Page 1
Accepted Manuscript
Small molecule fusion inhibitors: design, synthesis and biological evaluation of
(Z)-3-(5- (3-benzyl-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-car‐
boxy-4-hydroxy)phenyl-2,5- dimethylpyrroles and related derivatives targeting
HIV-1 gp41
Xiao-Yang He, Lu Lu, Jiayin Qiu, Peng Zou, Fei Yu, Xing-Kai Jiang, Lin Li,
Shibo Jiang, Shuwen Liu, Lan Xie
PII: S0968-0896(13)00372-6
DOI: http://dx.doi.org/10.1016/j.bmc.2013.04.046
Reference: BMC 10786
To appear in: Bioorganic & Medicinal Chemistry
Received Date: 2 February 2013
Revised Date: 16 April 2013
Accepted Date: 17 April 2013
Please cite this article as: He, X-Y., Lu, L., Qiu, J., Zou, P., Yu, F., Jiang, X-K., Li, L., Jiang, S., Liu, S., Xie, L.,
Small molecule fusion inhibitors: design, synthesis and biological evaluation of (Z)-3-(5- (3-benzyl-4-oxo-2-
thioxothiazolidinylidene)methyl)-N-(3-carboxy-4-hydroxy)phenyl-2,5- dimethylpyrroles and related derivatives
targeting HIV-1 gp41, Bioorganic & Medicinal Chemistry (2013), doi: http://dx.doi.org/10.1016/j.bmc.2013.04.046
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Page 2
ACCEPTED MANUSCRIPT
1
Small molecule fusion inhibitors: design, synthesis and biological evaluation of (Z)-3-(5-
(3-benzyl-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-4-hydroxy)phenyl-2,5-
dimethylpyrroles and related derivatives targeting HIV-1 gp41
Xiao-Yang Hea, #, Lu Lub, Jiayin Qiuc, Peng Zoud, Fei Yub,c,d, Xing-Kai Jianga, Lin Lic, Shibo Jiangb,
d, Shuwen Liuc, Lan Xiea*
a Beijing Institute of Pharmacology and Toxicology, Beijing 100850, China
b Key Laboratory of Medical Molecular Virology of Ministries of Education and Health, Shanghai
Medical College and Institute of Medical Microbiology, Fudan University, Shanghai 200032, China
c School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
d Lindsley F. Kimball Research Institute, New York Blood Center, NY 10065, USA
*Corresponding author, e-mail: [email protected] , (L. Xie); Tel/Fax: 86-10-6931690 # Current address: Beijing Institute of Radiation Medicine, Beijing, 100850, China
Page 3
ACCEPTED MANUSCRIPT
2
Abstract:
By a scaffold elongation strategy, a series of (Z)-3-(5-(3-benzyl-4-oxo-2-thioxothiazolidinyl
idene)methyl)-N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrroles and related derivatives with a
linear multi- aromatic ring skeleton were designed, synthesized, and evaluated in HIV-1 gp41 and
cellular assays. Among them, the most active compounds, 12e, 12g, and 12k with a one-carbon
linker (n = 1) between the rhodanine (C) and phenyl (D) rings, exhibited very promising inhibitory
potency with IC50 values of 1.8-2.6 µM and EC50 values of 0.3-1.5 µM against gp41 6-HB formation
and HIV-1 replication in MT-2 cells, respectively. Additionally, they were almost equally effective
against both T20-sensitive and resistant strains. The related SAR studies and molecular modeling
results provided potential for further developing a new class of non-peptide small molecular fusion
inhibitors targeting the HIV-1 gp41.
Key words: 3-Substituted N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrrole derivatives,
Small-molecule fusion inhibitors, HIV-1 gp-41, Anti-HIV agents
Graphic Abstract
COOHHO
N
S N
O
S
A
B
CD
COOHHO
N
IC50 37.4 µM (gp41- 6HB)EC50 0.7 µM (cells)
IC50 1.8-2.6 µM (gp41- 6HB)EC50 0.3-1.5 µM (cells)
2 12e, 12g, 12k
R
Page 4
ACCEPTED MANUSCRIPT
3
1. Introduction
The HIV-1 envelope glycoprotein subunit gp41 is considered one of the most attractive targets
for anti-HIV drugs because it plays a crucial role in the fusion process between virus and host cell
membranes. Gp41 is originally buried within glycoprotein gp120 by non-covalent bond, but it
becomes exposed when gp120 binds to receptor CD4 on the T cells. This causes the formation of a
stable six-helix bundle (6-HB) core structure by an N-terminal heptad repeat (NHR) trimer with three
C-terminal heptad repeats (CHR) in an anti-parallel manner, thus bringing the viral and host cell
membrane into proximity and fusion.1-4 By mimicking the CHR peptide sequence of gp41, the
synthetic peptide drug T-20,5, 6 as the first fusion inhibitor targeting gp41, has been successfully used
to treat HIV infection and AIDS patients. However, the clinical use of this peptide has been limited
by high cost, lack of oral availability and metabolic instability. Therefore, to overcome the
limitations of current peptide HIV-fusion inhibitors,7 new strategies for discovering nonpeptide
small-molecule fusion inhibitors with high potency are needed. Indeed, we have already identified
the binding site on HIV-1 gp41 for small molecule inhibitors as a highly conserved hydrophobic
pocket on the surface of inner NHR trimer of gp41.8, 9 It is a linear cavity (ca. 16 Å long, 7 Å wide
and 5 – 6 Å) and can be filled by three conserved hydrophobic CHR residues, W628, W631 and I635,
to form stable 6-HB of gp41. In particular, amino acid K574 on the binding site is demonstrated to
play a critical role in 6-HB stability10-12 by forming a “salt bridge” with D632 of CHR. Therefore,
some small molecules that can bind to this pocket with high affinity should be able to block the 6-HB
formation of gp41 and thereby inhibit HIV-1 entry and replication. So far, several series of
small-molecule compounds with inhibitory activities against 6-HB of HIV-1 gp41 and/or cell-cell
fusion (CCF) have been reported, such as N-phenyl pyrroles NB-2, NB-64,13 phenyl furan
derivatives,14, 15 indole derivatives,16, 17 benzamide,18 biphenyl ethylene ethers,19 �-helical
peptidomimetic compounds,20, 21 and plant natural products.22-24 However, their potency is mostly at
�M level, much less than those of T20 and other peptide derivatives at nM level. Thus, more research
Page 5
ACCEPTED MANUSCRIPT
4
is required to discover highly potent small molecules and design them as HIV-1 fusion inhibitors
targeting gp41.
In our hit-to-lead optimization process for small-molecule inhibitors targeting gp41 (Figure
1), the initial structural modification based on hit NB-64 discovered two lead compounds, 1 and 2,
both N-phenyl-2,5-dimethylpyrroles, which showed promising inhibitory potency against gp41 6-HB
formation (IC50 25.6 and 37.4 �M, respectively) and against HIV-1 replication (EC50 7.7 and 0.7 �M,
respectively) in cellular assay.25 Since it occupied only a small space in the gp41 binding pocket,
lead 1 was modified by elongating the molecular skeleton to increase molecular volume and shape
complement. Subsequently, a new lead 3 with a linear multi-aromatic-ring skeleton was discovered.
It exhibited improved inhibitory activity against gp41 6-HB formation (IC50 4.4 �M) and maintained
high potency against HIV-1 replication (EC50 3.2 �M) in cellular assay.26 Molecular modeling of 3
demonstrated that the new linear multi-aromatic-ring scaffold fit very well in the linear binding
cavity of gp41, suggesting that it would be beneficial in increasing the affinity between the small-
molecule inhibitor and its biologic target. Prior structure-activity relationship (SAR) analysis also
indicated that an introduced N-substituted rhodanine fragment was a preferable moiety for
maintaining and/or enhancing molecular inhibitory potency against gp41 6-HB formation and HIV-1
replication, thus offering a new linear scaffold that improves lead potency. In this continuing study,
the optimization of lead 2 was conducted to find even more potent small-molecule inhibitors
targeting gp41 and more SARs. As shown in Figure 1, a series of new 3-substituted
N-(3-carboxy)phenyl-2,5-dimethylpyrrole compounds with a linear multi-aromatic-ring scaffold
were designed, synthesized, and evaluated against gp41 6-HB formation and HIV-1 replication in
cellular assay. An N-aryl rhodanine moiety was first introduced at the 3-position of the pyrrole ring
in lead 2 to build up the required linear molecular scaffold, while the structure of 2 was maintained.
Subsequent modifications were performed on (1) the linker length (n = 0, 1, 2) between the
rhodanine ring (C-ring) and a phenyl ring (D-ring), (2) various substitutents (R1) and their positions
(p-, m-, o-) on the D ring, (3) heteroatom replacement X/Y on the rhodanine ring (C ring), and (4) the
ortho-substitutent of the carboxy group (R = OH, Cl, H ) on the A-ring, successively. Meanwhile,
molecular modeling was also used to assist us in designing the new compounds in the context of
Page 6
ACCEPTED MANUSCRIPT
5
their interaction with the hydrophobic pocket of HIV-1 gp41, especially for the key amino acid
residue K574. Related new results are reported herein.
Figure 1 Lead optimization strategy and new target compounds
2. Chemistry
All target compounds were synthesized as described in Schemes 1-2. For elongation of
molecular skeleton, lead 2 was first introduced an aldehyde group at the 3-position of the pyrrole
ring by using Vilsmeier-Hacck reaction with DMF/POCl3 to obtain N-(3-carboxy-4-hydroxy)
phenyl-2,5-dimethyl-3-formylpyrrole (4). Different N-substituted rhodanine building blocks,
including 8 (n = 0), 9 (n = 1), or 10 (n = 2), were synthesized from commercially available reagents
5-7, respectively, by using methods in our previous studies.25 The Knoevenagel condensation
reactions between 4 and 8, or 9 and 10, were then carried out in the presence of ammonium acetate in
toluene and methanol refluxed for 3-5 h to afford a series of new target compounds 11, 12 (a-e, g-k,
m-n), and 13. Compounds 12e and 12k with an ester group on the phenyl (D-ring) were hydrolyzed
in acidic condition to provide corresponding carboxylic compounds 12f and 12l, respectively.
