113
CADTH COMMON DRUG REVIEW Submission Guidelines for the CADTH Common Drug Review AUGUST 2014

CADTH COMMON DRUG REVIEW
Submission Guidelines for the CADTH Common Drug ReviewAUGUST 2014

CADTH Common Drug Review
RECORD OF UPDATES TO SUBMISSION GUIDELINES FOR THE CADTH COMMON DRUG REVIEW
Update Version of Submission Guidelines for the CADTH Common Drug Review Original June 2003
1 August 2003 2 September 2003 3 November 2003 4 April 2004 5 July 2004 6 December 2004 7 January 2005 8 July 2005 9 May 2006
10 September 2006 11 February 2007 12 July 2007 13 October 2007 14 November 2007 15 April 2008 16 April 2009 17 July 2009 18 May 2010 19 August 2010 20 December 2010 21 September 2011 22 November 2011 23 January 2013 24 August 2014
Submission Guidelines for the CADTH Common Drug Review August 2014 i

CADTH Common Drug Review
INQUIRIES All CADTH Common Drug Review–related inquiries should be directed in writing to:
Email: [email protected] Fax: 613 226 5392
Mail: Central Intake CADTH 600-865 Carling Avenue Ottawa, ON K1S 5S8
Submission Guidelines for the CADTH Common Drug Review August 2014 ii

CADTH Common Drug Review
TABLE OF CONTENTS ABBREVIATIONS ..................................................................................................................... vi 1. INTRODUCTION ................................................................................................................. 1
1.1 About This Document .................................................................................................. 1 1.2 Changes to These Guidelines ...................................................................................... 1
2. CADTH COMMON DRUG REVIEW – ELIGIBLE APPLICATIONS .................................. 2
2.1 CADTH Common Drug Review Submissions .............................................................. 2 2.1.1 New Drug ....................................................................................................... 2 2.1.2 Drug with a New Indication ............................................................................. 3 2.1.3 New Combination Product .............................................................................. 3 2.1.4 Subsequent Entry Biologic ............................................................................. 3
2.2 Notice of Compliance Status at the Time of Filing the Submission .............................. 4 2.2.1 Submissions Filed Pre-NOC or NOC/c ........................................................... 4 2.2.2 Submissions Filed Post-NOC or NOC/c ......................................................... 4
2.3 Drugs Not Eligible for Review Under the CADTH Common Drug Review Process ..... 4 2.4 CADTH Common Drug Review Resubmissions ........................................................... 5 2.5 Types of CDR Reviews Conducted for Submissions and Resubmissions ................... 5
3. PRE-SUBMISSION PROCEDURE ..................................................................................... 6
3.1 Pre-submission Meetings ............................................................................................ 6 3.1.1 Standard Pre-submission Meetings ................................................................ 6 3.1.2 Early Pre-submission Meetings ...................................................................... 6
3.2 CADTH Common Drug Review Advanced Notification Procedure ............................... 7 3.2.1 Voluntary Pipeline Notification of Pending CADTH Common Drug Review
Submissions ................................................................................................... 7 3.2.2 Mandatory Notification of Pending Submission or Resubmission ................... 7
4. APPLICATION AND SCREENING PROCESS ................................................................... 8
4.1 Filing an Application for a Submission or Resubmission .............................................. 8 4.2 Application Screening and Additional Copies ............................................................... 9
5. SUBMISSION REQUIREMENTS ...................................................................................... 10
5.1 Category 1 Requirements .......................................................................................... 10 5.1.1 Category 1 Requirements for a Standard CDR Review: New Drug,
Drug With a New Indication, or New Combination Product Submission ....... 11 5.1.2 Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission ........................................................................... 20
5.1.3 Category 1 Requirements for a Tailored CDR Review: Subsequent Entry Biologic Submission ........................................................................... 25
5.2 Category 2 Requirements .......................................................................................... 32 5.3 Additional Information ................................................................................................ 33
6. REQUESTING PRIORITY REVIEW STATUS ................................................................... 34
6.1 Priority Review Requests Based on Clinical Criteria .................................................. 34 6.2 Priority Review Requests Based on Economic Criteria .............................................. 34
Submission Guidelines for the CADTH Common Drug Review August 2014 iii

CADTH Common Drug Review
7. RESUBMISSION REQUIREMENTS ................................................................................. 35
7.1 Category 1 Requirements .......................................................................................... 37 7.2 Category 2 Requirements .......................................................................................... 43 7.3 Additional Information ................................................................................................ 44
8. RESUBMISSION BASED ON A REDUCED PRICE DURING EMBARGO PERIOD ........ 45
8.1 Eligibility .................................................................................................................... 45 8.2 Requirements ............................................................................................................ 45
9. GUIDELINES FOR THE TYPE OF ECONOMIC ANALYSIS TO BE SUBMITTED ........... 46
9.1 The Drug is the First Available for Treatment of the Disorder or Disease, or the First to be Listed by Drug Plans ....................................................................... 47 9.1.1 Suggested Content for Submission .............................................................. 48
9.2 The Drug Is Not the First Available Treatment for the Disorder or Disease ............... 49 9.2.1 Head-to-Head Trials Have Been Conducted Versus Other
Available Drugs ............................................................................................ 50 9.2.2 No Head-to-head Trial(s) Have Been Conducted Versus
Another Available Drug ................................................................................ 50 9.2.3 Suggested Content for Submission .............................................................. 51
9.3 Suggested Format of Cost Information ...................................................................... 51 APPENDIX 1: DRUG PLANS PARTICIPATING IN THE CADTH COMMON
DRUG REVIEW .............................................................................................. 55 APPENDIX 2: KEY DEFINITIONS ......................................................................................... 58 APPENDIX 3: CADTH CONTACT AND CADTH COMMON DRUG REVIEW
APPLICATION FILING INFORMATION ......................................................... 64 APPENDIX 4: CADTH COMMON DRUG REVIEW CONFIDENTIALITY GUIDELINES ........ 65 APPENDIX 5: EXAMPLE OF CONSORT DIAGRAM ............................................................ 69 APPENDIX 6: PREPARING LISTS OF REFERENCES ......................................................... 70 APPENDIX 7: LIST OF TEMPLATES .................................................................................... 71 APPENDIX 8: CHECKLISTS FOR PREPARING CADTH COMMON DRUG REVIEW
APPLICATIONS ............................................................................................. 72 APPENDIX 9: ELECTRONIC FILE STRUCTURE AND NAMING FORMAT ......................... 94
Submission Guidelines for the CADTH Common Drug Review August 2014 iv

