Defense during Infectious ColitisMediated Epithelial Host−Controls IL-22
STAT3 Activation in Th17 and Th22 Cells
Christoph Becker and Clemens NeufertMaximilian Waldner, Klaus Rajewsky, Markus F. Neurath, Hofmann, Kai Hildner, Nadine Wittkopf, Katrin Brecht,Kitowski, Ulrike Billmeier, Eva Martini, Katharina Ingo Backert, Sergei B. Koralov, Stefan Wirtz, Vera
STAT3 Activation in Th17 and Th22 Cells Controls IL-22–Mediated Epithelial Host Defense during Infectious Colitis
Ingo Backert,* Sergei B. Koralov,† Stefan Wirtz,* Vera Kitowski,* Ulrike Billmeier,*
Eva Martini,* Katharina Hofmann,* Kai Hildner,* Nadine Wittkopf,* Katrin Brecht,*
Maximilian Waldner,* Klaus Rajewsky,‡ Markus F. Neurath,* Christoph Becker,* and
Clemens Neufert*
The Citrobacter rodentium model mimics the pathogenesis of infectious colitis and requires sequential contributions from different
immune cell populations, including innate lymphoid cells (ILCs) and CD4+ lymphocytes. In this study, we addressed the role of
STAT3 activation in CD4+ cells during host defense in mice against C. rodentium. In mice with defective STAT3 in CD4+ cells
(Stat3DCD4), the course of infection was unchanged during the innate lymphoid cell–dependent early phase, but significantly
altered during the lymphocyte-dependent later phase. Stat3DCD4 mice exhibited intestinal epithelial barrier defects, including
downregulation of antimicrobial peptides, increased systemic distribution of bacteria, and prolonged reduction in the overall
burden of C. rodentium infection. Immunomonitoring of lamina propria cells revealed loss of virtually all IL-22–producing CD4+
lymphocytes, suggesting that STAT3 activation was required for IL-22 production not only in Th17 cells, but also in Th22 cells.
Notably, the defective host defense against C. rodentium in Stat3ΔCD4 mice could be fully restored by specific overexpression of IL-
22 through a minicircle vector–based technology. Moreover, expression of a constitutive active STAT3 in CD4+ cells shaped strong
intestinal epithelial barrier function in vitro and in vivo through IL-22, and it promoted protection from enteropathogenic
bacteria. Thus, our work indicates a critical role of STAT3 activation in Th17 and Th22 cells for control of the IL-22–mediated
host defense, and strategies expanding STAT3-activated CD4+ lymphocytes may be considered as future therapeutic options for
improving intestinal barrier function in infectious colitis. The Journal of Immunology, 2014, 193: 3779–3791.
Intestinal inflammation caused by pathogenic bacteria is afrequent and potentially life-threatening disease. Besides thefitness and diversity in the host’s immune system, there are
several influences that can modulate the colonization of pathogensand the course of disease, including bacterial virulence factors andcompetition with the resident gut flora (1). Moreover, compro-mised intestinal barrier function can render individuals susceptibleto intestinal inflammation and infection (2, 3). Antimicrobialpeptides are typically produced by crypt-residing Paneth cells inthe small intestine (3, 4). Upon challenge, however, intestinalepithelial cells (IECs) of the large intestine can also produce highamounts of antimicrobial peptides (4, 5).
Genome-wide association studies have connected the transcrip-tion factor STAT3, which is widely expressed by different tissues
and cell types, to intestinal pathology (6). In response to extra-
cellular stimuli, such as IL-22, IL-6, or IL-23, STAT3 molecules
are phosphorylated, translocate to the nucleus, and can influence
the transcription of target genes related to cellular key processes,
including proliferation, cell survival, activation, and differentiation
(7). STAT3 has cell type–specific functions that are regulated in
a context-dependent manner. On one hand, conditional deletion
of STAT3 activity in macrophages and neutrophils results in spon-
taneous enterocolitis, and defective STAT3 activation in IECs
impairs barrier function and mucosal healing (8, 9). On the other
hand, T cell–specific inactivation of STAT3 protects from intestinal
inflammation in a transfer model of colitis (10). Additionally, ac-
tivation of STAT3 is critically linked to the development of Th17
cells, which can promote intestinal pathology (11, 12). Conversely,
dominant negative mutations of STAT3 are causative for the hyper-
IgE syndrome, which is characterized by Th17 deficiency and im-
paired defense against extracellular bacteria (13, 14).Infection of mice with Citrobacter rodentium is a well-established
disease model mimicking infectious colitis by enterohemorrhagic
or enteropathogenic Escherichia coli in humans. All of these patho-
gens use attaching and effacing lesions to colonize the gastroin-
testinal tract of the host and share the type III secretion system (15).In wild-type mice, infection with C. rodentium is mostly self-
limiting and the bacterial levels in the colon peak around days 8–
11 with variations depending on the mouse strain and the resident
flora (15). The host defense against C. rodentium comprises se-
quential contributions from different immune cell populations,
including a variety of type 3 ILC (ILC3) subsets, dendritic cells,
and adaptive immune cells, including T and B lymphocytes (15–
*Medizinische Klinik 1, Universitatsklinikum Erlangen, Friedrich-Alexander-Universitat Erlangen-N€urnberg, D-91054 Erlangen, Germany; †Department ofPathology, New York University School of Medicine, New York, NY 10016; and‡Max Delbr€uck Center for Molecular Medicine, 13125 Berlin, Germany
Received for publication November 14, 2013. Accepted for publication August 4,2014.
This work was supported by the Interdisciplinary Center for Clinical ResearchErlangen (to C.N.), by Deutsche Forschungsgemeinschaft Grants NE1927 (to C.N.),CEDER KFO257 (to M.F.N. and C.B.), and SPP1656 (to S.W.), and by the Friedrich-Alexander-Universitat Emerging Fields Initiative (to C.N.) and the ErlangerLeistungsbezogene Anschubfinanzierung und Nachwuchsforderung-Fonds Erlangen(to C.N.).
Address correspondence and reprint requests to Dr. Clemens Neufert, First Depart-ment of Medicine, Universitatsklinikum Erlangen, Friedrich-Alexander-UniversitatErlangen-N€urnberg Ulmenweg, 18, 91054 Erlangen, Germany. E-mail address:[email protected]
20). Several studies have suggested an important role for Th17cells during the course of C. rodentium infection (21–23).Although CD4 expression is frequently found in lymphocytes,
other immune cell subsets such as dendritic cells or ILC3s can alsoexpress CD4. Indeed, CD4+ ILC3s are crucial mediators in hostdefense upon infection with enteropathogenic bacteria (24).Strikingly, several features critically important during the hostdefense against C. rodentium such as expression of IL-17, IL-22,IL-23R, AHR, and CD4 are shared by both Th17 cells and ILC3subsets (5, 16, 20, 21, 24–26). Thus, studies with mice deficient insuch molecules are restricted in the analysis of differential con-tributions by ILC3s or Th17 cells. However, ILC3s act rather earlyupon C. rodentium infection (16, 24, 25), whereas major con-tributions of T cells occur with delayed kinetics (15, 27). Forthe analysis of differential contributions of Th17 cells, it wouldbe interesting to study gene-modified mice with intact ILC3responses, but specific lack of Th17 cells.In this study, we have analyzed the role of STAT3 in CD4+ cells
during the course of C. rodentium infection, revealing a criticalcontribution of STAT3 activation in Th17 and Th22 cells forcontrol of the IL-22–mediated host defense against enteropatho-genic bacteria in vivo.
Materials and MethodsMice
Stat3fl/fl and Stat3ΔCD4 mice were previously described (28). R26Stat3Cstopfl/fl
mice were already published (29). In brief, they were generated bytransfecting Bruce4 embryonic stem cells with a vector targeting Stat3-CcDNA and a 59 loxP-flanked NeoR stop cassette into the Rosa26 gene. Themodified Rosa26 vector contained FLAG-tagged Stat3-C cDNA togetherwith a 39 FRT-flanked IRES-EGFP cloned downstream of the floxed NeoR
stop cassette and was fully sequenced and linearized before being elec-troporated into the C57BL/6-derived embryonic stem cells. For CD4-specific constitutive activation of STAT3, R26Stat3Cstopfl/fl mice werecrossed with CD4Cre mice. The analysis included animals with one or twoalleles expressing transgenic Stat3-C, as specified in the figure legends.
All mice were bred and maintained in individually ventilated cages.Experiments were performed with cohoused littermates. Animal care waswithin Institutional Care Committee guidelines, and all animal experimentswere performed in accordance with protocols approved by the governmentsof Rhineland-Palatinate and Middle Franconia, Germany.
C. rodentium infection and bioluminescence imaging
For our study, the bioluminescent strain ICC 169 of C. rodentium was used(30). Inoculation was performed via oral gavage of 8 3 109 CFU in a totalvolume of 200 ml PBS. Mice were starved for 8 h prior to infection. Bodyweights were measured daily and the bacterial burden per mouse and timepoint were quantified by bioluminescence imaging with an IVIS Lumina IIsystem and Living Image Software v3.0 (Xenogen). Per measurement,bioluminescent counts were recorded for 3 min from the abdomen as theregion of interest.
