136 SHAPE-MEMORY POLYMERS Vol. 4 SINGLE-SITE CATALYSTS Introduction In 1953, Karl Ziegler discovered that the activation of certain early transition metal halides with aluminum alkyls resulted in the formation of new organometal- lic catalysts capable of polymerizing ethylene at room temperature and ambi- ent pressure (1–3). In 1954, Giulio Natta and co-workers from the Politecnico di Milano and Montecatini for the first time prepared crystalline isotactic polypropylene (iPP) by using the heterogeneous TiCl 4 /(C 2 H 5 ) 3 Al Ziegler cata- lyst system (4–8). Three years later, in 1957, the first PP plant went on line at the Montecatini site in Italy. Only six years later, in 1963, Karl Ziegler and Giulio Natta shared the Nobel Prize in chemistry for their landmark discov- eries that laid the foundation for today’s multibillion-dollar-a-year polyolefin industry (see ZIEGLER–NATTA CATALYSTS). The industrial and academic impact of Ziegler–Natta catalysis has been tremendous (9–11). Several generations of catalysts and processes have been in- troduced on a commercial scale to produce a variety of polymeric materials ranging from fibers to commodity thermoplastics, engineering plastics, and elastomers. At the turn of the millennium, over 40 million tons of polyolefins per year worldwide are produced using the Ziegler–Natta technology. Modern MgCl 2 -supported Lewis- base-modified catalyst systems produce spherical pellet-sized polyolefin replicas with catalytic activities exceeding 1 ton of polymer per gram of transition metal, thus eliminating the need for pelletizing extrusion and additional polymer purifi- cation steps. Annual demand for polyethylene (PE), the number one commodity plastic, is approaching 50 million metric tons. Polypropylene (PP) is ranked third, after poly(vinyl chloride) (PVC), and has the fastest growing market (see ETHYLENE POLYMERS;PROPYLENE POLYMERS (PP)). Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.
Transcript
136 SHAPE-MEMORY POLYMERS Vol. 4
SINGLE-SITE CATALYSTS
Introduction
In 1953, Karl Ziegler discovered that the activation of certain early transitionmetal halides with aluminum alkyls resulted in the formation of new organometal-lic catalysts capable of polymerizing ethylene at room temperature and ambi-ent pressure (1–3). In 1954, Giulio Natta and co-workers from the Politecnicodi Milano and Montecatini for the first time prepared crystalline isotacticpolypropylene (iPP) by using the heterogeneous TiCl4/(C2H5)3Al Ziegler cata-lyst system (4–8). Three years later, in 1957, the first PP plant went on lineat the Montecatini site in Italy. Only six years later, in 1963, Karl Ziegler andGiulio Natta shared the Nobel Prize in chemistry for their landmark discov-eries that laid the foundation for today’s multibillion-dollar-a-year polyolefinindustry (see ZIEGLER–NATTA CATALYSTS).
The industrial and academic impact of Ziegler–Natta catalysis has beentremendous (9–11). Several generations of catalysts and processes have been in-troduced on a commercial scale to produce a variety of polymeric materials rangingfrom fibers to commodity thermoplastics, engineering plastics, and elastomers. Atthe turn of the millennium, over 40 million tons of polyolefins per year worldwideare produced using the Ziegler–Natta technology. Modern MgCl2-supported Lewis-base-modified catalyst systems produce spherical pellet-sized polyolefin replicaswith catalytic activities exceeding 1 ton of polymer per gram of transition metal,thus eliminating the need for pelletizing extrusion and additional polymer purifi-cation steps. Annual demand for polyethylene (PE), the number one commodityplastic, is approaching 50 million metric tons. Polypropylene (PP) is ranked third,after poly(vinyl chloride) (PVC), and has the fastest growing market (see ETHYLENE
POLYMERS; PROPYLENE POLYMERS (PP)).
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.
Vol. 4 SINGLE-SITE CATALYSTS 137
Fig. 1. General structure of single-site catalysts.
Fifty years after the initial discoveries, Ziegler–Natta catalysis remains asan outstandingly vital field. The recent rapid scientific and technological outburstof single-site transition metal catalysts is undoubtedly one of the most remark-able developments in the application of molecular organometallic chemistry tocatalysis and polymer science (for reviews and general references, see Refs. 12–38). These predominantly homogeneous catalyst systems consist of well-definedactive sites with the general formula [LnMR], where Ln represents an organic lig-and set bound to the transition metal M, and R is the growing polymer chain orinitiating group (Fig. 1).
By variation of the organic ligand and thus the steric and electronic envi-ronment of the metal center, these catalysts can be tailored to control the olefinpolymerization reaction in an unprecedented fashion. Almost any vinyl monomer,irrespective of its molecular weight or steric hindrance, can be polymerized bychoosing the proper catalyst and polymerization conditions. Virtually all feasiblepolymer microstructures ranging from atactic to isotactic, hemiisotactic, syndio-tactic, and stereoblock poly-α-olefins can be produced by rational modification ofthe catalyst structure. In addition, molecular weight, molecular weight distri-bution, comonomer content, and end-group composition of the produced polymercan be varied independently. Entirely new polyolefin materials, not accessiblewith traditional Ziegler–Natta catalysts, have also emerged through single-sitecatalyst (SSC) technologies. Notable examples include syndiotactic polystyrene,thermoplastic PP-based elastomers, as well as cycloaliphatic polymers obtainedby cyclopolymerization of nonconjugated dienes or direct polymerization of cy-cloolefins, such as norbornene and cyclopentene (see ELASTOMERS THERMOPLASTIC;ETHYLENE-NORBORNENE COPOLYMERS).
The first generations of SSCs were based on Group 4 metallocene complexesas pioneered by Ewen, Kaminsky, and Brintzinger. More recent examples includealternative ligand systems complexed with Group 4 metals, as well as several latetransition-metal-based catalyst systems developed by Brookhart, Gibson, Grubbs,and others. The less oxophilic late transition metals are even capable of incorpo-rating a variety of polar comonomers into polyolefin chains.
This article provides a broad overview of the current status and the enor-mous amount of literature on single-site olefin polymerization catalysts. For the
138 SINGLE-SITE CATALYSTS Vol. 4
sake of consistency, single-site catalyst is used in this article for describing a cata-lyst system or precursor consisting of essentially uniform active centers producingnarrow molecular weight distribution polymers (Mw/Mn ≈ 2), in accordance withSchulz–Flory statistics. Since several of these catalysts have two coordinationsites located at the same metal center, the term single-center catalyst is also incommon use. The references have, with a few exceptions, been selected from theopen literature with the main emphasis on the well-established Group 4 metal-locenes and the recently developed late transition metal catalysts. For detaileddescriptions of the various catalysts and processes as well as references to the ex-tensive patent literature, the reader is referred to the original publications citedin the bibliography and the available reviews and text books (12–38).
Evolution and Classification of SSC Systems
Classical Ziegler–Natta catalysts are multisited and heterogeneous with the poly-merization taking place on edges and dislocations of transition metal halide crys-tals. Development of homogeneous single-site olefin polymerization catalysts wasinitiated by efforts to model the reaction mechanisms of the heterogeneous cat-alysts. In 1955, Breslow and Newburg (39,40) and Natta and co-workers (41)discovered that titanocene dichloride (Cp2TiCl2) could be activated for ethylenepolymerization by addition of aluminum alkyls (C2H5)2AlCl or (C2H5)3Al. Thefirst soluble catalysts were inactive for polymerization of propylene and exhib-ited much lower activities in ethylene polymerization than their heterogeneouscounterparts. Nevertheless, these and other subsequent early studies (42–49) con-tributed significantly to the understanding of both heterogeneous and homoge-neous Ziegler–Natta catalysis.
In the mid-1970s, a series of serendipitious discoveries initiated the rapiddevelopment in single-site olefin polymerization catalysis, which in less than25 years has revolutionized modern polymer synthesis. In 1973, Reichert andMeyer observed a remarkable increase in polymerization activity by adding smallamounts of water to the Cp2TiCl2/C2H5AlCl2 metallocene catalyst system (50).Similar observations were reported by Long and Breslow for Cp2TiCl2/(CH3)2AlCl(51). In 1975, Sinn and Kaminsky discovered an enormous and unexpected in-crease in polymerization activity after addition of considerable amounts of waterto the otherwise inactive Cp2Ti(CH3)2/(CH3)3Al mixture (52). The suspected for-mation of methylaluminoxane (MAO) by partial hydrolysis of (CH3)3Al was subse-quently supported by its controlled synthesis. The direct activation of titanocenesand zirconocenes with preformed MAO gave exceedingly active catalysts for poly-merization of ethylene (53). For the first time, homogeneous metallocene-basedSSCs became more active than the commercially used heterogeneous Ziegler–Natta catalyst systems. In addition, the MAO-activated Group 4 metalloceneswere capable of polymerizing propylene and higher α-olefins (54,55), althoughonly atactic polymers of prochiral monomers were obtained (because of the achi-ral nature of the catalytic site).
Group 4 Metallocene-Based Catalysts. Revolutionary breakthroughsoccurred in the early 1980s when Ewen (56) and Kaminsky (57) reportedthat MAO-activated C2 symmetric chiral ethylene-bridged bis(tetrahydroindenyl)
Vol. 4 SINGLE-SITE CATALYSTS 139
Group 4 ansa-metallocenes rac-C2H4(IndH4)2MCl2 (M = Ti, Zr), developed byBrintzinger and co-workers (58–60), polymerized propylene to iPP. The prefix ansafrom Latin (for bent, handle) was first used for compounds with an alkane bridgeacross an arene ring, and later adopted by Brintzinger as a short notation formetallocene derivatives with an interannular bridge.
Since the mid-1980s billions of dollars have been invested worldwide in re-search focusing on modifying and improving the metallocene-based olefin poly-merization catalysts. Key structures are summarized in Figure 2.
In 1988, Ewen reported the first preparation of a Cs symmetric ansa-metallocene catalyst precursor [(CH3)2C(Cp)(Flu)]ZrCl2 producing syndiotacticpolypropylene (sPP) (61), and a few years later its C1 symmetric modification
[(CH3)2C(3-CH3Cp)(Flu)]ZrCl2 for hemiisospecific propylene polymerization (62).In 1995, Coates and Waymouth described an unbridged bis(2-phenylindenyl)zirconocene producing elastomeric isotactic–atactic stereoblock PP (63), presum-ably by oscillating stereocontrol in which the catalyst switches between isospecificand aspecific conformations during the growth of a single polymer chain.
In these pseudotetrahedral bis(cyclopentadienyl) metallocenes the d0 cen-tral transition metal (Ti, Zr, or Hf) is bound to two η5-cyclopentadienyl-type ringligands and two σ -ligands (usually Cl or CH3), one or both of which are removedupon formation of the active catalyst. The most commonly employed cyclopenta-dienyl ligands are based on plain or substituted cyclopentadienyl (Cp), indenyl(Ind), tetrahydroindenyl (IndH4), and fluorenyl (Flu) anions. Because of the highstructural diversity of the available ligand structures, the steric and electronic en-vironment of the active site can be modified by variation of the ligand size, shape,and substitution pattern (for representative examples of structural variations, seeRefs. 13, 33, and 58–93). The function of the interannular ansa-bridging group isto lock the ancillary ligand in the desired conformation or symmetry, thus allow-ing steric control over the enantiodifferentiating steps in stereoselective olefinpolymerization or differentiation of olefins of different sizes in copolymerizationreactions.
The most notable examples of monocyclopentadienyl-based catalyst pre-cursors are the “constrained geometry” ansa-monoCp-amido Group 4 complexes(25,94–105) (Fig. 3) developed by Dow Chemical Co. (106) and Exxon ChemicalCo. (107) (for further references to the patent literature, see Ref. 25). The originalligand design was introduced for organoscandium complexes (108). A remarkablefeature of these catalysts is the open nature of the active site which allows theincorporation of higher α-olefins, including styrene, in copolymerization with ethy-lene. Incorporation of vinyl-terminated macromonomers results in the formationof long-chain branched ultralow-density polyethylenes possessing unique rheo-logical properties. The titanium-based constrained geometry catalysts (CGCs) aregenerally preferred over their zirconium and hafnium analogues showing higheractivities and producing polymers with higher molecular weights. Several struc-tural modifications of the CGCs including monoCp-alkoxide analogues have beendescribed (109–111).
Monocyclopentadienyl and (monoindenyl)titanium trichlorides are generat-ing interest as catalyst precursors for syndiospecific polymerization of styrene(112–127). A class of highly active olefin polymerization catalysts based on a(monocyclopentadienyl)titanium dimethyl cation [CpTi(CH3)2]+, generated by
Fig. 3. Prototypical Group 4 constrained geometry catalysts (CGC). M = Ti, Zr; R = alkyl,aryl; Cp = cyclopentadienyl, tetramethylcyclopentadienyl.
Vol. 4 SINGLE-SITE CATALYSTS 141
methyl abstraction from CpTi(CH3)3 or Cp∗Ti(CH3)3 (Cp∗ = pentamethylcy-clopentadienyl), has been described (127–132).
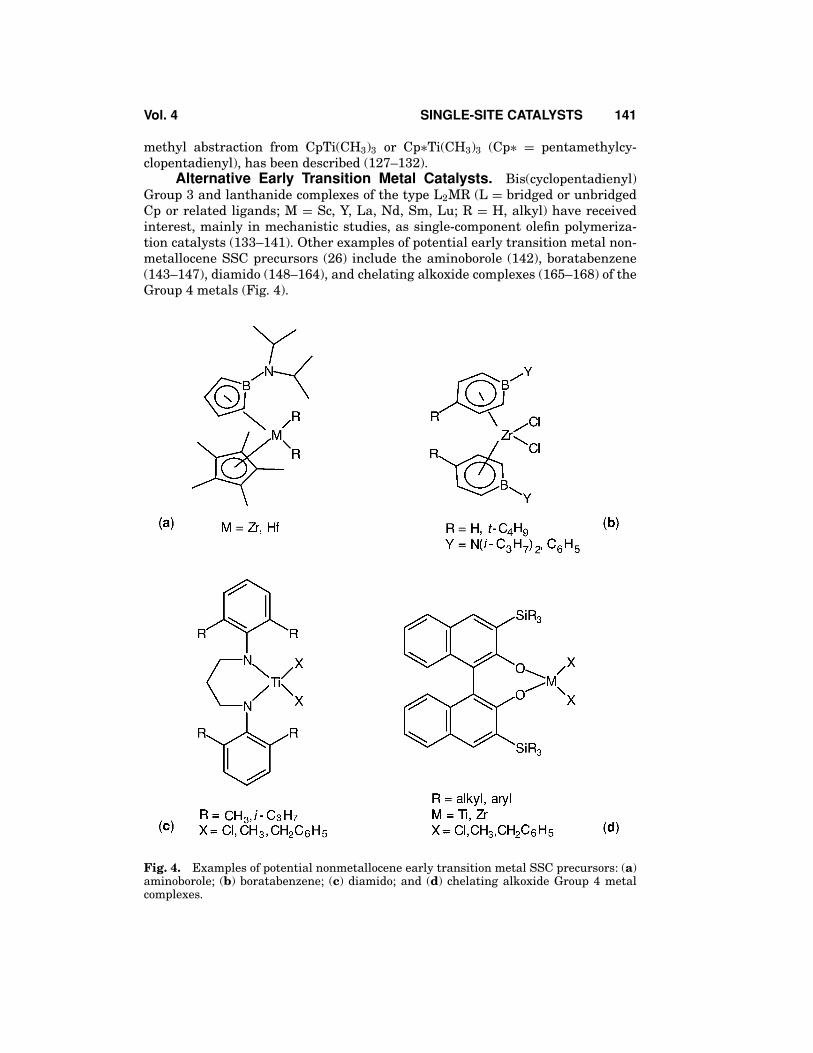
Alternative Early Transition Metal Catalysts. Bis(cyclopentadienyl)Group 3 and lanthanide complexes of the type L2MR (L = bridged or unbridgedCp or related ligands; M = Sc, Y, La, Nd, Sm, Lu; R = H, alkyl) have receivedinterest, mainly in mechanistic studies, as single-component olefin polymeriza-tion catalysts (133–141). Other examples of potential early transition metal non-metallocene SSC precursors (26) include the aminoborole (142), boratabenzene(143–147), diamido (148–164), and chelating alkoxide complexes (165–168) of theGroup 4 metals (Fig. 4).
Fig. 4. Examples of potential nonmetallocene early transition metal SSC precursors: (a)aminoborole; (b) boratabenzene; (c) diamido; and (d) chelating alkoxide Group 4 metalcomplexes.
