ORIGINAL RESEARCH Structural behavior of sugar radicals formed by proton transfer reaction of deoxycytidine cation radical: detailed view from NBO analysis Marjan Jebeli Javan • Zahra Aliakbar Tehrani • Alireza Fattahi Received: 25 June 2011 / Accepted: 27 December 2011 / Published online: 17 January 2012 Ó Springer Science+Business Media, LLC 2012 Abstract The cation radicals of DNA constituents gen- erated by the ionizing radiation initiate the alteration of the bases, which is one main type of cytotoxic DNA lesions. These cation radical spices are known for their role in producing nucleic acid strand break, and it is important to identify the cation radical formation at particular atomic site in these molecules so that the major pathway for the nucleic acid damage may be trapped. In the present study, we explored theoretically energetic, structural, and elec- tronic properties of the possible radicals formed via proton atom abstraction at various sites of sugar part of deoxy- cytidine cation radical by employing density functional theory at B3LYP/6-311??G (d,p) level. The computation revealed 0.0–22.6 kcal/mol energy disparity in these radi- cals. Radical-centered carbon increases the extent of bonding with its adjacent atoms. This tendency should be important in predicting the reactivity of sugar-based radi- cals. Based on DFT calculations, sugar radicals of deoxy- cytidine have following stability order: raH1 0 [ raH2 0 [ raH4 0 [ raH3 0 [ raH5 0 [ raO5 0 H [ raO3 0 H. Furthermore, influence of cation radical formation on acidities of mul- tiple sites in deoxycytidine nucleosides was investigated. For instance upon cation radical formation, DH acidity of O3 0 H and O5 0 H sites of deoxycytidine varies from 348.6 and 351.5 to 228.8 and 227.5 kcal/mol, respectively. Keywords Deoxycytidine Cation radical Hydrogen atom abstraction Sugar puckering mode NBO analysis Introduction In biological system, ionizing radiation causes several deleterious effects including reproductive cell death, mutagenesis and transformation. These affects are mainly dues to damages induced in cellular DNA, which is believed to be the prime target for the action of ionizing radiation [1–3]. Radiation damages in DNA and RNA occurs by direct or indirect action of high-energy photons or electrons on the nucleobase and, to a lesser extent, carbohydrate residues [4]. In the direct mechanism, the nucleobase is ionized by the radiation to form a cation radical [4, 5]. Carbon-centered neutral sugar radicals in the DNA deoxyribose backbone are known to lead to single strand- ing breaks in DNA. These lesions are among the most serious of DNA damages. Elucidation of the production and nature of DNA sugar radicals is, therefore, critical to understanding their damaging effects in DNA. The deoxyribose radical formed by hydrogen atom loss at the C1 0 position is known to result in an alkali-labile stranding break, whereas the C3 0 , C4 0 , and C5 0 sugar radicals can lead to frank strand breaks [6–8]. It was recently reported that irradiation of DNA by a high-energy Argon ion-beam [9] (high linear energy transfer, LET, radiation) produced a far greater yield of sugar radicals than was found by c-irradi- ation (a low LET radiation). Since these sugar radicals were formed predominantly along the ion track, where excitations and ionizations are in proximity, it was pro- posed that excited-state cation radicals could be the pre- cursors of the neutral sugar radicals [10, 11]. Shukla et al. [12] reported the formation of sugar radi- cals on photoexcitation of guanine cation radical (G •? ) in DNA. They proposed that excitation of guanine cation radical results in delocalization of a significant fraction of M. Jebeli Javan Z. Aliakbar Tehrani A. Fattahi (&) Department of Chemistry, Sharif University of Technology, P.O. Box 11365-9516, Tehran, Iran e-mail: [email protected]123 Struct Chem (2012) 23:1185–1192 DOI 10.1007/s11224-011-9942-5
Transcript
ORIGINAL RESEARCH
Structural behavior of sugar radicals formed by proton transferreaction of deoxycytidine cation radical: detailed view from NBOanalysis
Marjan Jebeli Javan • Zahra Aliakbar Tehrani •
Alireza Fattahi
Received: 25 June 2011 / Accepted: 27 December 2011 / Published online: 17 January 2012
� Springer Science+Business Media, LLC 2012
Abstract The cation radicals of DNA constituents gen-
erated by the ionizing radiation initiate the alteration of the
bases, which is one main type of cytotoxic DNA lesions.
These cation radical spices are known for their role in
producing nucleic acid strand break, and it is important to
identify the cation radical formation at particular atomic
site in these molecules so that the major pathway for the
nucleic acid damage may be trapped. In the present study,
we explored theoretically energetic, structural, and elec-
tronic properties of the possible radicals formed via proton
atom abstraction at various sites of sugar part of deoxy-
cytidine cation radical by employing density functional
theory at B3LYP/6-311??G (d,p) level. The computation
revealed 0.0–22.6 kcal/mol energy disparity in these radi-
cals. Radical-centered carbon increases the extent of
bonding with its adjacent atoms. This tendency should be
important in predicting the reactivity of sugar-based radi-
cals. Based on DFT calculations, sugar radicals of deoxy-
cytidine have following stability order: raH10[ raH20[raH40[ raH30[ raH50[ raO50H [ raO30H. Furthermore,
influence of cation radical formation on acidities of mul-
tiple sites in deoxycytidine nucleosides was investigated.