Similar to the preparation of 4, an aldehyde group was introduced in reagents
N-(4-chloro-3-carboxy)phenyl-2,5-dimethylpyrrole and N-(3-carboxy)phenyl-2,5-dimethylpyrrole to
produce intermediates 14 (R’ = Cl) and 15 (R’ = H), respectively. Subsequently, a coupling reaction
Page 7
ACCEPTED MANUSCRIPT
6
with 9a yielded compounds 16 and 17, respectively. The condensation of 4 with 18, which was
prepared from thiazolidine-2,4-dione and benzyl bromide in the presence of K2CO3 in DMF, yielded
compound 19, containing an isostere of rhodanine as the C-ring. Building block 21 with a
pyrrolin-2,4-dione ring was prepared by treatment of maleic anhydride with phenyl ethylamine,
followed by a condensation in acetic anhydride.27 After an esterification of 4, the produced
compound 20 was coupled with 21 in the presence of PPh3 in ethanol to afford product 22. Finally,
compound 22 was hydrolyzed under acidic condition to afford carboxylic compound 23.
All new target compounds were identified by 1H NMR and MS spectroscopic data.
Consistent with our prior studies, these new 5-arylidene rhodanine derivatives were also identified as
thermodynamically stable cis-isomers (Z-isomer) because their olefin proton signals of the exocyclic
C=C bond showed a range of 7.66-7.83 ppm (literature � 7.70-7.82 ppm) by the deshielding effect of
the cis-carbonyl (C=O). However, thiazolin-2,4-dione compound 23 was assigned as trans-isomer
(E-isomer) based on the group priority on the exocyclic C=C bond.
9,12 R a H b p-CF3 c p-Br d p-OCH3 e p-COOCH3 f p-COOH g p-t-Bu h m-CF3 i m-Br j m-OCH3 k m-COOCH3 l m-COOH
m o-CF3 n o-Br
�
Scheme 1. Reagents and conditions: (i) POCl3/DMF, <10 ºC; (ii) for 5, bis(carboxylmethyl)
trithiocarbonate, H2O, 100 ºC, 19 h; (iii) for 6 and 7: (1) CS2 in Et2O, 30 min and (2)
BrCH2CO2H/EtOH, reflux, 1 h; (iv) NH4OAc, toluene/CH3OH, reflux, 3-5 h; (v) conc. HCl/HOAc,
under N2, reflux, 24 h.
Page 8
ACCEPTED MANUSCRIPT
7
R' COOH
N
CHO
N
O
O
OO ROOC
HON
N
O
O
HO COOCH3
N
CHO
i
ii
4 R' = OH14 R' = Cl15 R' = H
2022 R = Me23 R = H
S N
O
OHOOC
HO N SN
O
O
4
18
19
COOH
N
R'
SN
O
S16 R' = Cl17 R' = H
9a
i
21
iii
iv
Scheme 2 Reagents and conditions: (i) NH4OAc, toluene/CH3OH, reflux, 5 h; (ii) CH3I, Cs2CO3,
DMF, rt, 24 h; (iii) Ph3P, EtOH, under N2, reflux, 12 h; (iv) conc. HCl/HOAc, under N2, reflux, 24 h.
3. Results and Discussion
Twenty newly synthesized target compounds (11-13, 16, 17, 19, and 23) with a linear
multi-aromatic-ring scaffold were evaluated against both HIV-1 gp41 6-HB formation (IC50) and
HIV replication in MT-2 cell line (EC50), respectively, by enzyme-linked immunosorbent assays
(ELISA). The colorimetric XTT assay was also performed to test the cytotoxicity of these
compounds on MT-2 cells (CC50). All assay data were summarized in Table 1 and NB-64 was
included as a reference in the same assays.
Based on the known binding site of gp41 for small-molecule inhibitors, we inferred that a
suitably elongated molecule would be the best fit to accommodate the space and shape
complementation requirements for binding the gp41 pocket in order to afford more interactions with
amino acids on the surface of the binding site. As the first attempt, we synthesized compounds 11 (n
= 0), 12a, and 13 with a one-atom (n = 1) or two-atom (n = 2) linker between the rhodanine (C-ring)
and its N-phenyl (D-ring) ring. In comparison to 11, compounds 12a and 13 exhibited significantly
improved potency against gp41 6-HB formation with IC50 values of 11.9 and 12.5 µM, respectively,
suggesting that a linker with at least one atom between C-ring and D-ring might favor ligand binding
to the gp41 binding cavity. Both 12a and 13 also showed high potency against HIV-1 replication
Page 9
ACCEPTED MANUSCRIPT
8
with EC50 values of 3.2 and 6.2 µM, respectively, in cellular assays. These results prompted us to
investigate a series of compounds 12b-n (n = 1) with diverse substituents at different positions on the
phenyl (D-ring). As shown in Table 1, most of the series 12 compounds, except 12f and 12n,
displayed improved potency in inhibiting gp41 6-HB formation with an IC50 value range of 0.9-22.6
µM. The most potent compound, 12k with an m-CO2CH3 on the D-ring, exhibited an IC50 value of
1.8µM and an EC50 value of 0.3 µM against gp41 6-HB formation and HIV-1 replication,
respectively. Compounds 12e and 12g are also very promising with IC50 values of 1.8 and 2.6 µM
and EC50 values of 1.5 and 0.9 µM in both molecular and cellular assays, respectively. The biologic
data of series 12 compounds revealed some relationships between the substituents on the D-ring and
anti-HIV activity. Compared with 12a, the presence of a bromo (12c), methoxy (12d), methyl ester
(12e), or tert-butyl group (12g) at the para-position on the D-ring obviously improved the inhibitory
activity (IC50 0.9-10.3 µM) for gp41 6-HB formation, but trifluoromethyl (12b) and very polar
carboxy (12f) groups decreased inhibitory potency (IC50 18.8 and 68.0 µM, respectively) in the same
assay. A similar potency pattern was also observed in compounds 12h-12l with an m-substituent on
the D-ring. These compounds were as potent as (IC50 1.8-20.8 µM), or a little better than, the
corresponding p-substituted compounds 12b-12g. Therefore, the presence of a suitable substituent on
the D-ring could increase the affinity of small-molecule inhibitors with gp41 6-HB binding site
regardless of p- or m-substituted position. On the other hand, compounds 12m (R = o-CF3) and 12n
(R = o-Br) with an o-substituted phenyl moiety (D-ring) exhibited obviously reduced or abolished
potency in the gp41 6-HB formation assay, suggesting that the o-position on the D-ring was not
modifiable. Consistent with the results of gp41-6HB formation assay, most of the above active 12
compounds also exhibited high anti-HIV potency in the cellular assay. In addition, when the
hydroxyl group on the A-ring was replaced with a chloro- or hydrogen, the resultant compounds 16
(R’ = Cl) and 17 (R’ = H) showed less potency in both molecular and cellular assays than 12a,
suggesting that the hydroxyl group on the A-ring might be better than others for anti-HIV activity.
Finally, the isostere replacement of the rhodanine ring (C ring) with pyrrolin-2,4-dione or
thiazolin-2,4-dione moiety resulted in compounds 19 and 23 with much less anti-HIV activities in
both assays (data not shown) than 12a and 13. These results supported our previous results25
suggesting that the rhodanine ring was preferred to another five-membered ring as the C-ring moiety.
Page 10
ACCEPTED MANUSCRIPT
9
Notably, the cytotoxicity, based on the CC50 value, of 12k is about 4-fold higher than that of
12a. However, the anti-HIV-1 potency, based on the IC50 value, of 12k is about 11-fold higher than
that of 12a. Therefore, the selectivity index of 12k (SI = 128) is much higher than that of 12a (SI =
50)(Table 1). We will select 12k as a lead for further structure optimization, in order to identify a
drug candidate with reduced cytotoxicity and increased anti-HIV-1 potency for future pre-clinical
study.
Table 1. Inhibitory activities against HIV-1 gp41 6-helix bundle (6-HB) formation and HIV-1
replication a
R'
HOOC
NS
N
S
O
nR
gp41 6-HB MT-2 cell line
# R’ n R IC50b (µM) EC50
c (µM) CC50d (µM) SIe
11 OH 0 H NA 35.6 ± 4.7 >100 >2.8
12a OH 1 H 11.9 ± 0.5 3.2 ± 0.1 158.7 ± 0.0 50.2
13 OH 2 H 12.5 ± 0.0 6.2 ± 0.6 26.0 ± 2.9 4.2
12b OH 1 p-CF3 18.8 ± 0.4 17.2 ± 1.8 45.9 ± 2.2 2.7
12c OH 1 p-Br 10.3 ± 0.8 42.7 ± 7.0 94.4 ± 6.4 2.2
12d OH 1 p-OCH3 0.9 ± 0.0 19.8 ± 3.5 75.3 ± 6.9 3.8
12e OH 1 p-CO2CH3 1.8 ± 1.1 1.5 ± 0.3 36.9 ± 3.4 24.6
12f OH 1 p-CO2H 68.0 ± 4.4 7.1 ± 2.4 105.1 ± 10.3 14.7
12g OH 1 p-t-Bu 2.6 ± 0.0 0.9 ± 0.1 55.2 ± 6.3 61.3
12h OH 1 m-CF3 9.2 ± 0.6 3.9 ± 0.4 26.3 ± 4.1 6.7
12i OH 1 m-Br 7.8 ± 0.3 32.4 ± 1.3 48.2 ± 5.1 1.5
12j OH 1 m-OCH3 2.1 ± 0.2 23.5 ± 3.9 102.3 ± 4.4 4.4
12k OH 1 m-CO2CH3 1.8 ± 0.8 0.3 ± 0.2 38.5 ± 30.2 128
12l OH 1 m-CO2H 20.8 ± 0.2 12.1 ± 1.6 >100 >8.3
Page 11
ACCEPTED MANUSCRIPT
10
12m OH 1 o-CF3 22.6 ± 2.2 29.1 ± 3.3 28.8 ± 4.3 1
12n OH 1 o-Br ND 41.1 ± 7.7 25.7 ± 1.2 0.6
16 Cl 1 H 34.3 ± 4.7 19.1 ± 2.3 68.3 ± 2.7 3.6
17 H 1 H 45.7 ± 8.6 15.3 ± 2.8 83.4 ± 11.5 5.5
NB-2 37.4 0.7 133.4 191
NB-64 58.7 2.4 335.7 140
a Compounds were tested in triplicate, and the data were presented as the mean ± SD; b Inhibiting
gp41 6-helix bundle formation ; c Inhibiting p24 production in the MT-2 cells; d Cytotoxicity; e SI: selectivity index (CC50/EC50); f
Not determined.
