Naringenin inhibits glucose uptake in MCF-7 breast cancer cells: a mechanism for impaired cellular proliferation. Breast Cancer Res Treat 85: 103-110.
Harridge SD. (2007) Plasticity of human skeletal muscle: gene expression to in vivo function. Exp Physiol 92: 783-797.
Heikkinen J, Kyllönen E, Kurttila-Matero E, Wilén-Rosenqvist G, Lankinen KS, Rita H, Väänänen HK. (1997) HRT and exercise: effects on bone density, muscle strength and lipid metabolism. A placebo controlled 2-year prospective trial on two estrogen–progestin regimens in healthy postmenopausal women. Maturitas 26: 139-149.
102
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson J-Å. (2007) Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 87: 905-931.
Henderson BE, Feigelson HS. (2000) Hormonal carcinogenesis. Carcinogenesis 21: 427-433.
Henderson VW, Paganini-Hill A, Emanuel CK, Dunn ME, Buckwalter JG. (1994) Estrogen replacement therapy in older women: comparisons between Alzheimer’s disease cases and nondemented control subjects. Arch Neurol 51: 896-900.
Herbert B, Truss M, Beato M, Muller R. (1994) Inducible regulatory elements in the human cyclin D1 promoter. Oncogene 9: 2105-2107.
Herrington DM, Braden GA, Williams JK, Morgan TM. (1994) Estrogen modulates coronary vasomotor responses in postmenopausal women with early atherosclerosis. Am J Cardiol 73: 951-952.
Herynk MH, Fuqua SA. (2004) Estrogen receptor mutations in human disease. Endocr Rev 25: 869-898.
Hiroi H, Inoue S, Watanabe T, Goto W, Orimo A, Momoeda M, Tsutsumi O, Taketani Y, Muramatsu M. (1999) Differential immunolocalization of estrogen receptor α and β in rat ovary and uterus. J Mol Endocrinol 22: 37-44.
Horvath LG, Henshall SM, Lee CS, Head DR, Quinn DI, Makela S, Delprado W, Golovsky D, Brenner PC, O'Neill G, Kooner R, Stricker PD, Grygiel JJ, Gustafsson J-Å, Sutherland RL. (2001) Frequent loss of estrogen receptor-β expression in prostate cancer. Cancer Res 61: 5331-5335.
Ikeda K, Inoue S. (2004) Estrogen receptors and their downstream targets in cancer. Arch Histol Cytol 67: 435-442.
Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. (1999) Estrogen-induced activation of mitogenactivated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci USA 96: 4686-4691.
Incerpi S, D’Arezzo S, Marino M, Musanti R, Pallottini V, Pascolini A, Trentalance A. (2003) Short-term activation by low 17β-estradiol concentrations of the Na+/H+ exchanger in rat aortic smooth muscle cells: physiopathological implications. Endocrinology 144: 4315-4324.
Ing NH. (2005) Steroid hormones regulate gene expression posttranscriptionally by altering the stabilities of messenger RNAs. Biol Reprod 72: 1290-1296.
Iwao K, Miyoshi Y, Egawa C, Ikeda N, Noguchi S. (2000) Quantitative analysis of estrogen receptor-β mRNA and its variants in human breast
103
cancers. Int J Cancer 88: 733-736. Jacobsen DE, Samson MM, Kezic S, Verhaar HJ. (2007) Postmenopausal
HRT and tibolone in relation to muscle strength and body composition. Maturitas 58: 7-18.
Jacquemin V, Furling D, Bigot A, Butler-Browne GS, Mouly V. (2004) IGF-1 induces human myotube hypertrophy by increasing cell recruitment. Exp Cell Res 299: 48-58.
Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. (2004) American Cancer Society. Cancer statistics, 2004. CA Cancer J Clin 54: 8-29.
Jessop HL, Sjoberg M, Cheng MZ, Zaman G, Wheeler-Jones CP, Lanyon LE. (2001) Mechanical strain and estrogen activate estrogen receptor α in bone cells. J Bone Miner Res 16: 1045-1055.
Ji LL. (1995) Oxidative stress during exercise: implication of antioxidants nutrients. Free Radic Biol Med 18: 1079-1086.
Ji LL. (2001) Exercise at old age: does it increase or alleviate oxidative stress? Ann. NY Acad. Sci. 928: 236-247.
Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NC, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. (2000) Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA 97: 12735-12740.
Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. (2000) Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J Biol Chem 275: 18447-18453.
Kampa M, Castanas E. (2006) Membrane steroid receptor signaling in normal and neoplastic cells. Mol Cell Endocrinol 246: 76-82.
Kanashiro CA, Khalil RA. (2001) Gender-related distinctions in protein kinases C activity in rat vascular smooth muscle. Am J Physiol Cell Physiol 280: C34-C35.
Kasahara K, Taguchi T, Yamasaki I, Kamada M, Yuri K, Shuin T. (2002) Detection of genetic alterations in advanced prostate cancer by comparative genomic hybridization. Cancer Genet Cytogenet 137: 59-63
Kawaii S, Tomono Y, Katase E, Ogawa K, Yano M. (1999) Antiproliferative effects of the readily extractable fractions prepared from various citrus juices on several cancer cell lines. J Agric Food Chem 47: 2509-2512.