For instance upon cation radical formation, DHacidity of
O30H and O50H sites of deoxycytidine varies from 348.6
and 351.5 to 228.8 and 227.5 kcal/mol, respectively.
where mi is dihedral angles i.e., m0 = C40–O40–C10–C20,m1 = O40–C10–C20–C30, m2 = C10–C20–C30–C40, m3 = C20–C30–C40–O40, and m4 = O30–C40–O40–C10. (3) The glyco-
sidic bond connecting these units. The glycosyl torsion angle
v is defined as v = O40–C10–N1–C2 in pyrimidine nucleo-
sides and as v = O40–C10–N9–C4 in purine nucleosides,
allowing the orientation of the base with respect to the sugar
to be determined. (4) Rotation around the C40–C50 bond
leads to three possible conformers: ‘‘gauche-trans’’ (gt),
‘‘trans-gauche’’ (tg), and ‘‘gauche–gauche’’ (gg) (Scheme 2)
concerning the intramolecular hydrogen bonds involving
hydroxyl groups. Their orientations depend on the endo or
exo character of the ribose part and on the nature of the
possible interaction of hydroxyl group with the cytosine
heterocycle.
The first step was to identify the minima on the con-
formational potential energy surface for deoxycytidine and
its ionized form starting form several low-lying confor-
mations. The optimized structures of neutral deoxycyti-
dine, its cation radical form obtained at B3LYP/6-311??G
(d,p) level of theory are sketched in Fig. 1, which high-
lights the most interesting structural features. The lowest
energy conformer in neutral deoxycytidine is C20-endo/syn
which consists of two (1.834 A) O2…HO50 and
O40…HO50 (2.782 A) intramolecular hydrogen bonds. The
syn orientation of the base unit with respect to the sugar
unit is strongly stabilized by formation of the intramolec-
ular hydrogen bond with participation of the O2…HO50
group [44–51]. As seen in Fig. 1, the obvious geometrical
variations during cation radical formation involve struc-
tural features in hydrogen bonding. In fact cytosine base
unit should turn around the glycosyl linkage to make for-
mation of cation radical and consequently cleavage of
intramolecular O2…HO50 hydrogen bond in neutral
deoxycytidine molecule.
The adiabatic ionization energy (AIE or IEa) was pre-
dicted as the difference between the total energy of the
appropriate neutral and cation radical at their respective
optimized geometries (i.e., AIE = E cation radical - E neutral).
The B3LYP/6-311??G (d,p) adiabatic ionization energy of
the deoxycytidine (8.3 eV) exhibits substantial decrease as
compared to the cytosine nucleobase 8.6 eV [52]. The
decrease of the AIP from cytosine nucleobase to deoxycyt-
idine nucleoside amounts to 0.3 eV, demonstrating the
addition of deoxyribose to cytosine nucleobase decreases the
ionization potential. The relatively high ionization energy of
neutral deoxycytidine (8.3 eV) makes the cation radical a
reactive species for charge-transfer ionization of neutral
molecules as well as cytosine nucleobase.
Abstraction of proton at various sites of deoxycytidine
cation radical leads to neutral radicals which may capture
electrons, forming closed shell anions. To differentiate the
reactivity of radicals formed at deoxycytidine cation radi-
cal, a number of atomic sites are arbitrarily chosen for
generating radicals. Two types of free radicals may be
found by hydrogen abstraction: carbon centered and oxy-
gen centered radicals. For simplification, the following
notations are adopted in this article to better clarifying
geometries and energetic aspects of radicals: The carbon
and oxygen centers (i.e., C10–H, C20–H, C30–H, C40–H,
C50–H, O30–H and O50–H) chosen for generating radical on
H
OHH
H
C3 O
H
HHO
H
C3 O
OH
HH
H
C3 O
gt tg gg
Scheme 2 Definition of three possible rotamers about C40–C50 bond
Struct Chem (2012) 23:1185–1192 1187
123
deoxycytidine cation radical are simplified as raH10,raH20, raH30, raH40, raH50, raO30H, and raO50H,
respectively. For example, raH10 and raO20H radicals
show that proton abstraction occurred through C10 and O20
atoms of deoxycytidine cation radical, respectively.
