Page 1
SP4: In silico methodsPartner 16A EMBL (Russell, Bork)Partner 1 (CRG Serrano)Partner 5 (NKI Perrakis)Partner 10 (HU Margalit)Partner 12 (CCNet)Partner 17 IRB (Aloy)Partner 3A (Paris-Sud, Janin)
Page 2
SP4 In silico methods
• WP4.1: Target identification & annotationPartners: EMBL-Bork/Russell, HU, CCNet, IRB
• WP4.2: Complex modelingPartners: EMBL-Russell, IRB, Gif, CRG
• WP4.3: Interface to the scientific community & scientific data managementPartners: NKI, EMBL-Russell, CCNet
Page 3
WP4.1: Target identification & annotation
Partners EMBL-HD, HU, CCN, IRB
Activities in:• Interaction prediction (HU, EMBL-Bork)• Complex prediction & ranking, the ‘list of 20’
(IRB)• Complex visualisation (CCNet/EMBL)• Data gathering (CCNet)
(e.g. protein-chemical interactions)• Gel processing (EMBL)
Page 4
Complex database, web interface & wiki
Matthew Betts (EMBL)
Page 5
The list of 20
Aloy group (IRB)
Page 6
Experimental tests on the 20
Aloy, Seraphin, van Tilburgh & Dziembowski groups
Page 7
WP4.2: Complex modelingPartners EMBL-HD, IRB, Gif, CRG
Activities in:• Complex modelling
– Automated procedures (EMBL-Russell/IRB)– Interaction prediction via structure (EMBL-Russell/IRB)
• New methods for modelling– FoldX (CRG/EMBL)– High-throughput Docking (IRB)
• Analyses, individual models (Everybody)
Page 8
Building a complex from pieces
Aloy et al, Curr. Opin. Struct. Biol, 2005.
For 636 complexes in yeast 3505 : proteins modelable 419 : complexes single subunit models 224 : 2+ subunit models 122 : 3+ subunit models
Page 9
Damien Devos (EMBL)
Example: MCM complex model agrees with existing EM data
Page 10
eIF2 /eIF2B complex
Preliminary reconstructions (Bettina Boettcher)
GCD7GCD2
GCN3
SUI2
SUI4
SUI3
GCN3
GCD6
GCD1
SUI4
GCD7
SUI4SUI3
GCD1
Sub-complexes
Intact complex MS (Carol Robinson)
Modelling (Damien Devos)
Page 11
The 3D-Repertoire Model Gallery
Page 12
New interactions and principles arising from 3DR SP4
Page 13
Defining new interfaces:424 candidate interfaces to date
Complex 1
Complex 2 Complex 3
New complex
Complex 1,2 & 3
Complex 1Complex 2
Complex 1 & 2
New complex
SuperimpositionSuperimposition
A common shape denotes a similar fold
Page 14
Example: Transcription factor SPX dimer
Domain 1, 1z3eA.c.47.1.12-1-trans3 (chain A) Transcriptional regulator SPX (B.subtilis) Domain 2, 1z3eA.c.47.1.12-1-trans4 (chain B) Transcriptional regulator SPX (B.subtilis) Domain 3, 1z3eB.a.60.3.1-1-trans1p (chain C) RNA polymerase alpha (B.subtilis) Domain 4, 1z3eB.a.60.3.1-1-trans2p (chain D) RNA polymerase alpha (B.subtilis) Domain 5, 1lb2E.a.60.3.1-1-trans1 (chain E) RNA polymerase alpha (E.coli) Domain 6, 1lb2B.a.60.3.1-1-trans2 (chain F) RNA polymerase alpha (E.coli)
E.coli dimer in one protein, forms nice interface in B.subtilis – good evidence from other sources (Myco TAP)
Complex 1
Complex 2
Complex 3
New interface
Page 15
Enabling/disabling loops can predict mulimerization state
Homodimer E. Coli Guanylate kinase
Monomer S. Cerevisiae
Guanylate kinase
E. Coli Guanylate kinase
V. Cholerae Guanylate kinase
Yeast Guanylate kinase
Mouse Guanylate kinase
Pig Guanylate kinase
Bovine Guanylate kinase
Homodimers
enabling loop
Monomers
Page 16
When modelling fails – docking?
