Lipopolysaccharides (LPS, endotoxin) with different coreoligosaccharide glycoforms from the same species are com-mon, but diversity among glycoforms reported to date hasbeen related to terminal residues only. In this work, contraryto the variety of known LPS structures, we observed a 30%replacement of glucosamine to glucose in the middle of theouter core region of the Plesiomonas shigelloides serotypeO33:H3 (CNCTC 34/89) lipopolysaccharide. Such atypicalmodification within the same serotype raises questions about
Introduction
Plesiomonas shigelloides is a Gram-negative, rod-shapedbacterium of the Enterobacteriaceae family, which causesgastrointestinal and localized infections with diarrhea asthe main symptom. P. shigelloides is found in freshwaterand surface water in tropical and temperate climates,[1] buthas also been identified in the Nordic countries.[2] Out-breaks of human infections of P. shigelloides are most oftenassociated with seafood, especially uncooked shellfish orraw oysters.[3] Incidences of P. shigelloides infections are ir-regularly reported and there is probably an underestimationof the number of infected subjects.[3] P. shigelloides has beenranked third as a cause of traveler’s diarrhea in Japan[3] andthe majority of travelers returning to Japan with diarrhealillness were carrying P. shigelloides.[4]
The pathogenesis of P. shigelloides is yet not understoodin detail. A number of possible virulence factors from P.shigelloides have been described, including a cholera-liketoxin,[5] a thermostable and a thermolabile enterotoxin,[6]
[a] Department of Chemistry, Swedish University of AgriculturalSciences,P. O. Box 7015, 75007 Uppsala, SwedenE-mail: [email protected]://www.slu.se/kemi
[b] Ludwik Hirszfeld Institute of Immunology and ExperimentalTherapy, Polish Academy of Sciences,R. Weigla 12, 53-114 Wroclaw, PolandSupporting information for this article is available on theWWW under http://dx.doi.org/10.1002/ejoc.201301399.
the biosynthesis of core structures. The core oligosaccharidewas an undecasaccharide with a high level of O-acetylationon the residue linked to the O-specific polysaccharide. Thecore oligosaccharide, the O-specific polysaccharide, and thelinkage between them were determined by 1H and 13C NMRspectroscopy, mass spectrometry and chemical analysis. Thepresence of the O-specific polysaccharide on the bacterialcell surface was confirmed by 1H high-resolution magicangle spinning NMR spectroscopy.
a β-hemolysin[7] and a cytotoxic protein,[8] which forms acomplex with lipopolysaccharides (LPS) on the outer bacte-rial membrane.[8] LPSs are the main components of theouter membrane of the cell wall of Gram-negative bacteria,and in addition to a protective function, they are potentvirulence factors. The LPS consists of an O-specific poly-saccharide, a core oligosaccharide, and lipid A, which canall be involved in the pathogenic reaction, and are thusstudied to gain insight into the mechanisms of pathogen–host interactions. Only a few structures of LPSs from dif-ferent strains of P. shigelloides have been published to date,and they were recently reviewed.[9] The complete LPS pro-files of serotypes O54:H2,[10] O74:H5[11] and O37[12] havebeen characterized, as well as the core oligosaccharide ofserotype O13,[13] and the core oligosaccharide substitutedto a repeating unit of the O-specific polysaccharide of sero-types O17[14] and O1,[15] and the O-specific polysaccharideof serotype O51,[16] strains 22074 and 12254,[17] and strainAM36565.[18]
In this paper we report structural studies of the coreoligosaccharide and the O-specific polysaccharide from theP. shigelloides strain CNCTC 34/89 LPS, associated to theserotype O33:H3,[19] which is a clinical isolate collectedfrom a patient from Cuba. NMR spectroscopy, mass spec-trometry and chemical analysis were used for structural elu-cidation. The crude bacteria and native LPS were also in-vestigated by high-resolution magic angle spinning (HR-MAS) NMR spectroscopy to confirm the structure of theO-specific polysaccharide in situ.
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPER
Results and Discussion
Isolation of O-Specific Polysaccharide and CoreOligosaccharides
The LPS of P. shigelloides CNCTC 34/89 was isolated bythe hot phenol/water method. The yields of LPSH2O and
Figure 1. 1H NMR (A) and SDS-PAGE (B) analysis of P. shigello-ides O33 LPSs. 1H NMR spectra of the O-antigen of P. shigelloidesO33 in situ on intact bacteria, in LPSPhOH and as isolated polysac-charide (PSPhOH) are shown. NMR spectra of bacteria and LPSwere acquired by HR-MAS NMR and the CPMG experiment withpresaturation of the water peak during relaxation delay. The up-percase letters refer to designation of carbohydrate residues(Table 1). The signal marked by asterisk (*) indicates residualamounts of acetate. LPS isolated from two batches were analyzedby SDS-PAGE (8 μg LPS per line): Batch 1 LPSPhOH (a) andLPSH2O (b); batch 2 LPSPhOH (c) and LPSH2O (d). A 2.5% stackinggel and a 15% resolving gel were used and were visualized by thesilver staining method.
Figure 2. 1H NMR and ESI mass spectra of oligosaccharides of P. shigelloides O33 LPS. OSIII, OSII and OSI represent unsubstitutedcore oligosaccharide, core oligosaccharide substituted with one and with two RU, respectively. Spectra were acquired using NOESY withpresaturation. The uppercase letters refer to designation of carbohydrate residues (Table 1). M*1, N*1, O*1 and P*1 refer to the terminalRU of OSI. Charge-deconvoluted ESI mass spectra obtained in negative mode are displayed as insets, where labeled peaks representmonoisotopic masses.
LPSPhOH extracted from bacteria grown in 1.5 L flasks(batch 1) were 0.18 % and 0.5% of dry bacterial mass,respectively. In contrast to batch 1, the yields of LPSH2O
and LPSPhOH extracted from bacteria grown in a 9 L fer-mentor with optimized conditions (batch 2, see experimen-tal procedures) were 1.2 % and 0.01%. LPSH2O andLPSPhOH from both batches were analyzed by SDS-PAGE,showing fractions of different degree of polymerization(Figure 1). LPSH2O exhibited a higher degree of polymeriza-tion compared to LPSPhOH and LPS isolated from bacteriagrown in the fermentor showed a higher yield compared tobacteria grown in flasks.
Mild acidic hydrolysis of LPSPhOH (batch 1) and LPSH2O
(batch 2) and subsequent size-exclusion chromatography ofthe released poly- and oligosaccharides yielded four main
Scheme 1. Isolation, degradation and fractionation procedure of P.shigelloides O33 LPS. One repeating unit of the O-specific polysac-charide is marked by a circle (�), and the core is marked by asquare (�). Yields are calculated from the fraction above the ar-rows.
Structure Elucidation of a Bacterial Lipopolysaccharide
fractions: PS (yield 8% of LPSPhOH and 20% of LPSH2O),OSIH2O (yield 1 % of LPSH2O), OSIIH2O (yield 2% ofLPSH2O) and OSIIIPhOH/OSIIIH2O (yield 7% of LPSPhOH
and 2 % of LPSH2O). The fractions were analyzed by NMRspectroscopy (Figures 1 and 2) and ESI MS (Figure 2). ThePS fractions consisted of the core oligosaccharide substi-tuted with the O-specific polysaccharide, OSIH2O consistedof the core oligosaccharide substituted with two repeatingunits (RU) of the O-specific polysaccharide, OSIIH2O con-sisted of the core oligosaccharide substituted with one RU,and OSIIIPhOH and OSIIIH2O, which were identical, con-sisted of the unsubstituted core oligosaccharide (Scheme 1).
