95
Studies on Syntheses of gem-Difluorovinylic Compounds and Their Applications Ken Oh February 2012

Studies on Syntheses of gem-Difluorovinylic
Compounds and Their Applications
Ken Oh
February 2012

Studies on Syntheses of gem-Difluorovinylic Compounds and Their Applications
Ken Oh
(Doctoral Program in Chemistry)
Submitted to Graduate School of
Pure and Applied Sciences
in Partial Fulfillment of Requirements
for Degree of Doctor of Philosophy in
Science
at
University of Tsukuba

Acknowledgement
This research has been accomplished at the Department of Chemistry, Graduate School of Pure and
Applied Sciences, University of Tsukuba during 2008-2011.
First I would like thank my supervisor, Professor Junji Ichikawa, for the opportunity of doing research
in his group. Without his continuous instructions and encouragement, this thesis would not be completed.
I am also indebt to Dr. Fuchibe who gave me invaluable comments, suggestions and personal helps.
I would like to thank all members of the Ichikawa group, especially Dr. Takeshi Fujita, Dr. Misaki
Yokota, Mr. Yousuke Chiba, Ms. Yuka Mayumi, Mr. Masahiro Hattori, Mr. Masahiko Shinjo, Mr. Toshiyuki
Morikawa, Ms. Yuka Nishikiori, Mr. Hiroki Takahashi, Ms. Mikiko Ueda, and Ms. Nan Zhao for their
helpful discussions and friendship.
Thanks to Du Pont-Mitsui Fluorochemicals Co. Ltd. for the constant support during this research. I
would like to thank my colleagues at Du Pont-Mitsui Fluorochemicals Co. Ltd. Especial thanks to Mr. Etsuo
Takenouji, Mr. Ichiro Imai, Mr. Yasunori Shibuta, Mr. Hidetaka Hiromatsu, Mr. Akito Abe, Dr. Jeong Chang
Lee, Mr. Hideyuki Takahashi, and Mr. Hideki Moriyama for their continuous support and encouragement.
Many thanks to Dr. Mureo Kaku (Du Pont K.K.) for his helpful instructions and support on preparation
of perfluoropolyethers.
I would like to express my grateful acknowledgement to Professor Kenji Uneyama (University of
Okayama) for his warm encouragement.
I would like to thank Professor Dr. Koichi Kitazawa (JST), my (master’s) supervisor at University of
Tokyo, for his recommendation and continuous instructions. I am also grateful to Mrs. Kuniko Kitazawa for
her warm hospitality throughout my family’s visits in Echigo-Yuzawa.
Many thanks to my father, mother and brother Jim for their continuous support and love. Finally,
special thanks to my wife Ayane, my son Mototaka and my daughter Yuika. I love you.
Ken Oh

I know nothing except the fact of my ignorance.
Socrates

Abbreviations
Ac ACF B3LYP BINAP Bn bp BuLi CFC dba DCM DFT DMF DMSO dppe dppf Eq. Equiv. Et Et2O FEP GC GC-MS HCFC HFC HFC-43-10mee HFIP HFP HMPA HOMO HPLC HRMS IR iPr KHMDS LDA LiHMDS LUMO Me mp NMR TFA
Acetyl Aluminum chlorofluoride (AlClxFy) Becke 3-parameter (exchange), Lee, Yang and Parr (correlation; density functional theory) 2,2'-Bis(diphenylphosphino)-1,1'-binaphthyl Benzyl Boiling point Butyllithium Chlorofluorocarbon Dibenzylideneacetone Dichloromethane Density functional theory N,N’-dimethylformamide Dimethyl sulfoxide 1,2-Bis(diphenylphosphino)ethane 1,1'-Bis(diphenylphosphino)ferrocene Equation Equivalent Ethyl Diethyl ether Tetrafluoroethylene-hexafluoropropylene copolymer Gas chromatography Gas chromatography – mass spectrum Hydrochlorofluorocarbon Hydrofluorocarbon 2,3-Dihydrodecafluoropentane Hexafluoro-iso-propanyl alcohol Hexafluoropropene Hexamethylphosphoric triamide Highest occupied molecular orbital High performance liquid chromatography High resolution mass spectrum Infrared iso-Propyl Potassium hexamethyldisilazane Lithium diisopropyl amide Lithium hexamethyldisilazane Lowest unoccupied molecular orbital Methyl Melt point Nuclear magnetic resonance Tetrafluoroethylene-perfluoroalkylvinylether copolymer

Ph PTFE PVDF PVF SN2’ SNV TASF TBAF TES Tf TFE TfO Tg THF TLC TMEDA TMS Ts VDF VF XANTPHOS
Phenyl Polytetrafluoroethylene Polyvinylidene fluoride Polyvinyl fluoride Bimolecular nucleophilic substitution (Prime) Nucleophilic vinylic substitution Tris(dimethylamino)sulfonium difluorotrimethylsilicate Tetrabutylammonium fluoride Triethylsilane Triflyl (Trifluoromethylsulfonyl) Tetrafluoroethylene Triflate (Trifluoromethylsulfonate) Glass transition temperature Tetrahydrofuran Thin layer chromatography Tetramethylethylenediamine Trimethylsilyl Tosyl (p-Toluenesulfonyl) Vinylidene fluoride Vinyl fluoride 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene

TABLE OF CONTENTS
Chapter 1. General Introduction page
1.1 Effects of Fluorine Substituents on Physical and Chemical Properties of Organofluorine Compounds
・・・ 1
1.2 gem-Difluorovinylic Compounds: Reactivity, Synthesis, and Applications ・・・ 5
1.3 Objectives and Summary ・・・ 15
References and Notes ・・・ 19
Chapter 2. Synthesis of 1,1-Difluoroallenes and Their Applications
2.1 Introduction ・・・ 23
2.2 Initial Exploration of Synthesis of 1,1-Difluoroallenes ・・・ 27
2.3 A General Synthetic Method from Commercially Available 1,1,1-Trifluoro-2-iodoethane
・・・ 29
2.4 Friedel–Crafts-type Cyclizations of 1,1-Difluoroallenes ・・・ 35
References and Notes ・・・ 37
Experimental Section ・・・ 39
Chapter 3. Syntheses of 2,2-Difluorovinylic Silanes and Their Applications
3.1 Introduction ・・・ 48
3.2 Synthesis of (2,2-Difluoro-1-iodovinyl)silanes from 1,1,1-Trifluoro-2-iodoethane ・・・ 49
3.3 Synthesis of (2,2-Difluoro-1-triflyloxyvinyl)silanes from 2,2,2-Trifluoroethanol ・・・ 51
3.4 Applications of 2,2-Difluorovinylsilanes ・・・ 53
References and Notes ・・・ 55
Experimental Section ・・・ 56

Chapter 4. Synthesis of 3,3-Difluoroallyl Arenes via Cationic SN2’-type Reactions of 2-Trifluoromethyl-1-alkenes
4.1 Introduction ・・・ 59
4.2 C–F Bond Cleavage of Trifluoromethyl Groups Promoted by Group 3 and Group 4 Metal Halides
・・・ 62
4.3 Catalytic C–F Bond Cleavage of Trifluoromethyl Groups by Aluminium Halides ・・・ 66
References and Notes ・・・ 67
Experimental Section ・・・ 69
Chapter 5. Conclusion ・・・ 71
List of publications ・・・ 76
Appendix. Reprints of original papers
A1. Oh, K.; Fuchibe, K.; Ichikawa, J. “A Facile Synthesis of 1,1-Difluoroallenes from Commercially Available 1,1,1-Trifluoro-2-iodoethane,” Synthesis 2011, 881.
・・・ 77
A2. Oh, K.; Fuchibe, K.; Yokota, M; Ichikawa, J. “Facile Synthesis of Substituted 1,1-Difluoroallenes via Carbonyl Difluorovinylidenation,” Synthesis, in press.
・・・ 83

1
Chapter 1. General Introduction
Organofluorine compounds play an important role in modern organic chemistry, biochemistry, and
chemical industries because of their unique properties.1 This chapter gives a brief overview of the effects of
fluorine substituent on properties of organofluorine compounds, especially fluorine-containing π-systems.
Reactivity, synthetic methods, and some applications of gem-difluorovinyl compounds that concern this
thesis are also provided. Objectives and summary of this thesis are described at the end of this chapter.
1.1 Effects of Fluorine Substituents on Physical and Chemical Properties of Organofluorine Compounds
Most of the physical and chemical properties of the organofluorine compounds are comprehensible and
predictable based on the nature of fluorine (Table 1.1).2–4 Some of the most important properties include:
(1) high electronegativity;
(2) relatively small atom size;
(3) lone pairs in the 2p orbital;
(4) extremely low polarizability.
For example, the high electronegativity makes C–F bonds polarized and the partial charge separation
contributes to the strong C–F bond. The high ionization potential and low polarizability imply very weak
inter- and intramolecular interactions in saturated fluorocarbons, for which the highly fluorinated compounds
have much lower refractive indexes, dielectric constants, and surface tensions. Other physical properties of
fluorocarbons, such as boiling points, miscibility, lipophilicity, and gas solubility, also differ from their
analogous, parent hydrocarbons because of the effects of fluorine.3,4
Table 1.1 Physical properties of halogenated and nonhalogenated compounds5
Atom Electronegativity
(Pauling) van der Waals
radius (Å) Atom
Polarizability (Å3) Ionization
Potential (eV)
C–X Bond Dissociation
Energya (kcal/mol) H 2.20 1.10 0.667 13.598 80.8 F 3.98 1.47 0.557 17.423 122.7 Cl 3.16 1.75 2.18 12.968 94.3 Br 2.96 1.83 3.05 11.814 80.0 I 2.66 1.98 4.7 10.451 60.5
a: In diatomic molecules.
The fluorine substituent also affects the chemical reactivity of fluorinated compounds. Figure 1.1 shows
the energy diagrams of fluorinated and fluorine-free compounds.4 Because the HOMO and LUMO levels of
fluorinated compounds (R–F) are lower than those of fluorine-free compounds (R–H), fluorinated

2
compounds have a high reactivity toward reducing agents (electron donor to the LUMO) and a low reactivity
toward oxidizing agents (electron acceptor from the HOMO).
R SOMO–13.6 eV
–18.6 eV
Energy Level
H 1s
R–H MOs
bonding
antibondingLUMO
HOMO
bonding
HOMO
antibonding
LUMO
F 2p
R–F MOs
Figure 1.1 MO diagrams of fluorinated and nonfluorinated compounds.
Interestingly, the fluorine substituent acts as not only an electron-withdrawing group but also an
electron-donating group. Major electronic effects of fluorine substituent on the chemical reactivity of
fluorinated molecules are summarized below.3,4
A. Effects on carbocations
(1) Stabilizing α-carbocations
Although the electron-withdrawing inductive effect of fluorine substituent destabilizes α-carbocations
(–I effect), the mesomeric, effective interaction of the lone pairs of fluorine in its 2p orbital with the vacant
2p orbital of carbon (+M effect) leads to a significant α-cation stabilization effect (Figure 1.2). The Shorter
C–F bond length than C–X bond lengths (X = Cl, Br, and I) also contributes to this effect.
Inductive withdrawal (–I effect)
destabilization
Mesomeric donation (+M effect)
stabilization>
C Fα
C F C Fα
Figure 1.2 Stabilizing effect on α-carbocations.

3
(2) Destabilizing β-carbocations
The carbocations bearing β fluorine are destabilized because of the −I effect of fluorine (Figure 1.3).
Inductive withdrawal (−I effect)
C C
Fβ
Figure 1.3 Destabilizing effect on β-carbocations.
B. Effects on carbanions
(1) Stabilizing sp3-hybridized α-carbanions
sp3-Hybridized α-carbanions are stabilized by the −I effect of fluorine (Figure 1.4).
Inductive withdrawal (–I effect)
C Fα
Figure 1.4 Stabilizing effect on sp3-hybridized α-carbanions.
(2) Stabilizing β-carbanions.
The β-carbanions are stabilized by the −I effect of fluorine (Figure 1.5a). “Negative hyperconjugation”
also contributes to the β-carbanion stabilizing effect (Figure 1.5b).
(a) Inductive withdrawal (–I effect)
C C
Fβ
C C
Fβ C C
Fβ
σ*C–F
(b) Negative hyperconjugation
Figure 1.5 Stabilizing effect on β-carbanions.
C. Effects on neutral systems
Electronic repulsion of fluorine lone pairs with alkene π electrons makes the C=C double bond
polarized, in which the Cα is electron-deficient and the Cβ is electron-rich (+Iπ effect, Figure 1.6a). The
fluorine substituent acts as a leaving group because of the stability of fluoride ion (Figure 1.6b).
Trifluoromethyl compounds readily undergo C–F bond cleavage because of this effect.

4
E C
F
(b) Elimination of F– (leaving group ability)
C CF
electronic repulsionδ – δ +
(a) Polarization of alkene (+ Iπ effect)
β α
Figure 1.6 Effects of fluorine on neutral systems.

5
1.2 gem-Difluorovinylic Compounds: Reactivity, Synthesis, and Applications
gem-Difluorovinyl compounds have two fluorines on the same sp2-hybridized carbon, which has a
geminal (“gem”) relationship. These compounds have been used in various fields such as synthetic,
medicinal, agricultural chemistries and material sciences because of their unique properties. In this section
reactivity, synthesis, and applications of gem-difluoroalkenes are briefly described.
1.2.1 Reactivity of gem-Difluorovinyl Compounds
gem-Difluorovinyl compounds exhibit unique reactivities6 in ionic,7,8 radical,9 and pericyclic10 reactions.
They behave in very different manners from their nonfluorinated analogues.11
A. Reactions with nucleophiles
gem-Difluorovinyl compounds are highly electron-deficient and polarized alkenes. The
electron-deficiency originates from the −I effect of fluorine, and the polarity originates from the +Iπ effect of
fluorine.12 Figure 1.7 shows calculated charges of carbons of parent, chlorinated, and fluorinated ethylenes.
Both of the carbons of parent ethylene exhibit negative values of –0.366 (–0.732 in total), which shows
ethylene is an electron-rich alkene. However, α and β carbons of difluoroethylene exhibit the values of
+0.781 and –0.562, respectively. These values suggest that Cα=Cβ double bond is highly polarized, and the
total value of the two carbons (+0.219) suggest that difluoroalkene is an electron-poor alkene. In this context,
dichloroethylene is an electron-rich and less polarized alkene.
C CH
H H
HCα Cβ
F
F H
HCα Cβ
Cl
Cl H
H
Cα: −0.366Cβ: −0.366Ctotal= −0.732
Cα: +0.781Cβ: −0.562Ctotal= +0.219
Cα: −0.118Cβ: −0.414Ctotal= −0.532
Figure 1.7 Calculated charges of carbons of (dihalo)ethylenes by ab initio calculations.12
The gem-difluorovinyl compounds thus readily react with nucleophiles, and the reaction takes place
selectively on the difluoromethylene carbon (Cα).13 For instance, 1,1-dichloro-2,2-difluoroethene reacts with
phenylmagnesium bromide or phenyllithium to give a substituted monofluoroalkene (Equation 1.1).14 A
tetrathiafulvalene derivative is synthesized by the nucleophilic substitution of a 1,1-difluoroalkene with a
1,2-bis(thiolate) (Equation 1.2).15 Recently, our group has revealed that gem-difluorovinyl compounds
bearing a nucleophilic N, O, S, and C center undergo 5-endo-trig cyclizations, which are, in general,
disfavored processes in the Baldwin’s rules (Scheme 1.1).16 In contrast to β,β-difluorostyrenes, β,β-dichloro-
and β,β-dibromostyrenes gave none or only 15% yield of the cyclization product, respectively.

6
F
F Cl
Cl
PhM
reflux, Et2O Ph
F Cl
Cl
M = MgBr: 60% Li : 85%
F
F
rt, HCONMe2S
S
SLi
SLi S
S S
S
70%
(1.1)
(1.2)
n-Bu
HO
NaH (1.2 eq)60 °C, DMF
O
XX
n-Bu– F–
O
n-Bu
FX = F: 80% (2 h)X = Cl: – (8 h)X = Br: 15% (5 h)
CX2
5-endo-trig cyclization
Scheme 1.1 Fluorine substituent as a promoter in “5-endo-trig” cyclizations.
B. Reactions with electrophiles
Because –I effect of fluorine makes gem-difluoroalkenes highly electron-deficient, only few reactions
of gem-difluorovinyl compounds with electrophiles have been reported. For instance, vinylidene fluoride
(VDF) undergoes a Friedel–Crafts-type reaction,17 a nitration reaction,18 and a sulfenylation reaction19 in the
presence of the corresponding electrophiles (Scheme 1.2). Tetrafluoroethylene (TFE) reacted with carbon
tetrachloride in the presence of a catalytic amount of aluminum chloride (Equation 1.3).20,21 Note that all the
reactions take place to form stabilized α-fluorocarbocations, which makes the addition regioselective.
HNO3 / HF
68%
F
F H
H
70%
93%
ClSPh
+CF2CH2–CO2i-Pr
+CF2CH2–NO2
+CF2CH2–SPh
Cl–
F–
Cl–
Cl–CF2CH2–CO2i-Pr
F–CF2CH2–NO2
Cl–CF2CH2–SPh
" +CO2i-Pr "
ClCO2i-Pr, AlCl3
" +NO2 "
" +SPh "
VDF
Scheme 1.2 Reactions of vinylidene fluoride (VDF) with electrophiles.
F
F F
F
ClCCl3cat. AlCl3 Cl–CF2CF2–CCl3
82%" +CCl3 "
+CF2CF2–CCl3Cl–
TFE
(1.3)

7
The α-fluorocarbocations generated by the reaction of difluoroalkenes with electrophiles (electrophilic
activators) are of special synthetic importance. Our group has found that protonation of difluoroalkenes was
realized with a super acid (FSO3H·SbF5, magic acid) and that the resulting difluoromethyl carbocations are
useful intermediates for domino Friedel–Crafts-type cyclizations (Equation 1.4). 22 This opened up a new
route to substituted [4]–[6]helicenes. Electrophilic, catalytic activation of difluoroalkenes with a cationic
palladium(II) complex was also developed by our group23 (Scheme 1.3). Treatment of difluoroalkenes with a
catalytic amount of [Pd(MeCN)4](BF4)2 afforded substituted tetralones.
CF2
FSO3H·SbF5(2.5 equiv)
(CF3)2CHOH0 °C, RT
CF2
H
+Domino
Friedel–CraftsPh3CBF4
(2.3 equiv)
[4]helicene87%80%
(1.4)
5 mol% [Pd(MeCN)4](BF4)21 equiv BF3·OEt2
(CF3)2CHOHCF2
CF2
F
Pd2+
2(BF4 )
PdL4(BF4)2
HBF4
Pd+
FFBF4
H2OO
BF3
R
R R
R R
65 89%
Scheme 1.3 Cationic Pd(II)-catalyzed Friedel–Crafts-type cyclization of gem-difluoroalkenes.
C. Reactions with radicals
Radical reactions are often used to functionalize fluoroalkenes. Most of the fluoropolymers are
produced industrially via radical processes. Especially noteworthy is that regioselectivity of the radical
additions is strongly influenced by the fluorine substituents.24,6b A nucleophilic methyl radical (·CH3 radical)
attacks the electron-deficient =CF2 carbon of trifluoroethylene, while an electrophilic trifluoromethyl radical
(·CF3 radical) attacks the electron-rich =CHF carbon (+Iπ effect, Scheme 1.4).

8
H
F F
F· CF3 CF3CHFCF2
· CH3CHFCF2CH3
δ – δ +
(nucleophilic)
(electrophilic)
Scheme 1.4 Regioselectivity of radical addition of ·CH3 and ·CF3 to trifluoroethylene.
Rates of radical addition is also influenced by the effect of fluorine. The relative rates of addition of
methyl and trifluoromethyl radicals show opposite trends as shown in Table 1.2.24,25 This phenomenon can
also be rationalized by considering that ·CH3 radical is electron-rich and ·CF3 radical is electron-deficient.
Table 1.2 Relative rates of addition of ·CH3 and ·CF3 radicals to ethylene and fluoroethylenes.
Radical CH2=CH2 CH2=CHF CHF=CH2 CHF=CF2 CF2=CHF
·CH3 1 0.9 0.2 1.9 3.9
·CF3 1 0.45 0.05 0.033 0.017

9
1.2.2 Synthesis of gem-Difluorovinyl Compounds
Representative synthetic methods of gem-difluorovinyl compounds are described in this section.6a,26
There are four practical strategies for the synthesis of a gem-difluoroalkenes. Namely, (A) introduction of a
difluoromethylene unit (C1); (B) Introduction of a difluorovinyl unit (C2); (C) Introduction of a nucleophile
to the 2-trifluoromethyl-1-alkenes by SN2’-type reactions (C3); and (D) β-elimination.
Strategy A: Introduction of a difluoromethylene (C1) unit
A wide variety of Wittig-type approaches have been developed to synthesize difluoroalkenes from
carbonyl compounds (mainly aldehydes, Scheme 1.5).27–33 Some Horner–Wadsworth–Emmons-type
reactions using a commercially available diethyl difluoromethylphosphonate32 or
difluoromethyldiphenylphosphine oxide33 are also known. An approach in which a fluorine-free ylides
reacted with a difluorocarbene was reported (Equation 1.5).34
R1 R2
O
R1 R2
CF2Conditions
a) ClCF2CO2Na/Ph3P/160 oC 27
b) CF2Br2 / Ph3P (2eq) 28
c) CF2Br2 / (Me2N)3P (2eq) 29
d) CF2Br2/Ph3P (1eq) / Zn 30
e) CF2Br2 / (Me2N)3P (1eq) / Zn 31
f) HCF2PO(OEt)2 / base 32
g) HCF2PO(Ph)2 / base 33
Scheme 1.5 Wittig-type approaches to gem-difluorovinyl compounds.
PPh3R1
HR2X n-BuLi
0 oC2 2 PPh3
R2
R1 HCF2Cl
R2
R1 F
F+ PPh3
R1
HR2Cl (1.5)
The direct difluoromethylenation of carbonyl compounds is a useful method to synthesize
1,1-difluoroalkenes, while these syntheses are often limited to the sterically less hindered substrates
(aldehydes) or require harsh reaction conditions. Decarboxylation of difluorinated β-lactones that can be
prepared from ketones and ethyl bromodifluoroacetate is an alternative method for difluoromethylenation of
ketones.26 A modified Julia-type approach is also known.35
Strategy B: Introduction of difluorovinyl (C2) units with difluorovinylmetals
Syntheses of difluoroalkenes can be accomplished straightforwardly with difluorovinylmetals.36
Difluorovinyllithiums are attractive reagents for this purpose because of their high reactivity. They are
generated by lithiation of difluoroethylenes37 or by elimination of trifluoroethyl compounds38 using
alkyllithiums or LDA (Scheme 1.6).

10
F
F X
Li
2 RLi (or 2 LDA)
F
F X
YXCF3
X = hydrogen, (pseudo)halogen, alkyl, aryl, etc.Y = hydrogen or halogen.
RLi– RY – 2 RH, – LiF
Scheme 1.6 Generation of gem-difluorovinyllithiums.
gem-Difluorovinylboranes, readily prepared from 2,2-difluoro-1-tosyloxyvinyllithium and
trialkylboranes, are versatile intermediates for the synthesis of difluoroalkenes. Our group revealed that the
2,2-difluoro-1-tosyloxyvinyllithium affords a wide variety of products upon protonation, cross-coupling with
a variety of organohalides, iodination, and so on (Scheme 1.7).39
CF2
CF2
NaOMe
I 2
PPh2
R
R'COCl
R
R'O
CuI
CF2I
R
ArICF2
R
ArPd
AcOH
CF2R
H
R
BR2
CF2 C
R' R''
R
PdXCR'=CHR''
CF2R
R'
Cl
R'
CF2OTs
Li
F
F
CuIPh2PCl
CuI
Scheme 1.7 Synthesis of gem-difluorovinyl compounds via gem-difluorovinylboranes.
Other useful difluorovinylmetal reagents are gem-difluorovinylzincs (Scheme 1.8).
gem-Difluorovinylzinc reagents can be generated by two methods: (i) transmetallation of the corresponding
vinyllithiums with zinc(II) halide and (ii) direct zincation of gem-difluorovinyl halides. Our group has
recently reported a Negishi cross-coupling reaction of difluorovinylzinc–TMEDA complex with aryl halides
(Equation 1.6).40,41
F
F R
ZnX
Zn(0)
F
F R
Li
ZnX2
– LiX F
F R
I
Scheme 1.8 Generation of gem-difluorovinylzinc reagents.

11
CF2 CH2
1) s-BuLi (1.0 equiv), –110 °C2) ZnCl2 (1.0 equiv) TMEDA (1.3 equiv), –100 °C
THF/ether F
F
ZnCl·TMEDA
2 mol% Pd(PPh3)41-NaphI (1.0 equiv)
(1.2 equiv)
reflux F
F
1-Naph
87%
(1.6)
Strategy C: Introduction of nucleophiles to 2-trifluoromethyl-1-alkenes (C3) by SN2’-type reactions
2-Trifluoromethyl-1-alkenes, possessing the low-lying LUMO, readily undergo nucleophiles attack. The addition usually takes place at the position γ to the fluorine.4,42 Organolithiums, Grignard reagents, lithium amide, and other strong nucleophiles react with 2-trifluoromethyl-1-alkenes to afford the corresponding gem-difluorovinyl compounds (Scheme 1.9).43–46 Note that SN2’-type reactions reported so far require strongly basic conditions.
R1R2CHCO2R3
F
F R
F
F R
CC
F
F
Baseor R'Li
R1 R
2 NLi LiAlH4
CH3
R
R'O
Li
F
F
F
F
R
R
CF3R1
CO2R3R2R'
R'MgX
NR1R2R
OR'
Scheme 1.9 Synthesis of gem-difluoroalkenes by SN2’-type reactions of 2-trifluoromethyl-1-alkenes.
Combination of the SN2’-type reaction of 2-silylated 2-trifluoromethyl-1-alkenes and the coupling
reaction of the remaining vinylsilane moiety with electrophiles furnished the synthesis of gem-difluoroallylic
compounds (Scheme 1.10):46 (i) the SN2’-type reaction of 2-silyl-2-trifluoromethyl-1-alkenes with
nucleophiles forms 2,2-difluorovinylsilanes, and (ii) the subsequent capture of the remaining vinylsilane
moiety with electrophiles affords the desired products. 2-Silylated trifluoromethylalkenes act as a synthon of
difluoropropyrenes that possess positive charge and negative charge on the 3- and 2-positions, respectively.
CF3SiR3
Nu–CF2
SiR3
Nu
– F–CF2
E
NuE+
CF2
Scheme 1.10 Synthesis of functionalized gem-difluoroallylic compounds by SN2’-type reactions of silylated 2-trifluoromethyl-1-alkenes.
Relatedly, trifluoromethylthioketones and trifluoromethylimines are also efficient acceptors for
SN2’-type reactions. Alkylmetals attack the S and N atoms to afford heteroatom substituted
gem-difuoroalkenes. (Scheme 1.11).47,48

12
– F–
NuM
F
F Z
R
Nu Z = S, NAr, NAlk, etc.Nu- = Alk-M, etc.CF3
R
Z
Scheme 1.11 Synthesis of functionalized gem-difluorovinyl compounds by hetero SN2’-type reactions.
Strategy D: β-Elimination of F–
β-Elimination of F– is an important process for constructing a difluoroalkene moiety. This method is
often used to prepare gem-difluorovinyl compounds from trifluoromethyl compounds. For instance,
treatment of dichlorotrifluoroethane (HCFC-123) with zinc and then with aldehydes leads to
3,3-difluoroallylic alcohols. This reaction takes place via an organozinc intermediate, which is stabilized by
–I effect of fluorine (Equation 1.12).49–51
F3C Cl
Cl Zn
F3C ZnCl
Cl2 2
R H
O
F3C ZnCl
Cl Cl
+ CF3CH2Cl
F3C
Cl ClOH
R
ZnF2C
ClOH
R1-22
Scheme 1.12 Synthesis of a 3,3-difluoroallyl alcohols from HCFC-123.
Some industrially important fluorinated alkenes are produced by β-elimination. For example,
perfluoropropyl vinyl ether (PPVE) is a co-monomer for production of a melt processible perfluoropolymer,
perfluoroalkoxyl (perfluoroalkoxyalkane, PFA). PPVE is produced by pyrrolitic β-elimination (Equation
1.7).52
CF3 CF
CF2
O2
dimerizationCF3 C
F2
F2C O
CFCF3
O
F 300 oCNa2CO3
CF3 CF2
F2C O
FC
CF2
PPVE
(1.7)

13
1.2.3 Applications of gem-Difluorovinyl Compounds
As described above, gem-difluorovinyl compounds are useful synthetic intermediates or building blocks
for construction of fluorine containing molecules. Difluorovinyl compounds are also of industrial
significance as summarized below.
Fluoropolymers
Most of commercial fluoropolymers are produced by free radical polymerizations of fluorovinyl
monomers such as tetrafluoroethylene (TFE), 1,1-difluoroethylene (vinylidene fluoride, VDF),
fluoroethylene (vinyl fluoride, VF), and chlorotrifluoroethylene (CTFE). In some cases, copolymers of
fluorovinyl monomers and (non)fluorovinyl monomers are produced to find wider application. Some
commercial fluoropolymers are listed in Table 1.3 along with their applications.
Table 1.3 Major commercial fluoropolymers produced from fluorovinyl compounds.
Polymer Monomers Applications
PTFE CF2=CF2 Cookware coatings; waterproof clothing; electrical
insulators; medical uses such as artificial blood vessels, etc.
FEP CF2=CF2 + CF2=CFCF3 Fabrication by conventional melt processing; wire and cable
insulators; heat-sealable film, tubing, etc.
PFA CF2=CF2 + CF2=CFORf
Tubing, injection or blow-molded articles, chemical linings (tanks, pipes, valves, pumps), fluid handling components for
critical, high-purity processes like semiconductor, pharmaceutical, and biotechnology, etc.
Teflon AF® (DuPont)
CF2=CF2 +
Optically clear, used in corrosive environments where glass is unsuitable, e.g. in computer chips manufacture.
Cytop® (AGC)
CF2=CFO(CF2)nCF=CF2 Same as above.
PCTFE CF2=CFCl Gaskets, seals, oils, coatings, transparent inert covers.
PVDF CF2=CH2 Weather-resistant coatings; cable insulation; piezo-electric
devices.
PVF CHF=CH2 Coatings, flexible films, e.g. surface protection uses in
photovoltaic modules and aerospace interiors.
Viton A® (DuPont)
CF2=CH2 + CF2=CFCF3 Elastomers used for sealants, O-rings, fuel-resistant seals for
aircraft and automobiles.
ETFE CF2=CF2 + CH2=CH2 Laminated films as construction materials, wire and cable
insulators. Nafion® (DuPont)
CF2=CF2 + CF2=CFOCF2CF(CF3)OCF2CF2SO2F
Membranes in chloralkali process, polymer electrolyte fuel cell (PEFC).
Flemion® (AGC)
CF2=CF2 + CF2=CFOCF2CF(CF3)OCF2CF2CO2Me
Same as above.

14
Pharmaceuticals and agrochemicals
Fluoroalkenes have been utilized as bioisosters of some functionalities.12,53 For instance,
monofluoroalkenes are bioisosters of amides (Figure 1.8a) and gem-difluoroalkenes are bioisosters of
carbonyl compounds (Figure 1.8b).53,54
N
O
N
O– F O FF
+
(a) (b)
amides monofluoroalkenes difluoroalkenescarbonyl compounds
Figure 1.8 Fluoroalkenes as bioisosteres.
An exo-difluoromethylene-artemisinins is a mimic of artemisinin. The biological studies indicated that
the replacement of the carbonyl group by a difluoroethylene moiety resulted in a higher antimalarial
activity.55
OO
OO
CF2
OO
OO
O
exo-Difluoromethylene artemisinin Artemisinin
Figure 1.9 Structures of exo-difluoromethylene-artemisinin and artemisinin.

15
1.3 Objectives and Summary
This thesis aims to:
(i) provide practical synthetic methods for functionalized gem-difluorovinyl compounds by using commercially available starting materials; and
(ii) develop novel applications of these gem-difluorovinyl compounds in synthetic organic chemistry.
In Chapter 2, a general and practical synthesis of 1,1-difluoroallenes and their application in
Friedel–Crafts-type cyclizations are described. 1,1-Difluoroallenes are attractive synthetic intermediates
because of the vinylic fluorine substituents and the cumulated double bonds. 1,1-Difluoroallenes also serve
as promising pharmaceuticals, because some of non-fluorinated allenes have been used for therapeutic
purposes. However, synthetic methods for substituted 1,1-difluoroallenes have not been fully explored.
The synthesis of 1,1-difluoroallenes has been achieved by the following two-step process: (i)
3,3-Difluoro-2-iodoallylic acetates are prepared by the reaction of aldehydes or ketones with
2,2-difluoro-1-iodovinyllithium, which is generated from commercially available
1,1,1-trifluoro-2-iodoethane and LDA (Scheme 1.13); (ii) 1,1-Difluoroallenes are synthesized under mild
conditions via zinc-promoted elimination of the iodo and acetoxy groups. A wide variety of
1,1-difluoroallenes were obtained in high yield by this sequence.
CF2
I
AcO
R1 CF2R2
R2
R1
CF3CH2ILDA (2 equiv)
–93 to –85 °C30 min, THF
CF2
I
Li
1) R1(C=O)R2 (1 equiv) –93 to –30 °C, 2 h
2) Ac2O (1.2equiv) –30 to 0 °C, 2 h
Zn (2 equiv)
RT, 3–12 hDMF or THF
80–87% 86–93%
Scheme 1.13 Synthesis of substituted 1,1-difluoroallenes.
The author has revealed that activation of 1,1-difluoroallenes bearing an arylmethyl group is effected by
an electrophilic reagent, that is a Lewis acid such as BF3·OEt2 and ZrCl4, to cause Friedel–Crafts-type
cyclizations (Scheme 1.14). This reaction is accompanied by 1,2-migration of the benzylic alkyl group to
provide an effective method for the synthesis of fluorinated naphthalenes.
CF2 FF FFLA
LA
F
and/or– HF
1,2-Me migration
LA (1 equiv)
RT, 30 minCH2Cl2/(CF3)2CHOH
(1:1)40% (BF3·OEt2)
32% (ZrCl4) Scheme 1.14. Synthesis of fluoronaphthalenes by Friedel–Crafts-type cyclizations of 1,1-difluoroallenes.