CADTH Common Drug Review
Tables Table 1: Summary of CADTH Common Drug Review Submission Types .................................. 2 Table 2: Types of CDR Reviews Conducted for Submissions and Resubmissions .................... 6 Table 3: Additional Copies of CDR Category 1 Requirements .................................................... 9 Table 4: Submission Requirement Categories ..........................................................................10 Table 5: Category 1 Requirements for a Standard CDR Review: New Drug,
Drug With a New Indication, or New Combination Product Submission .....................11 Table 6: Common Technical Document Module Sectionsa Required .........................................15 Table 7: CADTH Common Drug Review Pharmacoeconomic ...................................................18 Table 8: Category 1 Requirements for a Tailored CDR Review: New Combination Product
(Funded Components or CADTH-Designated Tailored CDR Review) .........................20 Table 9: Category 1 Requirements for a Tailored CDR Review: SEB Submission ....................25 Table 10: Common Technical Document Sectionsa Required for SEB Submissions .................29 Table 11: Summary of New Information Required for Resubmissions .......................................36 Table 12: Resubmission Requirement Categories ....................................................................37 Table 13: CADTH Common Drug Review Pharmacoeconomic .................................................41 Table 14: Guidelinesa for the Economic Analyses of Drugs .......................................................48 Table 15: Guidelines for Economic Analyses for Drugs That .....................................................49 Table 16: Price Comparison Table ............................................................................................52 Table 17: Direct Health Care Costs ...........................................................................................53 Table 18: Non–Health Care Resources and Costs ....................................................................54 Table 19: Contact Information and Requirements for CDR-Participating Drug Plans .................55 Table 20: How and Where to Direct CDR-Related Inquiries or CDR Applications .....................64 Table 21: Delivery Times ..........................................................................................................64 Table 22: Category 1 Requirements for a Standard CDR Review .............................................73 Table 23: Category 1 Requirements for a Standard CDR Review Filed on a
Post-NOC Basis ........................................................................................................76 Table 24: Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Pre-NOC Basis......................................................................79
Table 25: Category 1 Requirements for a Tailored CDR Review: New Combination Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Post-NOC Basis ....................................................................81
Table 26: Category 1 Requirements for a Tailored CDR Review: ..............................................83 Table 27: Category 1 Requirements for a Tailored CDR Review: ..............................................86 Table 28: Category 1 Requirements for a Resubmission ..........................................................88 Table 29: Priority Review Applications ......................................................................................90 Table 30: Category 2 Requirements for all Submission Types ..................................................92 Table 31: Category 2 Requirements for Resubmissionsa ..........................................................93 Figures Figure 1: Summary of the Guidelines for the Type of Economic Analysis to Submit ..................47 Figure 2: Example of CONSORT Flowchart ..............................................................................69
Submission Guidelines for the CADTH Common Drug Review August 2014 v

CADTH Common Drug Review
ABBREVIATIONS BIA budget impact analysis
BSEAR Biologics Safety and Efficacy Assessment Report
CDEC Canadian Drug Expert Committee
CDR CADTH Common Drug Review
CEDAC Canadian Expert Drug Advisory Committee
CPID Certified Product Information Document
F/P/T
ITC
federal, provincial, territorial
indirect treatment comparison
LYG life-year gained
NOC Notice of Compliance
NOC/c Notice of Compliance with conditions
pCODR pan-Canadian Oncology Drug Review
PSEA Pharmaceutical Safety and Efficacy Assessment
QALY quality-adjusted life-years
RCT randomized controlled trial
SEB subsequent entry biologic
Submission Guidelines for the CADTH Common Drug Review August 2014 vi

CADTH Common Drug Review
1. INTRODUCTION 1.1 About This Document The objective of this document is to provide guidance to manufacturers in the preparation of applications for the review of submissions and resubmissions for drugs through the CADTH Common Drug Review (CDR) process. The Submission Guidelines for the CADTH Common Drug Review is a companion document to the Procedure for the CADTH Common Drug Review, a document that describes CDR procedures to be followed by all participants involved in the CDR process. The Submission Guidelines for the CADTH Common Drug Review and the Procedure for the CADTH Common Drug Review must be read in conjunction with one another, as well as any issues of the CDR Update subsequent to CDR Update – Issue 108. Revisions from all applicable CDR Update issues 86 to 108 are included in this version of the Submission Guidelines for the CADTH Common Drug Review. All references to number of days in this document are in business days unless otherwise specified. Please refer to the CADTH website “Contact Us” section for a current listing of the CADTH holiday schedule and business hours. Key terms in this document are defined in Appendix 2. All section numbers are in reference to this guidance document, unless otherwise specified. Various document and letter templates are hyperlinked throughout this document, and are also available on the CADTH website. These templates are to be used by applicants, as directed in the requirement descriptions, when preparing a CDR submission or resubmission. A list of these templates is provided in Appendix 7. 1.2 Changes to These Guidelines From time to time, CADTH may amend the Submission Guidelines for the CADTH Common Drug Review and all matters related to CDR. The drug plans are consulted as required. Amendments to, and clarifications of, the Submission Guidelines for the CADTH Common Drug Review and all related documents may be effected by means of directives (called CDR Updates) issued by CADTH on an “as-needed” basis, between revisions of these documents. Generally, changes that are corrections or clarifications become effective immediately.
Submission Guidelines for the CADTH Common Drug Review August 2014 1

CADTH Common Drug Review
2. CADTH COMMON DRUG REVIEW – ELIGIBLE APPLICATIONS 2.1 CADTH Common Drug Review Submissions This section provides guidance regarding eligibility for the majority of CDR submissions. However, there may be situations where CADTH may consult with the drug plans to confirm CDR eligibility of a drug and make a decision on a case-by-case basis, if necessary. A manufacturer or the drug plans may file a submission for a new drug, a drug with a new indication, a new combination product, a new combination product (funded components), or a subsequent entry biologic (SEB) that: • has received a Notice of Compliance (NOC) or a Notice of Compliance with conditions
(NOC/c) for the indication(s) to be reviewed; or • has a pending NOC or NOC/c for the indication(s) to be reviewed. Table 1 provides an overview of each CDR-eligible submission type.
Table 1: Summary of CADTH Common Drug Review Submission Types Submission Type
Description
New drug
• A new active substance that has not been previously marketed in Canada. • See the full description (section 2.1.1) regarding new salts and line extensions.
Drug with a new indication
• A drug previously reviewed by CDR that has received an NOC or NOC/c for a new indication; or
• A drug marketed before the establishment of CDR that has received an NOC or NOC/c for a new indication.
New combination product
• Two or more drugs that have not been previously marketed in Canada in that combination. It may consist of: two or more new drugs two or more previously marketed drugs a combination of new drug(s) and previously marketed drug(s).
• See the full description (section 2.1.3) regarding a new combination product (funded components).
Subsequent entry biologic
• Biologic drug demonstrating a high degree of similarity to an already authorized biologic drug (i.e., a reference product).
CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions. 2.1.1 New Drug A new drug is a new active substance that has not been previously marketed in Canada, regardless of when the NOC or NOC/c was issued. A new drug submission includes a new salt of a marketed product, but does not include the following variations of existing products being funded by drug plans (line extensions) containing the same active substance(s): • New dosage form with the same route of administration (e.g., if a drug in tablet form
becomes available in capsule form, a submission for the capsule is not required). Note: New parenteral products or formulations (e.g., intravenous, intramuscular, subcutaneous dosage forms) are not considered line extensions of one another, for
Submission Guidelines for the CADTH Common Drug Review August 2014 2