Isolation of lamina propria cells
Colons were washed with ice-cold PBS, cut longitudinally, and separatedinto ∼1-cm-long pieces. The epithelium was removed by incubating thetissue for 20 min at 37˚C and gentle shaking twice in Ca2+- and Mg2+-freeHBSS containing 5 mM EDTA (Carl Roth), 5% FBS (PAA Laboratories),and 1 mM DTT (Carl Roth) and once in Ca2+- and Mg2+-free HBSScontaining 10 mM HEPES (PAA Laboratories). The remaining tissue wasfurther digested with a lamina propria dissociation kit (Miltenyi Biotec)according to the manufacturer’s instructions.
In vitro stimulation of CD4+ splenocytes
CD4+ T cells were freshly isolated from spleens with MACS isolation kits(Miltenyi Biotec). For polyclonal in vitro stimulation, hamster anti-mouseCD3 Ab (clone 145-2C11)–coated wells (10 mg/ml in PBS for 3 h at 37˚C)were used together with soluble hamster anti-mouse CD28 (2 mg/ml, clone37.51). For T cell polarizations, the following supplements were used:rat anti-mouse IFN-g (4 mg/ml, clone XMG1.2), rat anti-mouse IL-4(4 mg/ml, clone 11B11), TGF-b (5 ng/ml, R&D Systems), IL-6 (50 ng/ml,
ImmunoTools), and IL-23 (30 ng/ml, R&D Systems). TGF-b inhibitorSB431542 (10 nmol/ml) was purchased from Sigma-Aldrich; all Abs werefrom Bio X Cell.
Organoids
Organoids were cultured as described previously (31). In short, cryptswere isolated from the small intestine with ice-cold PBS containing 2 mMEDTA. The crypts were suspended in Matrigel (BD Biosciences), seededonto a culture plate, and grown in advanced DMEM/F12 medium(Invitrogen) containing 2 mM GlutaMAX (Invitrogen), N2 supplement(Invitrogen), B27 supplement (Invitrogen), 1 mM N-acetylcysteine (Sigma-Aldrich), 10 mM HEPES, 100 U/ml penicillin/streptomycin (Life Tech-nologies), 50 ng/ml murine epidermal growth factor (ImmunoTools), 1 mg/mlrecombinant human R-spondin (R&D Systems), and 100 ng/ml recombi-nant murine noggin (PeproTech). Organoids were cultured for 7–10 dbefore stimulation. Medium and Matrigel were changed every third day.For stimulation, medium was replaced for 12 h with supernatant frompolarized CD4+ T cells.
Construction of in vivo overexpression vectors
Expression constructs for sustained in vivo overexpression of IL-17A, IL-22, IFN-g, and IL-4 were generated by cloning cDNA fragments encodingfor murine full-length cDNAs as described (32). DNA was isolated withQiagen plasmid maxi kits including endotoxin removal. To further ensureefficient endotoxin removal, DNAwas treated with a MiraClean endotoxinremoval kit (Mirus Bio, Madison, WI). Five to 10 mg constructs was ad-ministered in Krebs–Ringer solution to mice via hydrodynamic tail veininjection as described previously (33).
Histopathological analyses and immunohistochemicalstainings
Histopathological analyses were performed on colon tissue that was eitherinstantly frozen in liquid nitrogen and embedded in OCT compound (SakuraFinetek) or fixed in 4.5% formaldehyde (Carl Roth) and embeddedin paraffin. Cross-sections were stained with H&E. Crypt lengths weremeasured with ImageJ software.
Immunostainings of cryosections were performed using the TSA Cy3system (PerkinElmer). Primary Abs were hamster anti-mouse CD11c (cloneHL3, BD Biosciences), rat anti-mouse CD4 (clone RM4-5, BD Bio-sciences), rabbit anti-mouse MPO (polyclonal, Abcam), rat anti-mouseF4/80 (clone BM8, eBioscience), rat anti-mouse B220 (clone RA3-6B2,BioLegend), and rabbit anti-mouse p-STAT3 (clone D3A7, Cell Signal-ing Technology). Nuclei were counterstained with DAPI (Life Technolo-gies), and fluorescence analysis was performed with DMI 4000B (Leica)or TCS SP5 (Leica). For quantitative analyses, four to five representativesections (103) were evaluated in a blinded fashion.
Flow cytometry analyses
For FACS analyses, the following directly conjugated Abs were used: ratanti-mouse CD4 (clone GK1.5), rat anti-mouse CD8 (clone 53-6.7), rat anti-mouse CD3e (clone 17A2), rat anti-mouse CD62L (clone MEL14-H2.100),rat anti-mouse CD44 (clone IM7), rat anti-mouse IL-17A (clone TC11-18H10), goat anti-mouse IL-22 (Poly5146), rat anti-mouse IFN-g (cloneXMG1.2), rat anti-mouse Ly6G (clone 1A8), rat anti-mouse Ly6C (cloneHK1.4), rat anti-mouse CD11b (clone M1/70), rat anti-mouse F4/80 (cloneBM8), rat anti-mouse B220 (clone RA3-6B2), hamster anti-mouse CD11c(clone HL3), rat anti-retinoic acid–related orphan receptor (ROR)gt (cloneB2D), and mouse anti T-bet (eBIO4B10). All FACS Abs were purchasedfrom BioLegend except for anti-CD8a and CD62L, which were fromMiltenyi Biotec, and anti-CD11c, which was from BD Biosciences. Cellswere stimulated with 50 ng/ml PMA (Merck), 500 ng/ml ionomycin(Sigma-Aldrich), and 1 mg/ml brefeldin A (Sigma-Aldrich) at 37˚C for4 h prior to intracellular cytokine staining with a Fix/Perm cell per-meabilization kit (An der Grub) according to the manufacturer’s instruc-tions. For FACS studies that included RORgt or T-bet, a staining set fortranscriptions factors from eBioscience was used. Cells were analyzed ona LSR Fortessa (BD Biosciences) and data analysis was performed withFlowJo v7.6.5 (Tree Star).
RNA isolation and quantitative real-time PCR
Total RNAwas isolated from the distal colon with the NucleoSpin RNA kit(Macherey Nagel) or from organoids with the RNeasy micro kit (Qiagen)and quantified and quality checked with a Nanodrop ND-1000 (ThermoScientific). cDNA was subsequently synthesized with the iScript cDNAsynthesis kit (Bio-Rad). Real-time quantitative PCR (qPCR) was performed
3780 STAT3 IN Th17/Th22 CONTROLS INFECTIOUS COLITIS THROUGH IL-22
with SsoFast EvaGreen supermix (Bio-Rad) according to the manu-facturer’s instructions on a CFX96 thermal cycler (Bio-Rad). Each samplewas run in duplicates and GAPDH was used as reference gene. Data wereanalyzed with the CFX Manager v2.1 (Bio-Rad) using the DDCT method.
Analyses of organ lysates
Organs were aseptically removed at the peak of the infection and ho-mogenized in 200 ml sterile PBS. Serial dilutions were plated on Luria–Bertani agar supplemented with 500 mg/ml erythromycin, and biolumi-nescent colonies were counted after 1 d in culture at 37˚C.
Statistical analysis
Quantitative data are shown as mean values, and error bars represent SDs.Unless otherwise specified, significance analyses were performed usinga Student t test, and p values ,0.05 or ,0.01 were considered significantor highly significant, respectively.
ResultsStat3DCD4 mice are compromised in limiting infection withC. rodentium
The expression of IL-17 in Th17 cells requires STAT3 (12),whereas IL-17 production from ILC3s was reported to occurpartly independent of STAT3 (34), which prompted us to studySTAT3 activation in the lamina propria upon infection with en-teropathogenic bacteria that are known to be controlled by bothILC3s and Th17 cells.Therefore, we analyzed colonic cross-sections from the
C. rodentium model by immunofluorescence for p-STAT3. In unin-fected animals, few cells stained positive for active STAT3 in thecolon of wild-type mice (Fig. 1A, 1B). Around days 6–8 after bac-terial challenge with C. rodentium, we observed a strong infiltrationwith lymphocytes, and the number of cells expressing p-STAT3highly increased in the colonic lamina propria of wild-type mice(Fig. 1A, 1B). Importantly, a large number of CD4+ cells stained alsopositive for p-STAT3 (Fig. 1C, upper panel). Of note, we observedonly a minor increase in p-STAT3+ mononuclear cells during theearly effector phase (days 1–5) upon infection with C. rodentium(data not shown). To test the functional relevance of STAT3 activationin CD4+ cells in this model, we used conditional knockout micedefective for STAT3 activation in CD4+ cells (Stat3ΔCD4). In contrastto CD4+ cells from Stat3fl/fl controls, there were no CD4+ cells inStat3ΔCD4 mice that stained positive for p-STAT3 (Fig. 1C, lowerpanel). Upon infection with bioluminescent C. rodentium, bothStat3ΔCD4 and Stat3fl/fl littermates showed bioluminescent signalsincreasing with similar kinetics and peaking at days 8–11 (Fig. 1D,1E). Of note, the bioluminescence top levels were similar in bothgroups. Importantly, however, whereas Stat3fl/flmice displayed a rapiddecrease in bioluminescent bacterial counts after the peak of infection(Fig. 1D, 1E), Stat3ΔCD4 mice did not manage to fight the entero-pathogenic bacteria efficiently. In detail, high bioluminescent signalscould still be detected in Stat3ΔCD4 animals at day 16, that is, at timepoints when the bacterial counts in controls had already decreased tobackground level (Fig. 1D, 1E). Interestingly, the great majority ofStat3ΔCD4 animals survived and reached a bioluminescent intensity atthe threshold level around day 30 (data not shown).Of note, when Stat3ΔCD4 mice on a Rag1-deficient background
(Stat3ΔCD4;Rag12/2) and their littermate controls (Stat3fl/fl;Rag12/2)were compared with each other, we observed similar bioluminescentsignals throughout the course of infection, providing additionalevidence that STAT3 activation is critically important in adaptiveimmune cells (data not shown).Thus, defective STAT3 activation in CD4+ cells does not sig-
nificantly alter the early phase of infection with C. rodentium, butis required for efficient limiting of bacteria during the lymphocyte-dependent phase.