16
In Chapter 3, synthesis of 2,2-difluorovinylic silanes bearing a (pseudo)halogen substituent on the 1
position is described. These vinylic silanes are promising bifunctional intermediates for the synthesis of
molecules containing a difluorovinylidene moiety (Scheme 1.15): The silanes would react with nucleophiles
(electrophiles) and then with electrophiles (nucleophiles) to give a variety of fully substituted
difluoroalkenes. Thus, the silanes might serve as a 2,2-difluorovinylidene synthon possessing both positive
and negative charges on the 1 position.
CF2X
SiR3E+
CF2X
E
Nu–
CF2Nu
E
CF2Nu
SiR3
Nu– E+
F
F
vinylidene
Scheme 1.15. Concept of 2,2-difluoro-1-(pseudo)halovinylsilanes as a difluorovinylidene synthon.
The author has developed the synthetic methods for the 2,2-difluorovinylsilanes bearing a
(pseudo)halogen substituent from commercially available compounds. Silylation of
2,2-difluoro-1-iodovinyllithium, described in Chapter 2, was effected with chlorosilanes to give the
corresponding iodinated difluorovinylsilanes in 83–84% yield (Scheme 1.16a). Triple deprotonation of
2,2,2-trifluoroethanol was conducted with LDA in the presence of chlorosilanes, followed by retro-Brook
rearrangement and triflylation. Thus, triflyloxy-substituted difluorovinylsilanes were obtained in good yield
(Scheme 1.16b).
CF3CH2ILDA (2 equiv)
CF2
I
SiR3
CF2
I
Li
R3SiCl (1 equiv)
CF3CH2OH
LDA (3.3 equiv)R3SiCl (1 equiv)
CF2
OSiR3
Li
retro BrookCF2
OLi
SiR3
CF2
OTf
SiR3
PhNTf2 (1 equiv)
84% (SiMe2Ph)83% (Sii-Pr3)84% (SiMe3) (19F NMR yield)
72% (SiEt3)61% (SiPh2t-Bu)*
(a)
(b)
–93 to –85 °C, 30 minTHF
–85 °C, 1 h
–93 °C to RT, 1 hTHF(–HMPA*)
0 °C
Scheme 1.16 Synthesis of 2,2-difluoro-1-(pseudo)halovinylsilanes.
The iododifluorovinylsilane reacted with a boronic acid (Nu–) in the presence of palladium catalyst to
give β,β-difluorostyrene derivative in 81% yield (Scheme 1.17a). The iododifluorovinylsilane also reacted
with an aldehydes (E+) in the presence of cesium fluoride to provide 3,3-difluoro-2-iodoallylic silyl ether in
77% yield (Scheme 1.17b). Combination of these results would open up a new route to fully substituted
1,1-difluoro-1-akenes.

17
20 mol% CsFn-C4H9CH(Et)CHO (1.0 equiv) CF2
I
CH(Et)n-C4H9i-Pr3SiO
CF2
I
SiMe2Ph
1 mol% PEPPSI-IPrPhB(OH)2 (1 equiv)
CF2
Ph
SiMe2Ph
81%
K2CO3 (2 equiv)60 °C, EtOH, 3 h
(b) Reactions with an electrophile
CF2
I
Sii-Pr3
Pd
NNR
R
R
R
N
Cl
PEPPSI-IPr (R = i-Pr)
Cl Cl
(a) Reaction with a nucleophile
77%
70 °C, diglyme, 4 h
Scheme 1.17 Reactions of 2,2-difluoro-1-iodovinylsilanes with nucleophiles or electrophiles.
Synthesis of 3,3-difluoroallylic compounds by SN2’-type reactions of 2-trifluoromethyl-1-alkenes is
described in Chapter 4. 3,3-Difluoroallylic compounds, in general, can be prepared by SN2’-type reactions of
2-trifluoromethyl-1-alkenes with nucleophiles (Scheme 1.18a), which are conducted under basic conditions.
A possible alternative to the anionic SN2’-type reactions is an acid-promoted cationic version (Scheme
1.18b).
R
CF3Nu–
– F–
R
CF2Nu
R
CF2
Nu–H– LA, – HF
FLA
+ +Previous reports This work
Basic conditions (Lewis) Acidic conditions
(a) (b)
Scheme 1.18. Concept of Lewis acid-promoted SN2’-type reactions of 3,3,3-trifluoropropenes.
The author has achieved novel Zr(IV)-promoted and Al(III)-catalyzed SN2’-type reactions of the
trifluoromethylalkenes with simple arenes. 2-Trifluoromethyl-1-alkenes were treated with an equimolar
amount of zirconium(IV) chloride (Scheme 1.19a) or with 10 mol% of aluminium chloride (bromide)
(Scheme 1.19b) in the presence of arenes. The desired SN2’-type reactions took place smoothly to afford
3,3-difluoroallylic arenes in good yield.

18
R
CF2Ar
R
CF3
ZrCl4 (1 equiv)
0 °C to RT, CH2Cl2+ Ar–H
(3 eq)16–95%R = Br or Ph
+
(10 eq)
neat, RT, 8 h+
10 mol% AlX3Br
CF3p-Xylene
X = Cl; 71% (40:60)X = Br; 61% (42:58)
Br
CF2
Br
CF3
(a)
(b)
Scheme 1.19 Zr(IV)-Promoted and Al(III)-catalyzed SN2’-type reactions of 2-trifluoromethyl-1-alkenes.
It must be mentioned that CF3 group is chemically robust and that activation and synthetic
transformation of the CF3 group have been a challenging task for chemists. The Lewis acid mediated
SN2’-type reaction provided the method to cleave the chemically stable C–F bond of the CF3 group on
alkenes.
In this thesis, the author describes practical syntheses and synthetic applications of gem-difluorovinylic
compounds (1,1-difluoroallenes, 2,2-difluoro-1-(pseudo)halovinylsilanes, and 3,3-difluoroallylic arenes).
These achievements definitely inspire potential industrial applications of functionalized gem-difluorovinylic
compounds to production of useful fluorine-containing materials.

19
References and Notes
1. (a) Johns, K.; Stead, G. J. Fluorine Chem. 2000, 104, 5. (b) Banks, R. E. (ed.) Preparation, Properties and Industrial Applications of Organofluorine Compounds, Ellis Horwood, Chichester, 1982. (c) Banks, R.E.; Sharp, D. W. A.; Tatlow, J.C. (eds) Fluorine: The First One Hundred Years, Elsevier Sequoia, New York, 1986. (d) Banks, R.E.; Smart, B.E.; Tatlow, J.C. (eds) Organofluorine Chemistry. Principles and Commercial Applications, Plenum, New York, 1994. (e) Kirsch, P. Modern Fluoroorganic Chemistry, WILEY-VCH, Weinheim, 2004.
2. (a) Hudlicky, M. Chemistry of Organic Fluorine Compounds, 2nd Ed. Ellis Horwood PTR, 1992. (b) Hudlicky, M.; Pavlath, A.E. (eds) Chemistry of Organic Fluorine Compounds II, ACS Monograph 187, American Chemical Society, Washington, DC. 1995.
3. (a) Chambers, R.D. Fluorine in Organic Chemistry, John Wiley and Sons, New York, 1973. (b) Chambers, R. D. Fluorine in Organic Chemistry, Blackwell Publishing, Oxford, 2004.
4. Uneyama, K. Organofluorine Chemistry, Blackwell Publishing, Oxford, 2006.
5. Haynes, W. M.; Lide, D. R. (eds) CRC Handbook of Chemistry and Physics, 91st Ed. CRC Press, Boca, Raton, 2010.
6. (a) Tozer, M.J.; Herpin, T.F.; Tetrahedron 1996, 52, 8619. (b) Chambers, R.D. (eds) Organofluorine Chemistry: Fluorinated Alkenes and Reactive Intermediates, Springer-Verlag, Berlin Heidelberg New York, 1997.
7. For anionic reactions, see: (a) Ichikawa, J.; Wada, Y.; Okauchi, T.; Minami, T. Chem. Commun.1997, 1537. (b) Kim, B.T.; Park, N.K.; Pak, C.S.; Kim, M.S;. Jeong, I.H. Heterocycles, 1997, 45, 37.
8. For cationic reactions, see: (a) Morikawa, T.; Kumadaki, I.; Shiro, M. Chem. Pharm. Bull. 1985, 33, 5144. (b) Kendrick, D.A.; Kolb, M. J. Fluorine Chem. 1989, 45, 273.
9. (a) Narita, T.; Hagiwara, T.; Hamana, H.; Tomooka, K.; Liu, Y.-Z.; Nakai, T. Tetrahedron Lett. 1995, 36, 6091. (b) Piettre, S.R. Tetrahedron Lett. 1996, 37, 2233. (c) Herpin, T.F.; Motherwell, W.B.; Roberts, B.P.; Roland, S.; Weibel, J.-M. Tetrahedron 1997, 53, 15085. (d) Herpin, T.F.; Motherwell, W.B.; Weibel, J.-M. Chem. Commun. 1997, 923.
10. (a) Patel, S.T.; Percy, J.M.; Wilkes, R.D. Tetrahedron 1995, 51, 11327. (b) Patel, S.T.; Percy, J.M.; Wilkes, R.D. J. Org. Chem. 1996, 61, 166.
11. Chambers, R.D.; James, S.R. in: Stoddart, J. F. (eds) Comprehensive Organic Chemistry, 1, Pergamon Press, Oxford, 1979, 545.
12. Ojima, I. (ed) Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing, Oxford, 2009, p12-13.
13. (a) Smart, B. E. Organofluorine Chemistry Principles and Commercial Applications, Banks, R. E. Smart, B. E.; Tatlow, J. C. (Eds) Plenum, New York, 1994, 57. (b) Lee, V. J. Comprehensive Organic Synthesis, 4, Trost, B. M. (ed) Pergamon, Oxford, 1991, 69.
14. Katz, T. J. Angew. Chem. 2000, 112, 1997; Angew. Chem. Int. Ed. 2000, 39, 1921.
15. Grimme, S.; Harren, L.; Sobanski, A.; Vögtle, F. Eur. J. Org. Chem. 1998, 1491.

20
16. Ichikawa, J.; Wada, Y.; Fujiwara, M.; Sakoda, K. Synthesis 2002, 13, 1917.
17. Archibald, T. G.; Baum, K. J. Org. Chem. 1990, 55, 3562.
18. Baasner, B.; Hagemann, H.; Klauke, E. Ger. Offen, DE. 3 305 201.
19. Feiring, A. E. J. Org. Chem. 1980, 45, 1958.
20. Joyce, R. M. U.S. 2,462,402, 1949, to DuPont; Chem. Abstr. 1949, 43, 3834e.
21. For a review on the chemistry of highly fluorinated carbocations, see: Krespan, C. G.; Petrov, V. A. Chem. Rev., 1996, 96, 3269.
22. (a) Ichikawa, J.; Jyono, H.; Kudo, T.; Fujiwara, M.; Yokota, M. Synthesis 2005, 39. (b) Ichikawa, J.; Yokota, M.; Kudo, T.; Umezaki, S. Angew. Chem. Int. Ed. 2008, 47, 4870.
23. Yokota, M.; Fujita, D.; Ichikawa, J. Org. Lett. 2007, 9, 4639.
24. (a) Tedder, J. M. Angew. Chem. Int. Ed. 1982, 21, 401. (b) Low, H. C.; Tedder, J. M.; Walton, J. C. J. Chem. Soc., Faraday Trans. 1, 1976, 72, 1707.
25. Hirunsit, P.; Balbuena, P. B. J. Phys. Chem. A. 2008, 112, 4483.
26. (a) Percy, J.M. Contemporary Organic Synthesis 1995, 2, 251. (b) Ocampo, R.; Dolbier, W. R. Jr.; Paredes, R. J. Fluorine Chem. 1998, 88, 41. (c) Dolbier, W. R. Jr.; Ocampo, R. J. Org. Chem. 1995, 60, 5378.
27. Fuqua, S. A.; Duncan, W.G.; Silverstein, R.M. Tetrahedron Lett. 1964, 1461.
28. Naae, D. G.; Burton, D.J. J. Fluorine Chem. 1971, 1, 123.
29. Naae, D. G.; Burton, D.J. Synth. Commun. 1973, 3, 197.
30. Hayashi, S.; Nakai, T.; Ishikawa, N.; Burton, D.J.; Naae, D. G.; Kesling, H.S. Chem. Lett. 1979, 983.
31. Naae, D. G.; Kesling, H.S.; Burton, D.J. Tetrahedron Lett. 1975, 44, 3789.
32. Obayashi, M.; Ito, E.; Matsui, K.; Kondo, K. Tetrahedron Lett. 1982, 23, 2323.
33. Edwards, M. L.; Stemerick, D.M.; Jarvi, E.T.; Matthews, D.P.; McCarthy, J. R. Tetrahedron Lett. 1990, 31, 5571.
34. (a) Wheaton, G. A.; Burton, D. J. Tetrahedron Lett. 1976, 43, 895. (b) Wheaton, G. A.; Burton, D. J. J. Org. Chem., 1983, 48, 917.
35. Sabol, J. S.; McCarthy, J.R. Tetrahedron Lett. 1992,33,3101.
36. Burton, D. J.; Yang, Z-Y.; Morken, P. A. Tetrahedron, 1994, 50, 2993.
37. Sauvêtre, R.; Normant, J. F. Tetrahedron Lett. 1981, 22, 957.
38. (a) (X = OPh), Nakai, T. Chem. Lett. 1976, 1263. (b) (X = OTs), Nakai, T. Tetrahedron Lett. 1978, 4809. (c) (X = O-phosphazene), Evans, T. L. Organometallics 1982, 1, 1443. (d) (X = O-ally), Jarvi, E. T. Tetrahedron Lett. 1985, 26, 2861. (e) (X = OMEM), Percy, J. M. Tetrahedron Lett. 1990, 31, 3931. (f) (X = OC(O)Net2), Percy, J. M. Synlett 1992, 483. (g) (X = F) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. Chem. Commun. 1996, 49; Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem.

21
1999, 99, 127. (h) (X = Cl) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1997, 85, 151. (i) (X = F or Cl) Coe, P. L.; Burdon, J.; Haslock, I. B. J. Fluorine Chem. 2000, 102, 43.
39. (a) Ichikawa, J. J. Fluorine Chem. 2000, 105, 257. (b) Ichikawa, J.; Sonoda, T.; Kobayashi, H. Tetrahedron Lett. 1989, 30, 1641. (c) Ichikawa, J.; Ikeura, C.; Minami, T. J. Fluorine Chem. 1993, 63, 281. (d) Ichikawa, J.; Sonoda, T.; Kobayashi, H. Tetrahedron Lett. 1989, 30, 6379. (e) Ichikawa, J.; Hamada, S.; Sonoda, T.; Kobayashi, H. Tetrahedron Lett. 1992, 33, 337. (f) Ichikawa, J.; Yonemaru, S.; Minami, T. Synlett. 1992, 833. (g) Ichikawa, 1.; Moriya, T.; Sonoda, T.; Kobayashi, H. Chem. Lett. 1991, 961. (h) Ichikawa, J.; Ikeura, C.; Minami, T. Synlett. 1992, 739.
40. For a review on fluorinated organozinc reagents, see: Davis, C. R.; Burton, D. J. In The Chemistry of Organozinc Compounds, Chapter 16, Rappoport, Z.; Marek, I. (eds) John Wiley & Sons, Ltd. 2006.
41. For the generation of 2,2-difluoro-1-halovinylzinc(II) chloride (CF2=CXZnCl) at room temperature, see: (a) (X = F) Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 2731. (b) (X = Cl) Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 6979. (c) (X = Br) Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2004, 125, 561. (d) (X = I) Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2005, 126, 455. (e) Fujita, T.; Ichitsuka, T.; Fuchibe, K.; Ichikawa, J. Chem. Lett. 2011, 40, 986.
42. Hiyama, T., Obayashi, M., and Sawahata, M. Tetrahedron Lett. 1983, 24, 4113.
43. For PhLi: (a) Fontanelli, R., and Sianesi, D. Ann. Chim. (Roma),1965, 55, 862. For PhMgBr: (b) Kendrick, D. A., and Kolb,M. J. Fluorine Chem. 1989, 45, 265. (c) Bergstrom, D. E.; Ng, M. W.; Wong, J. J. J. Org. Chem. 1983, 48, 1902.
44. (a) Fuchikami, T.; Shibata,Y.; Suzuki, Y. Tetrahedron Lett. 1986, 27, 3173. (b)Kitazume, T.; Ohnogi, T. Synthesis, 1988, 614. (c) Kitazume, T.; Ohnogi, T.; Miyauchi, H.; Yamazaki, T. J. Org. Chem. 1989, 54, 5630.
45. (a) Begue, J-P.; Bonnet-Delpon, D.; Rock, M. H. J. Chem. Soc.,PerkinTrans. 1, 1996, 1409. (b) Begue, J.-P.; Bonnet-Delpon, D.; Rock, M.H. Tetrahedron Lett. 1995, 36, 5003.
46. Ichikawa, J.; Fukui, H.; Ishibashi, Y. J. Org. Chem. 2003, 68, 7800.
47. Portella, C.; Shermolovich, Y. Tetrahedron Lett. 1997, 38, 4063.
48. Uneyama, K.; Yang, F-Y.; Hirama, S.; Katagiri, T. Tetrahedron Lett. 1996, 37, 2045.
49. Tamura, M.; Sekiya, A. J. Fluorine Chem. 1995, 71, 119.
50. Fujita, M.; Hiyama, T. Tetrahedron Lett. 1986, 27, 3655.
51. Fujita, M.; Hiyama, T. Tetrahedron Lett. 1986, 27, 3659.
52. (a) Fritz, C. G.; Moore, E. P.; Selman, S. USP 3,114,778, 1963. (b) Fritz, C. G.; Selman, S. USP 3,291,843, 1966.
53. (a) Bey, P.; McCarthy, J.R.; McDonald, I.A. in: Welch, J.T. (eds.) Selective Fluorination in Organic and Bioorganic Chemistry, ACS Symp. Ser. No. 456, Am. Chem. Soc., Washington, DC, 105, 1991. (b) Ojima, I.; McCarthy, J. R.; Welch, J.T. (eds.), Biomedical Frontiers of Fluorine Chemistry, ACS Symp. Ser. No. 639, Am. Chem. Soc. Washington DC, 1996.
54. Heidelberger, C.; Chaudhuri, N. K.; Danenberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer,

22
J.; Pleven, E.; Scheiner, J. Nature 1957, 179, 663.
55. Magueur, G.; Crousse, B.; Ourévitch, M.; Bonnet-Delpon, D.; Bégué, J. P. J. Fluorine Chem. 2006, 127, 637.

23
Chapter 2. Synthesis of 1,1-Difluoroallenes and Their Applications
2.1 Introduction
Allenes are important compounds both as intermediates and targets in organic chemistry. Nearly 50 natural products comprising an allene structure have been known, and many of them show interesting biological activities (Figure 2.1).1–4 Fluoroallenes are, therefore, promising materials for pharmaceutical utility.
Figure 2.1 Bioactive allenic natural products.
From a synthetic viewpoint, fluorinated allenes are useful C3 units to synthesize organofluorine
compounds. The reported reactions of fluoroallenes can be classified into two types: i) addition or
substitution with nucleophiles5,6 and ii) cycloaddition with 1,3-dienes or 1,3-dipoles.7
1,1-Difluoroallenes readily undergo nucleophilic addition with Grignard reagents in the presence of
Cu(I) salt. The addition occurred regioseletiviely at the α carbon, and a subsequent elimination of F– affords
substituted monofluoroallenes (SNV products, Equation 2.1).6a On the other hand, thiols readily react with
1,1-difluoroallenes on the γ carbon to form γ,γ-difluoroallyl sulfides (Equation 2.2).6b
OO
CuBr SMe2n-HexMgBr
THF, –78 oC
OO
63%
10 mol% KOHPhSH
THF, 60 oC F
F H
SPh89%
(2.1)
(2.2)
F
F
F
F
F
n-Hex
Calculated electrostatic charges and coefficients of the LUMO of parent 1,1-difluoroallene are shown in
Figure 2.2. The difluoromethylene carbon (Cα) has a positive charge, while the γ carbon has a large
coefficient of LUMO. On the basis of the results of calculation, the experimental results can be interpreted as
follows: The reactions with hard nucleophiles such as organometals proceed under charge control, and the
nucleophiles thus attack the α carbon. The reactions of soft nucleophiles such as thiolates proceed under
orbital control, and the nucleophiles thus attack the γ carbon.

24 24
Figure 2.2 Calculated electrostatic charges and coefficients of LUMO of 1,1-difluoroallene (DFT, B3LYP/6-31G*).
The other type of reactions of 1,1-difluoroallenes is cycloaddition reactions. The Diels–Alder reactions
with 1,3-dienes and [3+2] cycloaddition reactions with 1,3-dipoles proceed on the internal, non-fluorinated
alkene moiety to give the corresponding exo-difluoromethylene compounds.5,6 It is worthy to note that the
reactivity of allenes toward the Diels–Alder reaction is strongly affected by the substitution of fluorine. For
instance, 1,1-difluoroallene (CF2=C=CH2) gives the Diels–Alder products under very mild conditions (–20
ºC, 1 min, Scheme 2.1). The monofluorinated allene (CHF=C=CH2) requires a longer reaction time at higher
temperature (0 °C, 100 h) to complete the reaction, while the fluorine-free counterpart (CH2=C=CH2)
requires vigorous conditions (200 °C, 6 h).7 These reactivities can be rationalized by considering that the
LUMO levels of fluoroallenes are lower than those of the fluorine-free counterparts.
Scheme 2.1 The Diels–Alder reactions of (fluoro)allenes with cyclopentadiene.
Besides the two types of reactions, some reactions of 1,1-difluoroallenes have been reported recently:
Difluorohomoallenyl bromide undergoes a cross coupling reaction with aryl halides to provide
1,1-difluoro-1,3-dienes (Equation 2.3).8 Molybdenum-catalyzed intramolecular [2+2] cycloaddition reaction
is also reported (Equation 2.4).9

25
Although 1,1-difluoroallenes are promising as pharmaceuticals and synthetic intermediates, the
methods for their synthesis have not been fully developed: The parent 1,1-difluoroallene (CF2=C=CH2) has
been known since the 1950s.10 β-Elimination of LiF from trifluoromethylvinyllithiums, readily prepared
from 2-bromo-1,1,1-trifluoro-2-alkenes5a or trifluoromethyl substituted hydrazones,11 affords
1,1-difluoroallenes (Scheme 2.2). α,α-Difluoropropargyl bromides act as both nucleophiles and electrophiles
for the synthesis of difluoroallenes. Treatment of difluoropropargyl bromide with indium metal and then with
formaldehyde affords fluorinated homoallenylalcohols (Scheme 2.3).12 On treatment with Grignard reagents
in the presence of copper(I) salt, difluoropropargyl bromide gives the corresponding SN2’-type products.6a In
spite of these results, synthesis of difluoroallenes have been still limited, especially in terms of disubstituted
derivatives.
H
HCF3
Br
n-BuLiH
HCF3
Li
H
H
R
HCF3
N NHSO2Ar
RCF3
N NSO2Ar
n-BuLi(2 eq)
RCF3
Li R
CF2
CF2
45–80%
97%
Scheme 2.2 Synthesis of difluoroallenes via trifluoromethylvinyllithiums.

26 26
In(0) Si(i-Pr)3
RMgBr
OH
HCHO
Si(i-Pr)3BrCF2
CuXSN2'
Si(i-Pr)3
R
FF
Si(i-Pr)3
In
CF2
CF2
35–94%
67%
Scheme 2.3 Synthesis of difluoroallenes from difluoropropargyl bromide.
In this chapter, practical synthesis of 1,1-difluoroallenes from trifluoroiodoethane is described. This
method is of wide generality, and difluoroallenes bearing functionalities such as ester and pyridine ring are
synthesized. Novel Friedel–Crafts-type cyclizations of the prepared 1,1-difluoroallenes are also described.

27
2.2 Initial Exploration of Synthesis of 1,1-Difluoroallenes
There are three possible approaches to construct the C=C=C moiety of difluoroallenes.1b These include:
(A) Construction of the allene moiety with the degree of unsaturation in the reaction course remain two: eg. substitution reactions of difluoropropargyl compounds or conjugated 1,3-dienes.
(B) Construction of the allene moiety with the degree of unsaturation of the reactant(s) less than two: eg. elimination reactions of alkenes.
(C) Construction of the allene moiety with the degree of unsaturation of the reactant(s) larger than two: addition reactions to a conjugated enyne or related compounds.
Since the approach C is not practical because of the difficulty of the preparation of unsaturated
fluorine-containing substrates (starting materials), the approaches A and B were examined.
Approach A. via SN2’ reaction of 1,1-difluoro-1,3-dienes
The simple retrosynthetic analysis for 1,1-difluoroallenes based on the appoach A is shown in Scheme 2.4. This proposal was inspired by the successes of fluorine-free allene synthesis via SN2’-type reaction of 1,3-dienes.13
Scheme 2.4 A retrosynthetic analysis for 1,1-difluoroallenes via SN2’ reaction.
The required difluoro-1,3-butadiene 2-6a, the SN2’ acceptor, bearing a leaving group on the 3 position
was prepared with difluorovinylzinc reagent (Table 2.1): The commercially available 2,2,2-trifluoroethyl
tosylate (p-toluenesulfonate) was treated with 2.1 equivalents of butyllithium.14 The resulting
1-tosyloxy-2,2-difluorovinyllithium 2-4a was subjected to transmetallation with zinc(II) chloride, affording
the corresponding zinc reagent.15 1-Tosyloxy-2,2-difluorovinylzinc chloride 2-5a was coupled with vinyl
bromide in the presence of palladium catalyst to give the required difluoro-1,3-butadiene in moderato yield.
It is also worth noting that 2-5a was so stable even at room temperature in the presence of
tetramethylethylenediamine (TMEDA) that it was identified by 19F NMR spectroscopy.

28 28
Table 2.1 Synthesis of 1,1-difluoro-1,3-diene from CF3CH2OTs via the Negishi coupling reaction.
Entry x y 2-6a, Yield (%)
1 1.02 1.1 53
2 1.55 1.55 60
The reactions of difluorodiene 2-6a with nucleophiles were examined (Table 2.2). However, instead of
the desired 1,1-difluoroallenes 2-7, monofluoroalkenes 2-8 (E/Z mixtures) were selectively obtained in high
yield via SNV pathway. The Tsuji-Trost reaction using 2-6a was also examined in vain (Scheme 2.5).
Table 2.2 Reactions of 1,3-diene 2-6a with nucleophiles.
CF2 •Nu
2-7
CF2
OTs
2-6a
OTsF
Nu
2-8
NuM+
THF, RT, 12 h
Entry NuM 2-7 (%) 2-8 (%)
1 (1.5 eq)
– 83 (10/1)
2 (1.1 eq)
– 90 (3/2)
CF2 •NuCF2
PdTsO
2-7 –
CF2
OTs
2-6a
OTsF
Nu
2-8 83%
EtO2C CO2Et
Bn Na+
–
expected intermediate
10 mol% Pd(PPh3)4Cs2CO3 (5 eq)
MeCN, RT, 12h+
Nu = CBn(CO2Et)2
+
Scheme 2.5 An attempt for the synthesis of difluoroallenes 2-7 by the Tsuji–Trost reaction.

29
2.3 A General Synthetic Method from Commercially Available 1,1,1-Trifluoro-2-iodoethane
Approach B. via β elimination reaction
The retrosynthetic analysis for 1,1-difluoroallenes based on the appoach B is shown in Scheme 2.6. The
targeted difluoroallenes are obtained by a β-elimination of 2-matalo-1,1-difluoro-1-alkenes bearing a leaving
group on the 3 position. The metalodifluoroalkenes are synthesized with a 2,2-difluorovinylanion with
appropriate electrophiles.
Scheme 2.6 A retrosynthetic analysis for 1,1-difluoroallenes via β-elimination.
Our group has succeeded in synthesizing 1,1-difluoroallenes by this approach (Scheme 2.7):16
1,1-Dibromo-2,2-difluoroethylene was selected as the starting material. Their lithiation with butyllithium at
–100 °C generates 1-bromo-2,2-difluorovinyllithium 2-4b. Treatment of the generated vinyllithium with
aldehydes then with acetic anhydride afforded 2-bromo-3,3-difluoroallylic acetates 2-9 in good yield. The
second lithiation of 2-9, performed with butyllithium at 0 °C,16 gave the desired 1,1-difluoroallenes 2-10 in
good yield.
n-BuLi (1.2 eq)
0 oC, Hexane, 1 min•
F
F
2-10 72–90%
R2
R1
F
F Li
AcO R2R1
– LiOAc
CF2
Br1) R1C(=O)R2 (1 eq) –100 oC, 15 min
2-4b
F
F Br
Br
n-BuLi (1 eq)
F
F Br
AcO R2R1
2-9 80–93%
Li–100 oC, 15 minEt2O
2) Ac2O (1.5 eq) –100–0 oC, 2 h
Scheme 2.7 Synthesis of 1,1-difluoroallenes 2-10.
However, there are two drawbacks: (a) The starting material, CF2=CBr2, is a high-cost, potential
ozone-depleting substance, and is now unavailable because of the ban on its industrial manufacture. (b) The
reactive butyllithium is required, which restricts the choice of substrate.
To overcome these issues, the author considered as follows: First, the key intermediate,
1-bromo-2,2-difluorovinyl lithium 2-4b, would be replaced with the anion, generated from
1,1,1-trifluoro-2-haloethanes on treatment with base.6,7,10 1,1,1-Trifluoro-2-haloethanes are recognized to
have much lower ozone depletion potential (ODP) and manufactured industrially for the use as refrigerants
or as fluorinated intermediates. Second, elimination process of the allylic acetates 2-9 would be conducted

30 30
with a zero-valent metal. This alternation seemed to expand the scope of the substrates.
We selected 1,1,1-trifluoro-2-iodoethane as a starting material, because this compound is easy to handle
(bp. 55−56 °C/760 mmHg) and of no ozone depletion potential (ODP). The lithiation of
1,1,1-trifluoro-2-iodoethane with LDA (two equivalents) was performed at low temperature (–93 to –85 °C)
to generate 2,2-difluoro-1-iodovinyllithium successfully (2-4c, Table 2.3).17 The vinyllithium reacted with
Table 2.3 Preparation of 3,3-difluoro-2-iodoallylic acetates 2-9 from 1,1,1-trifluoro-2-iodoethane.
Entry Carbonyl Compound 3,3-Difluoro-2-iodoallylic Acetate 2-9 2-9 (%)
1
2-9a, 82
2
2-9b, 84
3
2-9c, 83
4
2-9d, 87
5
2-9e, 83
6
2-9f, 74
7
2-9g, 77
8
2-9h, 81

31
aldehydes, and then with acetic anhydride to afford the corresponding 3,3-difluoro-2-iodoallylic acetates 2-9
in good yield.
When methyl phenethyl ketone was used as a substrate, acetylation with acetic anhydride gave the
corresponding acetate only in 42% yield. The acetylation was, therefore, performed with isopropenyl acetate
and a catalytic amount of TsOH (Scheme 2.8).17,18 Thus, the yield increased dramatically to give acetate 2-9k
in 80% yield (two steps).
Scheme 2.8 Synthesis of 2-iodo-3,3-difluoroallylic acetate from ketone.
In order to promote the desired β-elimination of allylic acetates 2-9, zero-valent metals were examined.
Among the metals examined, only zinc gave the targeted 1,1-difluoroallenes (Table 2.4, Entries 7,8). No
reactions occurred when magnesium, aluminum, or iron was used. A complex mixture was obtained when a
Pd(0) complex was used (Table 2.4, Entries 9–12).
Table 2.4 Zero-valent-metal promoted β-elimination of 3,3-difluoroallylic acetates 2-9.
Entry Acetate 2-9 M0 2-10 (%)
1 2-9a Mg 2-10a, 0a 2 2-9c Mg 2-10c, 0a 3 2-9a Al 2-10a, 0a 4 2-9c Al 2-10c, 0a 5 2-9a Fe 2-10a, 0a 6 2-9c Fe 2-10c, 0a 7 2-9a Zn 2-10a, 72 8 2-9c Zn 2-10c, 92 9 2-9a Pd(PPh3)4 2-10a, traceb
10 2-9c Pd(PPh3)4 2-10c, traceb 11 2-9a Pd(PPh3)4 (0.1 eq) / Et2Zn (2 eq) 2-10a, traceb 12 2-9c Pd(PPh3)4 (0.1 eq) / Et2Zn (2 eq) 2-10c, traceb
a: No reaction. Most of acetates 2-9 were recovered. b: Complex mixture. Only a trace of 1,1-difluoroallenes 2-10 was observed in 19F NMR spectra.

