Structural investigation of ribosomally synthesized natural products by hypothetical structure enumeration and evaluation using tandem MS Qi Zhang a,b , Manuel Ortega a,c , Yanxiang Shi a,b , Huan Wang a,b , Joel O. Melby b,d , Weixin Tang a,b , Douglas A. Mitchell b,d,e , and Wilfred A. van der Donk a,b,c,d,1 a Howard Hughes Medical Institute, Departments of b Chemistry, c Biochemistry, and e Microbiology, and d Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, IL 61801 Edited by John C. Vederas, University of Alberta, Edmonton, AB, Canada, and accepted by the Editorial Board July 9, 2014 (received for review April 8, 2014) Ribosomally synthesized and posttranslationally modified pepti- des (RiPPs) are a growing class of natural products that are found in all domains of life. These compounds possess vast structural diversity and have a wide range of biological activities, promising a fertile ground for exploring novel natural products. One challenging aspect of RiPP research is the difficulty of structure determination due to their architectural complexity. We here describe a method for automated structural characterization of RiPPs by tandem mass spectrometry. This method is based on the combined analysis of multiple mass spectra and evaluation of a collection of hypothetical structures predicted based on the bio- synthetic gene cluster and molecular weight. We show that this method is effective in structural characterization of complex RiPPs, including lanthipeptides, glycopeptides, and azole-containing pep- tides. Using this method, we have determined the structure of a previously structurally uncharacterized lanthipeptide, prochlorosin 1.2, and investigated the order of the posttranslational modifica- tions in three biosynthetic systems. dehydration | genome mining | lantibiotics | directionality R ibosomally synthesized and posttranslationally modified peptides (RiPPs) are a major class of natural products as revealed by the genome-sequencing efforts of the past decade (1). RiPPs are biosynthesized from genetically encoded and ribosomally produced precursor peptides, which typically consist of a core peptide that is transformed to the final product and an N-terminal extension called the leader peptide that is usually important for recognition by the posttranslational modification (PTM) enzymes (1). Because of the highly diverse PTMs, these compounds possess vast structural diversity and have a wide range of biological activities, thus representing a fertile ground for exploration. Furthermore, the ribosomal origin of RiPPs makes them particularly well suited for genome mining efforts. By using genome mining to explicitly avoid species harboring biosynthetic gene clusters identical to those that produce known compounds, a combination of strain prioritization and mass spectrometry (MS)-based analysis offers a new route to discov- ering natural products that can overcome the burden of redis- covery that has increasingly hampered discovery efforts (2, 3). One challenging aspect of high-throughput genome mining for new natural products is the difficulty to determine their molec- ular structures in high throughput. We present here a method that allows automated RiPP structure elucidation. In contrast to nonribosomal peptides that have an average molecular weight of less than 1,000 Da, as documented in the NORINE database (4), RiPPs in many cases have molecular weights larger than 2,500 Da. Molecules of this size are difficult to rapidly analyze by NMR spectroscopy, rendering MS the most convenient tool for RiPP structural characterization. Even when the precursor peptide sequences are known and the types of PTMs can be predicted based on the sequences of the bio- synthetic enzymes (5–8), multiple possible PTM sites on the precursor peptide typically result in a myriad of structures that are often difficult to differentiate. This challenge is further ex- acerbated by the frequent occurrence of one or more cross-links in RiPPs, which complicates traditional tandem MS-based struc- ture elucidation. One of the main difficulties is that the spectra only contain a small fraction of informative signals among a large number of less diagnostic signals that cloud spectrum inter- pretation. As the spectra often also vary significantly with dif- ferent instrument settings (9), selection of the most suitable spectra for drawing conclusions is time consuming and sometimes introduces bias. Indeed, a number of incorrect structural assign- ments of RiPPs have been reported based on insufficient in- formation content of tandem MS data (10–15). Here, we report use of hypothetical structure enumeration and evaluation (HSEE) for automated and unbiased interpretation of tandem MS data. The method is based on the prediction of a collection of hypothetical structures for a RiPP of certain mass and known biosynthetic information. By listing all of the theoretical daughter ions from this enumeration and automated evaluation of their matches with one or several experimental spectra, the most probable RiPP structure can be determined. We demonstrate here for multiple classes of known RiPPs with complex structures that HSEE is highly effective in analyzing tandem MS data and predicting the correct structure. In addition, we used HSEE to characterize a lanthipeptide whose structure was elusive despite Significance Ribosomally synthesized and posttranslationally modified pep- tides (RiPPs) constitute a promising repertoire of natural prod- ucts with potentially useful properties. Several tools have been developed in recent years to facilitate RiPP discovery and to connect genetic information with the chemical entities produced by microorganisms. Structure elucidation remains challenging for the RiPP field and is a bottleneck for genome mining and/or synthetic biology efforts to identify and characterize new RiPP members. This study presents a method for automated struc- tural analysis of RiPPs, which is based on enumeration of a col- lection of hypothetical structures predicted based on genomic information and evaluation of each structure with multiple tandem mass spectra. We show that this approach provides a powerful method for structural characterization of complex RiPPs and their biosynthetic intermediates. Author contributions: Q.Z. and W.A.v.d.D. designed research; Q.Z., M.O., Y.S., H.W., J.O.M., and W.T. performed research; Q.Z., D.A.M., and W.A.v.d.D. analyzed data; and Q.Z., D.A.M., and W.A.v.d.D. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. J.C.V. is a guest editor invited by the Editorial Board. 1 To whom correspondence should be addressed. Email: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1406418111/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1406418111 PNAS | August 19, 2014 | vol. 111 | no. 33 | 12031–12036 BIOCHEMISTRY Downloaded by guest on March 26, 2020
Transcript
Structural investigation of ribosomally synthesizednatural products by hypothetical structure enumerationand evaluation using tandem MSQi Zhanga,b, Manuel Ortegaa,c, Yanxiang Shia,b, Huan Wanga,b, Joel O. Melbyb,d, Weixin Tanga,b,Douglas A. Mitchellb,d,e, and Wilfred A. van der Donka,b,c,d,1
aHoward Hughes Medical Institute, Departments of bChemistry, cBiochemistry, and eMicrobiology, and dInstitute for Genomic Biology, University of Illinois atUrbana–Champaign, Urbana, IL 61801
Edited by John C. Vederas, University of Alberta, Edmonton, AB, Canada, and accepted by the Editorial Board July 9, 2014 (received for review April 8, 2014)
Ribosomally synthesized and posttranslationally modified pepti-des (RiPPs) are a growing class of natural products that are foundin all domains of life. These compounds possess vast structuraldiversity and have a wide range of biological activities, promisinga fertile ground for exploring novel natural products. Onechallenging aspect of RiPP research is the difficulty of structuredetermination due to their architectural complexity. We heredescribe a method for automated structural characterization ofRiPPs by tandem mass spectrometry. This method is based on thecombined analysis of multiple mass spectra and evaluation of acollection of hypothetical structures predicted based on the bio-synthetic gene cluster and molecular weight. We show that thismethod is effective in structural characterization of complex RiPPs,including lanthipeptides, glycopeptides, and azole-containing pep-tides. Using this method, we have determined the structure of apreviously structurally uncharacterized lanthipeptide, prochlorosin1.2, and investigated the order of the posttranslational modifica-tions in three biosynthetic systems.