Loss of STAT3 activation in CD4+ cells does not affectleukocyte infiltration to the colon
Histopathological analysis at the top of infection (days 8–10)demonstrated crypt hyperplasia in both Stat3ΔCD4 and Stat3fl/fl
control mice. Although we did not detect a significant change, thehyperplasia appeared to be slightly less prominent in Stat3ΔCD4
mice as compared with Stat3fl/fl littermates (Fig. 2A, 2B). Addi-tionally, comparison of colon cross-sections did not reveal majorchanges regarding the recruitment and infiltration of immune cellsas assessed by markers for neutrophils, macrophages, dendriticcells, and B cells by immunofluorescence and flow cytometry(Fig. 2C–F). Moreover, the presence of CD4+ cells was not sig-nificantly changed in Stat3ΔCD4 animals, as demonstrated by im-munofluorescence and FACS, suggesting altered effector functionsrather than differences in cell recruitment, survival, or prolifera-tion in vivo (Fig. 2C–F). Crypt hyperplasia and immune cell in-filtration was also similar between Stat3ΔCD4and control mice atdays 15–16 upon infection with C. rodentium (data not shown).Hence, the loss of STAT3 activation in CD4+ cells did not
significantly affect the presence of leukocytes in the colon uponinfection with C. rodentium.
Defective STAT3 activation in CD4+ cells results in nearlycomplete loss of IL-22 production by CD3+CD4+ laminapropria cells
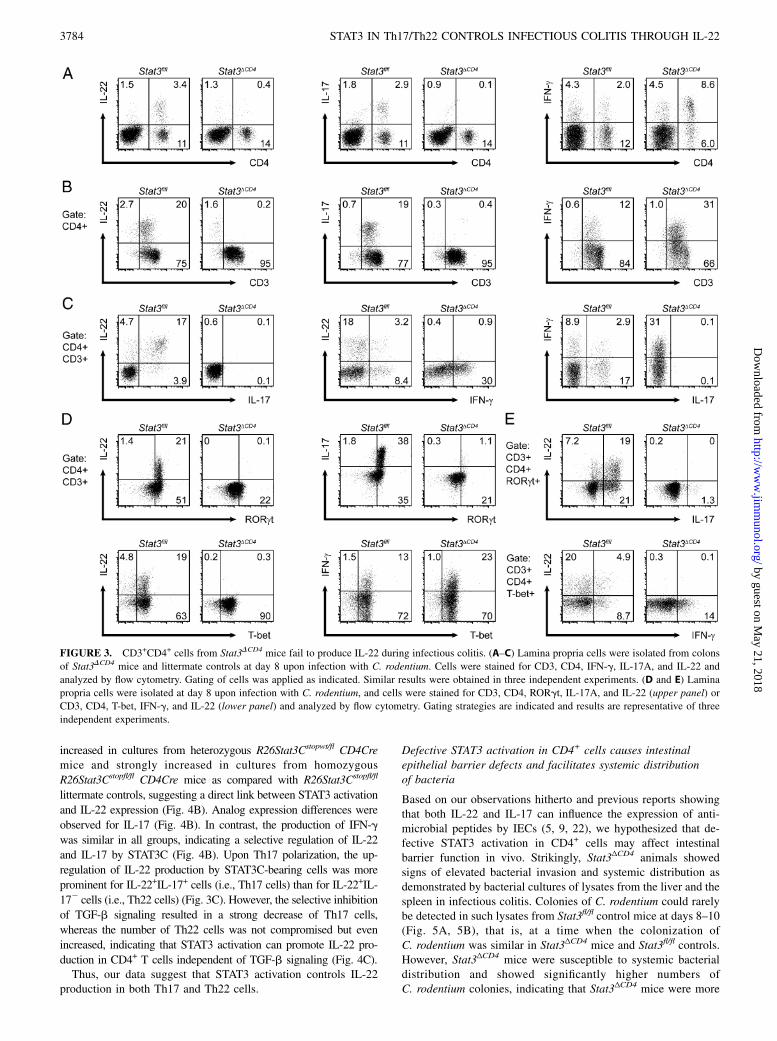
Taking into account previous work showing high upregulation ofIL-22 at the peak of infection in wild-type mice (27), we studiedthe cytokine production by lamina propria cells from colons atthat time. Interestingly, FACS analysis of those lamina propriacells revealed that in Stat3fl/fl control mice, the large majority of IL-22–producing cells were CD4+, suggesting that CD4+ cells are themajor source of IL-22 at the peak of C. rodentium infection(Fig. 3A). In contrast, IL-22 was strongly reduced in CD4+ cells ofStat3ΔCD4 mice (Fig. 3A). Similar data were obtained for the pro-duction of IL-17a, whereas IFN-g production was increased inCD4+ lamina propria cells of Stat3ΔCD4 mice (Fig. 3A). To excludea contribution of CD8+ T cells, which also lack STAT3 activation inStat3ΔCD4 mice, we tested the IL-22 production by CD8+ T cells,which was very low in wild-type mice at that time (data not shown).To further differentiate the relative contribution of CD3+CD4+
T cells versus CD32CD4+ cells, we studied the coexpression ofCD3 with IL-17a or IL-22 in CD4+ gated lamina propria cells. Inthis study, we could show that .90% of IL-17–producing CD4+
cells and .80% of IL-22–producing CD4+ cells also expressedCD3, demonstrating T lymphocytes as the dominant source ofIL-17 and IL-22 in wild-type mice at the peak of infection (Fig.3B). In accordance with STAT3 activation mandatorily needed forTh17 development, IL-17 production was strongly decreased inCD3+CD4+ cells of Stat3ΔCD4 mice (Fig. 3B). Importantly, IL-22production was also fully abrogated in CD3+CD4+ lymphocytes ofStat3ΔCD4 mice, suggesting that IL-22 production was highly de-pendent on STAT3 activation not only in Th17 cells, but also inTh22 cells (Fig. 3B). In contrast to IL-22 production from CD3+
CD4+ lymphocytes, including both Th22 and Th17 cells, IL-22production from CD32CD4+ cells was diminished in a smallproportion of cells only (Fig. 3B).Notably, we observed that more than 75% of the CD3+CD4+IL-
22+ cells stained positive for both IL-22 and IL-17, suggesting thatTh17 cells (IL-22+IL-17+), but not Th22 (IL-22+IL-172) cells,were the dominant source of IL-22 production at the peak of in-fection with C. rodentium in our setup (Fig. 3C). Similar resultswere obtained for the population of intraepithelial lymphocytes,in which CD3+CD4+IL-17+IL-22+ cells were also the dominantsubset expressing IL-22 (data not shown).
Next, we addressed the expression of the transcription factorsRORgt and T-bet in our setup by FACS. In this study we couldshow that the vast majority (.90%) of IL-22–producing CD4+
CD3+ lamina propria cells also expressed RORgt in Stat3fl/fl
control mice (Fig. 3D, upper panel). In contrast, CD4+CD3+
RORgt+IL-22+ cells were not detectable and CD4+CD3+RORgt+
IL-222 cells were strongly diminished in Stat3ΔCD4 mice (Fig. 3D,upper panel).Interestingly, most of the IL-22–producing CD4+CD3+ cells
(.75%) from control mice also expressed the transcription factorT-bet (Fig. 3D, lower panel). In Stat3ΔCD4 mice, the number of
T-bet+CD4+CD3+ lamina propria cells was increased (i.e., 82–90.3%), which is in accordance with results demonstrating ele-vated IFN-g production in Stat3ΔCD4 mice (Fig. 3B–D). FurtherFACS analysis revealed that most CD4+CD3+RORgt+IL-22+ cellsproduced IL-17 (Th17 cells), but we also detected a substantialpopulation of CD4+CD3+RORgt+IL-22+ cells (.25%) that did notshow IL-17 production (Th22 cells, Fig. 3E, upper panel). Bystudying the expression patterns of CD4+CD3+T-bet+ laminapropria cells, we observed some overlap of IL-22 production withIFN-g expression, but most of the cells stained positive for eitherIL-22 or IFN-g (Fig. 3E, lower panel).
FIGURE 1. Stat3DCD4 mice are compromised in restricting infection with C. rodentium. (A and B) Colon tissue from wild-type mice was snap-frozen at
day 8 postinfection with C. rodentium. Immunostainings were performed for p-STAT3 or Ab control. For quantitative analyses, more than five repre-
sentative sections (original magnification 310) per sample were scored for the number of p-STAT3+ cells. Data are mean values 6 SD. *p , 0.05. Scale
bars, 10 mm. (C) Cryosections were prepared from the colon of Stat3flfl and Stat3DCD4 mice at day 8 postinfection with C. rodentium and stained for
p-STAT3 and CD4. Arrows indicate the presence of double-positive cells in Stat3flfl controls but their absence in Stat3DCD4 mice. Scale bars, 10 mm. (D and
E) Stat3DCD4 mice and Stat3flfl littermate controls were infected with 8 3 109 CFU luminescent C. rodentium via gastric gavage. The course of colonization
and reduction in the overall burden of C. rodentium of luminescent bacteria was studied by serial analyses with a whole-body bioluminescent imager. The
definition of counts per second is the average number of bioluminescent counts per second. Bioluminescent counts were detected from the abdomen as
region of interest with an IVIS Lumina II system during a 3-min period. The course of infection was quantified by daily analyses of bioluminescent counts
in C. rodentium–infected Stat3DCD4 mice (n = 17) and littermate controls (n = 15). Data are mean values of individual mice6 SD and were pooled from five
independent experiments. *p , 0.05, **p , 0.01.