CADTH Common Drug Review
purposes of CDR, as they have different routes of administration and, as a result, there may be potential differences in pharmacokinetics and pharmacodynamics as well as differences in cost. Manufacturers are asked to contact CADTH at [email protected] for guidance on whether a submission is required for parenteral line extensions.
• New strength of the same dosage form (e.g., if a 200 mg tablet becomes available in addition to an already-marketed 100 mg tablet, a submission for the 200 mg tablet is not required).
All submissions for new drugs undergo a standard review by CDR (hereafter referred to as a “standard CDR review”), following the submission requirements outlined in section 5.1.1. 2.1.2 Drug with a New Indication A drug with a new indication is either: • a drug previously reviewed by CDR that has received an NOC or NOC/c for a new indication; or • a drug marketed before the establishment of CDR (in September 2003) that has received an
NOC or NOC/c for a new indication. All submissions for drugs with a new indication undergo a standard CDR review for which the submission requirements are outlined in section 5.1.1. 2.1.3 New Combination Product A new combination product consists of two or more drugs that have not been previously marketed in Canada in that combination. One or more of the components may be a non-prescription drug, but at least one component must be a prescription drug. New combination products may consist of: • two or more new drugs • two or more previously marketed drugs • a combination of new drug(s) and previously marketed drug(s). A new combination product (funded components) is a new combination product containing two or more drugs that are already funded by the drug plans. All submissions for new combination products (funded components) will undergo a tailored review by CDR (hereafter referred to as a “tailored CDR review”). All submissions for other new combination products will typically undergo a standard CDR review; however, a decision to conduct a tailored CDR review may be made by CADTH on a case-by-case basis. Manufacturers planning to file a submission for any new combination product are required to complete and submit the following template to CADTH ([email protected]) before filing the submission: • New Combination Product Considerations Form CADTH will review the provided information and, with input from the drug plans, determine if the new combination product should undergo a tailored or standard CDR review. 2.1.4 Subsequent Entry Biologic An SEB is a biologic drug (i.e., a drug derived from living sources versus a chemically synthesized drug), demonstrating a high degree of similarity to an already authorized biologic drug (i.e., a “reference product” that has been authorized in Canada, or in some circumstances can be an authorized non-Canadian biologic from a jurisdiction that has an established
Submission Guidelines for the CADTH Common Drug Review August 2014 3

CADTH Common Drug Review
relationship with Health Canada). Similarity between an SEB and the reference product is established in accordance with Health Canada’s Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs), for the authorized indications. All submissions for SEBs undergo a tailored CDR review and have specific submission requirements as outlined in section 5.1.3. 2.2 Notice of Compliance Status at the Time of Filing the Submission A CDR submission can be filed on either a pre-NOC or a post-NOC basis.1 2.2.1 Submissions Filed Pre-NOC or NOC/c When Health Canada is highly likely to issue an NOC or NOC/c for the indications to be reviewed by CDR within 90 calendar days, a submission may be filed on a pre-NOC basis for a new drug, drug with a new indication, new combination product, new combination product (funded components), or an SEB. This type of submission is accepted with the agreement that some submission requirements (e.g., product monograph) may not be finalized at the time of filing; however, they are to be provided as soon as finalized because the embargoed Canadian Drug Expert Committee (CDEC) recommendation will not be released until all required information is received. Although Health Canada cannot provide assurance that an NOC or NOC/c will be issued on a particular date or at all, manufacturers may consider filing a submission with CDR up to 90 calendar days in advance of the anticipated NOC or NOC/c if no significant issues have been raised by Health Canada to date during the review process. Please also refer to the Procedure for the CADTH Common Drug Review for information regarding finalized information during the review of a submission filed on a pre-NOC basis. 2.2.2 Submissions Filed Post-NOC or NOC/c A submission may be filed on a post-NOC or NOC/c basis after the drug has been granted an NOC or NOC/c by Health Canada for the indication(s) to be reviewed by CDR. 2.3 Drugs Not Eligible for Review Under the CADTH Common Drug Review Process
Applications for oncology drugs used for the active treatment of cancer should be filed through the pan-Canadian Oncology Drug Review (pCODR) process. Submissions should be made directly to drug plans for the following items until further notice: • Line extensions of marketed products, including new dosage forms with the same route of
administration and new strengths of the same dosage form. For other line extensions (including new parenteral products or formulations that are not administered through the same route of administration), contact CADTH for direction.
• Generic products.
1 Pre-NOC also includes pre-NOC/c and post-NOC includes post NOC/c submissions.
Submission Guidelines for the CADTH Common Drug Review August 2014 4

CADTH Common Drug Review
Whenever there is doubt as to whether a drug submission should be made to CDR, manufacturers are invited to contact CADTH by email at [email protected] for direction. CADTH may consult with the participating drug plans in those cases where drugs do not clearly fall into a category described above. 2.4 CADTH Common Drug Review Resubmissions A resubmission from a manufacturer or the drug plans may be filed for a new drug, drug with a new indication, new combination product, new combination product (funded components), or an SEB that has previously been reviewed through the CDR process and for which a CDEC Final Recommendation has been issued by CADTH. To be eligible for a resubmission, the applicant must submit new information that was not previously provided in the initial submission or previous resubmission(s). The new information must consist of one or both of the following: • new clinical information in support of improved efficacy or safety • new cost information that significantly affects the cost-effectiveness of the drug. If the new information is in support of improved efficacy, it must be from a randomized controlled trial. If the new information is in support of improved safety, case-control or cohort studies will be accepted if randomized controlled trials are unavailable. Manufacturers or drug plans are not limited in the number of resubmissions that they may file; however, resubmissions must meet the requirement of new information to be eligible for the CDR process. 2.5 Types of CDR Reviews Conducted for Submissions and Resubmissions The review team of CDR (hereafter referred to as “CDR review team”) conducts either a standard CDR review or a tailored CDR review, depending on the type of submission or resubmission filed by a manufacturer. • A standard CDR review consists of the CDR review team conducting a systematic review of
clinical evidence provided by the manufacturer along with studies identified through its independent, systematic literature search, and an appraisal of the manufacturer-provided pharmacoeconomic evaluation.
• A tailored CDR review consists of the CDR review team conducting an appraisal of the clinical evidence and pharmacoeconomic evaluation filed by the manufacturer using a CADTH-provided review template that is specific to the type of drug product to be reviewed.
Table 2 summarizes the type of CDR review conducted for the different submission and resubmission categories.
Submission Guidelines for the CADTH Common Drug Review August 2014 5

CADTH Common Drug Review
Table 2: Types of CDR Reviews Conducted for Submissions and Resubmissions
Type of CDR Review
Type of Submission or Resubmission
Standard CDR Review
• New drug submission • Drug with a new indication submission • New combination product submission • Resubmission based on new clinical information with or without new cost
information Tailored CDR Review
• New combination product (funded components or CADTH-designated tailored CDR review) submission
• Subsequent entry biologic submission • Resubmission based only on new cost information
CDR = CADTH Common Drug Review.
3. PRE-SUBMISSION PROCEDURE 3.1 Pre-submission Meetings 3.1.1 Standard Pre-submission Meetings To facilitate the efficient preparation and filing of submissions for review under the CDR process, manufacturers may request pre-submission meetings with CADTH for pending submissions to be filed within six months. These meetings provide an opportunity for the manufacturer to introduce a drug to CADTH and discuss submission requirements. Pre-submission meetings are intended to offer the opportunity for dialogue between CADTH staff and manufacturers and are not meant to be consultative in nature, outside of clarifying submission requirements. Manufacturers are also invited to provide information on drugs in their pipeline so that CADTH can plan for future submissions. Pre-submission meetings are scheduled for a maximum of one hour and applicants are limited to one meeting per pending drug submission. To request a pre-submission meeting, manufacturers are required to complete the following form and submit it to CADTH ([email protected]): Pre-submission Meeting Request Form. 3.1.2 Early Pre-submission Meetings CADTH offers opportunities for dialogue between CDR staff and manufacturers earlier in the pre-submission phase, 6 to 12 months in advance of filing, for drug submissions with all of the following characteristics: • the drug is indicated for a relatively small patient population • clinical data are limited to surrogate end points • natural history of the disease is poorly characterized • there is a limited number of clinical trials and they have small sample sizes • treatment has a high cost relative to appropriate comparators • the manufacturer has questions regarding the appropriate type of economic analysis to
submit.
Submission Guidelines for the CADTH Common Drug Review August 2014 6