142 SINGLE-SITE CATALYSTS Vol. 4
Group 5 metal catalysts based on various cyclopentadienyl and dianionicmixed ligand and related complexes of tantalum (169,170), niobium (171,172),and vanadium (173,174) have also been described.
Late Transition Metal Catalysts. In contrast to early transition metalcatalysts, late metal systems most often dimerize or oligomerize olefins due to com-peting β-hydride elimination (175,176). For example, soluble Ni(II)-based ethy-lene oligomerization catalysts form the basis for the Shell Higher Olefin Process(SHOP) (177,178). In 1995, a series of highly electrophilic nickel(II) and palla-dium(II) complexes were reported (179–182). These complexes incorporate bulkydiimine ligands (Fig. 5), which upon activation with MAO polymerize ethyleneand α-olefins with high activities comparable to those of metallocene catalysts. Inthese complexes, the presence of equatorial sterically demanding aryl substituentseffectively blocks the associative olefin exchange, retarding the chain transfer andresulting in high molecular weight polymers. Copolymerization of ethylene andpropylene with functionalized vinyl monomers was reported shortly after the ini-tial findings (193,194). The first reports stimulated an extensive search for otherlate transition metal olefin polymerization catalysts. Subsequent studies on theseand related nickel and palladium systems using neutral or anionic chelating N N,N O, and N P ligands have been described by several academic and industrialgroups (for reviews and references to the patent literature, see Refs. 26, 195, and196).
During 1997 and 1998, highly active iron(II)- and cobalt(II)-based ethylenepolymerization catalysts bearing 2,6-bis(imino)pyridyl ligands (Fig. 5) were de-scribed independently by Brookhart (183,184), and Gibson (187,188), as well asby DuPont (185,186). Oligomerization of ethylene to linear α-olefins (189) as well
Fig. 5. Late transition metal SSC precursors based on (a) diimine (179–182), (b) 2,6-bis(imino)pyridyl (183–190), and (c) salicylaldimine (191,192) ligands.
Vol. 4 SINGLE-SITE CATALYSTS 143
as polymerization of propylene (190), using the iron-based catalysts, was reportedin subsequent papers.
A family of neutral single-component salicylaldimine nickel(II) catalysts(Fig. 5) that produce high molecular weight PE, polymerize functionalizedolefins, and require no cocatalyst have been reported (191,192). These cata-lysts can be regarded as an extension of the original SHOP oligomerizationsystems where the P O chelate of the neutral SHOP catalyst has been re-placed with a sterically demanding N O ligand leading to high molecular weightproducts.
Catalyst Generation
Active Species and Function of the Cocatalyst. The active speciesin olefin polymerization is, in most cases, a coordinatively unsaturated cationicmetal alkyl [LnMR]+ (197). Activity and stereoregularity of the produced poly-mer are determined by the steric and electronic properties of the ancillary lig-and set Ln and the ion–ion interactions between the highly electrophilic metalcation and its counterion. Several methods exist for generating the active cat-alyst (for a review on cocatalysts for metal-catalyzed olefin polymerization, seeRef. 198). MAO is by far the most common cocatalyst (28), including large-scaleindustrial production. The exact composition, structure, and function of the alu-minoxane cocatalysts are still a matter of controversy (199–202); controlled hy-drolysis of Al(CH3)3 produces a complex mixture of cyclic and linear oligomericMAOs, possibly containing cluster-like or supramolecular aggregates with thegeneral formula (CH3AlO)n (Fig. 6). The degree of oligomerization varies from5 to 20.
In the case of Group 4 metallocene dihalide catalyst precursors L2MX2(eq. 1), addition of MAO generates first a monomethyl complex L2MXCH3(203,204). Excess MAO gives the dialkylated species L2M(CH3)2. Presumably,some of the Al centers in MAO have a high propensity to abstract a methyl an-ion from L2M(CH3)2 forming a weakly coordinating anion [CH3 MAO]− . Themetallocene cation [L2MCH3]+ is stabilized coordinatively by the [CH3 MAO]−
counterion, which in the presence of olefins gives way to olefin-separated ionpairs [L2M(olefin)CH3]+[CH3 MAO]− . Repeated olefin insertion to the metal–alkyl bond forms the polyolefin.
Fig. 6. Generalized structures of MAO: linear (a), and cyclic (b).
144 SINGLE-SITE CATALYSTS Vol. 4
(1)
A large excess of MAO is usually required to achieve high polymerizationactivities. For Group 4 metallocene/MAO catalyst systems, activity maxima aregenerally found at [Al]:[M] ratios ranging from 1000:1 to 10,000:1. The excessMAO is possibly required to shift the equilibrium involving the metal monoalkyland MAO to the cationic active species. In addition, MAO has been found to reacti-vate inactive complexes formed by hydrogen tranfer reactions and to prevent thedeactivation of the catalyst by bimolecular processes. Aluminoxanes also scavengeimpurities such as water and oxygen from the reaction medium.
Intensive work has been carried out to develop stoichiometric activators formetallocene and other SSCs (17,198). Early systems based on tetraphenylborateor other carborane anions exhibited only low polymerization activities as a re-sult of the strong interaction of the cationic metal center with the counterion(197,205–213). The introduction of perfluorinated tetraphenylborate [(C6F5)4B]−
(214,215) produced exceedingly active cationic metallocene catalysts for poly-merization of α-olefins. The application of [(C6F5)4B]− , [(C6F5)3BCH3]− , andrelated fluoroarylborate counterions in various homo- and copolymerization re-actions has been studied in detail (62,216–237). Methods for generating thecationic metal complex include mixing of metal dialkyl with dimethylanilin-ium tetrakis(perfluorophenyl)borate [NH(CH3)2C6H5]+[(C6F5)4B]− , alkyl aniontransfer with trityl tetrakis(perfluorophenyl)borate [(C6H5)3C]+[(C6F5)4B]− , andmethyl abstraction from the metal dialkyl complex with (C6F5)3B. Other base-freeor weakly stabilized metal alkyl cations, including metallocene–borate–betainecomplexes (238–242), other zwitterionic catalyst systems (230,243–247), and Al-,Nb-, and Ta-based perfluoroaryloxide anions (248) have been developed.
Polymerization Mechanism. The basic mechanistic steps of olefin co-ordination to the metal center and the subsequent insertion into the metal–carbon bond are not entirely understood. Several plausible hypotheses have been
Vol. 4 SINGLE-SITE CATALYSTS 145
suggested (249–257), most notably the Cossee-Arlman model based on cis-insertion of an α-olefin into the metal–carbon bond (eq. 2) (249–252). Both kineticisotope studies and molecular modeling suggest that the transition state of theinsertion step is stabilized by agostic interactions between the metal center andan α-hydrogen atom of the growing chain (for a review, see Ref. 20). The agosticinteraction apparently increases the rigidity of the transition state resulting inan increase in the rate of olefin insertion and, in the case of chiral catalysts, anincrease in stereospecificity.
(2)
The molecular weight of the produced polymer is determined by the relativerates of monomer insertion and chain termination (Fig. 7) (for a review on chaintransfer processes in propylene polymerization, see Ref. 258). The dominant chaintermination process in olefin polymerization involves an intramolecular β-H elim-ination (259,260) producing a vinylidene-terminated polymer chain and a metalhydride. Termination by β-elimination may occur either by direct hydrogen trans-fer from the polymer chain to the metal center, or by transfer of a β-H from thepolymer chain to a carbon atom of a coordinated olefin (261). The rate of the latterprocess increases with olefin concentration in parallel with increased rate of prop-agation, whereas the rate of the former is olefin-independent. Intramolecular β-alkyl elimination (261–263) produces a metal–alkyl and an allyl-terminated chainend. Reinsertion of terminally unsaturated polymer chains results in long-chainbranching. Saturated chain ends are obtained in the presence of chain-transferagents, such as H2 or aluminum alkyls (eg, complexed MAO). Hydrogen functionsas an effective means to control the molecular weight of polyolefins (264,265),
Fig. 7. Chain termination processes in olefin polymerization.
146 SINGLE-SITE CATALYSTS Vol. 4
producing an alkyl-terminated chain end and a metal hydride. Transalkylation tothe aluminum cocatalyst results in a metal–alkyl and an aluminum-terminatedpolymer chain, which for PP generates isopropyl end groups after hydrolysis ofthe carbon–aluminum bond (266–271). The barrier to exchange of alkyl groups be-tween the transition metal and aluminum compounds decreases with increasingelectron density at the metal center (267,271).
Mechanisms of Stereocontrol. Stereochemistry of the olefin inser-tion step can be controlled by both the steric environment of the active site(enantiomorphic-site control) as well as the growing polymer chain (chain end con-trol). In chain end stereocontrol, stereospecificity arises from the chiral β-carbonatom of the last enchained monomer unit, which in turn influences the stereo-chemistry of monomer addition. Chain-end control is usually less effective thansite control and has been observed for some achiral metallocenes at low polymer-ization temperatures. Partially iPP resulting from chain end stereocontrol hasbeen obtained with Cp2TiPh2/MAO (56,272). The syndiospecific polymerizationof 1-butene using the Cp∗2MCl2/MAO (M = Zr, Hf) catalyst systems has beendescribed (273). Predominantly sPP has been obtained under chain end control,using Brookhart’s diimine nickel catalysts (274–277).
Stereoselectivity of C2 symmetric Group 4 ansa-metallocenes in isospecificpropylene polymerization results from an enantiomorphic-site control mechanism.Chain propagation proceeds prevailingly via 1,2 (primary) monomer insertion.Both coordination sites of the C2 symmetric catalyst are homotopic and thus se-lective for the same olefin enantioface. Repulsive interactions force the coordinatedpropylene monomer into that enantiofacial approach to the metal–alkyl unit whichhas the methyl substituent of the monomer trans to the β-C atom of the growingpolymer chain (Fig. 8). To avoid repulsive interaction with the ligand, the grow-ing chain preferably obtains a conformation where the first C C bond is directedtoward the most open sector of the ligand framework. Repeated chain migratoryinsertion with the same olefin enantioface generates an isotactic polymer chain.
Experimental proof for the presented mechanism has been obtained bypropylene oligomerization studies with enantiomerically pure ansa-metallocenecatalysts (259,264,278,279). The participation of α-agostic interactions in the in-sertion process by polymerization of α-deuterated olefins has been demonstrated(280,281). Agostic interactions control the stabilities of the alternative transitionstates for propylene insertion. Of the two possible orientations of the polymerchain involving a three-membered Zr H(α) C(α) ring, one is sterically hinderedby collision of the growing chain with a β-substituent of the chiral ligand. Theother one is essentially unencumbered placing the β-C in the open position ofthe ligand framework. The stereochemistry of the propylene insertion step hasbeen studied using a s-cis-η4-butadiene complex derived from a C2 symmetricbis(indenyl) ansa-zirconocene rac-(CH3)2Si(Ind)2ZrCl2 (242).
Syndiospecific propylene polymerization with Cs symmetric ansa-metallocene catalysts proceeds in an analogous fashion (Fig. 8). The growingpolymer chain is directed away from the bulkier cyclopentadienyl ligand, andthe favored propylene enantioface places the methyl substituent trans to thegrowing chain. Migratory insertions result in regular alternation of the monomerapproach from the left and right side of the metallocene wedge, consequentlyproducing the syndiotactic polymer (61,76).
Vol. 4 SINGLE-SITE CATALYSTS 147
Fig. 8. Enantioselective coordination of propylene to the coordination sites of a C2 sym-metric isospecific metallocene catalyst (a) (only one enantiomer of the racemic pair isshown), and a Cs symmetric syndiospecific metallocene catalyst (b) [(CH3)2C bridge omittedfor clarity].
Theoretical Studies
Numerous theoretical studies on both early and late transition metal SSCs haveappeared in recent years covering nearly all aspects of the olefin polymerizationprocess (for reviews, see Refs. 282 and 283). The employed methodologies includemolecular mechanics (284–291), ab initio electronic structure methods (292–297),density functional studies (298–303), as well as various hybrid techniques (304–308), such as the combination of quantum and molecular mechanics (QM/MM). Adetailed description of these studies is outside the scope of this article; neverthe-less, these theoretical investigations have played a major role in elucidating theelementary steps of olefin complexation, chain propagation, and chain terminationas well as the mechanisms of stereocontrol in catalytic olefin polymerization.
Polymerization of Ethylene
The properties of polyethylene are strongly influenced by its molecular weight,molecular weight distribution, and branching content. Three major classes ofpolyethylene are commercially available. Conventional low density polyethylene(LDPE) is produced at high polymerization temperatures and pressures. As a
148 SINGLE-SITE CATALYSTS Vol. 4
consequence of the radical polymerization mechanism, LDPE contains both short-and long-chain branching. Heterogeneous Ziegler–Natta catalysts polymerizeethylene to highly linear high density polyethylene (HDPE) characterized byexcellent strength but poor melt processability. Linear low density polyethylene(LLDPE) is traditionally obtained by controlled incorporation of higher α-olefinssuch as 1-hexene or 1-octene into polyethylene, using Ziegler–Natta or chromiumcatalysts (see ETHYLENE POLYMERS, LLDPE).
In contrast to multisite heterogeneous Ziegler–Natta systems, metallocenesand other SSCs allow a precise control of the polymerization reaction resulting innarrow distribution of all molecular characteristics, including molecular weightdistribution, comonomer distribution and architecture, and content of branches.
Homopolymers. Both unbridged and stereorigid (bridged) metalloceneshave been employed in polymerization of ethylene. Cp2ZrCl2/MAO homopolymer-izes ethylene with 10–100 times higher activity compared to conventional Ziegler–Natta catalysts. The insertion rate is comparable to reaction rates observed inenzymatic systems, and polymerization activities reaching 40,000 kg PE/g Zr/hhave been reported for this catalyst system alone (18,22,55). Essentially linear ho-mopolyethylenes (HDPE) are obtained in most cases using bis(cyclopentadienyl),bis(indenyl), and related metallocene catalysts. The choice of metal and ligandsubstitution pattern highly influences the polymerization performance. Activitygenerally decreases in the order of Zr > Ti > Hf. Increasing electron-donatingability of the ligand substituents appears to increase the activity in the cyclopen-tadienyl series whereas steric crowding has a detrimental effect. Consequently(CpCH3)2ZrCl2 is more active than (CpC2H5)2ZrCl2 or Cp∗2ZrCl2 (266). Substi-tuted bis(indenyl) (71) and bis(fluorenyl) (90,309) ansa-metallocenes display, insome cases, very high activities exceeding those of Cp2ZrCl2. Molecular weightcan be controlled by addition of hydrogen or by increasing the polymerizationtemperature. Hafnium complexes produce higher molecular weight polymers thantheir zirconium analogues. The stronger Hf–carbon σ -bond slows down both bond-making and bond-breaking processes resulting in a lower activity and highermolecular weight.
The nickel and palladium diimine catalysts developed by Brookhart (Fig. 5a)display some unique properties; for example, they can produce highly branchedpolymers from a single ethylene feedstock. Polymers derived from the nickelcatalysts are generally less branched than those obtained with their palladiumanalogues, and products ranging from conventional LLDPE to amorphous, elas-tomeric, or hyperbranched materials can be obtained by variation of the metal,monomer concentration, polymerization temperature, and pressure. The branchformation is explained by a chain walking mechanism (179,185,186,310), wherethe growing polymer chain undergoes a series of β-hydride eliminations and rein-sertions resulting in migration of the metal species along the polymer backbone.
Activities of the MAO-activated nickel catalysts are comparable to thoseof metallocenes, and turnover frequencies of 3.9 × 105 h− 1 corresponding to11,000 kg PE/mol Ni/h were reported in the initial publications (179). Molecu-lar weights can be varied from oligomers to high polymers (Mw = 30,000 to >
1,000,000) depending on the catalyst structure and polymerization conditions(185). The palladium analogues display more moderate activities, but due tothe extensive “chain walking” the range of accessible materials is broadened
Vol. 4 SINGLE-SITE CATALYSTS 149
considerably. DuPont is currently developing its VersipolTM (E.I. du Pont deNemours & Co., Inc.) catalyst technology based on the Brookhart-type late tran-sition metal catalyst systems (186).