3782 STAT3 IN Th17/Th22 CONTROLS INFECTIOUS COLITIS THROUGH IL-22
Of note, we did not detect any major changes in Citrobacter-specific or total Ig production between Stat3ΔCD4 mice and Stat3fl/fl
controls, as evaluated in the serum and feces (data not shown).In summary, Stat3ΔCD4 mice show a nearly complete loss of IL-
22 production by CD3+CD4+ lamina propria cells during infec-tious colitis.
STAT3 activation in CD4+ splenocytes controls IL-22production by Th17 and Th22 cells
Stat3ΔCD4 mice showed compromised reduction in the overallburden of C. rodentium and loss of IL-22 production from CD4+
lamina propria lymphocytes. Similarly, IL-22 production was alsostrongly diminished upon in vitro polarization of CD4+ spleno-cytes from unchallenged mice, providing additional evidence that
STAT3 activation is required for IL-22 production in CD4+ T cells(Fig. 4A).To further evaluate the role of STAT3 for IL-22 expression in
T cells, we intended to analyze how elevated STAT3 activationin CD4+ T cells may affect cytokine production. Therefore,conditional transgenic mice were used expressing constitutivelyactive STAT3 protein (STAT3C) in CD4+ cells (heterozygousR26Stat3Cstopwt/fl CD4Cre and homozygous R26Stat3Cstopfl/fl
CD4Cre) (29). STAT3C protein is a STAT3 homodimer formed bydisulfide bonds between two cysteine substitutions, leading toactivation-independent tyrosine phosphorylation. To test the influ-ence of STAT3C expression on cytokine production in Th17 cells, weperformed Th17 polarization with freshly purified CD4+ splenocytes.Strikingly, the number of IL-22–expressing cells was moderately
FIGURE 2. Stat3DCD4 mice and littermate controls show similar crypt hyperplasia and immune cell infiltrations. (A and B) Cross-sections from the colons
of Stat3DCD4 mice and littermate controls at day 8 postinfection with C. rodentium were stained with H&E. Colon crypt lengths were quantified by the
analysis of.25 crypts per animal and three animals per group (mean values6 SD). Scale bars, 100 mm. (C and D) Cryosections with representative images
from day 8 postinfection with C. rodentium were immunostained for leukocyte surface markers (MPO, F4/80, CD11c, B220, CD4). For quantitative
analyses, four to five representative sections (original magnification 310) per sample were scored for the number of positive cells. Data are mean values
from three mice per group6 SD. Scale bars, 10 mm. (E and F) Lamina propria cells were purified from colons of Stat3DCD4 mice and Stat3fl/fl littermates at
day 8 upon infection with C. rodentium. Cells were stained for CD11b, Ly6C, F4/80, Ly6G, B220, CD11c, CD3, and CD4 analyzed by flow cytometry. Data
are representative of three independent experiments.
increased in cultures from heterozygous R26Stat3Cstopwt/fl CD4Cremice and strongly increased in cultures from homozygous
R26Stat3Cstopfl/fl CD4Cre mice as compared with R26Stat3Cstopfl/fl
littermate controls, suggesting a direct link between STAT3 activation
and IL-22 expression (Fig. 4B). Analog expression differences were
observed for IL-17 (Fig. 4B). In contrast, the production of IFN-g
was similar in all groups, indicating a selective regulation of IL-22
and IL-17 by STAT3C (Fig. 4B). Upon Th17 polarization, the up-
regulation of IL-22 production by STAT3C-bearing cells was more
prominent for IL-22+IL-17+ cells (i.e., Th17 cells) than for IL-22+IL-
172 cells (i.e., Th22 cells) (Fig. 3C). However, the selective inhibition
of TGF-b signaling resulted in a strong decrease of Th17 cells,
whereas the number of Th22 cells was not compromised but even
increased, indicating that STAT3 activation can promote IL-22 pro-
duction in CD4+ T cells independent of TGF-b signaling (Fig. 4C).Thus, our data suggest that STAT3 activation controls IL-22
production in both Th17 and Th22 cells.
Defective STAT3 activation in CD4+ cells causes intestinalepithelial barrier defects and facilitates systemic distributionof bacteria
Based on our observations hitherto and previous reports showingthat both IL-22 and IL-17 can influence the expression of anti-microbial peptides by IECs (5, 9, 22), we hypothesized that de-fective STAT3 activation in CD4+ cells may affect intestinalbarrier function in vivo. Strikingly, Stat3ΔCD4 animals showedsigns of elevated bacterial invasion and systemic distribution asdemonstrated by bacterial cultures of lysates from the liver and thespleen in infectious colitis. Colonies of C. rodentium could rarelybe detected in such lysates from Stat3fl/fl control mice at days 8–10(Fig. 5A, 5B), that is, at a time when the colonization ofC. rodentium was similar in Stat3ΔCD4 mice and Stat3fl/fl controls.However, Stat3ΔCD4 mice were susceptible to systemic bacterialdistribution and showed significantly higher numbers ofC. rodentium colonies, indicating that Stat3ΔCD4 mice were more
FIGURE 3. CD3+CD4+ cells from Stat3DCD4 mice fail to produce IL-22 during infectious colitis. (A–C) Lamina propria cells were isolated from colons
of Stat3DCD4 mice and littermate controls at day 8 upon infection with C. rodentium. Cells were stained for CD3, CD4, IFN-g, IL-17A, and IL-22 and
analyzed by flow cytometry. Gating of cells was applied as indicated. Similar results were obtained in three independent experiments. (D and E) Lamina
propria cells were isolated at day 8 upon infection with C. rodentium, and cells were stained for CD3, CD4, RORgt, IL-17A, and IL-22 (upper panel) or
CD3, CD4, T-bet, IFN-g, and IL-22 (lower panel) and analyzed by flow cytometry. Gating strategies are indicated and results are representative of three
independent experiments.
3784 STAT3 IN Th17/Th22 CONTROLS INFECTIOUS COLITIS THROUGH IL-22
prone to systemic dissemination despite equivalent C. rodentiumburden in the colon (Fig. 5A, 5B). Similar results with increased
dissemination to the liver and spleen in Stat3ΔCD4 mice were
obtained at days 15–16.Moreover, the production of some IEC-derived antimicrobial
peptides, including defensin b1, regenerating islet-derived 3 b and g
(RegIIIb, RegIIIg), and calgranulin A (S100A8), was reduced in
the colon of Stat3ΔCD4 mice at day 8 upon infection with C.
rodentium (Fig. 5C). In contrast, other molecules related to IEC-
mediated barrier function such as mucin 2 and 13 (Muc2, Muc13),
serum amyloid 1, or trefoil factor 3 did not show major changes
(Fig. 5C). At days 15–16, when the overall burden of C. rodentium
was strongly decreased in control mice already, antimicrobial
peptides were low in control mice as well (data not shown).Thus, our data suggest that defective STAT3 activation in CD4+
lymphocytes causes intestinal epithelial barrier defects and ren-ders individuals susceptible to systemic bacterial distributionduring infectious colitis.
Systemic overexpression of IL-22 restores the host defenseagainst C. rodentium in Stat3ΔCD4 mice
To further address the differential role of Th cell–derived cytokineson the intestinal epithelium, we used a recently established minicirclevector-based method in combination with hydrodynamic tail veininjection for the systemic overexpression of cytokines (32). Vectorswere constructed for IL-17A, IL-17F, IFN-g, and IL-4, and one in-jection of 10 mg vector DNA resulted in high expression (.5 ng/ml)of the respective cytokine as measured in the blood (data not shown).Next, we tested the expression of IEC-derived peptides in the distalcolon tissue upon systemic expression of those cytokines in unin-fected mice. In this study, we could directly demonstrate that deliveryof IL-22, but not IL-17A, IL-17F, IFN-g, or IL-4, caused high ex-pression of antimicrobial peptides, including RegIIIb and RegIIIgat day 3 upon vector injection, as analyzed by qPCR (Fig. 6A). Onthe contrary, other genes related to epithelial defense mechanisms inIECs, including S100A8, Muc2, defensin b1, or serum amyloid 1,remained without major changes (Fig. 6A and data not shown).
FIGURE 4. STAT3 activation in CD4+ splenocytes controls IL-22 production from Th17 and Th22 cells. (A) CD4+ T cells from spleens of Stat3DCD4
mice and Stat3fl/fl controls were stimulated with plate-bound anti-CD3 and soluble anti-CD28 and grown under Th17 polarizing conditions with anti–IFN-g,
anti–IL-4, TGF-b, IL-6, and IL-23. A specific TGF-b inhibitor (SB431542) was applied instead of rTGF-b where indicated. Intracellular staining and
FACS analysis for IL-22, IL-17, and IFN-g was done at day 4. Gating was performed on CD4+ cells. Similar results were obtained in two independent
experiments. (B and C) CD4+ T cells were freshly isolated from spleens of the indicated genotypes. Cells were stimulated with anti-CD3 and anti-CD28 and
polarized with anti–IFN-g, anti–IL-4, IL-6, IL-23, and TGF-b or TGF-b inhibitor as indicated. FACS analysis for IL-22, IL-17, and IFN-g was performed
at day 3. Gating was performed on CD4+ cells. Similar results were obtained in three independent experiments.