CADTH Common Drug Review
Manufacturers are advised to send CADTH supporting information for the points listed above and an overall rationale for requesting an early pre-submission meeting, as soon as possible after the drug submission has been accepted by Health Canada for review. A decision to accept a manufacturer’s request for an early pre-submission meeting will be made by CADTH on a case-by-case basis. Pre-submission meetings are intended to offer the opportunity for dialogue between CADTH staff and manufacturers and are not meant to be consultative in nature. To request an early pre-submission meeting, manufacturers are required to complete the following form and submit to CADTH ([email protected]): Pre-submission Meeting Request Form.
3.2 CADTH Common Drug Review Advanced Notification Procedure CADTH uses the following two-step process for obtaining information regarding pending CDR submissions and resubmissions, as applicable: 3.2.1 Voluntary Pipeline Notification of Pending CADTH Common Drug Review Submissions Manufacturers are encouraged to voluntarily provide advanced notification of a pending CDR submission at the time of regulatory filing (i.e., providing advanced notification of approximately 12 months). Manufacturers willing to participate in this voluntary process are asked to complete and submit the advanced notification template to [email protected]: • CADTH Common Drug Review Voluntary Pipeline Notification Template • CADTH Common Drug Review Advanced Notification Instructions.
3.2.2 Mandatory Notification of Pending Submission or Resubmission Manufacturers are required to provide CADTH with advanced notification of a pending submission or resubmission at least 20 business days before filing with CDR. All manufacturers must complete and submit the appropriate advanced notification template for a submission or resubmission by email to [email protected]. Failure to provide notification at least 20 business days in advance of filing may result in a delay in the processing and review of the submission or resubmission by CADTH. The date that the advanced notification template is received by CADTH is considered day zero (for purposes of counting back 20 business days in advance of the date on which CADTH will receive the submission). • CADTH Common Drug Review Mandatory Notification Submission Template • CADTH Common Drug Review Mandatory Notification Resubmission Template • CADTH Common Drug Review Advanced Notification Instructions.
Submission Guidelines for the CADTH Common Drug Review August 2014 7

CADTH Common Drug Review
4. APPLICATION AND SCREENING PROCESS An application filed with CADTH for the review of a drug submission or resubmission through the CDR process represents a submission or resubmission to all of the drug plans. The Submission Guidelines for the CADTH Common Drug Review consolidate the submission requirements of the drug plans and the requirements for the CDR process. All submission and resubmission requirements must be provided in English. 4.1 Filing an Application for a Submission or Resubmission a) The appropriate submission or resubmission requirements filed must adhere to the content,
format, and organization stipulated in the current version of the Submission Guidelines for the CADTH Common Drug Review.
b) Submissions and resubmissions must be delivered to CADTH by courier, registered mail, or in person (see Appendix 3 for further information about how and where to file applications). The only exceptions are resubmissions based on a reduced price during the embargo period, which may be submitted by email.
c) When filing a submission or resubmission, the manufacturer should initially deliver only one copy of all category 1 requirements2 to CADTH in electronic format on CDs, DVDs, or USB flash drives following the electronic file folder and file format specified in Appendix 9: Electronic File Structure and Naming Format.
As part of the category 1 requirements, three additional CDs, DVDs, or USB flash drives containing copies of the economic model in unlocked and fully executable format are also required as part of the initial information filed for all drugs undergoing a standard CDR review, drugs undergoing a tailored CDR review for a new combination product (funded components), or a resubmission based on new cost information.
d) Category 2 requirements may be filed at the same time as category 1 requirements, if available.
e) When not provided at the same time as category 1 requirements, category 2 requirements should be submitted at least 20 business days before the targeted Canadian Drug Expert Committee meeting at which the submission or resubmission will be considered. Incomplete category 2 requirements will not preclude CADTH CDR reviews from being placed on the agenda for the targeted Canadian Drug Expert Committee meeting; however, the CDEC Final Recommendation will not be issued until all category 2 requirements are complete.
f) When filing category 2 requirements for a submission or resubmission, the manufacturer should deliver one copy of all requirements as a single package to CADTH in electronic format on a CD, DVD, or USB flash drive, using the electronic file folder and file format specified in Appendix 9: Electronic File Structure and Naming Format. No further copies of category 2 requirements are required by CADTH.
2 Category 2 requirements may be filed at the same time, if they are available.
Submission Guidelines for the CADTH Common Drug Review August 2014 8

CADTH Common Drug Review
4.2 Application Screening and Additional Copies a) CADTH screens applications for submissions and resubmissions in accordance with
the Procedure for the CADTH Common Drug Review sections 4.3 and 4.4.
b) When category 1 requirements for a submission or resubmission have been accepted for review, CADTH sends an acknowledgement to the manufacturer and requests that the manufacturer provide additional copies of the category 1 requirements on separate CDs, DVDs, or USB flash drives, in accordance with Table 3.
Table 3: Additional Copies of CDR Category 1 Requirements
Application Type Number of Additional Copiesa
Submission 5 Resubmission based on new clinical information 5 Resubmission based on new cost information 3 Resubmission based on new clinical and new cost information 5
a CADTH may request additional copies if required.
Submission Guidelines for the CADTH Common Drug Review August 2014 9

CADTH Common Drug Review
5. SUBMISSION REQUIREMENTS The CDR submission requirements for all eligible submission types are grouped into the following categories: category 1 requirements, category 2 requirements, priority review request requirements, and additional information. A brief description of these requirements is provided in Table 4 and detailed descriptions are provided in subsequent sections. For all submission types, the clinical and pharmacoeconomic information provided in the category 1 and category 2 requirements should focus on the indication(s) to be reviewed under the CDR process, unless otherwise specified.3
Table 4: Submission Requirement Categories Requirement Category
Function in the CDR Process Due
Category 1 Used by the CDR review team and CDEC for the review and recommendation process.
At the time of filing the application.
Category 2 Used by the drug plans and are not considered as part of the CDR review or recommendation process. CADTH provides secretariat support to the drug plans by ensuring that category 2 requirements have been filed in accordance with the Submission Guidelines for the CADTH Common Drug Review.
≥ 20 business days before the targeted CDEC meeting.
Priority review request Used by CADTH, CDEC and the drug plans for determining whether a submission should be granted priority review status.
At the time of filing the submission.
Additional information Additional information that CADTH may require for completion of the review (e.g., Clinical Study Reports).
As soon as possible following a request by CADTH, to avoid delays in the review process.
CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review. To expedite the screening of submissions for completeness and to help with the efficient use of documents, manufacturers must organize all submission information in the order prescribed in the category 1 and category 2 requirements below and follow the electronic file folder format in Appendix 9: Electronic File Structure and Naming Format. The submission checklists used by CADTH for screening category 1 and category 2 requirements can be found in Appendix 8: Checklists for Preparing CADTH COMMON DRUG REVIEW Applications. These checklists may assist manufacturers in ensuring that all requirements have been included in the submission. 5.1 Category 1 Requirements Category 1 requirements are used by the CDR review team and the Canadian Drug Expert Committee for the review and recommendation process. One copy of all category 1 requirements4 must be filed with CADTH as a single submission package in electronic format on CDs, DVDs, or USB flash drives and accepted by CADTH before the review can proceed. When
3 An exception to this is the “table of studies” requirement. 4 Category 2 requirements may be filed at the same time, if they are available.
Submission Guidelines for the CADTH Common Drug Review August 2014 10