Structure Analysis of the O-Specific Polysaccharide
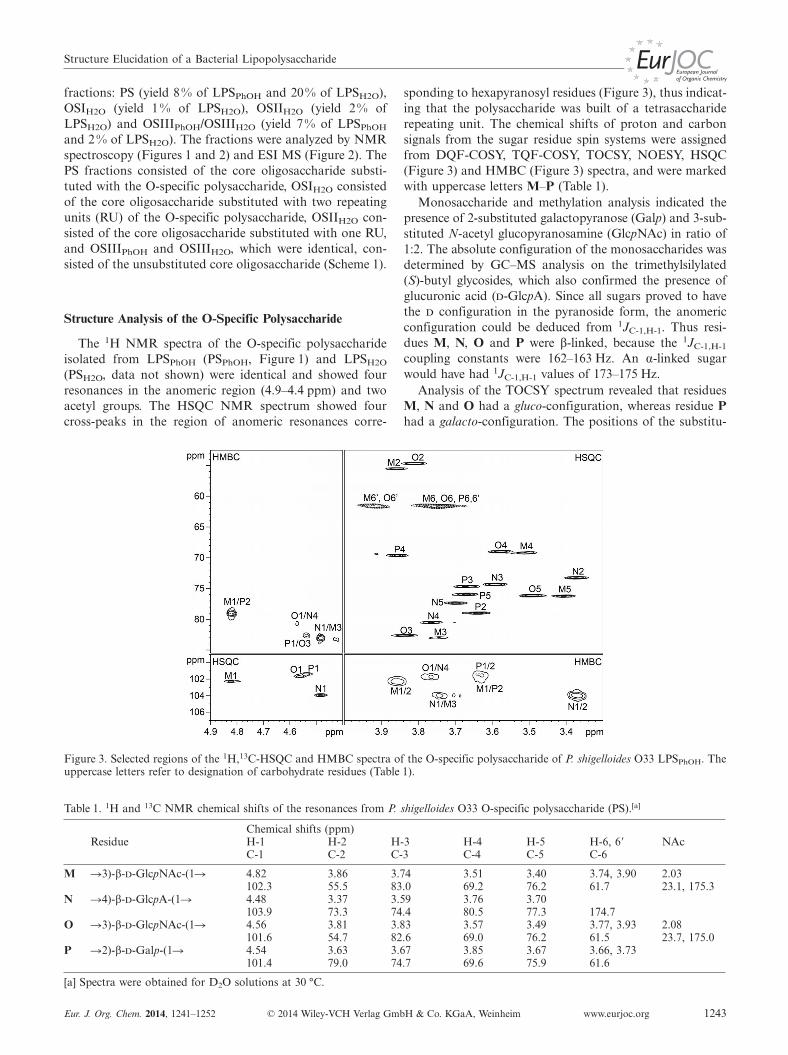
The 1H NMR spectra of the O-specific polysaccharideisolated from LPSPhOH (PSPhOH, Figure 1) and LPSH2O
(PSH2O, data not shown) were identical and showed fourresonances in the anomeric region (4.9–4.4 ppm) and twoacetyl groups. The HSQC NMR spectrum showed fourcross-peaks in the region of anomeric resonances corre-
Figure 3. Selected regions of the 1H,13C-HSQC and HMBC spectra of the O-specific polysaccharide of P. shigelloides O33 LPSPhOH. Theuppercase letters refer to designation of carbohydrate residues (Table 1).
Table 1. 1H and 13C NMR chemical shifts of the resonances from P. shigelloides O33 O-specific polysaccharide (PS).[a]
sponding to hexapyranosyl residues (Figure 3), thus indicat-ing that the polysaccharide was built of a tetrasacchariderepeating unit. The chemical shifts of proton and carbonsignals from the sugar residue spin systems were assignedfrom DQF-COSY, TQF-COSY, TOCSY, NOESY, HSQC(Figure 3) and HMBC (Figure 3) spectra, and were markedwith uppercase letters M–P (Table 1).
Monosaccharide and methylation analysis indicated thepresence of 2-substituted galactopyranose (Galp) and 3-sub-stituted N-acetyl glucopyranosamine (GlcpNAc) in ratio of1:2. The absolute configuration of the monosaccharides wasdetermined by GC–MS analysis on the trimethylsilylated(S)-butyl glycosides, which also confirmed the presence ofglucuronic acid (d-GlcpA). Since all sugars proved to havethe d configuration in the pyranoside form, the anomericconfiguration could be deduced from 1JC-1,H-1. Thus resi-dues M, N, O and P were β-linked, because the 1JC-1,H-1
coupling constants were 162–163 Hz. An α-linked sugarwould have had 1JC-1,H-1 values of 173–175 Hz.
Analysis of the TOCSY spectrum revealed that residuesM, N and O had a gluco-configuration, whereas residue Phad a galacto-configuration. The positions of the substitu-
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPERtion sites were identified by a downfield shift of the signalof the substituted carbon.
Residue M, which had H-1/C-1 signals at 4.82/102.3 ppm(1JC-1,H-1 = 162 Hz), was assigned as a 3-substituted β-d-GlcpNAc based on the upfield shift of the C-2 signal (δ =55.5 ppm), and the downfield shifts of the H-2 (δ =3.86 ppm) and C-3 (δ = 83.0 ppm) signals.
Residue O, which had H-1/C-1 signals at 4.56/101.6 ppm(1JC-1,H-1 = 163 Hz), was assigned, in analogy with residueM as a 3-substituted β-d-GlcpNAc based on the upfieldshift of the C-2 signal (δ = 54.7 ppm) and the downfieldshifts of the H-2 (δ = 3.81 ppm) and C-3 (δ = 82.6 ppm)signals. The assignments of the N-acetyl groups of the twoGlcNAc (M and O residues) were further supported bytwo- and three-bond heteronuclear connectivities in HMBCspectra between H-2 and C=O and between C=O andmethyl groups (Table 1).
Residue N, which had H-1/C-1 signals at 4.48/103.9 ppm(1JC-1,H-1 = 163 Hz), was assigned as a 4-substituted β-d-GlcpA based on the downfield shifts of C-4 (δ = 80.5 ppm)and C-6 (δ = 174.7 ppm), and the characteristic five-protonspin system with large vicinal couplings between all ringprotons.
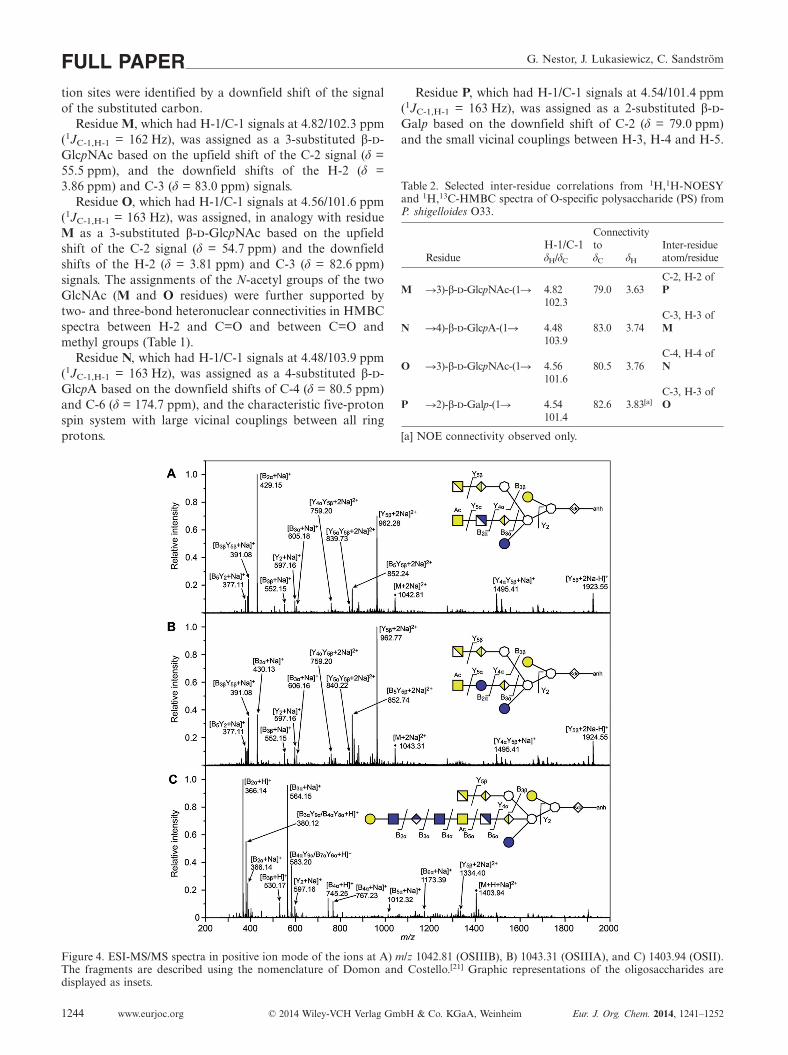
Figure 4. ESI-MS/MS spectra in positive ion mode of the ions at A) m/z 1042.81 (OSIIIB), B) 1043.31 (OSIIIA), and C) 1403.94 (OSII).The fragments are described using the nomenclature of Domon and Costello.[21] Graphic representations of the oligosaccharides aredisplayed as insets.