The 2,6-bis(imino)pyridyl iron catalysts (Fig. 5b) show likewise extremelyhigh activities in homopolymerization of ethylene. Upon activation with modifiedMAO, turnover frequencies of greater than 107 h− 1 corresponding to 330,000 kgPE/mol Fe/h have been reported (187). The highest molecular weights (Mw) exceed500,000 (192). The recently discovered single-component nickel catalysts (Fig. 5c)can produce homopolyethylenes with Mw > 250,000 and turnover frequenciesranging from 0.5 × 106 to 3.0 × 106 g PE/mol Ni/h at low temperatures andpressures (196). The total content of methyl, ethyl, propyl, and butyl branchesremains under 10 branches per 1000 carbon units under optimal polymerizationconditions. At slightly elevated polymerization temperatures, activities compara-ble to those of the first metallocenes and Brookhart’s diimino nickel catalysts wereobserved.
Copolymers with Propylene and Higher α-Olefins. Group 4 metal-locene catalyzed copolymerization of ethylene with higher α-olefins such as 1-butene, 1-pentene, 1-hexene, or 1-octene produces well-defined copolymers withrandom distribution of the comonomer (311–322). The copolymerization rates areoften higher than those of the corresponding homopolymerizations. Chiral C2 sym-metric ansa-metallocenes incorporate higher α-olefins more readily than the un-bridged complexes. Syndiospecific Cs symmetric metallocene catalysts show evenbetter copolymerization characteristics. Alternating copolymers of ethylene and 1-octene have been obtained with some meso-diastereomers of bridged bis(indenyl)catalysts (323). Among the various polyolefins produced using SSC technology theLLDPE-grades show perhaps the greatest industrial potential.
Copolymers of ethylene and propylene show, in some cases, elastic proper-ties (324,325). Further incorporation of dienes, such as ethylidenenorbornene or1,4-hexadiene, produces ethylene–propylene–diene monomer (EPDM) elastomers.Kaminsky (326) and Waymouth (327) have reported the predominantly alter-nating copolymerization of ethylene and propylene, using C1 and Cs symmetric(cyclopentadienyl)(fluorenyl) ansa-zirconocene-based catalysts (eq. 3), demon-strating yet another example of preparation of new materials through rationalexploitation of SSC systems.
(3)
150 SINGLE-SITE CATALYSTS Vol. 4
Long-Chain Branching. The constrained geometry Group 4 catalystsof the Dow and Exxon chemical companies allow the incorporation of, besidesC4–C20 α-olefins, even styrene (99,328), isobutylene (329), and vinyl-terminatedmacromonomers into HDPE or LLDPE polyethylene chains (see Ref. 25 and ref-erences to the patent literature therein). At high polymerization temperatures(typically Tp = 130–160◦C) these catalysts produce controlled amounts of vinyl-terminated polymers or oligomers that are reinserted to the growing polymerchain. Typical levels of long-chain branching (LCB) range from 1 to 3 branches per1000 carbon atoms. The resulting materials exhibit some unique properties sincethe presence of LCB significantly affects the melt viscosity, elasticity, shear thin-ning, and extension thickening of the produced polymer, thus improving its pro-cessability. Typically, the LCB-PEs exhibit the strength and toughness of LLDPEwhile having the melt processability of LDPE (25). Formation of very low lev-els of long-chain branched material has been reported also for ethylene bridgedbis(indenyl) ansa-zirconocene/MAO catalyst systems (330–332).
Polymerization of Propylene
Stereoselective polymerization of α-olefins, especially propylene, has received anenormous amount of attention in recent years (see PROPYLENE POLYMERS (PP)).Group 4 metallocene-based SSCs allow the production of PP in a considerablywider range of composition and microstructures than any other catalytic system(Fig. 9) (for reviews, see Refs. 14, 333, and 334). The physical properties of poly-olefins are strongly influenced by their microstructures. Highly stereoregular iPPsand sPPs are crystalline thermoplastics whereas stereorandom atactic polypropy-lene (aPP) is an amorphous elastomer. The most efficient tool for determining themicrostructure and stereoregularity of poly-α-olefins is 13C nmr spectroscopy. Tac-ticity of the polymer is commonly expressed in terms of its pentad content, which isthe fraction of stereosequences containing five adjacent stereocenters. An isotactic[mmmm] pentad consists of a sequence of five identical stereocenters, whereas inthe syndiotactic [rrrr] pentad the configuration of the stereocenters is regularlyalternating. The [mmmm] pentad content of highly iPP approaches 100% whereasperfectly atactic polymers consist of a statistical mixture of all 10 possible pentads.
Because of the steric constraints of the metallocene catalysts, propylene in-sertion mainly proceeds in a regioregular fashion (1,2-insertion), although occa-sional 2,1-regioinversions are observed. The amount of regioerrors depends on thestructure of the catalyst and affects the crystallinity and melting point of the pro-duced polymer (14,84,85,334). Insertion of propylene into a growing chain with a2,1 last-inserted unit is usually exceedingly slow because of the highly unfavor-able nonbonded interactions, and the chain end may isomerize to a more reactive3,1-unit, which would then be followed by a regioregular primary insertion.
Isolated stereoerrors result from regioregular 1,2-insertions of the wrongolefin enantioface. Alternatively, a regioregular last-inserted monomer unit mayundergo an epimerization reaction producing an inversion of configuration. Re-cent studies (280,281,335–342) suggest that the major part of stereoerrors in iPPsin fact results from the chain end epimerization reaction, rather than from regioir-regular 2,1-insertions or regioregular insertions with the incorrect enantioface.
Vol. 4 SINGLE-SITE CATALYSTS 151
Fig. 9. PP microstructures.
Homopolymers.Atactic Polypropylene. Low molecular weight aPP waxes can be obtained
from solvent extraction of iPP prepared with heterogeneous Ziegler–Natta cata-lysts. The product is neither completely atactic nor fully amorphous, and findslimited use as an additive in bitumen and adhesives (22). Achiral and unbridgedmetallocene catalysts such as Cp2ZrCl2 also produce mainly low molecular weightaPP, although at low temperatures partially isotactic material has been ob-tained by chain end control mechanism (56). High molecular weight, fully atactic
152 SINGLE-SITE CATALYSTS Vol. 4
elastomeric aPP has been obtained using a C2v symmetric bis(fluorenyl) ansa-zirconocene catalyst (CH3)2Si(Flu)2ZrX2/MAO (X = Cl, CH3) under technical pro-duction conditions (83,343). Molecular weights (Mw) ranging from 100,000 to400,000 and polymerization activities corresponding to 1300–11,200 kg PP/molZr/h were obtained at Tp = 50◦C at variable monomer concentrations. The lowdensities, high transparency, softness, low modulus, and high elongation of thesepolymers result from the totally amorphous state of the polymer chains (22).
Isotactic Polypropylene. IPP is generally obtained with C2 sym-metric ansa-metallocene catalysts. In most cases, bridged bis(indenyl)- orbis(tetrahydroindenyl)-derived Group 4 complexes are used (56,57,62,65–71,84,85,87,344,345), with a few exceptions based on C2 symmetric substitutedbis(cyclopentadienyl) analogues (346,347) and some sterically congested C1 sym-metric catalysts (72,73,78,348–354). A series of unbridged asymmetrically sub-stituted bis(indenyl) and bis(tetrahydroindenyl) zirconocenes that produce par-tially iPP under enantiomorphic site control at low polymerization temperatureshas been described (355–357). In these catalysts, the isospecific conformationalisomer is stabilized by partially hindered ligand rotation. An unbridged bis(1-methylfluorenyl)zirconium dichloride/MAO catalyst system reportedly producesiPP even at elevated polymerization temperatures (78,358). Chiral disposal of thesubstituted fluorenyl ligands results from a strongly hindered rotation around theZr C5 axis. Several other bridged or unbridged metallocene catalysts lacking C2symmetry have been employed in propylene polymerization producing PPs withvariable microstructures ranging from nearly atactic to partially isotactic ma-terials (63,64,88,89,93,359–379). Remarkable examples are the elastomeric PPs(vide infra) obtained with C1 symmetric ansa-metallocenes (89,366–373) and theisotactic–atactic stereoblock elastomeric PPs (63,64,374–379).
The original Brintzinger-type catalyst systems based on rac-C2H4(IndH4)2TiCl2 and its zirconocene analogue exhibit reasonably high activities in propy-lene polymerization; however, the molecular weights, melting points, and iso-tacticities of the produced iPPs are by far inferior to those of heterogeneousZiegler–Natta catalysts when employed under technical conditions. For exam-ple, bulk polymerization of propylene at 70◦C using rac-C2H4(IndH4)2ZrCl2/MAOproduces iPP with Mw = 15,000 and Tm = 125◦C (344), whereas commercialZiegler–Natta grades of iPP have molecular weights between 100,000 and 500,000and melting points ranging from 160 to 165◦C. Rational structural modifica-tions (68–71,344) of the original catalysts culminated in the 1994 publicationsof the Hoechst group (71) and Brintzinger (65) describing true high perfor-mance metallocene catalysts for isospecific propylene polymerization (Fig. 10). The2,2′-dialkyl-4,4′-diaryl-substituted dimethylsilylene bridged bis(indenyl) ansa-zirconocene catalysts polymerize propylene to highly iPP with stereoselectivities,polymer melting points, and molecular weights comparable to those obtained withheterogeneous Ziegler–Natta catalysts (Table 1) (65,67,71,344,380–382). Thesemetallocene-based catalysts are over 40 times more active than the conventionalsystems. Hydrogen gas has to be applied as a molecular weight regulator, evenunder technical polymerization conditions, in order to produce commercial iPPgrades.
The 2,2′-alkyl substitution at the C5 rings of the bis(indenyl) ansa-zirconocene catalysts dramatically improves the molecular weight of the produced
Vol. 4 SINGLE-SITE CATALYSTS 153
Fig. 10. High performance metallocene catalysts for isospecific polymerization of propy-lene. (a) Hoechst group (71); (b) Brintzinge (65).
PP (65,67,70,71,344). The observed enhancement has been explained by a com-bination of steric and electronic influence on the chain-termination reaction. En-hanced rigidity of the ligand framework combined with a direct steric interac-tion of the alkyl group with the growing polymer chain decreases the rate of β-elimination. Electron donation from the alkyl substituent may further decreasethe local Lewis acidity of the central metal atom, thus reducing its tendency forβ-H abstraction.
The role of the substituents in β-position to the interannular bridge, ie, 4,4′-positions of the six-membered rings of the indenyls, appears to be twofold. The gainin stereospecificity by enlarging the 4,4′-substituents from isopropyl to naphthyl,as shown in Table 1, results from indirect steric control (71). The high rotationalbarrier of the naphthyl substituent places it in an optimal position for interactionwith the growing chain. The synergistic effect of 2-alkyl-4-aryl substitution further
Table 1. Propylene Polymerization Performances of Various Bis(indenyl)ansa-zirconocene/MAO Catalyst Systemsa,b
A, kg [mmmm]Metallocene/catalyst PP/mmol M/h Mw Pentad, % Tm, ◦C
improves the catalyst rigidity and enhances the stereospecificity. Secondly, elec-tron donation from the aryl or alkyl substituents may stabilize the cationic activespecies or facilitate its formation (65,67,71). The resulting increase in polymeriza-tion activity also influences the polymer molecular weight by increasing the ratio ofthe rates of chain propagation vs. chain termination. Even higher polymerizationactivities have been reported by using the 2,2-di-n-propyl-4,4-di-9-phenanthrylsubstitution pattern (382).
The structure of the interannular bridge mainly influences the stereospeci-ficity and polymerization activity of the bis(indenyl) catalysts. Replacement ofthe ethylene bridge of rac-C2H4(Ind)2ZrCl2 by a one-membered silylene (65–71)or germylene (69) bridge improves the stereoselectivity whereas a one-memberedcarbon bridge decreases both the stereoselectivity and molecular weight of theproduced polymer (69,87,344). Longer than two-atom bridges decrease the ac-tivity and stereoselectivity because of the distortion of the molecular C2 sym-metry and narrowing of the coordination gap aperture (68,383,384). Aromaticsubstitution on the bridge enhances the molecular weight, although the size ofthe effect appears to be unpredictable (69,71,344). Electron withdrawing sub-stituents in the six-membered rings of the indenyl ligands decrease the polymer-ization activity and molecular weight (75,82). Dimethylamino-substituents in the2,2′-positions result in modest activities and stereospecificities; rac-(CH3)2Si(2-(CH3)2NInd)2ZrCl2/MAO polymerizes propylene to iPP with [mmmm] = 85%, Tm= 132◦C, and Mn = 30,000 (Tp = 50◦C; P = 2 bar), albeit suffering from long induc-tion periods apparently resulting from interaction of the amino groups with thecocatalyst (66). tert-Butyldimethylsiloxy-substituted analogues exhibit likewisemodest activities and stereospecificities but polymerize propylene and ethylenewithout the induction delay (91,271).
Alkyl or silyl substitution in the 3,3′-positions of the five-membered ringsof ethylene and dimethylsilylene bridged bis(indenyl) catalysts markedly de-creases the polymerization activity and results in a complete loss of stereose-lectivity (62,69,344,385). Bilateral coverage of the coordination sites by the 3,3′-substituents and the hydrogen atoms of the 4,4′-positions equalizes the energeticdifferences between the two enantiofacial orientations of the incoming propylenemonomer (386). An exception to this are the single carbon bridged 3,3′-substitutedcatalysts rac-R2C(3-(CH3)3C-Ind)2ZrCl2 (R = H, CH3) (Fig. 11) (85,345) that
Fig. 11. Single carbon bridged isospecific metallocene catalysts.
Vol. 4 SINGLE-SITE CATALYSTS 155
produce highly isotactic polymers. For example, the methylene bridged complexrac-H2C(3-(CH3)3C-Ind)2ZrCl2 and its dimethyl analogue produce, in combinationwith MAO, fully regioregular iPP ([mmmm] = 95–98%) with medium–high molec-ular weights (Mw = 70,000–780,000) and high melting points (Tm = 154–163◦C)in the Tp range of 30–70◦C (345). The polymer properties and polymerizationactivities are inferior to those obtained with the high-performance 2,2′-dialkyl-4,4′-diaryl-substituted Hoechst catalysts. An advantage with the Montell systemsis, however, their simple and inexpensive ligand and catalyst synthesis.
Syndiotactic Polypropylene. Before the report on the first syndiospe-cific ansa-metallocene catalyst (61), only partially sPP had been obtained withvanadium-based Ziegler–Natta catalysts at low polymerization temperatures(387). The Cs symmetric metallocene-based systems are the first examples of cat-alysts that are capable of producing highly sPP in high yield and are practical forindustrial scale-up. The properties of metallocene sPP differ substantially fromthose of iPP, especially in their considerably higher room temperature impactstrength, higher optical clarity, and resistance against uv radiation.
The polymerization behavior of the original Ewen catalyst has been stud-ied in detail (61,62,388). For example, bulk polymerization of propylene at 50◦Cusing (CH3)2C(Cp)(Flu)ZrCl2/MAO produces sPP having syndiotactic pentad con-tent [rrrr] = 82%, Mw = 133,000, and Tm = 140◦C (62). Higher syndiotacticitiesare obtained at lower polymerization temperatures. Several structural variationsof the original catalyst are reported in the literature (62,74,76–78,385,389–400)(Fig. 12). Hafnocene-based catalysts exhibit a lower activity and slightly lowersyndiospecificity but produce polymers with higher molecular weights. The tita-nium analogues suffer from both poor activities and poor stereoselectivities (389).Replacement of the isopropylidene bridge of (CH3)2C(Cp)(Flu)ZrCl2 with an aro-matic Ph2C bridge improves both the syndiotacticity and molecular weight of thepolymer (77), whereas considerably lower stereoselectivities are observed for theethylene (78,390,396) and (CH3)2Si (391) bridged analogues. Exceedingly activeregio- and syndiospecific metallocene catalysts have been obtained by replacingthe fluorenyl ligand of (C6H5)2C(Cp)(Flu)ZrCl2 with the sterically demanding oc-tamethyloctahydrodibenzofluorenyl (Oct) moiety. The produced sPPs have syn-diotactic pentad contents [rrrr] approaching 99% and melting temperatures ashigh as 153◦C have been reported (399,400), demonstrating for the first time thedramatic effect of distal ligand perturbations on the polymer stereochemistry.
Fig. 12. Metallocene catalysts for syndiospecific polymerization of propylene.
156 SINGLE-SITE CATALYSTS Vol. 4
The doubly bridged Cs symmetric bis(cyclopentadienyl)zirconocene catalystsdeveloped by Bercaw and co-workers are likewise highly regio- and syndiospecific(397,398). Bulk polymerization of propylene at 20–70◦C gave sPPs with pentadcontents [rrrr] = 45.4–97.5% and molecular weights Mw of 130,000–1,250,000depending on the catalyst substitution pattern and the employed polymerizationtemperature (76). The highest reported polymerization activity corresponding to16,900 kg PP/g Zr/h was obtained at 70◦C using the (1,2-(CH3)2Si)2(Cp)(3,5-i-(C3H7)2-Cp)ZrCl2/MAO catalyst system.