• The Aloy group (IRB) is currently running many tens of thousands of docking experiments using Mare nostrum, the largest supercomputer in Europe
• Aim is to identify promising docking candidates to help model key interactions of interest
Page 17
Modelling versus docking• We can model an interaction structure if there is a
previously determined structure containing parts homologous to the two interacting proteins
• We can predict an interaction structure by docking if we have structures or models for parts of the interacting proteins
homology
homology
Page 18
Large-scale Docking36 million possible yeast protein interactions
Page 19
Large-scale Docking
Proteins Interactions all vs all 100
CPUs 1000 CPUs
100 CPUs
1000 CPUs
pdb 90% 8899 9645 39596100 24747,5 2474,7 197980,5 19798,0 pdb 30% 5224 5836 13645088 8528,2 852,8 68225,4 6822,5
Interaction types 2782 3737 3869762 2418,6 241,8 19348,8 1934,8 yeast xray orfs 79 97 3120,5 1,9 0,2 15,6 1,6
Unrefined Refined
Page 20
Using FoldX to assess docked or modelled interactions
Good Interaction, but many clashes, Model is not so good but could be rescuedBy backbone moves/further docking
Page 21
WP4.3: Interface to the scientific community and scientific data management
Partners: EMBL-HD, NKI, [CCN]
Activities in:• Web site maintenance
– New data (copy number, structural annotation)– Various optimisation– YeastWiz
• Complex target DB– Resting period for software development– Needs data. Listen to Tassos.
Page 22
WP4.3: Web site
Matthew Betts (EMBL)
Page 23
Yeast Wiz (CCNet)
Interactions of known structure
www.3drepertoire.org/yeastwizWindows XPLinuxManual (rather beta)
Accounts enabled Monday for everybodySuggestions for new data promising
Matthew Betts (EMBL), Tomasz Ignasiak (CCNet)
Page 24
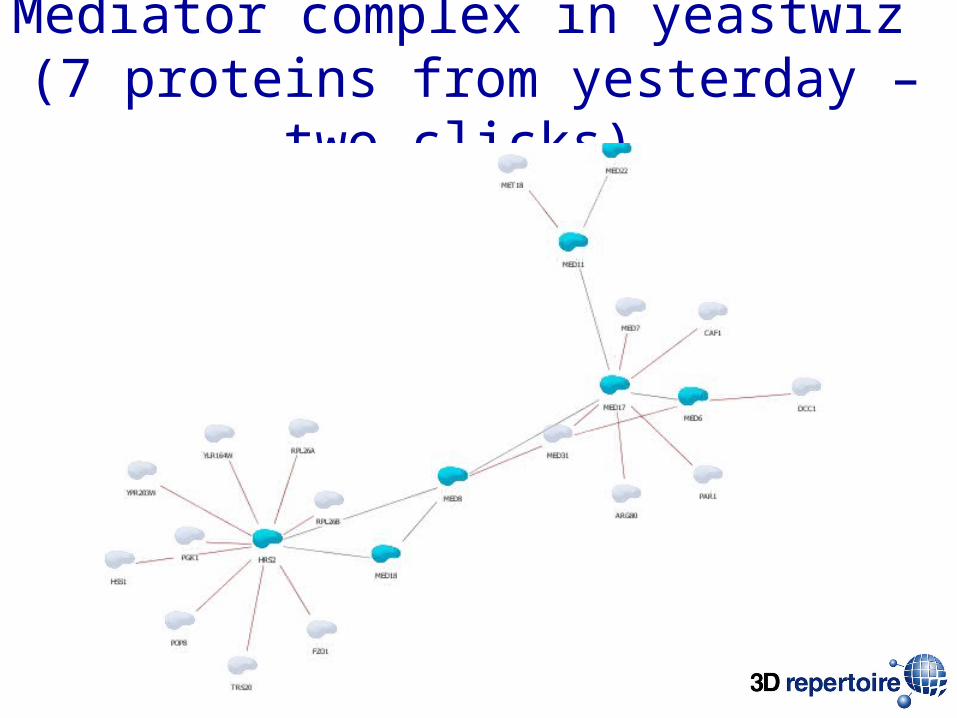
Mediator complex in yeastwiz (7 proteins from yesterday – two clicks)
3D
3D
Page 25
Current data contributions
CCN-DB
EMBL HU
IRB
STRING (interactions)SMART (orthologues)3D Interaction predictionsOrthologyModelsEtc.