Residue P, which had H-1/C-1 signals at 4.54/101.4 ppm(1JC-1,H-1 = 163 Hz), was assigned as a 2-substituted β-d-Galp based on the downfield shift of C-2 (δ = 79.0 ppm)and the small vicinal couplings between H-3, H-4 and H-5.
Table 2. Selected inter-residue correlations from 1H,1H-NOESYand 1H,13C-HMBC spectra of O-specific polysaccharide (PS) fromP. shigelloides O33.
ConnectivityH-1/C-1 to Inter-residue
Residue δH/δC δC δH atom/residue
C-2, H-2 ofM �3)-β-d-GlcpNAc-(1� 4.82 79.0 3.63 P
102.3C-3, H-3 of
N �4)-β-d-GlcpA-(1� 4.48 83.0 3.74 M103.9
C-4, H-4 ofO �3)-β-d-GlcpNAc-(1� 4.56 80.5 3.76 N
101.6C-3, H-3 of
P �2)-β-d-Galp-(1� 4.54 82.6 3.83[a] O101.4
[a] NOE connectivity observed only.
Structure Elucidation of a Bacterial Lipopolysaccharide
The connections between the monosaccharides were de-termined from NOESY and HMBC experiments (Table 2).The following sequence of the O-specific polysaccharidewas thus determined: �2)-β-d-Galp-(1�3)-β-d-GlcpNAc-(1�4)-β-d-GlcpA-(1�3)-β-d-GlcpNAc-(1�.
The structure was supported further by comparing the1H and 13C chemical shifts of the polysaccharide with pre-dictions made by the CASPER program. All experimentaland predicted chemical shifts were similar within �0.2 ppmfor 1H signals and within �1.5 ppm for 13C signals, exceptC-4 of N (δ = 80.5 ppm, 82.1 ppm predicted), C-3 of O (δ= 82.6 ppm, 85.2 ppm predicted), and C-1 of P (δ =101.4 ppm, 103.3 ppm predicted). These carbons were lo-cated at the glycosidic linkages, and the deviations wereprobably due to conformational effects.
The mass of the O-specific polysaccharide RU was deter-mined by comparison between the mass of the core oligo-saccharide with (OSII) and without (OSIII) one RU (Fig-ure 2), and by MS/MS analysis of OSII (vide infra and Fig-ure 4C).
Possible alterations of the structure during mild acidichydrolysis and purification should always be considered,and therefore HR-MAS NMR spectroscopy was used toconfirm the identical appearance of the O-antigen structuredirectly on bacteria and in extracted LPS, compared topurified O-specific polysaccharide. The chemical shifts ofsignals from polysaccharide components were in agreementwith those for extracted LPS and bacterial O-antigen (Fig-ure 1). TOCSY experiments were run on bacteria and ex-tracted LPS (Figure S1) to distinguish between signals fromthe lipopolysaccharide and other components. Additionalsignals, originating mainly from lipid components in lipidA and the cell membrane, were reduced by T2 filtering usingthe CPMG experiment.
Structure Analysis of Core Oligosaccharide OSIII
Oligosaccharide OSIIIH2O, corresponding to the unsub-stituted core oligosaccharide, was studied in detail. Mono-saccharide and methylation analysis of the N-acetylatedcore oligosaccharide OSIII indicated the presence of 2,3,7-substituted, 3,4-substituted and 7-substituted l,d-Hepp, 6-substituted GlcpN, and Galp, Glcp, GalpN, and GalpNAc.Additionally, minor amounts of 3,7-substituted l,d-Heppand 6-substituted Glcp were also identified, which indicatescore oligosaccharide heterogeneity. Determination of theabsolute configuration showed that all sugars had the d
configuration.The spin systems and sequence of OSIIIH2O were solved
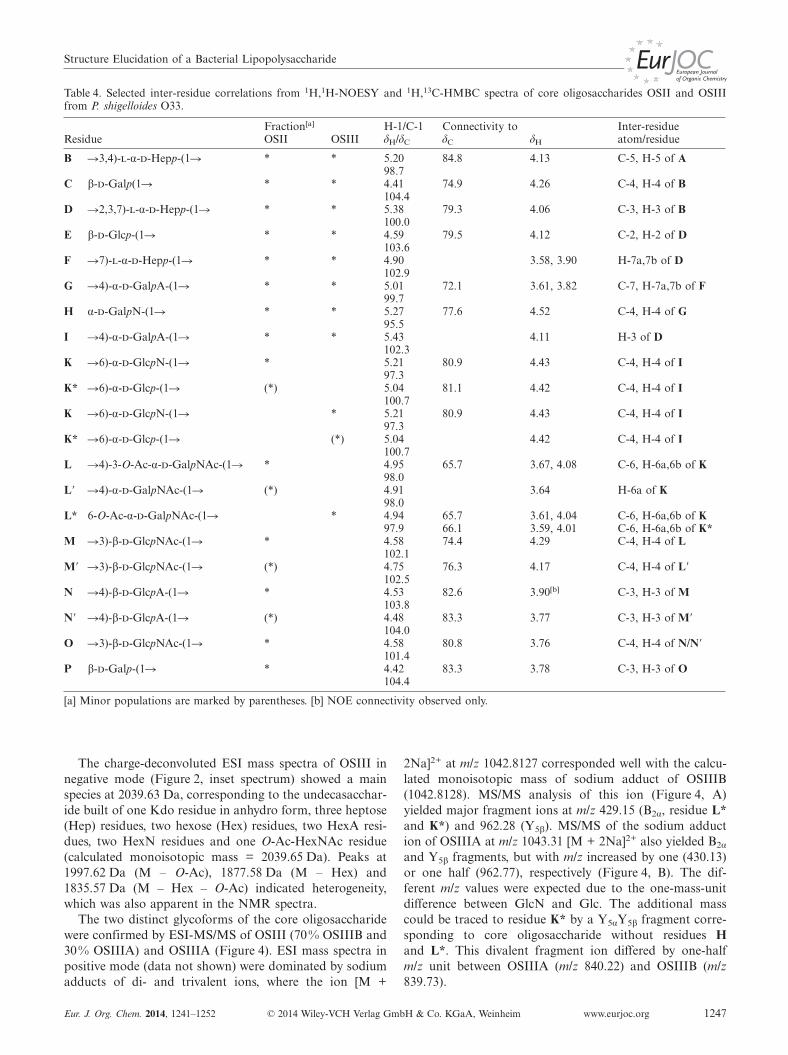
using a combination of 2D NMR spectra, and were markedwith uppercase letters (Table 3). The heterogeneous appear-ance of the OSIII fraction was due to: 1) different anhydroforms of Kdo (A), where 4,7-anhydro-Kdo is presented inTable 3, 2) partial lack of Glc E residue, 3) partial replace-ment of GlcN (K) by Glc (K*), 4) different positions of O-acetylation on residue L*/L**/L***, and 5) partial loss ofO-acetylation on residue L*. The chemical shifts in Table 3
are given for the most abundant form of each residue, al-though most residues occurred as a number of differentforms. The prevailing structure is shown in Scheme 2. Thedata were compared with a similar core oligosaccharideidentified previously for P. shigelloides serotype O17.[14a]
Since sugar residues A–K were the same as in the O17 coreoligosaccharide,[14a] only a distinctive outer core disac-charide element (residues K/K* and L*) and heterogeneityrelated to it is described in the text.