Stability and energetic aspects of deoxycytidine sugar
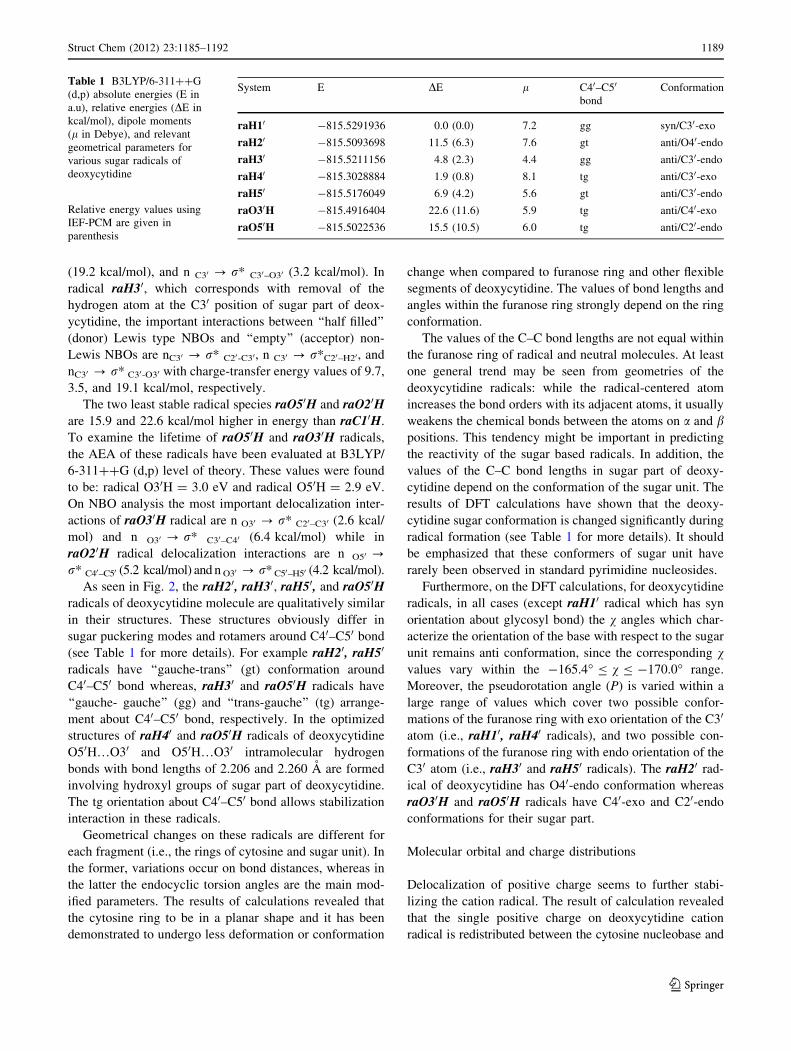
radicals
The optimized structures of the most stable conformers of
various radicals of deoxycytidine are depicted in Fig. 2
which highlights the most interesting structural features.
For an easier characterization of stability of radicals and
comparison geometrical changes after radical formation,
absolute energies (E in a.u), relative energies (DE in kcal/
mol), dipole moments (l in Debye), and relevant optimized
parameters for radicals of deoxycytidine were calculated at
B3LYP/6-311??G (d,p) level of theory which are shown
in Table 1. Bulk solvation effects were included in the
series of single-point energy calculations on the optimized
structures obtained from gas phase, through the integral
equation formalism of the polarized model (IEF-PCM)
[53]. The dielectric constant e = 78.4 was employed to
model aqueous solution. The aqueous solvation effects on
the stability order of these radicals are given in parentheses
as shown in Table 1. Results of calculation performed at
solution phase revealed that the stability order of deoxy-
cytidine radicals is the same in comparison with that in gas
phase. However, the energy gap and relative stability pre-
dicted by these methods are different.
As shown in Table 1, energies of deoxycytidine sugar
radicals are spread over a range of 23 kcal/mol. One gen-
eral trend is that radicals produced on the carbon center are
more stable than those produced on oxygen center. Struc-
ture raH10 from Fig. 2 is the most stable ribose radicals of
deoxycytidine from an energetic point of view. This radical
is stabilized by C = O…O20H hydrogen bond which sig-
nificantly weakened during radical formation as compared
with free deoxycytidine (for comparison see Figs. 1, 2).
As demonstrated by DE values reported in Table 1, for
deoxycytidine sugar radicals the energetic gap among the
most stable conformer (raH10) and structure raH40 is only
about 1.9 kcal/mol. Geometry optimization of raH40, leads
to the formation of O30H…O50 hydrogen bond with the
length 2.206 A. On DFT calculation, raH40 is followed by
raH30, raH50, and raH20 radicals. They are located at 4.8,
6.9, and 11.5 kcal/mol above raH10, respectively. It is
worth to mention that the C40 sugar radical (i.e., raH40) is
probably one of the most important DNA damaged radicals
due to its central role in the fabrication of detrimental
strand break.
The NBO analysis reveals that hyperconjugation inter-
action between the half-filled P orbital of the radical center
and the r* orbital of sugar or nucleoabse parts in radicals
can take place. For raH10 radical center of deoxycytidine
stabilizing interactions are as follow: n (1)C10 ? r* C20–H20
(4.3 kcal/mol), n (1)C10 ? r* C30–O30 (5.1 kcal/mol), and n
(1)C10 ? r* N1–C2 (6.0 kcal/mol). The most important
hyperconjugation interaction that stabilizes raH40 radical,
which corresponds with removal of the hydrogen atom at
the C40 position of sugar part of deoxycytidine, are n