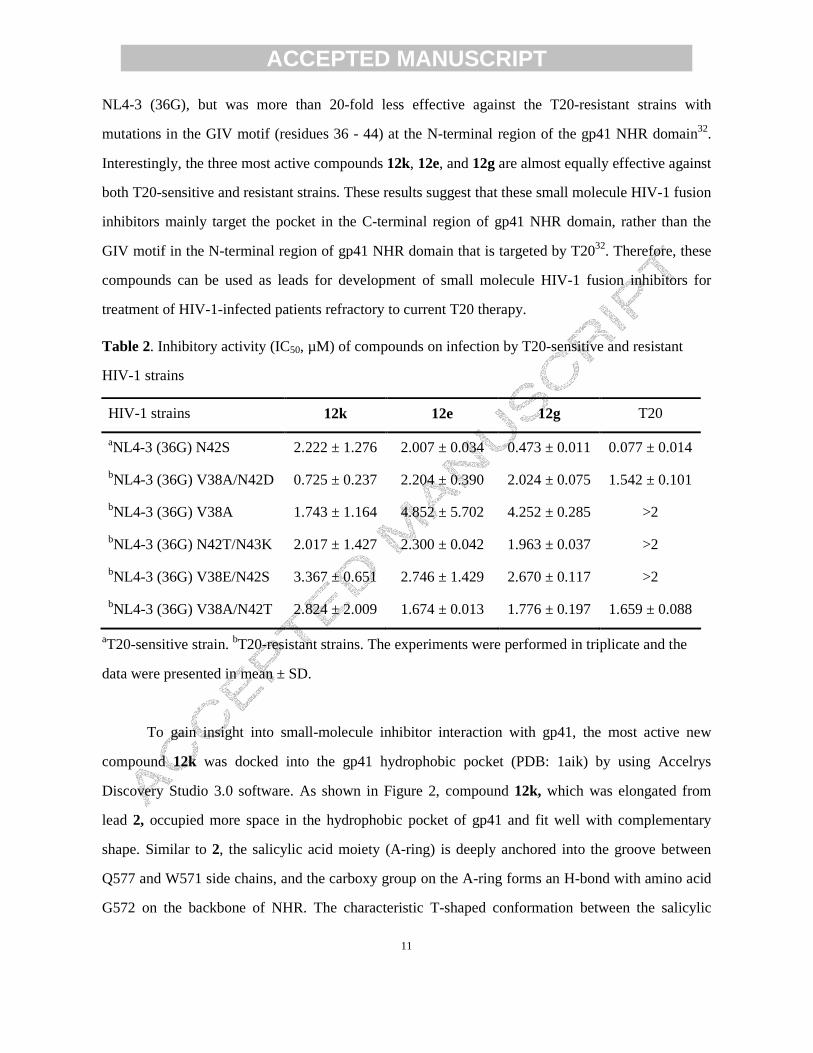
Figure 2 Docking small-molecule inhibitors 2 and 12k with gp41 (PDB: 1aik). (a): surface
representation of the gp41 core with lead 2; (b): surface representation of the gp41 binding site with
active compound 12k. (c): stereoview of 12k docked to gp41 with amino acids on the surface of
binding pocket. The hydrogen bond and cation-� interaction was shown in black dashed line and
yellow line, respectively. Compound 12k was shown by stick model with green color, and all
residues were shown by stick model with gray color, respectively.
To further assess the potential of these active compounds as leads for further development,
we tested the inhibitory activity of the most active compounds 12e, 12g, and 12k against HIV-1
strains resistant to T20 (enfuvirtide), the first HIV-1 fusion inhibitor drug for clinical use, and T20
was included as a control. As shown in Table 2, T20 was effective against the T20-senstive strain
Page 12
ACCEPTED MANUSCRIPT
11
NL4-3 (36G), but was more than 20-fold less effective against the T20-resistant strains with
mutations in the GIV motif (residues 36 - 44) at the N-terminal region of the gp41 NHR domain32.
Interestingly, the three most active compounds 12k, 12e, and 12g are almost equally effective against
both T20-sensitive and resistant strains. These results suggest that these small molecule HIV-1 fusion
inhibitors mainly target the pocket in the C-terminal region of gp41 NHR domain, rather than the
GIV motif in the N-terminal region of gp41 NHR domain that is targeted by T2032. Therefore, these
compounds can be used as leads for development of small molecule HIV-1 fusion inhibitors for
treatment of HIV-1-infected patients refractory to current T20 therapy.
Table 2. Inhibitory activity (IC50, µM) of compounds on infection by T20-sensitive and resistant
HIV-1 strains
HIV-1 strains 12k 12e 12g T20
aNL4-3 (36G) N42S 2.222 ± 1.276 2.007 ± 0.034 0.473 ± 0.011 0.077 ± 0.014
bNL4-3 (36G) V38A/N42D 0.725 ± 0.237 2.204 ± 0.390 2.024 ± 0.075 1.542 ± 0.101
bNL4-3 (36G) V38A 1.743 ± 1.164 4.852 ± 5.702 4.252 ± 0.285 >2
bNL4-3 (36G) N42T/N43K 2.017 ± 1.427 2.300 ± 0.042 1.963 ± 0.037 >2
bNL4-3 (36G) V38E/N42S 3.367 ± 0.651 2.746 ± 1.429 2.670 ± 0.117 >2
bNL4-3 (36G) V38A/N42T 2.824 ± 2.009 1.674 ± 0.013 1.776 ± 0.197 1.659 ± 0.088
aT20-sensitive strain. bT20-resistant strains. The experiments were performed in triplicate and the
data were presented in mean ± SD.
To gain insight into small-molecule inhibitor interaction with gp41, the most active new
compound 12k was docked into the gp41 hydrophobic pocket (PDB: 1aik) by using Accelrys
Discovery Studio 3.0 software. As shown in Figure 2, compound 12k, which was elongated from
lead 2, occupied more space in the hydrophobic pocket of gp41 and fit well with complementary
shape. Similar to 2, the salicylic acid moiety (A-ring) is deeply anchored into the groove between
Q577 and W571 side chains, and the carboxy group on the A-ring forms an H-bond with amino acid
G572 on the backbone of NHR. The characteristic T-shaped conformation between the salicylic
Page 13
ACCEPTED MANUSCRIPT
12
phenyl and pyrrole rings is maintained; thus, the 2,5-dimethylpyrrole is inserted into the hydrophobic
cavity, which was supposed to be occupied by residue W631 of CHR upon formation of 6-HB. The
newly introduced rhodanine moiety (C-ring) is located near the key amino acid K574 with a positive
charge. In addition, the presence of a one-carbon linker between the C-ring and D-ring increased
molecular flexibility, allowing the C and D rings to adjust binding conformation around key residue
K574 and produce a salt bridge between the thioxo and the side chain of K574 and the cation-�
interaction between the phenyl ring and K574, suggesting that a flexible linker between C and D
rings might be important to enhance biological activities. Meanwhile, the D-ring part filled a
hydrophobic hollow formed by residues Q567, V570, and W571 from the NHR, and the m-methoxy
carbonyl (m-CO2CH3) group was deeply inserted into this hollow to provide more interactions with
the target cavity. Therefore, this docking model of compound 12k illustrated that the
multi-aromatic-ring scaffold, alone with molecular flexibility, is favorable for increasing binding
affinity by more binding interaction points, especially the key residue K574. These results also
supported the improved inhibitory potency of 12k in both assays.
4. Conclusion
As a continuing structural optimization, a series of (Z)-3-(5-(3-benzyl-4-oxo-2-thioxothi
azolidinylidene)methyl)-N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrroles and related
derivatives were designed, synthesized, and evaluated in HIV-1 gp41 and cellular assays. Among
them, the most active compounds, 12k, 12e, and 12g with a one-carbon linker between the C and D
rings, exhibited very promising inhibitory potency with IC50 values of 1.8-2.6 µM and EC50 values of
0.3-1.5 µM against gp41 6-HB formation and HIV-1 replication, respectively, as well as an SI range
of 24-128. As such, they are more potent than our prior leads 1-3. The SAR studies revealed that (1)
the 3-carboxy-4-hydroxyphenyl (A-ring) is a favorable moiety, which might play an anchoring role
to maintain the binding orientation of small-molecule inhibitors; (2) a one-atom linker (n = 1)
between C and D rings provided the molecular flexibility to fit the gp41 binding pocket; (3)
substituents on the D-ring greatly affect anti-HIV potency, and an ester group at the p- or m-position
is preferable to others; (4) the p- and m-position on the D-ring were modifiable, whereas the
ortho-position was not; (5) the rhodanine ring was a preferable moiety compared with other hetero
Page 14
ACCEPTED MANUSCRIPT
13
five-membered rings. The molecular docking model of active compound 12k illustrated the ability of
this multi-aromatic-ring scaffold to fill the linear binding cavity with shape complementation and
interact with the key residue K574. These results show promise for the further development of
nonpeptide small-molecule fusion inhibitors targeting the gp41subunit.
5. Experimental section
5.1. Chemistry
The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400
(400MHz) spectrometer using tetramethylsilane (TMS) as internal standard. The solvent used was
DMSO-d6, unless otherwise indicated. Mass spectra (MS) were measured on an ABI Perkin-Elmer
Sciex API-150 mass spectrometer with electrospray ionization, and the relative intensity of each ion
peak is presented as a percent (%). Melting points were measured with an SGW X-4 Micro-Melting
apparatus without correction. The microwave reactions were performed on a microwave reactor from
Biotage, Inc. Thin-layer chromatography (TLC) on silica gel GF254 plates was used to monitor
reactions. Medium-pressure column chromatography was performed using a CombiFlash Companion
purification system from ISCO, Inc. All chemicals and solvents were obtained from Beijing
Chemical Works or Sigma-Aldrich, Inc. Purities of all target compounds were determined by an
Agilent 1200 HPLC system with an Eclipse XDB-C18 column, UV detector at 254 nm, mobile
MeOH/H2O/HCO2H (95/5/0.001) and flow rate of 1 mL/min.