Scheme 2. Structure of the OSII and OSIII (A and B) oligosac-charides of P. shigelloides O33 LPS. The capital letters refer tocarbohydrate residues. Residue A–L represent the core oligosac-charide, whereas residue M–P represent the repeating unit of theO-specific polysaccharide, framed with a dashed line. Residue Ewas present in ca. 70% and residue K was replaced by �6)-α-Glc-(1� (K*) in ca. 30%.
Residue L*, which had H-1/C-1 signals at 4.94/97.9 ppm,was assigned as a terminal α-d-GalpNAc residue. It had a1JC-1,H-1 value of 173 Hz, which showed that the sugar hadthe α-pyranoside configuration, and from the TOCSY spec-tra a galacto configuration could be deduced. The charac-teristic upfield shift of the C-2 signal (δ = 50.5 ppm) anddownfield shift of the H-2 signal (δ = 4.20 ppm) indicatedN-acetylation at position 2. The N-acetyl group was furthersupported by connectivities in the HMBC spectra betweenH-2 and C=O (δ = 175.7 ppm) and between C=O and themethyl group (δ = 2.05/22.8 ppm). The H-6/H-6� and C-6signals of L* were shifted downfield to δ = 4.25/4.33 ppmand δ = 64.9 ppm, consistent with O-acetylation at position6. This was confirmed by a 3JC,H correlation between H-6and C=O of the O-acetyl group (δ =175.0 ppm) and a 2JC,H
correlation between C=O and the methyl group (δ = 2.11/21.1 ppm). The presence of an O-acetyl group was furthersupported by de-O-acetylation with ammonia, which pro-duced an oligosaccharide without the signals at δ = 2.11/21.1 ppm and δ = 4.25, 4.33/64.9 ppm (data not shown).Residue L* was thus identified as 6-O-Ac-α-d-GalpNAc.Small amounts (�10 %) of 4-O-Ac-α-d-GalpNAc (L**) and3-O-Ac-α-d-GalpNAc (L***) were also determined(Table 3).
Residue K, which had H-1/C-1 signals at δ = 5.21/97.3 ppm (1JC-1,H-1 = 172 Hz), was assigned as a 6-substi-tuted α-d-GlcpN, based on the upfield shifts of the H-2/C-
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPERTable 3. 1H and 13C NMR chemical shifts of the resonances from P. shigelloides O33 core oligosaccharide OSII and OSIII.[a]
[a] Spectra were obtained for D2O solutions at 30 °C. n.d.: not determined. [b] Minor populations are marked by parenthesis.
2 signals, the downfield shift of the C-6 signal and the largevicinal couplings between all ring protons.
Residue K*, which had H-1/C-1 signals at δ = 5.04/100.7 ppm (1JC-1,H-1 = 172 Hz), was assigned as a 6-substi-tuted α-d-Glcp, based on chemical shifts in good agreementwith those of previously reported 6-substituted α-Glcp.[20]
The glycosidic linkages between all sugar residues weredetermined from inter-residue NOEs and 3JC,H couplings(Table 4), and the OSIII structure shown in Scheme 2 waselucidated. The H-1/C-1 signals of residue K and K*showed 3JC,H couplings and NOE connectivities to H-4/C-
4 of I (δ = 4.43/80.9 ppm). HMBC and NOESY spectraalso showed connectivity between H-1/C-1 of L* and H-6/C-6 of K, and between H-1/C-1 of L* and H-6/C-6 of K*.
The K and K* residues indicated unusual heterogeneityof the core oligosaccharide. The presence of the two glyco-forms was further supported by separating them on aDowex 50WX4 column, due to their different numbers ofpositive charges at neutral pH. The first-eluting component(OSIIIA) was characterized as the core oligosaccharidecontaining K*, whereas a second component (OSIIIB) wascharacterized as the core oligosaccharide containing K.
Structure Elucidation of a Bacterial Lipopolysaccharide
Table 4. Selected inter-residue correlations from 1H,1H-NOESY and 1H,13C-HMBC spectra of core oligosaccharides OSII and OSIIIfrom P. shigelloides O33.
B �3,4)-l-α-d-Hepp-(1� * * 5.20 84.8 4.13 C-5, H-5 of A98.7
C β-d-Galp(1� * * 4.41 74.9 4.26 C-4, H-4 of B104.4
D �2,3,7)-l-α-d-Hepp-(1� * * 5.38 79.3 4.06 C-3, H-3 of B100.0
E β-d-Glcp-(1� * * 4.59 79.5 4.12 C-2, H-2 of D103.6
F �7)-l-α-d-Hepp-(1� * * 4.90 3.58, 3.90 H-7a,7b of D102.9
G �4)-α-d-GalpA-(1� * * 5.01 72.1 3.61, 3.82 C-7, H-7a,7b of F99.7
H α-d-GalpN-(1� * * 5.27 77.6 4.52 C-4, H-4 of G95.5
I �4)-α-d-GalpA-(1� * * 5.43 4.11 H-3 of D102.3
K �6)-α-d-GlcpN-(1� * 5.21 80.9 4.43 C-4, H-4 of I97.3
K* �6)-α-d-Glcp-(1� (*) 5.04 81.1 4.42 C-4, H-4 of I100.7
K �6)-α-d-GlcpN-(1� * 5.21 80.9 4.43 C-4, H-4 of I97.3
K* �6)-α-d-Glcp-(1� (*) 5.04 4.42 C-4, H-4 of I100.7
L �4)-3-O-Ac-α-d-GalpNAc-(1� * 4.95 65.7 3.67, 4.08 C-6, H-6a,6b of K98.0
L� �4)-α-d-GalpNAc-(1� (*) 4.91 3.64 H-6a of K98.0
L* 6-O-Ac-α-d-GalpNAc-(1� * 4.94 65.7 3.61, 4.04 C-6, H-6a,6b of K97.9 66.1 3.59, 4.01 C-6, H-6a,6b of K*
M �3)-β-d-GlcpNAc-(1� * 4.58 74.4 4.29 C-4, H-4 of L102.1
M� �3)-β-d-GlcpNAc-(1� (*) 4.75 76.3 4.17 C-4, H-4 of L�102.5
N �4)-β-d-GlcpA-(1� * 4.53 82.6 3.90[b] C-3, H-3 of M103.8
N� �4)-β-d-GlcpA-(1� (*) 4.48 83.3 3.77 C-3, H-3 of M�104.0
O �3)-β-d-GlcpNAc-(1� * 4.58 80.8 3.76 C-4, H-4 of N/N�101.4
P β-d-Galp-(1� * 4.42 83.3 3.78 C-3, H-3 of O104.4
[a] Minor populations are marked by parentheses. [b] NOE connectivity observed only.