Kennedy AM, Shogren KL, Zhang M, Turner RT, Spelsberg TC, Maran A. (2005) 17β-estradiol-dependent activation of signal transducer and activator of transcription-1 in human fetal osteoblasts is dependent on Src kinase activity. Endocrinology 146: 201-207.
Keren A, Tamir Y, Bengal E. (2006) The p38 MAPK signaling pathway: a
104
major regulator of skeletal muscle development. Mol Cell Endocrinol 252: 224-230.
Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 21: 893-901.
Kim JK, Levin ER. (2006) Estrogen signaling in the cardiovascular system. Nucl Recept Signal 4: e013.
Kim KH, Bender JR. (2005) Rapid, estrogen receptor-mediated signaling: why is the endothelium so special? Sci STKE 14, pe28.
Klein-Hitpass L, Schorpp M, Wagner U, Ryffel GU. (1986) An estrogen-responsive element derived from the 50 flanking region of the Xenopus vitellogenin A2 gene functions in transfected human cells. Cell 46: 1053-1061.
Klinge CM, Blankenship KA, Risinger KE, Bhatnagar S, Noisin EL, Sumanasekera WK, Zhao L, Brey DM, Keynton RS. (2005) Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors α and β in endothelial cells. J Biol Chem 280: 7460-7468.
Klinge CM. (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 29: 2905-2919.
Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson J-Å. (2005) Reflections on the discovery and significance of estrogen receptor β. Endocr Rev 26: 465-478.
Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, ricopoulos P, Varakis I Sotiropoulou-Bonikou G, Papavassiliou AG. (2003) Oestrogen receptor β (ERβ) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour's dedifferentiation. Eur J Cancer 39: 1251-1258.
Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, Han K, Di Gregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. (2001) Nongenotropic, sex-nonspecific signaling through the estrogen or androgenreceptors: dissociation from transcriptional activity. Cell 104: 719-730.
Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, Plotkin L, Fu Q., Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. (2002) Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 298, 843-846. Erratum in: (2003) Science 299: 1184.
Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. (2003) Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest
105
111: 1651-1664. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT,
van der Burg B, Gustafsson J-Å. (1998) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 139: 4252-4263.
Kumar R, Johnson BH, Thompson EB. (2004) In: McEwan IJ. (Ed.), Essay in Biochemistry: The Nuclear Receptor Superfamily. Portland Press, London, pp. 27-39.
Kupzig S, Walker SA, Cullen PJ. (2005) The frequencies of calcium oscillations are optimized for efficient calcium-mediated activation of Ras and the ERK/MAPK cascade. Proc Natl Acad Sci USA 102: 7577-7582.
Lacey M, Bohday J, Fonseka S, Ullah A, Whitehead SA. (2005) Dose-response effects on phytoestrogens on the activity and expression of 3β-hydroxsteroid dehydrogenase and aromatase in human granulosa-luteal cells. J Steroid Biochem Mol Biol 96: 279-286.
Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, Stitt TN, Economides AN, Yancopoulos GD, Glass DJ. (2004) Conditional activation of AKT in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol 24: 9295-9304.
Lannigan DA. (2003) Estrogen receptor phosphorylation. Steroids 68: 1-9. Laughton MJ, Evans PJ, Moroney MA, Hoult JR, Halliwell B. (1991)
Inhibition of mammalian 5-lipoxygenase and cyclo-oxygenase by flavonoids and phenolic dietary additives. Relationship to antioxidant activity and to iron ionreducing ability. Biochem Pharmacol 42: 1673-1681.
Lawlor MA, Rotwein P. (2000) Coordinate control of muscle cell survival by distinct insulin-like growth factor activated signaling pathways. Mol Cell Biol 20: 8983-8995.
Leclercq G, Lacroix M, Laios I, Laurent G. (2006) Estrogen receptor α: impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr Cancer Drug Targets 6: 39-64.
Lee SY, Andoh T, Murphy DL, Chiueh CC. (2003) 17beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J 17: 947-8.
Lemoine S, Granier P, Tiffoche C, Rannou-Bekono F, Thieulant ML, Delamarche P. (2003) Estrogen receptor α mRNA in human skeletal muscles. Med Sci Sports Exerc 35: 439-443.
Leung SW, Teoh H, Keung W, Man RY. (2007) Non-genomic vascular actions of female sex hormones: physiological implications and
106
signalling pathways. Clin Exp Pharmacol Physiol 34: 822-826. Levin ER. (2005) Integration of the extra-nuclear and nuclear actions of
estrogen. Mol Endocrinol 19: 1951-1959. Linder M, Deschenes R. (2006) Protein palmitoylation. Methods 40: 125-
Am Coll Cardiol 35: 1403-1410. Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, Price RH
Jr, Pestell RG, Kushner PJ. (2002) Opposing action of estrogen receptors α and β on cyclin D1 gene expression. J Biol Chem 277: 24353-24360.