Hemiisotactic PP. As first demonstrated by Ewen (62), the C1 symmetricmetallocene (CH3)2C(3-CH3Cp)(Flu)ZrCl2 produces hemiisotactic polypropylene(hitPP) consisting of alternating isotactic and stereorandom monomer placements.The two coordination sites of this catalyst are different; the isospecific site isblocked by both the Cp-methyl substituent and the fluorenyl C6 ring whereas theaspecific site is unilaterally framed by the second fluorenyl moiety. As the growingchain occupies the more hindered site, the two conformations in which the firstC C bond is directed toward the substituted side of the Cp ring or toward thefluorenyl ring have equal probabilities. Consequently monomer insertion fromthe less hindered site is aspecific. At the less hindered site the polymer chain isoriented toward the open sector of the ligand framework resulting in stereospecificmonomer insertion from the more hindered site, thus forming the hemiisotacticmicrostructure.
Copolymers with Higher α-Olefins. Several researchers (381,401–404)have copolymerized propylene with 1-hexene (402–404) and 1-octene (381,401),using various MAO-activated metallocene catalysts. Random distribution of thecomonomer was observed in all cases. Comonomer incorporation is dependent onthe catalyst structure; combination of benzannelation and 2-methyl substitutionof silylene bridged bis(indenyl) ligands in rac-(CH3)2Si(2-CH3-benz[e]Ind)2ZrCl2was found to enhance both the molecular weight and comonomer content of thepropylene–octene copolymers (401). Poly(propylene)-graft-poly(styrene) copoly-mers have been prepared using the rac-(CH3)2Si(2-CH3-benz[e]Ind)2ZrCl2/MAOcatalyst in copolymerization of propylene with allyl-terminated polystyrenemacromonomers (405). Graft copolymers with a polystyrene content ranging from7 to 72% were obtained. The same catalyst system was used for copolymerizationof propylene with an aPP macromonomer (406).
Elastomeric Polypropylene (elPP). PPs consisting of blocks of atacticand isotactic stereosequences behave as thermoplastic elastomers. This materialwas identified for the first time by Natta in 1959 during fractionation studiesof first generation Ziegler–Natta iPP (407). The elastomeric properties of thesepolymers are believed to result from cocrystallization of isotactic stereoblocks inneighboring PP chains to form a physically cross-linked elastomer. More recently,elPP has been prepared using supported tetraalkyl Group 4 catalysts (408,409).
A number of Group 4 metallocene catalysts have been shown to produce PPelastomers (Fig. 13). The production of elPP has been reported using asymmet-rically substituted ansa-titanocene catalysts CH3CH((CH3)4Cp)(Ind)TiCl2/MAOunder a variety of polymerization conditions (366–370). The formation of shortisotactic sequences in predominantly atactic polymer chains was proposed to orig-inate from sequential monomer insertion at the aspecific and isospecific coordi-nation sites of the catalyst, although this mechanism has been challenged. elPPs
Vol. 4 SINGLE-SITE CATALYSTS 157
Fig. 13. Metallocene catalysts for preparation of elastomeric PP. (a) Titanocene Catalysts(366–370); (b) (371–373); (c) See also Figure 14 (63,64,374–379); (d) Zirconocene Catalysts(89).
have also been prepared using a related class of MAO-activated metallocenes(CH3)2X(Cp)(Ind)MCl2 (X = C, Si; M = Ti, Zr, Hf) (371,372). Microstructures of thePPs produced were found to be sensitive to both catalyst structure and polymer-ization conditions with the hafnium-based systems yielding the best elastomericproperties. The mechanism suggested (371,372) involves random monomer in-sertion at the isospecific and aspecific coordination sites, respectively. In a morerecent report, the preparation of high molecular weight semicrystalline elPPusing the asymmetrically substituted rac-like diastereomer of (CH3)2Si(Ind)(3-CH3Ind)ZrCl2 in combination with MAO has been described (373). The obtainedpolymers exhibited isotactic pentad contents [mmmm] = 30.4–41.9% and molecu-lar weights (Mw) of 37,000–115,000 depending on the monomer concentration andpolymerization temperature.
A series of C1 symmetric ansa-zirconocene catalysts based on an ethylenebridged mixed ligand (indenyl)(fluorenyl) framework has been described (89).These complexes, upon activation with MAO, retain their high activities evenat elevated polymerization temperatures producing PPs of variable isotacticities[mmmm] = 19.9–72.1% and molecular weights (Mw) up to 230,000 depending on
158 SINGLE-SITE CATALYSTS Vol. 4
Fig. 14. Oscillating stereocontrol for production of isotactic–atactic stereoblock PP.
the employed catalyst, polymerization temperature and monomer concentration.The presence of two different coordination sites allows precise control of stereo-error formation resulting in bulk properties ranging from flexible semicrystallinethermoplastics to excellent thermoplastic elastomers.
The oscillating catalysts that have been developed are based on unbridgedbis(2-arylindenyl) Group 4 metallocenes that presumably interconvert betweenan isospecific and aspecific conformer on the time scale of the polymerization re-action producing isotactic–atactic stereoblock polymers (Fig. 14) (63,64,374–379).The size of the stereoblocks is controlled by the relative rates of chain propa-gation and ligand isomerization. The nature of the substituent in 2-position ofthe indenyl ligand is crucial for obtaining elastomeric material. The prototypicalbis(2-phenylindenyl)zirconium dichloride/MAO system produces elPP having pen-tad contents [mmmm] = 6.3–28.1% as a function of the monomer concentrationand polymerization temperature (63). The 3,5-trifluoromethyl-substituted ana-logue [2-(3,5-(CF3)2-C6H5)Ind]2ZrCl2 produces PP of considerably higher isotac-ticity and crystallinity; values up to [mmmm] = 73% were reported for the MAO-activated catalyst system (64). Several other modifications generating mainly aPPhave been reported in subsequent papers (374–379).
A related strategy based on reversibly bridged donor–acceptor substitutedmetallocenes, potentially providing access to elastomeric polyolefins, has been de-scribed (93). In these complexes one of the cyclopentadienyl or indenyl ligands con-tains a donor group (eg, diethylphosphanyl or dimethylamino) whereas the otherone is substituted with an acceptor (eg, dichloroboranyl). Ligand rotation canbe controlled by temperature-dependent donor–acceptor interaction. For exam-ple, rac-[(C2H5)2P(2-CH3Ind)][Cl2B(2-CH3-Ind)]ZrCl2 activated with triisobutyla-luminum and [NH(CH3)2C6H5]+[(C6F5)4B]− produces at room temperature (bulkpolymerization of propylene) highly iPP with [mmmm] = 94%, Tm = 158◦C,and Mv = 2,000,000. At 50◦C the pentad content drops to [mmmm] = 82% and
Vol. 4 SINGLE-SITE CATALYSTS 159
molecular weight to Mv = 434,000, whereas the melting temperature remainshigh, Tm = 154◦C, indicating the formation of aPP sequences together with longisotactic blocks.
Other Monomers
Cycloolefins. A distinct feature of the Group 4 metallocene/MAO cat-alyst systems is their ability to polymerize cyclic olefins without ring-openingmetathesis, a reaction characteristic of heterogeneous Ziegler–Natta catalysts.Cyclobutene, cyclopentene, norbornene, and dimethanooctahydronaphthaleneare readily homopolymerized to isotactic polycycloalkenes, using chiral ansa-zirconocene catalysts (18,22,24,203,410–412) (eq. 4). Homopolymers of the cyclicolefins are not processable because of their high melting points (400–500◦C) andinsolubility in common organic solvents. Cycloolefin copolymers (COCs), producedby copolymerization of cyclic monomers with ethylene or propylene, are howeveramorphous thermoplastics exhibiting heat resistance up to 200◦C combined withhigh rigidity, toughness, and environmental stability (18,22,24,410,413–418). Theexcellent stiffness and low density of COCs combined with their stability againsthydrolysis and chemical degradation renders them as potential materials for com-pact discs, lenses, and optical fibers.
(4)
Dienes. Group 4 metallocene catalysts are capable of polymerizing bothconjugated and nonconjugated dienes. Polymerization of α,ω-dienes affords cy-clopolymers with remarkably high cyclo- and regioselectivities (269,270,419–425).Insertion of the terminal double bond of the nonconjugated diene into the metal–carbon bond is followed by intramolecular cyclization that produces the polymericring structure interspaced by methylene groups. Tacticity of the cyclopolymer isdetermined by the enantiofacial selectivity of the insertion step and is influencedby the symmetry and structure of the catalyst precursor. Diastereoselectivity ofthe cyclization step (ie, whether cis or trans rings are formed) is likewise in-fluenced by catalyst structure and sterical environment of the active site. Forexample, in the polymerization of 1,5-hexadiene, Cp2ZrX2/MAO (X = Cl, CH3)produces (at ambient temperature) predominantly atactic trans-poly(methylene-1,3-cyclopentane) (PMCP) whereas the sterically congested Cp∗2ZrCl2/MAOcatalyst produces mainly the atactic cis product (419–422). Interestingly, the cis-connected atactic PMCPs have melting points reaching Tm = 171◦C, whereas the
160 SINGLE-SITE CATALYSTS Vol. 4
trans-PMCP showed a considerably lower melting point of Tm = 86◦C (421). Thesynthesis of optically active cyclopolymers from 1,5-hexadiene using enantiomer-ically pure ansa-metallocene catalysts (423,424), the cyclopolymerization of func-tionalized α,ω-dienes (425), and the post-functionalization of hydroxy-terminatedPMCP (269,270) are discussed separately (vide infra).
Styrene. Syndiotactic polystyrene (sPS) is a high melting stereoregularpolymer first prepared in the mid-1980s using an undisclosed titanium-basedcatalyst in combination with aluminum activators (112). In contrast to isotacticpolystyrene (iPS), which shows a very low crystallization rate and is thus use-less for most commercial applications, sPS has a fast crystallization rate, highermelting point compared with iPS (270◦C vs 230◦C), superior heat and chemicalresistance, as well as low specific gravity, low dielectric constant, and high modu-lus of elasticity. These unique properties have resulted in remarkable interest insPS as an attractive low cost replacement for expensive engineering plastics (seeSYNDIOTACTIC POLYSTYRENE).
A variety of Group 4 metal complexes, in combination with common olefinpolymerization activators, have been evaluated as potential catalysts for syn-diospecific polymerization of styrene (for reviews, see Refs. 114, 115, 123, and426). Monocyclopentadienyl and monoindenyl titanocenes generally exhibit thehighest activities (eq. 5) (112–127). Curiously, half-sandwich titanium-trifluoride-based catalysts are more active than their trichloride analogues (124,427,428).The polymerization mechanism for sPS formation is under debate. Kinetic stud-ies and spectroscopic investigations of the catalytic systems suggest a cationicTi(III) complex as the active species (123).
(5)
Functional Polymers
Controlled in situ incorporation of polar or reactive functional groups into poly-olefins is a long-standing challenge (429–431). End uses of polyolefins are re-stricted by their chemical inertness and lack of adhesive properties. The absenceof polar groups results in a poor affinity for functional additives such as dyes,paints, and antioxidants, and limits the compatibility of polyolefins with functionalpolymers, biomaterials, metal, and glass. Traditional Ziegler–Natta catalystsare highly oxophilic and readily poisoned by Lewis-basic comonomers (alcohols,ethers, carboxylic acids, esters, and amines). Recently, considerable advances to-ward functionalized polyolefins have been made by direct polymerization and
Vol. 4 SINGLE-SITE CATALYSTS 161
copolymerization of functional monomers over SSCs as well as by various as-sociated indirect post-polymerization methods.
Homo- and Copolymerization of Polar Monomers.Metallocene Catalysts. Group 4 metallocene-based catalysts are, under
proper polymerization conditions, tolerant to selected polar comonomers. Variousfunctionalized dienes and α-olefins have been polymerized using cationic metal-locene catalysts (425,432,433). Polymerization activities were lower than thoseobserved for the analogous nonpolar monomers. Silyl-protected 1,6-heptadien-4-ols are readily cyclopolymerized by the sterically hindered [Cp∗2ZrCH3]+X−
catalyst [X = B(C6F5)4 or CH3B(C6F5)3] with turnovers reaching 280 (59% con-version) (eq. 6) (425). Deprotection of the silyl group generates the correspond-ing polyalcohol. Tertiary amines were homo- and copolymerized using both aspe-cific and isospecific metallocene catalysts (425,432,433). Borate cocatalysts gavehigher activities than MAO-based catalyst systems. Optimum activities wereachieved using diisopropyl-substituted amine with an olefin tether of three car-bon atoms. Catalyst stereospecificities were essentially insensitive to monomerfunctionality.
(6)
Other examples of polar comonomers incorporated by Group 4 metallocenesinclude silylamines (434), protected alcohols (435,436) and carboxylic acids (436),ω-chloroolefins (437), and primary long-chain hydroxyalkenes (438,439). Thecopolymerization of propylene with a hindered phenolic stabilizer 6-tert-butyl-2-(1,1-dimethylhept-6-enyl)-4-methylphenol over an isospecific metallocene/MAOcatalyst system has been described (440). A novel strategy for preparation of poly-olefin graft copolymers has been developed by combination of metallocene and“living” free radical polymerization techniques (441–443). Copolymerization ofan alkene-substituted hindered alkoxyamine with ethylene or propylene affordswell-defined macroinitiators with 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO)chain ends to which polystyrene, acrylate, and butadiene side chains can be graftedin a “living” fashion.
In addition, Group 4 metallocene-type catalyst systems have been employedin living ring-opening polymerization of lactones (444–446) and carbonates (447).Various metallocene or lanthanocene complexes have been used as initiators forhomo- and copolymerization of methacrylates, acrylates, and the synthesis of blockcopolymers (224,448–457). Single-site-type zinc alkoxide complexes have been de-scribed for ring-opening polymerization of lactides (458).
Late Transition Metal Catalysts. In contrast to early transition metals, latetransition metal catalysts (26,185) are considerably less oxophilic and are tolerantto a wide range of functional groups. The α-diimine palladium complexes devel-oped by Brookhart (179,180,183,184) copolymerize ethylene and propylene withalkyl acrylates (180,183) and protected alcohols (459) to high molecular weight
162 SINGLE-SITE CATALYSTS Vol. 4
random copolymers, albeit at reduced activities. The ethylene acrylate copoly-mers are amorphous, highly branched materials with ∼100 branches/1000 carbonatoms. Typical Tg values range from −67 to −77◦C.
The neutral salicylaldimine nickel complexes described by Grubbs and co-workers (195,196) show unprecedented functional group tolerance and are capableof incorporating substituted norbornenes, carbon monoxide, and α–ω functionalolefins into polyolefins with well-defined compositional distributions (eq. 7) (196).Furthermore, ethylene can be homopolymerized with these catalysts in the pres-ence of various functional additives including acetone, water, ethyl alcohol, andtriethyl amine. In the presence of 1500 equivalents of H2O, polyethylene was pro-duced at a rate of 5.4 × 104 g PE/mol Ni/h.
(7)
Modification of Preformed Polymers. Alkene end groups of poly-olefins can be functionalized in a post-polymerization process. PPs with olefinicend groups have been prepared using the isospecific (CH3)2Si(3-(CH3)3C-5-CH2Cp)2ZrCl2/MAO catalyst system (460). The vinylidene chain ends react fur-ther to give a range of monofunctional PPs with anhydride, silane, thiol, epoxy,borane, and carboxylic acid end groups. Thiol functionalized polymers were usedas chain-transfer agents in free radical polymerizations of styrene and acrylics toform PP block copolymers. A similar approach has been applied using aluminum(461) and borane (462) reagents. Ethylene or propylene have been copolymerizedwith unsymmetrical dienes, using MAO-activated metallocene catalysts (463–465). Selective incorporation of the α-olefinic double bonds of the comonomersprovides access to polymers containing internal unsaturations that can be con-verted to various functional groups.
Yet another methodology involves the metallocene-catalyzed copolymeriza-tion of olefins with borane containing monomers (466,467). The incorporated bo-rane group can be quantitatively converted to a hydroxy group or transformedinto a stable polymeric radical suitable for free radical grafting of acrylates.