The charge-deconvoluted ESI mass spectra of OSIII innegative mode (Figure 2, inset spectrum) showed a mainspecies at 2039.63 Da, corresponding to the undecasacchar-ide built of one Kdo residue in anhydro form, three heptose(Hep) residues, two hexose (Hex) residues, two HexA resi-dues, two HexN residues and one O-Ac-HexNAc residue(calculated monoisotopic mass = 2039.65 Da). Peaks at1997.62 Da (M – O-Ac), 1877.58 Da (M – Hex) and1835.57 Da (M – Hex – O-Ac) indicated heterogeneity,which was also apparent in the NMR spectra.
The two distinct glycoforms of the core oligosaccharidewere confirmed by ESI-MS/MS of OSIII (70% OSIIIB and30% OSIIIA) and OSIIIA (Figure 4). ESI mass spectra inpositive mode (data not shown) were dominated by sodiumadducts of di- and trivalent ions, where the ion [M +
2Na]2+ at m/z 1042.8127 corresponded well with the calcu-lated monoisotopic mass of sodium adduct of OSIIIB(1042.8128). MS/MS analysis of this ion (Figure 4, A)yielded major fragment ions at m/z 429.15 (B2α, residue L*and K*) and 962.28 (Y5β). MS/MS of the sodium adduction of OSIIIA at m/z 1043.31 [M + 2Na]2+ also yielded B2α
and Y5β fragments, but with m/z increased by one (430.13)or one half (962.77), respectively (Figure 4, B). The dif-ferent m/z values were expected due to the one-mass-unitdifference between GlcN and Glc. The additional masscould be traced to residue K* by a Y5αY5β fragment corre-sponding to core oligosaccharide without residues Hand L*. This divalent fragment ion differed by one-halfm/z unit between OSIIIA (m/z 840.22) and OSIIIB (m/z839.73).
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPERStructure Analysis of Oligosaccharide OSII
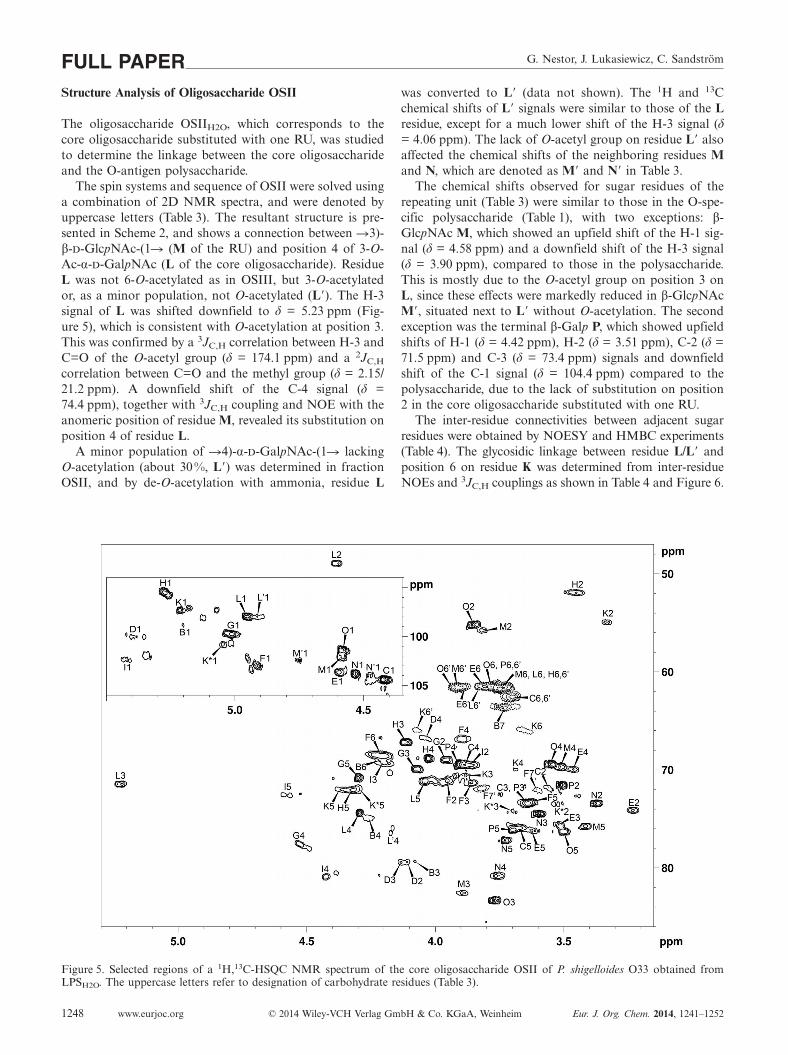
The oligosaccharide OSIIH2O, which corresponds to thecore oligosaccharide substituted with one RU, was studiedto determine the linkage between the core oligosaccharideand the O-antigen polysaccharide.
The spin systems and sequence of OSII were solved usinga combination of 2D NMR spectra, and were denoted byuppercase letters (Table 3). The resultant structure is pre-sented in Scheme 2, and shows a connection between �3)-β-d-GlcpNAc-(1� (M of the RU) and position 4 of 3-O-Ac-α-d-GalpNAc (L of the core oligosaccharide). ResidueL was not 6-O-acetylated as in OSIII, but 3-O-acetylatedor, as a minor population, not O-acetylated (L�). The H-3signal of L was shifted downfield to δ = 5.23 ppm (Fig-ure 5), which is consistent with O-acetylation at position 3.This was confirmed by a 3JC,H correlation between H-3 andC=O of the O-acetyl group (δ = 174.1 ppm) and a 2JC,H
correlation between C=O and the methyl group (δ = 2.15/21.2 ppm). A downfield shift of the C-4 signal (δ =74.4 ppm), together with 3JC,H coupling and NOE with theanomeric position of residue M, revealed its substitution onposition 4 of residue L.
A minor population of �4)-α-d-GalpNAc-(1� lackingO-acetylation (about 30 %, L�) was determined in fractionOSII, and by de-O-acetylation with ammonia, residue L
Figure 5. Selected regions of a 1H,13C-HSQC NMR spectrum of the core oligosaccharide OSII of P. shigelloides O33 obtained fromLPSH2O. The uppercase letters refer to designation of carbohydrate residues (Table 3).
was converted to L� (data not shown). The 1H and 13Cchemical shifts of L� signals were similar to those of the Lresidue, except for a much lower shift of the H-3 signal (δ= 4.06 ppm). The lack of O-acetyl group on residue L� alsoaffected the chemical shifts of the neighboring residues Mand N, which are denoted as M� and N� in Table 3.
The chemical shifts observed for sugar residues of therepeating unit (Table 3) were similar to those in the O-spe-cific polysaccharide (Table 1), with two exceptions: β-GlcpNAc M, which showed an upfield shift of the H-1 sig-nal (δ = 4.58 ppm) and a downfield shift of the H-3 signal(δ = 3.90 ppm), compared to those in the polysaccharide.This is mostly due to the O-acetyl group on position 3 onL, since these effects were markedly reduced in β-GlcpNAcM�, situated next to L� without O-acetylation. The secondexception was the terminal β-Galp P, which showed upfieldshifts of H-1 (δ = 4.42 ppm), H-2 (δ = 3.51 ppm), C-2 (δ =71.5 ppm) and C-3 (δ = 73.4 ppm) signals and downfieldshift of the C-1 signal (δ = 104.4 ppm) compared to thepolysaccharide, due to the lack of substitution on position2 in the core oligosaccharide substituted with one RU.
The inter-residue connectivities between adjacent sugarresidues were obtained by NOESY and HMBC experiments(Table 4). The glycosidic linkage between residue L/L� andposition 6 on residue K was determined from inter-residueNOEs and 3JC,H couplings as shown in Table 4 and Figure 6.
Structure Elucidation of a Bacterial Lipopolysaccharide
Figure 6. Selected regions of a 1H,1H-NOESY NMR spectrum ofthe core oligosaccharide OSII from P. shigelloides O33, with a mix-ing time of 50 ms. Different forms of the same sugar residues areframed with dashed lines.