To investigate whether IL-22 delivery might influence thephenotype of Stat3ΔCD4 mice, IL-22 vector was applied toStat3ΔCD4 mice at day 10 upon C. rodentium infection. Strikingly,Stat3ΔCD4 mice overexpressing IL-22 could clear the bacteria withsimilar kinetics as Stat3fl/fl control mice, indicating that reintro-duction of IL-22 can rescue the compromised host defense ofStat3ΔCD4 mice (Fig. 6B, 6C). In contrast, systemic overexpressionof IL-17A and IL-17F failed to rescue the phenotype of Stat3ΔCD4
mice regarding C. rodentium infection (data not shown). In linewith our observations in uninfected mice at day 3, we detectedincreased expression of RegIIIb and RegIIIg in mice infected withC. rodentium and overexpressing IL-22 (Fig. 6D).Thus, our data provide evidence that STAT3 activation in CD4+
lymphocyte subsets can shape strong epithelial defense mecha-nisms through IL-22 production and induction of antimicrobialpeptides in IECs.
Constitutive activation of STAT3 in CD4+ cells causes highinduction of antimicrobial peptides in IECs in vitro and in vivoand may offer a therapeutic approach for infectious colitis
To further evaluate the role of STAT3 in T cells for the cross-talk toIECs, intestinal epithelial organoids were grown from wild-typemice as previously described (31) and subsequently cultured inthe presence of conditioned medium derived from supernatants ofCD4+ splenocytes from R26Stat3Cstopfl/fl CD4Cre or control micepolarized under Th17 or Th22 conditions (Fig. 7A). Strikingly,conditioned medium from R26Stat3Cstopfl/fl CD4Cre mice, whichcontained elevated levels of IL-22, resulted in high expression ofRegIIIb and RegIIIg, but not of Muc2 or S100A8, by intestinalorganoids, providing direct evidence that activation of STAT3 inCD4+ T cells controls the production of some antimicrobial pep-tides through cross-talk to IECs (Fig. 7B, 7C, and data not shown).In line with our previous results (Figs. 5, 6), the induction ofRegIIIb and RegIIIg by conditioned medium was almost com-pletely abrogated by neutralizing anti–IL-22 Ab, indicating thatcross-talk from STAT3C-expressing CD4+ T cells to IECs in vitrowas mainly mediated through IL-22 production (Fig. 7D).Next, we hypothesized that an increase of active STAT3 in CD4+
cells may be useful as a therapeutic approach in infections with
enteropathogenic bacteria. Consistent with our hypothesis, weobserved that primary IECs purified from the colons of 6-wk-oldR26Stat3Cstopwt/fl CD4Cre mice and R26Stat3Cstopfl/fl CD4Cremice showed very high production of antimicrobial peptides evenunder unchallenged conditions (Fig. 7E). To further study the in vivorelevance and therapeutic potential for infections with enteropatho-genic bacteria, we tested the course of C. rodentium infection inmice expressing STAT3C. Remarkably, R26Stat3Cstopwt/fl CD4Cremice were protected and cleared the infection faster than did wild-type controls (Fig. 7F, 7G), suggesting that the specific elevation ofactive STAT3 in CD4+ cells may serve as a therapeutic strategy ininfections with enteropathogenic bacteria.Interestingly, some differences in the expression of antimi-
crobial peptides by IECs were still detectable at day 8 uponinfection with C. rodentium (Fig. 7H). Additionally, the analysis ofcolon tissue lysates from mice infected with C. rodentium con-firmed higher protein levels for IL-22 and antimicrobial peptidesin R26Stat3Cstopwt/fl CD4Cre mice compared with controls at day8, providing further support for the therapeutic approach (Fig. 7I).Of note, R26Stat3Cstopfl/fl CD4Cre mice could not be analyzed
in the C. rodentium model, as they suffer from chronic airwayinflammation and have a shortened lifespan with a mean survivalof ,40 d (29). Therefore, although STAT3 activation in CD4+ cellsmay act as a promising therapeutic strategy, the consequences ofits modulation need to be carefully controlled.In summary, our data suggest that the expression of STAT3C in
CD4+ cells increases the production of antimicrobial peptides inIECs through IL-22 and accelerates the reduction in the overallburden of enteropathogenic bacteria. Thus, STAT3 activation inCD4+ lymphocytes may offer a future therapeutic option forpromoting cross-talk to IECs and improving intestinal barrierfunction in infectious colitis.Finally, we propose a model for the role of STAT3 activation in
CD4+ cells during infection with enteropathogenic bacteria suchas C. rodentium (Fig. 8). In individuals with intact STAT3 phos-phorylation, such infections can be successfully counterbalancedthrough efficient cross-talk to IECs, which includes high produc-tion of IL-22 by CD4+ lamina propria lymphocytes shaping strongepithelial defense mechanisms in the colon. In individuals that
FIGURE 5. Stat3DCD4 mice display compromised intestinal epithelial barrier function and are susceptible to systemic distribution of bacteria. (A and B)
Livers and spleens from Stat3DCD4 mice and littermate controls were harvested under sterile conditions at day 8 upon infection with C. rodentium. Organ
lysates were cultured for 1 d. The number of CFU luminescent C. rodentium was counted (mean values 6 SD; n = 3/group). Similar results were measured
in three independent experiments. *p , 0.05. (C) Infectious colitis was induced by C. rodentium. The expression of various IEC-derived genes was
measured in colon bowel wall tissue from Stat3DCD4 mice and littermate controls (n = 4/group) by qPCR as indicated. Similar results were obtained in three
cannot activate STAT3 in CD4+ cells, the early phase of infectionis not altered because subsets of ILC3 and possibly other CD42
cells could provide substantial amounts of IL-22. Additionally,IL-22 production by CD32CD4+ cells might partly occur viaSTAT3-independent mechanisms. At the peak of infection, how-ever, individuals that cannot activate STAT3 in CD4+ cells fullylack their major source of IL-22, that is, CD4+ lymphocytes (Th17and Th22 cells), resulting in increased bacterial invasion anddelayed reduction of bacterial burden.Hence, individuals with defective STAT3 in CD4+ cells display
impaired intestinal epithelial barrier function, and protection fromenteropathogenic bacteria is severely compromised during theT cell–dependent phase of infection.
DiscussionOur work analyzed the specific contribution of STAT3 activationin CD4+ cells to host defense upon infection with enteropatho-genic bacteria. We found that, compared with littermate controls,Stat3ΔCD4 mice show a similar course of infection during the earlyphase, which is known to require critical contributions by ILC3s,including CD4+ lymphoid tissue inducer cells and NK22 cells (16,17, 20, 24). From the peak of infection on, however, Stat3ΔCD4
mice are heavily compromised in reducing the burden ofC. rodentium. Moreover, Stat3ΔCD4 animals were more susceptible
to bacterial distribution of C. rodentium to distant organs such asliver and spleen. Our study was performed with cohoused mice,indicating that the differences were stably related to genotypesand not driven by a dominant transmissible flora.In accordance with a mandatory role of STAT3 for Th17 cell
development (12), Stat3ΔCD4 mice are also lacking nearly all Th17cells in the C. rodentium model. Hence, our observations areconsistent with previous studies suggesting Th17 cells to be im-portant in the C. rodentium model (21, 23). However, previousstudies were often restricted in addressing the specific role ofTh17 cells in vivo, as they included mice deficient in genes thatare already critically important for the development or effectorfunctions of ILC3 subsets such as IL-22, IL-23R, or AHR (5, 16,21, 25). In contrast, our investigations using conditional knockoutmice allowed for more specific investigations into the role of Th17and Th22 cells at the peak of infection.It was demonstrated that STAT3 activation correlates with IL-22
transcript expression in CD4+ T cells (12, 35). Strikingly, our datastrongly suggest that STAT3 activation is mandatorily needed forIL-22 production from CD3+CD4+ lamina propria cells in theC. rodentium model. In line with previous reports showing thatSTAT3 is required for Th17 cell development and early Th17lineage differentiation (12, 36), our findings provide evidence thatthe lack of active STAT3 not only interferes with the expression of
FIGURE 6. Overexpression of IL-22 by a DNA vector rescues defective host defense of Stat3DCD4 mice in infectious colitis. (A) Minicircle vectors for
IL-17A, IL-17F, IL-22, IFN-g, IL-4, or empty control vector (10 mg each) were administered via hydrodynamic tail vein injection. The expression of IEC-
derived genes was measured in colon wall tissue by qPCR at day 3. Data are mean values 6 SD (n = 3/group). A second experiment gave similar results.
**p , 0.01. (B and C) Stat3DCD4 mice and littermate controls were infected with luminescent C. rodentium and the presence of bacteria was followed by
daily whole-body bioluminescent imaging. IL-22 vector or empty control vector (10 mg each) were applied at day 10. Data are mean values6 SD (n = 3–4/
group). Data are representative of three independent experiments. *p , 0.05. (D) IL-22 vector or empty control vector (10 mg each) were applied to
Stat3DCD4 mice 10 d upon infection with C. rodentium (8 3 109 CFU). Colon tissue was harvested 2 d later and analyzed by qPCR as indicated. Similar
results were obtained in three independent experiments. *p , 0.05.
