Specific solvent effect on lumazine photophysics: A combined fluorescence and intrinsic reaction coordinate analysis N. Shaemningwar Moyon, Pynsakhiat Miki Gashnga, Smritakshi Phukan, Sivaprasad Mitra ⇑ Department of Chemistry, North-Eastern Hill University, Shillong 793 022, India article info Article history: Received 11 December 2012 In final form 20 May 2013 Available online 29 May 2013 Keywords: Lumazine Fluorescence Hydrogen bonding DFT calculations Charge redistribution Intrinsic reaction coordinate analysis abstract The photophysical properties and tautomerization behavior of neutral lumazine were studied by fluores- cence spectroscopy and density functional theory calculation. A quantitative estimation of the contribu- tions from different solvatochromic parameters, like solvent polarizibility (p ⁄ ), hydrogen bond donation (a) and hydrogen bond accepting (b) ability of the solvent, was made using linear free energy relation- ships based on the Kamlet–Taft equation. The analysis reveals that the hydrogen bond acceptance ability of the solvent is the most important parameter characterizing the excited state behavior of lumazine. Theoretical calculations result predict an extensive charge redistribution of lumazine upon excitation corresponding to the N3 and N1 proton dissociation sites by solvents in the ground and excited states, respectively. Comparison of S 0 and S 1 state potential energy curves constructed for several water medi- ated tautomerization processes by intrinsic reaction coordinate analysis of lumazine-H 2 O cluster shows that (3,2) and (1,8) hydrogen migrations are the most favorable processes upon excitation. Ó 2013 Elsevier B.V. All rights reserved. 1. Introduction Lumazine (2,4-(1H,3H) pteridinedione) is a naturally occurring compound composed of fused pyrimidine and pyrazine rings (the so called pteridine derivative) and known to be involved in a wide variety of biological processes [1–3]. It is recognized as a substrate in the oxidative hydroxylation reaction catalyzed by xanthine oxi- dase resulting in the product violapterin (2,4,7-(1H,3H,8H)pteri- dinetrione) [4]. Lumazines and other structurally related flavins and pterins are also involved in forming coordination bonds with biologically essential metals in some metalloenzymes [5]. Based on this idea, several derivatives of lumazines were identified as selective sensors for several metal ions [6–8]. Lumazines have been of interest recently for their fluorescent properties and their poten- tial use as reporter groups in DNA probes [9,10]. The fluorescence behavior of lumazine has been well known for several decades [11–13], and also is a subject of renewed interest in recent times. Presiado et al. [14] reported that the deprotonation time of neutral lumazine (Lum) to form the corresponding anion (Lum-anion) is about 35 ps in the excited state; although, the ground and excited state pK a values of Lum mentioned by these authors are in sharp disagreement with the other reports [15]. De- spite the presence of several reports on Lum fluorescence in the lit- erature [11–15] and the use Lum in chemical as well as bio- chemical systems, to the best of our knowledge, a systematic study of Lum photophysics in a series of solvents with varying dielectric constants and hydrogen bonding properties has not been under- taken. Furthermore, Lum can exist in several tautomeric forms (Chart 1). The photophysical behavior of Lum is best understood by analyzing the relative stability of different tautomers and sol- vent mediated inter-conversions among them, more particularly in protic medium like water. Heterocyclic systems containing Lum and the structurally re- lated scaffold have also been studied intensively with a variety of theoretical models [16–18]. Emboldened by our previous investi- gations on the relative stability and water assisted inter-conver- sions among several structural isomers of biologically relevant heterocyclic systems like chemiluminescent luminol [19] and alloxazine derivatives [20], both in the ground and electronically excited states, we decided to begin a thorough investigation on lumazines also, particularly because of its structural similarity with alloxazine ring system of flavins. Multiple regression analysis of different spectroscopic proper- ties using Kamlet–Taft equation reveals the importance of solvent hydrogen bond acceptance (HBA) basicity in describing the solva- tochromic behavior of Lum. The details of these results are de- scribed in the first two sections of the results and discussion section. Theoretical calculations using Kohn–Sham density func- tional theory (KS-DFT) indeed predict the increased acidity of Lum upon electronic excitation. The energy parameters of different possible tautomers of Lum are compared in isolated condition as well as in presence of the solvated cluster with water molecule to predict the di-oxo conformer to be the most stable structure. 0301-0104/$ - see front matter Ó 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.chemphys.2013.05.012 ⇑ Corresponding author. Tel.: +91 364 2722634; fax: +91 364 2550076. E-mail addresses: [email protected], [email protected](S. Mitra). Chemical Physics 421 (2013) 22–31 Contents lists available at SciVerse ScienceDirect Chemical Physics journal homepage: www.elsevier.com/locate/chemphys
Transcript
Chemical Physics 421 (2013) 22–31
Contents lists available at SciVerse ScienceDirect
N. Shaemningwar Moyon, Pynsakhiat Miki Gashnga, Smritakshi Phukan, Sivaprasad Mitra ⇑Department of Chemistry, North-Eastern Hill University, Shillong 793 022, India
a r t i c l e i n f o
Article history:Received 11 December 2012In final form 20 May 2013Available online 29 May 2013
The photophysical properties and tautomerization behavior of neutral lumazine were studied by fluores-cence spectroscopy and density functional theory calculation. A quantitative estimation of the contribu-tions from different solvatochromic parameters, like solvent polarizibility (p⁄), hydrogen bond donation(a) and hydrogen bond accepting (b) ability of the solvent, was made using linear free energy relation-ships based on the Kamlet–Taft equation. The analysis reveals that the hydrogen bond acceptance abilityof the solvent is the most important parameter characterizing the excited state behavior of lumazine.Theoretical calculations result predict an extensive charge redistribution of lumazine upon excitationcorresponding to the N3 and N1 proton dissociation sites by solvents in the ground and excited states,respectively. Comparison of S0 and S1 state potential energy curves constructed for several water medi-ated tautomerization processes by intrinsic reaction coordinate analysis of lumazine-H2O cluster showsthat (3,2) and (1,8) hydrogen migrations are the most favorable processes upon excitation.