5.1.1. N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-formylpyrrole (4). Anhydrous DMF (20
mL) was placed in a dried 3-neck flask (250-mL) equipped with a thermometer. Then 7 mL of POCl3
(74.1 mmol) was added dropwise into the above flask with stirring and controlled below 10 °C in an ice
bath. After another 30 min stirring to ensure complete conversion to the Vilsmeier reagent, a solution
of N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrrole (2) (10.75 g, 50 mmol) in 40 mL of DMF was
added slowly for 30 min, keeping the temperature below 10 °C and then stirring for another 2 h at room
temperature. After quenching with ice, the solution was adjusted with 5% NaOH (aq) to pH 9-10 and
rapidly heated to reflux for 30 min. The mixture was slowly poured into ice water with stirring and left
Page 15
ACCEPTED MANUSCRIPT
14
to set overnight. The precipitated solid was filtered out, washed with water to neutral, and dried to
afford 10.7 g of 4, gray solid, 88% yield. mp 190 °C (dec.); 1H NMR (CDCl3) � ppm 11.18 (1H, br s,
COOH), 9.81 (1H, s, CHO), 7.92 (1H, d, J = 2.4 Hz, ArH-6), 7.30 (1H, dd, J = 2.4 & 8.8 Hz, ArH-4),
7.14 (1H, d, J = 8.8 Hz, ArH-3), 6.42 (1H, s, PyH), 2.31 & 2.00 (each 3H, s, CH3 × 2); MS m/z (%) 258
(M – 1, 100).
5.1.2. N-Phenyl-2-thioxothiazolidin-4-one (8). A suspension of aniline (465 mg, 5 mmol) in water
(10 mL) was heated to 95 °C until the aniline was fully dissolved. Then
bis(carboxylmethyl)trithiocarbonate (1.25 g, 5.5 mmol) was added and heated at 100 °C for 19 h.
After cooling to room temperature, the precipitated solid was filtered out and washed with water.
The dried solid was purified by a flash silica column (gradual eluent: EtOAc/petroleum ether, 0 ~
50%) to afford 730 mg of 8, pale-yellow solid, 70% yield. mp 184-186 °C; 1H NMR (CDCl3) � ppm
7.57~7.19 (5H, m, ArH × 5), 4.20 (2H, s, SCH2); MS m/z (%) 208 (M 1, 100).
5.1.3. General procedure for preparation of 3-substituted 2-thioxothiazolidin-4-one
intermediates (9 and 10). To a solution of substituted benzylamine (6) or phenylethylamine (7) (1.0
eq.) in ether was added CS2 (1.0 eq. in ether) dropwise in an ice bath with stirring for an additional 1 h.
The precipitated solid was filtered, washed with ether, and dried at room temperature. The collected
ammonium dithiocarbamate was then added into the solution of bromoacetic acid (0.5 eq.) in ethanol,
and the mixture was heated to reflux for 3-5 h. After cooling to room temperature, the mixture was
poured into ice water. The 3-substituted rhodanine product (9 or 10) was either filtered out or
extracted with EtOAc and purified by flash silica column (eluent: EtOAc/petroleum ether, 0 ~ 40%).
5.1.3.1. 3-Benzyl-2-thioxothiazolidin-4-one (9a). Starting from benzylamine 6a (5 mL, 45.8 mmol)
to afford 4.18 g of 9a, light yellow solid, 41% yield. mp 72-74 °C; 1H NMR (CDCl3) � ppm 7.22 ~
7.39 (5H, m, ArH), 4.59 (2H, s, NCH2Ar), 4.00 (2H, s, SCH2); MS m/z (%) 224 (M + 1, 50), 91
(PhCH2, 100).
Page 16
ACCEPTED MANUSCRIPT
15
5.1.3.2. 3-(4-(Trifluoromethyl)benzyl)-2-thioxothiazolidin-4-one (9b). Starting from
4-trifluoromethyl benzylamine 6b (1.75 g, 10 mmol) to afford 1.02 g of 9b, white solid, 35% yield.
mp 102-104 °C; 1H NMR (CDCl3) � ppm 7.53 ~ 7.59 (4H, m, ArH), 5.23 (2H, s, ArCH2N), 4.03 (2H,
s, SCH2); MS m/z (%) 290 (M – 1, 100).
5.1.3.3. 3-(4-Bromobenzyl)-2-thioxothiazolidin-4-one (9c). Starting from 4-bromobenzylamine 6c
(1.86 g, 10 mmol) to afford 745 mg of 9c, light yellow solid, 25% yield. mp 84-86 °C; 1H NMR
(CDCl3) � ppm 7.45 (2H, d, J = 8.4 Hz, ArH-3, 5), 7.34 (2H, d, J = 8.4 Hz, ArH-2, 6), 5.13 (2H, s,
ArCH2N), 4.00 (2H, s, SCH2); MS m/z (%) 169 (M – 132, p-BrBn, 100), 171 (169 + 2, 95).
5.1.3.4. 3-(4-Methoxybenzyl)-2-thioxothiazolidin-4-one (9d). Starting from
4-methoxybenzylamine 6d (1.37 g, 10 mmol) to afford 895 mg of 9d, white solid, 35% yield. mp
90-92 °C; 1H NMR (CDCl3) � ppm 7.43 (2H, d, J = 8.4 Hz, ArH-2, 6), 6.84 (2H, d, J = 8.4 Hz, ArH-3,
5), 5.12 (2H, s, ArCH2N), 3.96 (2H, s, SCH2), 3.78 (3H, s, OCH3); MS m/z (%) 254 (M + 1, 100).
5.1.3.5. 3-(4-(Methoxycarbonyl)benzyl)-2-thioxothiazolidin-4-one (9e). Starting from
4-methoxycarbonyl benzylamine 6e (3.0 g, 18 mmol) to afford 1.34 g of 9e, light yellow solid, 26%
yield. mp 154-156 °C; 1H NMR (CDCl3) � ppm 7.99 (2H, d, J = 8.4 Hz, ArH-3, 5), 7.48 (2H, d, J = 8.4
Hz, ArH-2, 6), 5.23 (2H, s, ArCH2N), 4.03 (2H, s, SCH2), 3.91 (3H, s, OCH3); MS m/z (%) 282 (M +
1, 100).
5.1.3.6. 3-(4-tert-Butylbenzyl)-2-thioxothiazolidin-4-one (9g). Starting from 4-chlorobenzylamine
6g (1.63 g, 10 mmol) to afford 1.07 g of 9g, light yellow oil, 41% yield. mp 100-102 °C; 1H NMR
(CDCl3) � ppm 7.40 (2H, d, J = 8.4 Hz, ArH-3, 5), 7.34 (2H, d, J = 8.4 Hz, ArH-2, 6), 5.15 (2H, s,
NCH2Ar), 3.97 (2H, s, SCH2), 1.30 (9H, s, CH3 × 3); MS m/z (%) 280 (M + 1, 40), 297 (M + 18, 100).
5.1.3.7. 3-(3-(Trifluoromethyl)benzyl)-2-thioxothiazolidin-4-one (9h). Starting from
3-trifluoromethyl benzylamine 6h (1.75 g, 10 mmol) to afford 1.02 g of 9h, light yellow solid, 35%
yield. mp 68-70 °C; 1H NMR (CDCl3) � ppm 7.70 (1H, s, ArH-2), 7.64 (1H, d, J = 8.0 Hz, ArH-4),
Page 17
ACCEPTED MANUSCRIPT
16
7.57 (1H, d, J = 8.0 Hz, ArH-6), 7.44 (1H, t, J = 8.0 Hz, ArH-5), 5.22 (2H, s, NCH2Ar), 4.03 (s, 2H,
SCH2); MS m/z (%) 290 (M 1, 100).
5.1.3.8. 3-(3-Bromobenzyl)-2-thioxothiazolidin-4-one (9i). Starting from 3-bromobenzylamine 6i
(1.86 g, 10 mmol) to afford 890 mg of 9i, white solid, 30% yield. mp 87-89 °C; 1H NMR (CDCl3) �
ppm 7.57 (1H, s, ArH-2), 7.44 (1H, d, J = 7.6 Hz, ArH-4), 7.38 (1H, d, J = 7.6 Hz, ArH-6), 7.19 (1H, t,
J = 7.6 Hz, ArH-5), 5.14 (2H, s, ArCH2N), 4.02 (2H, s, SCH2); MS m/z (%) 169 (M 132, BrBn,
100), 171 (169 + 2, 85).
5.1.3.9. 3-(3-Methoxybenzyl)-2-thioxothiazolidin-4-one (9j). Starting from 3-methoxybenzylamine
6j (1.37 g, 10 mmol) to afford 1 g of 9j, white solid, 39% yield. mp 78-80 °C; 1H NMR (CDCl3) �
ppm 7.23 (1H, t, J = 8.0 Hz, ArH-5), 6.99 ~7.02 (2H, m, ArH), 6.84 (1H, dd, J = 2.4 & 8.0 Hz, ArH-4),
5.16 (2H, s, ArCH2N), 3.99 (2H, s, SCH2), 3.79 (3H, s, OCH3); MS m/z (%) 254 (M + 1, 60), 276 (M
+ 23, 100).
5.1.3.10. 3-(3-(Methoxycarbonyl)benzyl)-2-thioxothiazolidin-4-one (9k). Starting from
3-methoxycarbonyl benzylamine 6k (1.0 g, 6.05 mmol) to afford 360 mg of 9k, light yellow solid,
21% yield. mp 126-128 °C; 1H NMR (CDCl3) � ppm 8.08 (1H, s, ArH-2), 7.98 (1H, d, J = 8.0 Hz,
ArH-4), 7.63 (1H, d, J = 8.0 Hz, ArH-6), 7.40 (1H, t, J = 8.0 Hz, ArH-5), 5.23 (2H, s, ArCH2N), 4.04
(2H, s, SCH2), 3.92 (3H, s, OCH3); MS m/z (%) 282 (M + 1, 80).
5.1.3.11. 3-(2-(Trifluoromethyl)benzyl)-2-thioxothiazolidin-4-one (9m). Starting from
2-trifluoromethyl benzylamine 6m (1.75 g 10 mmol) to afford 970 mg of 9m, white solid, 33%
yield. mp 119-122 °C; 1H NMR (CDCl3) � ppm 7.91 (1H, d, J = 7.6 Hz, ArH-3), 7.46 (1H, t, J = 7.6
Hz, ArH-5), 7.38 (1H, t, J = 7.6 Hz, ArH-4), 6.91 (1H, d, J = 7.6 Hz, ArH-6), 5.41 (2H, s, ArCH2N),
4.14 (2H, s, SCH2); MS m/z (%) 292 (M + 1, 60).