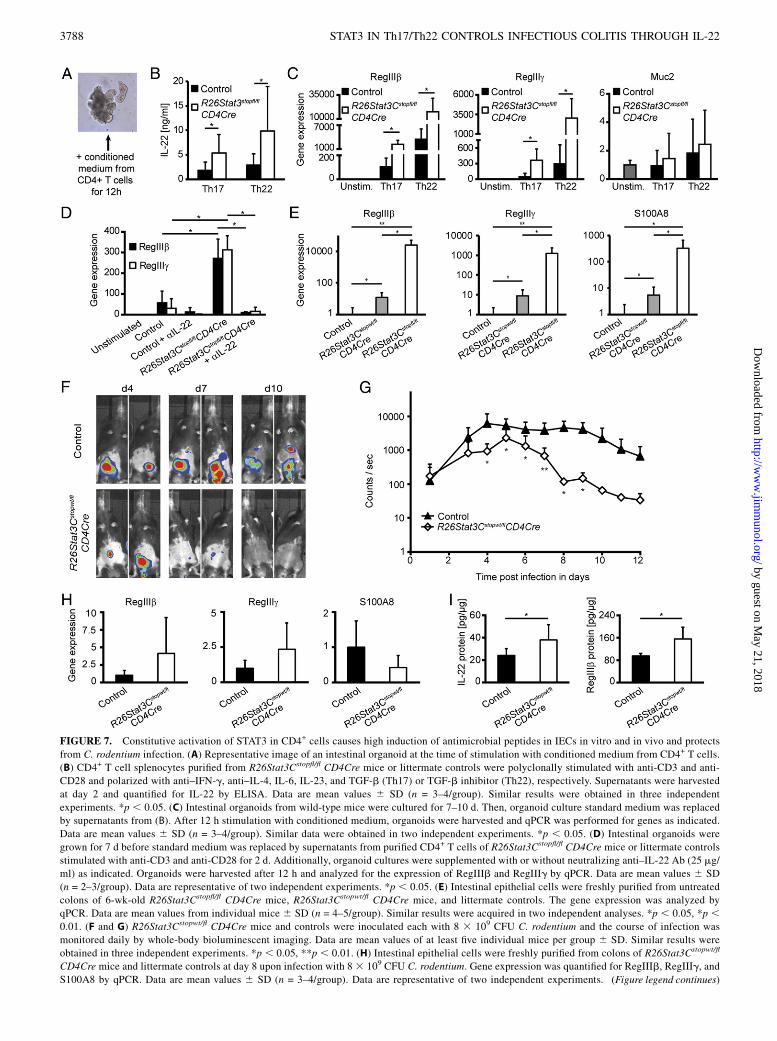
FIGURE 7. Constitutive activation of STAT3 in CD4+ cells causes high induction of antimicrobial peptides in IECs in vitro and in vivo and protects
from C. rodentium infection. (A) Representative image of an intestinal organoid at the time of stimulation with conditioned medium from CD4+ T cells.
(B) CD4+ T cell splenocytes purified from R26Stat3Cstopfl/fl CD4Cre mice or littermate controls were polyclonally stimulated with anti-CD3 and anti-
CD28 and polarized with anti–IFN-g, anti–IL-4, IL-6, IL-23, and TGF-b (Th17) or TGF-b inhibitor (Th22), respectively. Supernatants were harvested
at day 2 and quantified for IL-22 by ELISA. Data are mean values 6 SD (n = 3–4/group). Similar results were obtained in three independent
experiments. *p , 0.05. (C) Intestinal organoids from wild-type mice were cultured for 7–10 d. Then, organoid culture standard medium was replaced
by supernatants from (B). After 12 h stimulation with conditioned medium, organoids were harvested and qPCR was performed for genes as indicated.
Data are mean values 6 SD (n = 3–4/group). Similar data were obtained in two independent experiments. *p , 0.05. (D) Intestinal organoids were
grown for 7 d before standard medium was replaced by supernatants from purified CD4+ T cells of R26Stat3Cstopfl/fl CD4Cre mice or littermate controls
stimulated with anti-CD3 and anti-CD28 for 2 d. Additionally, organoid cultures were supplemented with or without neutralizing anti–IL-22 Ab (25 mg/
ml) as indicated. Organoids were harvested after 12 h and analyzed for the expression of RegIIIb and RegIIIg by qPCR. Data are mean values 6 SD
(n = 2–3/group). Data are representative of two independent experiments. *p , 0.05. (E) Intestinal epithelial cells were freshly purified from untreated
colons of 6-wk-old R26Stat3Cstopfl/fl CD4Cre mice, R26Stat3Cstopwt/fl CD4Cre mice, and littermate controls. The gene expression was analyzed by
qPCR. Data are mean values from individual mice 6 SD (n = 4–5/group). Similar results were acquired in two independent analyses. *p , 0.05, *p ,0.01. (F and G) R26Stat3Cstopwt/fl CD4Cre mice and controls were inoculated each with 8 3 109 CFU C. rodentium and the course of infection was
monitored daily by whole-body bioluminescent imaging. Data are mean values of at least five individual mice per group 6 SD. Similar results were
obtained in three independent experiments. *p , 0.05, **p , 0.01. (H) Intestinal epithelial cells were freshly purified from colons of R26Stat3Cstopwt/fl
CD4Cre mice and littermate controls at day 8 upon infection with 8 3 109 CFU C. rodentium. Gene expression was quantified for RegIIIb, RegIIIg, and
S100A8 by qPCR. Data are mean values 6 SD (n = 3–4/group). Data are representative of two independent experiments. (Figure legend continues)
3788 STAT3 IN Th17/Th22 CONTROLS INFECTIOUS COLITIS THROUGH IL-22
IL-22, but also with the population of CD4+CD3+RORgt+IL-17+
IL-22+ cells (Th17 cells). Additionally, our data might indicatethat STAT3 also interferes with the population of CD4+CD3+IL-172IL-22+ cells, which are usually referred to as Th22 cells.It remains unresolved whether Th22 cells represent a truly
unique lineage equivalent to other types of Th cells or rathera different activation state. Our analyses of primary lymphocytesfrom conditional STAT3-deficient mice and novel mice express-ing constitutively active STAT3 under control of the CD4 promoterare consistent with the concept that Th22 cells, in contrast to Th17cells, develop independently of TGF-b signaling (27, 37). Addi-tionally, a reciprocal need for RORgt and T-bet was recentlysuggested for Th17 cells and Th22 cells, respectively (27).However, whereas CD3+CD4+ lamina propria cells from Tbx-212/2
(encoding for T-bet) mice showed a diminished, but still clearlydetectable, population of IL-22–producing CD4+ T cells (27), ourwork could demonstrate that nearly all Th22 cells were absentin Stat3ΔCD4 mice, suggesting an even more important role forSTAT3 than for T-bet. On the basis of these findings and recentwork analyzing regulatory networks for Th17 specification (36), itmay be interesting to learn how other transcription factors that alsoguide early Th17 lineage differentiation such as BATF and IFNregulatory factor (IRF) 4 influence the development of Th22 cells.Both BATF and IRF4 were reported being similarly expressed in
Th17 cells and Th22 cells (27). Binding of BATF to the IL-22promoter was shown, suggesting modulation of IL-22 expressionindependent of promoting Th17 lineage specification (38). Inter-estingly, IRF4 deficiency was recently correlated with high IL-22expression (39), suggesting divergent roles for IRF4 and STAT3.In our experimental setup, IL-17+IL-22+CD3+CD4+ lympho-
cytes represented the most frequent IL-22–producing populationat the peak of infection in the lamina propria, which differs froma recent study arguing for Th22 cells as the predominant sourceof IL-22 (27). In contrast to Basu et al. (27), our ex vivo FACSanalyses of lamina propria cells were performed without preced-ing IL-23 restimulation. Additionally, differences may be causedby the resident gut microbiota, which can influence the course ofC. rodentium infection and the level of Th17 cells (23, 40).Although this study did not directly address the upstream sig-
naling responsible for STAT3 activation in Th17 and Th22 cellsin vivo, IL-6 seems to be a likely candidate molecule. It has beenpreviously linked to the production of IL-22 (41), development ofTh22 cells (42), and host defense upon C. rodentium infection (21,43). Other cytokines that are also highly expressed at the top ofinfection, including IL-23, may additionally contribute to STAT3activation.Our data demonstrate that Stat3ΔCD4 mice compared with
cohoused littermate controls show a similar course of infection
(I) Colons of R26Stat3Cstopwt/fl CD4Cre mice and littermate controls were harvested at day 8 upon infection with 8 3 109 CFU C. rodentium. The protein
levels of IL-22 and RegIIIb per colon tissue were quantified in colon tissue lysates by ELISA. Data are mean values 6 SD (n = 3–4/group). Similar results
were obtained in two independent experiments. *p , 0.05.