� 2013 Elsevier B.V. All rights reserved.
1. Introduction
Lumazine (2,4-(1H,3H) pteridinedione) is a naturally occurringcompound composed of fused pyrimidine and pyrazine rings (theso called pteridine derivative) and known to be involved in a widevariety of biological processes [1–3]. It is recognized as a substratein the oxidative hydroxylation reaction catalyzed by xanthine oxi-dase resulting in the product violapterin (2,4,7-(1H,3H,8H)pteri-dinetrione) [4]. Lumazines and other structurally related flavinsand pterins are also involved in forming coordination bonds withbiologically essential metals in some metalloenzymes [5]. Basedon this idea, several derivatives of lumazines were identified asselective sensors for several metal ions [6–8]. Lumazines have beenof interest recently for their fluorescent properties and their poten-tial use as reporter groups in DNA probes [9,10].
The fluorescence behavior of lumazine has been well known forseveral decades [11–13], and also is a subject of renewed interestin recent times. Presiado et al. [14] reported that the deprotonationtime of neutral lumazine (Lum) to form the corresponding anion(Lum-anion) is about �35 ps in the excited state; although, theground and excited state pKa values of Lum mentioned by theseauthors are in sharp disagreement with the other reports [15]. De-spite the presence of several reports on Lum fluorescence in the lit-erature [11–15] and the use Lum in chemical as well as bio-chemical systems, to the best of our knowledge, a systematic study
of Lum photophysics in a series of solvents with varying dielectricconstants and hydrogen bonding properties has not been under-taken. Furthermore, Lum can exist in several tautomeric forms(Chart 1). The photophysical behavior of Lum is best understoodby analyzing the relative stability of different tautomers and sol-vent mediated inter-conversions among them, more particularlyin protic medium like water.
Heterocyclic systems containing Lum and the structurally re-lated scaffold have also been studied intensively with a variety oftheoretical models [16–18]. Emboldened by our previous investi-gations on the relative stability and water assisted inter-conver-sions among several structural isomers of biologically relevantheterocyclic systems like chemiluminescent luminol [19] andalloxazine derivatives [20], both in the ground and electronicallyexcited states, we decided to begin a thorough investigation onlumazines also, particularly because of its structural similaritywith alloxazine ring system of flavins.
Multiple regression analysis of different spectroscopic proper-ties using Kamlet–Taft equation reveals the importance of solventhydrogen bond acceptance (HBA) basicity in describing the solva-tochromic behavior of Lum. The details of these results are de-scribed in the first two sections of the results and discussionsection. Theoretical calculations using Kohn–Sham density func-tional theory (KS-DFT) indeed predict the increased acidity ofLum upon electronic excitation. The energy parameters of differentpossible tautomers of Lum are compared in isolated condition aswell as in presence of the solvated cluster with water moleculeto predict the di-oxo conformer to be the most stable structure.
Chart 1. Possible tautomeric structures of neutral lumazine (Lum).
N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31 23
Finally, the water mediated hydrogen migration reaction leading tothe inter-conversions among several tautomers is checked byintrinsic reaction coordinate (IRC) analysis. The combined effectof relative increase in exothermicity and reduced barrier heightin the excited state Gibbs free energy surface makes (3,2) and(1,8) hydrogen migration reactions feasible in the S1 state in com-parison with the ground (S0) state. All these results are described atthe last part of the results and discussion section.
2. Materials and Methods
2.1. Chemicals
Lumazine (Lum) was received from Sigma-Aldrich ChemicalPvt. Ltd. (product no. L-3307, Lot no. #13619CZ) and used withoutany further purification. A series of spectroscopic grade (>99.5%)organic solvents with varying dielectric constants and hydrogenbonding properties (mentioned in Table 1) were used as receivedfrom Alfa Aesar and, in some cases, from Aldrich Chemical Com-pany. The analytical grade type – II water, also used as solvent,
Table 1Name of the solvents used with corresponding solvent parameters.
was obtained from an Elix 10 water purification system (MilliporeIndia Pvt. Ltd.). The chromophore concentration (�5 lM) was verylow to avoid any aggregation and kept constant during spectralmeasurements in different solvents.
2.2. Experimental procedure
Steady-state absorption spectra were recorded on a Perkin-El-mer model Lambda25 absorption spectrophotometer. Fluorescencespectra were taken with a Hitachi model FL4500 spectrofluorime-ter and all the spectra were corrected for the instrument responsefunction [20]. Quartz cuvettes of 10 mm optical path length re-ceived from PerkinElmer, USA (part no. B0831009) and Hellma,Germany (type 111-QS) were used for measuring absorption andfluorescence spectra, respectively. Fluorescence quantum yields(/f) were calculated by comparing the total fluorescence intensityunder the whole fluorescence spectral range with that of a stan-dard (quinine bisulfate in 0.5 M H2SO4 solution, /s
f ¼ 0:546 [21])with the following equation using adequate correction for solventrefractive index (n) [22]
/if ¼ /s
f �Fi
Fs �1� 10�As
1� 10�Ai �ni
ns
� �2
ð1Þ
where Ai and As are the optical density of the sample and standard,respectively, and ni is the refractive index of solvent at 293 K. Therelative experimental error of the measured quantum yield wasestimated to be ±10%.
2.3. Theoretical calculations
Density functional theory (DFT) within the framework of Kohn–Sham (KS) formalism has been successfully applied to study thehydrogen bonding in various systems and the recent results arefound to be in good agreement even with the results of severalother computationally costlier methods like Møller–Plesset (MP2)or coupled-cluster (CC) calculations [23–26]. Myshakina et al. usedB3LYP functional alongwith 6-311++G(d,p) basis set to estimatethe impact of hydrogen bonding on amide frequency for modelpeptides [27]; whereas, in a recent paper, the effectiveness ofDFT method using similar level of calculations to predict the dativebond energy of Lewis adducts were checked to be within �1 kcal/mol of the benchmark results [28]. The relationship between theformation and electrolytic dissociation of carbonic acid was stud-ied by DFT calculation in one of the pioneering works by Yamabeand Kwagishi [29]. A good number of recent reports are also avail-able in literature dealing with ab-initio and/or DFT calculation on
30)b Kamlet-Taft solvent parameters
a b p⁄
.4 0.98 0.66 0.6
.7 0.84 0.9 0.52
.7 0.84 0.84 0.47
.1 – – –
.4 0.76 0.95 0.48
.1 1.17 0.47 1.09
.6 0.19 0.4 0.75
.1 0 0.45 0.55
.1 0 0.76 0.1
.2 0 0.69 0.88
.4 0 0.55 0.58
.7 0 0.1 0.810 0.37 0.55
refractive indices are represented by e and n, respectively.