Lluìs F, Perdiguero E, Nebreda A R, Muñoz-Cànoves P. (2006) Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol 16: 36-44.
Lobenhofer EK, Huper G, Iglehart JD, Marks JR. (2000) Inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase activity in MCF-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ. Cell Growth Differ 11: 99-110.
Loveday RL, Greenman J, Simcox DL, Speirs V, Drew PJ, Monson JR, Kerin MJ. (2000) Genetic changes in breast cancer detected by comparative genomic hybridisation. Int J Cancer 15: 494-500
Loven MA, Wood JR, Nardulli AM. (2001) Interaction of estrogen receptors alpha and beta with estrogen response elements. Mol Cell Endocrinol 181: 151-163.
Luisi S, Galleri L, Marini F, Ambrosiani G, Brandi ML, Petraglia F. (2006) Estrogen receptor gene polymorphisms are associated with recurrence of endometriosis. Fertil Steril 85: 764-766.
Mabuchi S, Ohmichi M, Kimura A, Nishio Y, Arimoto-Ishida E, Yada-Hashimoto N, Tasaka K, Murata Y. (2004) Estrogen inhibits paclitaxel-induced apoptosis via the phosphorylation of apoptosis signal-regulating kinase 1 in human ovarian cancer cell lines. Endocrinology 145: 49-58.
Maki PM, Zonderman AB, Resnick SM. (2001) Enhanced verbal memory in nondemented elderly women receiving hormone-replacement therapy. Am J Psychiatry 158: 227-233.
Malyala A, Kelly MJ, Ronnekleiv OK. (2005) Estrogen modulation of hypothalamic neurons, activation of multiple signaling pathways and gene expression changes. Steroids 70: 397-406.
Manthey JA, Guthrie N. (2002) Antiproliferative activities of citrus flavonoids against six human cancer cell lines. J Agric Food Chem 50: 5837-5843.
Marino M, Acconcia F, Ascenzi P. (2005) Estrogen receptor signalling:
107
Bases for drug actions. Curr Drug Targets Immune Endocr Metabol Disord 5: 305-314.
Marino M, Acconcia F, Bresciani F, Weisz A, Trentalance A. (2002) Distinct non-genomic signal transduction pathways controlled by 17β-estradiol regulate DNA synthesis and cyclin D1 gene transcription in HepG2 cells. Mol Biol Cell 13: 3720-3729.
Marino M, Acconcia F, Trentalance A. (2003) Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol Biol Cell 14: 2583-2591.
Marino M, Ascenzi P, Acconcia F. (2006b) S-palmitoylation modulates estrogen receptor α localization and functions. Steroids 71: 298-303.
Marino M, Ascenzi P. (2006) Estrogen receptor-α: Plasma membrane localization and functions. Immunol Endoc Metab Agents in Med Chem 6: 281-289.
Marino M, Distefano E, Pallottini V, Caporali S, Bruscalupi G, Trentalance A. (2001a) Activation of IP3-protein kinase C-α signal transduction pathway precedes the changes of plasma cholesterol, hepatic lipid metabolism and induction of low-density lipoprotein receptor expression in 17-β estradiol-treated rats. Exp Physiol 86: 39-45.
Marino M, Distefano E, Pallottini V, Caporali S, Ceracchi G, Trentalance A. (2001b) β-estradiol stimulation of DNA synthesis requires different PKC isoforms in HepG2 and MCF7 cells. J Cell Physiol 188: 170-177.
Marino M, Galluzzo P, Ascenzi P. (2006a) Estrogen signaling multiple pathways to impact gene transcription. Current Genomics 7: 497-508
Marino M, Galluzzo P, Leone S, Acconcia F, Ascenzi P. (2006c) Nitric oxide impairs the 17β-estradiol-induced apoptosis in human colon adenocarcinoma cells. Endocr Relat Cancer 13: 559-569.
Marino M, Galluzzo P. (2007) The molecular basis underlying nutritional flavonoids anti-estrogenicity. In: Endocrine modulating substances Marino M and Mita DG Eds. Research Signpost, Trivandrum, Kerala, India.
Marino M, Galluzzo P. (2008) Molecular mechanism(s) at the root of the tumor soppressor role of estrogen receptor β: the case of colon cancer. Cancer Therapy 6: 149-162.
Marino M, Galluzzo P. (2009) Membrane localization of estrogen receptor Immun Endoc Metab Agents in Med Chem IEMAMC 9: In press
Marino M, Pallottini V, Trentalance A. (1998) Estrogens cause rapid activation of IP3-PKC-α signal transduction pathway in HepG2 cells. Biochem Biophys Res Commun 245: 254-258.
Matthews J, Gustafsson J-Å. (2003) Estrogen signaling: a subtle balance between ERα and ERβ. Mol Inter 3: 281-292.
108
McCormick KM, Burns KL, Piccone CM, Gosselin LE, Brazeau GA. (2004) Effects of ovariectomy and estrogen on skeletal muscle function in growing rats. J Muscle Res Cell Motil 25: 21-27.