32 32
The solvents and reaction times were optimized (Table 2.5). In most cases, 1,1-difluoroallenes 2-10
were obtained in good yield by treating the acetates 2-9 with two equivalents of zinc either in
N,N-dimethylformamide or in tetrahydrofuran at room temperature for 3–12 h (Entries 1–6). It should be
noted that 1,1-difluoroallene 2-10i was formed only in N,N-dimethylformamide (Entries 11,12). The yield of
1,1-difluoroallenes bearing a primary alkyl group decreased, when the reaction time was extended (Entries
1,2), whereas the yield of 1,1-difluoroallenes bearing a secondary or tertiary alkyl group at the 3-position
remained steady (Entries 3–5).19
Table 2.5 Optimization of solvents and reaction time.
Entry Acetate 2-9 x Solvent Time (h) 2-10 (%)
1 2-9a 2 DMF 3 2-10a, 86 2 2-9a 2 DMF 6 2-10a, 72a 3 2-9a 4 DMF 3 2-10a, 84 4 2-9a 2 THF 3 2-10a, 73 5 2-9a 2 THF 6 2-10a, 75 6 2-9c 2 DMF 3 2-10c, 83 7 2-9c 2 DMF 6 2-10c, 92 8 2-9c 2 DMF 12 2-10c, 90 9 2-9c 4 DMF 12 2-10c, 88
10 2-9c 2 THF 12 2-10c, 89 11 2-9i 2 DMF 8 h 2-10i, 71 12 2-9i 2 THF 12 h 2-10i, traceb
a: Allene 2-10a partly decomposed to form a complex mixture. b: Acetate 2-9i was recovered quantitatively.
1,1-Difluoroallenes 2-10 were synthesized from the prepared 3,3-difluoroallylic acetates 2-9 under the
optimized conditions (Table 2.6, Entries 1–8). Not only the aldehyde-derived difluoroallenes but also
ketone-derived difluoroallenes were synthesized efficiently (Entry 9).
It must be mentioned that 1,1-difluoroallenes bearing reactive functionality were successfully
synthesized because the procedures developed by the author are mild (Scheme 2.9). Difluoroallenes bearing
an ester functionality or a pyridine ring were obtained in good yield.

33
Table 2.6 Synthesis of 1,1-difluoroallenes 2-10 via zinc-mediated β-elimination of 3,3-difluoro-2-iodoallylic acetate 2-9.a
Entry Acetate 2-9 1,1-Difluoroallenes 2-10 Reaction time (h) 2-10 (%)
1 2-10a
3 86
2
2-10b 6 87
3 2-10c
6 82
4 2-10d
12 92
5
2-10e
12 93
6
2-10f
12 75
7
2-10g
6 89
8 2-10h
6 95
9 2-10k
8 86

34 34
Scheme 2.9 Synthesis of 1,1-difluoroallenes bearing an ester functionality or a pyridine ring.
In summary, the author has developed a practical and general method for the synthesis of
1,1-difluoroallenes from 1,1,1-trifluoro-2-iodoethane under mild reaction conditions. This facile and
low-cost synthesis allows 1,1-difluoroallenes to be used as practical building blocks for the synthesis of
various useful fluorinated molecules.

35
2.4 Friedel–Crafts-type Cyclizations of 1,1-Difluoroallenes
One of the most important synthetic utility of difluoromethyl cations is for the Friedel–Crafts-type
cyclization. For example, as shown in Equation 1.4 (Chapter 1), difluoroalkenes bearing two aryl groups are
protonated with Magic Acid (FSO3H·SbF5) to generate difluoromethyl cations, stabilized by two fluorine
atoms. The domino Friedel–Crafts-type cyclization and subsequent dehydrogeation afforded [4]–[6]helices
in good yield.20
Although being useful, the helicene synthesis required the use of super acid (Magic Acid), because the
difluoromethyl cation precursor, difluoroalkenes, are highly electron-deficient due to the –I effect of fluorine.
Since the 1,1-difluoroallene has an electron-rich C=C double bond adjacent to the 1,1-difluorovinyl moiety,
the author considered that difluoromethyl cation can be generated from difluoroallenes with the aide of mild
acid (Scheme 2.10).
F
F
E+ F
F E+
F
F E
F
F E(Brønsted or Lewis acid)
++
difluoroallylic cation
Scheme 2.10 Generation of difluoroallylic cations by electrophilic activation of 1,1-difluoroallenes.
Friedel–Crafts-type cyclization of 1,1-difluoroallenes was examined by using various Brønsted and
Lewis acids (Table 2.7). Interestingly, fluoronaphtharene 2-11a was obtained from 1,1-difluoroallene 2-10f
(Entries 1–6). A trifluoromethylpropene 2-12a and hexafluoroisopropyl ester 2-13a were also formed by the
capture of the key difluoromethyl cation A with HF or HFIP molecule. It must be mentioned that these
Friedel–Crafts-type cyclizations were conducted via difluoromethyl cations, generated on treatment with
rather mild acid such as triflic acid or TiCl4.
The detailed mechanism for the formation of fluoronaphthalene 2-11a is shown in Scheme 2.11.
Activation of the ‘normal’ double bond of 1,1-difluoroallene by a Brønsted acid or a Lewis acid leads to the
formation of the key difluoromethyl cation intermediate A. The Friedel–Crafts-type cyclization of A
provides bicyclic intermediate B. Elimination of fluoride ion from B forms the second cationic intermediate
C, whose 1,2-methyl migration affords the product.

36 36
Table 2.7 Activation of a 1,1-difluoroallene and its intramolecular cyclization.
Yield (%)a Entry E+ Solvent
2-11a 2-12a 2-13a
1 TsOH HFIP + DCM (1:1) trace – – 2 TfOH HFIP 14 43 – 3 TfOH HFIP + DCM (1:1) 27 40 –
4 TiCl4 HFIP + DCM (1:1) 32 3 trace
5 ZrCl4 HFIP + DCM (1:1) 32 – –
6 BF3·OEt2 HFIP + DCM (1:1) 40 6 38
a: 19F NMR yield
Scheme 2.11 Proposed mechanism for the formation of fluoronaphthalene.
In conclusion, the author has developed a practical synthesis of substituted 1,1-difluoroallenes. The
difluoroallenes are found to be useful difluoromethyl cation precursors, and the Friedel–Crafts-type
cyclization of the difluoroallenes took place with mild acids to afford fluoronaphthalenes.

37
References and Notes
1. For recent comprehensive coverage see: (a) Ma, S. Chem. Rev. 2005, 105, 2829. (b) Krause, N., Hashmi, A. S. K. (Eds) Modern Allene Chemistry, Vols. 1 and 2, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. (c) Hashmi, A. S. K. Angew. Chem., Int. Ed. 2000, 39, 3590.
2. (a) Johnson, E. A.; Burdon, K. L.; J. Bacteriol. 1947, 54, 281. (b) Celmer, W. D.; Solomons, I. A. J. Am. Chem. Soc. 1952, 74, 1870-1871, 2245-2248, 3838-3842.
3. (a) Bendz, G. Ark. Kemi 1959, 14, 305–321, 475–481. (b) Cambie, R. C.; Hirschberg. A.; Jones, E. R. H.; Lowe, G. J. Chem. Soc. 1963, 4120. Syntheses: (c) Bohlmann, F.; Herbst, P.; Gleinig, H. Chem. Ber. 1961, 94, 948. (c) Evans, R. J. D.; Landor, S. R.; Regan, J. P. J. Chem. Soc. Perkin Trans. 1 1974, 552. (d) Landor, S. R.; Miller, B. J.; Regan, J. P.; Tatchell, A. R. J. Chem. Soc., Perkin Trans. 1 1974, 557. (e) de Graaf, W.; Smits, A.; Boersma, J.; van Koten, G.; Hoekstra, W. P. Tetrahedron 1988, 44, 6699.
4. (a) Bew, R. E.; Chapman, J. R.; Jones, E. R. H.; Lowe, B. E.; Lowe, G. J. Chem. Soc. C 1966, 129. (b) Bew, R. E.; Cambie, R. C.; Jones, E. R. H.; Lowe, G. J. Chem. Soc. C 1966, 135. (c) Landor, P. D.; Landor, S. R.; Leighton, P. J. Chem. Soc., Perkin Trans.1 1975, 1628. (d) Bohlmann, F.; Sucrow, W.; Queck, I. Chem. Ber. 1964, 97, 2586. (e) Bu’Lock, J. D.; Jones, E. R.H.; Leeming, P. R. J. Chem. Soc. 1955, 4270. (f) Bu’Lock, J. D.; Jones, E. R.H.; Leeming, P. R.; J. Chem. Soc. 1957, 1097. (g) Bu’Lock, J. D.; Gregory, H. J. Chem. Soc. 1960, 2280. (h) Jones, E. R. H.; Leeming, P. R.; Remers, W. A. J. Chem. Soc. 1960, 2257. (i) Schlingmann, G.; Milne, L.; Pearce, C. J.; Borders, D. B.; Greenstein, M.; Maise, W. M.; Carter, G. T. J. Antibiot. 1995, 48, 375. (j) Omura, S.; Imai, H.; Takeshima, H.; Nakagawa, A. Chem. Pharm. Bull. 1976, 24, 3139.
5. (a) Dolbier, W. R., Jr.; Burkholder, C. R.; Piedrahita, C. A. J. Fluorine Chem. 1982, 20, 637; (b) Dolbier, W. R., Jr.; Burkholder, C. R.; Winchester, W. R. J. Org. Chem. 1984, 49, 1518; (c) Dolbier, W. R., Jr.; Burkholder, C. R. Israel J. Chem. 1985, 26, 115; (d) Dolbier, W. R., Jr.; Wicks, G. E.; Burkholder, C. R. J. Org. Chem. 1987, 52, 2196; (e) Dolbier, W. R., Jr.; Burkholder, C. R.; Wicks, G. E.; Palenik, G. J.; Gawron, M. J. Am. Chem. Soc. 1985, 107, 7183.
6. (a) Mae, M.; Hong, J. A.; Xu, B.; Hammond, G. B. Org. Lett. 2006, 8, 479; (b) Xu, Y.-Y.; Jin, F.-Q.; Huang, W.-Y. J. Fluorine Chem. 1995, 70, 5.
7. Dolbier, W. R., Jr. Acc. Chem. Res. 1991, 24, 63.
8. Wang, Z.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547.
9. Mae, M.; Hong, J. A.; Xu, B.; Hammond, G. B. Org. Lett. 2006, 8, 479.
10. Blomquist, A. T.; Longone, D. T. J. Am. Chem. Soc. 1957, 79, 4981; (b) Knoth, W. H.; Coffman, D. D. J. Am. Chem. Soc. 1960, 82, 3873. (c) Kuhnel, M. F.; Lentz, D. J. Chem. Soc. Dalton Trans.2009, 4747.
11. Wang, Z.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547.
12. (a) Shen, Q.; Hammond, G. B. J. Am. Chem. Soc. 2002, 124, 6534. (b) Shen, Q.; Hammond, G. B. Org. Lett. 2001, 3, 2213.
13. (a) Claesson, A.; Quader, A.; Sahlberg, C. Tetrahedron Lett. 1983, 24, 1297. (b) Nishiyama, T., Esumi, T.; Iwabuchi, Y.; Irie, H.; Hatakeyama, S. Tetrahedron Lett. 1998, 39, 43. (c) Ogasawara, M.; Ikeda, H.; Hayashi, T. Angew. Chem., Int. Ed. 2000, 39, 1042. (d) Djahanbini, D.; Cazes, B.; Gore, J. Tetrahedron

38 38
Lett. 1984, 25, 203. (e) Djahanbini, D.; Cazes, B;, Gore, J. Tetrahedron 1987, 43, 3441. (f) Ogasawara, M.; Ikeda, H.; Nagano, T.; Hayashi, T. J. Am. Chem. Soc. 2001, 123, 2089. (g) Ogasawara, M.; Ikeda, H.; Nagano, T.; Hayashi, T. Org. Lett. 2001, 3, 2615. (h) Ogasawara, M.; Ueyama, K.; Nagano, T.; Mizuhata, Y.; Hayashi, T. Org. Lett. 2003, 5, 217.
14. For the generation of 2,2-difluoro-1-tosyloxyvinyllithium (CF2=C(OTs)Li), see: (a) Tanaka, K.; Nakai, T.; Ishikawa, N. Tetrahedron Lett. 1978, 19, 4809; (b) Ichikawa, J.; Hamada, S.; Sonoda, T.; Kobayashi, H. Tetrahedron Lett. 1992, 33, 337.
15. For the generation of 2,2-difluoro-1-halovinylzinc(II) chloride (CF2=CXZnCl) at room temperature, see: (a) (X = F) Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 2731. (b) (X = Cl) Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 6979. (c) (X = Br) Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2004, 125, 561. (d) (X = I) Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2005, 126, 455.
16. Yokota, M.; Fuchibe, K.; Ueda, M.; Mayumi, Y.; Ichikawa, J. Org. Lett., 2009, 11, 3994.
17. For the generation of 2,2-difluoro-1-halovinyllithium (CF2=CXLi), see: (a) (X = F) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. Chem. Commun. 1996, 49. (b) (X = F) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1999, 99, 127. (c) (X = Cl) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1997, 85, 151. (d) (X = F or Cl) Coe, P. L.; Burdon, J.; Haslock, I. B. J. Fluorine Chem. 2000, 102, 43.
18. Jeffery, E. A.; Satchell, D. P. N. J. Chem. Soc., 1962, 1876.
19. Hammond et al.reported that decomposition with loss of fluorine alkyl- and silyl-substituted difluoroallenes was observed if they were stored at ambient temperatures for 24 h, while these difluoroallenes could be stored neat for a month at 0 oC without noticeable decomposition. For detailed information, see: ref. 6a.
20. Ichikawa, J.; Yokota, M.; Kudo, T.; Umezaki, S. Angew. Chem. Int. Ed. 2008, 47, 4870.

39
Experimental Section
NMR spectra were recorded in CDCl3 at 500 MHz (1H NMR), 126 MHz (13C NMR), and 470 MHz (19F
NMR) on a Bruker AVANCE-500 instrument, or at 400 MHz (1H NMR), 101 MHz (13C NMR), and 376
MHz (19F NMR) on a Bruker AVANCE-400 instrument. Chemical shift values were given in ppm relative to
internal SiMe4 (for 1H NMR: δ 0.00), CDCl3 (for 13C NMR: δ 77.0), and C6F6 (for 19F NMR: δ 0.0). HRMS
(EI-TOF or ESI-TOF) data were recorded on a JEOL AccuTOF GCv (JMS-T100GCv) instrument or a JEOL
AccuTOF CS (JMS-T100CS) instrument. IR spectra were recorded by ATR (attenuated total reflectance)
method on a Horiba FREEXACT-II FT-IR instrument.
Column chromatography and preparative thin layer chromatography (preparative TLC) were conducted
on silica gel (Silica Gel 60 N, Kanto Chemical Co., Inc. for column chromatography and Wakogel B-5F,
Wako Pure Chemical Industries for PTLC, respectively). All reactions were conducted under argon. THF
and DMF were dried by passing over a column of activated alumina followed by a column of Q-5 scavenger
(Engelhard). 1,1,1-Trifluoroethyl 2,2,2-trifluoroethyl toluene sulfonate (CF3CH2OTs) and zinc powder were
purchased from Tokyo Chemical Industry Co., Ltd. and used without any treatment.
1,1-Dibromo-2,2-difluoroethylene was purchased from SynQuest Labs, Inc. 1,1,1-Trifluoro-2-iodoethane
was obtained from Tosoh F-tech, Inc., and distilled from activated molecular sieves 4A.
Synthesis and spectral data of 3,3-difluoro-iodoallyl acetates 2-9a–j
Synthesis of 2-9a is described as a typical procedure. Precursors 2-9b–j were prepared by the same
method.
To a THF (10 mL) solution of diisopropylamine (2.8 mL, 20 mmol) was added butyllithium (12.0 mL,
1.67 M in hexane, 20.0 mmol) over 10 min at 0 oC under argon. The resulting solution was allowed to stir for
an additional 15 min, and then cooled to –93 °C using a cold hexane bath. To this cold LDA solution was
added a THF (5 mL) solution of CF3CH2I (2.10 g, 10.0 mmol) over 10 min, keeping the temperature between
–93 °C and –85 °C. After stirring for 20 min at the same temperature, a THF (5 mL) solution of
3-phenylpropanal (1.34 g, 10.0 mmol) was added over 5 min, keeping the temperature between –93 °C and
–85 °C. The mixture was stirred for an additional 30 min, then warmed to –30 °C over 90 min. After acetic
anhydride (1.23 g, 12.0 mmol) was added, the mixture was allowed to warm to 0 °C over 2 h. The reaction
was quenched with saturated aqueous ammonium chloride, and the products were extracted with Et2O. The
combined organic layers were washed with brine and dried over anhydrous sodium sulfate. After removal of
the solvent under reduced pressure, the residue was purified by column chromatography (hexane–AcOEt,
20:1). The acetate 2-9a was obtained as a colorless liquid (3.01 g, 82%).

40
3,3-Difluoro-2-iodo-5-phenylpent-1-en-3-yl acetate (2-9a)
1H NMR (500 MHz, CDCl3): δ 1.87–1.93 (m, 1H), 2.05–2.17 (m, 1H), 2.07 (s, 3H), 2.58 (t, J = 7.2 Hz, 2H),
4.98 (t, J = 7.2 Hz, 1H), 7.17–7.22 (m, 3H), 7.29 (dd, J = 7.3, 7.6 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 20.9, 30.9, 36.0, 53.8 (dd, JCF = 25, 26 Hz), 68.9 (d, JCF = 3 Hz), 126.2,
128.2, 128.5, 140.2, 154.0 (dd, JCF = 286, 286 Hz), 169.6. 19F NMR (470 MHz, CDCl3): δ 89.2 (d, JFF = 22 Hz, 1F), 90.2 (d, JFF = 22 Hz, 1F).
IR (ATR): 3028, 2954, 1743, 1716, 1267, 1219, 1024, 698 cm–1.
HRMS (ESI+): m/z calcd for C13H13F2IO2Na [M + Na]+: 388.9826; found: 388.9830.
3,3-Difluoro-2-iodo-1-nonylprop-2-en-1-yl acetate (2-9b)
1H NMR: δ 0.88 (t, J = 6.9 Hz, 3H), 1.19–1.35 (b, 14H), 1.52–1.61 (m, 1H), 1.65–1.74 (m, 1H), 2.07 (s, 3H),
4.94 (t, J = 7.2 Hz, 1H). 13C NMR: δ 14.0, 20.9, 22.6, 24.5, 28.9, 29.2, 29.30, 29.34, 31.8, 34.2, 54.1 (dd, JCF = 24, 26 Hz), 69.3 (d,
JCF = 3 Hz), 153.9 (dd, JCF = 286, 299 Hz), 169.6. 19F NMR: δ 88.3 (d, JFF = 24 Hz, 1F), 89.6 (d, JFF = 24 Hz, 1F).
IR (ATR): 2925, 2856, 1749, 1716, 1458, 1371, 1269, 1225, 1024, 962, 604 cm–1.
HRMS (EI) calcd for C14H23F2IO2 – C2H4O2 [M − AcOH] 328.0500, found 328.0478.
3,3-Difluoro-2-iodo-1-[2-(1-naphthyl)ethyl]prop-2-en-1-yl acetate (2-9c)
1H NMR: δ 1.87–1.96 (m, 1H), 1.97 (s, 3H), 2.04–2.13 (m, 1H), 2.92 (t, J = 8.1 Hz, 2H), 5.00 (tdd, J = 6.4,
2.2, 1.4 Hz, 1H), 7.19 (d, J = 6.9 Hz, 1H), 7.28 (dd, J = 7.1, 7.1 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.41 (d, J
= 7.0 Hz, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 8.6 Hz, 1H). 13C NMR: δ 20.9, 28.1, 35.4, 53.8 (t, JCF = 25 Hz), 69.2 (d, JCF = 3 Hz), 123.4, 125.5, 126.0 (2C), 127.1,
128.9, 131.5, 133.9, 136.3, 154.1 (dd, JCF = 299, 286 Hz), 169.6. 19F NMR: δ 89.4 (d, JFF = 22 Hz, 1F), 90.3 (d, JFF = 22 Hz, 1F).
IR (ATR): 3047, 2939, 1743, 1716, 1371, 1269, 1225, 1026, 966, 798 cm–1.
HRMS (EI) calcd for C17H15F2IO2 [M]+ 416.0085, found 416.0059.
3,3-Difluoro-2-iodo-1-[2-(4-tert-butylphenyl)-1-methylethyl]prop-2-en-1-yl acetate (2-9d)
1H NMR (1:1 diastereomeric mixture): δ 0.74 (d, J = 6.4 Hz, 1.5H), 0.91 (d, J = 6.2 Hz, 1.5H), 1.15–1.45 (m,
1H), 1.31 (s, 9H), 2.06 (s, 1.5H), 2.09 (s, 1.5H), 2.05–2.13 (m, 0.5H), 2.34 (dd, J = 13.5, 9.5 Hz, 0.5H), 2.67
(d, J = 12.2 Hz, 0.5H), 2.92 (d, J = 13.5 Hz, 0.5H), 4.70 (d, J = 10.0 Hz, 0.5H), 4.75 (d, J = 9.5 Hz, 0.5H),
7.08 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 8.3 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H). 13C NMR (1:1 diastereomeric mixture): δ 14.5, 14.8, 20.8, 31.4, 34.3, 37.6, 38.0, 38.4, 39.1, 53.3 (dd, JCF =

41
26, 26 Hz), 73.3 (d, JCF = 3 Hz), 73.4 (d, JCF = 3 Hz), 125.2, 128.7, 128.8, 136.0, 136.4, 148.9, 149.0, 154.3
(dd, JCF = 298, 286 Hz), 154.4 (dd, JCF = 297, 286 Hz), 169.7, 169.8. 19F NMR (470 MHz, CDCl3) (1:1 diastereomeric mixture): δ 88.5 (d, JFF = 23 Hz, 0.5F), 89.1 (d, JFF = 22 Hz,
0.5F), 89.8 (d, JFF = 23 Hz, 0.5F), 90.6 (d, JFF = 22 Hz, 0.5F);
IR (ATR) (1:1 diastereomeric mixture): 2962, 2871, 1741, 1716, 1510, 1462, 1369, 1269, 1225, 1020, 968,
606, 573 cm–1.
HRMS (ESI+) calcd for C18H23F2IO2Na [M + Na]+ 459.0608, found 459.0610.
3,3-Difluoro-2-iodo-1-[1-methyl-1-phenylethyl]prop-2-en-1-yl acetate (2-9e)
1H NMR: δ 1.46 (s, 3H), 1.48 (s, 3H), 2.05 (s, 3H), 5.14 (dd, J = 1.9, 1.0 Hz, 1H), 7.24 (t, J = 7.7 Hz, 1H),
7.32 (dd, J = 7.7, 7.7 Hz, 2H), 7.40 (d, J = 7.7 Hz, 2H). 13C NMR: δ 20.8, 24.9, 25.0 (d, JCF = 9 Hz), 42.8, 48.0 (dd, JCF = 25, 25 Hz), 74.5 (d, JCF = 2 Hz), 126.7,
127.0, 128.0, 144.4, 153.8 (dd, JCF = 298, 286 Hz), 169.3. 19F NMR: δ 91.1 (d, JFF = 23 Hz, 1F), 91.3 (d, JFF = 23 Hz, 1F).
IR (ATR): 2976, 1745, 1709, 1498, 1442, 1369, 1265, 1219, 1030, 980, 768, 698, 609 cm–1.
HRMS (ESI+) calcd for C14H15F2IO2Na [M + Na]+ 402.9982, found 403.0012.
3,3-Difluoro-2-iodo-1-[1-methyl-1-(4-methylphenyl)ethyl]prop-2-en-1-yl acetate (2-9f)
1H NMR (500 MHz, CDCl3): δ1.43 (s, 3H), 1.44 (s, 3H), 2.06 (s, 3H), 2.32 (s, 3H), 5.12 (s, 1H), 7.12 (d, J = 8.2
Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ20.79, 20.84, 24.97, 24.99, 42.4 (dd, JCF = 1.8, 1.8 Hz), 48.0 (dd, JCF = 24.6, 24.6
Hz), 74.5 (d, JCF = 2.3 Hz), 126.8, 128.7, 136.2, 141.4, 153.7 (dd, JCF = 298, 285 Hz), 169.4. 19F NMR (470 MHz, CDCl3): δ91.0 (d, JFF = 23 Hz, 1F), 91.2 (d, JFF = 23 Hz, 1F).
IR (CHCl3): 2976, 1745, 1709, 1265, 1221, 978, 816 cm‐1.
HRMS (EI): m/z calcd for C13H13F2I ([M–AcOH]+): 334.0030; found: 334.0021.
3,3-Difluoro-2-iodo-1-[1-(4-methoxylphenyl)ethyl]prop-2-en-1-yl acetate (2-9g)
1H NMR (500 MHz, CDCl3): δ1.32 (d, J = 6.5 Hz, 3H), 2.15 (s, 3H), 3.00-3.08 (m, 1H), 3.80 (s, 3H), 5.01 (ddd,
J = 11, 2.5, 1.0 Hz, 1H), 6.83 (d, J =8.5 Hz, 2H), 7.15 (d, J=9.0 Hz, 2H). 19F NMR (470 MHz, CDCl3): δ88.4 (1F, d, JFF=22 Hz), 89.0 (1F, dd, JFF = 23 Hz, JFH = 2.2 Hz).
3,3-Difluoro-2-iodo-1-(2-phenylpropyl)prop-2-en-1-yl acetate (2-9h)
1H NMR (6:4 diastereomeric mixture): δ 1.28 (d, J = 7.0 Hz, 1.8H), 1.29 (d, J = 7.0 Hz, 1.2H), 1.80–1.88 (m,
1H), 1.95 (s, 1.8H), 1.98–2.05 (m, 0.4H), 2.06 (s, 1.2H), 2.06–2.12 (m, 0.6H), 2.62–2.69 (m, 0.6H),

42
2.70–2.78 (ddq, J = 7.0, 7.0, 7.0 Hz, 0.4H), 4.78 (dd, J = 6.5, 6.5 Hz, 0.6H), 4.84 (dd, J = 7.5, 7.5 Hz, 0.4H),
7.16 (dd, J = 7.0, 5.0 Hz, 1.8H), 7.20 (dd, J = 7.5, 7.5 Hz, 1.2H), 7.28 (t, J = 7.5 Hz, 2H). 13C NMR (6:4 diastereomeric mixture): δ 20.8, 20.9, 21.7, 23.1, 35.8, 36.0, 42.3, 42.7, 54.0 (dd, JCF = 25, 25
Hz), 54.2 (dd, JCF = 26, 26 Hz), 68.0 (d, JCF = 3 Hz), 126.5, 126.7, 126.8, 128.58, 128.61, 145.2, 145.5, 153.7
(d, JCF = 299, 286 Hz), 153.9 (d, JCF = 300, 286 Hz), 169.4, 169.5. 19F NMR (6:4 diastereomeric mixture): δ 88.8 (d, JFF = 23 Hz, 0.4F), 89.6 (d, JFF = 21 Hz, 0.6F), 89.7 (d, JFF
= 23 Hz, 0.4F), 90.2 (d, JFF = 21 Hz, 0.6F).
IR (ATR) (6:4 diastereomeric mixture): 3028, 2962, 1747, 1716, 1495, 1452, 1371, 1269, 1225, 1020, 978,
700 cm–1.
HRMS (ESI+) calcd for C14H15F2IO2Na [M + Na]+ 402.9982, found 403.0000.
3,3-Difluoro-2-iodo-1-[1-methyl-2-(3-pyridyl)ethyl]prop-2-en-1-yl acetate (2-9i)
1H NMR (6:4 diastereomeric mixture): δ 0.75 (d, J = 7.0 Hz, 1.6H), 0.92 (d, J = 6.0 Hz, 1.4H), 2.10–2.11 (m,
4.4H), 2.40 (dd, J = 13.5, 9.5 Hz, 0.6H), 2.72 (d, J = 10.0 Hz, 0.4H), 2.99 (dd, J = 13.5, 4.5 Hz, 0.6H), 4.72
(d, J = 10.0 Hz, 0.6H), 4.78 (d, J = 9.5 Hz, 0.4H), 7.28 (dd, J = 8.0, 4.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H),
8.45 (s, 1H), 8.49 (d, J = 4.5 Hz, 1H). 13C NMR (6:4 diastereomeric mixture): δ 14.3, 14.6, 20.79, 20.83, 35.3, 35.6, 38.3, 39.0, 52.7 (dd, JCF = 25,
25 Hz), 72.9 (d, JCF = 3.5 Hz), 73.2 (d, JCF = 3.2 Hz), 123.4, 134.8, 135.2, 136.8, 136.9, 147.3, 147.5, 150.0,
150.1, 154.4 (dd, JCF = 299, 286 Hz), 154.5, (dd, JCF = 299, 286 Hz), 169.59, 169.62. 19F NMR (6:4 diastereomeric mixture): δ 89.0 (d, JFF = 22 Hz, 0.6F), 89.5 (d, JFF = 21 Hz, 0.4F), 90.3 (d, JFF
= 22 Hz, 0.6F), 91.2 (d, JFF = 21 Hz, 0.4F).
IR (ATR) (6:4 diastereomeric mixture): 2968, 2933, 1736, 1714, 1425, 1371, 1265, 1221, 1024, 968, 793,
715 cm–1.
HRMS (ESI+) calcd for C13H15F2INO2 [M + H]+ 382.0116, found 382.0117.
3,3-Difluoro-2-iodo-1-[2-(2-methoxycarbonylphenyl)ethyl]prop-2-en-1-yl acetate (2-9j)
1H NMR: δ 1.86–1.92 (m, 1H), 2.04–2.13 (m, 1H), 2.08 (s, 3H), 2.90 (ddd, J = 15.5, 10.0, 5.5 Hz, 1H), 2.98
(ddd, J = 15.5, 10.0, 5.5 Hz, 1H), 3.91 (s, 3H), 5.02 (ddt, J = 7.0, 2.0, 2.0 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H),
7.28 (dd, J = 7.5, 1.0 Hz, 1H), 7.44 (td, J = 7.5, 1.5 Hz, 1H), 7.91 (dd, J = 7.5, 1.4 Hz, 1H). 13C NMR: δ 20.9, 29.7, 36.2, 52.0, 53.9 (t, JCF = 26 Hz), 69.2 (d, JCF = 4 Hz), 126.4, 129.4, 130.9, 131.0,
132.2, 142.3, 154.0 (dd, JCF = 299, 286 Hz), 167.7, 169.7. 19F NMR: δ 89.1 (d, JFF = 23 Hz, 1F), 89.9 (d, JFF = 23 Hz, 1F);
IR (ATR): 3068, 2952, 1747, 1716, 1435, 1373, 1259, 1228, 1088, 1026, 966, 712 cm–1.
HRMS (ESI+) calcd for C15H15F2IO4Na [M + Na]+ 446.9881, found 446.9879.

43
Synthesis and spectral data of 3,3-difluoro-2-iodo-1-methyl-1-(2-phenylethyl)prop-2-en-1-yl acetate
2-9k
To a THF (5 mL) solution of diisopropylamine (1.1 mL, 8.0 mmol) was added butyllithium (4.8 mL,
1.67 M in hexane, 8.0 mmol) over 10 min at 0 oC under argon. The resulting solution was allowed to stir for
an additional 15 min, then cooled to –93 °C using a cold hexane bath. To this cold LDA solution was added
a THF (2 mL) solution of CF3CH2I (840 mg, 4.00 mmol) over 10 min, keeping the temperature between
–93 °C and –85 °C. After stirring for 20 min at the same temperature, a THF (2 mL) solution of
4-phenylbutan-2-one (593mg, 4.00 mmol) was added over 5 min, keeping the temperature between –93 °C
and –85 °C. The mixture was stirred for an additional 30 min, then warmed to –30 °C over 90 min. The
reaction was quenched with saturated aqueous ammonium chloride, and the products were extracted with
Et2O. The combined organic layers were washed with brine and dried over anhydrous sodium sulfate. After
removal of the solvent under reduced pressure, the residue was purified by column chromatography
(hexane–AcOEt, 10:1). This alcohol was used for the next step without further purification.
To a solution of the alcohol in isopropenyl acetate (3 mL) was added 4-methylbenzenesufonic acid
monohydrate (5 mg, 0.03 mmol). After refluxing for 4 h, the reaction was quenched with saturated aqueous
sodium hydrogen carbonate. The products were extracted with ether. The combined organic layers were
washed with brine and dried over anhydrous sodium sulfate. After removal of the solvent under reduced
pressure, the residue was purified by column chromatography (hexane–AcOEt, 30:1). Acetate 2-9k was
obtained as a colorless liquid (1.22 g, 80%, two steps).
3,3-difluoro-2-iodo-1-methyl-1-(2-phenylethyl)prop-2-en-1-yl acetate (2-9k)
1H NMR: δ 1.87 (d, J = 4.6 Hz, 1H), 2.04 (s, 3H), 2.11–2.23 (m, 2H), 2.58 (t, J = 8.6 Hz, 2H), 7.18–7.20 (m,
3H), 7.29 (dd, J = 7.0, 7.0 Hz, 2H). 13C NMR: δ 21.8, 22.9 (d, JCF = 7 Hz), 30.0, 42.8 (dd, JCF = 2 Hz), 59.7 (dd, JCF = 26, 22 Hz), 80.6 (d, JCF =
3 Hz), 126.1, 128.3, 128.5, 140.8, 152.5 (dd, JCF = 301, 281 Hz), 169.3. 19F NMR: δ 89.0 (dq, JFF = 33 Hz, JFH = 5 Hz, 1F), 97.3 (d, JFF = 33 Hz, 1F).
IR (ATR): 3028, 2931, 2862, 1790, 1741, 1712, 1496, 1454, 1369, 1238, 1196, 1068, 1020, 700 cm–1.
HRMS (ESI+) calcd for C14H15F2IO2Na [M + Na]+ 402.9982, found 402.9979.