24 N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31
isolated alloxazines and related compounds [4,16,18,30] contain-ing similar heterocyclic ring system to that of Lum along with someearlier calculation results involving quantum-mechanical (QM)/molecular-mechanics (MM) as well as other approximation meth-ods [31,32]. The usefulness of the KS-DFT method in elucidatingthe complex phenomenon like hydrogen bonding and successfulapplication of this technique to discuss specific hydrogen bondingof structurally related luminol and lumichrome with water fromour laboratory [19,20], prompts us to use it further in studyingthe energetic parameters of isolated Lum and its complex withwater molecules. To elucidate the lowest energy conformeramongst the structures given in Chart 1, full geometry optimiza-tion was performed for all the structures of Lum both in isolatedas well as in their hydrated complexes using hybrid exchange-cor-relation B3LYP functional in combination with 6-311++G(d,p) basisset as implemented in Gaussian09 program package [33]. Fre-quency calculations were done in each stationary point to charac-terize the minimum energy equilibrium structure. Verticaltransition energies up to the first ten singlet excited states and cor-responding oscillator strengths for all the structures were esti-mated using time dependent DFT method (TD-DFT) at the samelevel of calculation. The effect of solvent on the energy parameterswas incorporated by self consistent reaction field calculation usingpolarizable continuum model (SCRF-PCM) as implemented inGaussian09 software.
3. Results and discussion
3.1. Steady state spectral properties in pure solvents
Fig. 1 shows few absorption and emission spectra of Lum insome of the representative organic solvents like ethyl acetate,dichloromethane, acetonitrile, dimethylformamide, methanol, 1-butanol and water. It is observed that, in all the cases, Lum showsa broad absorption band within 290–350 nm spectral range with apeak position centered at �325 nm. From the relatively largeabsorption coefficient (emax � 9.0 � 104 dm3 mol�1 cm�1), it canbe concluded that this transition is S1(p) S0(p) in nature. Fluo-rescence emission obtained by exciting at this wavelength resultsin a strong fluorescence band within 350–450 nm range having apeak position at �380 nm. Interestingly, in all the alcoholic sol-vents studied here, an additional broad fluorescence emission bandat �480 nm is also observed. Furthermore, in the aqueous medium,although the absorption behavior is similar to other organic sol-vents, the fluorescence emission shows only one peak at
Fig. 1. Steady state absorption (a) and fluorescence emission (b) spectra of �5.0 � 10�6
325 nm in all the cases.
�460 nm with ca. 8–10 times higher fluorescence intensity in com-parison with other organic solvents. The excitation spectra (Fig. 1S)obtained by monitoring the principal emission peak at 380 nmresembles that of the 325 nm absorption band and appear with330–340 nm range; whereas, the excitation spectra correspondingto the 480 nm emission gives a peak at �375 nm. In analogy withthe previous reports on Lum photophysics, the normal absorptionand emission peaks at �325 nm and �380 nm in organic solventsare assigned to the neutral structure of lumazine. The assignmentof the absorption band as well as the origin of the electronic tran-sition in the neutral form of Lum is further confirmed from theoret-ical calculations described in the following sections. On the otherhand, the long wavelength absorption (�380 nm) and the corre-sponding emission (�480 nm) are considered to originate from aproton dissociated anionic structure, Lum-anion [34]. It has alreadybeen reported that the pKa value for the ground state deprotona-tion of Lum is about 8.0 ± 0.1 [15].
3.2. Solvatochromism of Lum photophysics: Importance of solventhydrogen bonding
The steady state spectral properties of Lum were studied in aseries of solvents with varying polarity and hydrogen bondingparameters given in Table 1. Table 2 summarizes the steady statespectral behavior of Lum in these solvents. To verify the effect ofsolvent polarity, the steady state spectral parameters (P) of Lumwere plotted against the solvent polarity parameter Df (e,n) withthe general form of Lippert-Mataga (LM) equation given below[35,36]
P ¼ P0 þ a� Df ðe;nÞ; ð2Þ
where, the orientation polarizibility, Df(e,n) is related to the solventdielectric constant (e) and refractive index (n) with the relation
Df ðe;nÞ ¼ e� 12eþ 1
� n2 � 12n2 þ 1
; ð3Þ
where P0 is the measured property in the gas phase or in the non-interacting solvents; the slope of the equation a represents a termcontaining the cavity radius and also the difference in dipole mo-ment between the ground and excited states. From the results givenin the supplementary section (Fig. 2S), it is clear that the spectro-scopic properties of Lum do not show any regular solvatochromismbehavior on the solvent polarity parameter. This observation pointsto the existence of specific solute–solvent interactions [35]; similarto those seen by our group in several nitrogen heterocyclic systems
mol dm�3 lumazine solution in different solvents. The excitation wavelengths are
Table 2Steady state spectral properties Lumazine in homogeneous solvents.a
a Abbreviations used: m = absorption and emission energy; Dmss = Stokes shift;/f = fluorescence quantum yield (error in measurement ±10%).
b The name of the solvents are listed in Table 1.c Values correspond only to the neutral form of Lumazine (see text for details).
N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31 25
[19,20], donor-acceptor charge transfer systems [37,38] and also bymany other groups in the cases of amino acids and peptides[39,40].Also, the empirical solvent polarity scale, ET(30), built witha betaine dye, was used to correlate the solvent dependence ofthe steady state spectral properties of Lum. The uni-parametricscale depends on both the solvent dielectric properties and hydro-gen bonding acidity (a), but it does not account for the solvent
Table 3Regression fit to solvatochromic parameters and their relative contribution towards the s
a The regression analysis was done using equation (4).