The charge-deconvoluted ESI mass spectra in negativemode of OSII (Figure 2, inset spectrum) showed a mainspecies at 2783.88 Da, corresponding to one Kdo in anhy-dro form, three Hep, three Hex, three HexA, two HexN,one O-Ac-HexNAc and two HexNAc residues (calculatedmonoisotopic mass 2783.89 Da). The heterogeneous ap-pearance was indicated by peaks at 2741.87 Da (M –O-Ac), 2621.83 Da (M – Hex) and 2579.82 (M – Hex – O-Ac).
ESI mass spectra of OSII in positive mode were domi-nated by sodium adducts of di- and trivalent ions (data notshown). The mass of [M + Na + H]2+ (1403.9484) corre-sponded with the calculated monoisotopic mass(1403.9436) of the structure presented in Figure 4 (C). MS/MS analysis of this ion (Figure 4, C) yielded major frag-ments at m/z 366.14 (B2α) and 564.15 (B3α). The protonatedand monosodiated ions of the complete RU were also ob-served as the B4α fragments at m/z = 745.25 and 767.23,respectively. Thus, ions B2α, B3α and B4α indicated the se-quence at the non-reducing end. The ion at m/z 1173.39(B6α) resulted from fragmentation of the predominantglycoform with residue K (GlcN) as the constituent (coreoligosaccharide of OSIIIB type).
Different O-Acetylation Patterns of OSII and OSIII
The O-acetylation patterns of core oligosaccharide frac-tions of the P. shigelloides O33 serotype showed importantdifferences. The core oligosaccharide substituted with oneRU (OSII) showed the presence of 3-O-Ac-α-d-GalpNAc(residue L), whereas the unsubstituted core oligosaccharide(OSIII) contained a 6-O-Ac-α-d-GalpNAc residue (L*) andminor amounts (�10 %) of 3-O-Ac and 4-O-Ac forms.Without substitution on position 4 of GalNAc (OSIII), theO-acetyl group situated on position 3 could migrate to posi-tion 4 and further to position 6. This is supported by theminor amounts of 3- and 4-O-Ac isomers in OSIII and the
absence of 6-O-Ac form in OSII. Migrations of O-acetylgroups in core oligosaccharides have previously been ob-served by Knirel et al. for 2-O-Ac-Rha.[22] Studies on acylgroup migrations in β-galactopyranosides have shown a ra-pid migration at alkaline pH[23] and acidic pH[24] and aslower migration at neutral pH.[23] Thus, the migrationcould probably occur during the mild acidic hydrolysis withacetic acid, but migrations already on the bacterial cellmembrane of rough-type LPS cannot be excluded. Minorfractions of core oligosaccharides of P. shigelloides O33without O-acetylation were also found (about 30% of OSIIand OSIII), but are probably resulting from partial hydroly-sis of the O-acetyl group during the acidic hydrolysis oflipid A.
Conclusions
This is the first report of the partial replacement of asugar residue in the outer core region of a lipopolysac-charide within the same serotype. The presence of differentglycoforms from the same O-serotype has been previouslyreported for LPS isolated from P. shigelloides,[10a,11a,14a] butwith diversity limited to terminal residues only. Core oligo-saccharide heterogeneity is usually restricted to non-stoichiometric substitution of terminal residues and non-sugar substitutions, for example partial substitution byacetyl groups, phosphate groups, and amino acids. In thecase of LPS O33, a structural difference resulted from re-placement of one sugar in the middle of the outer core re-gion from �6)-α-d-GlcpN-(1� (residue K) to �6)-α-d-Glcp-(1� (residue K*) in ca. 30% abundance, keeping link-age positions untouched (Scheme 2). Such diversity has notbeen previously observed for P. shigelloides O-serotypes orfor any other species.
Structural diversity among the core oligosaccharides ofLPS is otherwise common in nature, and is typically relatedto a few core oligosaccharide types for one species.[25] Todate, six different core oligosaccharides of P. shigelloidesLPSs have been published: one from serotypes O54:H2[10a]
and O37,[12] one from O74:H5,[11a] one from O1,[15a] twofrom O17,[14] and one from O13.[13] The core oligosaccha-rides from serotypes O1 and O17 only differ in one or tworesidues. They all lack substitution by phosphate groups,which is present in most enterobacterial LPSs, but the nega-tive charge is instead provided by GalA.
The core oligosaccharide of the P. shigelloides O33(Scheme 2) represents a new core structure. It is similar tothat of P. shigelloides O17 reported by Kubler-Kielb etal.[14a] with residues A–K identical in the two LPSs. Resi-dues A–I were also identical to the LPS O13 core oligosac-charide,[13] residues A–F, I and K were found in the O54and O37 core oligosaccharide[10a,12] and residue A–G, I andK were found in another strain of the O17 LPS core oligo-saccharides[14b] and in the O1 LPS core oligosaccharide.[15a]
The characteristic appearance of the P. shigelloides O33core oligosaccharide was determined as a partial lack of
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPERGlc E in ca. 30 % and a high degree of O-acetylation onGalNAc L/L*, except for the partial replacement of GlcNK to Glc K*.
O-Acetylation of core oligosaccharides of P. shigelloidesLPSs has not been observed before, and is rare in mostGram-negative bacteria. Core oligosaccharides with O-acetyl groups have been characterized from Neisseriagonorrhoeae,[26] Neisseria meningitidis,[27] Haemophilus influ-enza,[28] Vibrio cholera,[29] Legionella pneumophila,[30] andPseudomonas aeruginosa.[31] 3-O-Ac-α-d-GalNAc has pre-viously not been found as a residue in core oligosac-charides, but 3-O-Ac-α-d-GlcNAc has been identified inlipooligosaccharides of Neisseria meningitidis.[27] In this me-ningococcal lipooligosaccharide core, O-acetylation hasbeen shown to have an important role in determining innercore assembly and immunotype expression.[27] Moreover,�4)-3-O-Ac-α-d-GalpNAc(1� is a part of the Haemo-philus influenza type f capsular polysaccharide.[32] Further-more, it is known that O-acetyl groups on polysaccharidesare strong immunodominant sites, thus O-acetyl modifica-tions may significantly influence antimicrobial activity ofanti-core oligosaccharide antibodies. It is important, be-cause conservative core oligosaccharide-based antigens arebelieved to be effective constituents of vaccines againstGram-negative bacterial infections.[33] This ability of bacte-ria to modify core oligosaccharides makes efforts in makingLPS-based antimicrobial vaccines more challenging.
Experimental SectionP. shigelloides: Strain CNCTC 34/89, classified as serovarO33:H3,[19] was obtained from the collection of the National Insti-tute of Public Health, Prague, Czech Republic. The bacteria weregrown in Davis medium in 5 L flasks (12�1.5 L, batch 1) or 10 Lfermentors (2� 9 L, batch 2), killed with 0.5% phenol, centrifugedusing a CEPA flow laboratory centrifuge, suspended in water, andfreeze-dried. The following parameters were kept for fermentor cul-ture: agitation of 200 rpm, gas flow of 5 L/min and a foam and pH7.0–6.8 control.
Purification of LPS: The LPSs were extracted from bacterial cellsby the hot phenol/water extraction and purified as previously re-ported.[11a] LPSs from water and phenol phases (LPSH2O,LPSPhOH) were collected and dialyzed extensively against de-ion-ized water and purified by ultracentrifugation (3�6 h, 105,000�g).
Mild Acid Hydrolysis of the LPS: The LPS from phenol phase(LPSPhOH, batch 1, 56 mg) and water phase (LPSH2O, batch 2,207 mg) were separately degraded by treatment with 1.5% aceticacid containing 2% SDS at 100 °C for 15 min.[34] After lyophiliza-tion, the SDS was removed by repeated extraction with ethanol,and the remaining residue was suspended in water and centrifuged.The supernatants were collected and lyophilized.