Chain Transfer to Organometallic Compounds. Hydroxy-terminatedpoly(methylene-1,3-cyclopentane) has been prepared by selective chain trans-fer to aluminum in the cyclopolymerization of 1,5-hexadiene with the sterically
Vol. 4 SINGLE-SITE CATALYSTS 163
congested Cp2∗ZrCl2/MAO catalyst (269,270). Aluminum alkoxide of the pro-duced polymer was used as a macroinitiator for ring-opening polymerization ofε-caprolactone to produce diblock copolymers (270).
A similar strategy was used to prepare borane-terminated polyethyleneswith [Cp∗2ZrCH3]+X− [X = B(C6F5)4 or CH3B(C6F5)3] in the presence of 9-borabicyclo[3.3.1]nonane chain-transfer agent (468). The borane end group wasselectively oxidized and transformed to a polymeric radical for grafting of methylmethacrylate.
Silyl-capped polyolefins have been prepared by employing organotitanium-mediated silanolytic chain-transfer processes (469,470). Various monomers in-cluding ethylene, propylene, 1-hexene, and styrene were homo- and copolymerizedusing the cationic [((CH3)2Si((CH3)4Cp)(CH3)3CN)TiCH3]+X− , [rac-C2H4(Ind)2TiCH3]+X− , and [Cp∗2TiCH3]+X− [X = B(C6F5)4] metallocene catalyst sys-tems in the presence of organosilanes (C6H5SiH3, C6H5CH3SiH2), (CH3)2SiH2,(C2H5)2SiH2) (470). Silyl-linked linear A–A and star block copolymers as wellas A–B block copolymers were obtained in catalytic cycles using polyfunctionalsilabenzenes.
Enantioselective Polymerization
The characteristic functionalities of naturally occurring polymers are, in mostcases, related to their specific chiral structure. In nature, proteins, nucleic acids,and polysaccharides are constructed of readily available chiral monomers such assugars and amino acids. Both natural and synthetic chiral polymers are findingapplication as chromatographic supports, polymeric reagents and catalysts, chiralmembranes, and materials for preparation of cholesteric liquid crystal polymers(471,472).
High molecular weight stereoregular vinyl polymers contain mirror planesof symmetry perpendicular to the molecular axis (Fig. 15) and thus do not haveinherent chirality associated with the main chain. Synthesis of chiral polymersfrom vinyl monomers, with the exception of low molecular weight oligomers,
Fig. 15. Mirror planes of symmetry in stereoregular vinyl polymers.
164 SINGLE-SITE CATALYSTS Vol. 4
requires more complex polymer architectures to circumvent the symmetryconstraints.
Enantiomerically pure SSCs (79), readily available through resolution of theracemic C2-symmetric Brintzinger-type ansa-metallocenes (59,473–475), have at-tracted considerable interest in catalytic and enantioselective C C bond formationreactions (476). Despite the aforementioned symmetry concerns, these catalystshave opened new possibilities in synthesis and design of chiral macromolecules.
Polymerization and Oligomerization of Prochiral Olefins. The syn-thesis of optically active iPP and polybutene with the enantiomerically pure cat-alyst derived from (S,S)-C2H4t(IndH4)2ZrCl2 has been reported (311,477). Theobtained polymers showed fairly large optical activities in soaked suspensions([α]D = −123◦, −250 ◦ for PP, +130 ◦ for polybutene) attributed to a helical con-formation or predominant screw sense. As expected, racemization occurred whenthe polymer was heated or completely dissolved.
Oligomerization of α-olefins can be promoted by two methods: (1) reduc-ing the monomer concentration in the polymerization medium thus favoring β-Helimination over chain propagation and (2) conducting the polymerization in thepresence of H2 gas. Optically active low molecular weight oligomers of propyleneand butene have been prepared using the (S,S)-C2H4(IndH4)2ZrCl2 catalyst athigh temperature and low alkene concentration (478). The binaphtholate (R,R)-C2H4(IndH4)2Zr(BINOL) has been used for asymmetric oligomerization of propy-lene (479,480), 1-pentene (480), and 4-methyl-1-pentene (480) in the presence ofhydrogen and deuterium chain-transfer agents. A detailed microstructural analy-sis of the oligomeric fractions showed for the first time that the stereoregulation ofthe polymerization reaction with these catalyst systems results from preferentialaddition of one enantioface of the prochiral monomer to the growing polymer chain.
Cyclopolymerization. As discussed earlier, nonconjugated dienes can bepolymerized with metallocene-based catalysts to afford cyclopolymers. In contrastto linear polyolefins which have only two microstructures of maximum order (iso-tactic and syndiotactic), cyclic polymers have four microstructures due to the pos-sibility of configurational isomerism (cis vs trans) in the main chain (Fig. 16). Ofthese the trans-diisotactic structure contains no mirror planes of symmetry and ischiral by virtue of its main-chain stereochemistry (481). Two criteria for chiralityof this microstructure are the presence of trans rings and isotacticity (the same
Fig. 16. Poly(methylene-1,3-cyclopentane) structures of maximum order.
Vol. 4 SINGLE-SITE CATALYSTS 165
relative configuration of every other stereocenter). All other structures containmirror planes within the repeating subunits.
Using both enantiomers of the resolved C2H4(IndH4)2Zr(BINOL)/MAO cata-lyst 1,5-hexadiene has been polymerized to obtain optically active poly(methylene-1,3-cyclopentane) (eq. 8) (423,424). The polymer obtained with the (R,R) catalystdisplayed a molar optical rotation of [�]28
405 = +51.0◦ (c = 0.8 in CHCl3). Microstruc-tural analysis by 13C nmr indicated an enantiofacial selectivity of 91% for theolefin insertion step and 72% content of trans rings. Cyclopolymerization with the(S,S) catalyst gave the enantiomeric polymer with [�]28
405 = − 51.2◦. Interestingly,the measured optical rotations are considerably higher than that obtained for di-asteromerically pure model compound trans-(1R,3R)-1,3-dimethylcyclopentane,possibly resulting from a preferential helical conformation in solution.
(8)
Styrenic Copolymers. Alternating copolymerization of vinyl monomerswith carbon monoxide produces polyketones with stereogenic centers along thepolymer backbone (482). Brookhart and co-workers have reported the copolymer-ization of p-tert-butylstyrene and CO under the enantiomorphic site control mech-anism, using an enantiomerically pure cationic palladium catalyst based on aC2-symmetric bisoxazoline ligand (eq. 9) (483). The obtained copolymer showed ahigh molar optical rotation of [�]25
589 = − 536◦ (c = 0.5 in CH2Cl2) resulting frommain-chain chirality. 1H and 13C NMR analysis revealed a high degree of stereo-regularity (>98%) confirming the isotactic microstructure. The narrow molecularweight distribution (Mw/Mn = 1.4) of this polymer is consistent with the Schulz–Flory statistics for a SSC.
(9)
166 SINGLE-SITE CATALYSTS Vol. 4
Heterogenization of SSCs
Essentially homogeneous SSCs have to be supported for use in industrial gas-phase, slurry, and bulk-monomer polymerization processes (for reviews, seeRefs. 484–491). Not surprisingly, most of the information on the supported catalystsystems has been published only in the patent literature (for extensive coverage,see Ref. 490). The main problems are associated with catalyst feeding, reactorfouling, and controlled particle morphology of the produced polymer. Continuousoperation of gas-phase polymerization processes requires the use of free-flowingcatalyst particles that can be easily fed into the reactor. Controlled growth of thepolymer particles during the polymerization reaction (Fig. 17) is likewise essen-tial, since overheating and subsequent melting of the forming polymer grains mayresult in sheets and lumps that disturb the agitation, fluidization, and productremoval processes.
The most common inorganic supports used for heterogenization of metal-locenes are various types of silica, alumina, and magnesium compounds (381,484–502), although other less conventional carrier materials based on zeolites (503–505), mesoporous (MCM-41) silica (506–510), cyclodextrin (511,512), polystyrene(513–515), polysiloxanes (516,517), and sulfated zirconia (518) have been reported.Self-supporting catalyst systems based on olefin or alkyne functionalized metal-locenes have also been described (519). In this case, the initially homogeneousmetallocene is incorporated into the growing polymer chain, which upon precipi-tation from the reaction mixture functions as a heterogeneous catalyst system.
Immobilization of metallocenes on the carrier materials can be accomplishedby various methods, including (1) direct impregnation of the activated metalcomplex/aluminoxane mixture; (2) premodification of the support with MAO oraluminum alkyls followed by reaction with the metallocene; (3) anchoring of themetallocene to the support through covalent bonds followed by activation withthe cocatalyst. Similar approaches can be applied to the boron-activated catalystsystems (489,490).
In most cases, properties of the polymers obtained with the supported cat-alysts are similar to those obtained with their homogeneous analogues. The
single-site nature of the metallocene is generally preserved, although activitesare often remarkably reduced. The surrounding carrier material may increasethe steric hindrance around the active sites, thus contributing to decreased over-all productivities. On the other hand, enhanced site-isolation may suppress binu-clear chain transfer and deactivation pathways resulting in improved molecularweights and, most notably, satisfactory activities at considerably reduced cocat-alyst concentrations. Supported metallocene catalysts are commonly activatedwith [Al]:[M] ratios in the range of 50–400:1, far below those required for theirhomogeneous counterparts.
Despite the technological difficulties associated with catalyst preparation,leaching and process parameters, the number of announcements on commer-cial production of polyolefins with heterogeneous SSCs is, at the time of writing,rapidly increasing. The advanced supported SSCs are highly active and suitablefor use as drop-in catalysts in existing commercial plants. These catalysts producecoherent polymer grains that are enlarged replicas of the catalyst particles exhibit-ing the desired bulk densities and other material properties. Dow and Exxon havebeen the key players in commercialization of SSC technologies. Both companieslaunched their homogeneous high pressure and solution ethylene polymerizationprocesses in 1991. BASF commercialized its heterogeneous metallocene-basedpolyethylene in 1995. Several other companies, including Exxon, Mitsui Petro-chemical, Borealis Polymers and Elenac (BASF/Shell joint venture), are now pro-ducing commercial grades of metallocene polyethylenes in gas-phase or slurry pro-cesses (489). Isotactic metallocene PPs are limited to speciality polymers producedby Targor (BASF/Hoechst) and Exxon. sPP, EPDM-polymers, ethylene–styrenecopolymers, sPS, and cyclic olefin copolymers are, at the time of writing, in earlierdevelopment cycles.
BIBLIOGRAPHY
1. Ger. Pat. 973 626 (filed Nov. 17, 1953) K. Ziegler and co-workers, Chem. Abstr. 54,14794b (1960).
2. K. Ziegler and co-workers, Angew. Chem. 67, 426 (1955).3. K. Ziegler, Angew. Chem. 76, 545–553 (1964).4. Ital. Pat. 526 101 (filed June 8, 1954), G. Natta, P. Pino, and G. Mazzanti.5. G. Natta and co-workers, J. Am. Chem. Soc. 77, 1708–1710 (1955).6. G. Natta, J. Polym. Sci. 16, 143–154 (1955).7. G. Natta, Angew. Chem. 68, 393–403 (1956).8. G. Natta, Angew. Chem. 76, 553–566 (1964).9. H. Sinn and W. Kaminsky, Adv. Organomet. Chem. 18, 99–149 (1980).
10. T. Simonazzi and U. Giannini, Gazz. Chim. Ital. 124, 533–541 (1994).11. P. Galli, Macromol. Symp. 89, 13–16 (1995).12. K. B. Sinclair and R. B. Wilson, Chem. Ind. (London) 857–862 (1994).13. P. C. Mohring and N. J. Coville, J. Organomet. Chem. 479, 1–29 (1994).14. H. H. Brintzinger and co-workers, Angew. Chem., Int. Ed. Engl. 34, 1143–1170 (1995).15. G. W. Coates and R. M. Waymouth, in E. W. Abel, F. G. A. Stone, and G. Wilkinson,
eds., Comprehensive Organometallic Chemistry II, Pergamon, Oxford, 1995, pp. 1193–1208.
16. S. S. Reddy and S. Sivaram, Prog. Polym. Sci. 20, 309–367 (1995).17. M. Bochmann, J. Chem. Soc., Dalton Trans. 255–270 (1996).
168 SINGLE-SITE CATALYSTS Vol. 4
18. W. Kaminsky, Macromol. Chem. Phys. 197, 3907–3945 (1996).19. A. E. Hamielec and J. B. P. Soares, Prog. Polym. Sci. 21, 651–706 (1996).20. R. H. Grubbs and G. W. Coates, Acc. Chem. Res. 29, 85–93 (1996).21. M. Bochmann, Curr. Opin. Solid State Mater. Sci. 2, 639–646 (1997).22. W. Kaminsky and M. Arndt, Adv. Polym. Sci. 127, 143–187 (1997).23. A. A. Montagna, A. H. Dekmezian, and R. M. Burkhart, CHEMTECH 27(12), 26–31
(1997).24. W. Kaminsky, J. Chem. Soc., Dalton Trans. 1413–1418 (1998).25. A. L. McKnight and R. M. Waymouth, Chem. Rev. 98, 2587–2598 (1998).26. G. J. P. Britovsek, V. C. Gibson, and D. F. Wass, Angew. Chem., Int. Ed. Engl. 38,
428–447 (1999).27. I. Tritto and U. Giannini, eds., Macromol. Symp. 89, 1–586 (1995).28. H. Sinn and W. Kaminsky, eds., Macromol. Symp. 97, 1–246 (1995).29. R. F. Jordan, ed., J. Mol. Catal. A: Chem. 128, 1–337 (1998).30. T. J. Marks and J. C. Stevens, eds., Topics Catal. 7, 1–210 (1999). (Special Issue on
Advances in Polymerization Catalysis, Catalysts and Processes.)31. J. A. Gladysz, ed., Chem. Rev. 100, 1167–1682 (2000) (Special Issue on Frontiers in
Metal-Catalyzed Polymerization.)32. G. Fink, R. Mulhaupt, and H. H. Brintzinger, eds., Ziegler Catalysts: Recent Scientific
and Technological Improvements, Springer-Verlag, Berlin, 1995.33. A. Togni and R. L. Halterman, eds., Metallocenes, Wiley-VCH, Weinheim, 1998.34. G. M. Benedikt and B. L. Goodall, eds., Metallocene Catalyzed Polymers. Materials,
Properties, Processing & Markets, Plastics Design Library, New York, 1998.35. G. M. Benedikt, ed., Metallocene Technology in Commercial Applications, Plastics
Design Library, New York, 1999.36. W. Kaminsky, ed., Metalorganic Catalysts for Synthesis and Polymerization: Recent
Results by Ziegler–Natta and Metallocene Investigations, Springer-Verlag, Berlin,1999.
37. J. Scheirs and W. Kaminsky, eds., Metallocene-Based Polyolefins. Preparation, Prop-erties and Technology, Wiley, New York, 1999.
38. P. Arjunan, J. E. McGrath, and T. L. Hanlon, eds., Olefin Polymerization: EmergingFrontiers, American Chemical Society, Washington, DC, 2000. ACS Symposium Se-ries, Vol. 749.
39. D. S. Breslow and N. R. Newburg, J. Am. Chem. Soc. 79, 5072–5073 (1957).40. D. S. Breslow and N. R. Newburg, J. Am. Chem. Soc. 81, 81–86 (1959).41. G. Natta and co-workers, J. Am. Chem. Soc. 79, 2975–2976 (1957).42. H. Sinn and F. Patat, Angew. Chem. 70, 496–500 (1958).43. G. Natta, P. Corradini, and I. W. Bassi, J. Am. Chem. Soc. 80, 755–756 (1958).44. J. C. W. Chien, J. Am. Chem. Soc. 81, 86–92 (1959).45. W. P. Long and D. S. Breslow, J. Am. Chem. Soc. 82, 1953–1957 (1960).46. G. Mazzanti and G. Natta, Tetrahedron 8, 86–100 (1960).47. H. Sinn and F. Patat, Angew. Chem. 75, 805–813 (1963).48. G. Henrici-Olive and S. Olive, Angew. Chem. 79, 764–773 (1967).49. G. Henrici-Olive and S. Olive, J. Organomet. Chem. 16, 339–341 (1969).50. K. H. Reichert and K. R. Meyer, Makromol. Chem. 169, 163–176 (1973).51. W. P. Long and D. S. Breslow, Liebigs Ann. Chem. 463–469 (1975).52. A. Andersen and co-workers, Angew. Chem., Int. Ed. Engl. 15, 630–632 (1976).53. H. Sinn and co-workers, Angew. Chem., Int. Ed. Engl. 19, 390–392 (1980).54. J. Herwig and W. Kaminsky, Polym. Bull. 9, 464–469 (1983).55. W. Kaminsky and co-workers, Makromol. Chem., Rapid Commun. 4, 417–421 (1983).56. J. A. Ewen, J. Am. Chem. Soc. 106, 6355–6364 (1984).57. W. Kaminsky and co-workers, Angew. Chem., Int. Ed. Engl. 24, 507–508 (1985).