Ribosomally synthesized and posttranslationally modifiedpeptides (RiPPs) are a major class of natural products as
revealed by the genome-sequencing efforts of the past decade(1). RiPPs are biosynthesized from genetically encoded andribosomally produced precursor peptides, which typically consistof a core peptide that is transformed to the final product and anN-terminal extension called the leader peptide that is usuallyimportant for recognition by the posttranslational modification(PTM) enzymes (1). Because of the highly diverse PTMs, thesecompounds possess vast structural diversity and have a widerange of biological activities, thus representing a fertile groundfor exploration. Furthermore, the ribosomal origin of RiPPsmakes them particularly well suited for genome mining efforts.By using genome mining to explicitly avoid species harboringbiosynthetic gene clusters identical to those that produce knowncompounds, a combination of strain prioritization and massspectrometry (MS)-based analysis offers a new route to discov-ering natural products that can overcome the burden of redis-covery that has increasingly hampered discovery efforts (2, 3).One challenging aspect of high-throughput genome mining fornew natural products is the difficulty to determine their molec-ular structures in high throughput. We present here a methodthat allows automated RiPP structure elucidation.In contrast to nonribosomal peptides that have an average
molecular weight of less than 1,000 Da, as documented in theNORINE database (4), RiPPs in many cases have molecularweights larger than 2,500 Da. Molecules of this size are difficultto rapidly analyze by NMR spectroscopy, rendering MS the mostconvenient tool for RiPP structural characterization. Even whenthe precursor peptide sequences are known and the types ofPTMs can be predicted based on the sequences of the bio-synthetic enzymes (5–8), multiple possible PTM sites on the
precursor peptide typically result in a myriad of structures thatare often difficult to differentiate. This challenge is further ex-acerbated by the frequent occurrence of one or more cross-linksin RiPPs, which complicates traditional tandem MS-based struc-ture elucidation. One of the main difficulties is that the spectraonly contain a small fraction of informative signals among a largenumber of less diagnostic signals that cloud spectrum inter-pretation. As the spectra often also vary significantly with dif-ferent instrument settings (9), selection of the most suitablespectra for drawing conclusions is time consuming and sometimesintroduces bias. Indeed, a number of incorrect structural assign-ments of RiPPs have been reported based on insufficient in-formation content of tandem MS data (10–15). Here, we reportuse of hypothetical structure enumeration and evaluation(HSEE) for automated and unbiased interpretation of tandemMS data. The method is based on the prediction of a collection ofhypothetical structures for a RiPP of certain mass and knownbiosynthetic information. By listing all of the theoretical daughterions from this enumeration and automated evaluation of theirmatches with one or several experimental spectra, the mostprobable RiPP structure can be determined. We demonstratehere for multiple classes of known RiPPs with complex structuresthat HSEE is highly effective in analyzing tandem MS data andpredicting the correct structure. In addition, we used HSEE tocharacterize a lanthipeptide whose structure was elusive despite
Significance
Ribosomally synthesized and posttranslationally modified pep-tides (RiPPs) constitute a promising repertoire of natural prod-ucts with potentially useful properties. Several tools have beendeveloped in recent years to facilitate RiPP discovery and toconnect genetic information with the chemical entities producedby microorganisms. Structure elucidation remains challengingfor the RiPP field and is a bottleneck for genome mining and/orsynthetic biology efforts to identify and characterize new RiPPmembers. This study presents a method for automated struc-tural analysis of RiPPs, which is based on enumeration of a col-lection of hypothetical structures predicted based on genomicinformation and evaluation of each structure with multipletandem mass spectra. We show that this approach providesa powerful method for structural characterization of complexRiPPs and their biosynthetic intermediates.
Author contributions: Q.Z. andW.A.v.d.D. designed research; Q.Z., M.O., Y.S., H.W., J.O.M.,andW.T. performed research; Q.Z., D.A.M., and W.A.v.d.D. analyzed data; and Q.Z., D.A.M.,and W.A.v.d.D. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.C.V. is a guest editor invited by the EditorialBoard.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406418111/-/DCSupplemental.
our previous efforts, and to determine the directionality of thia-zole-forming enzymes and lanthipeptide synthetases.
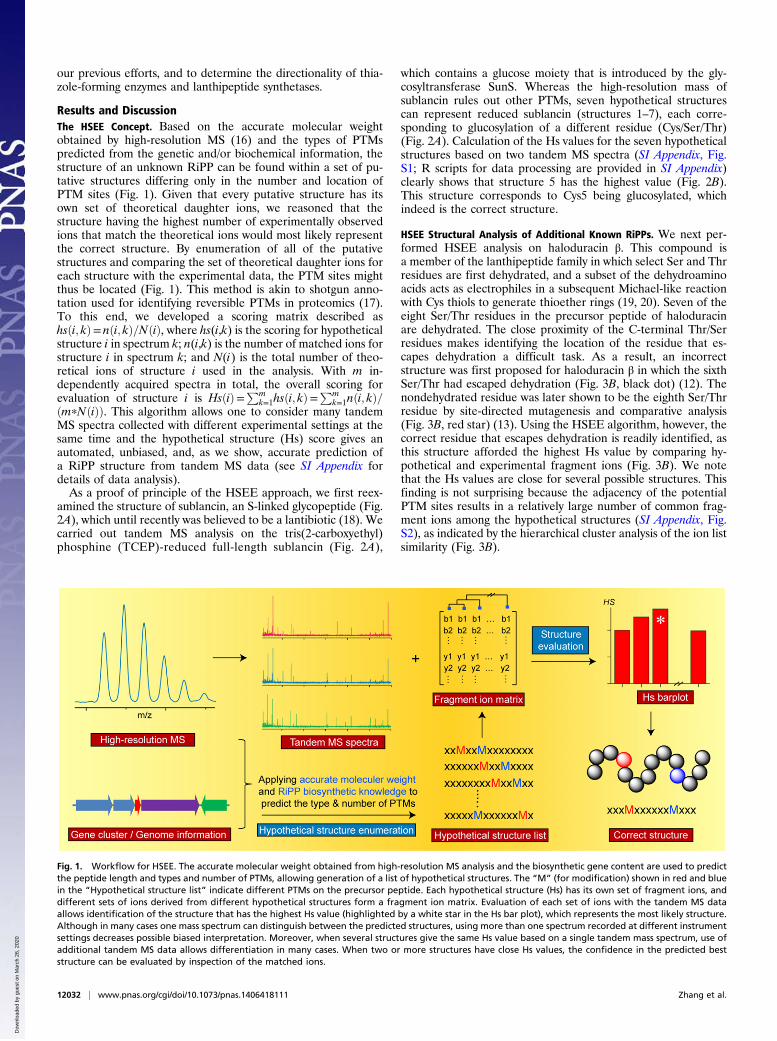