McCullough LD, Hurn PD. (2003) Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab 14: 228-235.
McEwan IJ. (2004) Sex, drugs and gene expression: signalling by members of the nuclear receptor superfamily. In: McEwan IJ. (Ed.), Essays in Biochemistry: The Nuclear Receptor Superfamily. Portland Press, London, pp. 1-10.
McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. (1998) Transcription activation by the human estrogen receptor subtype β (ERβ) studied with ERα and ERβ receptor chimeras. Endocrinology 139: 4513-4522.
McMichael AJ, Potter JD. (1980) Reproduction, endogenous and exogenous sex hormones, colon cancer: a review and hypothesis. J Natl Cancer Inst 65: 1201-1207.
Mendelsohn ME, Karas RH. (1999) The protective effects of estrogen on the cardiovascular system. N Engl J Med. 340: 1801-1811.
Mendelsohn ME. (2000) Mechanisms of estrogen action in the cardiovascular system. J Steroid Biochem Mol Biol 74: 337-343.
Mendez P, Wandosell F, Garcia-Segura LM. (2006) Cross-talk between estrogen receptors and insulin-like growth factor-I receptor in the brain: cellular and molecular mechanisms. Front Neuroendocrinol 27: 391-403.
Meydani MW, Evans G, Handelman RA, Fielding SN, Meydani MA, Fiatarone JB, Blumberg C, Cannon JG. (1992) Antioxidant response to exercise-induced oxidative stress and protection by vitamin E. Ann NY Acad Sci 669: 363-374.
Middleton E, Kandaswami C, Theoharides TC. (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev 52: 673-751.
Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Auricchio F. (2002) Src is an initial target of sex steroid hormone action. Ann NY Acad Sci 963: 185-190.
Migliaccio S, Newbold RR, Bullock BC, Jefferson WJ, Sutton FG, McLachlan JA, Korach KS. (1996) Alterations of maternal estrogen levels during gestation affect the skeleton of female offspring. Endocrinology 137: 2118-2125.
Miller JB, Stockdale FE. (1986) Developmental regulation of the multiple myogenic cell lineages of the avian embryo. J Cell Biol 103: 219-2208.
Moon YJ, Wang X, Morris ME. (2006) Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro 20: 187-210.
109
Mor G, Munoz A, Redlinger R Jr, Silva I, Song J, Lim C, Kohen F. (2001) The role of the Fas/Fas ligand system in estrogen-induced thymic alteration. Am J Reprod Immunol 46: 298-307.
Mori-Abe A, Tsutsumi S, Takahashi K, Toya M, Yoshida M, Du B, Kawagoe J, Nakahara K, Takahashi T, Ohmichi M, Kurachi H. (2003) Estrogen and raloxifene induce apoptosis by activating p38 mitogen-activated protein kinase cascade in synthetic vascular smooth muscle cells. Journal of Endocrinology 178: 417-426.
Morley P, Whitfield JF, Vanderhyden BC, Tsang BK, Schwartz JL. (1992) A new, nongenomic estrogen action: the rapid release of intracellular calcium. Endocrinology 131: 1305-1312.
Mosselman S, Polman J, Dijkema R. (1996) ERβ: identification and characterization of a novel human estrogen receptor. FEBS Lett. 392: 49-53.
New G, Timmins KL, Duffy SJ, Tran BT, O'Brien RC, Harper RW, Meredith IT. (1997) Long-term estrogen therapy improves vascular function in male to female transsexuals. J Am Coll Cardiol 29: 1437-1444.
Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson J-Å. (2001) Mechanisms of estrogen action. Physiol Rev 81: 1535-1565.
Nishihara E, Nagayama Y, Inoue S, Hiroi H, Muramatsu M, Yamashita S, Koji T. (2000) Ontogenetic changes in the expression of estrogen receptor α and β in rat pituitary gland detected by immunohistochemistry. Endocrinology 141: 615-620.
Norfleet AM, Thomas ML, Gametchu B, Watson CS. (1999) Estrogen receptor-α detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology 140: 3805-3814.
Nuedling S, Kahlert S, Loebbert K, Meyer R, Vetter H, Grohe C. (1999) Differential effects of 17β-estradiol on mitogen-activated protein kinase pathways in rat cardiomyocytes. FEBS Lett. 454: 271-276.
O’Lone R, Frith MC, Karlsson EK, Hansen U. (2004) Genomic targets of nuclear estrogen receptors. Mol Endocrinol 18: 1859-1875.
Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. (1998a) The complete primary structure of human estrogen receptor β (hERβ) and its heterodimerization with ER α in vivo and in vitro. Biochem Biophys Res Commun 243, 122-126.
Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. (1998b) Molecular cloning and characterization of human estrogen receptor βcx: a potential inhibitor ofestrogen action in human. Nucleic
110
Acids Res 26, 3505-3512. Ortı` E, Bodwell JE, Munck A. (1992) Phosphorylation of steroid hormone
Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC (2004) Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res 64: 423-428.
Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. (2007) A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem 282: 22278-22288.
