29
“SLE: Fighting Self Sabotage” Feb. 2005 Immunology in health and Disease Susan Manzi, MD, MPH Associate Professor of Medicine and Epidemiology Co-Director Lupus Center of Excellence

“SLE: Fighting Self Sabotage”
Feb. 2005Immunology in health and Disease
Susan Manzi, MD, MPH
Associate Professor of Medicine and Epidemiology
Co-Director Lupus Center of Excellence

ObjectivesI. Epidemiology
II. Pathogenesis
Genetic
Sources of autoantibodies
Environmental triggers
Defective immune regulation
Gender/hormonal factors
III. Clinical and laboratory features
Diagnosis/natural history
Autoantibodies
Treatment



Who Gets Lupus?
Females > Males 7:1– Childbearing 12:1– Children, elderly 2:1
African-American (3-4x) > CaucasianAsianHispanic

Female Preponderance of
Autoimmune Disease
25-50:1
4-7:1
4:1
6:1
8:1
4-8:1
2:1
Thyroid diseases
Diffuse lymphocytic thyroiditis
Goitrous, struma lymphomatosa (Hashimoto),
Hypercellular variant, adult onset
Hypercellular variant, juvenile onset
Fibrous variant
Non goitrous
Severe atrophic (myxedema)
Mild atrophic (asymptomatic)
Primary hyperthyroidism (Graves Basedow disease)
With benign or no exophthalmos
With progressive ophthalmopathy
Female/ Male Ratio
Disease

Female Preponderance of
Autoimmune Disease Disease Female/Male
Ratio
Systemic lupus erythematosus
Rheumatoid arthritis
Sjogren’s syndrome
Idiopathic adrenal insufficiency (autoimmune adrenal disease)
Scleroderma
Myasthenia gravis
Multiple sclerosis
9:1
2-4:1
9:1
2-3:1
3-4:1
2:1
1-5:1

Prevalence of SLE
• African-American women: 56-283• Caucasian women: 17-71• African-American men: 3-53• Caucasian men: 3-19
Range of prevalence figures per 100,000 persons
Systemic lupus erythematosus in “Women and Health”(Goldman and Hatch, eds.), pp 704-723, 2000

Observations to Support Genetic Factors in Lupus
1. Clustering in families
2. Concordance
- monozygotic (identical twins)
25-30%
- dizygotic 5%
3. Other autoimmune conditions in family members

Pathogenesis of SLE

Pathogenesis of SLE
Genetic
Environmental
Hormonal
THELPER
Ag Autoantibodies
Immune Complexes
Tissue Damage
B
B and T Cell Hyperactivity
Defective Immune Regulation

Mode of Inheritance
Polygenic (>95%) VS Monogenic (<5%)
Homozygous deficiency of:C1q 38/41 (93%) C4 14/16 (88%)C2 38/66 (58%)


Paradox
Complement activation plays a critical role in the inflammatory process and tissue damage in SLE, but early complement deficiencies cause SLE.

Possible Explanations
1. C1q clears immune complexes
2. C1q binds to and clears apoptotic blebs (sources of autoantigens)
3. Absence of C1q permits sustained infections that could trigger autoimmune response.

Genes increase susceptibility to SLE
In the major histocompatibility complex (MHC)C2,C4 deficiencyDR2,DR3TNF- polymorphisms
In non-MHCC1q deficiency (rare, but greatest risk!!)Chromosome 1 region 1q41-43 (PARP)
region 1q23 (FcRIIA,
RIIIA)Polymorphisms in IL-10, IL-6 and
mannose-binding protein

Sources of Autoantigens
1. Apoptotic cells
2. Activated cells (antigens move to cell
membrane)
3. Modification of proteins during apoptosis
4. Infectious agents


Sources of Autoantigens
4. Infectious agents
- molecular mimicry
- epitope spreading
- nonspecific activation of B/T cells
- infection induced apoptosis