Results and DiscussionThe HSEE Concept. Based on the accurate molecular weightobtained by high-resolution MS (16) and the types of PTMspredicted from the genetic and/or biochemical information, thestructure of an unknown RiPP can be found within a set of pu-tative structures differing only in the number and location ofPTM sites (Fig. 1). Given that every putative structure has itsown set of theoretical daughter ions, we reasoned that thestructure having the highest number of experimentally observedions that match the theoretical ions would most likely representthe correct structure. By enumeration of all of the putativestructures and comparing the set of theoretical daughter ions foreach structure with the experimental data, the PTM sites mightthus be located (Fig. 1). This method is akin to shotgun anno-tation used for identifying reversible PTMs in proteomics (17).To this end, we developed a scoring matrix described ashsði; kÞ= nði; kÞ=NðiÞ, where hs(i,k) is the scoring for hypotheticalstructure i in spectrum k; n(i,k) is the number of matched ions forstructure i in spectrum k; and N(i) is the total number of theo-retical ions of structure i used in the analysis. With m in-dependently acquired spectra in total, the overall scoring forevaluation of structure i is HsðiÞ=Pm
k=1hsði; kÞ=Pm
k=1nði; kÞ=ðmpNðiÞÞ. This algorithm allows one to consider many tandemMS spectra collected with different experimental settings at thesame time and the hypothetical structure (Hs) score gives anautomated, unbiased, and, as we show, accurate prediction ofa RiPP structure from tandem MS data (see SI Appendix fordetails of data analysis).As a proof of principle of the HSEE approach, we first reex-
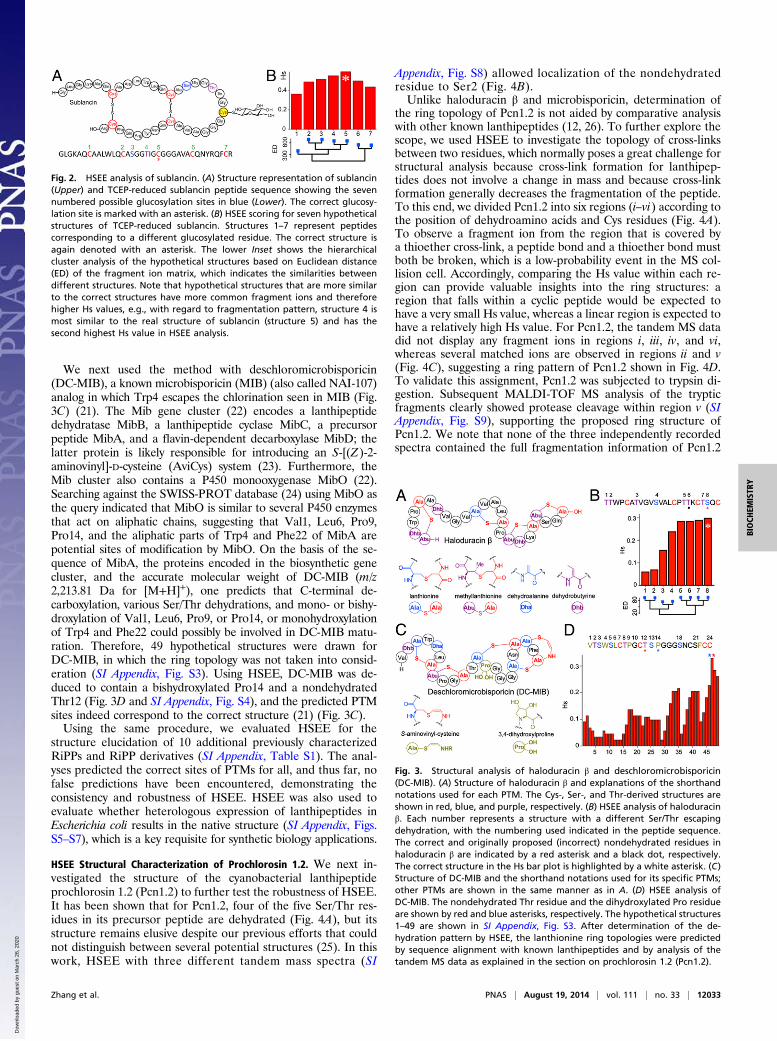
amined the structure of sublancin, an S-linked glycopeptide (Fig.2A), which until recently was believed to be a lantibiotic (18). Wecarried out tandem MS analysis on the tris(2-carboxyethyl)phosphine (TCEP)-reduced full-length sublancin (Fig. 2A),
which contains a glucose moiety that is introduced by the gly-cosyltransferase SunS. Whereas the high-resolution mass ofsublancin rules out other PTMs, seven hypothetical structurescan represent reduced sublancin (structures 1–7), each corre-sponding to glucosylation of a different residue (Cys/Ser/Thr)(Fig. 2A). Calculation of the Hs values for the seven hypotheticalstructures based on two tandem MS spectra (SI Appendix, Fig.S1; R scripts for data processing are provided in SI Appendix)clearly shows that structure 5 has the highest value (Fig. 2B).This structure corresponds to Cys5 being glucosylated, whichindeed is the correct structure.
HSEE Structural Analysis of Additional Known RiPPs. We next per-formed HSEE analysis on haloduracin β. This compound isa member of the lanthipeptide family in which select Ser and Thrresidues are first dehydrated, and a subset of the dehydroaminoacids acts as electrophiles in a subsequent Michael-like reactionwith Cys thiols to generate thioether rings (19, 20). Seven of theeight Ser/Thr residues in the precursor peptide of haloduracinare dehydrated. The close proximity of the C-terminal Thr/Serresidues makes identifying the location of the residue that es-capes dehydration a difficult task. As a result, an incorrectstructure was first proposed for haloduracin β in which the sixthSer/Thr had escaped dehydration (Fig. 3B, black dot) (12). Thenondehydrated residue was later shown to be the eighth Ser/Thrresidue by site-directed mutagenesis and comparative analysis(Fig. 3B, red star) (13). Using the HSEE algorithm, however, thecorrect residue that escapes dehydration is readily identified, asthis structure afforded the highest Hs value by comparing hy-pothetical and experimental fragment ions (Fig. 3B). We notethat the Hs values are close for several possible structures. Thisfinding is not surprising because the adjacency of the potentialPTM sites results in a relatively large number of common frag-ment ions among the hypothetical structures (SI Appendix, Fig.S2), as indicated by the hierarchical cluster analysis of the ion listsimilarity (Fig. 3B).
Fig. 1. Workflow for HSEE. The accurate molecular weight obtained from high-resolution MS analysis and the biosynthetic gene content are used to predictthe peptide length and types and number of PTMs, allowing generation of a list of hypothetical structures. The “M” (for modification) shown in red and bluein the “Hypothetical structure list” indicate different PTMs on the precursor peptide. Each hypothetical structure (Hs) has its own set of fragment ions, anddifferent sets of ions derived from different hypothetical structures form a fragment ion matrix. Evaluation of each set of ions with the tandem MS dataallows identification of the structure that has the highest Hs value (highlighted by a white star in the Hs bar plot), which represents the most likely structure.Although in many cases one mass spectrum can distinguish between the predicted structures, using more than one spectrum recorded at different instrumentsettings decreases possible biased interpretation. Moreover, when several structures give the same Hs value based on a single tandem mass spectrum, use ofadditional tandem MS data allows differentiation in many cases. When two or more structures have close Hs values, the confidence in the predicted beststructure can be evaluated by inspection of the matched ions.