CADTH Common Drug Review
the category 1 requirements have been screened and accepted for review by CADTH, the manufacturer and the drug plans are informed and steps commence to determine the order and timing for initiating the review. The manufacturer is responsible for ensuring that appropriate copyright permissions have been obtained for electronic copies of articles included in category 1 requirements of a submission, to be shared among the CDR review team, the Canadian Drug Expert Committee, and the drug plans for the review of the submission. 5.1.1 Category 1 Requirements for a Standard CDR Review: New Drug, Drug With a New Indication, or New Combination Product Submission Submissions for new drugs, drugs with a new indication, and new combination products that CADTH has not designated for a tailored CDR review undergo a standard CDR review. The category 1 requirements are summarized in Table 5. Detailed descriptions of the information that comprise each of the category 1 requirements for a standard CDR review submission are described in this section. Where there are specific requirements for a submission filed on a pre-NOC versus post-NOC basis, they are delineated in the descriptions that follow the table.
Table 5: Category 1 Requirements for a Standard CDR Review: New Drug, Drug With a New Indication, or New Combination Product Submission
Section Specific Items and Criteria General Information
• Completed application overview template • Signed cover letter • Completed executive summary template for a submission • Product monograph
Health Canada Documentation
• NOC or NOC/c • Health Canada clinical reviewers’ report • Table of Clarifaxes
Efficacy, Effectiveness, and Safety Information
• Common Technical Document sections 2.5, 2.7.1, 2.7.3, 2.7.4, 5.2, or statement indicating any section(s) not required for the Health Canada submission
• Reference list and copies of key clinical studies and errata • Table of studies • Reference list and copies of editorial articles (or statement that no editorials) • Literature search strategies • Signed declaration that all known unpublished studies have been disclosed • CONSORT diagrams • Reference list and copies of new data • Reference list and copies of articles for validity of outcome measure
Economic and Epidemiologic Information
• Pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR
• Three separate CDs, DVDs, or USB flash drives, each with a copy of the unlocked and fully executable economic model
• Number of patients accessing a new drug to within 20 business days of filing the submission
• Disease prevalence and incidence data, with specified breakdown if available
Submission Guidelines for the CADTH Common Drug Review August 2014 11

CADTH Common Drug Review
Table 5: Category 1 Requirements for a Standard CDR Review: New Drug, Drug With
a New Indication, or New Combination Product Submission Section Specific Items and Criteria Pricing and Distribution Information
• Submitted unit pricing to four decimal places • Method of distribution • Signed commitment to honour the submitted price
Sharing of information
• Signed letter authorizing unrestricted sharing of information
Pre-NOC Letters
• Letter for sending NOC or NOC/c to CADTH a • Letter for finalized category 1 requirements a
CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions. a This is only a requirement for a submission filed on a pre-NOC basis.
a) General Information i. Application Overview Template
• A completed Application Overview Template. ii. Signed Cover Letter
• A signed cover letter (an electronic signature is acceptable) from the applicant, providing the following information: A clear description of the submission being filed (e.g., category 1 requirements for a
new drug submission filed on a pre-NOC basis). If requesting priority review, notification that a priority review is being requested and
the justification for the priority review request (i.e., based on clinical and/or economic criteria) and confirmation that a completed priority review request template has been included.
Confirmation that all of the requirements have been provided in the submission. The indication(s) to be reviewed under the CDR process. The date the NOC or NOC/c was issued for the indication(s) to be reviewed or, in the
case of a submission filed on a pre-NOC basis, the anticipated date the NOC will be issued.
The requested listing criteria, if applicable. Intention to provide category 2 requirements at least 20 business days before the
targeted Canadian Drug Expert Committee meeting (if not being provided with category 1 requirements).
A statement confirming whether the submitted price is the anticipated or current marketed price, or the confidential price per unit that is submitted to CDR, and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation, irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer.
The names and contact information (email and phone number) for the primary and backup contact(s) that CADTH can contact regarding the submission. The manufacturer may designate the consultant(s) preparing the submission as primary and/or backup contact(s). Any changes in contacts should be communicated to CADTH as soon as possible, by emailing [email protected].
Submission Guidelines for the CADTH Common Drug Review August 2014 12

CADTH Common Drug Review
iii. Executive Summary
• A high-level summary of the submission (five pages maximum excluding reference list), following the Executive Summary Template for a Submission.
iv. Product Monograph
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a copy of the most recent draft product monograph
showing the company, drug brand, and non-proprietary names that correspond to the anticipated NOC.
• As soon as available, sent by email to [email protected]: a copy of the draft product monograph initially filed showing, in tracked changes, all
of the clinical and label review changes made up to the time of the product monograph being approved by Health Canada
a copy of the clean and dated product monograph approved by Health Canada. For drugs reviewed by Health Canada’s Therapeutics Products Directorate, the approved product monograph refers to the pristine product monograph to which the Letter of Certification applies.
All submissions filed on a post-NOC basis: • A copy of the most current version of the Health Canada-approved product monograph.
b) Health Canada Documentation i. Health Canada NOC or NOC/c
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating the anticipated
target date for receipt of an NOC or NOC/c for the indication(s) to be reviewed. • A copy of the granted NOC or NOC/c for the indication(s) under review by CDR, dated
and signed by Health Canada, must be sent by email to [email protected] as soon as it is available (i.e., on the day of, or next business day after, receipt from Health Canada).
• If the drug is granted an NOC/c for the indication(s) being reviewed by CDR: a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the clinical benefit, including an indication of time frames, must also be provided by email to [email protected] as soon as it is available.
All submissions filed on a post-NOC basis: • A copy of the NOC or NOC/c granted, dated, and signed by Health Canada. The NOC or
NOC/c must be for the indication(s) for which the drug is to be reviewed under the CDR process.
• If the drug in the submission has received an NOC/c for the indication(s) to be reviewed, the manufacturer must provide a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the drug’s clinical benefit, including an indication of time frames.
ii. Health Canada Clinical Reviewers’ Report
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating that a copy of
Health Canada’s Pharmaceutical Safety and Efficacy Assessment (PSEA) or Biologics
Submission Guidelines for the CADTH Common Drug Review August 2014 13