Separations of Poly- and Oligosaccharides: The LPS after mild acidhydrolysis was fractionated by size-exclusion chromatography ona Superdex75 prep grade HiLoad 16/60 column (GE Healthcare,Uppsala, Sweden), and yielded O-specific polysaccharides (PSPhOH,4.5 mg; PSH2O 42 mg) and oligosaccharides (OSPhOH, 8.1 mg;
OSH2O, 25 mg). The oligosaccharides were further fractionated bychromatography on a Superdex 30 prep grade HiLoad 16/60 col-umn (GE Healthcare, Uppsala, Sweden), yielding two core oligo-saccharides substituted by shorter chains of O-specific polysac-charide (OSIH2O, 2.4 mg; OSIIH2O, 3.5 mg) and unsubstituted coreoligosaccharides (OSIIIPhOH, 3.8 mg; OSIIIH2O, 5.0 mg). The sepa-rations were performed on an ÄKTA system consisting of a P-900pump, a UV-900 detector and Frac-900 fraction collector (Amer-sham Pharmacia Biotech, Uppsala, Sweden). Ammonium acetate(0.1 m) in water was used as a solvent (0.7 mL/ min) and the eluatewas monitored by UV detection at 230 and 255 nm. The fractionswere analyzed by electrospray ionization (ESI) mass spectrometry(MS) and NMR spectroscopy. Core oligosaccharide OSIIIH2O
(2.1 mg) was further fractionated on a Dowex 50WX4 cation ex-change column (10 cm�1 cm) in its H+ form. The flow rate was0.1 mL/ min and the column was initially eluted with water, yield-ing OSIIIA (0.7 mg). The eluent was changed to 1% acetic acidand then to 0.8 % TFA to yield OSIIIB (0.4 mg). The single frac-tions were analyzed by ESI-MS and the pooled fractions by NMRspectroscopy.
Analytical Procedures: The LPSs were analyzed by SDS-PAGE ac-cording to the method of Laemmli[35] with modifications as de-scribed previously,[36] and LPS bands were visualized by the silverstaining method.[37] Monosaccharides were analyzed as alditolacetates by GC–MS.[34,38] The absolute configuration of the sugarswas determined by GC–MS on the trimethylsilylated (S)-butylglycosides.[39] Partially methylated alditol acetates were preparedaccording to the method of Hakomori,[40] with modifications asdescribed previously.[34] GC–MS analysis was performed on a Hew-lett–Packard 5890/5970 system using a fused silica capillary column(0.25 mm�30 m, HP-5MS from Agilent) and a temperature pro-gram of 50�190 °C at 20 °C/min and 190�250 °C at 3 °C/min.
N-Acetylation of the Core Oligosaccharide: Core oligosaccharidesOSIIIPhOH and OSIIIH2O (0.3 mg) were N-acetylated with aceticanhydride (25 µL) in dry methanol at ambient temperature for onehour, prior to monosaccharide and methylation analysis.
De-O-Acetylation of the Core Oligosaccharide: Core oligosac-charide OSIIH2O and OSIIIH2O (0.8–3 mg) were treated with aque-ous 12.5% ammonia (1.5 mL) overnight at ambient temperature,followed by dilution and lyophilization. The product was analyzedby NMR spectroscopy.
Mass Spectrometry: ESI mass spectra were obtained in positive andnegative modes by using a Bruker maXis Impact Q-TOF massspectrometer. Samples were dissolved in methanol/H2O (1:1) andinjected via direct injection at a flow rate of 2 μL/min. Externalcalibration was obtained with Tunemix (Bruker Daltonics, Ger-many) in quadratic regression mode. MS/MS analysis was per-formed by collision-induced dissociation with a collision energy of50–60 eV.
NMR Spectroscopy: NMR spectra were recorded with Bruker600 MHz spectrometers using a 2.5-mm 1H/13C inverse detectionprobe, a 5 mm broadband observe detection SmartProbe and a5 mm 1H/13C/15N/31P inverse detection CryoProbe, all equippedwith z-gradient. NMR spectra of poly- and oligosaccharides wereobtained of D2O solutions at 30 °C. The residual HDO signal wassuppressed by presaturation during the recycle delay in one-dimen-sional NOESY-presaturation (noesygppr1d) and 1H,1H-TOCSYexperiments, and by excitation sculpting in 1H,1H-TOCSY and1H,1H-DQF-COSY experiments. The chemical shifts for NMR sig-nals were referenced by using acetone as an internal reference (δH =2.225 ppm, δC = 31.05 ppm). The 1H,1H-DQF-COSY, TQF-COSY,TOCSY, NOESY and ROESY spectra and the 1H,13C-HSQC with
Structure Elucidation of a Bacterial Lipopolysaccharide
and without proton decoupling, HMBC, HSQC-TOCSY andHSQC-NOESY spectra were recorded with 2 K to 8 K data pointsin t2 and 256 or 512 increments in t1, using a minimum of 16 scansper increment and a relaxation delay of 1.5–2.0 s. TOCSY andHSQC-TOCSY spectra were recorded with mixing times of 20–120 ms. NOESY, ROESY and HSQC-NOESY experiments wererun with mixing times (τm) ranging from 50 to 200 ms and the delaytime in HMBC spectra was set to 50–83 ms. The Bruker softwareTopspin 3.1 was used for processing. The data were zero-filled be-fore applying a π/4 or π/2 shifted sine-squared bell function in bothdimensions, except for HMBC spectra, where an exponential multi-plication of 3 Hz was applied. In some cases band-selective con-stant-time HMBC experiments[41] were run to improve the resolu-tion in the carbon dimension.
1H HR-MAS NMR spectra of bacteria and LPS suspensions wereobtained with a Bruker 600 MHz spectrometer using a 4 mm HR-MAS 1H/13C inverse detection probe, equipped with z gradient.Samples (4 mg of dried killed bacteria or 2 mg LPS) were sus-pended in 20 μL D2O and placed into ZrO2 rotors, which weresubjected to a spinning rate of 4 kHz at 30 °C. One-dimensionalspectra with water presaturation were recorded using the noes-ygppr1d pulse sequence. The water-suppressed spin-echo Carr–Pur-cell–Meiboom–Gill (CPMG) pulse sequence (relaxation delay-90-(t-180-t)n-acquire) was used to allow attenuation of broad signalsfrom macromolecules. A total delay time of 24–90 ms was used,and the water suppression was achieved by irradiation during therecycle delay of 5 s. 1H,1H-TOCSY experiments were run with amixing time of 60 ms.
Predictions of NMR Chemical Shifts: Predictions of 1H and 13Cchemical shifts of the polysaccharide were made by the use of theCASPER program.[42]
Supporting Information (see footnote on the first page of this arti-cle): Selected regions of TOCSY spectra from 1H HR-MAS NMRof bacteria, LPSPhOH, and isolated polysaccharide.
Acknowledgments
The Carl Tryggers Foundation is acknowledged for financial sup-port. Suresh Gohil is acknowledged for expert MS assistance.
[1] R. E. Levin, Food Biotechnol. 2008, 22, 189–202.[2] C. González-Rey, S. B. Svenson, L. Bravo, A. Siitonen, V. Pas-
quale, S. Dumontet, I. Ciznar, K. Krovacek, Comp. Immunol.Microbiol. Infect. Dis. 2004, 27, 129–139.
[3] I. Stock, Rev. Med. Microbiol. 2004, 15, 129–139.[4] M. Shigematsu, M. E. Kaufmann, A. Charlett, Y. Niho, T. L.
Pitt, Epidemiol. Infect. 2000, 125, 523–530.[5] S. E. Gardner, S. E. Fowlston, W. L. George, J. Infect. Dis.