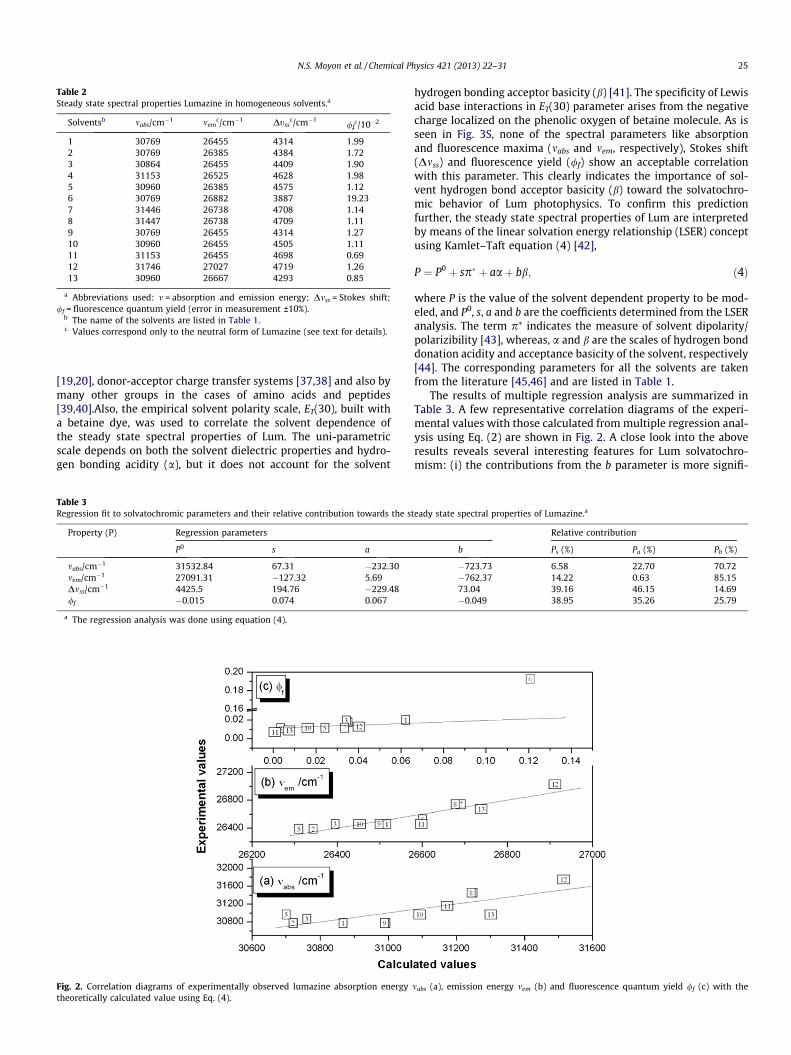
Fig. 2. Correlation diagrams of experimentally observed lumazine absorption energytheoretically calculated value using Eq. (4).
hydrogen bonding acceptor basicity (b) [41]. The specificity of Lewisacid base interactions in ET(30) parameter arises from the negativecharge localized on the phenolic oxygen of betaine molecule. As isseen in Fig. 3S, none of the spectral parameters like absorptionand fluorescence maxima (mabs and mem, respectively), Stokes shift(Dmss) and fluorescence yield (/f) show an acceptable correlationwith this parameter. This clearly indicates the importance of sol-vent hydrogen bond acceptor basicity (b) toward the solvatochro-mic behavior of Lum photophysics. To confirm this predictionfurther, the steady state spectral properties of Lum are interpretedby means of the linear solvation energy relationship (LSER) conceptusing Kamlet–Taft equation (4) [42],
P ¼ P0 þ sp� þ aaþ bb; ð4Þ
where P is the value of the solvent dependent property to be mod-eled, and P0, s, a and b are the coefficients determined from the LSERanalysis. The term p⁄ indicates the measure of solvent dipolarity/polarizibility [43], whereas, a and b are the scales of hydrogen bonddonation acidity and acceptance basicity of the solvent, respectively[44]. The corresponding parameters for all the solvents are takenfrom the literature [45,46] and are listed in Table 1.
The results of multiple regression analysis are summarized inTable 3. A few representative correlation diagrams of the experi-mental values with those calculated from multiple regression anal-ysis using Eq. (2) are shown in Fig. 2. A close look into the aboveresults reveals several interesting features for Lum solvatochro-mism: (i) the contributions from the b parameter is more signifi-
mabs (a), emission energy mem (b) and fluorescence quantum yield /f (c) with the
Table 4Relative energy parameters (in kJ mol�1) of different possible conformers of Lumazine calculated at the B3LYP/6-311++G(d,p) level in the gas phase as well as in somerepresentative solvents.a
Entryb Gas Phase Tetrahydrofuran Acetonitrile Dimethylsulfoxide Water
Lum-I 316.163(625.420)
110.140(434.940)
91.394(420.343)
90.448(419.450)
87.560(417.533)
Lum-II 148.551(504.831)
73.015(432.919)
55.451(416.562)
54.610(415.485)
51.932(413.516)
Lum-III 152.200(490.496)
73.934(421.209)
55.477(405.430)
54.663(404.511)
51.118(401.833)
Lum-IV 91.840(443.762)
19.770(382.877)
3.439(368.883)
2.731(368.069)
0.000(366.100)
a Ground state values are obtained by full geometry optimization and the corresponding values in the excited state (shown in parenthesis) are obtained by TD calculation.b The corresponding figures are shown in Chart 1.
Table 5The energy of absorption (nm), it’s oscillator strength (f) and coefficient of contribution from different orbitals in the transition to the corresponding excited states (ES) oflumazine (structure Lum-IV) in gas phase as well as in different solvents obtained by TD-DFT calculation at B3LYP/6-311++G(d,p) level. The highest occupied and lowestunoccupied MOs are represented by numbers 42 and 43, respectively.