12032 | www.pnas.org/cgi/doi/10.1073/pnas.1406418111 Zhang et al.
We next used the method with deschloromicrobisporicin(DC-MIB), a known microbisporicin (MIB) (also called NAI-107)analog in which Trp4 escapes the chlorination seen in MIB (Fig.3C) (21). The Mib gene cluster (22) encodes a lanthipeptidedehydratase MibB, a lanthipeptide cyclase MibC, a precursorpeptide MibA, and a flavin-dependent decarboxylase MibD; thelatter protein is likely responsible for introducing an S-[(Z)-2-aminovinyl]-D-cysteine (AviCys) system (23). Furthermore, theMib cluster also contains a P450 monooxygenase MibO (22).Searching against the SWISS-PROT database (24) using MibO asthe query indicated that MibO is similar to several P450 enzymesthat act on aliphatic chains, suggesting that Val1, Leu6, Pro9,Pro14, and the aliphatic parts of Trp4 and Phe22 of MibA arepotential sites of modification by MibO. On the basis of the se-quence of MibA, the proteins encoded in the biosynthetic genecluster, and the accurate molecular weight of DC-MIB (m/z2,213.81 Da for [M+H]+), one predicts that C-terminal de-carboxylation, various Ser/Thr dehydrations, and mono- or bishy-droxylation of Val1, Leu6, Pro9, or Pro14, or monohydroxylationof Trp4 and Phe22 could possibly be involved in DC-MIB matu-ration. Therefore, 49 hypothetical structures were drawn forDC-MIB, in which the ring topology was not taken into consid-eration (SI Appendix, Fig. S3). Using HSEE, DC-MIB was de-duced to contain a bishydroxylated Pro14 and a nondehydratedThr12 (Fig. 3D and SI Appendix, Fig. S4), and the predicted PTMsites indeed correspond to the correct structure (21) (Fig. 3C).Using the same procedure, we evaluated HSEE for the
structure elucidation of 10 additional previously characterizedRiPPs and RiPP derivatives (SI Appendix, Table S1). The anal-yses predicted the correct sites of PTMs for all, and thus far, nofalse predictions have been encountered, demonstrating theconsistency and robustness of HSEE. HSEE was also used toevaluate whether heterologous expression of lanthipeptides inEscherichia coli results in the native structure (SI Appendix, Figs.S5–S7), which is a key requisite for synthetic biology applications.
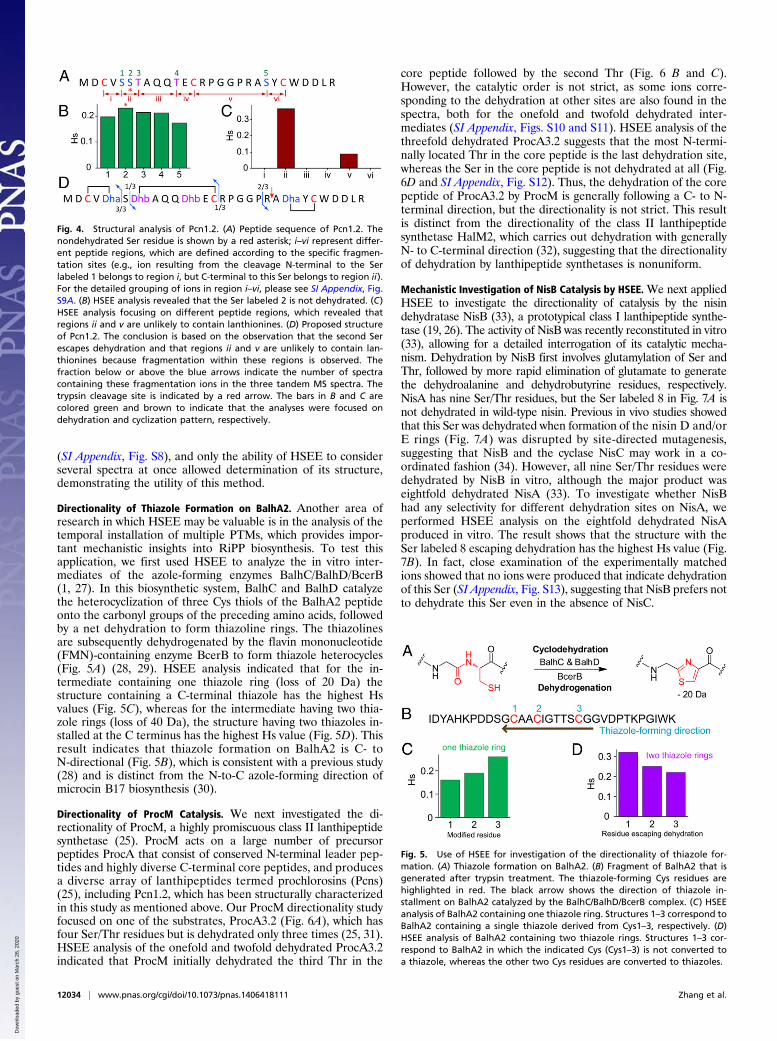
HSEE Structural Characterization of Prochlorosin 1.2. We next in-vestigated the structure of the cyanobacterial lanthipeptideprochlorosin 1.2 (Pcn1.2) to further test the robustness of HSEE.It has been shown that for Pcn1.2, four of the five Ser/Thr res-idues in its precursor peptide are dehydrated (Fig. 4A), but itsstructure remains elusive despite our previous efforts that couldnot distinguish between several potential structures (25). In thiswork, HSEE with three different tandem mass spectra (SI
Appendix, Fig. S8) allowed localization of the nondehydratedresidue to Ser2 (Fig. 4B).Unlike haloduracin β and microbisporicin, determination of
the ring topology of Pcn1.2 is not aided by comparative analysiswith other known lanthipeptides (12, 26). To further explore thescope, we used HSEE to investigate the topology of cross-linksbetween two residues, which normally poses a great challenge forstructural analysis because cross-link formation for lanthipep-tides does not involve a change in mass and because cross-linkformation generally decreases the fragmentation of the peptide.To this end, we divided Pcn1.2 into six regions (i–vi) according tothe position of dehydroamino acids and Cys residues (Fig. 4A).To observe a fragment ion from the region that is covered bya thioether cross-link, a peptide bond and a thioether bond mustboth be broken, which is a low-probability event in the MS col-lision cell. Accordingly, comparing the Hs value within each re-gion can provide valuable insights into the ring structures: aregion that falls within a cyclic peptide would be expected tohave a very small Hs value, whereas a linear region is expected tohave a relatively high Hs value. For Pcn1.2, the tandem MS datadid not display any fragment ions in regions i, iii, iv, and vi,whereas several matched ions are observed in regions ii and v(Fig. 4C), suggesting a ring pattern of Pcn1.2 shown in Fig. 4D.To validate this assignment, Pcn1.2 was subjected to trypsin di-gestion. Subsequent MALDI-TOF MS analysis of the trypticfragments clearly showed protease cleavage within region v (SIAppendix, Fig. S9), supporting the proposed ring structure ofPcn1.2. We note that none of the three independently recordedspectra contained the full fragmentation information of Pcn1.2
Fig. 2. HSEE analysis of sublancin. (A) Structure representation of sublancin(Upper) and TCEP-reduced sublancin peptide sequence showing the sevennumbered possible glucosylation sites in blue (Lower). The correct glucosy-lation site is marked with an asterisk. (B) HSEE scoring for seven hypotheticalstructures of TCEP-reduced sublancin. Structures 1–7 represent peptidescorresponding to a different glucosylated residue. The correct structure isagain denoted with an asterisk. The lower Inset shows the hierarchicalcluster analysis of the hypothetical structures based on Euclidean distance(ED) of the fragment ion matrix, which indicates the similarities betweendifferent structures. Note that hypothetical structures that are more similarto the correct structures have more common fragment ions and thereforehigher Hs values, e.g., with regard to fragmentation pattern, structure 4 ismost similar to the real structure of sublancin (structure 5) and has thesecond highest Hs value in HSEE analysis.