Peng B, Lu B, Leygue E, Murphy LC. (2003) Putative functional characteristics of human estrogen receptor-β isoforms. J Mol Endocrinol 30: 13-29.
Persky AM, Green PS, Stubley L, Howell CO, Zaulyanov L, Brazeau GA, Simpkins JW. (2000) Protective effect of estrogens against oxidative damage to heart and skeletal muscle in vivo and in vitro. Proc Soc Exp Biol Med 223: 59-66.
Pettersson K, Delaunay F, Gustafsson J-Å. (2000) Estrogen receptorβ acts as a dominant regulator of estrogen signaling. Oncogene 19: 4970-4978.
Pettersson K, Gustafsson J-Å. (2001) Role of estrogen receptor β in estrogen action. Annu Rev Physiol 63: 165-192.
Phillips GB, Pinkernell BH, Jing T-Y. (1997) Relationship between serum sex hormones and coronary artery disease in postmenopausal women. Arterioscler Thromb Vasc Biol 17: 695-701.
Phillips SK, Rook KM, Siddle NC, Bruce SA, Woledge RC. (1993) Muscle weakness in women occurs at an earlier age than men, but strength is preserved by hormone replacement therapy. Clin Sci 84: 95-98.
111
Piccone CM, Brazeau GA, McCormick KM. (2005) Effect of oestrogen on myofibre size and myosin expression in growing rats. Exp Physiol 90: 87-93.
Picotto G, Massheimer V, Boland R. (1996) Acute stimulation of intestinal cell calcium influx induced by 17β-estradiol via the cAMP messenger system. Mol Cell Endocrinol 119: 129-134.
Picotto G, Vazquez G, Boland R. (1999) 17β-estradiol increases intracellular Ca2+ concentration in rat enterocytes. Potential role of phospholipase C-dependent store-operated Ca2+ influx. Biochem J 339: 71-77.
Pietras RJ, Ma´rquez DC, Chen H-W, Tsai E, Weinberg O, Fishbein M. (2005) Estrogen and growth factor receptor interactions in human breast and non-small cell lung cancer cells. Steroids 70: 372-381.
Ponglikitmongkol M, White JH, Chambon P. (1990) Synergistic activation of transcription by the human estrogen receptor bound to tandem responsive elements. EMBO J 9: 2221-2231.
Potter JD. (1999) Colorectal cancer: molecules and populations. J Natl Cancer Inst 91: 916-932.
Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. (1997) Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 272: 10882-10894.
Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. (2003) Identification of a structural determinant necessary for the localization and function of estrogen receptor α at the plasma membrane. Mol Cell Biol 23: 1633-1646.
Razandi M, Pedram A, Greene GL, Levin ER. (1999) Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol 13: 307-319.
Razandi M, Pedram A, Levin ER. (2000) Estrogen signals to preservation of endothelial cell form and function. J Biol Chem 275: 38540-38546.
Resh MD. (1999) Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys ACTA 1451: 1-16.
Resnick SM, Maki PM, Golski S, Kraut MA, Zonderman AB. (1998) Effects of estrogen replacement therapy on PET cerebral blood flow and neuropsychological performance. Horm Behav 34: 171-182.
Resnick SM, Maki PM. (2001) Effects of hormone replacement therapy on cognitive and brain aging. Ann N Y Acad Sci 949: 203-214.
112
Robinson LJ, Busconi L, Michel T. (1995) Agonist modulated palmitoylation of endothelial nitric oxide synthase. J Biol Chem 270: 995-998.
Roger P, Sahla ME, Makela S, Gustafsson J-Å, Baldet P, Rochefort H. (2001) Decreased expression of receptor β protein in proliferative preinvasive mammary tumors. Cancer Res 61: 2537-2541.
Ronda AC, Buitrago C, Colicheo A, de Boland AR, Roldán E, Boland R. (2007) Activation of MAPKs by 1α,25(OH)2-Vitamin D3 and 17β-estradiol in skeletal muscle cells leads to phosphorylation of Elk-1 and CREB transcription factors. J Steroid Biochem Mol Biol 103: 462-466.
Roodman GD. (1996) Advances in bone boilogy: the osteoclast. Endocr Rev 17: 308-332.
Ropero AB, Soria B, Nadal A. (2002) A nonclassical estrogen membrane receptor triggers rapid differential actions in the endocrine pancreas. Mol Endocrinol 16: 497-505.
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Writing Group for the Women’s Health Initiative Investigators. (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results. From the Women’s Health Initiative randomized controlled trial. JAMA 288: 321-333.
Ruggiero RJ, Likis FE. (2002) Estrogen: Physiology, pharmacology, and formulations for replacement therapy. J Midwifery Women’s Health 47: 130-138.
Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR. (2000) Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA. 97: 5930-5935.
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399-412.
Sar M, Welsch F. (1999) Differential expression of estrogen receptor-β and estrogen receptor-α in the rat ovary. Endocrinol 140: 963-971.
Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson J-Å, Safe S. (2000) Ligand-, cell-, estrogen receptor subtype
Sheffield-Moore M, Urban RJ. (2004) An overview of the endocrinology of skeletal muscle. Trends Endocrinol Metab 15: 110-115.