ES Gas Phase Tetrahydrofuran Acetonitrile Dimethylsulfoxide Water
26 N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31
cant than those from both the a and s parameters, particularly fordefining the absorption and fluorescence peak position, indicatingthe importance of solvent hydrogen bonding basicity for Lum, asindeed pointed out in the discussion above; (ii) the importanceof solvent hydrogen bonding basicity in Lum spectroscopy pointstoward the acidic behavior of the probe, more so upon excitation(see the DFT calculation results in the following section) in sharpcontrast to other structurally similar systems like luminol andlumichrome [19,20]; and finally, (iii) the calculated value of thespectral parameters correlates quite well (correlation coefficientgreater than 0.93 ± 0.03 in each case) with the experimentallymeasured values for all the solvents, although a large deviation isobserved for fluorescence yield in the case of water as a solvent.This large deviation in fluorescence yield in aqueous medium ispossibly due to the formation of highly solvated microstructureinvolving Lum and several water molecules. Similar structuresare already reported in the literature for different systems[47,48]. These hydrated structures are expected to be more stableupon electronic excitation due to extensive charge redistribution(also, see below for DFT calculation results) that causes a substan-tial decrease in the relative energy difference between the groundand excited states.
3.3. Calculation results using density functional theory
3.3.1. Energetic of different conformers in the ground stateFull geometry optimization of different conformers of neutral
Lum (structures are given in Chart 1) in the isolated condition aswell as in different solvents with the SCRF/PCM model was doneusing B3LYP/6-311++G(d,p) methodology. The fully optimizedstructures are shown in the supplementary section (Fig. 4S) andthe corresponding energies are listed in Table 4.
It is clear that, in the ground state, the structure represented byLum-IV (the di-oxo conformer) is the most stable conformer in theisolated condition as well as in different solvent systems. Themono-hydroxy derivatives (Lum-II and Lum-III) are less stableby about 50–60 kJ mol�1 energy in comparison with this structure;whereas, the energy difference with the di-hydroxy derivative is�228 and �88 kJ mol�1 in the isolated state and water, respec-tively. It is also to be noted that the energy difference among dif-ferent conformers decreases substantially in aqueous medium.Therefore, the ground state population of Lum is dominated bystructure Lum-IV and we will confine our discussion of spectralproperties to this structure only. To understand the absorptionspectral behavior, we have carried out TD-DFT calculation of con-former Lum-IV in different solvents for the first ten lowest excitedsinglet states. The contributions of different orbitals in the corre-sponding excited states within the experimentally observed spec-tral range (290–350 nm) are given in Table 5. The calculatedexcitation wavelengths are in very good agreement with theabsorption spectral peak positions listed in Table 2. Also, carefulanalysis of the electron density distributions in all these orbitals(see below) confirms the p⁄ p nature of the transition observedin optical absorption spectroscopy of Lum. Further, from Table 5, itis clear that although several occupied levels, namely highest occu-pied molecular orbital (HOMO), HOMO-1 and/or HOMO-2 contrib-ute in the electronic excitation of Lum; the excited state is mainlydescribed by the lowest unoccupied molecular orbital (LUMO).
3.3.2. Analysis of frontier orbitals: Enhanced acidity in the excited stateFig. 3 shows the electron density distributions in the frontier
molecular orbitals of lumazine in the most stable configuration.Careful analyses of these orbitals reveal extensive charge redistri-bution on excitation. It has already been noted from the results
Fig. 3. Frontier orbital diagrams obtained by population analysis calculation of fully optimized Lum-IV structure at B3LYP/6-311++G(d,p) level. The arrow marks indicate thepoint of specific interaction through solvent hydrogen bond acceptance (HBA) basicity.
N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31 27
in Table 3 that both the absorption and emission maxima of Lumare strongly dependent on the solvent hydrogen bond accepting(HBA) basicity (b), where Lum behaves as a hydrogen bond donor(HBD). From the orbital diagram, it is quite clear that the point ofHBD is the N3 proton in the ground state; whereas, the N1 protonbecomes more pertinent in the excited state. This observation is inexcellent agreement with the predictions made by Klein and Tati-scheff from their pH dependent study on the ground and excitedstate proton dissociation equilibrium of Lum and its derivatives[13]. Therefore, the strongly basic solvents like DMSO etc. involvedin the specific hydrogen bond formation at these sites mainly par-ticipate through the proton abstraction by forming the correspond-ing anionic structure. However, the polar protic solvents like waterand alcohols, due to their amphoteric nature, might form cyclicintramolecular hydrogen bonded complexes with the neighboringatom(s) after the initial hydrogen bond formation through solventhydrogen bond basicity. The process would lead to the formation ofmultiple species through solvent mediated tautomerization, bothin the ground and excited states, and contribute toward the broadnature of the absorption and emission spectra.
3.3.3. Water mediated inter-conversion among several speciesSpecific hydrogen bond formation through solvent basicity at
the N1 and N3 protons with protic solvents like water do result
Scheme 1. Formation of several hydrated complex on specific
in the formation of several structures as depicted in Scheme 1. Itis indeed necessary to mention here that only primary hydrogen-bonded structure with water was considered to give different com-plexes. However, it is obvious that additional solvent moleculeswill combine to give secondary water cluster and that the actualhydrated structures are far more complex than what is consideredfor these calculations. Nevertheless, the effect of internal hydrogenbonding (IHB) is expected to diminish very fast from the center oforigin and consequently, it is reasonable to believe that any furtheraddition of water molecules as a secondary layer will have insignif-icant contribution toward the relative energy parameters. There-fore, we have followed the formation of all these microstructureswith a single water molecule specifically hydrogen bonded at dif-ferent positions of the most stable Lum structure and constructedthe corresponding potential energy curves by intrinsic reactioncoordinate (IRC) calculation; both in the ground as well as in firstexcited states. The results are depicted in Figs. 4 and 5 along withthe corresponding fully optimized transition state structure. Theenergy parameters for the terminal structures and also the transi-tion states are given in Table 6. It is observed that water mediatedcatalytic conversion to both the Lum-II and Lum-III structure bysolvent HBA at N3 and N1 protons of Lum-IV through the forma-tion of respective transition state structures, Lum-IV-H2O(3,4)and Lum-IV-H2O(1,2) in Scheme 1, are highly unlikely from the
hydrogen bond formation at N1 and N3 atoms of Lum-IV.