Fig. 3. Structural analysis of haloduracin β and deschloromicrobisporicin(DC-MIB). (A) Structure of haloduracin β and explanations of the shorthandnotations used for each PTM. The Cys-, Ser-, and Thr-derived structures areshown in red, blue, and purple, respectively. (B) HSEE analysis of haloduracinβ. Each number represents a structure with a different Ser/Thr escapingdehydration, with the numbering used indicated in the peptide sequence.The correct and originally proposed (incorrect) nondehydrated residues inhaloduracin β are indicated by a red asterisk and a black dot, respectively.The correct structure in the Hs bar plot is highlighted by a white asterisk. (C)Structure of DC-MIB and the shorthand notations used for its specific PTMs;other PTMs are shown in the same manner as in A. (D) HSEE analysis ofDC-MIB. The nondehydrated Thr residue and the dihydroxylated Pro residueare shown by red and blue asterisks, respectively. The hypothetical structures1–49 are shown in SI Appendix, Fig. S3. After determination of the de-hydration pattern by HSEE, the lanthionine ring topologies were predictedby sequence alignment with known lanthipeptides and by analysis of thetandem MS data as explained in the section on prochlorosin 1.2 (Pcn1.2).
Zhang et al. PNAS | August 19, 2014 | vol. 111 | no. 33 | 12033
(SI Appendix, Fig. S8), and only the ability of HSEE to considerseveral spectra at once allowed determination of its structure,demonstrating the utility of this method.
Directionality of Thiazole Formation on BalhA2. Another area ofresearch in which HSEE may be valuable is in the analysis of thetemporal installation of multiple PTMs, which provides impor-tant mechanistic insights into RiPP biosynthesis. To test thisapplication, we first used HSEE to analyze the in vitro inter-mediates of the azole-forming enzymes BalhC/BalhD/BcerB(1, 27). In this biosynthetic system, BalhC and BalhD catalyzethe heterocyclization of three Cys thiols of the BalhA2 peptideonto the carbonyl groups of the preceding amino acids, followedby a net dehydration to form thiazoline rings. The thiazolinesare subsequently dehydrogenated by the flavin mononucleotide(FMN)-containing enzyme BcerB to form thiazole heterocycles(Fig. 5A) (28, 29). HSEE analysis indicated that for the in-termediate containing one thiazole ring (loss of 20 Da) thestructure containing a C-terminal thiazole has the highest Hsvalues (Fig. 5C), whereas for the intermediate having two thia-zole rings (loss of 40 Da), the structure having two thiazoles in-stalled at the C terminus has the highest Hs value (Fig. 5D). Thisresult indicates that thiazole formation on BalhA2 is C- toN-directional (Fig. 5B), which is consistent with a previous study(28) and is distinct from the N-to-C azole-forming direction ofmicrocin B17 biosynthesis (30).
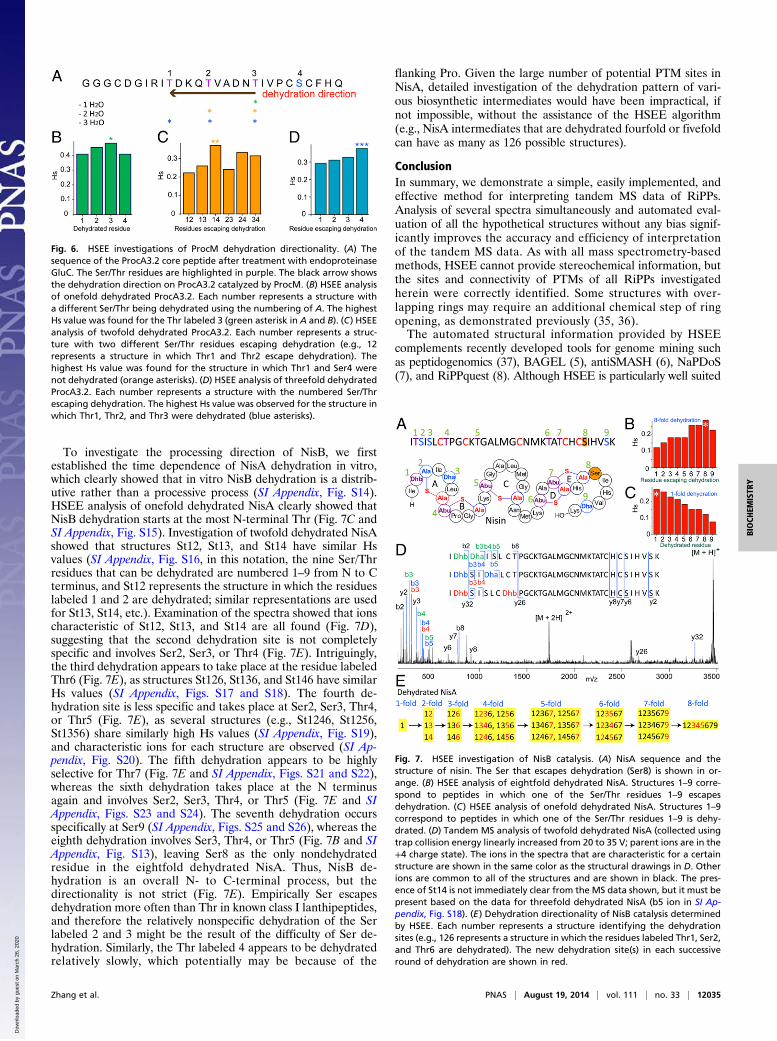
Directionality of ProcM Catalysis. We next investigated the di-rectionality of ProcM, a highly promiscuous class II lanthipeptidesynthetase (25). ProcM acts on a large number of precursorpeptides ProcA that consist of conserved N-terminal leader pep-tides and highly diverse C-terminal core peptides, and producesa diverse array of lanthipeptides termed prochlorosins (Pcns)(25), including Pcn1.2, which has been structurally characterizedin this study as mentioned above. Our ProcM directionality studyfocused on one of the substrates, ProcA3.2 (Fig. 6A), which hasfour Ser/Thr residues but is dehydrated only three times (25, 31).HSEE analysis of the onefold and twofold dehydrated ProcA3.2indicated that ProcM initially dehydrated the third Thr in the
core peptide followed by the second Thr (Fig. 6 B and C).However, the catalytic order is not strict, as some ions corre-sponding to the dehydration at other sites are also found in thespectra, both for the onefold and twofold dehydrated inter-mediates (SI Appendix, Figs. S10 and S11). HSEE analysis of thethreefold dehydrated ProcA3.2 suggests that the most N-termi-nally located Thr in the core peptide is the last dehydration site,whereas the Ser in the core peptide is not dehydrated at all (Fig.6D and SI Appendix, Fig. S12). Thus, the dehydration of the corepeptide of ProcA3.2 by ProcM is generally following a C- to N-terminal direction, but the directionality is not strict. This resultis distinct from the directionality of the class II lanthipeptidesynthetase HalM2, which carries out dehydration with generallyN- to C-terminal direction (32), suggesting that the directionalityof dehydration by lanthipeptide synthetases is nonuniform.