44
Synthesis of 1,1-difluoroallenes 2-10a-l
Synthesis of 2-10a is described as a typical procedure. 2-10b–k were synthesized by the same
procedure.
To a suspension of zinc (powder, 131 mg, 2.00 mmol) in DMF or THF (3.0 mL) was added a DMF or
THF (2.0 mL) solution of 2-9a (366 mg, 1.00 mmol) at room temperature under argon. After stirring for 3 h,
the resulting reaction mixture was filtered to remove the excess zinc and then diluted with Et2O and brine.
The products were extracted with Et2O. The combined organic layer were washed with brine and dried over
anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the residue was purified by
column chromatography (pentane). Allene 2-10a was obtained as a colorless liquid (155 mg, 86%).
1,1-Difluoro-5-phenylpenta-1,2-diene (2-10a)
1H NMR (500 MHz, CDCl3): δ 2.53–2.61 (m, 2H), 2.81 (t, J = 7.5 Hz, 2H), 6.47 (tt, J = 6.1 Hz, JHF = 2.4 Hz,
1H), 7.17–7.22 (m, 3H), 7.30 (dd, J = 7.3, 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 33.76, 33.77, 121.4 (t, JCF = 6 Hz), 126.2, 128.4, 128.5, 140.6, 152.8 (t, JCF =
261 Hz), 170.1 (t, JCF = 36 Hz). 19F NMR (470 MHz, CDCl3): δ 60.0 (td, JFH = 5, 2 Hz, 2F).
IR (ATR): 3030, 2929, 2362, 2013, 1462, 1196, 744, 698 cm–1.
HRMS (EI): m/z calcd for C11H10F2 [M]+: 180.0751; found: 180.0749.
1,1-difluorododeca-1,2-diene (2-10b)
1H NMR (500 MHz, CDCl3): δ 0.88 (t, J = 7.0 Hz, 3H), 1.27–1.30 (m, 12H), 1.49 (tq, J = 7.5, 7.0 Hz, 2H),
2.23 (ttd, J = 7.0, 6.3, 6.0 Hz, 2H), 6.42 (tt, J = 6.3 Hz, JHF = 2.3 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 14.1, 22.7, 27.6, 28.9, 29.3, 29.4, 29.5, 31.9, 32.3 (t, JCF = 2 Hz), 122.5 (t,
JCF = 6 Hz), 152.5 (t, JCF = 261 Hz), 169.3 (t, JCF = 36 Hz). 19F NMR (470 MHz, CDCl3): δ 59.4 (td, JFH = 6, 3 Hz, 2F).
IR (ATR): 2925, 2856, 2011, 1462, 1246, 1194, 721 cm–1.
HRMS (EI): m/z calcd for C12H20F2 [M]+: 202.1533, found: 202.1516.
1,1-Difluoro-5-(1-naphthyl)panta-1,2-diene (2-10c)
1H NMR (500 MHz, CDCl3): δ 2.48–2.54 (m, 2H), 3.09 (t, J = 7.4 Hz, 2H), 6.36 (tt, J = 6.1 Hz, JHF = 2.4 Hz,
1H), 7.16 (d, J = 6.6 Hz, 1H), 7.25 (dd, J = 7.6 Hz, 1H), 7.32–7.39 (m, 2H), 7.59 (d, J = 7.5 Hz, 1H), 7.72 (d,
J = 8.0 Hz, 1H), 7.83 (d, J = 7.5 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 30.8, 33.0, 121.6 (t, JCF = 5.5 Hz), 123.4, 125.5, 125.6, 126.0, 126.1, 127.1,
128.9, 131.6, 133.9, 136.6, 152.9 (t, JCF = 261 Hz), 170.0 (t, JCF = 36 Hz).

45
19F NMR (470 MHz, CDCl3): δ 60.4 (dt, JFH = 2, 5 Hz, 2F).
IR (ATR): 3062, 2941, 2009, 1745, 1458, 1186, 791 cm–1.
HRMS (EI): m/z calcd for C15H12F2 [M]+: 230.0907; found: 230.0906.
5-(4-tert-Butylphenyl)-1,1-difluoro-4-methylpenta-1,2-diene (2-10d)
1H NMR (500 MHz, CDCl3): δ 1.05 (d, J = 6.6 Hz, 3H), 1.30 (s, 9H), 2.57 (dd, J = 13.0, 6.6 Hz, 1H),
2.59–2.68 (m, 1H), 2.75 (dd, J = 13.0, 7.6 Hz, 1H), 6.41 (ddd, J = 5.0 Hz, JHF = 2.5, 2.5 Hz, 1H), 7.08 (d, J =
8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 18.5, 31.4, 34.3, 38.4, 41.6 (d, JCF = 2 Hz), 125.2, 127.1 (dd, JCF = 6, 6 Hz),
128.8, 136.4, 149.1, 153.5 (dd, JCF = 261, 261 Hz), 168.6 (dd, JCF = 36, 36 Hz). 19F NMR (470 MHz, CDCl3): δ 60.0 (ddd, JFF = 127 Hz, JFH = 3, 3 Hz, 1F), 60.3 (ddd, JFF = 127 Hz, JFH = 3,
3 Hz, 1F).
IR (ATR): 2964, 2870, 2009, 1446, 1238, 1190, 937, 858 cm–1.
HRMS (EI): m/z calcd for C16H20F2 [M]+: 250.1533; found: 250.1532.
1,1-Difluoro-4-methyl-4-phenylpenta-1,2-diene (2-10e)
1H NMR (500 MHz, CDCl3): δ 1.47 (s, 6H), 6.55 (t, JHF = 2.8 Hz, 1H), 7.23–7.25 (m, 1H), 7.31–7.36 (m,
4H). 13C NMR (126 MHz, CDCl3): δ 28.0, 42.4 (dd, JCF = 2 Hz), 125.9, 126.6, 128.5, 131.0 (t, JCF = 6 Hz), 146.2
(d, JCF = 2 Hz), 153.3 (dd, JCF = 262, 262 Hz), 167.1 (dd, JCF = 36, 36 Hz). 19F NMR (470 MHz, CDCl3): δ 61.1 (d, JHF = 2 Hz, 2F).
IR (ATR): 2972, 2931, 2873, 2009, 1601, 1495, 1435, 1192, 958, 854, 760, 696 cm–1.
HRMS (EI): m/z calcd for C12H12F2 [M]+ 194.0907, found: 194.0903.
1,1-Difluoro-4-methyl-4-(4-methylphenyl)penta-1,2-diene (2-10f)
1H NMR : δ 1.45 (s, 6H), 2.33 (s, 3H,), 6.54 (t, JHF = 2.4 Hz, 1H), 7.14 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0
Hz, 2H). 13C NMR: δ 20.9, 28.1, 42.1 (t, JCF = 2 Hz), 125.8, 129.1, 131.2 (t, JCF = 6 Hz), 136.2, 143.3 (t, JCF = 2 Hz),
153.3 (t, JCF = 261 Hz), 166.9 (t, JCF = 36 Hz). 19F NMR: δ 61.1 (d, JFH = 2 Hz, 2F).
IR (neat): 2972, 2927, 2009, 1514, 1437, 1190, 958, 816 cm–1;
Anal. Calcd for C13H14F2: C, 74.98; H, 6.78. Found: C, 75.07; H, 7.04.

46
1,1-Difluoro-4-(4-methoxyphenyl)-4-methylpenta-1,2-diene (2-10g)
1H NMR: δ 1.45 (s, 6H), 3.79 (s, 3H), 6.52 (t, JHF = 2.4 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.8 Hz,
2H). 13C NMR: δ 28.1, 41.8 (t, JCF = 2 Hz), 55.2, 113.7, 127.0, 131.3 (t, JCF = 5 Hz), 138.3 (t, JCF = 2 Hz), 153.2 (t,
JCF = 261 Hz), 158.2, 166.7 (t, JCF = 36 Hz). 19F NMR: δ 61.1 (d, JFH = 2 Hz, 2F).
IR (neat): 2968, 2009, 1512, 1437, 1250, 1182, 1036, 829 cm–1.
Anal. Calcd for C13H14F2O: C, 69.63; H, 6.29. Found: C, 69.73; H, 6.52.
1,1-Difluoro-5-phenylhexa-1,2-diene (2-10h)
1H NMR (500 MHz, CDCl3): δ 1.30 (d, J = 7.0 Hz, 3H), 2.44–2.60 (m, 2H), 2.95 (qdd, J = 7.1, 7.1, 7.1 Hz,
1H), 6.28 (dddd, J = 6.9, 6.9 Hz, JHF = 2.4, 2.4 Hz, 1H), 7.18–7.23 (m, 3H), 7.30 (t, J = 6.5 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 21.6, 38.8, 40.7, 120.4 (dd, JCF = 6, 6 Hz), 126.4, 126.9, 128.5, 145.7, 152.4
(dd, JCF = 260, 260 Hz), 170.9 (dd, JCF = 36, 36 Hz). 19F NMR (470 MHz, CDCl3): δ 59.1 (br ddd, JFF = 122 Hz, JFH = 6, 4 Hz, 1F), 59.6 (br ddd, JFF = 122 Hz,
JFH = 7, 4 Hz, 1F).
IR (ATR): 3030, 2964, 2009, 1726, 1603, 1495, 1458, 1240, 1190, 760, 698 cm–1.
HRMS (EI): m/z calcd for C12H12F2 [M]+: 194.0907; found: 194.0906.
3-(5,5-Difluoro-2-methylpenta-3,4-dien-1-yl)pyridine (2-10i)
1H NMR (400 MHz, CDCl3): δ 1.09 (d, J = 8.0 Hz, 3H), 2.60–2.72 (m, 2H), 2.81 (dd, J = 12.7, 6.0 Hz, 1H),
6.42 (dt, J = 8.0 Hz, JHF = 2.6 Hz, 1H), 7.23 (ddd, J = 7.8, 4.8, 0.6 Hz, 1H), 7.49 (ddd, J = 7.8, 2.1, 1.9 Hz,
1H), 8.45 (d, J = 1.9 Hz, 1H), 8.48 (dd, J = 4.8, 1.5 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 18.5, 38.0 (t, JCF = 2 Hz), 38.9 (t, JCF = 2 Hz), 123.3, 126.0 (t, JCF = 6 Hz),
134.8, 136.5, 147.7, 150.3, 153.4 (t, JCF = 262 Hz), 169.4 (t, JCF = 36 Hz). 19F NMR (376 MHz, CDCl3): δ 60.5 (d, J = 120 Hz, 1F), 60.7 (d, J = 120 Hz, 1F).
IR (ATR): 2970, 2931, 2009, 1576, 1446, 1188, 1026, 939, 856, 796, 714 cm–1.
HRMS (ESI+): m/z calcd for C11H12F2N [M + H]+: 196.0938; found: 196.0947.
Methyl 2-(5,5-difluoropenta-3,4-dien-1-yl)benzoate (2-10j)
1H NMR (500 MHz, CDCl3): δ 2.54–2.59 (m, 2H), 3.13 (t, J = 7.5 Hz, 2H), 3.88 (s, 3H), 6.49 (tt, J = 6.0 Hz,
JHF = 2.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 7.27 (dt, J = 7.5, 1.0 Hz, 1H), 7.42 (td, J = 7.5, 1.5 Hz, 1H), 7.92
(dd, J = 7.5, 1.5 Hz, 1H).

47
13C NMR (126 MHz, CDCl3): δ 32.6, 33.9, 51.9, 121.6 (t, JCF = 6 Hz), 126.3, 129.3, 130.9, 131.1, 132.1,
142.6, 152.6 (t, JCF = 261 Hz), 167.7, 169.7 (t, JCF = 36 Hz). 19F NMR (470 MHz, CDCl3): δ 60.0 (s, 2F).
IR (ATR): 2952, 2009, 1716, 1460, 1254, 1186, 1130, 1082, 962, 748, 708 cm–1.
HRMS (EI): m/z calcd for C13H12F2O2 [M]+: 238.0805; found: 238.0805.
1,1-Difluoro-3-methyl-5-phenylpenta-1,2-diene (2-10k)
1H NMR (500 MHz, CDCl3): δ 1.91 (t, J = 5.0 Hz, 3H), 2.40–2.48 (m, 2H), 2.74 (t, J = 8.2 Hz, 2H),
7.13–7.18 (m, 3H), 7.25 (t, J = 7.6 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 22.8, 33.4, 38.6, 126.1, 128.3, 128.4, 132.3 (t, JCF = 6 Hz), 141.0, 150.4 (t,
JCF = 260 Hz), 163.0 (t, JCF = 35 Hz). 19F NMR (470 MHz, CDCl3): δ 61.5 (tq, J = 5, 5 Hz, 2F).
IR (ATR): 3064, 2922, 2360, 2004, 1801, 1604, 1481, 1173, 1043, 995, 696 cm–1.
HRMS (EI): m/z calcd for C12H12F2 [M]+: 194.0907; found: 194.0909.
Friedel–Crafts Type Cyclizations of 1,1-Difluoroallenes
BF3•OEt2 (0.03 mL, 0.24 mmol) was added to a solution of 2-10f (50 mg, 0.24 mmol) in
(CF3)2CHOH–CH2Cl2 (1:1, 3.0 mL) at room temperature under argon. After stirring for 30 min at the same
temperature, the reaction was quenched with aq. NH4Cl, and organic materials were extracted with Et2O
three times. The combined extracts were washed with brine and dried over Na2SO4. After removal of the
solvent under reduced pressure, the residue was purified by column chromatography on silica gel (pentane)
to give 2-11a (18 mg, 40%) as a pale yellow liquid.
4-Fluoro-1,2,6-trimethylnaphthalene (2-11a)
1H NMR (500 MHz, CDCl3): δ 2.44 (s, 3H), 2.52 (s, 3H), 2.52 (s, 3H), 6.95 (d, JHF = 11.5 Hz, 1H), 7.36 (dd,
J = 8.7 Hz, JHF = 1.8 Hz, 1H), 7.83 (s, 1H), 7.90 (dd, J = 8.7 Hz, JHF = 2.0 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 14.1, 20.6 (d, JCF = 1 Hz), 21.6, 112.2 (d, JCF = 19 Hz), 119.6 (d, JCF = 5 Hz),
122.4 (d, JCF = 16 Hz), 123.7 (d, JCF = 3 Hz), 126.6 (d, JCF = 31 Hz), 128.8, 132.1 (d, JCF = 7 Hz), 132.2 (d,
JCF = 5 Hz), 134.3 (d, JCF = 2 Hz), 156.4 (d, JCF = 248 Hz). 19F NMR (470 MHz, CDCl3): δ 33.2 (br d, JFH = 12 Hz, 1F).
IR (ATR): 2921, 1612, 1444, 1384, 1355, 1236, 1076, 809 cm–1.
HRMS (FAB): m/z calcd for C13H14F ([M+H]+): 189.1080; found: 189.1092.

48
Chapter 3. Syntheses of 2,2-Difluorovinylic Silanes and Their Applications
3.1 Introduction
Vinylsilanes are stable and widely used reagents in organic synthesis.1–3 They readily react with
electrophiles in two ways under basic and acidic conditions. In the presence of a Lewis base (typically, F–),
the reaction proceeds via vinylsilicate intermediates. The other pathway is the reaction via β-silyl
carbocation intermediates, in which the cationic center is stabilized by an interaction of C–Si σ bond with a
vacant p orbital (hyperconjugation).
2,2-Difluorovinylic silanes 3-1, bearing a (pseudo)halogen substituent on the 1 position, are therefore
promising bifunctional intermediates for the synthesis of molecules containing a difluorovinylidene moiety
(CF2=C). Silanes 3-1 would react with electrophiles (nucleophiles) and then with nucleophiles (electrophiles)
to give a variety of fully substituted difluoroalkenes (Scheme 3.1). Thus, 3-1 would serve as a
2,2-difluorovinylidene synthon possessing both positive and negative charges on the same sp2-hybridized
carbon. Free 2,2-difluorovinylidene (CF2=C:) is a very useful intermediate as a “super electrophilic carbene”
for the synthesis of a variety of 1,1-difluoroalkenes.4 However, free 2,2-difluorovinylidene is extremely
unstable and only exists at very low temperature (< 40K) in argon matrix.
CF2X
SiR3
Nu–CF2
Nu
SiR3 E+
CF2E
Nu
CF2X
EE+ Nu–
3-1
F
F
Scheme 3.1 Concept of 2,2-difluoro-1-(pseudo)halovinylsilanes as a difluorovinylidene synthon.
In this section, two groups were selected as substituent X in 3-1. Syntheses of
(2,2-difluoro-1-iodovinyl)silane (X = I) and (2,2-difluoro-1-triflyloxyvinyl)silane (X = OTf,
trifluoromethanesulfonyloxy group) are described. Their synthetic applications including the reaction based
on a umpolung of the iodine substituent in the former vinylsilane are also described.

49
3.2 Synthesis of (2,2-Difluoro-1-iodovinyl)silanes from 1,1,1-Trifluoro-2-iodoethane from
1,1,1-Trifluoro-2-iodoethane
As described in Chapter 2, 1,1,1-trifluoro-2-iodoethane (CF3CH2I) is an environmentally preferable
starting material for the synthesis of organofluorine compounds. The author has revealed that
(2,2-difluoro-1-iodovinyl)silanes (CF2=C(I)SiR3) can be synthesized from CF3CH2I by a one-pot process
(Table 3.1): Lithiation of CF3CH2I was performed with LDA (2 equivalents) at low temperatures (–93 to
–85 °C). The generated 2,2-difluoro-1-iodovinyllithium (CF2=C(I)Li) then reacted with chlorosilanes (1
equivalent) to afford the desired vinylsilanes in high yield (Entries 1–4). These vinylsilanes were stable
enough to be isolated by column chromatography (silica gel). They can be stored at 0 °C for several months
without any decomposition, whereas they became discolored slightly when exposed to air at room
temperature for several days.
Table 3.1 Synthesis of (2,2-difluoro-1-iodovinyl)silanes 3-1 from 1,1,1-trifluoro-2-iodoethane.
LDA (2 eq)
–93 to –85 oC, 30 min, THFCF2
I
Li
ClSiR3 (1.05 eq)
–85 oC, 1 hCF2
I
SiR3
3-1
CF3CH2I
Entry ClSiR3 Product Yield (%)
1 ClSiMe3
3-1a 84 a
2 ClSiEt3
3-1b 85
3 ClSi(i-Pr)3 CF2I
Si(i-Pr)3 3-1c 83
4 ClSiMe2Ph
3-1d 84
5 ClSiMePh2
3-1e 39 b
a:19F NMR yield. Isolation resulted in only a partial success because of its low boiling point. b: Non-optimized yield.
While being useful, this procedure requires a low temperature (–85 °C) for the generation of the
unstable 2,2-difluoro-1-iodovinyllithium, which precludes scale-up for industrial application. In general,
fluorine-containing vinyllithiums CF2=CXLi decompose readily to form fluoroacetylene even at –70 °C.5
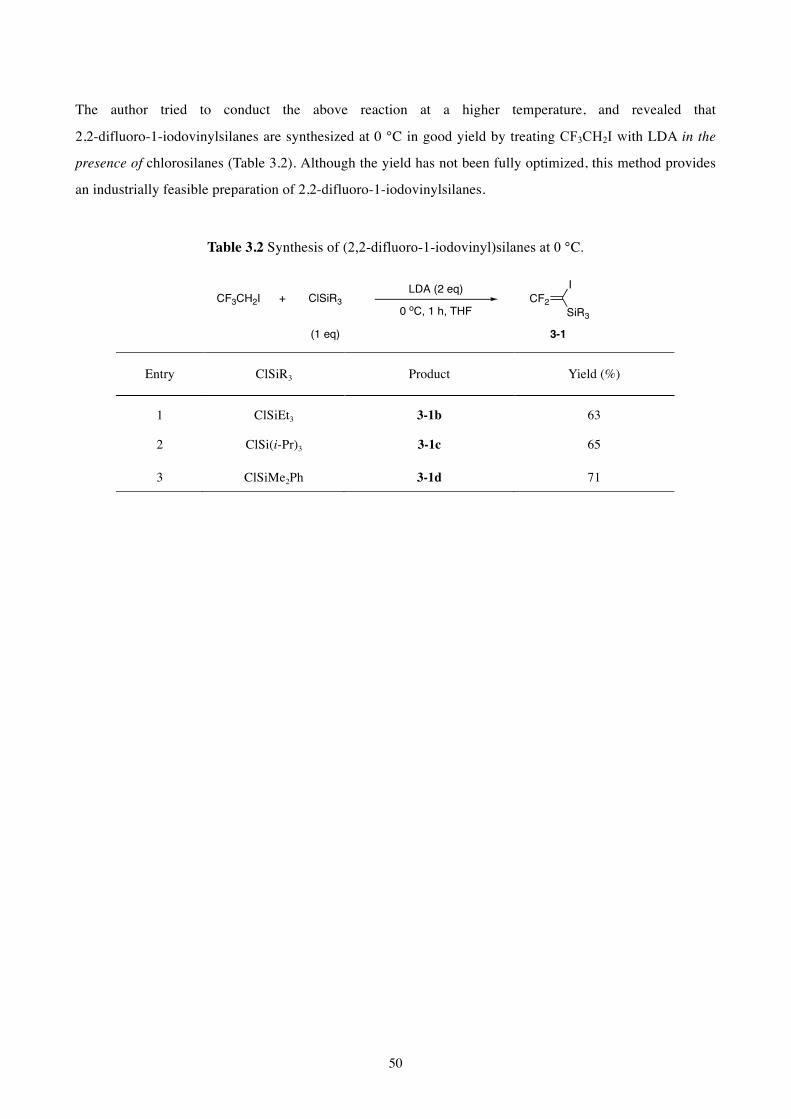
50
The author tried to conduct the above reaction at a higher temperature, and revealed that
2,2-difluoro-1-iodovinylsilanes are synthesized at 0 °C in good yield by treating CF3CH2I with LDA in the
presence of chlorosilanes (Table 3.2). Although the yield has not been fully optimized, this method provides
an industrially feasible preparation of 2,2-difluoro-1-iodovinylsilanes.
Table 3.2 Synthesis of (2,2-difluoro-1-iodovinyl)silanes at 0 °C.
LDA (2 eq)
0 oC, 1 h, THFCF2
I
SiR3
3-1
+ ClSiR3CF3CH2I
(1 eq)
Entry ClSiR3 Product Yield (%)
1 ClSiEt3 3-1b 63
2 ClSi(i-Pr)3 3-1c 65
3 ClSiMe2Ph 3-1d 71

51
3.3 Synthesis of (2,2-Difluoro-1-triflyloxyvinyl)silanes from 2,2,2-Trifluoroethanol
Aryl and alkenyl triflates are, generally, more reactive than the corresponding bromides in
cross-coupling reactions,6 and thus frequently used as pseudohalides in organic synthesis. Triflyloxy
2,2-difluorovinylsilanes have the potential to introduce a variety of difluorovinyl moieties to organic
molecules. For the synthesis of (2,2-difluoro-1-triflyloxyvinyl)silanes, two methods were examined.
Route A: Synthesis of (2,2-difluoro-1-triflyloxyvinyl)silanes from 2,2,2-trifluoroethyl triflate
The process used for the synthesis of (2,2-difluoro-1-iodovinyl)trialkylsilanes (Tables 3.1 and 3.2) is a
possible route to their triflyloxy analogues. Namely, treatment of 2,2,2-trifluoroethyl triflate, readily
prepared from 2,2,2-trifluoroethanol and triflic anhydride, with 2 equivalents of base, followed by silylation
with chlorosilanes might provide the targeted (2,2-difluoro-1-triflyloxyvinyl)silanes (Scheme 3.2).
CF2OTf ClSiR3 CF2
OTf
SiR3
Base (2 eq)CF3CH2OTf
Scheme 3.2 Possible route to (2,2-difluoro-1-triflyloxyvinyl)silanes from trifluoroethyl triflates.
In order to synthesize (2,2-difluoro-1-triflyloxyvinyl)silanes by the route A, several bases were
examined at low temperature in ethereal solvent (Scheme 3.3). However, when LDA or t-BuOK was
employed, complex mixture was obtained. When lithium hexamethyldisilazide (LHMDS) was employed,
SN2 replacement of the OTf group with the amide ion and subsequent silylation took place to give 3-2 in
79% yield.
ClSiMe3 (1 eq)LDA or t-BuOK (2 eq)CF3CH2OTf
–110 °C, ether, 30 min –110 to 0 °C, 1 hcomplex mixture
LHMDS (2 eq)CF3CH2OTf
–110 °C, ether, 30 minCF3CH2N(SiMe3)2
CF3 N(SiMe3)2
SiMe3
3-2 79%
Scheme 3.3 Attempts to synthesize triflyloxivinylsilane by the route A.
Route B: Synthesis of (2,2-difluoro-1-triflyloxyvinyl)silanes from 2,2,2-trifluoroethanol
Welch and his colleagues reported that 2,2-difluoro-1-silylethenol derivatives were synthesized from
2,2,2-trifluoroethanol and chlorosilanes via retro-Brook rearrangement (Scheme 3.4).7 When
2,2,2-trifluoroethanol was treated with 3 equivalents of LDA, sequential silylation, elimination, and

52
deprotonation occurred to give 2,2-difluoro-1-silylvinyllithium, which in turn underwent retro-Brook
rearrangement to generate difluoroenolate A. Treatment of the resulting A with water afforded
difluoroacetylsilanes 3-3 (Scheme 3.4a). Treatment with chlorosilanes instead of water led to the formation
of 2,2-difluoro-1-siloxyvinylsilanes 3-4 in good yield (Scheme 3.4b). The author considered that A would act
as an intermediate to synthesize (2,2-difluoro-1-triflyloxyvinyl)silanes by triflation.
LDA (3.3 eq)ClSiR3 (1 eq)
CF2
OSiR3
CF2
O
SiR3
retro-BrookTHF
ClSiR'3
HCF2
O
SiR3
CF2
OSiR'3
SiR3A
3-3
3-4
CF3CH2OH
H3O+(a)
(b)
Scheme 3.4 Welch Synthesis of 2,2-difluoro-1-trialkylsilylethenols based on retro-Brook rearrangement.
Although the attempt to trap the intermediate A with triflic anhydride (Tf2O) resulted in the formation
of a complex mixture, N-phenyltrifluoromethanesulfonimide (PhNTf2)8 successfully reacted with A to afford
the desired silanes (Table 3.3). When ClSiEt3 was used as a silylating agent, triethylsilylated difluorovinyl
triflate 3-1f was obtained in 72% yield (Entry 1). In the case of sterically more hindered ClSi(i-Pr)3 and
ClSi(t-Bu)Ph2, only a trace amount of the corresponding vinyl triflates 3-1g,h were obtained (Entries 2 and
3). The yield increased dramatically, when the reaction was performed at elevated temperature in the
presence of HMPA (Entry 4).
Table 3.3 Synthesis of triflyloxyvinylsilanes from 2,2,2-trifluoroethanol by retro-Brook rearrangement.
LDA (3.3 eq)ClSiR3 (1 eq)
CF2
O
SiR3 CF2
OTf
SiR3
A 3-1
PhNTf2 (1 eq)
Conditions BCF3CH2OH
THF, AdditiveConditions A
Entry ClSiR3 Additive Conditions A Conditions B Product Yield (%)
1 ClSiEt3 – –93 oC, 1 h then 0 oC, 3 h 0 oC, 0.5 h 3-1f 72
2 ClSi(i-Pr)3 – –93 oC, 1 h then 0 oC, 3 h 0 oC, 0.5 h 3-1g trace
3 ClSi(t-Bu)Ph2 – –93 oC, 1 h then 0 oC, 3 h 0 oC, 0.5 h 3-1h trace
4 ClSi(t-Bu)Ph2 HMPA –93 oC, 1 h then RT, 12 h 0 oC, 3 h 3-1h 61
HMPA: Hexamethylphosphoric triamide.
2,2,2-Trifluoroethanol is one of the most inexpensive fluorinated building blocks. This sequence
provides, therefore, a facile and practical method for the synthesis of (2,2-difluoro-1-triflyloxyvinyl)silanes.

53
3.4 Applications of 2,2-Difluorovinylsilanes
2,2-Difluorovinylsilanes bearing an iodo or a triflyloxy group at the 1 position were successfully
synthesized in the former sections (3.2 and 3.3). Then, they were applied to the reactions with electrophiles
and with nucleophiles.
Reactions with aldehydes
2,2-Difluoro-1-iodovinylsilanes reacted with aldehydes in the presence of cesium fluoride to form
3,3-difluoroallyl silyl ethers 3-5 (Table 3.4, Entries 1,2). This reaction also proceeded in a catalytic fashion
in terms of CsF (Entry 3). The plausible catalytic cycle is shown in Scheme 3.5.
Table 3.4 Reactions of 2,2-Difluoro-1-iodovinylsilanes with aldehydes.
CF2 Si(i-Pr)3
I RCHO (1.0–1.2 eq), CsF
Conditions CF2
IOSi(i-Pr)3
R3-1c 3-5
Entry RCHO (eq) CsF (eq) Conditions Product Yield (%)a
1 CHO
t-Bu 1 1.1 THF, RT, 5h 3-5a 81
2 2-ethylhexanal 1.2 1.5 diglyme, RT, overnight 3-5b 83
3 2-ethylhexanal 1.2 0.2 diglyme, 70 oC, 4 h 3-5c 77
a: 19F NMR yield.
FCF2 Si(i-Pr)3
I
CF2 Si(i-Pr)3
I
F
OR
CF2
I
R
O + F–Si(i-Pr)3
CF2
I
R
OSi(i-Pr)3
––
Scheme 3.5 Proposed catalytic cycle.

54
Reactions with boronic acids: Suzuki coupling reaction
The Suzuki coupling reaction of 2,2-difluoro-1-iodovinylsilanes took place smoothly by using a
palladium NHC complex as a catalyst (Table 3.5). The coupling reaction of vinyl iodide 3-1d with
phenylboronic acid was catalyzed by PdCl2(PPh3)2 to afford the corresponding β,β-difluorostyrene 3-6, albeit
in low yield (41%), and the starting 3-1d was recovered in 37% yield (Entry 1). In contrast, 3-1d was
consumed completely by using PEPPSI-IPr as a catalyst, which led to the production of 3-6 in 81% yield
(Entry 2).9
Table 3.5 Suzuki coupling reaction of 2,2-difluoro-1-iodovinylsilane 3-1d.
CF2
I
SiMe2Ph
3-1d
K2CO3 (2 eq)
3-6
1 mol% Pd complex+ PhB(OH)2 CF2
Ph
SiMe2Ph(1 eq)
Pd
NNR
R
R
R
N
Cl
PEPPSI-IPr (R = i-Pr)
Cl Cl
Entry Pd complex Conditions Yield (%) a
1 PdCl2(PPh3)2 THF/H2O (10/1), RT, 72 h 41 b
2 PEPPSI-IPr EtOH, 60 oC, 3 h 81
a:19F NMR yield. b: 3-1d was recovered in 37% yield.
Reaction with electrophiles: An umpolung of the iodo group
The reactions of difluorovinylsilanes with electrophiles on the silyl group or with nucleophiles on the
iodo group are so far descried as shown in Scheme 3.1. Apart from these reactions, the reaction with
electrophiles on the iodo group was realized by adopting the concept of umpolung (Scheme 3.6).
Iodovinylsilane 3-1c was treated with zinc metal (2 equivalents) in diglyme at room temperature, and then
the reaction was quenched with water. A protonated product of the generated vinylzinc species B, 3-7, was
obtained in 68% yield, accompanied by the formation of 32% yield of fluoroacetyene 3-8. This result
suggests that vinylzinc B was successfully generated in situ and would be used as a 2,2-difluoro-1-silylvinyl
anion equivalent.
F2C
I
Si(i-Pr)3
Zn (2 eq)
diglyme, RTovernight
F2C
H
Si(i-Pr)3+
F
Si(i-Pr)3
3-1c 3-7
CF2
ZnI
Si(i-Pr)3
H3O+
68% 32%3-8B
CF2 Si(i-Pr)3 Scheme 3.6 Activation of 2,2-difluoro-1-iodovinylsilanes by zinc at room temperature.

55
References and Notes
1. (a) For the comprehensive review on organosilicon chemistry, see: Colvin, E. W. Chem. Soc. Rev.. 1978, 7, 15. (b) For a review covering general organosilicon chemistry and a detailed summary of the early literature, see: Eaborn, C.; Bott, R. W. “Organometallic Compounds of the Group IV Elements”, Part I, MacDiarmid, A. G.. (Ed) Marcel Dekker, New York, 1968.
2. For some general methods of preparing vinylsilanes see: (a) Stork, G.; Jung, M. E.; Colvin, E.; Noel, Y. J. Am. Chem. Soc. 1974, 96, 3684. (b) Chan, T. H.; Baldassarre, A.; Massuda, D. Synthesis 1976, 801. (c) Taylor, R. T.; Degenhardt, C. R.; Melega, W. P.; Paquette, L. A. Tetrahedron Lett. 1977, 159. (d) Chamberlin, A. R.; Stemke, J. E.; Bond, F. T. J. Org. Chem. 1978, 43, 147.
3. (a) Fristad, W. E.; Dime, D. S.; Bailey, T. R.; Paquette, L. A. Tetrahedron Lett. 1979, 22, 1999. (b) Cooke, F.; Moerck, R.; Schwindeman, J.; Magnus, P. J. Org. Chem. 1980, 45, 1046. (c) Blumenkopf, T. A.; Overman, L. E. Chem. Rev. 1986, 86, 857.
4. Sander, W.; Kotting, C. Chem. Eur. J. 1999, 5, 24.
5. (a) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. Chem. Commun. 1996, 49. (b) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1997, 85, 151. (c) Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1999, 99, 127.
6. (a) Espino, G.; Kurbangalieva, A.: Brown, J. M. Chem. Commun. 2007, 1742. (b) Kamikawa, T.; Hayashi T. J. Org. Chem. 1998, 63, 8922. (c) Kamikawa, T.; Hayashi T. Tetrahedron Lett. 1997, 38, 7087.
7. Higashiya, S.; Chung, W. J.; Lim, D. S.; Ngo, S. C.; Kelly IV, W. H.; Toscano, P. J.; Welch, J. T. J. Org. Chem. 2004, 69, 6323.
8. (a) Hendrickson, J. B.; Bergeron, R. Tetrahedron Lett. 1973, 4607. (b) MC Murry, J. E.; Scott, W. J. Tetrahedron Lett. 1983, 24, 979.
9. O’Brien, C. J.; Kantchev, E. A. B.; Hadei, N.; Valente, C. Chass, G. A.; Nasielski, J. C.; Lough, A.; Hopkinson, A. C.; Organ, M. G. Chem. Eur. J. 2006, 12, 4743.