Sherwin BB. (1997) Estrogen effects on cognition in menopausal women. Neurology 48: S21–S26.
Sherwin BB. (1998) Estrogen and cognitive functioning in women. Proc Soc Exp Biol Med 217: 17-22.
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase Nature 407: 538-41.
Simpkins JW, Singh M. (2008) More than a decade of estrogen neuroprotection. Alzheimers Dement. 4: S131-S136.
Sinoncini T, Fornari L, Mannella P, Varone G, Caruso A, Liao JK, Genazzani AR. (2002) Novel non-transcriptional mechanisms for estrogen receptor signaling in the cardiovascular system: Interaction of estrogen receptor α with phosphatidylinositol-3-OH kinase. Steroids 67: 935-939.
Sipila S, Taaffe DR, Cheng S, Puolakka J, Toivanen J. Suominen H. (2001) Effects of hormone replacement therapy and high-impact physical exercise on skeletal muscle in post-menopausal women: a randomized placebo-controlled study. Clin Sci (Lond) 101: 147-157.
Sirola J, Rikkonen T. (2005) Muscle performance after the menopause. J Br Menopause Soc. 11: 45-50.
Slattery ML, Potter JD, Curtin K, Edwards S, Ma KN, erson K, Schaffer D, Samowitz WS. (2001) Estrogens reduce and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res 61: 126-130.
Smith CL, O'Malley BW. (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25: 45-71.
Smotrys JE, Linder ME. (2004) Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev of Biochem 73: 559-587.
So FV, Guthrie N, Chambers AF, Moussa M, Carroll KK. (1996) Inhibition of human breast cancer cell proliferation and delay of mammary tumorigenesis by flavonoids and citrus juices. Nutr Cancer 26: 167-181.
Solomon AM, Bouloux PM. (2006) Modifying muscle mass - the endocrine perspective. J Endocrinol 191: 349-360.
Song RX, Fan P, Yue W, Chen Y, Santen RJ. (2006) Role of receptor complexes in the extranuclear actions of estrogen receptor α in breast cancer. Endocr Relat Cancer 13 (Suppl 1): S3-S13.
114
Song RXD, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. (2004) The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma Proc Natl Acad Sci USA. 101: 2076-2081.
Song RXD, Santen RJ. (2003) Apoptotic action of estrogen. Apoptosis 8: 55-60.
Song RXD, Zhang Z, Santen RJ. (2005) Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol Metab 16: 347-353.
Stallone JN, Crofton JT, Share L. (1991) Sexual dimorphism in vasopressinnduced contraction of rat aorta. Am J Physiol 260: H453-H458.
Stampfer MJ, Colditz GA, Willett WC Manson JE, Rosner B, Speizer FE, Hennekens CH. (1991) Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurses’ health study. N Engl J Med 325: 756-762.
Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson J-Å. (2004) Estrogen receptor β inhibits 17β-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA 101: 1566-1571.
Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. (2003) Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem 278: 33753-33762.
Thomas P, Pang Y, Filardo EJ, Dong J. (2005) Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146: 624-632.
Tiidus PM. (2005) Can oestrogen influence skeletal muscle damage, inflammation, and repair? Br J Sports Med 39: 251-253.
Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES. Jr, Nethrapalli IS, Tinnikov AA. (2002) ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci 22: 8391-8401.
Totta P, Acconcia F, Leone S, Cardillo I, Marino M (2004) Mechanisms of naringenin-induced apoptotic cascade in cancer cells: involvement of estrogen receptor α and β signalling. IUBMB Life 56: 491-499.
Tremblay A, Tremblay GB, Labrie F, Giguère V. (1999) Ligand-independent recruitment of SRC-1 to estrogen receptor α through phosphorylation of activation function AF-1. Mol Cell 3: 513-519.
Turner RT, Wakley GK, AND HAannon KS. (1990) Differential effects of
115
androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Orthop Res 8: 612–617.
Turner RT. (1999) Mice, estrogen and postmenopausal osteoporosis. J Bone Miner Res 14: 187-191.
Van der Schouw YT, Van der Graaf Y, Steyerberg EW, Eijkemans MJC, Banga JD. (1996) Age at menopause as a risk factor for cardiovascular mortality. Lancet 347: 714-718.
Vandromme M, Rochat A, Meier R, Carnac G, Besser D, Hemmings BA, Fernandez A, Lamb NJ. (2001) Protein kinase B β/Akt2 plays a specific role in muscle differentiation. J Biol Chem 276: 8173-8179.
Varner AS, Ducker CE, Xia Z, Zhuang Y, De Vos ML, Smith CD. (2003) Characterization of human palmitoylacyl transferase activity using peptides that mimic distinct palmitoylation motifs. Biochem J 373: 91-99.
Virgili F, Acconcia F, Ambra R, Rinna A, Totta P, Marino M. (2004) Nutritional flavonoids modulate estrogen receptor a signaling. IUBMB Life 56: 145-151.
Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M. (2006) The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 20: 631-646.
Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, Warner M, Gustafsson J-Å. (2006a) Role of estrogen receptor β in colonic epithelium. Proc Natl Acad Sci USA 103: 2959-2964.
Wada-Hiraike O, Warner M, Gustafsson J-Å. (2006b) New developments in oestrogen signalling in colonic epithelium. Biochem Soc Trans 34: 1114-1116.
Waliszewski P, Blaszczyk M, Wolinska-Witort E, Drews M, Snochowski M, Hurst RE. (1997) Molecular study of sex steroid receptor gene expression in human colon and in colorectal carcinomas. J Surg Oncol 64: 3-11.
Warner M, Gustafsson J-Å. (2006) Nongenomic effects of estrogen: why all the uncertainty? Steroids 71: 91-95.
Watanabe T, Akishita M, Nakaoka T, Kozaki K, Miyahara Y, He H, Ohike Y, Ogita T, Inoue S, Muramatsu M, Yamashita N, Ouchi Y. (2003) Estrogen receptor β mediates the inhibitory effect of estradiol on vascular smooth muscle cell proliferation. Cardiovasc Res 59: 734-744.
Weihua Z, Andersson S, Cheng G, Simpson ER, Warner M, Gustafsson J-Å. (2003) Update on estrogen signaling. FEBS Lett 546: 17-24.
116
Weyant MJ, Carothers AM, Mahmoud NN, Bradlow HL, Remotti H, Bilinski RT, Bertagnolli MM. (2001) Reciprocal expression of ERα and ERβ is associated with estrogen mediated modulation of intestinal tumorigenesis. Cancer Res 61: 2547-2551.
Wiik A, Glenmark B, Ekman M, Esbjörnsson-Liljedahl M, Johansson O, Bodin K, Enmark E, Jansson E. (2003) Oestrogen receptor beta is expressed in adult human skeletal muscle both at the mRNA and protein level. Acta Physiol Scand 179: 381-387.
Wiik A, Hellsten Y, Berthelson P, Lundholm L, Fischer H, Jansson E. (2009) Activation of estrogen response elements is mediated both via estrogen and muscle contractions in rat skeletal muscle myotubes. Am J Physiol Cell Physiol 296: C215-C220.
Williams JK, Wagner JD, Li Z, Golden DL, Adams MR. (1997) Tamoxifen inhibits arterial accumulation of LDL degradation products and progression of coronary artery atherosclerosis in monkeys. Arterioscler Thromb Vasc Biol 17: 403-408.
Wilson EM, Rotwein P. (2006) Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by Insulin-like Growth Factor-II. J Biol Chem 281: 29962-29971.
Wong NA, Malcomson RD, Jodrell DI, Groome NP, Harrison DJ, Saunders PT. (2005) ERβ isoform expression in colorectal carcinoma: an in vivo and in vitro study of clinicopathological and molecular correlates. J Pathol 207: 53-60.
Woo CH, Lim JH, Kim JH. (2005) VCAM-1 upregulation via PKCδ- p38 kinase-linked cascade mediates the TNF-α-induced leukocyte adhesion and emigration in the lung airway epithelium. Am J Physiol Lung Cell Mol Physiol 288: L307-L316.
Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL. (2000) p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 20: 3951-3964.
Wuttke W, Jarry H, Becker T, Schultens A, Christoffel V, Gorkow C, Seidlova´-Wuttke D. (2003) Phytoestrogens: endocrine disrupter or replacement for hormone replacement therapy? Maturitas 44: S9–S20.
Young J, Legget B, Gustafson C, Ward M, Searle J, Thomas L, Buttenshaw R, Chenevix-Trench G. (1993) Genomic instability occurs in colorectal carcinomas but not in adenomas. Hum Mutat 2: 351-354.
Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ. (2003) AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J Biol Chem 278: 23432-23440.
117
Zhang Z, Kumar R, Santen RJ, Song RXD. (2004) The role of adapter protein Shc in estrogen non-genomic action. Steroids 69: 523-529.
Zhou W, Liu Z, Wu J, Liu JH, Hyder SM, Antoniou E, Lubahn DB. (2006) Identification and characterization of two novel splicing isoforms of human estrogen-related receptor β. J Clin Endocrinol Metab 91: 569-579.
Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson J-Å, Mendelsohn ME. (2002) Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science 295: 505-508.
118
ACKNOWLEDGEMENTS The experimental work of this Ph.D. thesis was carried out in the
Laboratory of Cellular Physiology under the supervision of Prof. Maria Marino (Department of Biology of ‘RomaTre’ University).
I am very grateful to Prof. Maria Marino who has been my supervisor since the beginning of my study. She provided me with many helpful suggestions, important advice and constant encouragement during the course of this work. She gave me the opportunity to undertake my Ph.D. studies and guided me with her expertise. Her motivation, contagious enthusiasm, new exiting ideas and immense knowledge in cell physiology that, taken together, make her a great mentor, gave me the opportunity to grow as a scientist.