5.1.3.12. 3-(2-Bromobenzyl)-2-thioxothiazolidin-4-one (9n). Starting from 2-bromobenzylamine
6n (930 mg, 5 mmol) to afford 590 mg of 9n, white solid, 39% yield. mp 86-88 °C; 1H NMR (CDCl3)
Page 18
ACCEPTED MANUSCRIPT
17
� ppm 7.59 (1H, d, J = 8.0 Hz, ArH-3), 7.24 (1H, t, J = 8.0 Hz, ArH-5), 7.14 (1H, t, J = 8.0 Hz, ArH-4),
6.86 (1H, d, J = 8.0 Hz, ArH-6), 5.26 (2H, s, ArCH2N), 4.11 (2H, s, SCH2); MS m/z (%) 169 (M
132, BrBn, 100), 171 (169 + 2, 100).
5.1.3.13. 3-Phenethyl-2-thioxothiazolidin-4-one (10). Starting from 2-phenyl ethylamine 7 (1 mL,
8 mmol) to afford 700 mg of 10, light yellow solid, 37% yield. mp 86-88 °C; 1H NMR (CDCl3) �
ppm 7.22 ~ 7.35 (5H, m, ArH), 4.22 (2H, t, J = 8.0 Hz, NCH2), 3.93 (2H, s, SCH2), 2.93 (2H, t, J =
8.0 Hz, CH2Ar); MS m/z (%) 238 (M + 1, 100).
5.1.4. General procedure for Knoevenagel condensation reaction to prepare target compounds
11-13, 16, and 17. A mixture of an aldehyde compound (4, 15, or 16) and a rhodanine derivative (8, 9
or 10, mole ratio 1:1) in the presence of NH4OAc (100 mg) in toluene (20 mL/mmol) and methanol (10
mL/mmol) was heated to reflux for 3-5 h. After the reaction was finished, ethyl acetate was added to
the solution, and organic phase was washed with water and brine, successively, and then dried over
Na2SO4. After removal of solvent under reduced pressure, the residue was purified by flash silica
column (gradual eluent: EtOAc/petroleum ether with AcOH (3%), 0 ~ 60%) to give the pure product.
5.1.4.1. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(4-oxo-3-phenyl-2-thioxothiazol
idinylidene)methyl)pyrrole (11). Starting from 4 (50 mg, 0.19 mmol) and 8 (41 mg, 0.19 mmol) to
afford 60 mg of 11, yellow solid, 69% yield. mp 200 °C (dec.); 1H NMR � ppm 7.71 (1H, s, =CH),
7.67 (1H, d, J = 2.8 Hz, ArH-6), 7.49 ~ 7.59 (4H, m, ArH), 7.34 ~ 7.39 (2H, m, ArH), 7.13 (1H, d, J =
8.8 Hz, ArH-3), 6.31 (1H, s, PyH), 2.17 & 2.02 (each 3H, s, CH3 × 2); MS m/z 449 (M – 1, 30), 405 (M
– 44, 100); HPLC purity 98.62%.
5.1.4.2. (Z)-3-(5-(3-Benzyl-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-4-hydroxy)
phenyl-2,5-dimethylpyrrole (12a). Starting from 4 (130 mg, 0.5 mmol) and 9a (112 mg, 0.5 mmol)
to afford 162 mg of 12a, yellow solid, 69% yield. mp 226-228 °C (dec.); 1H NMR � ppm 7.74 (1H, s,
=CH), 7.66 (1H, d, J = 2.4 Hz, ArH-6), 7.32 ~ 7.52 (6H, m, ArH-4, ArH on D-ring), 7.14 (1H, d, J =
8.8 Hz, ArH-3), 6.22 (1H, s, PyH), 4.69 (2H, s, ArCH2N), 2.16 & 1.98 (each 3H, s, CH3); MS m/z (%)
463 (M – 1, 100); HPLC purity 96.49%.
Page 19
ACCEPTED MANUSCRIPT
18
5.1.4.3. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(3-(4-trifluoromethylbenzyl)-4-
oxo-2-thioxothiazolidinylidene)methyl)pyrrole (12b). Starting from 4 (130 mg, 0.5 mmol) and 9b
(145 mg, 0.5 mmol) to afford 188 mg of 12b, yellow solid, 71% yield. mp 144-146 °C; 1H NMR �
ppm 7.71 ~ 7.75 (3H, m, =CH, ArH-3’, 5’), 7.66 (1H, d, J = 2.8 Hz, ArH-6), 7.50 ~ 7.53 (3H, m, ArH),
7.14 (1H, d, J = 8.8 Hz, ArH-3), 6.27 (1H, s, PyH), 5.32 (2H, s, ArCH2N), 2.17 & 2.00 (each 3H, s,
CH3); MS m/z (%) 531 (M – 1, 100); HPLC purity 95.62%.
5.1.4.4. (Z)-3-(5-(3-(4-Bromobenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-4-
hydroxy)phenyl-2,5-dimethylpyrrole (12c). Starting from 4 (100 mg, 0.39 mmol) and 9c (117 mg,
0.39 mmol) to afford 130 mg of 12c, yellow solid, 55% yield. mp 252-254 °C; 1H NMR � ppm 7.73
(1H, s, =CH), 7.67 (1H, d, J = 2.8 Hz, ArH-6), 7.55 (2H, d, J = 8.4 Hz, ArH-3’, 5’), 7.52 (1H, dd, J =
2.8 & 8.8 Hz, ArH-4), 7.29 (2H, d, J = 8.4 Hz, ArH-2’, 6’), 7.14 (1H, d, J = 8.8 Hz, ArH-3), 6.26 (1H,
s, PyH), 5.20 (2H, s, NCH2Ar), 2.16 & 2.00 (each 3H, s, CH3); MS m/z (%) 541 (M – 1, 80), 543 (M +
1, 100); HPLC purity 98.14%.
5.1.4.5. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-(3-(5-(3-(4-methoxybenzyl)-4-oxo-2-
thioxothiazolidinylidene)methyl)pyrrole (12d). Starting from 4 (130 mg, 0.5 mmol) and 9d (127
mg, 0.5 mmol) to afford186 mg of 12d, yellow solid, 75% yield. mp 248-250 °C (dec.); 1H NMR �
ppm 7.72 (1H, s, =CH), 7.66 (1H, d, J = 2.8 Hz, ArH-6), 7.50 (1H, dd, J = 2.8 & 8.8 Hz, ArH-4), 7.31
(2H, d, J = 8.4 Hz, ArH-2’, 6’), 7.13 (1H, s, J = 8.8 Hz, ArH-3), 6.90 (2H, d, J = 8.4 Hz, ArH-3’, 5’),
6.24 (1H, s, PyH-4), 5.16 (2H, s, ArCH2N), 3.72 (3H, s, OCH3), 2.16 & 1.99 (each 3H, s, CH3 × 2); MS
m/z (%) 493.2 (M – 1, 100); HPLC purity 96.58%.
5.1.4.6. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-(3-(5-
(3-(4-(methoxycarbonyl)benzyl) -4-oxo-2-thioxothiazolidinylidene)methyl)pyrrole (12e).
Starting from 4 (130 mg, 0.5 mmol) and 9e (140 mg, 0.5 mmol) to afford 204 mg of 12e, yellow solid,
78% yield. mp 158-160 °C; 1H NMR � ppm 7.95 (2H, d, J = 8.4 Hz, ArH-3’, 5’), 7.75 (1H, s, =CH),
7.66 (1H, d, J = 2.8 Hz, ArH-6), 7.48 (1H, dd, J = 2.8 & 8.4 Hz, ArH-4), 7.44 (2H, d, J = 8.4 Hz,
Page 20
ACCEPTED MANUSCRIPT
19
ArH-2’, 6’), 7.12 (1H, d, J = 8.4 Hz, ArH-3), 6.27 (1H, s, PyH), 5.31 (2H, s, ArCH2N), 3.84 (2H, s,
OCH3), 2.17 & 2.00 (each 3H, s, CH3 × 2); MS m/z (%) 522 (M – 1, 100); HPLC purity 95.59%.
5.1.4.7. (Z)-3-(5-(3-(4-Carboxybenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-
4-hydroxy)phenyl-2,5-dimethylpyrrole (12f). To a mixture of the methyl ester of 12e (100 mg, 0.19
mmol) in 10 mL glacial acetic acid was added 2 mL 36% HCl under nitrogen atmosphere, and the
mixture was heated to reflux for 24 h. After cooling to room temperature, the solution was poured into
40 mL ice water, and the precipitate was filtered out and purified by flash silica column [gradual
eluent: EtOAc/petroleum ether with AcOH (3%), 0 ~ 50%] to afford 67 mg of 12f, yellow solid, 65%
yield. mp 298-300 °C (dec.); 1H NMR � ppm 7.92 (2H, d, J = 7.6 Hz, ArH-2’, 6’), 7.75 (1H, s, =CH),
7.66 (1H, d, J = 2.8 Hz, ArH-6), 7.51 (1H, dd, J = 2.8 & 8.8 Hz, ArH-4), 7.41 (2H, d, J = 7.6 Hz,
ArH-3’, 5’), 7.14 (d, 1H, J = 8.8 Hz, ArH-3), 6.27 (1H, s, PyH), 5.30 (2H, s, ArCH2N), 2.17 & 2.00
(each 3H, s, CH3 × 2); MS m/z (%) 507 (M – 1, 100); HPLC purity 96.04%.
5.1.4.8. (Z)-3-(5-(3-(4-tert-Butylbenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy
-4-hydroxy)phenyl-2,5-dimethylpyrrole (12g). Starting from 4 (50 mg, 0.19 mmol) and 9g (54 mg,
0.19 mmol) to afford 67 mg of 12g, yellow solid, 67% yield. mp 140-142 °C; 1H NMR � ppm 7.72
(1H, s, =CH), 7.67 (1H, d, J = 2.8 Hz, ArH-6), 7.50 (1H, dd, J = 2.8 & 8.8 Hz, ArH-4), 7.37 (2H, d, J =
8.0 Hz, ArH-2’, 6’), 7.27 (2H, d, J = 8.0 Hz, ArH-3’, 5’), 7.15 (1H, d, J = 8.8 Hz, ArH-3), 6.26 (1H, s,
PyH-4), 5.20 (2H, s, ArCH2N), 2.16 & 1.99 (each 3H, s, CH3 × 2), 1.25 (9H, s, t-Bu); MS m/z (%) 519
(M – 1, 100); HPLC purity 95.37%.