Mechanistic Investigation of NisB Catalysis by HSEE.We next appliedHSEE to investigate the directionality of catalysis by the nisindehydratase NisB (33), a prototypical class I lanthipeptide synthe-tase (19, 26). The activity of NisB was recently reconstituted in vitro(33), allowing for a detailed interrogation of its catalytic mecha-nism. Dehydration by NisB first involves glutamylation of Ser andThr, followed by more rapid elimination of glutamate to generatethe dehydroalanine and dehydrobutyrine residues, respectively.NisA has nine Ser/Thr residues, but the Ser labeled 8 in Fig. 7A isnot dehydrated in wild-type nisin. Previous in vivo studies showedthat this Ser was dehydrated when formation of the nisin D and/orE rings (Fig. 7A) was disrupted by site-directed mutagenesis,suggesting that NisB and the cyclase NisC may work in a co-ordinated fashion (34). However, all nine Ser/Thr residues weredehydrated by NisB in vitro, although the major product waseightfold dehydrated NisA (33). To investigate whether NisBhad any selectivity for different dehydration sites on NisA, weperformed HSEE analysis on the eightfold dehydrated NisAproduced in vitro. The result shows that the structure with theSer labeled 8 escaping dehydration has the highest Hs value (Fig.7B). In fact, close examination of the experimentally matchedions showed that no ions were produced that indicate dehydrationof this Ser (SI Appendix, Fig. S13), suggesting that NisB prefers notto dehydrate this Ser even in the absence of NisC.
Fig. 4. Structural analysis of Pcn1.2. (A) Peptide sequence of Pcn1.2. Thenondehydrated Ser residue is shown by a red asterisk; i–vi represent differ-ent peptide regions, which are defined according to the specific fragmen-tation sites (e.g., ion resulting from the cleavage N-terminal to the Serlabeled 1 belongs to region i, but C-terminal to this Ser belongs to region ii).For the detailed grouping of ions in region i–vi, please see SI Appendix, Fig.S9A. (B) HSEE analysis revealed that the Ser labeled 2 is not dehydrated. (C)HSEE analysis focusing on different peptide regions, which revealed thatregions ii and v are unlikely to contain lanthionines. (D) Proposed structureof Pcn1.2. The conclusion is based on the observation that the second Serescapes dehydration and that regions ii and v are unlikely to contain lan-thionines because fragmentation within these regions is observed. Thefraction below or above the blue arrows indicate the number of spectracontaining these fragmentation ions in the three tandem MS spectra. Thetrypsin cleavage site is indicated by a red arrow. The bars in B and C arecolored green and brown to indicate that the analyses were focused ondehydration and cyclization pattern, respectively.
Fig. 5. Use of HSEE for investigation of the directionality of thiazole for-mation. (A) Thiazole formation on BalhA2. (B) Fragment of BalhA2 that isgenerated after trypsin treatment. The thiazole-forming Cys residues arehighlighted in red. The black arrow shows the direction of thiazole in-stallment on BalhA2 catalyzed by the BalhC/BalhD/BcerB complex. (C) HSEEanalysis of BalhA2 containing one thiazole ring. Structures 1–3 correspond toBalhA2 containing a single thiazole derived from Cys1–3, respectively. (D)HSEE analysis of BalhA2 containing two thiazole rings. Structures 1–3 cor-respond to BalhA2 in which the indicated Cys (Cys1–3) is not converted toa thiazole, whereas the other two Cys residues are converted to thiazoles.
12034 | www.pnas.org/cgi/doi/10.1073/pnas.1406418111 Zhang et al.
To investigate the processing direction of NisB, we firstestablished the time dependence of NisA dehydration in vitro,which clearly showed that in vitro NisB dehydration is a distrib-utive rather than a processive process (SI Appendix, Fig. S14).HSEE analysis of onefold dehydrated NisA clearly showed thatNisB dehydration starts at the most N-terminal Thr (Fig. 7C andSI Appendix, Fig. S15). Investigation of twofold dehydrated NisAshowed that structures St12, St13, and St14 have similar Hsvalues (SI Appendix, Fig. S16, in this notation, the nine Ser/Thrresidues that can be dehydrated are numbered 1–9 from N to Cterminus, and St12 represents the structure in which the residueslabeled 1 and 2 are dehydrated; similar representations are usedfor St13, St14, etc.). Examination of the spectra showed that ionscharacteristic of St12, St13, and St14 are all found (Fig. 7D),suggesting that the second dehydration site is not completelyspecific and involves Ser2, Ser3, or Thr4 (Fig. 7E). Intriguingly,the third dehydration appears to take place at the residue labeledThr6 (Fig. 7E), as structures St126, St136, and St146 have similarHs values (SI Appendix, Figs. S17 and S18). The fourth de-hydration site is less specific and takes place at Ser2, Ser3, Thr4,or Thr5 (Fig. 7E), as several structures (e.g., St1246, St1256,St1356) share similarly high Hs values (SI Appendix, Fig. S19),and characteristic ions for each structure are observed (SI Ap-pendix, Fig. S20). The fifth dehydration appears to be highlyselective for Thr7 (Fig. 7E and SI Appendix, Figs. S21 and S22),whereas the sixth dehydration takes place at the N terminusagain and involves Ser2, Ser3, Thr4, or Thr5 (Fig. 7E and SIAppendix, Figs. S23 and S24). The seventh dehydration occursspecifically at Ser9 (SI Appendix, Figs. S25 and S26), whereas theeighth dehydration involves Ser3, Thr4, or Thr5 (Fig. 7B and SIAppendix, Fig. S13), leaving Ser8 as the only nondehydratedresidue in the eightfold dehydrated NisA. Thus, NisB de-hydration is an overall N- to C-terminal process, but thedirectionality is not strict (Fig. 7E). Empirically Ser escapesdehydration more often than Thr in known class I lanthipeptides,and therefore the relatively nonspecific dehydration of the Serlabeled 2 and 3 might be the result of the difficulty of Ser de-hydration. Similarly, the Thr labeled 4 appears to be dehydratedrelatively slowly, which potentially may be because of the
flanking Pro. Given the large number of potential PTM sites inNisA, detailed investigation of the dehydration pattern of vari-ous biosynthetic intermediates would have been impractical, ifnot impossible, without the assistance of the HSEE algorithm(e.g., NisA intermediates that are dehydrated fourfold or fivefoldcan have as many as 126 possible structures).