Protein-protein (>10 sources, manual)Protein-chemical (Manual, TM)
Dom-dom profilesContext interactionsEnabling loops
Docking solutionsList of 20
3DR
Page 26
WP4.3: Interface to the scientific community and scientific data management
WIKI
TargetDB
Page 27
Need target db stuff
Page 29
The exosome
Damien Devos (w Carol Robinson)
Page 30
Analysis of a “gold set” of 61 models of known interactions by FoldX
Easy case : Good Interaction Energy, few clashesGood Model
Page 31
Analysis of a “gold set” of 61 models of known interactions by FoldX
Bad Interaction, loads of clashes, interpenetrating mainchainsBad Model
Page 32
Analysis of a “gold set” of 61 models of known interactions by
FoldX
Bad Interaction, many clashes, but Model could be rescued by some backbone moves/ further docking
Page 33
Analysis of a “gold set” of 61 models of known interactions by
FoldX
Good Interaction, many clashes, interpenetrating mainchains, gaps in the structureBad Model
Page 34
Analysis of a “gold set” of 61 models of known interactions by FoldX
Easy case : Good Interaction Energy, few clashes : Good Model
Bad Interaction, many clashes - interpenetrating mainchains, gaps in the structure : Bad Model- mainchains too close on a large region but this can be solved bybackbone moves/further docking (could improve the model?)
Good Interaction, many clashes: - interpenetrating mainchains, gaps in the structure : Bad Model- mainchains too close on a large region but this can be solved bybackbone moves/further docking
The magnitude of the local clashes correlate with the possibility to rescue or not a model (mild clashes on a lot of residues), but still there are exceptions.
Could we really skip a step of visualization?
Page 35
From protein-protein interactions to domain-domain interactions and back
Hanah MargalitThe Hebrew University of Jerusalem
Page 36
domain pairs
protein-protein interactions
Modularity in protein-protein interactions
fine tuners Yes YesNo No
Page 37
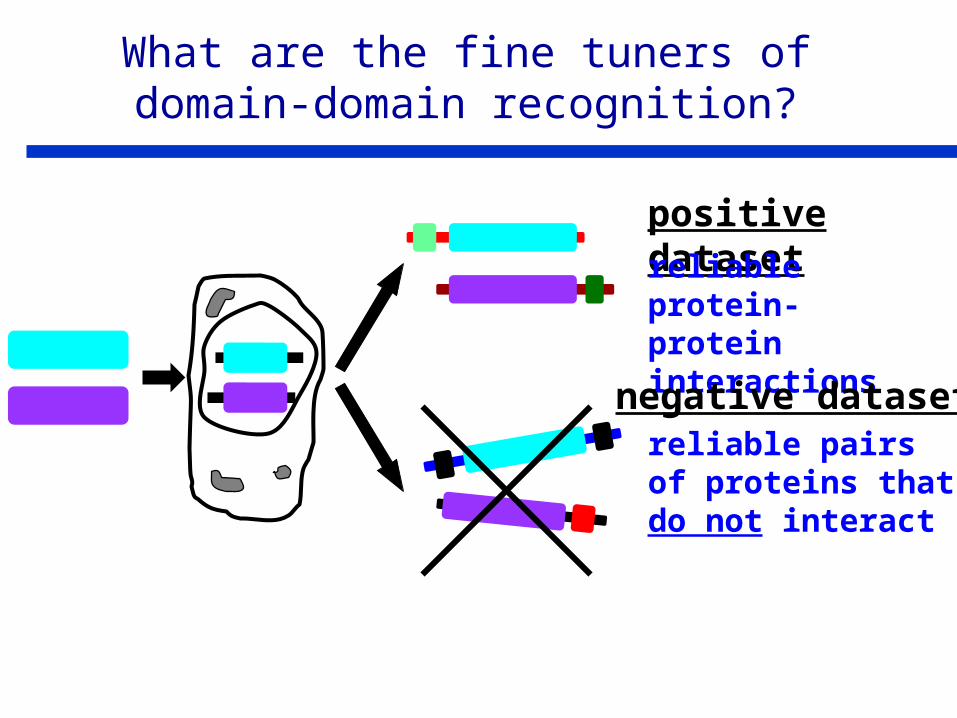
positive datasetreliable protein-protein interactions
reliable pairs of proteins that do not interact
negative dataset
What are the fine tuners of domain-domain recognition?