56
Experimental Section
Synthesis of (2,2-Difluoro-1-iodovinyl)silanes from 1,1,1-Trifluoro-2-iodoethane at –93 °C
Synthesis of 3-1b is described as a typical procedure. Silanes 3-1a and 3-1c–e were synthesized by the
same method.
To a THF (10 mL) solution of diisopropylamine (2.8 mL, 20 mmol) was added butyllithium (12.0 mL,
1.67 M in hexane, 20.0 mmol) over 10 min at 0 °C under argon. The resulting solution was allowed to stir
for an additional 15 min, and then cooled to –93 °C using a cold hexane bath. To this cold LDA solution was
added a THF (5 mL) solution of CF3CH2I (2.10 g, 10.0 mmol) over 10 min, keeping the temperature between
–93 °C and –85 °C. After stirring for 20 min at the same temperature, a THF (5 mL) solution of triethylsilyl
chloride (1.51 g, 10.0 mmol) was added over 5 min, keeping the temperature between –93 °C and –85 °C.
The mixture was stirred for an additional 1 h, then quenched with water, and the products were extracted
with Et2O. The combined organic layers were washed with brine and dried over anhydrous sodium sulfate.
After removal of the solvent under reduced pressure, the residue was purified by column chromatography
(hexane). The vinylsilane 3-1b was obtained as a colorless liquid (2.58 g, 85%).
Synthesis of 2,2-difluoro-1-iodovinylsilanes from 1,1,1-trifluoro-2-iodoethane at 0 oC
Synthesis of 3-1b is described as a typical procedure. Silanes 3-1c,d were synthesized by the same
method.
To a THF (10 mL) solution of diisopropylamine (2.8 mL, 20 mmol) was added butyllithium (12.0 mL,
1.67 M in hexane, 20.0 mmol) over 10 min at 0 °C under argon. The resulting solution was allowed to stir for
an additional 30 min at 0 °C. To a THF (30 mL) solution of CF3CH2I (2.10 g, 10.0 mmol) and triethylsilyl
chloride (1.51 g, 10.0 mmol) was added the prepared LDA solution at 0 °C over 10 min. After further stirring
for 5 min at 0 °C, the solution was quenched with water (50 mL), and the products were extracted with Et2O
(50 mL × 3). The combined organic layers were washed with brine (50 mL) and dried over anhydrous
sodium sulfate (5 g). After removal of the solvent under reduced pressure, the residue was purified by
column chromatography (pentane). The vinylsilane 3-1b was obtained as a colorless liquid (1.92 g, 63 %).
(2,2-Difluoro-1-iodovinyl)triethylsilane 3-1b
1H NMR (500 MHz, CDCl3): δ 0.75 (q, J = 7.9 Hz, 6H), 0.97 (t, J = 7.9 Hz, 9H). 13C NMR (126 MHz, CDCl3): δ 3.6, 7.0, 40.7 (d, JCF = 47 Hz), 156.1 (dd, JCF = 292, 300 Hz). 19F NMR (470 MHz, CDCl3): δ 95.0 (d, JFF = 22 Hz, 1F), 111.8 (d, JFF = 22 Hz, 1F).
IR (ATR): 2956, 2877, 2360, 1699, 1660, 1458, 1415, 1223, 1001, 845, 771, 721, 696, 600 cm–1.
HRMS (EI): m/z calcd for C8H15F2ISi: 303.9956; found: 303.9950.

57
(2,2-Difluoro-1-iodovinyl)triisopropylsilane 3-1c
1H NMR (500 MHz, CDCl3): δ 1.11 (d, J = 7.5 Hz, 18H), 1.35–1.44 (m, 3H). 13C NMR (126 MHz, CDCl3): δ 18.3, 156.1 (dd, JCF = 292, 300 Hz). 19F NMR (470 MHz, CDCl3): δ 96.7 (d, JFF = 22 Hz, 1F), 113.9 (d, JFF = 22 Hz, 1F).
IR (ATR): 2945, 2866, 2360, 1689, 1653, 1462, 1223, 1022, 985, 881, 771, 679, 648, 509 cm–1.
HRMS (EI): m/z calcd for C11H21F2ISi: 346.0425; found: 346.0421.
(2,2-Difluoro-1-iodovinyl)dimethyl(phenyl)silane 3-1d
1H NMR (500 MHz, CDCl3): δ 0.55 (s, 6H), 7.36–7.40 (m, 3H), 7.54 (d, J = 6.4 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ –1.8, 42.1 (d, JCF = 44 Hz), 127.9, 129.8, 133.8, 135.6, 156.1 (dd, JCF = 293,
301 Hz). 19F NMR (470 MHz, CDCl3): δ 96.9 (d, JFF = 18 Hz, 1F), 111.7 (d, JFF = 18 Hz, 1F).
IR (ATR): 2360, 1662, 1427, 1228, 1112, 1003, 860, 833, 806, 779, 731, 694, 656, 602 cm–1.
HRMS (EI): m/z calcd for C10H11F2ISi: 323.9643; found: 323.9651.
(2,2-Difluoro-1-iodovinyl)(methyl)diphenylsilane 3-1e
1H NMR (500 MHz, CDCl3): δ 0.81 (d, JHF = 1.6 Hz, 3H), 7.33–7.42 (m, 6H), 7.54–7.56 (m, 4H). 13C NMR (126 MHz, CDCl3): δ −2.0, 40.2 (d, JCF = 37 Hz), 128.0, 130.1, 133.5, 134.9, 156.8 (dd, JCF = 296,
301 Hz). 19F NMR (470 MHz, CDCl3): δ 99.6 (d, JFF = 12 Hz, 1F), 113.6 (d, JFF = 12 Hz, 1F).
IR (ATR): 3070, 2358, 1709, 1660, 1427, 1232, 1109, 999, 854, 791, 721, 694, 669, 604 cm–1.
HRMS (EI): m/z calcd for C15H13F2ISi: 385.9799; found: 385.9804.
Synthesis of 2,2-difluoro-1-(triethylsilyl)vinyl trifluoromethanesulfonate 3-1f
To a THF (5 mL) solution of diisopropylamine (2.0 mL, 14 mmol) was added butyllithium (8.4 mL,
1.67 M in hexane, 14 mmol) over 10 min at 0 °C under argon. The resulting solution was allowed to stir for
an additional 30 min at 0 °C. To a THF (3 mL) of CF3CH2OH (428 mg, 4.28 mmol) and triethylsilyl chloride
(645 mg, 4.28 mmol) was added the LDA solution at –93 °C over 10 min. After stirring for 1 h at the same
temperature, the solution was allowed to warm to 0 °C and stirred for 3 h. To the resulting solution was
added a THF (1 mL) solution of N-phenyl-trifluoromethanesulfonimide (PhNTf2) (1.61 g, 4.50 mmol) was
added at 0 °C. The mixture was stirred for an additional 30 min at 0 °C, then quenched with water. The
products were extracted with Et2O. The combined organic layers were washed with brine and dried over
anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the residue was purified by
column chromatography (hexane). The vinylsilane 3-1f was obtained as a colorless liquid (1.01 g, 72%).

58
1H NMR (500 MHz, CDCl3): δ 0.81 (q, 8.0 Hz, 6H), 1.0 (t, J = 7.9 Hz, 9H). 13C NMR (126 MHz, CDCl3): δ 2.1, 6.7, 115.1 (dd, JCF = 6, 69 Hz) 118.5 (q, JCF = 321 Hz) 162.4 (dd, JCF =
286, 318 Hz). 19F NMR (470 MHz, CDCl3): δ 65.0 (d, JFF = 33 Hz, 1F), 88.0 (d, J = 11 Hz, 3F), 90.7 (dq, JFF = 33, 11 Hz,
1F).
Synthesis of 2,2-difluoro-1-(tert-Butyldiphenylsilyl)vinyl trifluoromethanesulfonate 3-1h
To a THF (5 mL) solution of diisopropylamine (4.6 mL, 32 mmol) was added butyllithium (19.2 mL,
1.67 M in hexane, 32.0 mmol) over 10 min at 0 °C under argon. The resulting solution was allowed to stir
for an additional 30 min at 0 °C. To a THF (3 mL) of CF3CH2OH (1.00 g, 10.0 mmol), triethylsilyl chloride
(2.75 g, 10.0 mmol), and HMPA (1.03 g) was added the prepared the LDA solution at –93 °C over 10 min.
After stirring for 1 h at the same temperature, the solution was allowed to warm to room temperature and
stirred for 12 h. To the resulting solution was added a THF (5 mL) solution of PhNTf2 (3.57 g, 10.0 mmol) at
0 oC. The mixture was stirred for an additional 30 min at 0 °C, then quenched with water, and the products
were extracted with Et2O. The combined organic layers were washed with brine and dried over anhydrous
sodium sulfate. After removal of the solvent under reduced pressure, the residue was purified by column
chromatography (hexane). The vinylsilane 3-1h was obtained as a colorless liquid (2.76 g, 61%).
1H NMR (500 MHz, CDCl3): δ 1.19 (s, 9H), 7.38–7.40 (m, 4H), 7.42–7.45 (m, 2H), 7.64 (dd, J = 1.2, 6.0 Hz,
4H). 13C NMR (126 MHz, CDCl3): δ 28.1 (d, J = 1.3 Hz), 114.4 (dd, JCF = 4, 30 Hz), 119.7 (q, JCF = 321 Hz),
128.1, 129.7, 130.4, 136.0, 163.0 (dd, JCF = 290, 318 Hz). 19F NMR (470 MHz, CDCl3): δ 72.8 (d, JFF = 24 Hz, 1F), 88.0 (3F, d, J = 13 Hz), 93.2 (1F, dq, JFF = 38, 13
Hz).

59
Chapter 4. Synthesis of 3,3-Difluoroallyl Arenes via Cationic SN2’-type Reactions of 2-Trifluoromethyl-1-alkenes
4.1 Introduction
3,3,3-Trifluoropropene derivatives are manufactured industrially for several purposes. For example,
hexafluoropropylene (HFP) is a comonomer for the production of a melt processable fluoropolymer, FEP
(Figure 4.1).1 2,3,3,3-Tetrafluoropropene (HFO-1234yf) is a next generation refrigerant with a low global
warming potential (GWP).2 2-Bromo-3,3,3-trifluoropropene (Halon-1323) is a low-cost, fluorinated
synthetic intermediate, which is included in this section.3
CF3F
F
F
Hexafluoropropylene(HFP)
CF3
F
2,3,3,3-Tetrafluoropropylene(HFO-1234yf)
CF3
Br
2-Bromo-3,3,3-trifluoropropylene(Halon1323)
Figure 4.1 Industrially manufactured 3,3,3-trifluoropropene derivatives.
The synthetic utility of 2-trifluoromethyl-1-alkenes (3,3,3-trifluoropropene derivatives) is well
illustrated in their SN2’-type reactions (Scheme 4.1). Because of the negative hyperconjugation and the
β-carbanion stabilizing effect of fluorine substituent, nucleophiles attack the γ carbon of the
trifluoromethylalkenes in a regioselective manner. Subsequent elimination of the fluorine substituent as F–
results in migration of the C=C double bond to afford the SN2’-type products. Organolithiums, Grignard
reagents, lithium amides, etc. have been used for this transformation and a wide variety of 3,3-difluoroallylic
compounds are synthesized.4
Nu– = ester enolates CF2
EWG
CF2
EWG
CF2
EWG
CF2
EWG
EWGCF3
R1
CO2R3
R2
R
Nu– = RLi or RMgX
NR1R2
2-Trifluoromethy-1-alkenes
H
EWGCF3
Nu–
Nu– = R1R2NLi
Nu– = LiAlH4
Nu–
Scheme 4.1 Anionic SN2’-type reactions of 2-trifluoromethy-1-alkenes.

60
Although being useful, the SN2’-type reactions of 2-trifluoromethy-1-alkenes have a drawback in their
rather low reactivity: The reactions are conducted with reactive, anionic nucleophiles under strongly basic
conditions especially in the case of the trifluoromethylalkenes without an electron-withdrawing group at the
position β to the fluorines.5 These facts lead to limitations on the trifluoromethylalkenes and nucleophiles.
Transition metal-catalyzed reactions have been developed to overcome this problem. Our group
reported the palladium-catalyzed intramolecular insertion–elimination reaction of
2-trifluoromethyl-1-alkenes bearing oxime moieties, in which reaction proceeded via
alkylideneaminopalladium intermediates to provide difluoromethylene-substituted pyrrolines.6 Following our
palladium chemistry, Murakami has reported the rhodium(I)-catalyzed insertion–elimination reaction of
2-trifluoromethyl-1-alkenes with aryl boronates to afford substituted 1,1-difluoropropenes (Scheme 4.2).7
CF3 Ph
N OCOC6F5
CF3 Ph
N
OCH2OC6F5Pd N
CF2 Phβ-F eliminationPPh3 (1 eq)
Pd(PPh3)4 (0.1 eq)
ArCF3 +
OB
OPh
[RhCl(cod)]2 (2.5 mol%)MeMgCl (3 eq)
100 oC, 12 h
ArCF2
Ph
Scheme 4.2 Pd- and Rh-catalyzed SN2’-type reaction of 2-trifluoromethyl-1-alkenes.
Another possible alternative to the SN2’-type reactions is an acid-promoted cationic version of the
reactions (Scheme 4.3). Elimination of a fluoride ion from 2-trifluoromethyl-1-alkenes would be promoted
by Lewis acids to generate fluorine-stabilized allylic cations.8 With these cationic species, even less reactive
nucleophiles such as neutral arenes might undergo the SN2’-type reactions through a Friedel–Crafts-type
mechanism.
LA, – [LA–F]–RCF3
RCF2+
R'
Friedel–Crafts-type reaction
RCF2
R'
Scheme 4.3 Concept of Lewis acid-promoted, cationic SN2’-type reaction of 2-trifluoromethyl-1-alkenes.
So far, such attempts have led to only a partial success (Scheme 4.4). Kobayashi9 reported that BF3·OEt2
and AlCl3 promoted the reactions of 3,3,3-trifluoropropene with benzene to afford an addition product,
3,3,3-trifluoropropylbenzene, and a cyclized product, 1,1-difluoroindane, respectively. Okazaki10 reported

61
that the modified Al2O3 promoted reaction of the same compounds gave no 3,3-difluoroallylbenzene
(SN2’-type product) but again 3,3,3-trifluoropropylbenzene (addition product).
+ CF3
FFAlCl3
BF3·OEt2 CF358%
8%
CF3 CF2
17–27% –
modified Al2O3
Scheme 4.4 Earlier attempts of acid-promoted SN2’-type reaction of 2-trifluoromethyl-1-alkenes.
The author considered that the key to achieve the cationic SN2’-type reactions of
2-trifluoromethyl-1-alkenes is to find an appropriate reagent that strongly abstracts F–. In this chapter, a
survey on Lewis acids and the synthesis of 3,3-difluoroallylated benzenes by the SN2’-type reaction of
2-trifluoromethyl-1-alkenes with simple arenes are descried.

62
4.2 C–F Bond Cleavage of Trifluoromethyl Groups Promoted by Group 3 and Group 4 Metal Halides
A screening to find an efficient “fluorophilic” Lewis acid was performed by using
2-bromo-3,3,3-trifluoropropene and p-xylene as model substrates. The results of the survey are summarized
in Table 4.1. Although almost all of the Lewis acids examined were not effective, AlCl3, AlBr3, ZrCl4 and
HfCl4 did promote the desired SN2’-type reaction to afford 3,3-difluoroallyl-p-xylene in 60%, 55%, 97% and
47% yield, respectively (Entries 5, 6, 11, and 14).
Table 4.1 Screening of Lewis acids in the cationic SN2’-type reaction.
Entry Lewis acid 4-1a (%)a
1 MgCl2 0b
2 MgI2 0b
3 CaCl2 0b
4 BF3·OEt2 0b
5 AlCl3 60
6 AlBr3 55
7 Sc(OTf)3 0b
8 LaCl3 0b
9 YbCl3 0b
10 TiCl4 0b
11 ZrCl4 97
12 Cp2ZrCl2–AgClO4 (1:1) 0b
13 Cp2ZrCl2–AgBF4 (1:1) 0b
14 HfCl4 47
15 VCl3 0b
16 NbCl5 0b
17 FeCl2 0b

63
Table 4.1 (Cont.)
Entry Lewis acid 4-1a (%)a
18 RuCl3 0
19 PdCl2 0 c
20 CuCl2 0
21 AgOTf 0
22 AuCl 0
23 AuCl3 0
24 ZnCl2 0
a: 19F NMR yield based on C6F6 as an internal standard.
b: No reaction was observed.
c: 3,3,3-Trifluoropropene was observed in the presence of PdCl2.
ZrCl4 and AlX3 (X = Cl, Br) are less toxic and low-cost Lewis acids utilized in organic synthesis.11 The
bond dissociation energy of ZrF4 (Zr–F, 627 kJ/mol) and AlF3 (Al–F, 675 kJ/mol) are larger than that of CF4
(C–F, 514 kJ/mol).12 The author considered that the high affinity of Zr(IV) and Al(III) to fluorine played an
important role in the above mentioned SN2’-type reaction. It is worth noting that zirconocene dichloride in
combination with silver perchlorate is an efficient promoter in the glycosidation of fluorosugars (Scheme
4.5),13 whereas the Cp2ZrCl2/AgClO4 system was not effective for this SN2’-type reaction.
OAcO
OMeOMe
Me
FCp2MCl2-AgClO4 O
AcO
MeOMeO
Me
O
M = Ti, 0 °C, 30 min; 90% (α:β = 36:64)M = Zr, –20 °C, 5 min; 90% (α:β = 55:45)M = Hf, –20 °C, 30 min; 86% (α:β = 63:37)
HO+
(1:1)
Scheme 4.5 Zirconocene-promoted glycosidation of cyclohexylcarbinol.
Solvents were also optimized (not shown). The SN2’-type reaction of 2-bromo-3,3,3-trifluoropropene
with p-xylene proceeded smoothly in CH2Cl2, whereas the activity of ZrCl4 was completely suppressed in
THF, DMF, dioxane, MeCN, and even in Et2O. It is probably due to the Lewis basicity of these solvents.
HFIP (1,1,1,3,3,3-hexafluoropropan-2-ol) is often used to stabilize carbocations but did not work in this
reaction.

64
The synthesis of 2-bromo-3,3-difluoroallylbenzenes by the SN2’-type reaction of
2-bromo-3,3,3-trifluoropropene with simple arenes was accomplished (Table 4.2). Mesitylene afforded the
corresponding product in 35% yield (Entry 1). p-, m-, and o-Xylenes underwent the SN2’-type reaction to
give the difluoroallylated xylenes 4-1a–c in 72–83% yield (Entries 2–4). Toluene, benzene, and naphthalene
also worked well to afford the corresponding products in good to moderate yield (Entries 5–7). It must be
emphasized that not only electron-rich arenes but also electron-deficient arenes underwent the cationic
SN2’-type reaction. Chlorobenzene and bromobenzene afforded the corresponding products in 67% and 56%
yield, respectively (Entries 8,9). Even dihalobenzenes gave the corresponding products, albeit in low yield
(Entries 10,11).
Table 4.2 Synthesis of 2-bromo-3,3-difluoroallylbenzenes by the cationic SN2’-type reaction of 2-bromo-3,3,3-trifluoropropene.
Br
CF3 +ZrCl4 (1 eq)
(3 eq)CH2Cl2, 0 oC, 1 h, RT, time
Ar–HBr
CF2 Ar
Entry Ar–H time (h) 4-1 (%)
1 mesitylene 24 4-1i, 35
2 p-xylene 4 4-1a, 83, 98a
3 m-xylene 4 4-1c, 72b
4 o-xylene 4 4-1b,75c
5 toluene 4 4-1e, 95b
6 benzene 6 4-1d, 81
7 naphthalene 24 4-1h, 28d
8 chlorobenzene 4 4-1f, 67b
9 bromobenzene 4 4-1g, 56b
10 1,4-dichlorobenzene 12 4-1j, 17a
11 1,4-difluorobenzene 12 4-1k, 16a
a: 19F NMR yield based on C6F6 as an internal standard. b: Three isomers were observed on 19F NMR spectrum. c: o/p=64/36. d: α/β=72/28.

65
The SN2’-type reactions of 2-phenylated 3,3,3-trifluoropropene were also examined (Table 4.3).
Mesitylene, xylenes, and other arenes readily underwent the SN2’-type reaction to give the corresponding
3,3-difluoro-2-phenylallylated benzenes in good yield.
Table 4.3 Synthesis of 2-phenyl-3,3-difluoroallylbenzenes by the cationic SN2’-type reaction of 2-phenyl-3,3,3-trifluoropropene.
+ZrCl4 (1 eq)
(x eq)
CH2Cl2, 0 oC, 1 h, RT, 6 hAr
CF2
Ph
4-2
Ar–HPh
CF3
Entry x Ar-H 4-2 (%)a
1 5 mesitylene 41
2 1 p-xylene 80
3 5 m-xylene 72
4 10 o-xylene 71
5 10 toluene 69c
6 10 benzene 60
7 10 naphthalene 61d
a: 19F NM R yield based on C6F6. b: Three isomers were observed on 19F NMR spectrum. c: α/β = 88/12. d: A mixture of difluoroalkene and chlorofluoroalkene (87/13).
Since CF3 group is chemically robust, the activation of CF3 group for synthetic applications is a
challenging task for chemists. The Lewis acid-mediated SN2’-type reaction provides a method to form a new
C–C bond though cleavage of the chemically stable C–F bond of CF3 group.

66
4.3 Catalytic C–F Bond Cleavage of Trifluoromethyl Groups by Aluminium Halides
The author has found that the cationic SN2’-type reaction of 2-trifluoromethyl-1-alkenes proceeds with
a catalytic amount of AlX3 (Table 4.4): Treatment of 2-bromo-3,3,3-trifluoropropene with 10 mol% of
aluminium chloride or bromide afforded 3,3-difluoroallyl-p-xylene 4-1a in good yield, whereas ZrCl4 and
HfCl4 were not effective (not shown). The difluoroallylated p-xylene 4-1a was accompanied by the
formation of 2-bromo-1,1,1-trifluoropropane derivative 4-4a, which suggests that the elimination step of F–
was somewhat affected when the reaction was conducted with a catalytic amount of AlX3. Although this
procedure has not been fully optimized yet, the aluminium-catalyzed SN2’-type reaction is of potential
importance as a catalytic method for the introduction of 3,3-difluoroallyl moiety to less nucleophilic
compounds.
Table 4.4 Aluminium halide-catalyzed SN2’-type reaction of 2-trifluoromethyl-1-alkenes.
CF3
Br+
(10 eq)
neat, RT, 8 h
CF2
Br
CF3
Br+
4-1a 4-4a
AlX3
Entry AlX3 (mol%) 4-1a + 4-4a (%)a (Ratio)
1b AlCl3 100 60 (100:0)
2c AlBr3 100 55 (100:0)
2 AlCl3 10 71 (40:60)
3 AlBr3 10 61 (42:58)
a: 19F NMR yield.
b: Table 4.1, Entry 5.
c: Table 4.1, Entry 6.
In this chapter, the author described a novel Zr(IV)-promoted and Al-catalyzed cationic SN2’-type
reaction of 2-trifluoromethyl-1-alkenes with simple arenes. A wide variety of benzene derivatives readily
underwent the nucleophilic addition–elimination sequence and 3,3-difluoroallylated arenes were obtained in
good yield.

67
References and Notes
1. (a) Downing, F. B.; Benning, A. F.; and McHarness, R. C. US Patent 2,384,821, assigned to DuPont, 1945. (b) Henne, A. L.; Woalkes, T. P. J. Am Chem Soc. 1946, 68, 496.
2. (a) Spatz, M.; Minor, B. HFO-1234yf, a Low GWP Refrigerant for MAC. Presented at the VDA Alternative Refrigerant Winter Meeting, Saalfelden, Austria, Feb 13-14, 2008. (b) Ikegami, T.; Aoki, K.; Lijima, K. New Refrigerant Evaluation results. Presented at the VDA Alternative Refrigerant Winter Meeting, Saalfelden, Austria, Feb 13-14, 2008. (c) Leck, T. J. K. Proceedings of the 3rd Conference on Thermophysical Properties and Transfer Processes of Refrigerants, Colorado, June 23-16, 2009; IIF-IIR: France, 2009. (d) Schuster, P.; Bertermann, R.; Snow, T. A.; Han, X.; Rusch, G. M.; Jepson, G. W.; Dekant, W. Toxicol. Appl. Pharmacol. 2008, 233, 323. (e) Nielsen, O. J.; Javadi, M. S.; Sulbaek Andersen, M. P.; Hurley, M. D.; Wallington, T. J.; Singh, R. Chem. Phys. Lett. 2007, 439, 18. (f) Papadimitriou, V. C.; Talukdar, R. K.; Portmann, R. W.; Ravishankara, A. R.; Burkholder, J. B. Phys. Chem. Chem. Phys. 2008, 10, 808.
3. (a) Fuchikami, T.; Yamanouchi, A.; Ojima, I. Synthesis 1984, 766. (b) Jiang, B.; Xu, Y. J. Org. Chem. 1991, 56, 7336. (c) Hu, C.-M.; Hong, F.; Xu, Y.-Y. J. Fluorine Chem. 1993, 64, 1. (d) Xu, Y.; Jin, F.; Huang, W. J. Org. Chem. 1994, 59, 2638. (e) Shi, G.-Q.; Huang, X.-H.; Hong, F. R. J. Org. Chem. 1996, 61, 3200. (f) Ichikawa, J.; Fujiwara, M.; Okauchi, T.; Minami, T. Synlett 1998, 927. (g) Peng, S.; Qing, F.-L. J. Chem. Soc., Perkin Trans. 1999, 3345. (h) Pan, P.-Q.; Liu, X.-X.; Deng, M.-Z. J. Fluorine Chem. 1999, 95, 167. (i) Jiang, B.; Wang, Q.-F.; Yang, C.-G.; Xu, M. R. Tetrahedron Lett. 2001, 42, 4083. (j) Morioka, M.; Sasaki, M.; Morisaki, K.; Mori, S. Japan Kokai Tokkyo Koho 2001, JP 2001-288138; Chem. Abstr. 2001, 135, 288521.
4. (a) Fontanelli, R., and Sianesi, D. Ann. Chim. (Roma), 1965, 55, 862. (b) Kendrick, D. A., and Kolb,M. J. Fluorine Chem. 1989, 45, 265. (c) Bergstrom, D. E.; Ng, M. W.; Wong, J. J. J. Org. Chem. 1983, 48, 1902. (d) Fuchikami, T.; Shibata,Y.; Suzuki, Y. Tetrahedron Lett. 1986, 27, 3173. (e)Kitazume, T.; Ohnogi, T. Synthesis, 1988, 614. (f) Kitazume, T.; Ohnogi, T.; Miyauchi, H.; Yamazaki, T. J. Org. Chem. 1989, 54, 5630. (g) Begue, J-P.; Bonnet-Delpon, D.; Rock, M. H. J. Chem. Soc.,PerkinTrans. 1, 1996, 1409. (h) Begue, J.-P.; Bonnet-Delpon, D.; Rock, M.H. Tetrahedron Lett. 1995, 36, 5003. (i) Ichikawa, J.; Fukui, H.; Ishibashi, Y. J. Org. Chem. 2003, 68, 7800.
5. (a) Fuchikami, T.; Shibata, Y.; Suzuki, Y. Tetrahedron Lett. 1986, 27, 3173. (b) Fujita, M.; Obayashi, M.; Hiyama, T. Tetrahedron 1988, 44, 4135. (c) Kendrick, D. A.; Kolb, M.; J. Fluorine Chem. 1989, 45, 265. (d) Kitazume, T.; Ohnogi, T.; Miyauchi, H.; Yamazaki, T.; Watanabe, S. J. Org. Chem. 1989, 54, 5630. (e) Watanabe, S.; Sugahara, K.; Fujita, T.; Sakamoto, M.; Kitazume, T. J. Fluorine Chem. 1993, 62, 201. (f) Be´gue´, J.-P.; Bonnet-Delpon, D.; Rock, M. H. Synlett 1995, 659. (g) Be´gue´, J.-P.; Bonnet-Delpon, D.; Rock, M. H. Tetrahedron Lett. 1995, 36, 5003. (h) Be´gue´, J.-P.; Bonnet-Delpon, D.; Rock, M. H. J. Chem. Soc. Perkin Trans. 1, 1996, 1409. (i) Ichikawa, J.; Fukui, H.; Ishibashi, Y. J. Org. Chem. 2003, 68, 7800. (j) Ichikawa, J.; Miyazaki, H.; Sakoda, K.; Wada, Y.; J. Fluorine Chem.2004, 125, 585. (k) Mori, T.; Iwai, Y.; Ichikawa, J. Chem. Lett. 2005, 34, 778. (l) Cunico, R. F.; Motta, A. R. Org. Lett. 2005, 7, 771.
6. Ichikawa, J.; Nadano, R.; Ito, N. Chem. Commun. 2006, 4425.
7. Miura, T.; Ito, Y.; Murakami, M. Chem. Lett. 2008, 37, 1006.

68
8. (a) Belen’kii, G.G. J. Fluorine Chem. 1996, 77, 107. (b) Krespan, C. G.; Petrov, V. A. Chem. Rev. 1996, 96, 3269. (c) Krespan, C. G. U.S. Patent 5,162,594, 1992, to DuPont; Chem. Abstr. 1992, 117, 69439.
9. Kobayashi, Y.; Nagai, T.; Kumadaki, I.; Takahashi, M.; Yamauchi, T. Chem. Pharm. Bull. 1984, 32, 4382.
10. Takusari, H.; Okazaki, S. Chem. Lett. 1984, 13, 885.
11. (a) Firouzabadi, H.; Iranpoor, N.; Karimi, B. Synlett 1999, 321. (b) Kumar, V.; Kaur, S.; Kumar, S. Tetrahedron Lett. 2006, 47, 7001. (c) Khodaei, M. M.; Bahrami, K.; Khedri, M. J. Chin. Chem. Soc. 2007, 54, 807.
12. Luo, Y. R. CRC Handbook of Chemistry and Physics, 91st Ed. CRC Press, Boca, Raton, 2010.
13. (a) Matsumoto, T.; Maeta, H.; Suzuki, K.; Tsuchihashi, G. Tetrahedron Lett. 1988, 29, 3567. (b) Suzuki, K.; Maeta, H.; Matsumoto, T.; Tsuchihashi, G. Tetrahedron Lett. 1988, 29, 3571. (c) Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 4853. (d) Suzuki, K.; Maeta, H.; Suzuki, T.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 6879. (e) Matsumoto, T.; Katsuki, M.; Suzuki, K. Chem. Lett. 1989, 437.

69
Experimental Section
ZrCl4-Promoted SN2’-type reactions 2-trifluoromethyl-1-alkenes with arenes
The synthesis of 4-1a is described as a typical procedure. Compound 4-1b-l was prepared by the same
procedure.
To a CH2Cl2 (5 mL) solution of 2-bromo-3,3,3-trifluoropropene (175 mg, 1.0 mmol) and p-xylene (319
mg, 3.0 mmol) was added ZrCl4 powder (233 mg, 1.0 mmol) at 0 °C under argon. After stirred at 0 °C for 1h,
the resulting mixture was allowed to warm to room temperature (20–25 °C) and stirred for additional 4 h.
The mixture was then quenched with a saturated sodium tartrate aqueous solution. The product was extracted
with Et2O (30 mL x 3). The combined organic layers were washed with brine (50 mL) and dried over
anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the residue was purified by
column chromatography (hexane) and GPC (CHCl3 eluent). The desired 4-1a was obtained as a colorless
liquid (216 mg, 83%).
2-(2-Bromo-3,3-difluoroallyl)-1,4-dimethylbenzene 4-1a
1H NMR (500 MHz, CDCl3): δ 2.25 (s, 3H), 2.31 (s, 3H), 3.64 (s, 2H), 6.96 (s, 1H), 6.99 (d, J = 7.7 Hz, 1H),
7.04 (d, J = 7.6 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 18.8, 21.0, 34.2, 79.6 (dd, JCF = 21, 39 Hz), 128.0, 130.1, 130.3, 133.3,
134.1, 135.5, 153.3 (dd, JCF = 285, 289 Hz). 19F NMR (470 MHz, CDCl3): δ 72.3 (d, JFF = 42 Hz, 1F), 77.7 (d, JFF = 42 Hz, 1F).
IR (ATR): 2924, 1738, 1504, 1448, 1269, 1196, 1124, 999, 876, 810, 561 cm–1.
HRMS (EI): m/z calcd for C11H11BrF2: 260.0012, found: 260.0022.
1-(2-Bromo-3,3-difluoroallyl)-2,3-dimethylbenzene 4-1b (o) (64%) and
4-(2-bromo-3,3-difluoroallyl)-1,2-dimethylbenzene 4-1b (p) (36%)
1H NMR (500 MHz, CDCl3): δ 2.18 (s, 1.1H), 2.23 (s, 1.9H), 2.25 (s, 1.9H), 2.29 (s, 1.1H), 3.61 (s, 1.3H),
3.69 (s, 0.7H), 6.93–7.08 (m, 3H). 13C NMR (126 MHz, CDCl3): δ 14.9, 19.4, 19.7, 20.7, 34.8, 36.6, 79.9 (dd, JCF = 21, 39 Hz), 80.2 (dd, JCF =
21, 39 Hz), 125.4, 125.9, 129.1, 129.8, 133,8, 135.4, 136.8, 137.1, 128.0, 130.1, 130.3, 133.3, 134.1, 135.5,
153.2 (dd, JCF = 285, 288 Hz), 153.5 (dd, JCF = 285, 289 Hz). 19F NMR (470 MHz, CDCl3): δ 71.5 (d, JFF = 42 Hz, 0.64F), 72.5 (d, JFF = 42 Hz, 0.36F), 77.3 (d, JFF = 42
Hz, 0.64F), 77.6 (d, JFF = 42 Hz, 0.36F).
IR (ATR): 2921, 1738, 1504, 1456, 1269, 1201, 1119, 1009, 993, 866, 818, 729, 565 cm–1.
HRMS (EI): m/z calcd for C11H11BrF2: 260.0012, found: 260.0017.