ConclusionIn summary, we demonstrate a simple, easily implemented, andeffective method for interpreting tandem MS data of RiPPs.Analysis of several spectra simultaneously and automated eval-uation of all the hypothetical structures without any bias signif-icantly improves the accuracy and efficiency of interpretationof the tandem MS data. As with all mass spectrometry-basedmethods, HSEE cannot provide stereochemical information, butthe sites and connectivity of PTMs of all RiPPs investigatedherein were correctly identified. Some structures with over-lapping rings may require an additional chemical step of ringopening, as demonstrated previously (35, 36).The automated structural information provided by HSEE
complements recently developed tools for genome mining suchas peptidogenomics (37), BAGEL (5), antiSMASH (6), NaPDoS(7), and RiPPquest (8). Although HSEE is particularly well suited
Fig. 6. HSEE investigations of ProcM dehydration directionality. (A) Thesequence of the ProcA3.2 core peptide after treatment with endoproteinaseGluC. The Ser/Thr residues are highlighted in purple. The black arrow showsthe dehydration direction on ProcA3.2 catalyzed by ProcM. (B) HSEE analysisof onefold dehydrated ProcA3.2. Each number represents a structure witha different Ser/Thr being dehydrated using the numbering of A. The highestHs value was found for the Thr labeled 3 (green asterisk in A and B). (C) HSEEanalysis of twofold dehydrated ProcA3.2. Each number represents a struc-ture with two different Ser/Thr residues escaping dehydration (e.g., 12represents a structure in which Thr1 and Thr2 escape dehydration). Thehighest Hs value was found for the structure in which Thr1 and Ser4 werenot dehydrated (orange asterisks). (D) HSEE analysis of threefold dehydratedProcA3.2. Each number represents a structure with the numbered Ser/Threscaping dehydration. The highest Hs value was observed for the structure inwhich Thr1, Thr2, and Thr3 were dehydrated (blue asterisks).
Fig. 7. HSEE investigation of NisB catalysis. (A) NisA sequence and thestructure of nisin. The Ser that escapes dehydration (Ser8) is shown in or-ange. (B) HSEE analysis of eightfold dehydrated NisA. Structures 1–9 corre-spond to peptides in which one of the Ser/Thr residues 1–9 escapesdehydration. (C) HSEE analysis of onefold dehydrated NisA. Structures 1–9correspond to peptides in which one of the Ser/Thr residues 1–9 is dehy-drated. (D) Tandem MS analysis of twofold dehydrated NisA (collected usingtrap collision energy linearly increased from 20 to 35 V; parent ions are in the+4 charge state). The ions in the spectra that are characteristic for a certainstructure are shown in the same color as the structural drawings in D. Otherions are common to all of the structures and are shown in black. The pres-ence of St14 is not immediately clear from the MS data shown, but it must bepresent based on the data for threefold dehydrated NisA (b5 ion in SI Ap-pendix, Fig. S18). (E) Dehydration directionality of NisB catalysis determinedby HSEE. Each number represents a structure identifying the dehydrationsites (e.g., 126 represents a structure in which the residues labeled Thr1, Ser2,and Thr6 are dehydrated). The new dehydration site(s) in each successiveround of dehydration are shown in red.
Zhang et al. PNAS | August 19, 2014 | vol. 111 | no. 33 | 12035
for genome mining for new RiPPs because of the direct link be-tween final structure and the gene-encoded precursor peptide, themethod in principle can also be used for structural analysis of othercompounds such as nonribosomal peptides, and is complementaryto other tandem MS-based methods that aim to ultimately de-termine structures in high throughput, including blind search (38),network analysis (39–41), and fragmentation tree construction (42,43). Moreover, HSEE greatly facilitates investigations of RiPPbiosynthetic mechanisms, as illustrated by several examples inthis study.
Materials and MethodsHigh-resolution liquid chromatography (LC)-MS and LC-MS/MS were carriedout either using a Synapt electrospray ionization quadrupole TOF massspectrometry system (Waters) equipped with an Acquity Ultra PerformanceLiquid Chromatography (UPLC) system (Waters), or using a ThermoFisherScientific LTQ-FT hybrid linear ion trap connected directly to an Agilent1200 HPLC system with an autosampler. Deisotoping and deconvolution of
tandem MS spectra were performed using the MaxEnt3 program (Waters)or the Qualbrowser application of Xcalibur (ThermoFisher Scientific). Theresulting raw data were first processed to list all of the picked ions, whichwere selected if their intensities were above a certain level (S/N threshold)defined based on the average ion intensity in a certain range. The pickedion list was then compared with the fragment ion matrix, allowing forsumming of the experimentally observed ions for each hypothetical struc-ture and generation of the Hs bar plot. For details of instrumental settings,procedures for gene cloning, peptide expression and purification, in vitrobiochemical assays, lists of hypothetical structures, and methods for dataprocessing, please see SI Appendix. SI Appendix also contains the R scriptsfor HSEE analysis.
ACKNOWLEDGMENTS. We thank Stefano Donadio (New Anti-InfectivesConsortium Srl) for a sample of deschloromicrobisporicin and Mr. SubhaMukherjee (University of Illinois at Urbana–Champaign) for providing theMS/MS data of Pcn2.8 and Pcn3.3 derivatives. This work was supported byNational Institutes of Health Grants GM58822 (to W.A.v.d.D.) and GM097142(to D.A.M.).
1. Arnison PG, et al. (2013) Ribosomally synthesized and post-translationally modifiedpeptide natural products: Overview and recommendations for a universal nomen-clature. Nat Prod Rep 30(1):108–160.
2. Baltz RH (2006) Marcel Faber Roundtable: Is our antibiotic pipeline unproductivebecause of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol33(7):507–513.
3. Li JW, Vederas JC (2009) Drug discovery and natural products: End of an era or anendless frontier? Science 325(5937):161–165.
4. Caboche S, et al. (2008) NORINE: A database of nonribosomal peptides. Nucleic AcidsRes 36(Database issue):D326–D331.
5. van Heel AJ, de Jong A, Montalban-Lopez M, Kok J, Kuipers OP (2013) BAGEL3: Auto-mated identification of genes encoding bacteriocins and (non-)bactericidal post-translationally modified peptides. Nucleic Acids Res 41(Web Server issue):W448–W453.
6. Blin K, et al. (2013) antiSMASH 2.0—a versatile platform for genome mining of sec-ondary metabolite producers. Nucleic Acids Res 41(Web Server issue, W1):W204–W212.
7. Ziemert N, et al. (2012) The natural product domain seeker NaPDoS: A phylogenybased bioinformatic tool to classify secondary metabolite gene diversity. PLoS One7(3):e34064.
8. Mohimani H, et al. (2014) Automated genome mining of ribosomal peptide naturalproducts. ACS Chem Biol 9(7):1545–1551.
9. Duncan MW, Aebersold R, Caprioli RM (2010) The pros and cons of peptide-centricproteomics. Nat Biotechnol 28(7):659–664.