Page 38
Homodimers and monomers provide an ideal dataset
Domains that mediate homodimerization Domains that mediate homodimerization are found also in monomersare found also in monomers
homodimers: co-localized co-expressed interact
monomers: co-localized co-expressed do not interact
Database of 50 homodimers/monomerswith the same domain for which structural data is available
Page 39
Phosphorylations PP
monomersInterface residuesubstitutions
Different fine-tuners determine theself-interaction potential of domains
homodimers
Page 40
Enabling loops mediate homodimerization
Homodimer E. Coli Guanylate kinase
Monomer S. Cerevisiae
Guanylate kinase
E. Coli Guanylate kinase
V. Cholerae Guanylate kinase
Yeast Guanylate kinase
Mouse Guanylate kinase
Pig Guanylate kinase
Bovine Guanylate kinase
Homodimers
enabling loop
Monomers
Page 41
Disabling loops prevent homodimerization
Monomer: Bovine inositol polyphosphate 1-phosphatase
Homodimer: Bovine inositol monophosphatase
DL
Monomers
Homodimers
Page 42
Loop profiles
A multiple-sequence alignment with locations of potential loops
Presence AND absence are informative
monomerprotein 2
homodimerprotein 1
enablingloop
disablingloop
Page 43
The ‘core set’
64 / 73 are consistent (88%, p-value ≤ 3.2•10-6)
Page 44
Test set
experimental oligomeric state
loop profile
monomer
dimer
72/80 are consistent (90%, p-value ≤ 5•10-6)
80 proteins with documented oligomeric state based on experimental data