Special thanks to Prof. Paolo Ascenzi (Department of Biology of ‘RomaTre’ University) for sequence and kinetic analysis but also for his numerous ideas and his invaluable contributions.
Thanks to Prof. Daniela Caporossi (Department of Experimental Medicine and Biochemical Sciences, University of Rome ‘Tor Vergata’) for the gift of C2C12 cells
Thanks to Dr. Stefano Leone (Department of Biology of ‘RomaTre’ University) for cytofluorimetric analysis and to Dr. Sandra Moreno (Department of Biology of ‘RomaTre’ University) for providing information and discussion on confocal image analysis.
Thanks to Dr. Valentina Pallottini (Department of Biology of ‘RomaTre’ University) for helping discussions on ROS assay but also for her moral encouragement and support.
Many thanks to Dr. Filippo Acconcia for his fresh view on science and his critical daily suggestions.
I whould like to thank present and past members of the Lab 3.4: Dr. Chiara Martini, Dr Pamela Bulzomi, Dr. Alessandro Bolli, Dr. Chiara Rastelli, Dr. Laura Trapani, Dr. Elisabetta de Marinis, Dr. Marco Nardozzi, Valentina Bordoni, Marco Pellegrini, Marco Segatto neighboring colleagues and friends. Thanks for your helpful presence and for your daily support. I wish to thank you for for many hours of stimulating discussion and continued moral support.


































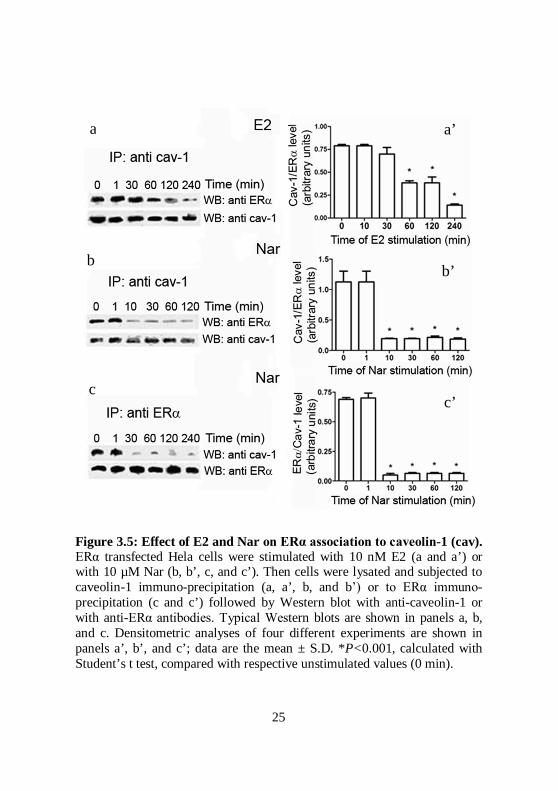























































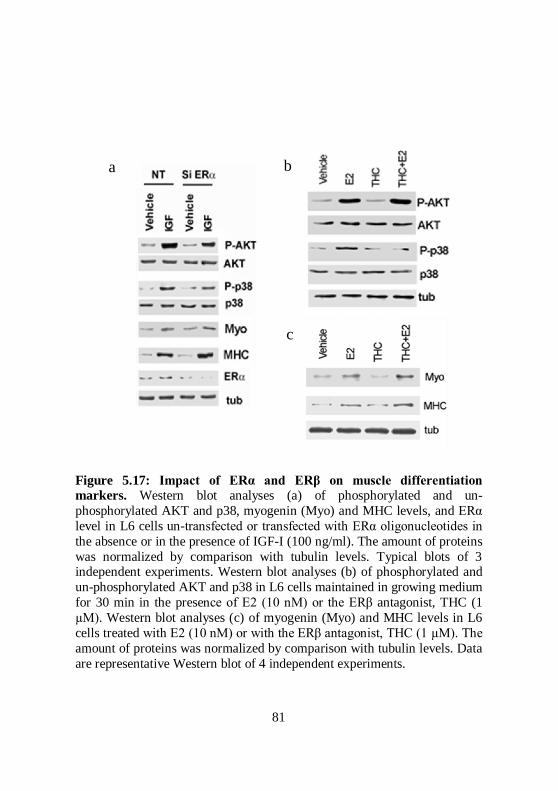








































![[ INTERVISTA ] [ SPECIALE: SEMIOTICA APPLICATA ALLA ... · [ SPECIALE: SEMIOTICA APPLICATA ALLA TELEVISIONE] STRISCIA LA NOTIZIA Il telegiornale che dà spettacolo [ INTERVISTA ]](https://static.documents.pub/doc/80x56/5c68de9209d3f2f5638c49bf/-intervista-speciale-semiotica-applicata-alla-speciale-semiotica.jpg)