Vol. 4 SINGLE-SITE CATALYSTS 169
58. H. Schnutenhaus and H. H. Brintzinger, Angew. Chem., Int. Ed. Engl. 18, 777–778(1979).
59. F. R. W. P. Wild and co-workers, J. Organomet. Chem. 232, 233–247 (1982).60. F. R. W. P. Wild and co-workers, J. Organomet. Chem. 288, 63–67 (1985).61. J. A. Ewen and co-workers, J. Am. Chem. Soc. 110, 6255–6256 (1988).62. J. A. Ewen and co-workers, Makromol. Chem., Macromol. Symp. 48/49, 253–295
(1991).63. G. W. Coates and R. M. Waymouth, Science 267, 217–219 (1995).64. E. Hauptman, R. M. Waymouth, and J. W. Ziller, J. Am. Chem. Soc. 117, 11586–11587
(1995).65. U. Stehling and co-workers, Organometallics 13, 964–970 (1994).66. E. Barsties and co-workers, J. Organomet. Chem. 520, 63–68 (1996).67. N. Schneider and co-workers, Organometallics 16, 3413–3420 (1997).68. W. A. Herrmann and co-workers, Angew. Chem., Int. Ed. Engl. 28, 1511–1512 (1989).69. W. Spaleck and co-workers, New. J. Chem. 14, 499–503 (1990).70. W. Spaleck and co-workers, Angew. Chem., Int. Ed. Engl. 31, 1347–1350 (1992).71. W. Spaleck and co-workers, Organometallics 13, 954–963 (1994).72. J. A. Ewen and M. J. Elder, in Ref. 32, pp. 99–109.73. J. A. Ewen, Macromol. Symp. 89, 181–196 (1995).74. J. A. Ewen and co-workers, J. Am. Chem. Soc. 120, 10786–10787 (1998).75. I.-K. Lee and co-workers, Organometallics 11, 2115–2122 (1992).76. D. Veghini and co-workers, J. Am. Chem. Soc. 121, 564–573 (1999).77. A. Razavi and J. L. Atwood, J. Organomet. Chem. 459, 117–123 (1995).78. A. Razavi and co-workers, in Ref. 32, pp. 111–147.79. R. L. Halterman, Chem. Rev. 92, 965–994 (1992).80. Z. Chen and R. L. Halterman, Organometallics 13, 3932–3942 (1994).81. R. L. Halterman, D. Combs, and M. A. Khan, Organometallics 17, 3900–3907 (1998).82. N. Piccolrovazzi and co-workers, Organometallics 9, 3098–3105 (1990).83. L. Resconi and co-workers, Organometallics 15, 998–1005 (1996).84. L. Resconi and co-workers, Organometalllics 15, 5046–5059 (1996).85. L. Resconi and co-workers, J. Am. Chem. Soc. 120, 2308–2321 (1998).86. C. J. Schaverien and co-workers, J. Am. Chem. Soc. 120, 9945–9946 (1998).87. V. A. Dang and co-workers, Organometallics 18, 3781–3791 (1999).88. B. Rieger and co-workers, Organometallics 13, 647–653 (1994).89. U. Dietrich and co-workers, J. Am. Chem. Soc. 121, 4348–4355 (1999).90. H. G. Alt, W. Milius, and S. J. Palackal, J. Organomet. Chem. 472, 113–118 (1994).91. R. Leino and co-workers, Organometallics 15, 2450–2453 (1996).92. H. J. G. Luttikhedde and co-workers, Organometallics 15, 3092–3094 (1996).93. K. A. Ostoja Starzewski and co-workers, Angew. Chem., Int. Ed. Engl. 38, 2439–2443
(1999).94. J. Okuda, Chem. Ber. 123, 1649–1651 (1990).95. J. Okuda and co-workers, Organometallics 14, 789–795 (1995).96. F. Amor and J. Okuda, J. Organomet. Chem. 520, 245–248 (1996).97. D. W. Carpenetti and co-workers, Organometallics 15, 1572–1581 (1996).98. S. Ciruelos and co-workers, Organometallics 15, 5577–5585 (1996).99. F. G. Sernetz, R. Mulhaupt, and R. M. Waymouth, Macromol. Chem. Phys. 197, 1071–
1083 (1996).100. F. G. Sernetz and co-workers, J. Polym. Sci., Part A: Polym. Chem. 35, 1571–1578
(1997).101. A. L. McKnight and co-workers, Organometallics 16, 2879–2885 (1997).102. Y.-X. Chen and T. J. Marks, Organometallics 16, 3649–3657 (1997).103. P.-J. Sinnema and co-workers, Organometallics 16, 4245–4247 (1997).
170 SINGLE-SITE CATALYSTS Vol. 4
104. L. Duda and co-workers, Eur. J. Inorg. Chem. 1153–1162 (1998).105. A. Bertuleit and co-workers, Top. Catal. 7, 37–44 (1999).106. Eur. Pat. Appl. EP 416815 (1991); U.S. Pat. Appl. 401345 (1989), J. C. Stevens and
co-workers (to The Dow Chemical Company); Chem. Abstr. 115, 93163m (1991).107. Eur. Pat. Appl. EP 420436 (1991); U.S. Pat. Appl. 406945 (1989), J. A. M. Canich (to
Exxon Chemical Co.); Chem. Abstr. 115, 184145y (1991).108. P. J. Shapiro and co-workers, Organometallics 9, 867–869 (1990).109. B. Rieger, J. Organomet. Chem. 420, C17–C20 (1991).110. Y.-X. Chen and co-workers, Organometallics 16, 5958–5963 (1997).111. E. E. C. G. Gielens and co-workers, Organometallics 17, 1652–1654 (1998).112. N. Ishihara and co-workers, Macromolecules 19, 2464–2465 (1986).113. N. Ishihara, M. Kuramoto, and M. Uoi, Macromolecules 21, 3356–3360 (1988).114. N. Ishihara, Macromol. Symp. 89, 553–562 (1995).115. K. Yokota and co-workers, in Ref. 36, pp. 435–445.116. T. E. Ready and co-workers, Macromolecules 26, 5822–5823 (1993).117. A. Kucht and co-workers, Organometallics 12, 3075–3078 (1993).118. T. E. Ready, J. C. W. Chien, and M. D. Rausch, J. Organomet. Chem. 519, 21–28 (1996).119. P. Foster, J. C. W. Chien, and M. D. Rausch, Organometallics 15, 2404–2409 (1996).120. A. Zambelli, L. Oliva, and C. Pellechia, Macromolecules 22, 2129–2130 (1989).121. C. Pellechia and co-workers, J. Am. Chem. Soc. 117, 6593–6594 (1995).122. A. Grassi and co-workers, Macromolecules 31, 5588–5591 (1998).123. C. Pellechia and A. Grassi, Top. Catal. 7, 125–132 (1999).124. W. Kaminsky and co-workers, Macromolecules 25, 7647–7650 (1997).125. N. Schneider, M.-H. Prosenc, and H.-H. Brintzinger, J. Organomet. Chem. 545/546,
291–295 (1997).126. G. Xu, Macromolecules 31, 586–591 (1998).127. E. F. Williams, M. C. Murray, and M. C. Baird, Macromolecules 33, 261–268 (2000).128. R. Quyoum and co-workers, J. Am. Chem. Soc. 116, 6435–6436 (1994).129. Q. Wang and co-workers, Organometallics 15, 693–703 (1996).130. T. L. Tremblay and co-workers, Chem. Commun. 831–832 (1997).131. S. W. Ewart and M. C. Baird, Top. Catal. 7, 1–8 (1999).132. M. C. Baird, Chem. Rev. 100, 1471–1478 (2000).133. P. L. Watson, J. Am. Chem. Soc. 104, 337–339 (1982).134. P. L. Watson, J. Am. Chem. Soc. 104, 6471–6473 (1982).135. D. Stern, M. Sabat, and T. J. Marks, J. Am. Chem. Soc. 112, 9558–9575 (1990).136. P. J. Shapiro and co-workers, Organometallics 9, 867–869 (1990).137. P. J. Shapiro and co-workers, J. Am. Chem. Soc. 116, 4623–4640 (1994).138. J. P. Mitchell and co-workers, J. Am. Chem. Soc. 118, 1045–1053 (1996).139. J. H. Gilchrist and J. E. Bercaw, J. Am. Chem. Soc. 118, 12021–12028 (1996).140. M. B. Abrams and co-workers, Organometallics 18, 1389–1401 (1999).141. E. Ihara and co-workers, Macromol. Chem. Phys. 197, 1909–1917 (1996).142. A. Pastor and co-workers, J. Organomet. Chem. 528, 65–75 (1997).143. G. C. Bazan and co-workers, J. Am. Chem. Soc. 118, 2291–2292 (1996).144. G. C. Bazan and co-workers, Organometallics 16, 2492–2494 (1997).145. A. J. Ashe III and co-workers, Organometallics 17, 3883–3888 (1998).146. A. J. Ashe III, S. Al-Ahmad, and X. G. Fang, J. Organomet. Chem. 581, 92–97 (1999).147. A. J. Ashe III, X. G. Fang, and J. W. Kampf, Organometallics 18, 1363–1365 (1999).148. S. A. A. Shah and co-workers, Organometallics 15, 3176–3181 (1996).149. J. D. Scollard, D. H. McConville, and J. J. Vittal, Organometallics 14, 5478–5480
(1995).150. J. D. Scollard and co-workers, Macromolecules 29, 5241–5243 (1996).151. J. D. Scollard and D. H. McConville, J. Am. Chem. Soc. 118, 10008–10009 (1996).
Vol. 4 SINGLE-SITE CATALYSTS 171
152. J. D. Scollard, D. H. McConville, and S. J. Rettig, Organometallics 16, 1810–1812(1997).
153. J. D. Scollard and co-workers, J. Mol. Catal. A: Chem. 128, 201–214 (1998).154. L.-C. Liang and co-workers, J. Am. Chem. Soc. 121, 5797–5798 (1999).155. S. Tinkler and co-workers, Chem. Commun. 2623–2624 (1996).156. H. Mack and M. Eisen, J. Organomet. Chem. 525, 81–87 (1996).157. A. D. Horton and J. de With, Chem. Commun. 1375–1376 (1996).158. A. D. Horton and co-workers, Organometallics 15, 2672–2674 (1996).159. N. A. H. Male, M. Thornton-Pett, and M. Bochmann, J. Chem. Soc., Dalton Trans.
2487–2494 (1997).160. R. Baumann, W. M. Davis, and R. R. Schrock, J. Am. Chem. Soc. 119, 3830–3831
(1997).161. R. R. Schrock and co-workers, Organometallics 18, 3649–3670 (1999).162. B. Tsuie and co-workers, Organometallics 16, 1392–1400 (1997).163. Y. M. Jeon and co-workers, Organometallics 17, 3161–3163 (1998).164. C. H. Lee, Y.-H. La, and J. W. Park, Organometallics 19, 344–351 (2000).165. A. van der Linden and co-workers, J. Am. Chem. Soc. 117, 3008–3021 (1995).166. S. Fokken and co-workers, Organometallics 15, 5069–5072 (1996).167. S. Fokken and co-workers, Organometallics 16, 4240–4242 (1997).168. F. G. Sernetz and co-workers, Macromolecules 30, 1562–1569 (1997).169. K. Mashima, S. Fujikawa, and A. Nakamura, J. Am. Chem. Soc. 115, 10990–10991
(1993).170. K. Hakala and co-workers, Macromol. Rapid Commun. 18, 635–638 (1997).171. J. Jaffart and co-workers, Eur. J. Inorg. Chem. 425–428 (1998).172. A. Spannenberg and co-workers, Angew. Chem., Int. Ed. Engl. 37, 3363–3365 (1998).173. P. T. Witte, A. Meetsma, and B. Hessen, Organometallics 18, 2944–2946 (1999).174. M. P. Coles and co-workers, J. Organomet. Chem. 591, 78–87 (1999).175. M. Peuckert and W. Keim, Organometallics 2, 594–597 (1983).176. G. Wilke, Angew. Chem., Int. Ed. Engl. 27, 185–206 (1988).177. W. Keim and co-workers, Angew. Chem., Int. Ed. Engl. 17, 466–467 (1978).178. W. Keim, Angew. Chem., Int. Ed. Engl. 29, 235–244 (1990).179. L. K. Johnson, C. M. Killian, and M. Brookhart, J. Am. Chem. Soc. 117, 6414–6415
(1995).180. C. M. Killian and co-workers, J. Am. Chem. Soc. 118, 11664–11665 (1996).181. S. A. Svejda, L. K. Johnson, and M. Brookhart, J. Am. Chem. Soc. 121, 10634–10635
(1999).182. D. P. Gates and co-workers, Macromolecules 33, 2320–2334 (2000).183. M. Brookhart and B. L. Small, in 9th IUPAC Symposium on Organometallic Chemistry
Directed Towards Organic Synthesis, Gottingen, Germany, July 20–25, 1997. Book ofAbstracts, p. 447.
184. B. L. Small, M. Brookhart, and A. M. A. Bennett, J. Am. Chem. Soc. 120, 4049–4050(1998).
185. WO 98/27124 (1998), A. M. A. Bennett (to E.I. du Pont de Nermours & Co., Inc.);Chem. Abstr. 129, 122973x (1998).
186. A. M. A. Bennett, CHEMTECH 29(7), 24–28 (1999).187. G. J. P. Britovsek and co-workers, Chem. Commun. 849–850 (1998).188. G. J. P. Britovsek and co-workers, J. Am. Chem. Soc. 121, 8728–8740 (1999).189. B. L. Small and M. Brookhart, J. Am. Chem. Soc. 120, 7143–7144 (1998).190. B. L. Small and M. Brookhart, Macromolecules 32, 2120–2130 (1999).191. C. Wang and co-workers, Organometallics 17, 3149–3151 (1998).192. T. R. Younkin and co-workers, Science 287, 460–462 (2000).193. L. K. Johnson, S. Mecking, and M. Brookhart, J. Am. Chem. Soc. 118, 267–268 (1996).
172 SINGLE-SITE CATALYSTS Vol. 4
194. S. Mecking and co-workers, J. Am. Chem. Soc. 120, 888–899 (1998).195. S. D. Ittel, L. K. Johnson, and M. Brookhart, Chem. Rev. 100, 1169–1204 (2000).196. S. D. Ittel, in Ref. 36, pp. 616–628.197. R. F. Jordan, Adv. Organomet. Chem. 32, 325–387 (1991).198. E. Y.-X. Chen and T. J. Marks, Chem. Rev. 100, 1391–1434 (2000).199. J. Bliemeister and co-workers, in Ref. 32, pp. 57–82.200. H. Sinn and co-workers, in Ref. 36, pp. 105–122.201. D. W. Imhoff and co-workers, Organometallics 17, 1941–1945 (1998).202. D. W. Imhoff and co-workers, Organometallics, 1, 177–191 (1998).203. W. Kaminsky and R. Steiger, Polyhedron 7, 2375–2381 (1988).204. D. Cam and U. Giannini, Makromol. Chem. 193, 1049–1055 (1992).205. M. Bochmann and L. M. Wilson, J. Chem. Soc., Chem. Commun. 1610–1611 (1986).206. M. Bochmann and co-workers, Organometallics 7, 1148–1154 (1988).207. M. Bochmann, A. J. Jaggar, and J. C. Nicholls, Angew. Chem., Int. Ed. Engl. 29, 780–
782 (1990).208. R. F. Jordan and co-workers, J. Am. Chem. Soc. 108, 7410–7411 (1986).209. R. F. Jordan and co-workers, J. Am. Chem. Soc. 109, 4111–4113 (1987).210. R. F. Jordan and co-workers, Organometallics 8, 2892–2903 (1989).211. G. G. Hlatky, H. W. Turner, and R. E. Eckman, J. Am. Chem. Soc. 111, 2728–2729
(1989).212. A. D. Horton and J. H. G. Frijns, Angew. Chem., Int. Ed. Engl. 30, 1152–1154 (1991).213. J. J. W. Eshuis and co-workers, Organometallics 11, 362–369 (1992).214. Eur. Pat. Appl. EP 277,004 (1988), H. W. Turner (to Exxon Chemical Co.); Chem. Abstr.