CADTH Common Drug Review
Safety and Efficacy Assessment Report (BSEAR), as applicable to the submission filed, will be provided as soon as it is available.
• After the NOC or NOC/c is issued: a copy of Health Canada’s PSEA or BSEAR, as applicable to the submission filed, must be provided by email to [email protected] as soon as available (i.e., on the day of, or next business day after, receipt from Health Canada). If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
• To avoid delays in providing the report to CADTH, manufacturers are encouraged to request it from Health Canada as soon as they are assured that an NOC or NOC/c will be issued, and forward it immediately upon receipt to CADTH.
All submissions filed on a post-NOC basis: • A copy of Health Canada’s PSEA or BSEAR, as applicable to the submission filed. • If the PSEA or BSEAR is unavailable from Health Canada at the time of filing the
submission, a placeholder document must be included, indicating that a copy of the report will be provided as soon as it is available.
• As soon as available (i.e., on the day of, or next business day after, receipt from Health Canada), a copy of Health Canada’s PSEA or BSEAR, as applicable to the submission filed, must be provided by email to [email protected]. If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
iii. Clarifaxes
All submissions filed on a pre-NOC basis: • At time of filing the submission: a summary table of Clarifaxes relating to any clinical
aspects of the Health Canada review of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the time of filing with CADTH. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
• On an ongoing basis up to the point of the NOC or NOC/c being issued, the manufacturer must provide CADTH with revised Clarifax summary tables to reflect any additional Clarifaxes as delineated above. The revised table(s) should be sent to CADTH by email ([email protected]).
All submissions filed on a post-NOC basis: • A summary table of Clarifaxes relating to any clinical aspects of the Health Canada
review of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the point of the NOC or NOC/c being issued. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
c) Efficacy, Effectiveness, and Safety Evidence i. Common Technical Document
• As shown in Table 6, a copy of selected sections from the Common Technical Document modules. If any of these sections of the Common Technical Document were not a requirement for filing the regulatory submission with Health Canada, a placeholder document with a statement confirming this is required.
Submission Guidelines for the CADTH Common Drug Review August 2014 14

CADTH Common Drug Review
Table 6: Common Technical Document Module Sectionsa Required
for Standard CDR Review Submissions Section Title 2.5 Clinical Overview 2.7.1 Summary of Biopharmaceutical Studies and Associated Analytical Methods 2.7.3 Summary of Clinical Efficacy 2.7.4 Summary of Clinical Safety 5.2 Tabular Listing of All Clinical Studies
CDR = CADTH Common Drug Review. a If any of these sections were not a requirement for filing the regulatory submission with Health Canada for the drug to be reviewed through the CADTH Common Drug Review, a placeholder document with a statement confirming this is required.
ii. Clinical Studies
• Copies of published and unpublished studies that address key clinical issues. It is preferred that unpublished data are submitted in manuscript format; however, if
unavailable in manuscript format, the information should be provided in accordance with the CONSORT 2010 Statement Checklist, using clearly labelled sections as outlined (i.e., title, abstract, introduction, methods, results, discussion, other information).
Include copies of any errata related to any published studies provided. If there are no errata, a placeholder document with a statement confirming this must be provided.
Should an unpublished study submitted as a category 1 requirement become published during the review process of CDR, manufacturers must email a copy of the published study to [email protected], indicating that it is the published version of a previously unpublished study included in the category 1 requirements initially submitted.
As specified in Appendix 9: Electronic File Structure and Naming Format, the first file in the folder must be a reference list of the articles and errata included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6.
iii. Table of Studies
• A tabulated list of all published and unpublished clinical studies using the Table of Studies Template.
iv. Editorials
• Copies of editorials relating to published clinical studies provided in the submission (i.e., studies listed in the first section of the “table of studies” requirement template). As specified in Appendix 9: Electronic File Structure and Naming Format, the first file
in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6.
If there are no editorial articles available, a placeholder document with a statement confirming this must be provided.
v. Search Strategies
• Search strategies used to locate published studies in medical literature databases. All search terms that were used (i.e., MeSH headings and keywords) and the names of databases (e.g., MEDLINE, Embase, Cochrane, etc.) that were searched are required. Search results are not required.
Submission Guidelines for the CADTH Common Drug Review August 2014 15

CADTH Common Drug Review
vi. Declaration for Disclosure of Known Unpublished Clinical Studies
• A signed declaration that all known unpublished clinical studies have been disclosed using the Letter Confirming Disclosure of all Known Unpublished Studies template. If CADTH discovers undisclosed unpublished trials through other sources, this may result in the submission being considered at a later Canadian Drug Expert Committee meeting to allow time for the retrieval and review of the trials.
vii. CONSORT Diagrams
• Diagrams following the CONSORT flow diagram reporting standards or similar diagrams that document the flow of patients through trials identified as pivotal trials in Health Canada documentation, as well as any other key trials included in the submission (i.e., studies listed in the first section of the “table of studies” requirement template).
• All information for the four stages of a trial (i.e., enrolment, intervention allocation, follow-up, and analysis) of the CONSORT flow diagram is required. Please consult Appendix 5 and/or the CONSORT web page (http://www.consort-statement.org/consort-statement/flow-diagram) for additional details regarding the structure and content of flow diagrams.
viii. New Data
• Copies of new data, generated since the last date that data were reported in the studies included in the Health Canada submission. Typically, the clinical studies submitted to CDR are the same as those submitted to Health Canada, and sometimes these studies are ongoing, with data collected after submission to Health Canada. The data that become available after the study has been submitted to Health Canada are required. These data will be accepted in a variety of formats, including late draft, Clinical Study Report, synopsis, abstract, or conference proceedings. As specified in Appendix 9: Electronic File Structure and Naming Format, the first
file in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6.
If no new data are available, a placeholder document with a statement confirming this must be provided.
ix. Validity of Outcome Measures
• Copies of references supporting the validity of primary outcome measures in clinical studies. As specified in Appendix 9: Electronic File Structure and Naming Format, the first file
in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6.
If no references are available, a placeholder document is required with a statement confirming that a search was undertaken but no references were located.
d) Economic and Epidemiologic Information i. Pharmacoeconomic Evaluation
• An appropriate pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR. If there are subgroups that may benefit from the drug or specific reimbursement criteria requested by the manufacturer, additional analyses should be provided. For example, if a manufacturer is requesting the review of one of several Health Canada–approved indications, the base-case analysis for the pharmacoeconomic evaluation must include the full population for
Submission Guidelines for the CADTH Common Drug Review August 2014 16

CADTH Common Drug Review
the indication to be reviewed. In addition, if the applicant decides to make a specific listing request for one or more subgroups of the patient population that may benefit from the drug, additional analyses for each subgroup must also be provided in the pharmacoeconomic submission.
• Only one type of pharmacoeconomic analysis is to be submitted, and the type of analysis must be in accordance with section 9 of this document as well as CADTH’s Guidelines for the Economic Evaluation of Health Technologies: Canada. Should the applicant decide to submit more than one pharmacoeconomic analysis, they must clearly indicate which of the analyses submitted should be reviewed. The submission will not be accepted by CADTH for review until the applicant confirms which submitted economic analysis is to be reviewed.
• Only the price submitted to CDR is to be used in all base-case pharmacoeconomic evaluations.
ii. The Economic Model • Three separate CDs, DVDs, or USB flash drives containing copies of the economic
model in unlocked and fully executable format provided only in the manufacturer’s initial submission sent for screening of acceptability. No additional copies are required by CADTH once the submission is accepted for review by CADTH.
• The submitted model must be used as the basis for the economic evaluation. • The preferred economic model software platforms are Excel, TreeAge, or Arena. Before using other specialized program software, manufacturers must contact
CADTH in advance to ensure that they meet CADTH’s requirements and to receive direction on how the model and software should be provided as part of the submission information. Manufacturers are expected to provide CADTH with the necessary requirements to run the model (i.e., licences, laptop with model, and software), which will be returned to the manufacturer at the end of the review process, at the manufacturer’s expense.
• The economic model must be provided in its entirety, meaning CADTH must have full access to the programming code and be able to fully execute the model based on modifications to parameters of interest. The CDR review team must be able to vary individual parameters, view the calculations, and run the model to generate results. The type of information that CDR requires for its examination of the model and the preferred format for receiving it are described in Table 7.
• Where the clinical inputs are based on an indirect treatment comparison (ITC), the full technical report of the ITC must be provided as part of the filed material.
• If statistical analyses of data sets are included in the model, the manufacturer must provide a description of the data sources and analyses conducted, and results from the analyses.
• Copies of any supporting materials that are used as part of the modelling exercise must be provided.
• Documentation detailing the methods used in the modelling exercise and basic user information must be included.
Submission Guidelines for the CADTH Common Drug Review August 2014 17