5.1.4.9. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(4-oxo-2-thioxo-3-(3-trifluoro
methylbenzyl)thiazolidinylidene)methyl)pyrrole (12h). Starting from 4 (130 mg, 0.5 mmol) and 9h
(145 mg, 0.5 mmol) to afford 100 mg of 12h, yellow solid, 38% yield. mp 124-126 °C; 1H NMR �
ppm 7.75 (1H, s, =CH), 7.59 ~ 7.71 (5H, m, ArH-6, ArH on D-ring), 7.50 (1H, dd, J = 2.8 & 8.8 Hz,
ArH-4), 7.13 (1H, d, J = 8.8 Hz, ArH-3), 6.26 (1H, s, PyH), 5.32 (2H, s, ArCH2N), 2.17 & 2.00 (each
3H, s, CH3 × 2); MS m/z (%) 531 (M – 1, 100), 487 (M – 44, 50); HPLC purity 96.02%.
Page 21
ACCEPTED MANUSCRIPT
20
5.1.4.10. (Z)-3-(5-(3-(3-Bromobenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy
-4-hydroxy)phenyl-2,5-dimethylpyrrole (12i). Starting from 4 (100 mg, 0.39 mmol) and 9i (117 mg,
0.39 mmol) to afford 130 mg of 12i, red solid, 62% yield. mp 208-210 °C; 1H NMR � ppm 7.74 (1H,
s, =CH), 7.67 (1H, d, J = 2.8 Hz, ArH-6), 7.48 ~ 7.53 (3H, m, ArH), 7.30 ~ 7.32 (2H, m, ArH), 7.14
(1H, d, J = 8.8 Hz, ArH-3), 6.26 (1H, s, PyH), 5.22 (2H, s, ArCH2N), 2.17 & 2.00 (each 3H, s, CH3 ×
2); MS m/z (%) 543 (M + 1, 100), 541 (M – 1, 80); HPLC purity 99.13%.
5.1.4.11. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(3-(3-methoxybenzyl)-4-oxo-2-
thioxothiazolidinylidene)methyl)pyrrole (12j). Starting from 4 (130 mg, 0.5 mmol) and 9j (127 mg,
0.5 mmol) to afford 163 mg of 12j, 66% yield. mp 244-246 °C; 1H NMR � ppm 7.73 (1H, s, =CH),
7.62 (1H, d, J = 2.4 Hz, ArH-6), 7.52 (1H, dd, J = 2.4 & 8.4 Hz, ArH-4), 7.25 (1H, t, J = 8.0 Hz,
ArH-5’), 7.14 (1H, d, J = 8.4 Hz, ArH-3), 6.84 ~ 6.88 (3H, m, ArH on D-ring), 6.26 (1H, s, PyH), 5.20
(2H, s, ArCH2N), 3.72 (3H, s, OCH3), 2.16 & 2.00 (each 3H, s, CH3 × 2); MS m/z (%) 493 (M – 1,
100); HPLC purity 96.28%.
5.1.4.12. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(3-(3-(methoxycarbonyl)
benzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)pyrrole (12k). Starting from 4 (100 mg, 0.39
mmol) and 9k (110 mg, 0.39 mmol) to afford 189 mg of 12k, yellow solid, 93% yield. mp 214-216
°C; 1H NMR � ppm 7.92 (1H, s, ArH-2’), 7.89 (1H, d, J = 7.6 Hz, ArH-4’), 7.75 (1H, s, =CH), 7.67
(1H, d, J = 2.4 Hz, ArH-6), 7.63 (1H, d, J = 7.6 Hz, ArH-6’), 7.50 ~ 7.54 (2H, m, ArH-4, ArH-5’), 7.15
(1H, d, J = 8.4 Hz, ArH-3), 6.27 (1H, s, PyH-4), 5.30 (2H, s, ArCH2N), 3.85 (3H, s, OCH3), 2.17 &
2.00 (each 3H, s, CH3 × 2); MS m/z (%) 521 (M – 1, 100); HPLC purity 96.08%.
5.1.4.13. (Z)-3-((3-(3-Carboxybenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-4
-hydroxy)phenyl-2,5-dimethylpyrrole (12l). The preparation was the same as 12f. 12k (100 mg,
0.19 mmol) was hydrolyzed to afford 70 mg of 12l, yellow solid, 72% yield. mp 292-294 °C (dec.);
1H NMR � ppm 7.89 (1H, s, ArH-2’), 7.87 (1H, d, J = 7.6 Hz, ArH-4’), 7.76 (1H, s, =CH), 7.68 (1H, d,
J = 2.8 Hz, ArH-6), 7.61 (1H, d, J = 7.6 Hz, ArH-6’), 7.47 ~ 7.53 (2H, m, ArH-4, ArH-5’), 7.15 (1H, d,
Page 22
ACCEPTED MANUSCRIPT
21
J = 8.4 Hz, ArH-3), 6.27 (1H, s, PyH), 5.30 (2H, s, ArCH2N), 2.17 & 2.00 (each 3H, s, CH3 × 2); MS
m/z (%) 507 (M – 1, 80), 463 (M- – 44, 100); HPLC purity 98.95%.
5.1.4.14.
(Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-(3-(5-(3-(2-(Trifluoromethyl)benzyl)
-4-oxo-2-thioxothiazolidinylidene)methyl)pyrrole (12m). Starting from 4 (100 mg, 0.39 mmol) and
9m (112 mg, 0.39 mmol) to afford 138 mg of 12m, yellow solid, 67% yield. mp 250-252 °C; 1H NMR
� ppm 7.81 (1H, d, J = 8.0 Hz, ArH-4’), 7.76 (1H, s, =CH), 7.68 (1H, d, J = 2.4 Hz, ArH-6), 7.62 (1H,
t, J = 8.0 Hz, ArH-5’), 7.49 ~ 7.53 (2H, m, ArH-4, ArH-5’), 7.15 (1H, d, J = 8.8 Hz, ArH-3), 6.99 (1H,
d, J = 8.0 Hz, ArH-6’), 6.31 (1H, s, PyH), 5.39 (2H, s, ArCH2N), 2.17 & 2.02 (each 3H, s, CH3 × 2);
MS (%) m/z 531 (M – 1, 100); HPLC purity 98.95%
5.1.4.15. (Z)-3-(5-(3-(2-Bromobenzyl)-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-
4-hydroxy)phenyl-2,5-dimethylpyrrole (12n). Starting from 4 (50 mg, 0.19 mmol) and 9n (60 mg,
0.19 mmol) to afford 130 mg of 12n, yellow solid, 62% yield. mp 286-288 °C (dec.); 1H NMR � ppm
7.76 (1H, s, =CH), 7.67 ~ 7.70 (2H, m, ArH-6, ArH-2’), 7.53 (1H, dd, J = 2.4 & 8.8 Hz, ArH-4), 7.33
(1H, t, J = 7.6 Hz, ArH-4’), 7.25 (1H, t, J = 7.6 Hz, ArH-5’), 7.15 (1H, d, J = 8.4 Hz, ArH-3), 6.86 (1H,
d, J = 7.6 Hz, ArH-6’), 6.31 (1H, s, PyH-4), 5.22 (2H, s, ArCH2N), 2.18 & 2.01 (each 3H, s, CH3 × 2);
MS m/z (%) 541 (M – 1, 40), 543 (M + 1, 50); 497 (M – 44, 100), 499 (497 + 2, 80); HPLC purity
98.92%.
5.1.4.17. (Z)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(5-(4-oxo-3-phenethyl-2-thioxo
thiazolidinylidene)methyl)pyrrole (13). Starting from 4 (130 mg, 0.5 mmol) and 10 (120 mg, 0.5
mmol) to afford 140 mg of 13, yellow solid, 58% yield. mp 228-230 °C; 1H NMR � ppm 7.66 ~ 7.67
(2H, m, ArH-6, =CH), 7.51 (1H, dd, J = 2.8 & 8.8 Hz, ArH-4), 7.20 ~ 7.31 (5H, m, ArH × 5 on
D-ring), 7.14 (1H, d, J = 8.8 Hz, ArH-3), 6.23 (1H, s, PyH), 4.23 (2H, t, J = 8.0 Hz, CH2N), 2.95(t, J
= 8.0 Hz, ArCH2), 2.15 & 1.99 (each 3H, s, CH3 × 2); MS m/z (%) 477 (M – 1, 100); HPLC purity
96.71%.
Page 23
ACCEPTED MANUSCRIPT
22
5.1.5. N-(3-Carboxy-4-chloro)phenyl-2,5-dimethyl-3-formylpyrrole (14). The preparation was the
same as 4. Starting with POCl3 (6 mL, 63.5 mmol), DMF (20 mL) and
2-chloro-3-(2,5-dimethyl-1H-pyrrol-1-yl)benzoic acid (10 g, 40 mmol) to afford 8.17 g of 14, gray
solid, 73% yield. mp 186-188 °C (dec.); 1H NMR (CDCl3) � ppm 9.87 (1H, s, CHO), 7.88 (1H, d, J =
2.4 Hz, ArH-6), 7.68 (1H, d, J = 8.4 Hz, ArH-3), 7.35 (1H, dd, J = 2.4 & 8.4 Hz, ArH-4), 6.43 (1H, s,
PyH), 2.32 & 2.02 (each 3H, s, CH3 × 2); MS m/z (%) 276 (M – 1, 100), 278 (M 1, 30).
5.1.6. N-(3-Carboxy)phenyl-2,5-dimethyl-3-formylpyrrole (15). The preparation was the same as
4. Starting with POCl3 (6 mL, 63.5 mmol), DMF (20 mL) and
3-(2,5-dimethyl-1H-pyrrol-1-yl)benzoic acid (9.25 g, 40 mmol) to afford 8.2 g of 15, gray solid, 79%
yield. mp 198-200 °C (dec.); 1H NMR (CDCl3) � ppm 9.89 (1H, s, CHO), 8.27 (1H, d, J = 8.0 Hz,
ArH-6), 7.98 (1H, s, ArH-2), 7.67 (1H, t, J = 8.0 Hz, ArH-5), 7.49 (1H, d, J = 8.0 Hz, ArH-4), 6.43
(1H, s, PyH), 2.31 & 2.01 (each 3H, s, CH3 × 2); MS m/z (%) 242 (M – 1, 100).