10. Begley M, Cotter PD, Hill C, Ross RP (2009) Identification of a novel two-peptidelantibiotic, lichenicidin, following rational genome mining for LanM proteins. ApplEnviron Microbiol 75(17):5451–5460.
11. Shenkarev ZO, et al. (2010) Isolation, structure elucidation, and synergistic antibac-terial activity of a novel two-component lantibiotic lichenicidin from Bacillus lichen-iformis VK21. Biochemistry 49(30):6462–6472.
12. McClerren AL, et al. (2006) Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc Natl Acad Sci USA 103(46):17243–17248.
13. Cooper LE, McClerren AL, Chary A, van der Donk WA (2008) Structure-activity re-lationship studies of the two-component lantibiotic haloduracin. Chem Biol 15(10):1035–1045.
14. Castiglione F, et al. (2007) A novel lantibiotic acting on bacterial cell wall synthesisproduced by the uncommon actinomycete Planomonospora sp. Biochemistry 46(20):5884–5895.
15. Maffioli SI, et al. (2009) Structure revision of the lantibiotic 97518. J Nat Prod 72(4):605–607.
16. Böcker S, Letzel MC, Lipták Z, Pervukhin A (2009) SIRIUS: Decomposing isotope pat-terns for metabolite identification. Bioinformatics 25(2):218–224.
17. Pesavento JJ, Kim YB, Taylor GK, Kelleher NL (2004) Shotgun annotation of histonemodifications: A new approach for streamlined characterization of proteins by topdown mass spectrometry. J Am Chem Soc 126(11):3386–3387.
18. Oman TJ, Boettcher JM, Wang H, Okalibe XN, van der Donk WA (2011) Sublancin isnot a lantibiotic but an S-linked glycopeptide. Nat Chem Biol 7(2):78–80.
19. Knerr PJ, van der Donk WA (2012) Discovery, biosynthesis, and engineering of lan-tipeptides. Annu Rev Biochem 81:479–505.
20. Piper C, Cotter PD, Ross RP, Hill C (2009) Discovery of medically significant lantibiotics.Curr Drug Discov Technol 6(1):1–18.
21. Castiglione F, et al. (2008) Determining the structure and mode of action of micro-bisporicin, a potent lantibiotic active against multiresistant pathogens. Chem Biol15(1):22–31.
22. Foulston LC, Bibb MJ (2010) Microbisporicin gene cluster reveals unusual features oflantibiotic biosynthesis in actinomycetes. Proc Natl Acad Sci USA 107(30):13461–13466.
23. Sit CS, Yoganathan S, Vederas JC (2011) Biosynthesis of aminovinyl-cysteine-con-taining peptides and its application in the production of potential drug candidates.Acc Chem Res 44(4):261–268.
24. Bairoch A, Apweiler R (1996) The SWISS-PROT protein sequence data bank and itsnew supplement TREMBL. Nucleic Acids Res 24(1):21–25.
25. Li B, et al. (2010) Catalytic promiscuity in the biosynthesis of cyclic peptide secondarymetabolites in planktonic marine cyanobacteria. Proc Natl Acad Sci USA 107(23):10430–10435.
26. Zhang Q, Yu Y, Vélasquez JE, van der Donk WA (2012) Evolution of lanthipeptidesynthetases. Proc Natl Acad Sci USA 109(45):18361–18366.
27. Melby JO, Nard NJ, Mitchell DA (2011) Thiazole/oxazole-modified microcins: Complexnatural products from ribosomal templates. Curr Opin Chem Biol 15(3):369–378.
28. Melby JO, Dunbar KL, Trinh NQ, Mitchell DA (2012) Selectivity, directionality, andpromiscuity in peptide processing from a Bacillus sp. Al Hakam cyclodehydratase.J Am Chem Soc 134(11):5309–5316.
29. Dunbar KL, Melby JO, Mitchell DA (2012) YcaO domains use ATP to activate amidebackbones during peptide cyclodehydrations. Nat Chem Biol 8(6):569–575.
30. Kelleher NL, Hendrickson CL, Walsh CT (1999) Posttranslational heterocyclization ofcysteine and serine residues in the antibiotic microcin B17: Distributivity and di-rectionality. Biochemistry 38(47):15623–15630.
31. Shi Y, Yang X, Garg N, van der Donk WA (2011) Production of lantipeptides in Es-cherichia coli. J Am Chem Soc 133(8):2338–2341.
32. Lee MV, et al. (2009) Distributive and directional behavior of lantibiotic synthetasesrevealed by high-resolution tandem mass spectrometry. J Am Chem Soc 131(34):12258–12264.
33. Garg N, Salazar-Ocampo LM, van der Donk WA (2013) In vitro activity of the nisindehydratase NisB. Proc Natl Acad Sci USA 110(18):7258–7263.
34. Lubelski J, Khusainov R, Kuipers OP (2009) Directionality and coordination of de-hydration and ring formation during biosynthesis of the lantibiotic nisin. J Biol Chem284(38):25962–25972.
35. Martin NI, et al. (2004) Structural characterization of lacticin 3147, a two-peptidelantibiotic with synergistic activity. Biochemistry 43(11):3049–3056.
37. Kersten RD, et al. (2011) A mass spectrometry-guided genome mining approach fornatural product peptidogenomics. Nat Chem Biol 7(11):794–802.
38. Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA (2005) Identification of post-trans-lational modifications by blind search of mass spectra. Nat Biotechnol 23(12):1562–1567.
39. Nguyen DD, et al. (2013) MS/MS networking guided analysis of molecule and genecluster families. Proc Natl Acad Sci USA 110(28):E2611–E2620.
40. Guthals A, Watrous JD, Dorrestein PC, Bandeira N (2012) The spectral networks par-adigm in high throughput mass spectrometry. Mol Biosyst 8(10):2535–2544.
41. Yang JY, et al. (2013) Molecular networking as a dereplication strategy. J Nat Prod76(9):1686–1699.
42. Rasche F, Svatos A, Maddula RK, Böttcher C, Böcker S (2011) Computing fragmenta-tion trees from tandem mass spectrometry data. Anal Chem 83(4):1243–1251.
43. Rasche F, et al. (2012) Identifying the unknowns by aligning fragmentation trees.Anal Chem 84(7):3417–3426.
12036 | www.pnas.org/cgi/doi/10.1073/pnas.1406418111 Zhang et al.


![Controlled sampling of ribosomally active protistan diversity in … · 2021. 3. 16. · environmental RNA sampling, for example from: rainforest surface soils [20] and spring sediments](https://static.documents.pub/doc/80x56/612d844f1ecc515869423d18/controlled-sampling-of-ribosomally-active-protistan-diversity-in-2021-3-16.jpg)











![Investigation of carbon dioxide adsorption by …62-66]-10.pdf · 62 Investigation of carbon dioxide adsorption by nitrogen-doped carbons synthesized from cubic MCM-48 mesoporous](https://static.documents.pub/doc/80x56/5b6965737f8b9af23e8e07bf/investigation-of-carbon-dioxide-adsorption-by-62-66-10pdf-62-investigation.jpg)