CADTH Common Drug Review
Table 7: CADTH Common Drug Review Pharmacoeconomic
Model Information Requirements Information Elements Format Basis for the pharmacoeconomic model
A modela that is unlocked and fully executable. The user should be able to specify inputs, view calculations, run analyses, and have full access to the programming code.
Media CD, DVD, USB flash drive, or laptop (see software requirements below regarding when laptop is required)
Software requirements
The submitted model must be in one of the following formats: Excel, TreeAge, or Arena. Where manufacturers are using specialized program software, they must contact CADTH in advance to ensure that the programs meet CADTH’s requirements and to receive direction on how the model and software should be provided in the submission. Where the model platform is agreed upon, the manufacturers must provide CADTH with the necessary requirements to run the model (i.e., licences, laptop with model, and software), which will be returned to the manufacturer at the end of the review process at the manufacturer’s expense.
Basic user guide to the model
Electronic format
Model documentation (manuscripts or a summary of the model report may be provided)
Electronic format
Description of the statistical analyses included in the model (e.g., data sources, methods, and results)
Electronic format
Copies of any supporting materials that are used as part of the modelling exercise
Electronic format
a The pharmacoeconomic model will be examined by internal and external CDR review team members. The pharmacoeconomic model will not be released by CADTH to any third parties.
iii. Number of Patients Accessing a New Drug The following information is required only for a new drug submission, or a new combination product submission if one of the components is a “new drug” (as defined in section 2.1.1): • For the indication(s) to be reviewed through CDR, the number of patients in Canada
currently accessing the new drug to within 20 business days of filing the submission. • This information must include the number of patients accessing the drug through each of
the different possible mechanisms, such as: compassionate use Health Canada’s Special Access Program participation in a clinical trial.
• Use the Number of Patients Accessing New Drug template for providing this information.
Submission Guidelines for the CADTH Common Drug Review August 2014 18

CADTH Common Drug Review
iv. Disease Prevalence and Incidence
• The prevalence and incidence of the disease(s) or condition(s) for the indication(s) to be reviewed provided for the Canadian population, with a breakdown by participating province, territory, and First Nations populations where available.
• References must be provided for this document in the following format: in-text citations numbered in their order of appearance a numbered reference list in the Citing Medicine format (Appendix 6).
e) Pricing and Distribution Information i. Submitted Price
• The submitted price for the drug, reported to four decimal places, as follows: price per smallest unit price per smallest dispensable unit for all dosage forms and strengths available in
Canada price for all packaging formats available in Canada.
• The submitted price is the price per unit that is submitted to CDR and that must not be
exceeded for any of the drug plans following release of a CDEC Final Recommendation,5 irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer. The submitted price can be an anticipated or current market price in Canada, or a confidential price.
• Only one price (anticipated or current market price, or confidential price) to four decimal places per dispensable unit is to be submitted per drug that is to be reviewed under the CDR process (i.e., only one price for all indications undergoing review by CDR concurrently).
• The submitted price must be used in the pharmacoeconomic evaluation included in the submission and in the budget impact analyses (BIAs).
ii. Method of Distribution
• Method of distribution to pharmacies (e.g., wholesale, direct, or other arrangements).
iii. Commitment to Honour Submitted Price • A signed commitment to honour the submitted price for all drug plans using
the Commitment to Honour Submitted Price Letter template.
f) Letter Authorizing Unrestricted Sharing of Information • A letter from the holder of the NOC or NOC/c (or from the manufacturer applying for an
NOC, in the case of a submission filed on a pre-NOC basis), using the Unrestricted Sharing of Information Letter template, printed on company letterhead and signed by an appropriate senior official, permitting unrestricted sharing of information regarding the drug product under review through the CDR process, between CADTH and: Federal, provincial, territorial governments, including their agencies and departments Patented Medicine Prices Review Board.
5 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 19

CADTH Common Drug Review
g) Additional Letters for Submissions Filed on Pre-NOC Basis i. Letter for Sending NOC or NOC/c to CADTH
• After the NOC or NOC/c has been issued, the manufacturer must provide a signed letter, using the Letter for Sending NOC or NOC/c to CADTH template, indicating whether or not any wording changes to the Health Canada–approved final product monograph, as compared with the draft product monograph filed in the initial package of category 1 requirements, result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis. The letter should be sent to CADTH by email ([email protected]).
ii. Letter for Finalized Category 1 Requirements
• A signed letter, using the Letter for Finalized Category 1 Requirements template, confirming that all finalized versions of category 1 requirements for the submission filed on a pre-NOC basis have been provided to CADTH. The letter should be sent to CADTH by email ([email protected]).
5.1.2 Category 1 Requirements for a Tailored CDR Review: New Combination Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission All submissions for new combination products (funded components) undergo a tailored CDR review. In addition, as outlined in section 2.1.3, CADTH may, on a case-by-case basis, designate new combination products without funded components to undergo a tailored CDR review. The category 1 requirements are summarized in Table 8. Detailed descriptions of the information that comprise each of the category 1 requirements for a new combination product (funded components or CADTH-designated tailored CDR review) submission are provided in this section. Where there are specific requirements for a submission filed on a pre-NOC versus post-NOC basis, they are delineated in the descriptions that follow the table.
Table 8: Category 1 Requirements for a Tailored CDR Review: New Combination Product (Funded Components or CADTH-Designated Tailored CDR Review)
Section Specific Items and Criteria General Information • Completed application overview template
• Signed cover letter • Completed executive summary template for a submission • Completed new combination product submission template • Product monograph
Health Canada Documentation
• NOC or NOC/c • Health Canada clinical reviewers’ report • Table of Clarifaxes
Clinical Study • One clinical study using the new combination product, not the individual components; this can be a pharmacokinetic study
Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
Pricing and Distribution Information
• Submitted unit pricing to four decimal places • Method of distribution • Signed commitment to honour the submitted price
Sharing of Information
• Signed letter authorizing unrestricted sharing of information
Submission Guidelines for the CADTH Common Drug Review August 2014 20

CADTH Common Drug Review
Table 8: Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Section Specific Items and Criteria Pre-NOC Letters • Letter for sending NOC or NOC/c to CADTHa
• Letter for finalized category 1 requirementsa
CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions. a This is only a requirement for a submission filed on a pre-NOC basis. a) General Information i. Application Overview Template
• A completed Application Overview Template. ii. Signed Cover Letter
• A signed cover letter (an electronic signature is acceptable) from the applicant, providing the following information: A clear description of the submission being filed (e.g., category 1 requirements for a
new combination product [funded components] submission filed on a pre-NOC basis; category 1 requirements for a new combination product designated by CADTH as a tailored CDR review submission filed on a post-NOC basis).
If requesting priority review, notification that a priority review is being requested and the justification for the priority review request (i.e., based on clinical and/or economic criteria) and confirmation that a completed priority review request template has been included.
Confirmation that all of the requirements have been provided in the submission. The indication(s) to be reviewed under the CDR process. The date the NOC or NOC/c was issued for the indication(s) to be reviewed or, in
the case of a submission filed on a pre-NOC basis, the anticipated date the NOC will be issued.
The requested listing criteria, if applicable. Intention to provide category 2 requirements at least 20 business days before the
targeted CDEC meeting (if not being provided with category 1 requirements). A statement confirming whether the submitted price is the anticipated or current
marketed price, or the confidential price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation, irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer.
The names and contact information (email and phone number) for the primary and backup contact(s) that CADTH can contact regarding the submission. The manufacturer may designate the consultant(s) preparing the submission as primary and/or backup contact(s). Any changes in contacts should be communicated to CADTH as soon as possible, by emailing [email protected].
iii. Executive Summary • A high-level summary of the submission (five pages maximum excluding reference list),
following the Executive Summary Template for a Submission.
Submission Guidelines for the CADTH Common Drug Review August 2014 21