70
(2-Bromo-3,3-difluoroallyl)benzene 4-1d
1H NMR (500 MHz, CDCl3): δ 3.69 (t, J = 2.5 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 7.26-7.29 (m, 1H),
7.32-7.35 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 37.0, 79.9 (dd, JCF = 21, 39 Hz), 127.2, 128.6, 128.6, 136.4 (t, JCF = 2.6 Hz),
153.6 (dd, JCF = 286, 289 Hz). 19F NMR (470 MHz, CDCl3): δ 71.8 (dt, J = 42, 3 Hz, 1F), 72.5 (d, JFF = 42 Hz, 1F).
IR (ATR): 2925, 1732, 1496, 1456, 1271, 1201, 1140, 1080, 1032, 764, 698 cm–1.
HRMS (EI): m/z calcd for C9H7BrF2: 231.9699, found: 231.9691.
Reactions between 2-bromo-3,3,3-trifluoropropene and p-xylene in the presence of AlX3
To a mixture of 2-bromo-3,3,3-trifluoropropene (175 mg, 1.0 mmol) and p-xylene (1.06 g, 10.0 mmol)
was added AlCl3 (13 mg, 0.10 mmol) at room temperature under argon. After stirred at room temperature for
8 h, the mixture was quenched with a saturated sodium tartrate aqueous solution. The product was extracted
with Et2O (30 mL x 3). The combined organic layers were washed with brine (50 mL) and dried over
anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the residue was purified by
column chromatography (hexane eluent). A mixture of 4-1a and 4-4a was obtained as a colorless liquid
(191 mg, 4-1a/4-4a=40/60). These compounds were isolated by GPC (CHCl3 eluent) to give pure samples.
2-(2-Bromo-3,3,3-trifluoropropyl)-1,4-dimethylbenzene 4-4 1H NMR (500 MHz, CDCl3): δ 2.29 (s, 3H), 2.31 (s, 3H), 3.07 (dd, J = 10.9, 25.8 Hz, 1F), 3.44 (dd, J = 3.5,
15.0 Hz, 1F), 4.23-4.28 (m, 1H), 6.99-7.02 (m, 2H), 7.07 (d, J = 7.7 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 18.8, 20.9, 35.0, 47.5 (dd, JCF = 32, 63 Hz), 124.1 (q, 37.0, 79.9 (dd, JCF = 21,
39 Hz), 127.2, 128.6, 128.6, 136.4 (t, JCF = 2.6 Hz), 153.6 (dd, JCF = 286, 289 Hz). 19F NMR (470 MHz, CDCl3): δ 89.5 (d, JHF = 7 Hz, 3F).
IR (ATR): 2925, 1506, 1442, 1362, 1308, 1271, 1244, 1174, 1153, 1103, 1039, 926, 810, 671, 592, 521 cm–1.
HRMS (EI): m/z calcd for C11H12BrF3: 280.0074, found: 280.0066.

71
Chapter 5. Conclusion
In this thesis, the author describes practical syntheses and synthetic applications of gem-difluorovinylic
compounds (1,1-difluoroallenes, 2,2-difluoro-1-(pseudo)halovinylsilanes, and 3,3-difluoroallylic arenes).
In Chapter 2, a general and practical synthesis of 1,1-difluoroallenes and their application in
Friedel–Crafts-type cyclizations are described. 1,1-Difluoroallenes bear one more carbon–carbon double
bond adjacent to the difluorovinylidene moieties. They are attractive synthetic intermediates because of the
vinylic fluorine substituents and the cumulated double bonds. 1,1-Difluoroallenes also serve as promising
pharmaceuticals, because some of non-fluorinated allenes have been used for therapeutic purposes. To date,
however, synthetic methods for substituted 1,1-difluoroallenes have not been fully explored.
The author has developed a general and practical synthesis of 1,1-difluoroallenes and their application
to Friedel–Crafts-type cyclizations. The synthesis of 1,1-difluoroallenes 2-10 was achieved by the following
two-step process: (i) 3,3-Difluoro-2-iodoallylic acetates 2-9 are prepared by the reaction of aldehydes or
ketones with 2,2-difluoro-1-iodovinyllithium 2-4c, which is generated from commercially available
1,1,1-trifluoro-2-iodoethane and LDA; (ii) 1,1-Difluoroallenes 2-10 are synthesized under mild conditions
via zinc-promoted elimination of the iodo and acetoxy groups. A wide variety of 1,1-difluoroallenes were
obtained in high yield by this sequence (Table 5.1, selected).
Table 5.1 Synthesis of 1,1-difluoroallenes 2-10 via 3,3-difluoro-2-iodoallylic acetates 2-9
CF2
I
AcO
R1 CF2R2
R2
2-10
R1
CF3CH2ILDA (2 equiv)
–93 to –85 °C30 min, THF
CF2
I
Li
2-4c
1) R1(C=O)R2 (1 equiv) –93 to –30 °C, 2 h
2) Ac2O (1.2equiv) –30 to 0 °C, 2 h
2-9
Zn (2 equiv)
RT, 3–12 hDMF or THF
Entry Carbonyl compound 3,3-Difluoro-2-iodoallylic acetate 2-9
2-9 (%) 1,1-Difluoroallene 2-10 2-10 (%) (time)
1
82 2-10a 86 (3 h)
2
87
2-10d 92 (6 h)
3
83
2-10e 93 (12 h)
4
80a
2-10k 86 (8 h)
aAcetylation was performed with isopropenyl acetate/TsOH.

72
This method is also applicable to the synthesis of functionalized difluoroallenes. Aldehydes, bearing an
ester moiety or a pyridine ring, were transformed to the corresponding difluoroallenes 2-10k and 2-10l in
61% and 52% yield in two steps, respectively (Scheme 5.1).
Activation of 1,1-difluoroallenes 2-10 bearing an arylmethyl group is affected by an electrophilic
reagent, typically Lewis acid such as BF3·OEt2 and ZrCl4, to cause Friedel–Crafts-type cyclizations (Scheme
5.2). This reaction is accompanied by 1,2-migration of the benzylic alkyl group to provide an effective
method for the synthesis of fluorinated naphthalenes.
ON
CF2
NO
CO2MeCF2
CO2Me
2-10m 52% (two steps)2-10l 61% (two steps)
As inTable 5.1
As inTable 5.1
Scheme 5.1 Synthesis of functionalized 1,1-difluoroallenes.
CF2
2-10f
FF FFLA
LA
F
and/or– HF
1,2-Me migration
LA (1 equiv)
RT, 30 minCH2Cl2/(CF3)2CHOH
(1:1)40% (BF3·OEt2)
32% (ZrCl4)
Scheme 5.2 Synthesis of fluoronaphthalenes by Friedel–Crafts-type cyclizations of 1,1-difluoroallenes.
In Chapter 3, synthesis of 2,2-difluorovinylic silanes bearing a (pseudo)halogen substituent on the 1
position is described. These silanes are promising bifunctional intermediates for the synthesis of molecules
containing a difluorovinylidene moiety. The silanes would react with nucleophiles (electrophiles) and then
with electrophiles (nucleophiles) to give a variety of fully substituted difluoroalkenes (Scheme 5.3). Thus,
the silanes might serve as a 2,2-difluorovinylidene synthon possessing both positive and negative charges on
the 1 position. Free 2,2-difluorovinylidene is a very useful intermediate as a “super electrophilic carbene” for
the synthesis of a variety of 1,1-difluoroalkenes. However, free difluorovinylidene is extremely unstable and
just exist at very low temperature (< 40K) even under inert atomosphere.
CF2X
SiR3E+
CF2X
E
Nu–
CF2Nu
E
CF2Nu
SiR3
Nu– E+
F
F
vinylidene
Scheme 5.3 Concept of 2,2-difluoro-1-(pseudo)halovinylsilanes as a difluorovinylidene synthon.

73
The author has developed the synthetic methods for 2,2-difluorovinylic silanes bearing a
(pseudo)halogen substituent on the 1 position from commercially available compounds. Silylation of
2,2-difluoro-1-iodovinyllithium 2-4c, which is used for the synthesis of 1,1-difluoroallenes in Chapter 2, was
effected with chlorosilanes to give the corresponding iodovinylsilanes 3-1a,c and 3-1d in satisfactory yield
(Scheme 5.4a). Triple deprotonation of 2,2,2-trifluoroethanol was conducted with LDA in the presence of
chlorosilanes, followed by retro-Brook rearrangement and triflylation. Thus, triflyloxyvinylsilanes 3-1f,h
were obtained in good yield (Scheme 5.4b). These vinylic silanes are stable and can be purified by silica gel
column chromatography.
CF3CH2ILDA (2 equiv)
CF2
I
SiR3
CF2
I
Li
R3SiCl (1 equiv)
CF3CH2OH
LDA (3.3 equiv)R3SiCl (1 equiv)
CF2
OSiR3
Li
retro BrookCF2
OLi
SiR3
CF2
OTf
SiR3
PhNTf2 (1 equiv)
3-1a 84% (SiMe3) (19F NMR yield)3-1c 83% (Sii-Pr3)3-1d 84% (SiMe2Ph)
3-1f 72% (SiEt3)3-1h 61% (SiPh2t-Bu)*
(a)
(b)
–93 to –85 °C, 30 minTHF
–85 °C, 1 h
–93 °C to RT, 1 hTHF(–HMPA*)
0 °C
2-4c
Scheme 5.4 Synthesis of 2,2-Difluoro-1-(pseudo)halovinylsilanes.
Iododifluorovinylsilane 3-1d reacted with a boronic acid (Nu–) in the presence of palladium catalyst to
give β,β-difluorostyrene derivative 3-6 in 81% yield (Scheme 5.5a). Iododifluorovinylsilane 3-1c underwent
cesium fluoride-catalyzed or -promoted difluorovinylation of aldehydes (E+) to provide
3,3-difluoro-2-iodoallylic silyl ethers 3-5c,a in 77% and 81% yield, respectively (Scheme 5.5b).
Combination of these results would open up a new route to fully substituted 1,1-difluoro-1-akenes.
CsF, RCHO (1.0–1.2 equiv) CF2
I
Ri-Pr3SiO
CF2
I
SiMe2Ph
1 mol% PEPPSI-IPrPhB(OH)2 (1 equiv)
CF2
Ph
SiMe2Ph
3-6 81%
K2CO3 (2 equiv)60 °C, EtOH, 3 h
(b) Reactions with an electrophile
3-1d
3-1c
CF2
I
Sii-Pr3
Pd
NNR
R
R
R
N
Cl
PEPPSI-IPr (R = i-Pr)
Cl Cl
For 3-5c: 20 mol% CsF, 70 °C, diglyme, 4 h For 3-5a: CsF 1.1 equiv, RT, THF, 5 h
Conditions
(a) Reaction with a nucleophile
3-5c 77%, R = CH(Et)n-C4H93-5a 81%, R = CH(Me)CH2C6H4t-Bu
Scheme 5.5 Reactions of 2,2-difluoro-1-iodovinylsilanes with nucleophiles or electrophiles.

74
Synthesis of 3,3-difluoroallylic compounds by SN2’-type reactions of 2-trifluoromethyl-1-alkenes is
described in Chapter 4. 3,3-Difluoroallylic compounds, in general, can be prepared by SN2’-type reactions of
3,3,3-trifluoropropene derivatives with nucleophiles (Scheme 5.6, left). However, the SN2’-type reactions
reported to date have been conducted under basic conditions, requiring reactive nucleophiles such as
alkyllithiums and lithium amides.
Scheme 5.6 Concept of Lewis acid-promoted SN2’-type reactions of 3,3,3-trifluoropropenes.
One possible alternative to the above-mensioned anionic SN2’-type reactions is an acid-promoted
cationic version (Scheme 5.6, right). Elimination of a fluoride ion from 2-trifluoromethyl-1-alkenes could be
promoted by Lewis acids to form fluorine-stabilized allylic CF2 carbocations. Even less nucleophilic,
nonlithiated arenes (Nu–H) might undergo difluoroallylation with the allylic CF2 carbocations by a
Friedel–Crafts-type mechanism, although such attempts had led to only a partial success.
The author has achieved a novel Lewis acid-promoted SN2’-type reaction of trifluoropropenes with
simple arenes: As shown in Table 5.2, zirconium(IV) chloride efficiently promoted the desired SN2’-type
reactions, where 3,3-difluoroallylic arenes 4-1, the SN2’-type products, were obtained in good yield. It is
noteworthy that electron-deficient chlorobenzene also gave the corresponding products in good yield.
Table 5.2. ZrCl4-promoted SN2’-type reactions of 3,3,3-trifluoropropenes with arenes.
Br
CF2Ar
Br
CF3
ZrCl4 (1 equiv)
0 °C to RT, CH2Cl2+ Ar–H
4-1(3 equiv)
Entry Arene 3,3-Difluoroallylic arene 4-1 Yield of 4-1 (%) (time)
1 p-xylene
4-1a 83 (4 h)
2 benzene
4-1d 81 (6 h)
3 toluene
4-1e 95 (4 h)
4 chlorobenzene
4-1f 67 (4 h)

75
It must be mentioned that CF3 group is chemically robust and that activation and synthetic application
of the CF3 group have been a challenging task for chemists. The Lewis acid mediated SN2’-type reaction
provided the method to cleave the chemically stable C–F bond of the CF3 group on alkenes.
The author describes practical syntheses and synthetic applications of gem-difluorovinylic compounds
(1,1-difluoroallenes, 2,2-difluoro-1-(pseudo)halovinylsilanes, and 3,3-difluoroallylic arenes). These
achievements definitely inspire potential industrial applications of functionalized gem-difluorovinylic
compounds to production of useful fluorine-containing materials.

76
List of Publications
(For the author’s doctoral program)
1. Oh, K.; Fuchibe, K.; Ichikawa, J. “A Facile Synthesis of 1,1-Difluoroallenes from Commercially Available 1,1,1-Trifluoro-2-iodoethane,” Synthesis 2011, 881 (Synfacts 2011, 609).
2. Oh, K.; Fuchibe, K.; Yokota, M; Ichikawa, J. “Facile Synthesis of Substituted 1,1-Difluoroallenes via Carbonyl Difluorovinylidenation,” Synthesis, in press.
(For the author’s master program)
3. “First Unsymmetrical Bisfullerene, C121: Evidence for the Presence of Both Homofullerene and Methanofullerene Cages in One Molecule”
Dragoe, N.; Shimotani, H.; Wang, J.; Iwaya, M.; de Bettencourt-Dias, A.; Balch, A. L.; Kitazawa, K. J. Am. Chem. Soc. 2001, 123, 1294.
4. “Synthesis and Electrochemical Behavior of Regioisomeric Bismethanofullerene Derivatives”
Xiao, L.; Ozawa, M.; Iwaya, M.; Wang, J.; Shimotani, H.; Dragoe, N.; Tanibayashi, S.; Kitazawa, K. Fullerene Science and Technology, 2000, 8, 77.
5. “Synthesis and Structure of All-carbon Bisfullerene C121”
Shimotani, H.; Wang, J.; Dragoe, N.; Kitazawa, K. AIP conference proceedings, 2001, 590.

PAPER 881
A Facile Synthesis of 1,1-Difluoroallenes from Commercially Available 1,1,1-Trifluoro-2-iodoethaneSynthesis of 1,1-DifluoroallenesKen Oh, Kohei Fuchibe, Junji Ichikawa*Department of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba, 305-8571, JapanFax +81(29)8534237; E-mail: [email protected] 30 November 2010; revised 19 January 2011
SYNTHESIS 2011, No. 6, pp 0881–0886xx.xx.2011Advanced online publication: 14.02.2011DOI: 10.1055/s-0030-1258438; Art ID: F21510SS© Georg Thieme Verlag Stuttgart · New York
Abstract: 1,1-Difluoroallenes are synthesized in good yield viazinc-promoted 1,2-elimination of 3,3-difluoro-2-iodoallylic ace-tates, which are prepared by the reaction of aldehydes or ketoneswith 1-iodo-2,2-difluorovinyllithium, generated from commerciallyavailable 1,1,1-trifluoro-2-iodoethane.
Key words: fluorinated allenes, metalation, carbanions, elimina-tion, difluorovinylidenation
1,1-Difluoroallenes have attracted much attention be-cause of their unusual reactivities, entailing them to beused as synthetic building blocks for fluorinated mole-cules. The Diels–Alder and [3+2]-cycloaddition reactionsof 1,1-difluoroallenes with 1,3-dienes and 1,3-dipolesreadily take place on the internal, nonfluorinated alkenemoiety to give the corresponding exo-difluoromethylenecompounds.1 For example, 1,1-difluoroallene(F2C=C=CH2), with a low LUMO energy level, gives anexcellent yield (>99%) of the cyclized product with cyclo-pentadiene under very mild conditions (–20 °C, 1 min),while the nonfluorinated counterpart allene(H2C=C=CH2) requires vigorous conditions (200–230 °C) to give the product in a modest yield (49%).1a,f
The [2+2]-cycloaddition reactions with alkenes andalkynes occur on the terminal, fluorinated alkene moietyto give ring fluorinated cyclobutane2a and cyclobutene2b
derivatives. 1,1-Difluoroallenes also react with variousnucleophiles to afford CF2-terminal or internal additionproducts selectively, depending on the character of the nu-cleophile.3
Although the parent 1,1-difluoroallene has been knownsince the 1950s,4 very few synthetic methods for the 3-substituted 1,1-difluoroallenes have been reported.3a,5 Re-cently, we have developed a versatile synthetic methodfor 3-substituted 1,1-difluoroallenes 1 using two steps: (i)lithiation of 1,1-dibromo-2,2-difluoroethene with butyl-lithium generates 1-bromo-2,2-difluorovinyllithium(F2C=CBrLi), which in turn, reacts with aldehydes or ke-tones to form 2-bromo-3,3-difluoroallylic acetates, and(ii) treatment of the bromoacetates with butyllithiumgives 1,1-difluoroallenes via the 1,2-elimination of lithi-um acetate.6
However, there are two factors that limit the scope of thismethod: (a) the starting material, F2C=CBr2, is a high-cost, potential ozone-depleting substance, and is now un-available because of the ban on its industrial manufacture,and (b) highly nucleophilic alkyllithiums are required inthe preparation of 1,1-difluoroallenes, which restricts thechoice of substrate. Here, we report an improved syntheticmethod for 1,1-difluoroallenes to overcome these issuesusing 1) an environmentally friendly and commerciallyavailable compound as the starting material, and 2) an ef-fective process for carrying out a 1,2-elimination reactionunder mild and tolerant reaction conditions.
First, we considered that the key intermediate, a 1-haloge-nated 2,2-difluorovinyl anion 2 (Scheme 1), would begenerated by the addition of two equivalents of a strongbase to 1,1,1-trifluoro-2-haloethanes,7–9 which bear twohydrogen atoms and are recognized to have much lowerozone depletion potential (ODP). These compounds aremanufactured industrially for use as refrigerants or asfluorinated intermediates. Second, we proposed a differ-ent route to access the desired 1,1-difluoroallenes 1 from3,3-difluoro-2-haloallylic acetates 3 (Scheme 1) on treat-ment with a zerovalent metal instead of highly reactivealkyllithiums, which would promote the 1,2-eliminationfrom acetates 3 to form one more double bond under mildconditions. This sequence would expand the scope of thesubstrates.
Scheme 1 A synthetic plan for 1,1-difluoroallenes from a 1,1,1-trifluoro-2-haloethane
1,1,1-Trifluoro-2-iodoethane was selected as the startingmaterial because of its ease of handling (bp 55–56 °C/760Torr). The lithiation of 1,1,1-trifluoro-2-iodoethane withtwo equivalents of lithium diisopropylamide at low tem-peratures (–93 to –85 °C) successfully gave 2,2-difluoro-1-iodovinyllithium (2, Table 1).10 Lithium species 2 then
base (2 equiv)O
R1
R2
– HF, – H+ CF2
X
1)
1
2
CF3CH2X2) acetylation
– M(OAc)X
3
F2C
X
AcO R2
R1F2C
R2
R1
–
M0
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
77
Appendix. Reprints of original papersA1. Oh, K.; Fuchibe, K.; Ichikawa, J. Synthesis 2011, 881.

882 K. Oh et al. PAPER
Synthesis 2011, No. 6, 881–886 © Thieme Stuttgart · New York
reacted with one equivalent of either an aldehyde or a ke-tone, and was subsequently acetylated with acetic anhy-dride (Table 1, entries 1–8) or with isopropenyl acetateand p-toluenesulfonic acid (entry 9) to afford 3,3-difluo-ro-2-iodoallylic acetates 3 in good yield.
Then, a facile and effective route by the zinc-promoted1,2-elimination of acetates 3 under mild and tolerant reac-tion conditions (e.g., at room temperature for severalhours) was found compared with the previously used n-
butyllithium-promoted 1,2-elimination.6,11,12 The condi-tions of the zinc-promoted 1,2-elimination were opti-mized, as shown in Table 2. In most cases, 1,1-difluoroallenes 1 were obtained in good yield on treatmentof acetates 3 with two equivalents of zinc, either in N,N-dimethylformamide or in tetrahydrofuran, at room tem-perature for 3–12 hours (Table 2, entries 1–6). However,1,1-difluoroallene 1g was only formed in N,N-dimethyl-formamide, and not in tetrahydrofuran (Table 2, entries 7
Table 1 Synthesis of 1,1-Difluoroallenes 1 via Difluorovinylidenation of Carbonyl Compounds
Entry Carbonyl compound 3,3-Difluoro-2-iodoallylic acetate 3 Yield of 3 (%) 1,1-Difluoroallene 1 Yield of 1 (%) (time)
1 3a: 82 1a: 86 (3 h)
2 3b: 84 1b: 87 (6 h)
3 3c: 83 1c: 82 (6 h)
4 3d: 87 1d: 92 (6 h)
5 3e: 83 1e: 93 (12 h)
6 3f: 81 1f: 95 (6 h)
7 3g: 73 1g: 71 (12 h)
8 3h: 82 1h: 74 (6 h)
9 3i: 80a 1i: 86 (8 h)
a Acetylation was performed with isopropenyl acetate and TsOH.6
LDA (2 equiv)
O
R1
R2
F2C
I
Li
THF, –93 to –30 °C, 2 h
1)
F2C
I
AcOR2
R1
F2C
R2
R1
1a–i2
CF3CH2IZn (2 equiv)
(1 equiv)
3a–i
2) Ac2O (1.2 equiv) –30 to 0 °C, 2 h
DMF or THF r.t., 3–12 h
THF–93 to –85 °C30 min
O
F2C
I
AcO
F2C •
O
CH2(CH2)7MeF2C
I
AcO
CH2(CH2)7Me
F2C •CH2(CH2)7Me
O
F2C
I
AcOF2C •
O
t-Bu
AcO
t-BuF2C
I
F2C •
t-Bu
OF2C
I
AcO
F2C •
OF2C
I
AcO
F2C •
O
NF2C
I
AcO
NF2C •
N
OCO2Me
F2C
I
AcO MeO2C
F2C •
CO2Me
O
F2C
I
AcOF2C •
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
78

PAPER Synthesis of 1,1-Difluoroallenes 883
Synthesis 2011, No. 6, 881–886 © Thieme Stuttgart · New York
and 8), although the reason for this is not clear at present.13
3-Substituted 1,1-difluoroallenes with a primary alkylgroup were produced readily using this method, while theyield decreased when the reaction period was extended byseveral hours (Table 2, entries 1 and 2). In contrast, theyield of 1,1-difluoroallenes with a secondary or tertiaryalkyl group at the 3-position remained steady, even afteran extended reaction time (Table 2, entries 3–5). This maybe the result of the stability of 1,1-difluoroallenes.
Owing to the mild conditions of the zinc-promoted 1,2-elimination, 1,1-difluoroallenes bearing a pyridine ring oran ester functionality were synthesized in good yield(Table 1, entries 7 and 8). A 3,3-disubstituted 1,1-difluo-roallene was obtained in high yield from a 1,1-disubstitut-ed 2-iodo-3,3-difluoroallylic acetate, which was preparedfrom the corresponding ketone (Table 1, entry 9).
In summary, we have developed a general and efficientmethod for the synthesis of 1,1-difluoroallenes from com-mercially available and environmentally friendly 1,1,1-trifluoro-2-iodoethane under mild reaction conditions.This facile and low-cost synthesis allows 1,1-difluoroal-lenes to be used as practical building blocks for the syn-thesis of a various useful fluorinated molecules. Theirapplication is in progress in our laboratory and will be re-ported in due course.
NMR spectra were recorded on a Bruker Avance 500 or a BrukerAvance 400 spectrometer in CDCl3. Chemical shift values weregiven in ppm relative to internal SiMe4 (for 1H NMR: d = 0.00),CDCl3 (for 13C NMR: d = 77.0), and C6F6 (for 19F NMR: d = 0.0).Mass spectra (EI-TOF or ESI-TOF) were recorded on Jeol JMS-T100GCv or JMS-T100CS mass spectrometer. IR spectra were re-corded by ATR (attenuated total reflectance) method on a HoribaFT-720 spectrometer. Column chromatography and preparativeTLC were conducted on silica gel (Silica Gel 60 N, Kanto ChemicalCo., Inc. for column chromatography and Wakogel B-5F, WakoPure Chemical Industries for PTLC, respectively). All reactionswere conducted under argon. THF and DMF were dried by passingover a column of activated alumina followed by a column of Q-5scavenger (Engelhard). 1,1,1-Trifluoro-2-iodoethane was obtained
from Tosoh F-tech, Inc., and distilled from activated 4 Å molecularsieves. This compound can also be purchased from Tokyo ChemicalIndustry Co., Ltd. or Sigma-Aldrich Co. NMR and IR Spectra ofcompounds 1a, 1c, 1d, and 1f are in agreement with the publisheddata.6
1,1-Difluoro-2-iodo-5-phenylpent-1-en-3-yl Acetate (3a); Typi-cal ProcedureTo a THF (10 mL) solution of (i-Pr)2NH (2.8 mL, 20 mmol) wasadded BuLi (12.0 mL, 1.67 M in hexane, 20.0 mmol) over 10 minat 0 °C under argon. The resulting solution was allowed to stir foran additional 15 min, and then cooled to –93 °C using a cold hexanebath. To this cold LDA solution was added a THF (5 mL) solutionof CF3CH2I (2.10 g, 10.0 mmol) over 10 min, keeping the tempera-ture between –93 and –85 °C. After stirring for 20 min at the sametemperature, a THF (5 mL) solution of 3-phenylpropanal (1.34 g,10.0 mmol) was added over 5 min, keeping the temperature be-tween –93 and –85 °C. The mixture was stirred for an additional 30min, then warmed to –30 °C over 90 min. After the addition ofAc2O (1.23 g, 12.0 mmol), the mixture was allowed to warm to 0 °Cover 2 h. The reaction was quenched with sat. aq NH4Cl (20 mL),and the product was extracted with Et2O (3 × 20 mL). The combinedorganic layers were washed with brine (20 mL) and dried (Na2SO4).After removal of the solvent under reduced pressure, the residuewas purified by column chromatography (hexane–EtOAc, 20:1).The acetate 3a was obtained as a colorless liquid (3.01 g, 82%).
IR (ATR): 3028, 2954, 1743, 1716, 1267, 1219, 1024, 698 cm–1.1H NMR (500 MHz, CDCl3): d = 1.87–1.93 (m, 1 H), 2.05–2.17 (m,1 H), 2.07 (s, 3 H), 2.58 (t, J = 7.2 Hz, 2 H), 4.98 (t, J = 7.2 Hz, 1H), 7.17–7.22 (m, 3 H), 7.29 (dd, J = 7.3, 7.6 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 20.9, 30.9, 36.0, 53.8 (dd,JC,F = 25, 26 Hz), 68.9 (d, JC,F = 3 Hz), 126.2, 128.2, 128.5, 140.2,154.0 (dd, JC,F = 286, 286 Hz), 169.6.19F NMR (470 MHz, CDCl3): d = 89.2 (d, JF,F = 22 Hz, 1 F), 90.2(d, JF,F = 22 Hz, 1 F).
HRMS (ESI+): m/z calcd for C13H13F2IO2 + Na [M + Na]+:388.9826; found: 388.9830.
1,1-Difluoro-2-iodododec-1-en-3-yl Acetate (3b) Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 84%; colorless liquid.
IR (ATR): 2925, 2856, 1749, 1716, 1458, 1371, 1269, 1225, 1024,962, 604 cm–1.1H NMR (500 MHz, CDCl3): d = 0.88 (t, J = 6.9 Hz, 3 H), 1.19–1.35(br, 14 H), 1.52–1.61 (m, 1 H), 1.65–1.74 (m, 1 H), 2.07 (s, 3 H),4.94 (t, J = 7.2 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 14.0, 20.9, 22.6, 24.5, 28.9, 29.2,29.30, 29.34, 31.8, 34.2, 54.1 (dd, JC,F = 24, 26 Hz), 69.3 (d, JC,F = 3Hz), 153.9 (dd, JC,F = 286, 299 Hz), 169.6.19F NMR (470 MHz, CDCl3): d = 88.3 (d, JF,F = 24 Hz, 1 F), 89.6(d, JF,F = 24 Hz, 1 F).
HRMS (EI): m/z calcd for C12H19F2I [M AcOH]+: 328.0500; found:328.0478.
1,1-Difluoro-2-iodo-5-(1-naphthyl)pent-1-en-3-yl Acetate (3c)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 83%; pale yellow liquid.
IR (ATR): 3047, 2939, 1743, 1716, 1371, 1269, 1225, 1026, 966,798 cm–1.1H NMR (500 MHz, CDCl3): d = 1.87–1.96 (m, 1 H), 1.97 (s, 3 H),2.04–2.13 (m, 1 H), 2.92 (t, J = 8.1 Hz, 2 H), 5.00 (tdd, J = 6.4, 2.2,1.4 Hz, 1 H), 7.19 (d, J = 6.9 Hz, 1 H), 7.28 (dd, J = 7.1, 7.1 Hz, 1
Table 2 Optimization of the Zinc-Promoted 1,2-Elimination
Entry Acetate 3 Zn (equiv) Solvent Time (h) Yield of 1 (%)
1 3a 2 DMF 3 1a: 86
2 3a 2 DMF 6 1a: 72a
3 3d 2 DMF 3 1d: 83
4 3d 2 DMF 6 1d: 92
5 3d 2 DMF 12 1d: 90
6 3d 2 THF 6 1d: 88
7 3g 2 DMF 8 1g: 71
8 3g 4 THF 12 1g: traceb
a Allene 1a partly decomposed to a complex mixture.b Acetate 3g was recovered quantitatively.
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
79