FIGURE 8. Model for the role of STAT3 ac-
tivation in CD4+ cells during infection with
C. rodentium. When STAT3 activation is intact
in CD4+ cells, infectious colitis can be success-
fully counterbalanced through efficient cross-talk
to IECs, which includes high expression of IL-22
shaping epithelial defense mechanisms in the
colon. In case of defective STAT3 activation in
CD4+ cells, IL-22 production by nearly all CD4+
lymphocytes, that is, Th17 and Th22 cells, is
absent, resulting in inadequate epithelial barrier
during the early phase upon inoculation with C. rodentium.Whereas our work demonstrated a critical role for STAT3 in CD4T cells for efficient reduction in the overall burden of C. rodentiumfrom the peak of infection on, a very recent study by Guo et al.(44) reported that STAT3 activation in ILC3s is very important forprotection during the early phase of infectious colitis. Notably,ILC3s comprise various subsets, and it appears possible thatsubpopulations of ILC3 might not require STAT3 activation forIL-22 production. In line with that hypothesis, our molecularanalyses could suggest that CD32CD4+ cells might partly produceIL-22 independent of STAT3. However, the efficiency of the CD4-driven Cre recombinase in ILCs was challenged (45). Neverthe-less, differential requirements for the transcription factor IRF4were recently reported between Th17 cells and ILC3s for theproduction of both IL-22 and IL-17 (46). Moreover, previous workdemonstrated that IL-17 production in ILC3s can occur in botha STAT3-dependent and STAT3-independent manner (34).Collectively, our observations are in line with sequential waves
of IL-22 expression produced by different immune cell populationsupon infection with enteropathogenic bacteria (27). Our work isconsistent with studies demonstrating that IL-22–producing ILC3sare critical during the early phase of immune defense in theC. rodentium model (17, 24, 25).Stat3ΔCD4 mice show decreased levels of IEC-derived antimi-
crobial peptides in the colon at the peak of C. rodentium infection,which is in accordance with IL-22, IL-17A, and IL-17F as potentinducers of antimicrobial peptides (5, 9, 22). Importantly, systemicoverexpression of IL-22, but not IL-17A and IL-17F, could restoreefficient reduction in the overall burden of C. rodentium inStat3ΔCD4 mice. Hence, our data argue for an important role ofSTAT3-dependent IL-22 release from CD4+ lymphocytes, whichis consistent with a more severe phenotype in IL-222/2 mice thanin IL-17A2/2 and IL-17F2/2 mice (5, 22).We and others have previously shown that IL-22 signaling
induces STAT3 activation in IECs upon mucosal damage, therebypromoting restoration of intestinal homeostasis (9, 47, 48). StrongSTAT3 activation in IECs is also found upon infection withC. rodentium (49). Thus, our data support a model in which STAT3activation in CD4+ lymphocytes links STAT3 activation in IECs viaIL-22, thereby shaping the host defense in the colon during infec-tion with enteropathogenic bacteria.As this work did not involve human studies, potential con-
clusions regarding implications for human diseases remain spec-ulative. In contrast to mice, no CD4+ ILC3s have been identified inhumans so far (20). Our work revealed a key role for STAT3 ac-tivation in CD4+ lymphocytes for efficient reduction in the overallburden of enteropathogenic bacteria in a well-established diseasemodel mimicking infections by enterohemorrhagic and entero-pathogenic E. coli in humans. As in mice, STAT3 activation isessential for Th17 cell biology in humans (13). Consistently, ge-netic defects in STAT3 have been connected to human diseasesrendering individuals susceptible to infections (14). Of note, sin-gle nucleotide polymorphisms of STAT3 and numerous othergenes related to STAT3 signaling in CD4+ T cells have been as-sociated with chronic intestinal pathology in inflammatory boweldisease (IBD) (6), and a pathogenic role of bacteria has beenproposed for the onset and perpetuation of IBD (50, 51). Addi-tionally, the contribution of a defective intestinal barrier functionto the multifactorial pathogenesis of IBD has become increasinglyacknowledged in the past few years. Thus, STAT3 activation inhuman CD4+ cells may guide host defense mechanisms by IECs ina similar way as in mice.In conclusion, our data suggest that approaches specifically
elevating STAT3 activation in CD4+ lymphocytes can shape strong
epithelial defense mechanisms against infections with entero-pathogenic bacteria. It is intriguing to speculate that the promotionof STAT3 phosphorylation in mucosal CD4+ T cells or the supplyof colon-tropic STAT3 activated CD4+ cells might serve as a fu-ture therapeutic option for individuals suffering from infectiouscolitis or intestinal diseases associated with compromised epi-thelial defense mechanisms.
AcknowledgmentsBioluminescent C. rodentium were provided by C.U. Riedel (University of
Ulm, Ulm, Germany). We thank K. Enderle, I. Zoeller-Utz, C. Lindner,
and A. Taut for excellent technical assistance and M. McLaughlin for
critically reading the manuscript.
DisclosuresThe authors have no financial conflicts of interest.
References1. Kamada, N., G. Y. Chen, N. Inohara, and G. Nunez. 2013. Control of pathogens
and pathobionts by the gut microbiota. Nat. Immunol. 14: 685–690.2. Van der Sluis, M., B. A. De Koning, A. C. De Bruijn, A. Velcich, J. P. Meijerink,
J. B. Van Goudoever, H. A. B€uller, J. Dekker, I. Van Seuningen, I. B. Renes, andA. W. Einerhand. 2006. Muc2-deficient mice spontaneously develop colitis,indicating that MUC2 is critical for colonic protection. Gastroenterology 131:117–129.
3. Wilson, C. L., A. J. Ouellette, D. P. Satchell, T. Ayabe, Y. S. Lopez-Boado,J. L. Stratman, S. J. Hultgren, L. M. Matrisian, and W. C. Parks. 1999. Regu-lation of intestinal a-defensin activation by the metalloproteinase matrilysin ininnate host defense. Science 286: 113–117.
4. Cunliffe, R. N., and Y. R. Mahida. 2004. Expression and regulation of antimi-crobial peptides in the gastrointestinal tract. J. Leukoc. Biol. 75: 49–58.
5. Zheng, Y., P. A. Valdez, D. M. Danilenko, Y. Hu, S. M. Sa, Q. Gong,A. R. Abbas, Z. Modrusan, N. Ghilardi, F. J. de Sauvage, and W. Ouyang. 2008.Interleukin-22 mediates early host defense against attaching and effacing bac-terial pathogens. Nat. Med. 14: 282–289.
6. Jostins, L., S. Ripke, R. K. Weersma, R. H. Duerr, D. P. McGovern, K. Y. Hui,J. C. Lee, L. P. Schumm, Y. Sharma, C. A. Anderson, et al. 2012. Host-microbeinteractions have shaped the genetic architecture of inflammatory bowel disease.Nature 491: 119–124.
7. O’Shea, J. J., and R. Plenge. 2012. JAK and STAT signaling molecules in im-munoregulation and immune-mediated disease. Immunity 36: 542–550.
8. Takeda, K., B. E. Clausen, T. Kaisho, T. Tsujimura, N. Terada, I. Forster, andS. Akira. 1999. Enhanced Th1 activity and development of chronic enterocolitisin mice devoid of Stat3 in macrophages and neutrophils. Immunity 10: 39–49.
9. Pickert, G., C. Neufert, M. Leppkes, Y. Zheng, N. Wittkopf, M. Warntjen,H. A. Lehr, S. Hirth, B. Weigmann, S. Wirtz, et al. 2009. STAT3 links IL-22signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med.206: 1465–1472.
10. Durant, L., W. T. Watford, H. L. Ramos, A. Laurence, G. Vahedi, L. Wei,H. Takahashi, H. W. Sun, Y. Kanno, F. Powrie, and J. J. O’Shea. 2010. Diversetargets of the transcription factor STAT3 contribute to T cell pathogenicity andhomeostasis. Immunity 32: 605–615.
11. Leppkes, M., C. Becker, I. I. Ivanov, S. Hirth, S. Wirtz, C. Neufert, S. Pouly,A. J. Murphy, D. M. Valenzuela, G. D. Yancopoulos, et al. 2009. RORg-expressing Th17 cells induce murine chronic intestinal inflammation via re-dundant effects of IL-17A and IL-17F. Gastroenterology 136: 257–267.
12. Yang, X. O., A. D. Panopoulos, R. Nurieva, S. H. Chang, D. Wang,S. S. Watowich, and C. Dong. 2007. STAT3 regulates cytokine-mediated gen-eration of inflammatory helper T cells. J. Biol. Chem. 282: 9358–9363.
13. Milner, J. D., J. M. Brenchley, A. Laurence, A. F. Freeman, B. J. Hill,K. M. Elias, Y. Kanno, C. Spalding, H. Z. Elloumi, M. L. Paulson, et al. 2008.Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452: 773–776.
14. Holland, S. M., F. R. DeLeo, H. Z. Elloumi, A. P. Hsu, G. Uzel, N. Brodsky,A. F. Freeman, A. Demidowich, J. Davis, M. L. Turner, et al. 2007. STAT3mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357: 1608–1619.
15. Mundy, R., T. T. MacDonald, G. Dougan, G. Frankel, and S. Wiles. 2005. Cit-robacter rodentium of mice and man. Cell. Microbiol. 7: 1697–1706.
16. Ota, N., K. Wong, P. A. Valdez, Y. Zheng, N. K. Crellin, L. Diehl, andW. Ouyang. 2011. IL-22 bridges the lymphotoxin pathway with the maintenanceof colonic lymphoid structures during infection with Citrobacter rodentium. Nat.Immunol. 12: 941–948.
17. Satoh-Takayama, N., C. A. Vosshenrich, S. Lesjean-Pottier, S. Sawa,M. Lochner, F. Rattis, J. J. Mention, K. Thiam, N. Cerf-Bensussan,O. Mandelboim, et al. 2008. Microbial flora drives interleukin 22 production inintestinal NKp46+ cells that provide innate mucosal immune defense. Immunity29: 958–970.