5.1.7. (Z)-3-(5-(3-Benzyl-4-oxo-2-thioxothiazolidin-5-ylidene)methyl)-N-(3-carboxy-4-chloro)
phenyl-2,5-dimethylpyrrole (16). Starting from 14 (140 mg, 0.5 mmol) and 9a (112 mg, 0.5 mmol)
to afford 180 mg of 16, red solid, 75% yield. mp 214-216 °C; 1H NMR � ppm 7.74 ~ 7.75 (3H, m,
ArH-3, 6, =CH), 7.58 (1H, dd, J = 2.4 & 8.4 Hz, ArH-4), 7.32 ~ 7.51 (5H, m, ArH × 5 on D-ring),
6.25 (1H, s, PyH), 4.70 (2H, s, ArCH2N), 2.18 & 2.01(each 3H, s, CH3 × 2); MS m/z (%) 481 (M – 1,
30), 483 (M + 1, 10), 437 (M- – 45, 100), 439 (437 + 2, 40); HPLC purity 98.15%.
5.1.7. (Z)-3-(5-(3-Benzyl-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy)phenyl-2,5-
dimethylpyrrole (17). Starting from 15 (97 mg, 0.4 mmol) and 9a (90 mg, 0.4 mmol) to afford 118
mg of 17, 66% yield. mp 220-222 °C (dec.); 1H NMR � ppm 8.10 (1H, d, J = 8.0 Hz, ArH-6), 7.80
(1H, s, ArH-2), 7.76 (1H, s, =CH), 7.72 (1H, t, J = 8.0 Hz, ArH-5), 7.67 (1H, d, J = 8.0 Hz, ArH-4),
7.32 ~ 7.51 (m, 5H, ArH × 5 on D-ring), 6.26 (1H, s, PyH), 4.70 (2H, s, ArCH2N), 2.17 & 2.00 (each
3H, s, CH3 × 2); MS m/z (%) 447 (M – 1, 100); HPLC purity 97.12%.
5.1.9. 3-Benzylthiazolin-2,4-dione (18). To a solution of thiazolin-2,4-dione (500 mg, 4.27 mmol)
and benzyl bromide (800 mg, 4.7 mmol) in 15 mL DMF was added K2CO3 (1.18 g, 8.55 mmol). The
Page 24
ACCEPTED MANUSCRIPT
23
reaction was stirred for 3 h under room temperature, the solid filtered off, and the filter extracted by
EtOAc. After the removal of the solvent, the residue was purified by flash silica column [gradual
eluent: EtOAc/petroleum ether, 0 ~ 30%] to afford 793 mg of 18, colorless oil, 90% yield. 1H NMR
(CDCl3) � ppm 7.31 ~ 7.39 (5H, m, ArH × 5), 4.77 (2H, s, ArCH2N), 3.94(2H, s, SCH2).
5.1.10. (Z)- 3-(5-(3-Benzyl-2,4-dioxothiazolidinylidene)methyl)-N-(3-carboxy-4-hydroxy)phenyl
-2,5-dimethylpyrrole (19). The preparation was the same as 12. Starting with 9 (50 mg, 0.19 mmol)
and 18 (40 mg, 0.19 mmol) to afford 53 mg of 19, light yellow solid, 61% yield. Amorphous; 1H
NMR � ppm 7.83 (1H, s, =CH), 7.63 (1H, d, J = 2.4 Hz, ArH-6), 7.46 (1H, dd, J = 2.4 & 8.8 Hz,
ArH-4), 7.38 ~ 7.29 (5H, m, ArH × 5 on D-ring), 7.10 (1H, d, J = 8.8 Hz, ArH-3), 6.20 (1H, s, PyH),
4.82 (2H, s, ArCH2N), 2.13 & 1.99 (each 3H, s, CH3 × 2); MS m/z (%) 447 (M – 1, 40), 404 (M – 44,
100); HPLC purity 96.19%.
5.1.11. N-(3-Carbmethoxy-4-hydroxy)phenyl-2,5-dimethyl-3-formyl-pyrrole (20). To a solution
of 4 (260 mg, 1 mmol) and Cs2CO3 (163 mg, 0.5 mmol) in 10 mL DMF was added CH3I (142 mg, 1
mmol). The solution was stirred at room temperature for 24 h and then poured into ice water. The
precipitate was filtered out and dried to afford 206 mg of 20, white needle, 75% yield. mp 136-138 °C;
1H NMR (CDCl3) � ppm 10.96 (1H, s, COOH), 9.87 (1H, s, CHO), 7.71 (1H, d, J = 2.4 Hz, ArH-6),
7.29 (1H, dd, J = 2.4 & 8.8 Hz, ArH-4), 7.13 (1H, d, J = 8.8 Hz, ArH-3), 6.37 (1H, s, PyH), 3.98 (3H,
s, OCH3), 2.18 & 1.99 (each 3H, s, CH3 × 2); MS m/z (%) 274 (M + 1, 40).
5.1.12. 2,5-Dioxo-1-phenethylpyrrolidin-3-yl acetate (21). To a solution of maleic anhydride (0.98
g, 10 mmol) in 15 mL ether, phenylethylamine (1.21 g, 10 mmol) was added dropwise in 15 mL ether,
and the reaction was stirred for 1.5 h at room temperature. Next, the white precipitate was filtered out,
washed with ether, and dried to afford 2.08 g of N-phenylethylmaleamic acid. Then the mixture of
maleamic acid and 100 mg NaOAc in 20 mL acetic anhydride was heated to reflux for 12 h. The
reaction was cooled to room temperature and poured into 100 mL ice water. The precipitate was
collected, washed with water, and dried to afford 1.44 g of 21, white solid, 55% yield. mp 108-110 °C;
1H NMR (CDCl3) � ppm 7.20 ~ 7.31 (5H, m, ArH × 5), 5.36 (1H, dd, J = 4.8 & 8.8 Hz, 3-H), 3.79 (2H,
Page 25
ACCEPTED MANUSCRIPT
24
t, J = 8.0 Hz, CH2N), 3.12 (1H, dd, J = 8.8 & 14.4 Hz, 4-H), 2.91 (2H, t, J = 8.0 Hz, ArCH2), 2.63 (1H,
dd, J = 4.8 & 8.8 Hz, 4-H), 2.16 (3H, s, COCH3); MS m/z (%) 262 (M + 1, 30).
5.1.13. (E)-N-(3-Carbmethoxy-4-hydroxy)phenyl-2,5-dimethyl-3-(3-(2,5-dioxo-1-phenethyl
pyrrolidinylidene)methyl)pyrrole (22). The mixture of 20 (136 mg, 0.5 mmol) and 21 (260 mg, 1
mmol) in 10 mL anhydrous ethanol in the presence of PPh3 (262 mg, 1 mmol) was heated to reflux
under nitrogen atmosphere for 12 h. After cooling to room temperature, the precipitate was filtered out,
washed with cold ethanol, and dried to afford 195 mg of 22, gray solid, 85% yield. mp 184-186 °C
(dec.); 1H NMR (CDCl3) � ppm 10.94 (1H, s, OH), 7.70 (1H, d, J = 2.8 Hz, ArH-6), 7.59 (1H, t, J = 2.0
Hz, =CH), 7.22 ~ 7.30 (6H, m, ArH-4, ArH × 5), 7.12 (1H, d, J = 8.8 Hz, ArH-3), 6.10 (1H, s, PyH),
3.97 (3H, s, OCH3), 3.85 (2H, t, J = 8.0 Hz, CH2N), 3.45 (2H, d, J = 2.0 Hz, COCH2), 2.95 (2H, t, J =
8.0 Hz, ArCH2), 2.15 & 2.02 (each 3H, s, CH3 × 2); MS m/z 459 (M + 1, 100).
5.1.14. (E)-N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethyl-3-(3-(2,5-dioxo-1-phenethylpyrroli
dinylidene)methyl)pyrrole (23). Compound 22 (200 mg, 0.44 mmol) in 10 mL glacial acetic acid and
2 mL 36% HCl under nitrogen atmosphere was heated to reflux for 24 h. The solution was then poured
into ice water. The precipitate was filtered out and purified by flash silica column [gradual eluent:
EtOAc/petroleum ether with AcOH (3%), 0 ~ 50%] to afford 60 mg of 23, gray solid, 33% yield.
Amorphous; 1H NMR � ppm 7.60 (1H, d, J = 2.8 Hz, ArH-6), 7.47 (1H, dd, J = 2.8 & 8.8 Hz,
ArH-4), 7.36 (1H, s, =CH), 7.19 ~ 7.31 (5H, m, ArH × 5), 7.13 (1H, d, J = 8.8 Hz, ArH-3), 6.25 (1H,
s, PyH), 3.69 (2H, t, J = 8.0 Hz, CH2N), 3.48 (2H, d, J = 2.0 Hz, COCH2), 2.85 (2H, t, J = 8.0 Hz,
ArCH2), 2.09 & 1.97 (each 3H, s, CH3 × 2); MS m/z (%) 443 (M – 1, 70), 399 (M – 44, 100); HPLC
purity 98.13%.
5.2.1. Sandwich ELISA to detect gp41 6-HB formation
A sandwich ELISA, as previously described,28 was used to test the inhibitory activity of the
compounds on gp41 six-helix bundle formation. Briefly, peptide N36 (2 �M) was preincubated with
the compound at graded concentrations at 37 °C for 30 min, followed by addition of C34 (2 �M). In
the control experiments, N36 was preincubated with C34 or PBS at 37 °C for 30 min in the absence
Page 26
ACCEPTED MANUSCRIPT
25
of the compounds tested. After incubation at 37 °C for 30 min, the mixture was added to wells of a
96-well polystyrene plate which were precoated with IgG (10 �g/mL) purified from rabbit antisera
directed against the gp41 six-helix bundle. Then, the mAb NC-1 recognizing gp41-6HB specifically,
biotin-labeled goat-anti-mouse IgG (Santa Cruz Biotechnolgy, Santa Cruz, CA), streptavidin-labeled
horseradish peroxidase (SA-HRP, Zymed, San Francisco, CA), and the substrate
3,3’,5,5’-tetramethylbenzidine (TMB, Sigma Chemical Co., St. Louis, MO), were added
sequentially. Absorbance at 450 nm was read in an ELISA reader (Ultra 386, TECAN Research Triangle
Park, NC), and the percentage of inhibition by the compounds was calculated as previously
described28. IC50 values were calculated using the computer program CalcuSyn, kindly provided by
Dr. T. C. Chou (Sloan-Kettering Cancer Center, New York). All the samples were tested in triplicate.