110, 58290a (1989).215. U.S. Pat. Appl. 459,921 (1990), G. G. Hlatky, D. J. Upton, and H. W. Turner (to Exxon
Chemical Co.); Chem. Abstr. 115, 256897v (1991).216. X. Yang, C. L. Stern, and T. J. Marks, J. Am. Chem. Soc. 113, 3623–3625 (1991).217. X. Yang, C. L. Stern, and T. J. Marks, J. Am. Chem. Soc. 116, 10015–10031 (1994).218. P. A. Deck and T. J. Marks, J. Am. Chem. Soc. 117, 6128–6129 (1995).219. M. A. Giardello and co-workers, J. Am. Chem. Soc. 117, 12114–12129 (1995).220. L. Jia and co-workers, Organometallics 14, 3135–3137 (1995).221. Y.-X. Chen and co-workers, J. Am. Chem. Soc. 118, 12451–12452 (1996).222. Y.-X. Chen, C. L. Stern, and T. J. Marks, J. Am. Chem. Soc. 119, 2582–2583 (1997).223. L. Jia and co-workers, Organometallics 16, 842–857 (1997).224. Y.-X. Chen and co-workers, J. Am. Chem. Soc. 120, 6287–6305 (1998).225. L. Luo and T. J. Marks, Top. Catal. 7, 97–106 (1999).226. M. V. Metz and co-workers, Angew. Chem., Int. Ed. Engl. 39, 1312–1316 (2000).227. M. Bochmann and S. J. Lancaster, Organometallics 12, 633–640 (1993).228. M. Bochmann and co-workers, Organometallics 13, 2235–2243 (1994).229. M. Bochmann and S. J. Lancaster, J. Organomet. Chem. 497, 55–59 (1995).230. M. Bochmann, Top. Catal. 7, 9–22 (1999).231. V. C. Williams and co-workers, J. Am. Chem. Soc. 121, 3244–3245 (1999).232. V. C. Williams and co-workers, Angew. Chem., Int. Ed. Engl. 38, 3695–3698 (1999).233. V. C. Williams and co-workers, Organometallics 19, 1619–1621 (2000).234. J. C. W. Chien, W.-M. Tsai, and M. D. Rausch, J. Am. Chem. Soc. 113, 8570–8571
(1991).235. J. C. W. Chien, W. Song, and M. D. Rausch, Macromolecules 26, 3239–3240 (1993).236. A. R. Siedle and R. A. Newmark, J. Organomet. Chem. 497, 119–125 (1995).237. G. Kehr and co-workers, Chem. Eur. J. 6, 258–266 (2000).238. B. Temme and co-workers, Angew. Chem., Int. Ed. Engl. 34, 1755–1757 (1995).239. B. Temme, J. Karl, and G. Erker, Chem. Eur. J. 2, 919–924 (1996).240. J. Karl, G. Erker, and R. Frohlich, J. Am. Chem. Soc. 119, 11165–11173 (1997).
Vol. 4 SINGLE-SITE CATALYSTS 173
241. J. Karl and G. Erker, Chem. Ber. 130, 1261–1267 (1997).242. M. Dahlmann and co-workers, J. Am. Chem. Soc. 121, 2820–2828 (1999).243. M. Bochmann, S. J. Lancaster, and O. B. Robinson, J. Chem. Soc., Chem. Commun.
2081–2082 (1995).244. X. Song and M. Bochmann, J. Organomet. Chem. 545/546, 597–600 (1997).245. Y. Sun and co-workers, J. Am. Chem. Soc. 119, 5132–5143 (1997).246. W. E. Piers, Chem. Eur. J. 4, 13–18 (1998).247. W. E. Piers, Y. Sun, and L. M. Lee, Top. Catal. 7, 133–143 (1999).248. Y. Sun and co-workers, Organometallics 19, 1625–1627 (2000).249. P. Cossee, Tetrahedron Lett. 17, 12–16 (1960).250. P. Cossee, Tetrahedron Lett. 17, 17–21 (1960).251. P. Cossee, J. Catal. 3, 80–88 (1964).252. E. J. Arlman and P. Cossee, J. Catal. 3, 99–104 (1964).253. M. L. H. Green, Pure Appl. Chem. 50, 27–35 (1978).254. K. J. Ivin and co-workers, J. Chem. Soc., Chem. Commun. 604–606 (1978).255. Z. Dawoodi and co-workers, J. Chem. Soc., Chem. Commun. 1410–1411 (1982).256. D. T. Laverty and J. J. Rooney, J. Chem. Soc., Faraday Trans. 1 79, 869–878 (1983).257. M. Brookhart and M. L. H. Green, J. Organomet. Chem. 250, 395–408 (1983).258. L. Resconi, I. Camurati, and O. Sudmeijer, Top. Catal. 7, 145–163 (1999).259. W. Kaminsky, A. Ahlers, and N. Moller-Lindenhof, Angew. Chem., Int. Ed. Engl. 28,
1216–1218 (1989).260. B. J. Burger and co-workers, J. Am. Chem. Soc. 112, 1566–1577 (1990).261. T. Tsutsui, A. Mizuno, and N. Kashiwa, Polymer 30, 428–431 (1989).262. J. J. W. Eshuis and co-workers, J. Mol. Catal. 62, 277–287 (1990).263. L. Resconi and co-workers, J. Am. Chem. Soc. 114, 1025–1032 (1992).264. W. Kaminsky and H. Luker, Macromol. Chem., Rapid Commun. 5, 225–228 (1989).265. T. Tsutsui, N. Kashiwa, and A. Mizuno, Macromol. Chem., Rapid Commun. 11, 565–
570 (1990).266. J. C. W. Chien and B.-P. Wang, J. Polym. Sci., Part A: Polym. Chem. 28, 15–38 (1990).267. A. R. Siedle and co-workers, Polyhedron 9, 301–308 (1990).268. L. Resconi, S. Bossi, and L. Abis, Macromolecules 23, 4489–4491 (1990).269. A.-L. Mogstad and R. M. Waymouth, Macromolecules 25, 2282–2284 (1992).270. A.-L. Mogstad and R. M. Waymouth, Macromolecules 27, 2313–2315 (1994).271. R. Leino and co-workers, Macromolecules 30, 3477–3483 (1997).272. G. Erker and C. Fritze, Angew. Chem., Int. Ed. Engl. 31, 199–202 (1992).273. L. Resconi, L. Abis, and G. Franciscono, Macromolecules 25, 6814–6817 (1992).274. C. Pellechia and A. Zambelli, Macromol. Rapid Commun. 17, 333–338 (1996).275. C. Pellechia and co-workers, Macromolecules 29, 6990–6993 (1996).276. C. Pellechia and co-workers, J. Mol. Catal. A: Chem. 128, 229–237 (1998).277. G. Milano and co-workers, Organometallics 19, 1343–1349 (2000).278. P. Pino, P. Cioni, and J. Wei, J. Am. Chem. Soc. 109, 6189–6191 (1987).279. R. Waymouth and P. Pino, J. Am. Chem. Soc. 112, 4911–4914 (1990).280. M. K. Leclerc and H. H. Brintzinger, J. Am. Chem. Soc. 117, 1651–1652 (1995).281. M. K. Leclerc and H. H. Brintzinger, J. Am. Chem. Soc. 118, 9024–9032 (1996).282. A. K. Rappe, W. M. Skiff, and C. J. Casewit, Chem. Rev. 100, 1435–1456 (2000).283. K. Angermund and co-workers, Chem. Rev. 100, 1457–1470 (2000).284. L. Cavallo, G. Guerra, and P. Corradini, Gazz. Chim. Ital. 126, 463–467 (1988).285. P. Corradini and co-workers, Gazz. Chim. Ital. 118, 173–177 (1988).286. L. Cavallo and co-workers, Macromolecules 24, 1784–1790 (1991).287. G. Guerra and co-workers, J. Am. Chem. Soc. 116, 2988–2995 (1994).288. L. Cavallo and G. Guerra, Macromolecules 29, 2729–2737 (1996).289. G. Guerra and co-workers, J. Am. Chem. Soc. 119, 4394–4403 (1997).
174 SINGLE-SITE CATALYSTS Vol. 4
290. Z. Yu and J. C. W. Chien, J. Polym. Sci., Part A: Polym. Chem. 33, 125–135 (1995).291. Y. van der Leek and co-workers, Chem. Eur. J. 3, 585–591 (1997).292. L. A. Castonguay and A. K. Rappe, J. Am. Chem. Soc. 114, 5832–5842 (1992).293. E. P. Bierwagen, J. E. Bercaw, and W. A. Goddard III, J. Am. Chem. Soc. 116, 1481–
1489 (1994).294. R. J. Meier and co-workers, J. Am. Chem. Soc. 116, 7274–7281 (1994).295. T. Yoshida, N. Koga, and K. Morokuma, Organometallics 14, 746–758 (1995).296. R. D. J. Froese and co-workers, J. Am. Chem. Soc. 119, 7190–7196 (1997).297. E. A. H. Griffiths and co-workers, Chem. Commun. 1333–1334 (1999).298. D. G. Musaev and co-workers, J. Am. Chem. Soc. 119, 367–374 (1997).299. J. C. W. Lohrenz, T. K. Woo, and T. Ziegler, J. Am. Chem. Soc. 117, 12793–12800 (1995).300. T. K. Woo and co-workers, J. Am. Chem. Soc. 118, 13021–13030 (1996).301. M. S. W. Chan and co-workers, Organometallics 18, 4624–4636 (1999).302. M.-H. Prosenc and H.-H. Brintzinger, Organometallics 16, 3889–3894 (1997).303. S. Lieber, M.-H. Prosenc, and H.-H. Brintzinger, Organometallics 19, 377–387 (2000).304. T. Yoshida, N. Koga, and K. Morokuma, Organometallics 15, 766–777 (1996).305. R. D. J. Froese, D. G. Musaev, and K. Morokuma, J. Am. Chem. Soc. 120, 1581–1587
(1998).306. L. Deng and co-workers, J. Am. Chem. Soc. 119, 6177–6186 (1997).307. L. Dengl, P. Margl, and T. Ziegler, J. Am. Chem. Soc. 121, 6479–6487 (1999).308. P. Margl, L. Dengl, and T. Ziegler, Organometallics 18, 5701–5708 (1999).309. H. G. Alt and A. Koppl, Chem. Rev. 100, 1205–1222 (2000).310. Z. Guan and co-workers, Science 283, 2059–2062 (1999).311. W. Kaminsky, K. Kulper, and S. Niedoba, Makromol. Chem., Macromol. Symp. 3,
377–387 (1986).312. H. Drogemuller, K. Heiland, and W. Kaminsky, in W. Kaminsky and H. Sinn,
eds., Transition Metals and Organometallics as Catalysts for Olefin Polymerization,Springer-Verlag, Berlin, 1988, pp. 303–308.
313. K. Heiland and W. Kaminsky, Makromol. Chem. 193, 601–610 (1992).314. J. C. W. Chien and D. He, J. Polym. Sci., Part A: Polym. Chem. 29, 1585–1593
(1991).315. J. C. W. Chien and D. He, J. Polym. Sci., Part A: Polym. Chem. 29, 1595–1601
(1991).316. J. C. W. Chien and T. Nozaki, J. Polym. Sci., Part A: Polym. Chem. 31, 227–237 (1993).317. T. Uozumi and K. Soga, Makromol. Chem. 193, 823–831 (1992).318. N. Herfert, P. Montag, and G. Fink, Makromol. Chem. 194, 3167–3182 (1993).319. J. Koivumaki, G. Fink, and J. V. Seppala, Macromolecules 27, 6254–6258 (1994).320. J. Suhm, M. J. Schneider, and R. Mulhaupt, J. Polym. Sci., Part A: Polym. Chem. 35,
735–740 (1997).321. M. J. Schneider and co-workers, Macromolecules 30, 3164–3168 (1997).322. M. Miri, D. Hetzer, A. Miles, M. Pecak, and B. Riscili, in Ref. 36, pp. 509–515.323. T. Uozumi and co-workers, Macromol. Rapid Commun. 18, 883–889 (1997).324. M. Galimberti and co-workers, Macromol. Symp. 89, 259–275 (1995).325. R. Kravchenko and R. M. Waymouth, Macromolecules 31, 1–6 (1998).326. M. Arndt and co-workers, Macromol. Chem. Phys. 199, 1135–1152 (1998).327. M. K. Leclerc and R. M. Waymouth, Angew. Chem., Int. Ed. Engl. 37, 922–925 (1998).328. G. Xu, Macromolecules 31, 2395–2402 (1998).329. T. D. Shaffer, J. A. M. Canich, and K. R. Squire, Macromolecules 31, 5145–5147 (1998).330. J. F. Vega and co-workers, Macromolecules 29, 960–965 (1996).331. A. Malmberg and co-workers, Macromolecules 31, 8448–8454 (1998).332. E. Kokko and co-workers, J. Polym. Sci., Part A: Polym. Chem. 38, 376–388 (2000).333. G. W. Coates, Chem. Rev. 100, 1223–1252 (2000).
Vol. 4 SINGLE-SITE CATALYSTS 175
334. L. Resconi and co-workers, Chem. Rev. 100, 1253–1346 (2000).335. L. Resconi and co-workers, Macromolecules 28, 6667–6676 (1995).336. L. Resconi, J. Mol. Catal. A: Chem. 146, 167–178 (1999).337. V. Busico and R. Cipullo, J. Am. Chem. Soc. 116, 9329–9330 (1994).338. V. Busico and co-workers, Macromolecules 27, 7538–7543 (1994).339. V. Busico and R. Cipullo, J. Organomet. Chem. 497, 113–118 (1995).340. V. Busico and co-workers, J. Am. Chem. Soc. 118, 2105–2106 (1996).341. V. Busico and co-workers, Macromolecules 30, 3971–3977 (1997).342. V. Busico and co-workers, J. Mol. Catal. A: Chem. 128, 53–64 (1998).343. L. Resconi, F. Piemontesi, and R. L. Jones, in G. M. Benedikt and B. L. Goodall, eds.,
Metallocene Catalyzed Polymers. Materials, Properties, Processing & Markets, PlasticsDesign Library, New York, 1998, pp. 43–55.
344. W. Spaleck and co-workers, in Ref. 32, pp. 83–97.345. L. Resconi and co-workers, Organometallics 19, 420–429 (2000).346. T. Mise, S. Miya, and H. Yamazaki, Chem. Lett. 1853–1856 (1989).347. S. Collins and co-workers, Organometallics 10, 2061–2068 (1991).348. M. A. Giardello and co-workers, J. Am. Chem. Soc. 115, 3326–3327 (1993).349. M. A. Giardello and co-workers, J. Am. Chem. Soc. 117, 12114–12129 (1995).350. Y. Obora and co-workers, Organometallics 16, 2503–2505 (1997).351. S. Miyake, Y. Okumura, and S. Inazawa, Macromolecules 28, 3074–3079 (1995).352. H. Deng and co-workers, Macromolecules 29, 6371–6376 (1996).353. W. Spaleck and co-workers, Macromol. Symp. 89, 237–247 (1995).354. A. Razavi and co-workers, Macromol. Symp. 89, 345–367 (1995).355. G. Erker and co-workers, J. Am. Chem. Soc. 115, 4590–4601 (1993).356. G. Erker and co-workers, J. Organomet. Chem. 450, 1–7 (1993).357. M. Knickmeier, G. Erker, and T. Fox, J. Am. Chem. Soc. 118, 9623–9630 (1996).358. A. Razavi and J. L. Atwood, J. Am. Chem. Soc. 115, 7529–7530 (1993).359. G. Erker and co-workers, J. Am. Chem. Soc. 114, 10983–10984 (1992).360. G. Erker and co-workers, Chem. Ber. 127, 1551–1553 (1994).361. B. Rieger and co-workers, Macromolecules 23, 3559–3568 (1990).362. R. Fierro, J. C. W. Chien, and M. D. Rausch, J. Polym. Sci., Part A: Polym. Chem. 32,
2817–2824 (1994).363. M. L. H. Green and N. Ishihara, J. Chem. Soc., Dalton Trans. 657–665 (1994).364. B. Rieger, Polym. Bull. 32, 41–46 (1994).365. W. Spaleck and co-workers, J. Mol. Catal. A: Chem. 128, 279–287 (1998).366. D. T. Mallin and co-workers, J. Am. Chem. Soc. 112, 2030–2031 (1990).367. J. C. W. Chien and co-workers, J. Am. Chem. Soc. 113, 8569–8570 (1991).368. G. H. Llinas and co-workers, Macromolecules 25, 1242–1253 (1992).369. J. C. W. Chien and co-workers, J. Polym. Sci., Part A: Polym. Chem. 30, 2601–2617
(1992).370. G. H. Llinas and co-workers, Organometallics 12, 1283–1288 (1993).371. W. J. Gauthier and co-workers, Macromolecules 28, 3771–3778 (1995).372. W. J. Gauthiuer and S. Collins, Macromolecules 28, 3779–3786 (1995).373. A. M. Bravakis and co-workers, Macromolecules 31, 1000–1009 (1998).374. R. Kravchenko, A. Masood, and R. M. Waymouth, Organometallics 16, 3635–3639
(1997).375. J. L. Maciejewski Petoff and co-workers, Organometallics 16, 5909–5916 (1997).376. M. D. Bruce and co-workers, J. Am. Chem. Soc. 119, 11174–11182 (1997).377. S. Lin and co-workers, J. Mol. Catal. A: Chem. 136, 23–33 (1998).378. R. Kravchenko and co-workers, J. Am. Chem. Soc. 120, 2039–2046 (1998).379. P. Witte, T. K. Lal, and R. M. Waymouth, Organometallics 18, 4147–4155 (1999).380. S. Jungling and co-workers, J. Polym. Sci., Part A: Polym. Chem. 33, 1305–1317 (1995).