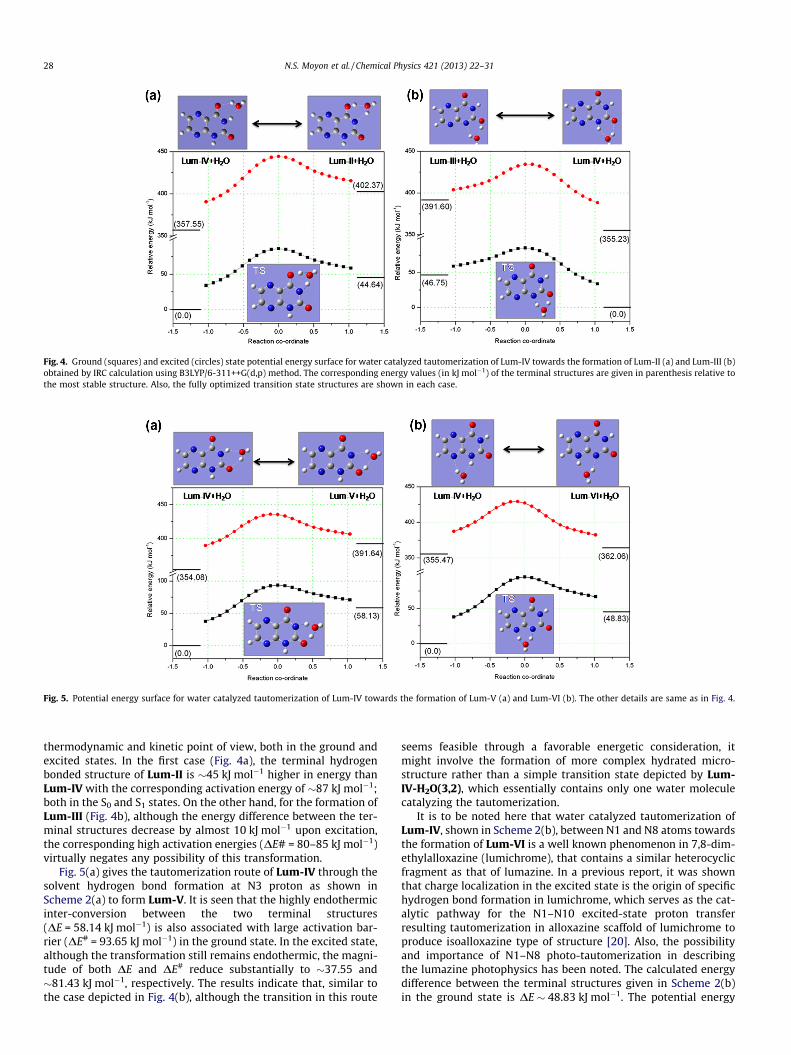
Fig. 4. Ground (squares) and excited (circles) state potential energy surface for water catalyzed tautomerization of Lum-IV towards the formation of Lum-II (a) and Lum-III (b)obtained by IRC calculation using B3LYP/6-311++G(d,p) method. The corresponding energy values (in kJ mol�1) of the terminal structures are given in parenthesis relative tothe most stable structure. Also, the fully optimized transition state structures are shown in each case.
Fig. 5. Potential energy surface for water catalyzed tautomerization of Lum-IV towards the formation of Lum-V (a) and Lum-VI (b). The other details are same as in Fig. 4.
28 N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31
thermodynamic and kinetic point of view, both in the ground andexcited states. In the first case (Fig. 4a), the terminal hydrogenbonded structure of Lum-II is �45 kJ mol�1 higher in energy thanLum-IV with the corresponding activation energy of �87 kJ mol�1;both in the S0 and S1 states. On the other hand, for the formation ofLum-III (Fig. 4b), although the energy difference between the ter-minal structures decrease by almost 10 kJ mol�1 upon excitation,the corresponding high activation energies (DE# = 80–85 kJ mol�1)virtually negates any possibility of this transformation.
Fig. 5(a) gives the tautomerization route of Lum-IV through thesolvent hydrogen bond formation at N3 proton as shown inScheme 2(a) to form Lum-V. It is seen that the highly endothermicinter-conversion between the two terminal structures(DE = 58.14 kJ mol�1) is also associated with large activation bar-rier (DE# = 93.65 kJ mol�1) in the ground state. In the excited state,although the transformation still remains endothermic, the magni-tude of both DE and DE# reduce substantially to �37.55 and�81.43 kJ mol�1, respectively. The results indicate that, similar tothe case depicted in Fig. 4(b), although the transition in this route
seems feasible through a favorable energetic consideration, itmight involve the formation of more complex hydrated micro-structure rather than a simple transition state depicted by Lum-IV-H2O(3,2), which essentially contains only one water moleculecatalyzing the tautomerization.
It is to be noted here that water catalyzed tautomerization ofLum-IV, shown in Scheme 2(b), between N1 and N8 atoms towardsthe formation of Lum-VI is a well known phenomenon in 7,8-dim-ethylalloxazine (lumichrome), that contains a similar heterocyclicfragment as that of lumazine. In a previous report, it was shownthat charge localization in the excited state is the origin of specifichydrogen bond formation in lumichrome, which serves as the cat-alytic pathway for the N1–N10 excited-state proton transferresulting tautomerization in alloxazine scaffold of lumichrome toproduce isoalloxazine type of structure [20]. Also, the possibilityand importance of N1–N8 photo-tautomerization in describingthe lumazine photophysics has been noted. The calculated energydifference between the terminal structures given in Scheme 2(b)in the ground state is DE � 48.83 kJ mol�1. The potential energy
Table 6Characterization of different water catalyzed tautomerization path for Lum-IV obtained by IRC calculation at B3LYP/6-31++G(d,p) level in the ground as well as in firstelectronically excited state.a
Catalytic point Product Terminal structures Energy Transition state DE DE#
Energy Frequency
3,4 Lum-II Lum-IV + H2OLum-II + H2O
0.000(357.553)44.645(402.364)
86.374(444.253)
1429.02 44.64(44.81)
86.37(86.70)
1,2 Lum-III Lum-IV + H2OLum-III + H2O
0.000(355.230)46.752(391.604)
85.449(434.196)
1661.24 46.75(36.37)
85.45(78.97)
3,2 Lum-IV Lum-IV + H2OLum-V + H2O
0.000(354.089)58.138(391.643)
93.659(435.520)
1357.56 58.14(37.55)
93.66(81.43)
1,8 Lum-VI Lum-IV + H2OLum-VI + H2O
0.000(355.471)(369.382b)48.83242.424b
(362.069)(358.231b)
94.970100.271b
(427.155)(433.423b)
1486.61 48.8342.42b
(6.60)(-11.15b)
94.97100.27b
(71.68)(64.04b)
All relative energy parameters and the frequencies are given in kJ mol�1 and cm�1, respectively. DE and DE# represent the energy difference between the terminal structuresand the activation barrier, respectively.