18. Hirata, Y., L. Egea, S. M. Dann, L. Eckmann, and M. F. Kagnoff. 2010. GM-CSF-facilitated dendritic cell recruitment and survival govern the intestinal mucosalresponse to a mouse enteric bacterial pathogen. Cell Host Microbe 7: 151–163.
3790 STAT3 IN Th17/Th22 CONTROLS INFECTIOUS COLITIS THROUGH IL-22
19. Bry, L., M. Brigl, and M. B. Brenner. 2006. CD4+-T-cell effector functions andcostimulatory requirements essential for surviving mucosal infection with Cit-robacter rodentium. Infect. Immun. 74: 673–681.
20. Spits, H., D. Artis, M. Colonna, A. Diefenbach, J. P. Di Santo, G. Eberl,S. Koyasu, R. M. Locksley, A. N. McKenzie, R. E. Mebius, et al. 2013. Innatelymphoid cells: a proposal for uniform nomenclature. Nat. Rev. Immunol. 13:145–149.
21. Mangan, P. R., L. E. Harrington, D. B. O’Quinn, W. S. Helms, D. C. Bullard,C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006.Transforming growth factor-b induces development of the TH17 lineage. Nature441: 231–234.
22. Ishigame, H., S. Kakuta, T. Nagai, M. Kadoki, A. Nambu, Y. Komiyama,N. Fujikado, Y. Tanahashi, A. Akitsu, H. Kotaki, et al. 2009. Differential roles ofinterleukin-17A and -17F in host defense against mucoepithelial bacterial in-fection and allergic responses. Immunity 30: 108–119.
23. Ivanov, I. I., K. Atarashi, N. Manel, E. L. Brodie, T. Shima, U. Karaoz, D. Wei,K. C. Goldfarb, C. A. Santee, S. V. Lynch, et al. 2009. Induction of intestinalTh17 cells by segmented filamentous bacteria. Cell 139: 485–498.
24. Sonnenberg, G. F., L. A. Monticelli, M. M. Elloso, L. A. Fouser, and D. Artis.2011. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut.Immunity 34: 122–134.
25. Qiu, J., J. J. Heller, X. Guo, Z. M. Chen, K. Fish, Y. X. Fu, and L. Zhou. 2012.The aryl hydrocarbon receptor regulates gut immunity through modulation ofinnate lymphoid cells. Immunity 36: 92–104.
26. Hirota, K., B. Martin, and M. Veldhoen. 2010. Development, regulation andfunctional capacities of Th17 cells. Semin. Immunopathol. 32: 3–16.
27. Basu, R., D. B. O’Quinn, D. J. Silberger, T. R. Schoeb, L. Fouser, W. Ouyang,R. D. Hatton, and C. T. Weaver. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37: 1061–1075.
28. Takeda, K., T. Kaisho, N. Yoshida, J. Takeda, T. Kishimoto, and S. Akira. 1998.Stat3 activation is responsible for IL-6-dependent T cell proliferation throughpreventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161: 4652–4660.
29. Fogli, L. K., M. S. Sundrud, S. Goel, S. Bajwa, K. Jensen, E. Derudder, A. Sun,M. Coffre, C. Uyttenhove, J. Van Snick, et al. 2013. T cell-derived IL-17mediates epithelial changes in the airway and drives pulmonary neutrophilia.J. Immunol. 191: 3100–3111.
30. Riedel, C. U., P. G. Casey, H. Mulcahy, F. O’Gara, C. G. Gahan, and C. Hill.2007. Construction of p16Slux, a novel vector for improved bioluminescentlabeling of Gram-negative bacteria. Appl. Environ. Microbiol. 73: 7092–7095.
31. Sato, T., R. G. Vries, H. J. Snippert, M. van de Wetering, N. Barker,D. E. Stange, J. H. van Es, A. Abo, P. Kujala, P. J. Peters, and H. Clevers. 2009.Single Lgr5 stem cells build crypt-villus structures in vitro without a mesen-chymal niche. Nature 459: 262–265.
32. McHedlidze, T., M. Waldner, S. Zopf, J. Walker, A. L. Rankin, M. Schuchmann,D. Voehringer, A. N. McKenzie, M. F. Neurath, S. Pflanz, and S. Wirtz. 2013.Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Im-munity 39: 357–371.
33. Liu, F., Y. Song, and D. Liu. 1999. Hydrodynamics-based transfection in animalsby systemic administration of plasmid DNA. Gene Ther. 6: 1258–1266.
34. Takatori, H., Y. Kanno, W. T. Watford, C. M. Tato, G. Weiss, I. I. Ivanov, II,D. R. Littman, and J. J. O’Shea. 2009. Lymphoid tissue inducer-like cells are aninnate source of IL-17 and IL-22. J. Exp. Med. 206: 35–41.
35. Nurieva, R., X. O. Yang, G. Martinez, Y. Zhang, A. D. Panopoulos, L. Ma,K. Schluns, Q. Tian, S. S. Watowich, A. M. Jetten, and C. Dong. 2007. Essential
autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature448: 480–483.
36. Ciofani, M., A. Madar, C. Galan, M. Sellars, K. Mace, F. Pauli, A. Agarwal,W. Huang, C. N. Parkurst, M. Muratet, et al. 2012. A validated regulatory net-work for Th17 cell specification. Cell 151: 289–303.
37. Volpe, E., M. Touzot, N. Servant, M. A. Marloie-Provost, P. Hupe, E. Barillot,and V. Soumelis. 2009. Multiparametric analysis of cytokine-driven human Th17differentiation reveals a differential regulation of IL-17 and IL-22 production.Blood 114: 3610–3614.
38. Schraml, B. U., K. Hildner, W. Ise, W. L. Lee, W. A. Smith, B. Solomon,G. Sahota, J. Sim, R. Mukasa, S. Cemerski, et al. 2009. The AP-1 transcriptionfactor Batf controls TH17 differentiation. Nature 460: 405–409.
39. Valdez, P. A., P. J. Vithayathil, B. M. Janelsins, A. L. Shaffer, P. R. Williamson,and S. K. Datta. 2012. Prostaglandin E2 suppresses antifungal immunity byinhibiting interferon regulatory factor 4 function and interleukin-17 expressionin T cells. Immunity 36: 668–679.
40. Kamada, N., Y. G. Kim, H. P. Sham, B. A. Vallance, J. L. Puente, E. C. Martens,and G. Nunez. 2012. Regulated virulence controls the ability of a pathogen tocompete with the gut microbiota. Science 336: 1325–1329.
41. Rutz, S., R. Noubade, C. Eidenschenk, N. Ota, W. Zeng, Y. Zheng, J. Hackney,J. Ding, H. Singh, and W. Ouyang. 2011. Transcription factor c-Maf mediates theTGF-b-dependent suppression of IL-22 production in TH17 cells. Nat. Immunol.12: 1238–1245.
42. Duhen, T., R. Geiger, D. Jarrossay, A. Lanzavecchia, and F. Sallusto. 2009.Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 10: 857–863.
43. Dann, S. M., M. E. Spehlmann, D. C. Hammond, M. Iimura, K. Hase, L. J. Choi,E. Hanson, and L. Eckmann. 2008. IL-6-dependent mucosal protection preventsestablishment of a microbial niche for attaching/effacing lesion-forming entericbacterial pathogens. J. Immunol. 180: 6816–6826.
44. Guo, X., J. Qiu, T. Tu, X. Yang, L. Deng, R. A. Anders, L. Zhou, and Y. X. Fu.2014. Induction of innate lymphoid cell-derived interleukin-22 by the tran-scription factor STAT3 mediates protection against intestinal infection. Immunity40: 25–39.
45. Eberl, G., and D. R. Littman. 2004. Thymic origin of intestinal alphabeta T cellsrevealed by fate mapping of RORgt+ cells. Science 305: 248–251.
46. Raifer, H., A. J. Mahiny, N. Bollig, F. Petermann, A. Hellhund, K. Kellner,A. Guralnik, K. Reinhard, E. Bothur, M. Huber, et al. 2012. Unlike ab T cells,gd T cells, LTi cells and NKT cells do not require IRF4 for the production of IL-17A and IL-22. Eur. J. Immunol. 42: 3189–3201.
47. Neufert, C., G. Pickert, Y. Zheng, N. Wittkopf, M. Warntjen, A. Nikolaev,W. Ouyang, M. F. Neurath, and C. Becker. 2010. Activation of epithelial STAT3regulates intestinal homeostasis. Cell Cycle 9: 652–655.
48. Sugimoto, K., A. Ogawa, E. Mizoguchi, Y. Shimomura, A. Andoh, A. K. Bhan,R. S. Blumberg, R. J. Xavier, and A. Mizoguchi. 2008. IL-22 ameliorates in-testinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118:534–544.
49. Gibson, D. L., M. Montero, M. J. Ropeleski, K. S. Bergstrom, C. Ma, S. Ghosh,H. Merkens, J. Huang, L. E. Mansson, H. P. Sham, et al. 2010. Interleukin-11reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinalSTAT3 signaling. Gastroenterology 139: 1277–1288.
50. Darfeuille-Michaud, A., J. Boudeau, P. Bulois, C. Neut, A. L. Glasser,N. Barnich, M. A. Bringer, A. Swidsinski, L. Beaugerie, and J. F. Colombel.2004. High prevalence of adherent-invasive Escherichia coli associated withileal mucosa in Crohn’s disease. Gastroenterology 127: 412–421.
51. Sartor, R. B. 2008. Microbial influences in inflammatory bowel diseases. Gas-troenterology 134: 577–594.