884 K. Oh et al. PAPER
Synthesis 2011, No. 6, 881–886 © Thieme Stuttgart · New York
H), 7.39 (d, J = 8.0 Hz, 1 H), 7.41 (d, J = 7.0 Hz, 1 H), 7.61 (d,J = 8.2 Hz, 1 H), 7.74 (d, J = 8.8 Hz, 1 H), 7.90 (d, J = 8.6 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 20.9, 28.1, 35.4, 53.8 (t, JC,F = 25Hz), 69.2 (d, JC,F = 3 Hz), 123.4, 125.5, 126.0 (2 C), 127.1, 128.9,131.5, 133.9, 136.3, 154.1 (dd, JC,F = 299, 286 Hz), 169.6.19F NMR (470 MHz, CDCl3): d = 89.4 (d, JF,F = 22 Hz, 1 F), 90.3(d, JF,F = 22 Hz, 1 F).
HRMS (EI): m/z calcd for C17H15F2IO2 [M]+: 416.0085; found:416.0059.
5-(4-tert-Butylphenyl)-1,1-difluoro-2-iodo-4-methylpent-1-en-3-yl Acetate (3d)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 87% (diastereomer ratio = 1:1); pale yellow liquid.
IR (ATR): 2962, 2871, 1741, 1716, 1510, 1462, 1369, 1269, 1225,1020, 968, 606, 573 cm–1.1H NMR (500 MHz, CDCl3): d = 0.74 (d, J = 6.4 Hz, 1.5 H), 0.91(d, J = 6.2 Hz, 1.5 H), 1.15–1.45 (m, 1 H), 1.31 (s, 9 H), 2.06 (s, 1.5H), 2.09 (s, 1.5 H), 2.05–2.13 (m, 0.5 H), 2.34 (dd, J = 13.5, 9.5 Hz,0.5 H), 2.67 (d, J = 12.2 Hz, 0.5 H), 2.92 (d, J = 13.5 Hz, 0.5 H),4.70 (d, J = 10.0 Hz, 0.5 H), 4.75 (d, J = 9.5 Hz, 0.5 H), 7.08 (d,J = 8.4 Hz, 1 H), 7.09 (d, J = 8.3 Hz, 1 H), 7.30 (d, J = 8.4 Hz, 1 H),7.31 (d, J = 8.3 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 14.5, 14.8, 20.8, 31.4, 34.3, 37.6,38.0, 38.4, 39.1, 53.3 (dd, JC,F = 26, 26 Hz), 73.3 (d, JC,F = 3 Hz),73.4 (d, JC,F = 3 Hz), 125.2, 128.7, 128.8, 136.0, 136.4, 148.9,149.0, 154.3 (dd, JC,F = 298, 286 Hz), 154.4 (dd, JC,F = 297, 286Hz), 169.7, 169.8.19F NMR (470 MHz, CDCl3): d = 88.5 (d, JF,F = 23 Hz, 0.5 F), 89.1(d, JF,F = 22 Hz, 0.5 F), 89.8 (d, JF,F = 23 Hz, 0.5 F), 90.6 (d,JF,F = 22 Hz, 0.5 F).
HRMS (ESI+): m/z calcd for C18H23F2IO2 + Na [M + Na]+:459.0608; found: 459.0610.
1,1-Difluoro-2-iodo-4-methyl-4-phenylpent-1-en-3-yl Acetate (3e)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 83%; colorless liquid.
IR (ATR): 2976, 1745, 1709, 1498, 1442, 1369, 1265, 1219, 1030,980, 768, 698, 609 cm–1.1H NMR (500 MHz, CDCl3): d = 1.46 (s, 3 H), 1.48 (s, 3 H), 2.05(s, 3 H), 5.14 (dd, J = 1.9, 1.0 Hz, 1 H), 7.24 (t, J = 7.7 Hz, 1 H),7.32 (dd, J = 7.7, 7.7 Hz, 2 H), 7.40 (d, J = 7.7 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 20.8, 24.9, 25.0 (d, JC,F = 9 Hz),42.8, 48.0 (dd, JC,F = 25, 25 Hz), 74.5 (d, JC,F = 2 Hz), 126.7, 127.0,128.0, 144.4, 153.8 (dd, JC,F = 298, 286 Hz), 169.3.19F NMR (470 MHz, CDCl3): d = 91.1 (d, JF,F = 23 Hz, 1 F), 91.3(d, JF,F = 23 Hz, 1 F).
HRMS (ESI+): m/z calcd for C14H15F2IO2 + Na [M + Na]+:402.9982; found: 403.0012.
1,1-Difluoro-2-iodo-5-phenylhex-1-en-3-yl Acetate (3f)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 81% (diastereomer ratio = 6:4); colorless liquid.
IR (ATR): 3028, 2962, 1747, 1716, 1495, 1452, 1371, 1269, 1225,1020, 978, 700 cm–1.1H NMR (500 MHz, CDCl3): d = 1.28 (d, J = 7.0 Hz, 1.8 H), 1.29(d, J = 7.0 Hz, 1.2 H), 1.80–1.88 (m, 1 H), 1.95 (s, 1.8 H), 1.98–2.05(m, 0.4 H), 2.06 (s, 1.2 H), 2.06–2.12 (m, 0.6 H), 2.62–2.69 (m, 0.6H), 2.70–2.78 (ddq, J = 7.0, 7.0, 7.0 Hz, 0.4 H), 4.78 (dd, J = 6.5,6.5 Hz, 0.6 H), 4.84 (dd, J = 7.5, 7.5 Hz, 0.4 H,), 7.16 (dd, J = 7.0,
5.0 Hz, 1.8 H), 7.20 (dd, J = 7.5, 7.5 Hz, 1.2 H), 7.28 (t, J = 7.5 Hz,2 H).13C NMR (126 MHz, CDCl3): d = 20.8, 20.9, 21.7, 23.1, 35.8, 36.0,42.3, 42.7, 54.0 (dd, JC,F = 25, 25 Hz), 54.2 (dd, JC,F = 26, 26 Hz),68.0 (d, JC,F = 3 Hz), 126.5, 126.7, 126.8, 128.58, 128.61, 145.2,145.5, 153.7 (d, JC,F = 299, 286 Hz), 153.9 (d, JC,F = 300, 286 Hz),169.4, 169.5.19F NMR (470 MHz, CDCl3): d = 88.8 (d, JF,F = 23 Hz, 0.4 F), 89.6(d, JF,F = 21 Hz, 0.6 F), 89.7 (d, JF,F = 23 Hz, 0.4 F), 90.2 (d,JF,F = 21 Hz, 0.6 F).
HRMS (ESI+): m/z calcd for C14H15F2IO2 + Na [M + Na]+:402.9982; found: 403.0000.
1,1-Difluoro-2-iodo-4-methyl-5-(3-pyridyl)pent-1-en-3-yl Ace-tate (3g)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 73% (diastereomer ratio = 6:4); colorless liquid.
IR (ATR): 2968, 2933, 1736, 1714, 1425, 1371, 1265, 1221, 1024,968, 793, 715 cm–1.1H NMR (500 MHz, CDCl3): d = 0.75 (d, J = 7.0 Hz, 1.6 H), 0.92(d, J = 6.0 Hz, 1.4 H), 2.10–2.11 (m, 4.4 H), 2.40 (dd, J = 13.5, 9.5Hz, 0.6 H), 2.72 (d, J = 10.0 Hz, 0.4 H), 2.99 (dd, J = 13.5, 4.5 Hz,0.6 H), 4.72 (d, J = 10.0 Hz, 0.6 H), 4.78 (d, J = 9.5 Hz, 0.4 H), 7.28(dd, J = 8.0, 4.0 Hz, 1 H), 7.53 (d, J = 8.0 Hz, 1 H), 8.45 (s, 1 H),8.49 (d, J = 4.5 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 14.3, 14.6, 20.79, 20.83, 35.3,35.6, 38.3, 39.0, 52.7 (dd, JC,F = 25, 25 Hz), 72.9 (d, JC,F = 3.5 Hz),73.2 (d, JC,F = 3.2 Hz), 123.4, 134.8, 135.2, 136.8, 136.9, 147.3,147.5, 150.0, 150.1, 154.4 (dd, JC,F = 299, 286 Hz), 154.5, (dd,JC,F = 299, 286 Hz), 169.59, 169.62.19F NMR (470 MHz, CDCl3): d = 89.0 (d, JF,F = 22 Hz, 0.6 F), 89.5(d, JF,F = 21 Hz, 0.4 F), 90.3 (d, JF,F = 22 Hz, 0.6 F), 91.2 (d,JF,F = 21 Hz, 0.4 F).
HRMS (ESI+): m/z calcd for C13H15F2INO2 [M + H]+: 382.0116;found: 382.0117.
Methyl 2-(3-Acetoxy-5,5-difluoro-4-iodopent-4-en-1-yl)ben-zoate (3h)Prepared from 1,1,1-trifluoro-2-iodoethane (840 mg, 4.00 mmol);yield: 82%; pale yellow liquid.
IR (ATR): 3068, 2952, 1747, 1716, 1435, 1373, 1259, 1228, 1088,1026, 966, 712 cm–1.1H NMR (500 MHz, CDCl3): d = 1.86–1.92 (m, 1 H), 2.04–2.13 (m,1 H), 2.08 (s, 3 H), 2.90 (ddd, J = 15.5, 10.0, 5.5 Hz, 1 H), 2.98 (ddd,J = 15.5, 10.0, 5.5 Hz, 1 H), 3.91 (s, 3 H), 5.02 (ddt, J = 7.0, 2.0, 2.0Hz, 1 H), 7.24 (d, J = 7.5 Hz, 1 H), 7.28 (dd, J = 7.5, 1.0 Hz, 1 H),7.44 (td, J = 7.5, 1.5 Hz, 1 H), 7.91 (dd, J = 7.5, 1.4 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 20.9, 29.7, 36.2, 52.0, 53.9 (t,JC,F = 26 Hz), 69.2 (d, JC,F = 4 Hz), 126.4, 129.4, 130.9, 131.0,132.2, 142.3, 154.0 (dd, JC,F = 299, 286 Hz), 167.7, 169.7.19F NMR (470 MHz, CDCl3): d = 89.1 (d, JF,F = 23 Hz, 1 F), 89.9(d, JF,F = 23 Hz, 1 F).
HRMS (ESI+): m/z calcd for C15H15F2IO4 + Na [M + Na]+:446.9881; found: 446.9879.
1,1-Difluoro-2-iodo-3-methyl-5-phenylpent-1-en-3-yl Acetate (3i)To a THF (5 mL) solution of (i-Pr)2NH (1.1 mL, 8.00 mmol) wasadded BuLi (4.8 mL, 1.67 M in hexane, 8.0 mmol) over 10 min at0 °C under argon. The resulting solution was allowed to stir for anadditional 15 min, then cooled to –93 °C using a cold hexane bath.To this cold LDA solution was added a THF (2 mL) solution of
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
80

PAPER 881
A Facile Synthesis of 1,1-Difluoroallenes from Commercially Available 1,1,1-Trifluoro-2-iodoethaneSynthesis of 1,1-DifluoroallenesKen Oh, Kohei Fuchibe, Junji Ichikawa*Department of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba, 305-8571, JapanFax +81(29)8534237; E-mail: [email protected] 30 November 2010; revised 19 January 2011
SYNTHESIS 2011, No. 6, pp 0881–0886xx.xx.2011Advanced online publication: 14.02.2011DOI: 10.1055/s-0030-1258438; Art ID: F21510SS© Georg Thieme Verlag Stuttgart · New York
Abstract: 1,1-Difluoroallenes are synthesized in good yield viazinc-promoted 1,2-elimination of 3,3-difluoro-2-iodoallylic ace-tates, which are prepared by the reaction of aldehydes or ketoneswith 1-iodo-2,2-difluorovinyllithium, generated from commerciallyavailable 1,1,1-trifluoro-2-iodoethane.
Key words: fluorinated allenes, metalation, carbanions, elimina-tion, difluorovinylidenation
1,1-Difluoroallenes have attracted much attention be-cause of their unusual reactivities, entailing them to beused as synthetic building blocks for fluorinated mole-cules. The Diels–Alder and [3+2]-cycloaddition reactionsof 1,1-difluoroallenes with 1,3-dienes and 1,3-dipolesreadily take place on the internal, nonfluorinated alkenemoiety to give the corresponding exo-difluoromethylenecompounds.1 For example, 1,1-difluoroallene(F2C=C=CH2), with a low LUMO energy level, gives anexcellent yield (>99%) of the cyclized product with cyclo-pentadiene under very mild conditions (–20 °C, 1 min),while the nonfluorinated counterpart allene(H2C=C=CH2) requires vigorous conditions (200–230 °C) to give the product in a modest yield (49%).1a,f
The [2+2]-cycloaddition reactions with alkenes andalkynes occur on the terminal, fluorinated alkene moietyto give ring fluorinated cyclobutane2a and cyclobutene2b
derivatives. 1,1-Difluoroallenes also react with variousnucleophiles to afford CF2-terminal or internal additionproducts selectively, depending on the character of the nu-cleophile.3
Although the parent 1,1-difluoroallene has been knownsince the 1950s,4 very few synthetic methods for the 3-substituted 1,1-difluoroallenes have been reported.3a,5 Re-cently, we have developed a versatile synthetic methodfor 3-substituted 1,1-difluoroallenes 1 using two steps: (i)lithiation of 1,1-dibromo-2,2-difluoroethene with butyl-lithium generates 1-bromo-2,2-difluorovinyllithium(F2C=CBrLi), which in turn, reacts with aldehydes or ke-tones to form 2-bromo-3,3-difluoroallylic acetates, and(ii) treatment of the bromoacetates with butyllithiumgives 1,1-difluoroallenes via the 1,2-elimination of lithi-um acetate.6
However, there are two factors that limit the scope of thismethod: (a) the starting material, F2C=CBr2, is a high-cost, potential ozone-depleting substance, and is now un-available because of the ban on its industrial manufacture,and (b) highly nucleophilic alkyllithiums are required inthe preparation of 1,1-difluoroallenes, which restricts thechoice of substrate. Here, we report an improved syntheticmethod for 1,1-difluoroallenes to overcome these issuesusing 1) an environmentally friendly and commerciallyavailable compound as the starting material, and 2) an ef-fective process for carrying out a 1,2-elimination reactionunder mild and tolerant reaction conditions.
First, we considered that the key intermediate, a 1-haloge-nated 2,2-difluorovinyl anion 2 (Scheme 1), would begenerated by the addition of two equivalents of a strongbase to 1,1,1-trifluoro-2-haloethanes,7–9 which bear twohydrogen atoms and are recognized to have much lowerozone depletion potential (ODP). These compounds aremanufactured industrially for use as refrigerants or asfluorinated intermediates. Second, we proposed a differ-ent route to access the desired 1,1-difluoroallenes 1 from3,3-difluoro-2-haloallylic acetates 3 (Scheme 1) on treat-ment with a zerovalent metal instead of highly reactivealkyllithiums, which would promote the 1,2-eliminationfrom acetates 3 to form one more double bond under mildconditions. This sequence would expand the scope of thesubstrates.
Scheme 1 A synthetic plan for 1,1-difluoroallenes from a 1,1,1-trifluoro-2-haloethane
1,1,1-Trifluoro-2-iodoethane was selected as the startingmaterial because of its ease of handling (bp 55–56 °C/760Torr). The lithiation of 1,1,1-trifluoro-2-iodoethane withtwo equivalents of lithium diisopropylamide at low tem-peratures (–93 to –85 °C) successfully gave 2,2-difluoro-1-iodovinyllithium (2, Table 1).10 Lithium species 2 then
base (2 equiv)O
R1
R2
– HF, – H+ CF2
X
1)
1
2
CF3CH2X2) acetylation
– M(OAc)X
3
F2C
X
AcO R2
R1F2C
R2
R1
–
M0
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
PAPER Synthesis of 1,1-Difluoroallenes 885
Synthesis 2011, No. 6, 881–886 © Thieme Stuttgart · New York
CF3CH2I (840 mg, 4.00 mmol) over 10 min, keeping the tempera-ture between –93 and –85 °C. After stirring for 20 min at the sametemperature, a THF (2 mL) solution of 4-phenylbutan-2-one (593mg, 4.00 mmol) was added over 5 min, keeping the temperature be-tween –93 and –85 °C. The mixture was stirred for an additional 30min, then warmed to –30 °C over 90 min. The reaction wasquenched with sat. aq NH4Cl (15 mL), and the product was extract-ed with Et2O (3 × 15 mL). The combined organic layers werewashed with brine (15 mL) and dried (Na2SO4). After removal ofthe solvent under reduced pressure, the residue was purified by col-umn chromatography (hexane–EtOAc, 10:1). This alcohol was usedin the next step without further purification. To a solution of the al-cohol in isopropenyl acetate (3 mL) was added 4-methylbenzene-sulfonic acid monohydrate (5 mg, 0.03 mmol). After refluxing for 4h, the reaction was quenched with sat. aq NaHCO3 (15 mL). Theproducts were extracted with Et2O (3 × 15 mL). The combined or-ganic layers were washed with brine (15 mL) and dried (Na2SO4).After removal of the solvent under reduced pressure, the residuewas purified by column chromatography (hexane–EtOAc, 30:1).Acetate 3i was obtained as a colorless liquid (1.22 g, 80%, twosteps).
IR (ATR): 3028, 2931, 2862, 1790, 1741, 1712, 1496, 1454, 1369,1238, 1196, 1068, 1020, 700 cm–1.1H NMR (500 MHz, CDCl3): d = 1.87 (d, J = 4.6 Hz, 1 H), 2.04 (s,3 H), 2.11–2.23 (m, 2 H), 2.58 (t, J = 8.6 Hz, 2 H), 7.18–7.20 (m, 3H), 7.29 (dd, J = 7.0, 7.0 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 21.8, 22.9 (d, JC,F = 7 Hz), 30.0,42.8 (dd, JC,F = 2 Hz), 59.7 (dd, JC,F = 26, 22 Hz), 80.6 (d, JC,F = 3Hz), 126.1, 128.3, 128.5, 140.8, 152.5 (dd, JC,F = 301, 281 Hz),169.3.19F NMR (470 MHz, CDCl3): d = 89.0 (dq, JF,F = 33 Hz, JF,H = 5 Hz,1 F), 97.3 (d, JF,F = 33 Hz, 1 F).
HRMS (ESI+): m/z calcd for C14H15F2IO2 + Na [M + Na]+:402.9982; found: 402.9979.
1,1-Difluoro-5-phenylpenta-1,2-diene (1a); Typical ProcedureTo a suspension of Zn powder (131 mg, 2.00 mmol) in DMF (3 mL)was added a DMF (2 mL) solution of 3a (366 mg, 1.00 mmol) at r.t.under argon. After stirring for 3 h, the resulting reaction mixturewas filtered to remove the excess Zn and then diluted with Et2O (20mL) and brine (15 mL). The products were extracted with Et2O(3 × 15 mL). The combined organic layers were washed with brine(15 mL) and dried (Na2SO4). After removal of the solvent under re-duced pressure, the residue was purified by column chromatogra-phy (pentane). Allene 1a was obtained as a colorless liquid (155 mg,86%).
IR (ATR): 3030, 2929, 2362, 2013, 1462, 1196, 744, 698 cm–1.1H NMR (500 MHz, CDCl3): d = 2.53–2.61 (m, 2 H), 2.81 (t,J = 7.5 Hz, 2 H), 6.47 (tt, J = 6.1 Hz, JH,F = 2.4 Hz, 1 H), 7.17–7.22(m, 3 H), 7.30 (dd, J = 7.3, 7.3 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 33.76, 33.77, 121.4 (t, JC,F = 6Hz), 126.2, 128.4, 128.5, 140.6, 152.8 (t, JC,F = 261 Hz), 170.1 (t,JC,F = 36 Hz).19F NMR (470 MHz, CDCl3): d = 60.0 (td, JF,H = 5, 2 Hz, 2 F).
HRMS (EI): m/z calcd for C11H10F2 [M]+: 180.0751; found:180.0749.
1,1-Difluorododeca-1,2-diene (1b)Prepared from 3b (388 mg, 1.00 mmol); yield: 87%; colorless liq-uid.
IR (ATR): 2925, 2856, 2011, 1462, 1246, 1194, 721 cm–1.
1H NMR (500 MHz, CDCl3): d = 0.88 (t, J = 7.0 Hz, 3 H), 1.27–1.30(m, 12 H), 1.49 (tq, J = 7.5, 7.0 Hz, 2 H), 2.23 (ttd, J = 7.0, 6.3, 6.0Hz, 2 H), 6.42 (tt, J = 6.3 Hz, JH,F = 2.3 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 14.1, 22.7, 27.6, 28.9, 29.3, 29.4,29.5, 31.9, 32.3 (t, JC,F = 2 Hz), 122.5 (t, JC,F = 6 Hz), 152.5 (t,JC,F = 261 Hz), 169.3 (t, JC,F = 36 Hz).19F NMR (470 MHz, CDCl3): d = 59.4 (td, JF,H = 6, 3 Hz, 2 F).
HRMS (EI): m/z calcd for C12H20F2 [M]+: 202.1533; found:202.1516.
1,1-Difluoro-5-(1-naphthyl)penta-1,2-diene (1c)Prepared from 3c (416 mg, 1.00 mmol); yield: 82%; colorless liq-uid.
IR (ATR): 3062, 2941, 2009, 1745, 1458, 1186, 791 cm–1.1H NMR (500 MHz, CDCl3): d = 2.48–2.54 (m, 2 H), 3.09 (t, J = 7.4Hz, 2 H), 6.36 (tt, J = 6.1 Hz, JH,F = 2.4 Hz, 1 H), 7.16 (d, J = 6.6Hz, 1 H), 7.25 (dd, J = 7.6 Hz, 1 H), 7.32–7.39 (m, 2 H), 7.59 (d,J = 7.5 Hz, 1 H), 7.72 (d, J = 8.0 Hz, 1 H), 7.83 (d, J = 7.5 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 30.8, 33.0, 121.6 (t, JC,F = 5.5Hz), 123.4, 125.5, 125.6, 126.0, 126.1, 127.1, 128.9, 131.6, 133.9,136.6, 152.9 (t, JC,F = 261 Hz), 170.0 (t, JC,F = 36 Hz).19F NMR (470 MHz, CDCl3): d = 60.4 (dt, JF,H = 2, 5 Hz, 2 F).
HRMS (EI): m/z calcd for C15H12F2 [M]+: 230.0907; found:230.0906.
5-(4-tert-Butylphenyl)-1,1-difluoro-4-methylpenta-1,2-diene (1d)Prepared from 3d (436 mg, 1.00 mmol); yield: 92%; colorless liq-uid.
IR (ATR): 2964, 2870, 2009, 1446, 1238, 1190, 937, 858 cm–1.1H NMR (500 MHz, CDCl3): d = 1.05 (d, J = 6.6 Hz, 3 H), 1.30 (s,9 H), 2.57 (dd, J = 13.0, 6.6 Hz, 1 H), 2.59–2.68 (m, 1 H), 2.75 (dd,J = 13.0, 7.6 Hz, 1 H), 6.41 (ddd, J = 5.0 Hz, JH,F = 2.5, 2.5 Hz, 1H), 7.08 (d, J = 8.0 Hz, 2 H), 7.30 (d, J = 8.0 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 18.5, 31.4, 34.3, 38.4, 41.6 (d,JC,F = 2 Hz), 125.2, 127.1 (dd, JC,F = 6, 6 Hz), 128.8, 136.4, 149.1,153.5 (dd, JC,F = 261, 261 Hz), 168.6 (dd, JC,F = 36, 36 Hz).19F NMR (470 MHz, CDCl3): d = 60.0 (ddd, JF,F = 127 Hz, JF,H = 3,3 Hz, 1 F), 60.3 (ddd, JF,F = 127 Hz, JF,H = 3, 3 Hz, 1 F).
HRMS (EI): m/z calcd for C16H20F2 [M]+: 250.1533; found:250.1532.
1,1-Difluoro-4-methyl-4-phenylpenta-1,2-diene (1e)Prepared from 3e (380 mg, 1.00 mmol); yield: 93%; colorless liq-uid.
IR (ATR): 2972, 2931, 2873, 2009, 1601, 1495, 1435, 1192, 958,854, 760, 696 cm–1.1H NMR (500 MHz, CDCl3): d = 1.47 (s, 6 H), 6.55 (t, JH,F = 2.8 Hz,1 H), 7.23–7.25 (m, 1 H), 7.31–7.36 (m, 4 H).13C NMR (126 MHz, CDCl3): d = 28.0, 42.4 (dd, JC,F = 2 Hz),125.9, 126.6, 128.5, 131.0 (t, JC,F = 6 Hz), 146.2 (d, JC,F = 2 Hz),153.3 (dd, JC,F = 262, 262 Hz), 167.1 (dd, JC,F = 36, 36 Hz).19F NMR (470 MHz, CDCl3): d = 61.1 (d, JH,F = 2 Hz, 2 F).
HRMS (EI): m/z calcd for C12H12F2 [M]+: 194.0907; found:194.0903.
1,1-Difluoro-5-phenylhexa-1,2-diene (1f)Prepared from 3f (380 mg, 1.00 mmol); yield: 95%; colorless liquid.
IR (ATR): 3030, 2964, 2009, 1726, 1603, 1495, 1458, 1240, 1190,760, 698 cm–1.
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
81

886 K. Oh et al. PAPER
Synthesis 2011, No. 6, 881–886 © Thieme Stuttgart · New York
1H NMR (500 MHz, CDCl3): d = 1.30 (d, J = 7.0 Hz, 3 H), 2.44–2.60 (m, 2 H), 2.95 (qdd, J = 7.1, 7.1, 7.1 Hz, 1 H), 6.28 (dddd,J = 6.9, 6.9 Hz, JH,F = 2.4, 2.4 Hz, 1 H), 7.18–7.23 (m, 3 H), 7.30 (t,J = 6.5 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 21.6, 38.8, 40.7, 120.4 (dd,JC,F = 6, 6 Hz), 126.4, 126.9, 128.5, 145.7, 152.4 (dd, JC,F = 260,260 Hz), 170.9 (dd, JC,F = 36, 36 Hz).19F NMR (470 MHz, CDCl3): d = 59.1 (br ddd, JF,F = 122 Hz,JF,H = 6, 4 Hz, 1 F), 59.6 (br ddd, JF,F = 122 Hz, JF,H = 7, 4 Hz, 1 F).
HRMS (EI): m/z calcd for C12H12F2 [M]+: 194.0907; found:194.0906.
3-(5,5-Difluoro-2-methylpenta-3,4-dien-1-yl)pyridine (1g)Prepared from 3g (381 mg, 1.00 mmol); yield: 71%; colorless liq-uid.
IR (ATR): 2970, 2931, 2009, 1576, 1446, 1188, 1026, 939, 856,796, 714 cm–1.1H NMR (400 MHz, CDCl3): d = 1.09 (d, J = 8.0 Hz, 3 H), 2.60–2.72 (m, 2 H), 2.81 (dd, J = 12.7, 6.0 Hz, 1 H), 6.42 (dt, J = 8.0 Hz,JH,F = 2.6 Hz, 1 H), 7.23 (ddd, J = 7.8, 4.8, 0.6 Hz, 1 H), 7.49 (ddd,J = 7.8, 2.1, 1.9 Hz, 1 H), 8.45 (d, J = 1.9 Hz, 1 H), 8.48 (dd,J = 4.8, 1.5 Hz, 1 H).13C NMR (101 MHz, CDCl3): d = 18.5, 38.0 (t, JC,F = 2 Hz), 38.9 (t,JC,F = 2 Hz), 123.3, 126.0 (t, JC,F = 6 Hz), 134.8, 136.5, 147.7,150.3, 153.4 (t, JC,F = 262 Hz), 169.4 (t, JC,F = 36 Hz).19F NMR (376 MHz, CDCl3): d = 60.5 (d, J = 120 Hz, 1 F), 60.7 (d,J = 120 Hz, 1 F).
HRMS (ESI+): m/z calcd for C11H12F2N [M + H]+: 196.0938; found:196.0947.
Methyl 2-(5,5-Difluoropenta-3,4-dien-1-yl)benzoate (1h)Prepared from 3h (424 mg, 1.00 mmol); yield: 74%; colorless liq-uid.
IR (ATR): 2952, 2009, 1716, 1460, 1254, 1186, 1130, 1082, 962,748, 708 cm–1.1H NMR (500 MHz, CDCl3): d = 2.54–2.59 (m, 2 H), 3.13 (t,J = 7.5 Hz, 2 H), 3.88 (s, 3 H), 6.49 (tt, J = 6.0 Hz, JH,F = 2.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1 H), 7.27 (dt, J = 7.5, 1.0 Hz, 1 H), 7.42(td, J = 7.5, 1.5 Hz, 1 H), 7.92 (dd, J = 7.5, 1.5 Hz, 1 H).13C NMR (126 MHz, CDCl3): d = 32.6, 33.9, 51.9, 121.6 (t, JC,F = 6Hz), 126.3, 129.3, 130.9, 131.1, 132.1, 142.6, 152.6 (t, JC,F = 261Hz), 167.7, 169.7 (t, JC,F = 36 Hz).19F NMR (470 MHz, CDCl3): d = 60.0 (s, 2 F).
HRMS (EI): m/z calcd for C13H12F2O2 [M]+: 238.0805; found:238.0805.
1,1-Difluoro-3-methyl-5-phenylpenta-1,2-diene (1i)Prepared from 3i (380 mg, 1.00 mmol); yield: 86%; colorless liquid.
IR (ATR): 3064, 2922, 2360, 2004, 1801, 1604, 1481, 1173, 1043,995, 696 cm–1.1H NMR (500 MHz, CDCl3): d = 1.91 (t, J = 5.0 Hz, 3 H), 2.40–2.48 (m, 2 H), 2.74 (t, J = 8.2 Hz, 2 H), 7.13–7.18 (m, 3 H), 7.25 (t,J = 7.6 Hz, 2 H).13C NMR (126 MHz, CDCl3): d = 22.8, 33.4, 38.6, 126.1, 128.3,128.4, 132.3 (t, JC,F = 6 Hz), 141.0, 150.4 (t, JC,F = 260 Hz), 163.0(t, JC,F = 35 Hz).19F NMR (470 MHz, CDCl3): d = 61.5 (tq, J = 5, 5 Hz, 2 F).
HRMS (EI): m/z calcd for C12H12F2 [M]+: 194.0907; found:194.0909.
Acknowledgment
This research was supported by a Grant-in-Aid for ScientificResearch from the Japan Society for the Promotion of Science, TheAsahi Glass Foundation, and Du Pont–Mitsui Fluorochemicals Co.,Ltd. We are grateful to Tosoh F-Tech, Inc. for a generous gift of1,1,1-trifluoro-2-iodoethane.
References
(1) (a) Dolbier, W. R. Jr.; Burkholder, C. R.; Piedrahita, C. A. J. Fluorine Chem. 1982, 20, 637. (b) Dolbier, W. R. Jr.; Burkholder, C. R.; Winchester, W. R. J. Org. Chem. 1984, 49, 1518. (c) Dolbier, W. R. Jr.; Burkholder, C. R. Israel J. Chem. 1985, 26, 115. (d) Dolbier, W. R. Jr.; Wicks, G. E.; Burkholder, C. R. J. Org. Chem. 1987, 52, 2196. (e) Dolbier, W. R. Jr.; Burkholder, C. R.; Wicks, G. E.; Palenik, G. J.; Gawron, M. J. Am. Chem. Soc. 1985, 107, 7183. (f) Dolbier, W. R. Jr. Acc. Chem. Res. 1991, 24, 63.
(2) (a) Dolbier, W. R. Jr.; Wicks, G. E. J. Am. Chem. Soc. 1985, 107, 3626. (b) Shen, Q.; Hammond, G. B. J. Am. Chem. Soc. 2002, 124, 6534.
(3) (a) Mae, M.; Hong, J. A.; Xu, B.; Hammond, G. B. Org. Lett. 2006, 8, 479. (b) Xu, Y.-Y.; Jin, F.-Q.; Huang, W.-Y. J. Fluorine Chem. 1995, 70, 5.
(4) (a) Blomquist, A. T.; Longone, D. T. J. Am. Chem. Soc. 1957, 79, 4981. (b) Knoth, W. H.; Coffman, D. D. J. Am. Chem. Soc. 1960, 82, 3873.
(5) (a) Shi, G.; Xu, Y. J. Fluorine Chem. 1989, 44, 161. (b) Wang, Z. G.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547. (c) Shen, Q.; Hammond, G. B. Org. Lett. 2001, 3, 2213. (d) Xu, B.; Hammond, G. B. Angew. Chem. Int. Ed. 2008, 47, 689.
(6) Yokota, M.; Fuchibe, K.; Ueda, M.; Mayumi, Y.; Ichikawa, J. Org. Lett. 2009, 11, 3994.
(7) For the generation of 2,2-difluoro-1-tosyloxyvinyllithium [F2C=C(OTs)Li], see: (a) Tanaka, K.; Nakai, T.; Ishikawa, N. Tetrahedron Lett. 1978, 19, 4809. (b) Ichikawa, J.; Hamada, S.; Sonoda, T.; Kobayashi, H. Tetrahedron Lett. 1992, 33, 337.
(8) For the generation of 2,2-difluoro-1-halovinyllithium (F2C=CXLi), see: (a) (X = F): Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. Chem. Commun. 1996, 49. (b) (X = F): Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1999, 99, 127. (c) (X = Cl): Burdon, J.; Coe, P. L.; Haslock, I. B.; Powell, R. L. J. Fluorine Chem. 1997, 85, 151. (d) (X = F or Cl): Coe, P. L.; Burdon, J.; Haslock, I. B. J. Fluorine Chem. 2000, 102, 43.
(9) For the generation of 2,2-difluoro-1-halovinylzinc(II) chloride (F2C=CXZnCl) at r.t., see: (a) (X = F): Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 2731. (b) (X = Cl): Anilkumar, R.; Burton, D. J. Tetrahedron Lett. 2002, 43, 6979. (c) (X = Br): Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2004, 125, 561. (d) (X = I): Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2005, 126, 455.
(10) The lithiation of CF3CH2I with BuLi instead of LDA at –93 to –85 °C led to a low yield of 2,2-difluoro-1-iodovinyl-lithium, probably due to I–Li exchange reaction.
(11) For 2-bromo-3,3-difluoroallylic acetates, zinc-promoted 1,2-elimination also took place readily at r.t. and led to the formation of the corresponding 1,1-difluoroallenes.
(12) Mg was employed in THF to promote the 1,2-elimination of acetate 3d in vain.
(13) When THF was used as a solvent, only a trace amount of 1g was observed by the 19F NMR measurement in spite of a large excess amount of Zn and an extended reaction time. A similar behavior was exhibited by some other acetates 3 without a heteroaromatic ring.
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
82

PRACTICAL SYNTHETIC PROCEDURES A
Facile Synthesis of Substituted 1,1-Difluoroallenes via Carbonyl DifluorovinylidenationSynthesis of Substituted 1,1-DifluoroallenesKen Oh, Kohei Fuchibe, Misaki Yokota, Junji Ichikawa*Department of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba 305-8571, JapanFax 81(29)8534237; E-mail: [email protected] 22 November 2011; revised 11 December 2011
SYNTHESIS 2012, No. x, pp 000A–000Exx.xx.2012Advanced online publication: xx.xx.2012DOI: 10.1055/s-0031-1290157; Art ID: Z109011SS© Georg Thieme Verlag Stuttgart · New York
Abstract: Two methods for the difluorovinylidenation of carbonyl compounds have been developed to synthesize 1,1-difluoroallenes bear-ing various substituents. The reaction of 1-bromo-2,2-difluorovinyllithium, generated from 1,1-dibromo-2,2-difluoroethylene and n-butyl-lithium, with aldehydes or ketones, and subsequent acetylation, gives 2-bromo-3,3-difluoroallylic acetates. Elimination of these acetateswith n-butyllithium affords 1,1-difluoroallenes in high yield. 3,3-Difluoro-2-iodoallylic acetates are similarly prepared from aldehydes orketones on treatment with 2,2-difluoro-1-iodovinyllithium, generated from 1,1,1-trifluoro-2-iodoethane and lithium diisopropylamide, fol-lowed by acetylation. These acetates readily undergo elimination with zinc metal to afford 1,1-difluoroallenes in high yield.
Key words: carbonyl compounds, metalation, difluorovinylidenation, 1,1-difluoroallenes
Introduction
1,1-Difluoroallenes are highly attractive synthetic inter-mediates because of their fluorine substituents and cumu-lative double bonds. In addition, 1,1-difluoroallenes canserve as promising pharmaceuticals, as some non-fluori-nated allenes have been used for therapeutic purposes.1 Todate, however, the synthetic methodology for 1,1-difluo-
roallenes bearing substituents has not been completely ex-plored.2–4
We have recently reported facile methods for the synthe-sis of substituted 1,1-difluoroallenes via difluorovinyli-denation of carbonyl compounds (Scheme 1).5 A widevariety of 1,1-difluoroallenes were efficiently synthesizedusing these methods.
Our synthesis consists of two steps (Scheme 2): (i) The re-action of an aldehyde or ketone with a 2,2-difluoro-1-ha-lovinyllithium 1 and subsequent acetylation to give the3,3-difluoro-2-haloallylic acetate 2. (ii) Metalation of
Scheme 1 Synthesis of 1,1-difluoroallenes by difluorovinylidenation of aldehydes or ketones
n-BuLi (1 equiv)
Zn (2 equiv)
O Ph
method A
F2C
Br
AcO
F2CPh Ph
O Ph
method B
F2C
I
AcO
F2CPh Ph
then Ac2O (1.2 equiv), 0 °C, 2 h
CF2=CBr2 (1 equiv)n-BuLi (1 equiv)
Et2O, –100 °C, 15 min
CF3CH2I (1 equiv)LDA (2 equiv)
THF, –93 to –30 °C, 2 h
then Ac2O (1 equiv), 0 °C, 2 h
2Aa 93% 3a 87%
2Ba 82% 3a 86%
O
n-Bu
F2C
Br
AcO
F2C
CF2=CBr2 (1 equiv)n-BuLi (1 equiv)
Et2O, –100 to –40 °C, 2 h
2Ak 80% 3k 85%
PhPh
n-Bu n-Bu
Ph
thenisopropenyl acetate
TsOH (2 mol%), reflux, 5 h
OF2C
I
AcO
F2C
2Bm 80% 3m 86%
thenisopropenyl acetate
TsOH (1 mol%), reflux, 4 h
PhCF3CH2I (1 equiv)
LDA (2 equiv)THF, –93 to –30 °C, 2 h
DMF, r.t., 3 h
hexane, 0 °C, 1 min
PhPh
n-BuLi (1 equiv)
hexane, 0 °C, 1 min
Zn (2 equiv)
DMF, r.t., 3 h
���
�����
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
83
A2. Oh, K.; Fuchibe, K.; Yokota, M; Ichikawa, J. Synthesis, in press.