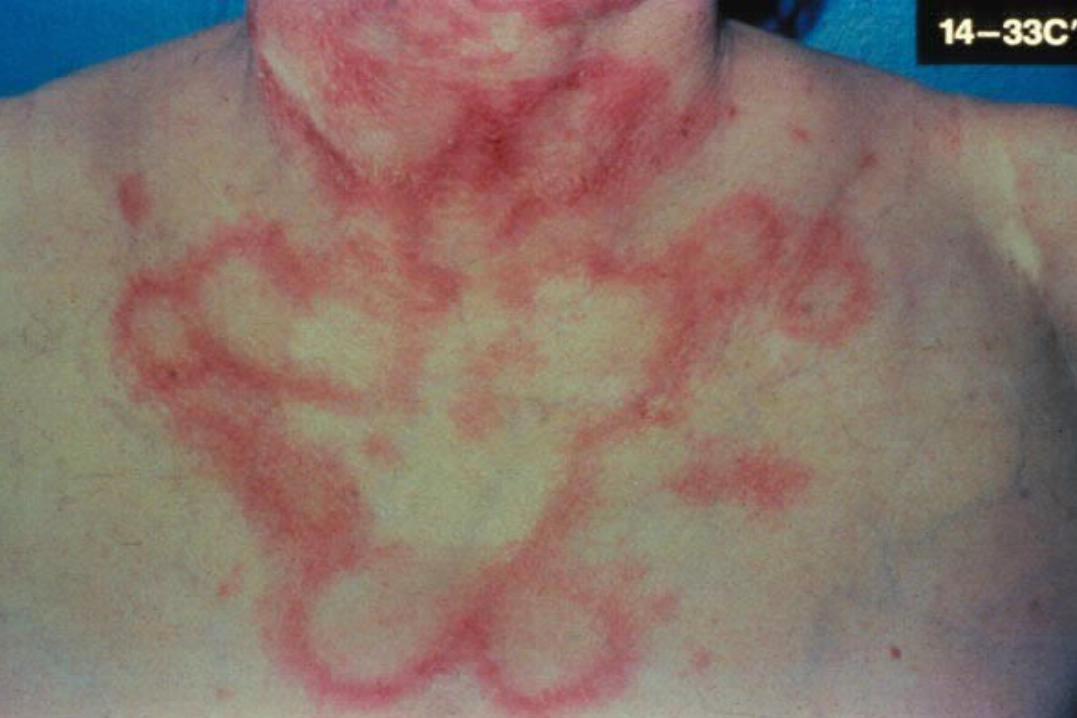
Environmental Triggers
Ultraviolet light (photosensitivity)
Drug-induced lupus
milder, male =female, older ages
Infectious agents (EBV, CMV)
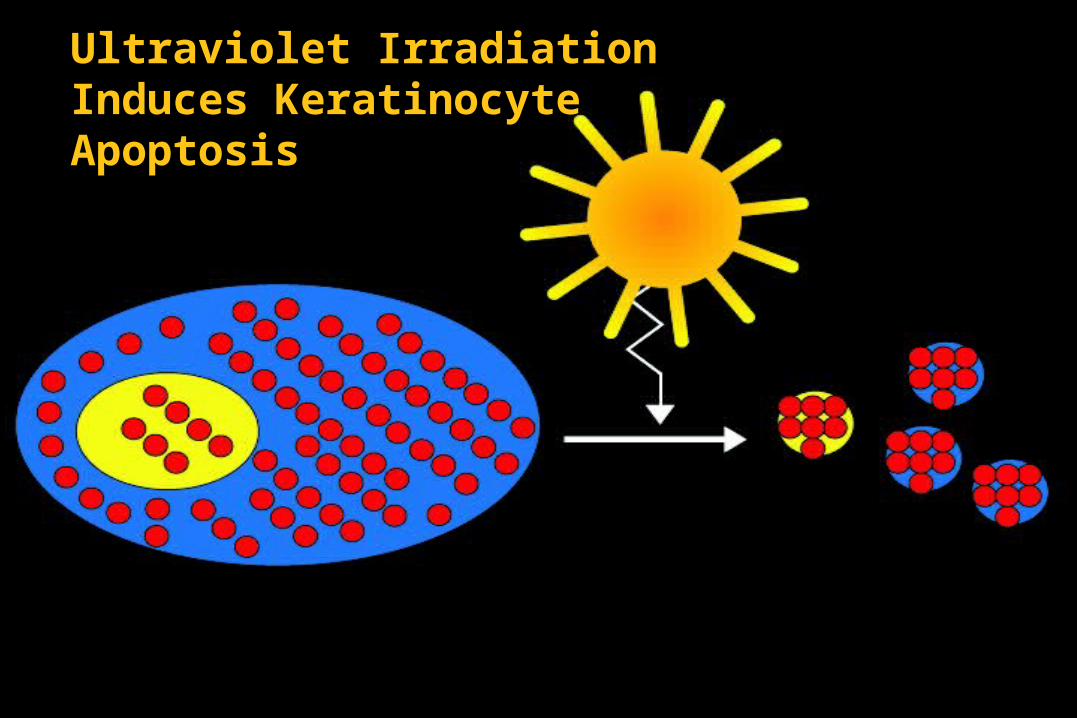
Ultraviolet Irradiation Induces Keratinocyte Apoptosis

Ultraviolet Irradiation Induces Pro-Inflammatory Cytokines
TNFIL1GMCSFIL8PGE2LTB4

Complement Mediates Clearance of Apoptotic Blebs
TNFIL1GMCSFIL8PGE2LTB4
Tolerance
C3C3
C3
C3
C3
C4
C4
C4
C4
C4 C4
C4
C4
C4
C3
C3C3
C3C4
C4

Impaired Clearance of Apoptotic Blebs in SLE
Lymph NodesSpleenAuto-Antibody
TNFIL1GMCSFIL8PGE2LTB4
C3C3
C3
C3
C3
C4
C4
C4
C4
C4 C4
C4
C4
C4
C3
C3C3
C3C4
C4

Defective Immune Regulation
B and T-cell hyperactivity
Sustained autoantigens/impaired clearance of apoptotic cells
Epitope spreading due to lack of “turn off”
Exaggerated intracellular response to activation

Defective Immune Regulation
B and T-cell hyperactivity
Increased production of pro-inflammatory cytokines
Decreased clearance of immune complexes
Increased expression of surface molecules that increase B/T- cell activation
(CD40L)

CD40L-CD40 Interactions
T-cellB
Cell
CD40 CD40L (gp39)
TCR
CD3
CD40: B-cells, endothelial cells, macrophages, Ag-presenting cells, renal parenchymal, tubular, etc cells
CD40L: T-cells, platelets

