176 SINGLE-SITE CATALYSTS Vol. 4
381. S. Jungling, S. Koltzenburg, and R. Mulhaupt, J. Polym. Sci., Part A: Polym. Chem.35, 1–8 (1997).
382. N. Kashiwa, S. Kojoh, J. Imuta, and T. Tsutsui, in Ref. 36, pp. 30–37.383. J. A. Bandy and co-workers, J. Chem. Soc., Dalton Trans. 2207–2216 (1991).384. T. K. Han and co-workers, Macromolecules 28, 4801–4805 (1995).385. J. A. Ewen, Macromol. Symp. 89, 181–196 (1995).386. L. Cavallo and co-workers, Polymer 32, 1329–1335 (1991).387. G. Natta, I. Pasquon, and A. Zambelli, J. Am. Chem. Soc. 84, 1488–1490 (1962).388. J. A. Ewen and co-workers, Stud. Surf. Sci. Catal. 56, 439–482 (1990).389. F. Grisi and co-workers, J. Mol. Catal. A: Chem. 140, 225–233 (1999).390. B. Rieger, T. Repo, and G. Jany, Polym. Bull. 35, 87–94 (1995).391. K. Patsidis and co-workers, J. Organomet. Chem. 509, 63–71 (1996).392. H. G. Alt, R. Zenk, and W. Milius, J. Organomet. Chem. 514, 257–270 (1996).393. H. G. Alt and R. Zenk, J. Organomet. Chem. 518, 7–15 (1996).394. H. G. Alt and R. Zenk, J. Organomet. Chem. 522, 39–54 (1996).395. H. G. Alt and E. Samuel, Chem. Soc. Rev. 27, 323–329 (1998).396. H. Lee and co-workers, J. Organomet. Chem. 561, 37–47 (1998).397. T. A. Herzog, D. L. Zubris, and J. E. Bercaw, J. Am. Chem. Soc. 118, 11988–11989
(1996).398. S. Miyake and J. E. Bercaw, J. Mol. Catal. A: Chem. 128, 29–39 (1998).399. S. A. Miller and J. E. Bercaw, Abstr. Papers Am. Chem. Soc. 217, 151–INOR (1999).400. S. A. Miller, Metallocene-Mediated Olefin Polymerization: The Effects of Distal Lig-
and Perturbations on Polymer Stereochemistry, Ph.D. Thesis, California Institute ofTechnology, Pasadena, Calif., 2000.
401. M. J. Schneider and R. Mulhaupt, Macromol. Chem. Phys. 198, 1121–1129 (1997).402. F. Forlini and co-workers, Macromol. Chem. Phys. 198, 2397–2408 (1997).403. F. Forlini and co-workers, Macromol. Chem. Phys. 201, 401–408 (2000).404. I. Kim and Y. J. Kim, Polym. Bull. 40, 415–421 (1998).405. O. Henschke, A. Neubauer, and M. Arnold, Macromolecules 30, 8097–8100 (1997).406. T. Shiono, S. M. Azad, and T. Ikeda, Macromolecules 32, 5723–5727 (1999).407. G. Natta, J. Polym. Sci. 34, 531–549 (1959).408. J. W. Collette and co-workers, Macromolecules 22, 3851–3858 (1989).409. J. W. Collette and co-workers, Macromolecules 22, 3858–3866 (1989).410. W. Kaminsky and R. Spiehl, Makromol. Chem. 190, 515–526 (1989).411. W. Kaminsky, A. Bark, and M. Arndt, Makromol. Chem., Macromol. Symp. 47, 83–93
(1991).412. M. Arndt and W. Kaminsky, Macromol. Symp. 97, 225–246 (1995).413. W. Kaminsky, R. Engehausen, and J. Kopf, Angew. Chem., Int. Ed. Engl. 34, 2273–
2275 (1995).414. W. Kaminsky and A. Noll, in Ref. 32, pp. 149–158.415. B. A. Harrington and D. J. Crowther, J. Mol. Catal. A: Chem. 128, 79–84 (1998).416. M. Arndt and I. Beulich, Macromol. Chem. Phys. 199, 1221–1232 (1998).417. I. Tritto and co-workers, in Ref. 36, pp. 493–501.418. A. L. McKnight and R. M. Waymouth, Macromolecules 32, 2816–2825 (1999).419. L. Resconi and R. M. Waymouth, J. Am. Chem. Soc. 112, 4953–4954 (1990).420. L. Cavallo and co-workers, Macromolecules 26, 260–267 (1993).421. O. R. de Ballesteros and co-workers, Macromolecules 28, 2383–2388 (1995).422. S. A. Miller and R. M. Waymouth, in Ref. 32, pp. 441–454.423. G. W. Coates and R. M. Waymouth, J. Am. Chem. Soc. 113, 6270–6271 (1991).424. G. W. Coates and R. M. Waymouth, J. Am. Chem. Soc. 115, 91–98 (1993).425. M. R. Kesti, G. W. Coates, and R. M. Waymouth, J. Am. Chem. Soc. 114, 9679–9680
(1992).
Vol. 4 SINGLE-SITE CATALYSTS 177
426. N. Tomotsu and co-workers, J. Mol. Catal. A: Chem. 128, 167–190 (1998).427. G. Xu and E. Ruckenstein, J. Polym. Chem., Part A: Polym. Chem. 37, 2481–2488
(1999).428. W. Kaminsky, D. Arrowsmith, and C. Strubel, J. Polym. Chem., Part A: Polym. Chem.
37, 2959–2968 (1999).429. U. Giannini and co-workers, J. Polym. Sci.: Part C 22, 157–175 (1968).430. J. Boor Jr., Ziegler–Natta Catalysts and Polymerizations, Academic Press, New York,
1979.431. T. Simonazzi, A. De Nicola, M. Aglietto, and G. Ruggeri, in G. Allan, S. L. Aggar-
wal, and S. Russo, eds., Comprehensive Polymer Science, First Supplement, PergamonPress, Oxford, 1992, pp. 133–158.
432. U. M. Stehling and co-workers, Macromolecules 31, 2019–2027 (1998).433. U. M. Stehling and co-workers, Macromolecules 32, 14–20 (1999).434. M. J. Schneider, R. Schafer, and R. Mulhaupt, Polymer 38, 2455–2459 (1997).435. R. Goretzki and G. Fink, Macromol. Rapid Commun. 19, 511–515 (1998).436. M. M. Marques and co-workers, J. Polym. Sci., Part A: Polym. Chem. 37, 2457–2469
(1999).437. S. Bruzard and co-workers, Macromol. Chem. Phys. 198, 291–303 (1997).438. P. Aaltonen and B. Lofgren, Macromolecules 28, 5353–5357 (1995).439. P. Aaltonen and co-workers, Macromolecules 29, 5255–5260 (1996).440. C.-E. Wilen and J. H. Nasman, Macromolecules 27, 4051–4057 (1994).441. U. M. Stehling and co-workers, Macromolecules 31, 4396–4398 (1998).442. P. A. Fox, R. M. Waymouth, and C. Hawker, Polym. Prepr. (Am. Chem. Soc., Div. Polym.
Chem.) 40, 872–873 (1999).443. W. Wiyatno and co-workers, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 41(1),
85–86 (2000).444. J. Okuda and I. L. Rushkin, Macromolecules 26, 5530–5532 (1993).445. J. Okuda and co-workers, J. Organomet. Chem. 501, 37–39 (1995).446. M. Hayakawa and co-workers, Macromol. Chem. Phys. 198, 1305–1317 (1997).447. M. Hayakawa and co-workers, Macromol. Rapid Commun. 17, 865–870 (1996).448. H. Yasuda and co-workers, J. Am. Chem. Soc. 114, 4908–4910 (1992).449. D. G. Ward and S. Collins, J. Am. Chem. Soc. 114, 5460–5462 (1992).450. Y. Li and co-workers, Macromolecules 30, 1875–1883 (1997).451. H. Nguyen and co-workers, Macromolecules 33, 1508–1510 (2000).452. M. A. Giardello and co-workers, J. Am. Chem. Soc. 117, 3276–3277 (1995).453. K. Soga and co-workers, Macromolecules 27, 7938–7940 (1994).454. H. Deng, T. Shiono, and K. Soga, Macromolecules 28, 3067–3073 (1995).455. H. Deng and K. Soga, Macromolecules 29, 1847–1848 (1996).456. L. S. Boffa and B. M. Novak, Macromolecules 27, 6993–6995 (1994).457. L. S. Boffa and B. M. Novak, Macromolecules 30, 3494–3506 (1997).458. M. Cheng and co-workers, J. Am. Chem. Soc. 121, 11583–11584 (1999).459. S. G. Correia and co-workers, J. Polym. Sci., Part A: Polym. Chem. 37, 2471–2480
(1999).460. R. Mulhaupt, T. Duschek, and B. Rieger, Makromol. Chem., Macromol. Symp. 48/49,
317–332 (1991).461. T. Shiono and K. Soga, Macromolecules 25, 3356–3361 (1992).462. T. Shiono and co-workers, Macromolecules 26, 2085–2089 (1993).463. T. C. Chung, H. L. Lu, and C. L. Li, Macromolecules 27, 7533–7537 (1994).464. W. Kaminsky, D. Arrowsmith, and H. R. Winkelbach, Polym. Bull. 36, 577–584 (1996).465. M. Hackmann and co-workers, Macromol. Chem. Phys. 199, 1511–1517 (1998).466. T. C. Chung and H. L. Lu, J. Mol. Catal. A: Chem. 115, 115–127 (1997).467. T. C. Chung and W. Janvikul, J. Organomet. Chem. 581, 176–187 (1999).
178 SINGLE-SITE CATALYSTS Vol. 4
468. G. Xu and T. C. Chung, J. Am. Chem. Soc. 121, 6763–6764 (1999).469. K. Koo and T. J. Marks, J. Am. Chem. Soc. 120, 4019–4020 (1998).470. K. Koo and T. J. Marks, J. Am. Chem. Soc. 121, 8791–8802 (1999).471. Y. Okamoto and T. Nakano, Chem. Rev. 94, 349–372 (1994).472. F. Ciardelli, in J. I. Kroschwitz, ed., Encyclopedia of Polymer Science and Engineering,
Vol. 10, Wiley, New York, 1987, pp. 463–493.473. A. Schafer and co-workers, J. Organomet. Chem. 328, 87–99 (1987).474. S. Collins, B. A. Kuntz, and Y. Hong, J. Org. Chem. 54, 4154–4158 (1989).475. B. Chin and S. L. Buchwald, J. Org. Chem. 62, 2267–2268 (1997).476. A. H. Hoveyda and J. P. Morken, Angew. Chem., Int. Ed. Engl. 35, 1262–1284 (1996).477. W. Kaminsky, Angew. Makromol. Chem. 145/146, 149–160 (1986).478. W. Kaminsky, A. Ahlers, and N. Moller-Lindenhof, Angew. Chem., Int. Ed. Engl. 28,
1216–1218 (1989).479. P. Pino, P. Cioni, and J. Wei, J. Am. Chem. Soc. 109, 6189–6191 (1987).480. P. Pino and co-workers, Makromol. Chem. 191, 1677–1688 (1990).481. M. Farina, Top. Stereochem. 17, 1–111 (1987).482. A. Sen, Acc. Chem. Res. 26, 303–310 (1993).483. M. Brookhart and co-workers, J. Am. Chem. Soc. 116, 3641–3642 (1994).484. M. R. Ribeiro, A. Deffieux, and M. F. Portela, Ind. Eng. Chem. Res. 36, 1224–1237
(1997).485. C. Jenny and P. Maddox, Curr. Opin. Solid State Mater. Sci. 3, 94–103 (1998).486. H. C. L. Abbenhuis, Angew. Chem., Int. Ed. Engl. 38, 1058–1060 (1999).487. J. C. W. Chien, Top. Catal. 7, 23–26 (1999).488. W. Kaminsky and H. Winkelbach, Top. Catal. 7, 61–67 (1999).489. M. O. Kristen, Top. Catal. 7, 89–95 (1999).490. G. G. Hlatky, Chem. Rev. 100, 1347–1376 (2000).491. G. Fink and co-workers, Chem. Rev. 100, 1377–1390 (2000).492. S. Jungling, S. Koltzenburg, and R. Mulhaupt, J. Polym. Sci., Part A: Polym. Chem.
35, 1–8 (1997).493. U.S. Pat. 4,808,561 (1989), H. Welborn (to Exxon Chemical Co.); Chem. Abstr. 106,
157033 (1987).494. J. C. W. Chien and D. He, J. Polym. Sci., Part A: Polym. Chem. 29, 1603–1607 (1991).495. S. Collins, W. M. Kelly, and D. A. Holden, Macromolecules 25, 1780–1785 (1992).496. W. Kaminsky and F. Renner, Makromol. Chem., Rapid Commun. 14, 239–243 (1993).497. K. Soga and M. Kaminaka, Macromol. Chem. Phys. 195, 1369–1379 (1994).498. K. Soga, H. J. Kim, and T. Shiono, Macromol. Chem. Phys. 195, 3347–3360 (1994).499. K. Soga and co-workers, Macromol. Symp. 97, 53–62 (1995).500. M. C. Sacchi and co-workers, Macromol. Rapid Commun. 16, 581–590 (1995).501. G. G. Hlatky and D. J. Upton, Macromolecules 29, 8019–8020 (1996).502. D. Harrison and co-workers, J. Mol. Catal. A: Chem. 128, 65–77 (1998).503. S. I. Woo, Y. S. Ko, and T. K. Han, Macromol. Rapid Commun. 16, 489–494 (1995).504. M. Michelotti and co-workers, J. Mol. Catal. A: Chem. 129, 241–248 (1998).505. M. Michelotti and co-workers, J. Mol. Catal. A: Chem. 152, 167–177 (2000).506. T. Maschmeyer and co-workers, Nature 378, 159–162.507. Y. S. Ko and co-workers, Macromol. Rapid Commun. 17, 749–758 (1996).508. J. Tudor and D. O’Hare, Chem. Commun. 603–604 (1997).509. L. K. Van Looveren and co-workers, Angew. Chem., Int. Ed. Engl. 37, 517–520 (1998).510. K. Kageyama, J. Tamazava, and T. Aida, Science 285, 2113–2115 (1999).511. D. Lee and K. Yoon, Macromol. Rapid Commun. 15, 841–843 (1994).512. D. Lee and K. Yoon, Macromol. Symp. 97, 185–193 (1995).513. H. Nishida and co-workers, Macromol. Rapid Commun. 16, 821–830 (1995).514. T. Kitagawa and co-workers, Polymer 38, 615–620 (1997).
Vol. 4 STABILIZATION 179
515. S. B. Roscoe and co-workers, Science 280, 270–273 (1998).516. T. Arai and co-workers, Macromol. Chem. Phys. 198, 229–237 (1997).517. K. Soga and co-workers, Macromol. Chem. Phys. 198, 2779–2787 (1997).518. H. Ahn and T. J. Marks, J. Am. Chem. Soc. 120, 13533–13534 (1998).519. H. G. Alt, J. Chem. Soc., Dalton Trans. 1703–1709 (1999).