B K. Oh et al. PRACTICAL SYNTHETIC PROCEDURES
Synthesis 2012, No. x, A–E © Thieme Stuttgart · New York
acetate 2 with an appropriate reducing agent which causeselimination of the halide ion (X–) and the acetate ion tofurnish the desired 1,1-difluoroallene 3. Depending on thereagents used for the difluorovinylation (the first step) andthe elimination (the second step), two methods are avail-able; namely, method A and B (see Scheme 1). Method Bshould have wide application because it includes a readilyavailable starting material and facilitates the synthesis of1,1-difluoroallenes bearing functionalities that are sensi-tive to n-butyllithium.
Scheme 2 Difluorovinylation–elimination sequence in 1,1-difluo-roallene synthesis
Method A: Synthesis of 1,1-Difluoroallenes from 1,1-Dibromo-2,2-difluoroethylene (First-Generation Syn-thesis)5a
Initially, 1,1-dibromo-2,2-difluoroethylene (CF2=CBr2)was used as a difluorovinylation agent. 1,1-Dibromo-2,2-difluoroethylene was lithiated with 1 equivalent of n-bu-tyllithium at –100 °C. Aldehydes or ketones were treatedwith the resulting vinyllithium, and then with acetic anhy-dride or with isopropenyl acetate/p-toluenesulfonic acid,respectively. Next, the isolated 2-bromo-3,3-difluoroal-lylic acetates 2A (X = Br) were lithiated with n-butyllith-ium in hexane; elimination of lithium acetate occurred toafford the 1,1-difluoroallenes 3.
Note that control of the temperature in the vinylation withdibromodifluoroethylene is crucial. The first lithiation ata temperature higher than –100 °C led to undesired 1,2-elimination of lithium fluoride from the intermediate 1-bromo-2,2-difluorovinyllithium. The second lithiation ofallylic acetates 2A must be performed in hexane to realizethe elimination in high yield.
The resulting 1,1-difluoroallenes were purified by stan-dard column chromatography (silica gel). The isolated di-fluoroallenes can be stored in a refrigerator (0 °C) for atleast one month.
Method B: Synthesis of 1,1-Difluoroallenes from 1,1,1-Trifluoro-2-iodoethane (Second-Generation Synthe-sis)5b
1,1-Dibromo-2,2-difluoroethylene, used in our first-gen-eration synthesis, is an expensive, potential ozone-deplet-ing substance and is now difficult to purchase because ofthe ban on its industrial manufacture. In addition, highly
reactive n-butyllithium is required in both metalationsteps of the alkenyl bromides.
The second-generation synthesis of 1,1-difluoroalleneswas realized using 1,1,1-trifluoro-2-iodoethane(CF3CH2I) as a difluorovinylation agent.6 This material isreadily available because it is manufactured industriallyfor use as a refrigerant or fluorinated intermediate. 1,1,1-Trifluoro-2-iodoethane was treated with two equivalentsof lithium diisopropylamide to generate 2,2-difluoro-1-io-dovinyllithium. Aldehydes react with the iodovinyllithi-um, and then the formed alkoxides are trapped with aceticanhydride in a one-pot operation. In the case of ketones,the isolated allylic alcohols were acetylated with isopro-penyl acetate and p-toluenesulfonic acid. The isolated 3,3-difluoro-2-iodoallylic acetates 2B (X = I) were reducedwith zinc metal (2 equiv). Elimination proceeds smoothlyin N,N-dimethylformamide or tetrahydrofuran at roomtemperature, and the desired 1,1-difluoroallenes were ob-tained in high yield.
Reaction Scope
Yields of the synthesized acetates 2 and 1,1-difluoroal-lenes 3 using methods A and B are summarized inTable 1. A wide variety of monosubstituted 1,1-difluoro-allenes were obtained from aldehydes, while the synthesisof disubstituted 1,1-difluoroallenes was accomplished bythe difluorovinylidenation of ketones.
Although yields obtained in the two methods are nearlyequal, method B is synthetically more favorable becauseit enables the synthesis of 1,1-difluoroallenes bearingfunctionalities that are sensitive to n-butyllithium. Alde-hyde 4, bearing an ester moiety, and aldehyde 5, bearinga pyridine ring, were transformed to the corresponding1,1-difluoroallenes 3n and 3o in 61% and 52% yield, re-spectively (Scheme 3).
Scheme 3 Synthesis of 1,1-difluoroallenes with an ester moiety anda pyridine ring
Summary
Difluorovinylation of aldehydes or ketones is readily real-ized with 1-bromo-2,2-difluorovinyllithium (method A)and 2,2-difluoro-1-iodovinyllithium (method B), which isfollowed by acetylation to give 2-bromo-3,3-difluoroal-lylic acetates and 3,3-difluoro-2-iodoallylic acetates, re-spectively. The formed allylic acetates undergo facileelimination on treatment with n-butyllithium (method A)or zinc metal (method B) to afford 1,1-difluoroallenes in
F2C
R2
R1
then acetylation
F2C
X
AcO
R1
R2
F2C
AcO
R1
R2 – MOAc
O
R2
R1 CF2=C(X)Li 1
M (metal)M
– X–
(i) difluorovinylation
(ii) elimination
2
3
O
N
F2C
N
OCO2Me
F2C
CO2Me
method B
3o 52% (two steps)
3n 61% (two steps)4
5
method B
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
84

PRACTICAL SYNTHETIC PROCEDURES Synthesis of Substituted 1,1-Difluoroallenes C
Synthesis 2012, No. x, A–E © Thieme Stuttgart · New York
high yield. Method B starts from the readily available1,1,1-trifluoro-2-iodoethane, and allows the synthesis of1,1-difluoroallenes bearing functionalities that are sensi-tive to n-butyllithium.
NMR spectra were recorded in CDCl3 on a Bruker Avance 500 orAvance 400 spectrometer. Chemical shift values are given in ppmrelative to internal TMS (for 1H NMR: d = 0.00 ppm), CDCl3 (for13C NMR: d = 77.0 ppm), and C6F6 (for 19F NMR: d = 0.0 ppm).Mass spectra (EI–TOF or ESI–TOF) were measured on a JEOLJMS-T100GCV or JMS-T100CS mass spectrometer. IR spectrawere recorded using the ATR (attenuated total reflectance) methodon a Horiba FT-720 spectrophotometer. Column chromatographyand preparative TLC were conducted on silica gel (Silica Gel 60 N,Kanto Chemical Co., Inc. for column chromatography and Wakogel
B-5F, Wako Pure Chemical Industries for PTLC, respectively). Allreactions were conducted under argon. THF, DMF, hexane, andEt2O were dried by passage over a column of activated alumina fol-lowed by a column of Q-5 scavenger (Engelhard). Pentane was dis-tilled from CaH2.
2-Bromo-1,1-difluoro-5-phenylpent-1-en-3-yl Acetate (2Aa); Typical Procedure for Method A (from Aldehydes; Table 1, Entry 1)To a soln of 1,1-dibromo-2,2-difluoroethylene (444 mg, 2.0 mmol)in Et2O (16 mL) was added a Et2O soln (2.0 mL) of n-BuLi (1.60 Min hexane; 1.28 mL, 2.0 mmol) at –100 °C under argon. The mixturewas stirred for 15 min at that same temperature, then 3-phenylpro-panal (0.28 mL, 2.0 mmol) was added. The mixture was stirred foran additional 15 min. After Ac2O (0.19 mL, 2.0 mmol) was added,the mixture was allowed to warm to 0 °C over 2 h. The reaction was
Table 1 Synthesis of 1,1-Difluoroallenes from 1,1-Dibromo-2,2-difluoroethylene or 1,1,1-Trifluoro-2-iodoethane
Entry Carbonyl compound Difluoroallene Yield (%) (method A) Yield (%) (method B)
2A 3 2B 3
12
Ar = PhAr = 1-Naph
93 (2Aa)84 (2Ab)
87 (3a)73a (3b)
82 (2Ba)83 (2Bb)
86 (3a)82 (3b)
3 n.a.b n.a.b 84 (2Bc) 87 (3c)
4 83 (2Ad) 85 (3d) 81 (2Bd) 95 (3d)
5 86 (2Ae) 84 (3e) 87 (2Be) 92 (3e)
6 85 (2Af) 84 (3f) n.a.b n.a.b
7 85 (2Ag) 83 (3g) n.a.b n.a.b
89
10
R = HR = MeR = OMe
n.a.b
87 (2Ai)87 (2Aj)
n.a.b
82 (3i)81 (3j)
83 (2Bh)n.a.b
n.a.b
93 (3h)n.a.b
n.a.b
11 80c (2Ak) 85a (3k) n.a.b n.a.b
1213
R = (CH2)2PhR = Me
84c (2Al)n.a.b
90 (3l)n.a.b
n.a.b
80c (2Bm)n.a.b
86 (3m)
a 19F NMR yield based on PhCF3.b n.a. = reaction not attempted.c Acetylation was performed with isopropenyl acetate and p-TsOH.
F2C
X
AcO
R1 F2C
R2R2
2A (X = Br)2B (X = I)
3
R1
O
R2
R1
then Ac2O (1 equiv), 0 °C
method ACF2=CBr2 (1 equiv)
n-BuLi (1 equiv)Et2O, –100 °C
method BCF3CH2I (1 equiv)
LDA (2 equiv)THF, –93 to –30 °C
method An-BuLi (1 equiv)Hexane, 0 °C
method BZn (2 equiv)
DMF or THF, r.t.
O Ar F2C Ar
OCH2(CH2)7Me
F2C
CH2(CH2)7Me
O Ph F2C Ph
O
t-Bu
F2C
t-Bu
OBr
F2C
Br
OPh
F2C
Ph
OR
F2C
R
O
n-Bu
PhF2C
n-Bu
Ph
O
R
Ph
F2C
R
Ph
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
85

D K. Oh et al. PRACTICAL SYNTHETIC PROCEDURES
Synthesis 2012, No. x, A–E © Thieme Stuttgart · New York
quenched with sat. aq NH4Cl (10 mL), and the products were ex-tracted with Et2O (3 × 15 mL). The combined organic layer waswashed with brine (20 mL) and dried (Na2SO4). After the solventwas removed under reduced pressure, the residue was purified bycolumn chromatography (hexane–EtOAc, 20:1) to give 2Aa as acolorless liquid; yield: 593 mg (93%).
IR: 3028, 2949, 1747, 1731, 1282, 1221, 1026, 700 cm–1.1H NMR (500 MHz, CDCl3): d = 1.98–2.08 (m, 1 H, CH2), 2.07 (s,3 H, CH3CO), 2.11–2.17 (m, 1 H, CH2), 2.58–2.62 (m, 2 H, CH2),5.44 (dddd, J = 7.3, 7.3 Hz, JHF = 2.1, 2.1 Hz, 1 H, CH), 7.17 (dd,J = 7.8, 0.8 Hz, 2 H, ArH), 7.21 (tt, J = 7.8, 0.8 Hz, 1 H, ArH), 7.29(dd, J = 7.8, 7.8 Hz, 2 H, ArH).13C NMR (126 MHz, CDCl3): d = 20.8, 31.1, 34.1, 68.3 (d, JCF = 3Hz), 81.0 (dd, JCF = 35, 21 Hz), 126.3, 128.2, 128.6, 140.2, 154.2(dd, JCF = 294, 289 Hz), 169.7.19F NMR (470 MHz, CDCl3): d = 80.7 (br d, JFF = 27 Hz, 1 F), 82.3(br d, JFF = 27 Hz, 1 F).
Anal. Calcd for C13H13BrF2O2: C, 48.92; H, 4.11. Found: C, 49.14;H, 4.34.
1,1-Difluoro-5-phenylpenta-1,2-diene (3a)n-BuLi (1.60 M in hexane; 0.12 mL, 0.19 mmol) was added to asoln of acetate 2Aa (60 mg, 0.19 mmol) in hexane (2.6 mL) at 0 °Cunder argon. The mixture was stirred for 1 min at that same temper-ature, then the reaction was quenched with aq NH4Cl (3 mL), andthe products were extracted with Et2O (3 × 5 mL). The combinedorganic layer was washed with brine (10 mL) and dried (Na2SO4).After the solvent was removed under reduced pressure, the residuewas purified by column chromatography (pentane) to give 3a as acolorless liquid; yield: 29 mg (87%).
IR: 3030, 2927, 2856, 2011, 1462, 1192, 744, 698 cm–1.1H NMR (500 MHz, CDCl3): d = 2.53–2.61 (m, 2 H, CH2), 2.81 (t,J = 7.5 Hz, 2 H, CH2), 6.47 (tt, J = 6.1 Hz, JHF = 2.4 Hz, 1 H, =CH),7.17–7.22 (m, 3 H, ArH), 7.30 (dd, J = 7.3, 7.3 Hz, 2 H, ArH).13C NMR (126 MHz, CDCl3): d = 33.8, 33.8, 121.4 (t, JCF = 6 Hz),126.2, 128.4, 128.5, 140.6, 152.8 (t, JCF = 261 Hz), 170.1 (t,JCF = 36 Hz).19F NMR (470 MHz, CDCl3): d = 59.9 (td, JFH = 5, 2 Hz).
Anal. Calcd for C11H10F2: C, 73.32; H, 5.59. Found: C, 73.16; H,5.77.
2-Bromo-1,1-difluoro-3-phenylhept-1-en-3-yl Acetate (2Ak); Typical Procedure for Method A (from Ketones; Table 1, Entry 11)To a soln of 1,1-dibromo-2,2-difluoroethylene (442 mg, 2.0 mmol)in Et2O (16 mL) was added a Et2O soln (2.0 mL) of n-BuLi (1.60 Min hexane; 1.28 mL, 2.0 mmol) at –100 °C under argon. The mixturewas stirred for 15 min at that same temperature, then 1-phenylpen-tan-1-one (0.30 mL, 2.0 mmol) in Et2O (3 mL) was added. The mix-ture was stirred for an additional 1 h, and then allowed to warm to–40 °C over 1 h. The reaction was quenched with sat. aq NH4Cl(10 mL), and the products were extracted with Et2O (3 × 15 mL).The combined organic layer was washed with brine (20 mL) anddried (Na2SO4). After the solvent was removed under reduced pres-sure, the residue was purified by column chromatography (hexane–EtOAc, 10:1) to give 2-bromo-1,1-difluoro-3-phenylhept-1-en-3-ol. To a soln of the alcohol in isopropenyl acetate (3 mL) was addedp-TsOH·H2O (5 mg, 0.03 mmol). The mixture was refluxed for 5 h,then the reaction was quenched with sat. aq NaHCO3 (2 mL). Theproducts were extracted with Et2O (3 × 3 mL). The combined or-ganic layer was washed with brine (3 mL) and dried (Na2SO4). Afterthe solvent was removed under reduced pressure, the residue was
purified by column chromatography (hexane–EtOAc, 30:1) to give2Ak as a colorless liquid; yield: 555 mg (80%, two steps).
IR: 2958, 2871, 1741, 1726, 1275, 1217, 962, 708 cm–1.1H NMR (500 MHz, CDCl3): d = 0.78 (t, J = 7.2 Hz, 3 H, CH3),0.81–0.91 (m, 1 H, CH2), 1.04–1.29 (m, 3 H, CH2), 2.11 (dddd,J = 14.0, 11.9, 4.4 Hz, JHF = 4.4 Hz, 1 H, CH2), 2.20 (s, 3 H,CH3CO), 2.94 (dddd, J = 14.0, 12.2, 4.4 Hz, JHF = 3.4 Hz, 1 H,CH2), 7.28–7.41 (m, 5 H, ArH).13C NMR (126 MHz, CDCl3): d = 13.8, 21.6, 22.5, 25.8 (d, JCF = 2Hz), 36.6 (d, JCF = 7 Hz), 84.1 (dd, JCF = 4, 2 Hz), 87.2 (dd,JCF = 29, 23 Hz), 125.2, 127.7, 128.2, 141.4 (dd, JCF = 2, 2 Hz),153.4 (dd, JCF = 297, 284 Hz), 168.6.19F NMR (470 MHz, CDCl3): d = 83.7 (ddd, JFF = 34 Hz, JFH = 4, 3Hz, 1 F), 87.0 (d, JFF = 34 Hz, 1 F).
Anal. Calcd for C15H17BrF2O2: C, 51.87; H, 4.94. Found: C, 51.83;H, 4.99.
1,1-Difluoro-3-phenylhepta-1,2-diene (3k)The acetate 2Ak was converted into the corresponding difluoroal-lene 3k by the same procedure as used for acetate 2Aa; yield: 85%.
IR: 2958, 2927, 1990, 1718, 1464, 1261, 1182, 769 cm–1.1H NMR (500 MHz, CDCl3): d = 0.95 (t, J = 7.4 Hz, 3 H, CH3),1.42 (tq, J = 7.5, 7.4 Hz, 2 H, CH2), 1.59 (tt, J = 7.5, 7.5 Hz, 2 H,CH2), 2.69 (tt, J = 7.5 Hz, JHF = 5.7 Hz, 2 H, CH2), 7.33–7.40 (m, 3H, ArH), 7.50–7.53 (m, 2 H, ArH).13C NMR (126 MHz, CDCl3): d = 14.0, 22.2, 29.7 (d, JCF = 5 Hz),33.0, 127.2, 128.5, 129.3, 134.8 (t, JCF = 6 Hz), 135.7, 152.6 (t,JCF = 259 Hz), 166.2 (t, JCF = 36 Hz).19F NMR (470 MHz, CDCl3): d = 60.0 (t, JFH = 6 Hz).
HRMS–FAB: m/z [M + H]+ calcd for C13H15F2: 209.1142; found:209.1130.
1,1-Difluoro-2-iodo-5-phenylpent-1-en-3-yl Acetate (2Ba); Typical Procedure for Method B (from Aldehydes; Table 1, Entry 1)To a soln of (i-Pr)2NH (2.8 mL, 20 mmol) in THF (10 mL) was add-ed n-BuLi (1.67 M in hexane; 12.0 mL, 20.0 mmol) over 10 min at0 °C under argon. The resulting solution was allowed to stir for anadditional 15 min, and then cooled to –93 °C in a cold hexane bath.To the cold LDA soln was added a soln of CF3CH2I (2.10 g, 10.0mmol) in THF (5 mL) over 10 min, keeping the temperature be-tween –93 °C and –85 °C. After the mixture was stirred for 20 minat that same temperature, a soln of 3-phenylpropanal (1.34 g, 10.0mmol) in THF (5 mL) was added over 5 min, while keeping the tem-perature between –93 °C and –85 °C. The mixture was stirred for anadditional 30 min, then warmed to –30 °C over 90 min. After Ac2O(1.23 g, 12.0 mmol) was added, the mixture was allowed to warmto 0 °C over 2 h. The reaction was quenched with sat. aq NH4Cl(20 mL), and the products were extracted with Et2O (3 × 20 mL).The combined organic layer was washed with brine (20 mL) anddried (Na2SO4). After the solvent was removed under reduced pres-sure, the residue was purified by column chromatography (hexane–EtOAc, 20:1). The acetate 2Ba was obtained as a colorless liquid;yield: 3.01 g (82%).
IR: 3028, 2954, 1743, 1716, 1267, 1219, 1024, 698 cm–1.1H NMR (500 MHz, CDCl3): d = 1.87–1.93 (m, 1 H, CH2), 2.05–2.17 (m, 1 H, CH2), 2.07 (s, 3 H, CH3CO), 2.58 (t, J = 7.2 Hz, 2 H,CH2), 4.98 (t, J = 7.2 Hz, 1 H, CH), 7.17–7.22 (m, 3 H, ArH), 7.29(dd, J = 7.3, 7.6 Hz, 2 H, ArH).13C NMR (126 MHz, CDCl3): d = 20.9, 30.9, 36.0, 53.8 (dd,JCF = 25, 26 Hz), 68.9 (d, JCF = 3 Hz), 126.2, 128.2, 128.5, 140.2,154.0 (dd, JCF = 286, 286 Hz), 169.6.
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.

PRACTICAL SYNTHETIC PROCEDURES A
Facile Synthesis of Substituted 1,1-Difluoroallenes via Carbonyl DifluorovinylidenationSynthesis of Substituted 1,1-DifluoroallenesKen Oh, Kohei Fuchibe, Misaki Yokota, Junji Ichikawa*Department of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba 305-8571, JapanFax 81(29)8534237; E-mail: [email protected] 22 November 2011; revised 11 December 2011
SYNTHESIS 2012, No. x, pp 000A–000Exx.xx.2012Advanced online publication: xx.xx.2012DOI: 10.1055/s-0031-1290157; Art ID: Z109011SS© Georg Thieme Verlag Stuttgart · New York
Abstract: Two methods for the difluorovinylidenation of carbonyl compounds have been developed to synthesize 1,1-difluoroallenes bear-ing various substituents. The reaction of 1-bromo-2,2-difluorovinyllithium, generated from 1,1-dibromo-2,2-difluoroethylene and n-butyl-lithium, with aldehydes or ketones, and subsequent acetylation, gives 2-bromo-3,3-difluoroallylic acetates. Elimination of these acetateswith n-butyllithium affords 1,1-difluoroallenes in high yield. 3,3-Difluoro-2-iodoallylic acetates are similarly prepared from aldehydes orketones on treatment with 2,2-difluoro-1-iodovinyllithium, generated from 1,1,1-trifluoro-2-iodoethane and lithium diisopropylamide, fol-lowed by acetylation. These acetates readily undergo elimination with zinc metal to afford 1,1-difluoroallenes in high yield.
Key words: carbonyl compounds, metalation, difluorovinylidenation, 1,1-difluoroallenes
Introduction
1,1-Difluoroallenes are highly attractive synthetic inter-mediates because of their fluorine substituents and cumu-lative double bonds. In addition, 1,1-difluoroallenes canserve as promising pharmaceuticals, as some non-fluori-nated allenes have been used for therapeutic purposes.1 Todate, however, the synthetic methodology for 1,1-difluo-
roallenes bearing substituents has not been completely ex-plored.2–4
We have recently reported facile methods for the synthe-sis of substituted 1,1-difluoroallenes via difluorovinyli-denation of carbonyl compounds (Scheme 1).5 A widevariety of 1,1-difluoroallenes were efficiently synthesizedusing these methods.
Our synthesis consists of two steps (Scheme 2): (i) The re-action of an aldehyde or ketone with a 2,2-difluoro-1-ha-lovinyllithium 1 and subsequent acetylation to give the3,3-difluoro-2-haloallylic acetate 2. (ii) Metalation of
Scheme 1 Synthesis of 1,1-difluoroallenes by difluorovinylidenation of aldehydes or ketones
n-BuLi (1 equiv)
Zn (2 equiv)
O Ph
method A
F2C
Br
AcO
F2CPh Ph
O Ph
method B
F2C
I
AcO
F2CPh Ph
then Ac2O (1.2 equiv), 0 °C, 2 h
CF2=CBr2 (1 equiv)n-BuLi (1 equiv)
Et2O, –100 °C, 15 min
CF3CH2I (1 equiv)LDA (2 equiv)
THF, –93 to –30 °C, 2 h
then Ac2O (1 equiv), 0 °C, 2 h
2Aa 93% 3a 87%
2Ba 82% 3a 86%
O
n-Bu
F2C
Br
AcO
F2C
CF2=CBr2 (1 equiv)n-BuLi (1 equiv)
Et2O, –100 to –40 °C, 2 h
2Ak 80% 3k 85%
PhPh
n-Bu n-Bu
Ph
thenisopropenyl acetate
TsOH (2 mol%), reflux, 5 h
OF2C
I
AcO
F2C
2Bm 80% 3m 86%
thenisopropenyl acetate
TsOH (1 mol%), reflux, 4 h
PhCF3CH2I (1 equiv)
LDA (2 equiv)THF, –93 to –30 °C, 2 h
DMF, r.t., 3 h
hexane, 0 °C, 1 min
PhPh
n-BuLi (1 equiv)
hexane, 0 °C, 1 min
Zn (2 equiv)
DMF, r.t., 3 h
���
�����
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
PRACTICAL SYNTHETIC PROCEDURES Synthesis of Substituted 1,1-Difluoroallenes E
Synthesis 2012, No. x, A–E © Thieme Stuttgart · New York
19F NMR (470 MHz, CDCl3): d = 89.2 (d, JFF = 22 Hz, 1 F), 90.2 (d,JFF = 22 Hz, 1 F).
HRMS (ESI+): m/z [M + Na]+ calcd for C13H13F2IO2Na: 388.9826;found: 388.9830.
1,1-Difluoro-5-phenylpenta-1,2-diene (3a)To a suspension of zinc powder (131 mg, 2.00 mmol) in DMF (3mL) was added a soln of 2Ba (366 mg, 1.00 mmol) in DMF (2 mL)at r.t. under argon, and the mixture was stirred for 3 h. The resultingmixture was filtered to remove the excess zinc and then diluted withEt2O (20 mL) and brine (15 mL). The products were extracted withEt2O (3 × 15 mL). The combined organic layer was washed withbrine (15 mL) and dried (Na2SO4). After the solvent was removedunder reduced pressure, the residue was purified by column chro-matography (pentane). Difluoroallene 3a was obtained as a color-less liquid; yield: 155 mg (86%).
1,1-Difluoro-2-iodo-3-methyl-5-phenylpent-1-en-3-yl Acetate (2Bm); Typical Procedure for Method B (from Ketones; Table 1, Entry 13)To a soln of (i-Pr)2NH (1.1 mL, 8.00 mmol) in THF (5 mL) was add-ed n-BuLi (1.67 M in hexane; 4.8 mL, 8.0 mmol) over 10 min at0 °C under argon. The resulting solution was allowed to stir for anadditional 15 min, and then cooled to –93 °C in a cold hexane bath.To the cold LDA soln was added a soln of CF3CH2I (840 mg, 4.00mmol) in THF (2 mL) over 10 min, keeping the temperature be-tween –93 °C and –85 °C. After the mixture was stirred for 20 minat that same temperature, a soln of 4-phenylbutan-2-one (593 mg,4.00 mmol) in THF (2 mL) was added over 5 min, while keeping thetemperature between –93 °C and –85 °C. The mixture was stirredfor an additional 30 min, then warmed to –30 °C over 90 min. Thereaction was quenched with sat. aq NH4Cl (15 mL), and the prod-ucts were extracted with Et2O (3 × 15 mL). The combined organiclayer was washed with brine (15 mL) and dried (Na2SO4). After thesolvent was removed under reduced pressure, the residue was puri-fied by column chromatography (hexane–EtOAc, 10:1) to give 1,1-difluoro-2-iodo-3-methyl-5-phenylpent-1-en-3-ol. To a soln of thealcohol in isopropenyl acetate (3 mL) was added p-TsOH·H2O (5mg, 0.03 mmol). The mixture was refluxed for 4 h, then the reactionwas quenched with sat. aq NaHCO3 (15 mL). The products were ex-tracted with Et2O (3 × 15 mL). The combined organic layer waswashed with brine (15 mL) and dried (Na2SO4). After the solventwas removed under reduced pressure, the residue was purified bycolumn chromatography (hexane–EtOAc, 30:1) to give 2Bm as acolorless liquid; yield: 1.22 g (80%, two steps).
IR: 3028, 2931, 2862, 1790, 1741, 1712, 1496, 1454, 1369, 1238,1196, 1068, 1020, 700 cm–1.1H NMR (500 MHz, CDCl3): d = 1.87 (d, J = 4.6 Hz, 3 H, CH3),2.04 (s, 3 H, CH3CO), 2.11–2.23 (m, 2 H, CH2), 2.58 (t, J = 8.6 Hz,2 H, CH2), 7.18–7.20 (m, 3 H, ArH), 7.29 (dd, J = 7.0, 7.0 Hz, 2 H,ArH).13C NMR (126 MHz, CDCl3): d = 21.8, 22.9 (d, JCF = 7 Hz), 30.0,42.8 (d, JCF = 2 Hz), 59.7 (dd, JCF = 26, 22 Hz), 80.6 (d, JCF = 3 Hz),126.1, 128.3, 128.5, 140.8, 152.5 (dd, JCF = 301, 281 Hz), 169.3.
19F NMR (470 MHz, CDCl3): d = 89.0 (dq, JFF = 33 Hz, JFH = 5 Hz,1 F), 97.3 (d, JFF = 33 Hz, 1 F).
HRMS (ESI+): m/z [M + Na]+ calcd for C14H15F2IO2Na: 402.9982;found: 402.9979.
1,1-Difluoro-3-methyl-5-phenylpenta-1,2-diene (3m)The acetate 2Bm was converted into the corresponding difluoroal-lene 3m by the same procedure as used for acetate 2Ba; yield: 86%.
IR: 3064, 2922, 2360, 2004, 1801, 1604, 1481, 1173, 1043, 995,696 cm–1.1H NMR (500 MHz, CDCl3): d = 1.91 (t, J = 5.0 Hz, 3 H, CH3),2.40–2.48 (m, 2 H, CH2), 2.74 (t, J = 8.2 Hz, 2 H, CH2), 7.13–7.18(m, 3 H, ArH), 7.25 (t, J = 7.6 Hz, 2 H, ArH).13C NMR (126 MHz, CDCl3): d = 22.8, 33.4, 38.6, 126.1, 128.3,128.4, 132.3 (t, JCF = 6 Hz), 141.0, 150.4 (t, JCF = 260 Hz), 163.0 (t,JCF = 35 Hz).19F NMR (470 MHz, CDCl3): d = 61.5 (tq, JFH = 5, 5 Hz).
HRMS (EI): m/z [M]+ calcd for C12H12F2: 194.0907; found:194.0909.
Acknowledgment
This research was partly supported by a Grant-in-Aid for Explora-tory Research from MEXT, Japan. We thank Tosoh F-Tech, Inc. forproviding 1,1,1-trifluoro-2-iodoethane.
References
(1) Krause, N.; Hoffmann-Röder, A. In Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, 2004, 997.
(2) For the synthesis of non-fluorinated allenes, see: Brummond, M.; DeForrest, J. E. Synthesis 2007, 795.
(3) For the synthesis of fluorinated allenes, see: (a) Dolbier, W. R. Jr.; Burkholder, C. R.; Piedrahita, C. A. J. Fluorine Chem. 1982, 20, 637. (b) Shi, G.; Xu, Y. J. Fluorine Chem. 1989, 44, 161. (c) Wang, Z.; Hammond, G. B. J. Org. Chem. 2000, 65, 6547. (d) Mae, M.; Hong, J. A.; Xu, B.; Hammond, G. B. Org. Lett. 2006, 8, 479.
(4) See also: (a) Zens, A. P.; Ellis, P. D.; Ditchfield, R. J. Am. Chem. Soc. 1974, 96, 1309. (b) Castelhano, A. L.; Krantz, A. J. Am. Chem. Soc. 1987, 109, 3491. (c) Lu, H.; Friedrich, H. B.; Burton, D. J. J. Fluorine Chem. 1995, 75, 83. (d) Xu, B.; Hammond, G. B. Angew. Chem. Int. Ed. 2008, 47, 689.
(5) (a) Yokota, M.; Fujita, D.; Ichikawa, J. Org. Lett. 2007, 9, 4639. (b) Oh, K.; Fuchibe, K.; Ichikawa, J. Synthesis 2011, 881.
(6) Anilkumar, R.; Burton, D. J. J. Fluorine Chem. 2005, 126, 455.
Dow
nloa
ded
by: T
akah
iko
Akiy
ama.
Cop
yrig
hted
mat
eria
l.
87

