a The values in parenthesis indicate the corresponding parameters for the excited state.b The corresponding values under fully hydrated condition, see text for details.
Scheme 2. Tautomerization scheme of Lum-IV through (3,2) and (1,8) hydrogentransfer.
N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31 29
surface (PES) given in Fig. 5(b) indicates that the water-assistedtautomerization is associated with large ground state activationbarrier of DE# � 94.97 kJ mol�1. However, in the first excited state,both the parameters decrease substantially and were calculated tobe �6.59 and �71.68 kJ mol�1, respectively. As a result, the water-catalyzed proton transfer process is believed to be highly feasiblein the excited state. Interestingly, a similar result was also calcu-lated for the water catalyzed excited state tautomerization processof lumichrome. However, in contrast to the case of lumichrome,where charge localization in the excited state and formation ofhydrogen-bonded cluster through solvent hydrogen bond donation(HBD) acidity on the N10 atom of alloxazine moiety were predictedto be the key step toward the water-catalyzed tautomerizationprocess, hydrogen bond accepting (HBA) basicity of the solvent isbelieved to be the primary step in case of lumazine.
3.3.4. Modeling the water catalyzed reaction under fully hydratedcondition
As mentioned before, the quantitative magnitude of differentthermodynamic parameters associated with several water cata-lyzed isomerization reactions are expected to vary significantly un-der fully hydrated condition in comparison with the single waterbound probe cluster. As a test case, we have analyzed the tauto-merization reaction of Lum involving N1 and N8 atoms, whichcan closely be related with the precursor state of riboflavin biosyn-thesis, under fully hydrated condition [49].
The primary question in modeling the chemical reaction underfully hydrated question is to ascertain the number of water mole-cules involved in first hydration shell and their distribution aroundthe probe. The most commonly used techniques to quantify the ef-fect of solvation is to hypothesize the solvent structure and its spe-cific interaction with the probe stems from chemical intuition and/or classical molecular dynamics (MD) simulation using molecularmechanics force fields [50–52]. A more comprehensive way to ob-tain these parameters is to use DFT based Born-Oppenheimer MDsimulation, as discussed in detail by Jalkanen et al. [53] for thedescription of structure and vibrational spectra of L-alanine andits derivatives. Nevertheless, we have added three more watermolecules, except the one primarily involved in the catalytic path-way, to complete the first hydration shell. As the cyclic hydrogenbonded conformation of Lum-IV (Scheme 2) with one water mole-cule at (3, 4) position (EH = �677.18337178) is somewhat more sta-ble than the corresponding (2, 3) isomer (EH = �677.1830118), wepreferred to put a cyclic water molecule specifically hydrogenbonded between N(3)–H and C(4)@O groups. The other two watermolecules were also chosen to form hydrogen bonds with C(2)@Oand N(5), respectively to complete the first hydration layer. Each ofthe resultant hydrated specie was then embedded in polarizablecontinuum model (SCRF-PCM) taking water as a solvent to con-sider the macroscopic solvent polarization effect. The fully opti-mized geometries of the two terminal structures and also the TSare depicted in Fig. 6, whereas, the corresponding energy parame-ters are listed in Table 6. It is interesting to note that although thequalitative pattern of the relative energy parameters remain same,the (1,8) tautomerization reaction becomes more exothermic
Fig. 6. Optimized structures of fully hydrated terminal species and also the transition (TS) corresponding to (1,8) hydrogen migration in Lum-IV leading to the formation ofLum-VI. The corresponding energy values are mentioned in Table 6.
30 N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31
(DE � �11.15 kJ mol�1) with reduced activation barrier (DE# -� 64.04 kJ mol�1) under fully hydrated condition in the excitedstate.
4. Conclusions
The fluorescence behavior and water catalyzed excited statetautomerization process of neutral form of lumazine have beenstudied by fluorescence spectroscopy in combination with DFT cal-culations. The observed solvatochromic property of Lum photo-physics are rationalized by multi-parametric approach usingKamlet–Taft equation. It has been found that the photolumines-cence behavior of lumazine is strongly modulated by specific sol-ute–solvent interaction through solvent hydrogen bondacceptance at N3 and N1 atoms of lumazine in the ground and ex-cited states, respectively. Intrinsic reaction coordinate analysispredict that water assisted (1,8) and (3,2) hydrogen migration pro-cesses is feasible upon electronic excitation form thermodynamicpoint of view. The results are interesting in view of understandingthe role of water towards several biochemical catalytic pathways.
Acknowledgement
NSM thanks Council of Scientific and Industrial Research (CSIR),Govt. of India for a fellowship. The Gaussian09 software was madeavailable to the Chemistry Department through a generous fundfrom NEHU Computer Center.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.chemphys.2013.05.012.
References
[1] S. Lilavivat, D. Sardar, S. Jana, G.C. Thomas, K.J. Woycechowsky, J. Am. Chem.Soc. 134 (2012) 13152.