monomer
95
dimer
363
Page 45
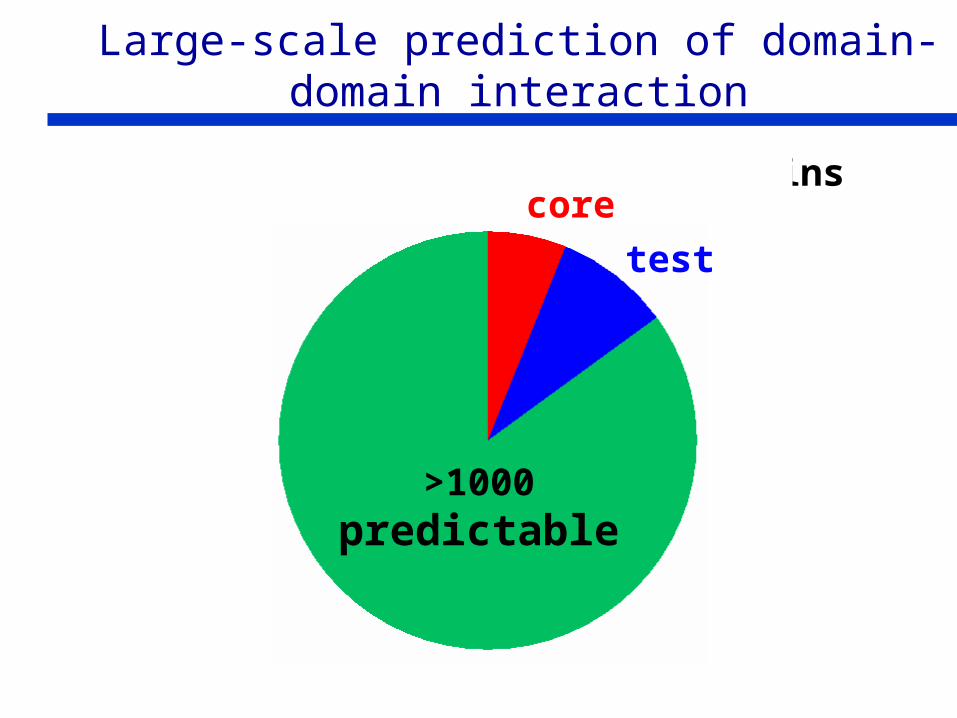
Large-scale prediction of domain-domain interaction
pfkB carbohydrate kinase domain proteins
EL
DL DL DL
108 homodimers
31 monomers
monomer
homodimer
core
Homodimer
Monomertest
>1000predictable
Page 46
Boundary loops
There are enabling/disabling loops that are located outside domain boundaries
Page 47
Metallo-beta-lactamase domain
Dominance of disabling over enabling loops
RNase Z (B. Subtilis)ccrA (B. Fragilis)
Page 48
Summary
1. Enabling/disabling loops are newly discovered fine-tuners of domain-domain interaction
2. Their presence/absence is highly preserved in evolution, implying that prevention of unwanted interactions is an evolutionary constraint
3. Prediction of self-interaction potential of domains according to loop profiles is highly
accurate (~90%)
Page 49
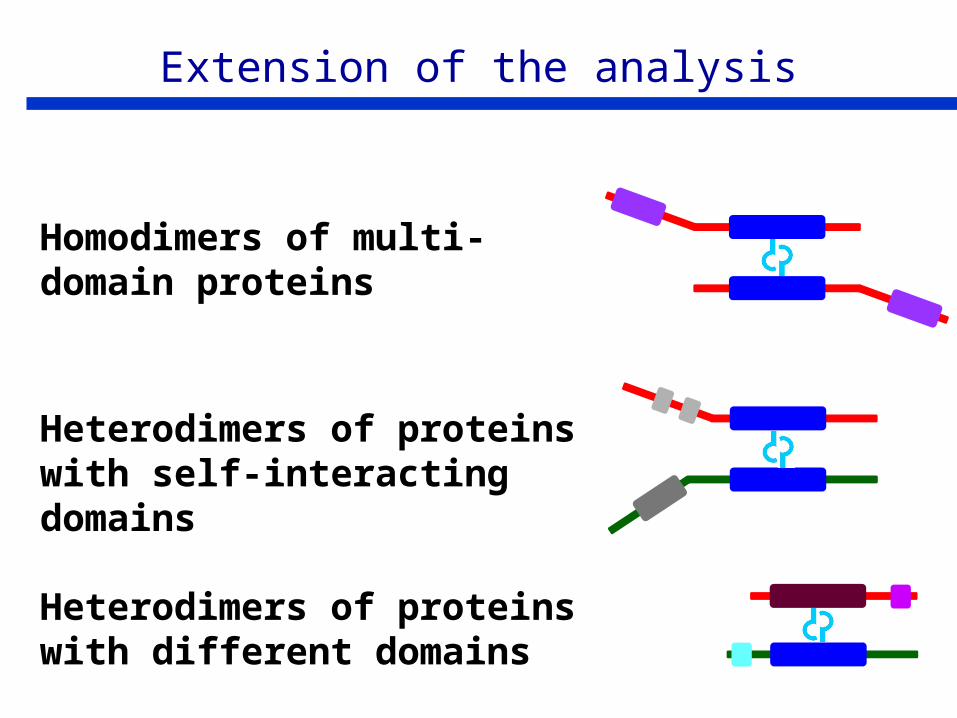
Extension of the analysis
Homodimers of multi-domain proteins
Heterodimers of proteins with self-interacting domains
Heterodimers of proteins with different domains













































![INDIVIDUAL PREDICTIVE CAPACITIES OF IN SILICO …2)_1900x1000_def[1].pdf · IN SILICO METHODS : PRINCIPLE AND PERFORMANCES C Piroird • A Del Bufalo • S Ringeissen • J Eilstein](https://static.documents.pub/doc/80x56/5b601bfd7f8b9ac1478b7766/individual-predictive-capacities-of-in-silico-21900x1000def1pdf-in-silico.jpg)
