1987, 156, 720–722.[6] C. L. Sears, J. B. Kaper, Microbiol. Rev. 1996, 60, 167–215.[7] J. M. Janda, S. L. Abbott, J. Clin. Microbiol. 1993, 31, 1206–
1208.[8] a) Y. Okawa, Y. Ohtomo, H. Tsugawa, Y. Matsuda, H. Kobaya-
shi, T. Tsukamoto, FEMS Microbiol. Lett. 2004, 239, 125–130;b) H. Tsugawa, A. Ogawa, S. Takehara, M. Kimura, Y. Okawa,FEMS Microbiol. Lett. 2008, 281, 10–16.
[9] E. L. Nazarenko, R. J. Crawford, E. P. Ivanova, Mar. Drugs2011, 9, 1914–1954.
[10] a) T. Niedziela, J. Lukasiewicz, W. Jachymek, M. Dzieciatkow-ska, C. Lugowski, L. Kenne, J. Biol. Chem. 2002, 277, 11653–11663; b) J. Czaja, W. Jachymek, T. Niedziela, C. Lugowski, E.
Aldova, L. Kenne, Eur. J. Biochem. 2000, 267, 1672–1679; c) J.Lukasiewicz, T. Niedziela, W. Jachymek, L. Kenne, C. Lugow-ski, Glycobiology 2006, 16, 538–550.
[11] a) T. Niedziela, S. Dag, J. Lukasiewicz, M. Dzieciatkowska,W. Jachymek, C. Lugowski, L. Kenne, Biochemistry 2006, 45,10422–10433; b) J. Lukasiewicz, M. Dzieciatkowska, T. Niedzi-ela, W. Jachymek, A. Augustyniuk, L. Kenne, C. Lugowski,Biochemistry 2006, 45, 10434–10447.
[12] M. Kaszowska, W. Jachymek, J. Lukasiewicz, T. Niedziela, L.Kenne, C. Lugowski, Carbohydr. Res. 2013, 378, 98–107.
[13] M. Kaszowska, W. Jachymek, T. Niedziela, S. Koj, L. Kenne,C. Lugowski, Carbohydr. Res. 2013, 380, 45–50.
[14] a) J. Kubler-Kielb, R. Schneerson, C. Mocca, E. Vinogradov,Carbohydr. Res. 2008, 343, 3123–3127; b) A. Maciejewska, J.Lukasiewicz, M. Kaszowska, A. Man-Kupisinska, W. Jachy-mek, C. Lugowski, Mar. Drugs 2013, 11, 440–454.
[15] a) G. Pieretti, S. Carillo, B. Lindner, R. Lanzetta, M. Parrilli,N. Jimenez, M. Regué, J. M. Tomás, M. M. Corsaro, Car-bohydr. Res. 2010, 345, 2523–2528; b) G. Pieretti, M. M.Corsaro, R. Lanzetta, M. Parrilli, R. Canals, S. Merino, J. M.Tomás, Eur. J. Org. Chem. 2008, 3149–3155; c) G. Pieretti,M. M. Corsaro, R. Lanzetta, M. Parrilli, S. Vilches, S. Merino,J. M. Tomás, Eur. J. Org. Chem. 2009, 1365–1371.
[16] A. Maciejewska, J. Lukasiewicz, T. Niedziela, Z. Szewczuk, C.Lugowski, Carbohydr. Res. 2009, 344, 894–900.
[17] M. Linnerborg, G. Widmalm, A. Weintraub, M. J. Albert, Eur.J. Biochem. 1995, 231, 839–844.
[18] E. Säwén, J. Östervall, C. Landersjö, M. Edblad, A. Weintraub,M. Ansaruzzaman, G. Widmalm, Carbohydr. Res. 2012, 348,99–103.
[19] T. Shimada, R. Sakazaki, Jpn. J. Med. Sci. Biol. 1985, 38, 73–76.
[20] B. Jann, A. S. Shashkov, D. S. Gupta, K. Jann, Eur. J. Biochem.1992, 210, 241–248.
[21] B. Domon, C. E. Costello, Glycoconjugate J. 1988, 5, 397–409.[22] Y. A. Knirel, H. Moll, U. Zähringer, Carbohydr. Res. 1996, 293,
223–234.[23] M. U. Roslund, O. Aitio, J. Wärnå, H. Maaheimo, D. Y. Mur-
zin, R. Leino, J. Am. Chem. Soc. 2008, 130, 8769–8772.[24] T. Horrobin, C. H. Tran, D. Crout, J. Chem. Soc. Perkin Trans.
1 1998, 1069–1080.[25] O. Holst, FEMS Microbiol. Lett. 2007, 271, 3–11.[26] C. M. John, J. M. Griffiss, M. A. Apicella, R. E. Mandrell,
B. W. Gibson, J. Biol. Chem. 1991, 266, 19303–19311.[27] C. M. Kahler, S. Lyons-Schindler, B. Choudhury, J. Glushka,
R. W. Carlson, D. S. Stephens, J. Biol. Chem. 2006, 281, 19939–19948.
[28] E. K. H. Schweda, J. R. Brisson, G. Alvelius, A. Martin, J. N.Weiser, D. W. Hood, E. R. Moxon, J. C. Richards, Eur. J. Bio-chem. 2000, 267, 3902–3913.
[29] Y. A. Knirel, S. N. Senchenkova, P. E. Jansson, A. Weintraub,Carbohydr. Res. 1998, 310, 117–119.
[30] O. Kooistra, E. Lüneberg, B. Lindner, Y. A. Knirel, M. Frosch,U. Zähringer, Biochemistry 2001, 40, 7630–7640.
[31] D. Kocincova, J. S. Lam, Biochemistry (Moscow) 2011, 76,755–760.
[32] a) W. Egan, F. P. Tsui, R. Schneerson, Carbohydr. Res. 1980,79, 271–277; b) P. Branefors-Helander, L. Kenne, B. Lindqvist,Carbohydr. Res. 1980, 79, 308–312.
[33] a) C. Lugowski, W. Jachymek, T. Niedziela, S. Rowinski,FEMS Immunol. Med. Microbiol. 1996, 16, 21–30; b) S.Müller-Loennies, L. Brade, H. Brade, Int. J. Med. Microbiol.2007, 297, 321–340.
[34] C. Petersson, T. Niedziela, W. Jachymek, L. Kenne, P. Zarzecki,C. Lugowski, Eur. J. Biochem. 1997, 244, 580–586.
[35] U. K. Laemmli, Nature 1970, 227, 680–685.[36] T. Niedziela, C. Petersson, A. Helander, W. Jachymek, L.
Kenne, C. Lugowski, Eur. J. Biochem. 1996, 237, 635–641.[37] C. M. Tsai, C. E. Frasch, Anal. Biochem. 1982, 119, 115–119.
G. Nestor, J. Lukasiewicz, C. SandströmFULL PAPER[38] J. S. Sawardeker, J. H. Sloneker, A. Jeanes, Anal. Chem. 1965,
37, 1602–1604.[39] a) G. J. Gerwig, J. P. Kamerling, J. F. G. Vliegenthart, Car-
bohydr. Res. 1978, 62, 349–357; b) G. J. Gerwig, J. P. Kamer-ling, J. F. G. Vliegenthart, Carbohydr. Res. 1979, 77, 1–7.
[40] S. I. Hakomori, J. Biochem. 1964, 55, 205–208.[41] T. D. W. Claridge, I. Pérez-Victoria, Org. Biomol. Chem. 2003,
[42] a) M. U. Roslund, E. Säwén, J. Landström, J. Rönnols,K. H. M. Jonsson, M. Lundborg, M. V. Svensson, G. Wid-malm, Carbohydr. Res. 2011, 346, 1311–1319; b) M. Lundborg,G. Widmalm, Anal. Chem. 2011, 83, 1514–1517.
Received: September 13, 2013Published Online: December 19, 2013