[2] M.A.C. Neves, M. Yeager, R. Abagyan, J. Phys. Chem. B 116 (2012) 7006.[3] B. Wörsdörfer, Z. Pianowski, D. Hilvert, J. Am. Chem. Soc. 134 (2012) 909.[4] C. Hemann, P. Ilich, R. Hille, J. Phys. Chem. B 107 (2003) 2139.[5] B. Fischer, S.J.N. Burgmayer, in: A. Sigel, H. Sigel (Eds.), Metal Ions in Biological
Systems, vol. 39, Marcel Dekker, New York, 2002, p. 265.[6] N. Saleh, A.M. Rawashdeh, Y.A. Yousef, Y.A. Al-Soud, Spectrochim. Acta. 68A
(2007) 728.[7] T. Kohzuma, A. Odani, Y. Morita, M. Takani, O. Yamauchi, Inorg. Chem. 27
(1988) 3854.
[8] A. Crispini, D. Pucci, A. Bellusci, G. Barberio, M. La Deda, A. Cataldi, M. Ghedini,Cryst. Growth Des. 5 (2005) 1597.
[9] W. Bamwarth, F. Müller, Helv. Chim. Acta 74 (1991) 2000.[10] L. Lu, D. Xing, N. Ren, Environ. Sci. Technol. 46 (2012) 6874.[11] A. Albert, D.J. Brown, G. Cheeseman, J. Chem. Soc. (1951) 474.[12] E. Lippert, H. Prigge, Z. Elektrochem. 64 (1960) 662.[13] R. Klein, I. Tatischeff, Photochem. Photobiol. 45 (1987) 55.[14] I. Presiado, Y. Erez, R. Gepshtein, D. Huppert, J. Phys. Chem. C 114 (2010) 3634.[15] M.P. Denofrio, A.H. Thomas, A.M. Braun, E. Oliveros, C. Lorente, J. Phys. Chem. C
114 (2010) 14307.[16] W.J. Schreier, I. Pugliesi, F.O. Koller, T.E. Schrader, W. Zinth, M. Braun, S.
Kacprzak, S. Weber, W. RÖmisch-Margl, A. Bacher, B. Illarionov, M. Fischer, J.Phys. Chem. B 115 (2011) 3689.
[17] P.A. Faria, X. Chen, J.R. Lombardi, R.L. Birke, Langmuir 16 (2000) 3984.[18] D. Prukała, E. Sikorska, J. Koput, I. Khmelinskii, J. Karolczak, M. Gierszewski, M.
Sikorski, J. Phys. Chem. A 116 (2012) 7474.[19] N.S. Moyon, A.K. Chandra, S. Mitra, J. Phys. Chem. A 114 (2010) 60.[20] N.S. Moyon, S. Mitra, J. Phys. Chem. A 115 (2011) 2456.[21] S.R. Meech, D. Phillips, J. Photochem. 23 (1983) 193.[22] N. Mataga, T. Kubota, Molecular Interactions and Electronic Spectra, Marcel
Dekker Inc., New York, 1970.[23] J.W. Blanchard, T.L. Groy, J.L. Yarger, G.P. Holland, J. Phys. Chem. C 116 (2012)
18824.[24] V. Kocevski, L. Pejov, J. Phys. Chem. A 116 (2012) 1939.[25] E. Iype, S.V. Nedea, C.C.M. Rindt, A.A. van Steenhoven, H.A. Zondag, A.P.J.
Jansen, J. Phys. Chem. C 116 (2012) 18584.[26] M.E. Reish, A.J. Kay, A. Teshome, I. Asselberghs, K. Clays, K.C. Gordon, J. Phys.
Chem. A 116 (2012) 5453.[27] N.S. Myshakina, Z. Ahmed, S.A. Asher, J. Phys. Chem. B 112 (2008) 11873.[28] R.G. Potter, D.M. Camaioni, M. Vasiliu, D.A. Dixon, Inorg. Chem. 49 (2010)
10512.[29] S. Yamabe, N. Kawagishi, Theor. Chem. Acc. 130 (2011) 909.[30] Y. Zhang, B. Illarionov, A. Bacher, M. Fischer, G.I. Georg, J. Org. Chem. 72 (2007)
2769.[31] A.L. Michaud, J.A. Herrick, J.E. Duplain, J.L. Manson, C. Hemann, P. Ilich, R.J.
Donohoe, R. Hille, W.A. Oertling, Biospectroscopy 4 (1998) 235.[32] S. Metz, W. Thiel, J. Am. Chem. Soc. 131 (2009) 14885.[33] M.J. Frisch et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT,
2009.[34] N.S. Moyon, M.M. Islam, S. Phukan, S. Mitra, J. Photochem. Photobiol. B 121
(2013) 37.[35] N. Mataga, Y. Kaifu, M. Koizumi, Bull. Chem. Soc. Jpn. 29 (1956) 465.[36] J.R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York,
2006.[37] T.S. Singh, S. Mitra, A.K. Chandra, N. Tamai, S. Kar, J. Photochem. Photobiol. A
197 (2008) 295.[38] T.S. Singh, N.S. Moyon, S. Mitra, Spectrochim. Acta. 73A (2009) 630.[39] B.F. Hermenegildo, G. Pereira, A.S. Abreu, E.M.S. Castanheira, P.M.T. Ferreira,
M.-J.R.P. Queiroz, J. Photochem. Photobiol. 221 (2011) 47.[40] M. Venanzia, G. Bocchinfuso, A. Palleschi, A.S. Abreu, P.M.T. Ferreira, M.-R.P.
N.S. Moyon et al. / Chemical Physics 421 (2013) 22–31 31
[48] T. van Mourik, V.I. Danilov, V.V. Dailidonis, N. Kurita, H. Wakabayashi, T.Tsukamoto, Theor. Chem. Acc. 125 (2010) 233.
[49] The authors thank one of the anonymous referees for noting this point to us.[50] K. Frimand, H. Bohr, K.J. Jalkanen, S. Suhai, Chem. Phys. 255 (2000) 165.[51] R. Misra, A. Mandal, M. Mukhopadhyay, D.K. Maity, S.P. Bhattacharyya, J. Phys.
Chem. B 113 (2009) 10779.
[52] M. Knapp-Mohammady, K.J. Jalkanen, F. Nardi, R.C. Wade, S. Suhai, Chem.Phys. 240 (1999) 63.