5.2.2. Determination of the inhibitory activity of the compounds on HIV-1 replication
The inhibitory activity of compounds on HIV-1IIIB replication in MT-2 cells was determined as
previously described29, 30. Briefly, 1 × 104 MT-2 cells were infected with an HIV-1IIIB strain or
T20-sensitive and resistant HIV-1 strains (100 TCID50) in 200 �L of RPMI 1640 medium containing
10% PBS in the presence or absence of a test compound at graded concentrations overnight. Next,
the culture supernatants were removed, and fresh media containing no test compounds were added.
On the fourth day post-infection, 100 �L of culture supernatants were collected from each well,
mixed with equal volumes of 5% Triton X-100, and assayed for p24 antigen, which was quantitated
by ELISA. Briefly, wells of polystyrene plates (Immulon 1B, Dynex Technology, Chantilly, VA)
were coated with HIV immunoglobulin (HIVIG), which was prepared from plasma of
HIV-seropositive donors with high neutralizing titers against HIV-1IIIB, in 0.085 M
carbonate-bicarbonate buffer (pH 9.6) at 4 °C overnight, followed by washing with buffer (0.01 M PBS
containing 0.05% Tween-20) and blocking with PBS containing 1% dry fat-free milk (Bio-Rad, Inc.,
Hercules, CA). Virus lysates were added to the wells and incubated at 37 °C for 1 h. After extensive
Page 27
ACCEPTED MANUSCRIPT
26
washes, anti-p24 mAb (183-12H-5C), biotin-labeled anti-mouse IgG1, SA-HRP, and TMB were
added sequentially. Reactions were terminated by addition of 1 N H2SO4. Absorbance at 450 nm was
recorded in an ELISA reader (Ultra 386, TECAN Research, Triangle Park, NC). Recombinant
protein p24 purchased from United States Biological (Swampscott, MA) was included to establish
standard dose-response curves. Each sample was tested in triplicate. The percentage of inhibition of
p24 production was calculated as previously described31. EC50 values were calculated using the
computer program CalcuSyn program.
5.2.3. Assessment of in Vitro Cytotoxicity
The in vitro cytotoxicity of compounds on MT-2 cells was measured by XTT assay.30 Briefly, 100
µL of the test compound at graded concentrations were added to equal volumes of cells (5
105/mL) in wells of 96-well plates. After incubation at 37 for 4 days, 50 µL of XTT solution (1
mg/mL) containing 0.02 µM of phenazine methosulphate (PMS, Sigma) was added. After 4 h, the
absorbance at 450 nm was measured with an ELISA reader. The CC50 (concentration for 50%
cytotoxicity) values were calculated using the CalcuSyn program.
5.3. Molecular docking of 14m onto the hydrophobic pocket
Molecular docking was performed by using Discovery Studio 3.0 (Accelrys Inc.). The default atom
types and parameters supplied with Discovery Studio 3.0 were used throughout the docking. The
ligand was prepared using the ligand preparation tool available in the software and then minimized
using the minimization tool with CHARMM Force Field. The X-ray crystal structure of the gp41
core (1aik) was available from the Protein Data Bank (PDB) at the Research Collaboratory for
Structural Bioinformatics (RCSB). The protein was prepared using the protein preparation tool; then
one of the three CHRs was deleted to expose the hydrophobic pocket. The binding sphere was
defined from the selection of the residues K574, Q577, L568, and W571, as shown in Figure 2, with
Page 28
ACCEPTED MANUSCRIPT
27
the radius value of 11 Å. The docking was performed using the CDOCKER tool in Discovery Studio,
and the pose was selected on the basis of the score and hydrophobic and hydrogen bond interactions.
Acknowledgement
This investigation was supported by grants 2006AA02Z319, 2006DFA33560, and 2009ZX09103-011
from the Ministry of Science and Technology in China, and grants U0832001/L02 and 81173098 from
the National Science Foundation of China (NSFC).
References and notes
1. Ashkenazi, A.; Shai, Y. Eur. Biophys. J. 2011, 349.
2. Harrison, S. C. Nat. Struct. Mol. Biol. 2008, 15, 690.
3. Eckert, D. M.; Kim, P. S. Annu. Rev. Biochem. 2001, 70, 777.
4. Chan, D. C.; Fass, D.; Berger, J. M.; Kim, P. S. Cell 1997, 89, 263.
5. Lazzarin, A. Exp. Opin. Pharm. 2005, 6, 453.
6. Hardy, H.; Skolnik, P. R. Pharmacotherapy 2004, 24, 198.
7. Liu, S. W.; Wu, S. G.; Jiang, S. B. Curr. Pharm. Des. 2007, 13, 143.
8. Esté, J. A.; Telenti, A. Lancet 2007, 370, 81.
9. Salzwedel, K.; West, J. T.; Hunter, E. J.Virol. 1999, 73, 2469.
10. He, Y. X.; Liu, S. W.; Jing, W. G.; Lu, H.; Cai, D. M.; Chin, D. J.; Debnath, A. K.; Kirchhoff,
F.; Jiang, S. B. J. Biol. Chem. 2007, 282, 25631.
11. He, Y. X.; Liu, S. W.; Li, J. J.; Lu, H.; Qi, Z.; Liu, Z. H.; Debnath, A. K.; Jiang, S. B. J. Virol.
2008, 82, 11129.
12. Jiang, S. B.; Debnath, A. K. Biochem. Biophys. Res. Commun. 2000, 270, 153.
13. Debnath, A. K.; Radigan, L.; Jiang, S. B. J. Med. Chem. 1999, 42, 3203.
Page 29
ACCEPTED MANUSCRIPT
28
14. Jiang, S.; Tala, S. R.; Lu, H.; Abo-dya, N. E.; Avan, I.; Gyanda, K.; Lu, L. J. Med. Chem. 2011,
54, 572.
15. Katritzky, A. R.; Tala, S. R.; Lu, H.; Vakulenko, A. V.; Chen, Q.-y.; Sivapackiam, J.; Pandya,
K.; Jiang, S.; Debnath, A. K. J. Med. Chem. 2009, 52, 7631.
16. Zhou, G.; Wu, D.; Hermel, E.; Balogh, E.; Gochin, M. Bioorg. Med. Chem. Lett. 2010, 20,
1500.
17. Zhou, G.; Wu, D.; Snyder, B.; Ptak, R. G.; Kaur, H.; Gochin, M. J. Med. Chem. 2011, 54, 7220.
18. Stewart, K. D.; Huth, J. R.; Ng, T. I.; Mcdaniel, K.; Newlin, R.; Stoll, V. S.; Mendoza, R. R.;
Matayoshi, E. D.; Carrick, R.; Mo, H.; Severin, J.; Walter, K.; Richardson, P. L.; Barrett, L. W.;
Meadows, R.; Anderson, S.; Kohlbrenner, W.; Maring, C.; Kempf, D. J.; Molla, A.; Olejniczak,
E. T. Bioorg. Med. Chem. Lett. 2010, 20, 612.
19. Liu, B.; Joseph, R. W.; Dorsey, B. D.; Schiksnis, R. A.; Northrop, K.; Bukhtiyarova, M.;
Springman, E. B. Bioorg. Med. Chem. Lett. 2009, 19, 5693.
20. Whitby, L. R.; Boyle, K. E.; Cai, L.; Yu, X.; Gochin, M.; Boger, D. L. Bioorg. Med. Chem.
Lett. 2012, 22, 2861.
21. Ernst, J. T.; Kutzki, O.; Debnath, A. K.; Jiang, S.; Lu, H.; Hamilton, A. D. Angew. Chem. 2002,
114, 288.
22. Yang, J.; Zhang, F.; Li, J.; Chen, G.; Wu, S.; Ouyang, W.; Pan, W.; Yu, R.; Yang, J.; Tien, P.
Bioorg. Med. Chem. Lett. 2012, 22, 1415.
23. Liu, S.; Lu, H.; Zhao, Q.; He, Y.; Niu, J.; Debnath, A. K. Bioch. Biophys. Acta 2005, 1723, 270
24. Bao, J.; Zhang, D. W.; Zhang, J. Z. H.; Huang, P. L.; Huang, P. L.; Lee-huang, S. FEBS Lett.
2007, 581, 2737.
25. He, X. Y.; Zou, P.; Qiu, J.; Hou, L.; Jiang, S.; Liu, S.; Xie, L. Bioorg. Med. Chem. 2011, 19,
6726.
Page 30
ACCEPTED MANUSCRIPT
29
26. Liu, K.; Lu, H.; Hou, L.; Qi, Z.; Teixeira, C.; Barbault, F.; Fan, B. T.; Liu, S. W.; Jiang, S. B.;
Xie, L. J. Med. Chem. 2008, 51, 7843.
27. Sortino, M.; Cechinel Filho, V.; Corrêa, R.; Zacchino, S. Bioorg. Med. Chem. 2008, 16, 560.
28. Jiang, S.; Lin, K.; Zhang, L.; Debnath, A. K. J. Virol. Methods 1999, 80, 85.
29. Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A. K. Antimicrob. Agents Chemother.
2004, 48, 4349.
30. Debnath, A. K.; Radigan, L.; Jiang, S. B. J. Med. Chem. 1999, 42, 3203.
31. Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A. K.
Virology 2005, 339, 213.
32. Rimsky, L. T.; Shugars, D. C.; Matthews, T. J. J. Virol. 1998, 72, 986.
Page 31
ACCEPTED MANUSCRIPT
30
Graphic Abstract
COOHHO
N
S N
O
S
A
B
CD
COOHHO
N
IC50 37.4 µM (gp41- 6HB)EC50 0.7 µM (cells)
IC50 1.8-2.6 µM (gp41- 6HB)EC50 0.3-1.5 µM (cells)
2 12e, 12g, 12k
R