CADTH Common Drug Review
iv. New Combination Product Submission Template
• A completed New Combination Product Submission Template for the required clinical and economic information.
v. Product Monograph
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a copy of the most recent draft product monograph
showing the company, drug brand, and non-proprietary names that correspond to the anticipated NOC.
• As soon as available, sent by email to [email protected]: a copy of the draft product monograph initially filed showing, in tracked changes, all
of the clinical and label review changes made up to the time of the product monograph being approved by Health Canada
a copy of the clean and dated product monograph approved by Health Canada. For drugs reviewed by Health Canada’s Therapeutics Products Directorate, the approved product monograph refers to the pristine product monograph to which the Letter of Certification applies.
All submissions filed on a post-NOC basis: • A copy of the most current version of the Health Canada-approved product monograph.
b) Health Canada Documentation i. Health Canada NOC or NOC/c
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating the anticipated
target date for receipt of an NOC or NOC/c for the indication(s) to be reviewed. • A copy of the granted NOC or NOC/c for the indication(s) under review by CDR, dated
and signed by Health Canada, must be sent by email to [email protected] as soon as it is available (i.e., on the day of, or next business day after, receipt from Health Canada).
• If the drug receives an NOC/c for the indication(s) being reviewed by CDR: a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the clinical benefit, including an indication of time frames, must also be provided by email to [email protected] as soon as it is available.
All submissions filed on a post-NOC basis: • A copy of the NOC or NOC/c granted, dated, and signed by Health Canada. The NOC or
NOC/c must be for the indication(s) for which the drug is to be reviewed under the CDR process.
• If the drug in the submission has received an NOC/c for the indication(s) to be reviewed, the manufacturer must provide a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the drug’s clinical benefit, including an indication of time frames.
ii. Health Canada Clinical Reviewers’ Report
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating that a copy of
Health Canada’s PSEA or BSEAR, as applicable to the submission filed, will be provided as soon as it is available.
Submission Guidelines for the CADTH Common Drug Review August 2014 22

CADTH Common Drug Review
• After NOC or NOC/c is issued: a copy of Health Canada’s PSEA or BSEAR, as
applicable to the submission filed, must be provided by email to [email protected] as soon as available (i.e., on the day of, or next business day after, receipt from Health Canada). If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
• To avoid delays in providing the report to CADTH, manufacturers are encouraged to request it from Health Canada as soon as they are assured that an NOC or NOC/c will be issued, and forward it immediately upon receipt to CADTH.
All submissions filed on a post-NOC basis: • A copy of Health Canada’s PSEA or BSEAR, as applicable to the submission filed. • If the PSEA or BSEAR is unavailable from Health Canada at the time of filing the
submission, a placeholder document must be included, indicating that a copy of the report will be provided as soon as it is available.
• As soon as available (i.e., on the day of, or next business day after, receipt from Health Canada), a copy of Health Canada’s PSEA or BSEAR, as applicable to the submission filed, must be provided by email to [email protected]. If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
iii. Clarifaxes
All submissions filed on a pre-NOC basis: • At time of filing the submission: a summary table of Clarifaxes relating to any clinical
aspects of the Health Canada review of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the time of filing with CADTH. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
• On an ongoing basis up to the point of the NOC or NOC/c being issued, the manufacturer must provide CADTH with revised Clarifax summary tables to reflect any additional Clarifaxes as delineated above. The revised table(s) should be sent to CADTH by email ([email protected]).
All submissions filed on a post-NOC basis: • A summary table of Clarifaxes relating to any clinical aspects of the Health Canada review
of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the point of the NOC or NOC/c being issued. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
c) Clinical Study A copy of one clinical study using the new combination product (funded components or CADTH-designated for a tailored CDR review) to be reviewed, not the individual components. The study can be a pharmacokinetic study.
d) Disease Prevalence and Incidence
• The prevalence and incidence of the disease(s) or condition(s) for the indication to be reviewed provided for the Canadian population, with a breakdown by participating province, territory, and First Nations populations where available.
Submission Guidelines for the CADTH Common Drug Review August 2014 23

CADTH Common Drug Review
• References must be provided for this document in the following format: in-text citations numbered in their order of appearance a numbered reference list in the Citing Medicine format (Appendix 6).
e) Pricing and Distribution Information i. Submitted Price
• The submitted price for the drug, reported to four decimal places, as follows: price per smallest unit price per smallest dispensable unit for all dosage forms and strengths available in
Canada price for all packaging formats available in Canada.
• The submitted price is the price per unit that is submitted to CDR and that must not be
exceeded for any of the drug plans following release of a CDEC Final Recommendation,6 irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer. The submitted price can be an anticipated or current market price in Canada, or a confidential price.
• Only one price (anticipated or current market price, or confidential price) to four decimal places per dispensable unit is to be submitted per drug that is to be reviewed under the CDR process (i.e., only one price for all indications undergoing review by CDR concurrently).
• The submitted price must be used in the pharmacoeconomic evaluation included in the submission and in the BIAs.
ii. Method of Distribution
• Method of distribution to pharmacies (e.g., wholesale, direct, or other arrangements).
iii. Commitment to Honour Submitted Price • A signed commitment to honour the submitted price for all drug plans using
the Commitment to Honour Submitted Price Letter template.
f) Letter Authorizing Unrestricted Sharing of Information • A letter from the holder of the NOC or NOC/c (or from the manufacturer applying for an
NOC in the case of a submission filed on a pre-NOC basis), using the Unrestricted Sharing of Information Letter template, printed on company letterhead and signed by an appropriate senior official, permitting unrestricted sharing of information regarding the drug product under review through the CDR process, between CADTH and: Canadian Drug Expert Committee members? Federal/provincial/territorial governments, including their agencies and departments Patented Medicine Prices Review Board.
6 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 24

CADTH Common Drug Review
g) Additional Letters for Submissions Filed on Pre-NOC Basis
iv. Letter for Sending NOC or NOC/c to CADTH • After the NOC or NOC/c has been issued, the manufacturer must provide a signed letter,
using the Letter for Sending NOC or NOC/c to CADTH template, indicating whether or not any wording changes to the Health Canada–approved final product monograph, as compared with the draft product monograph filed in the initial package of category 1 requirements, result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis. The letter should be sent to CADTH by email ([email protected]).
v. Letter for Finalized Category 1 Requirements
• A signed letter, using the Letter for Finalized Category 1 Requirements template, confirming that all finalized versions of category 1 requirements for the submission filed on a pre-NOC basis have been provided to CADTH. The letter should be sent to CADTH by email ([email protected]).
5.1.3 Category 1 Requirements for a Tailored CDR Review: Subsequent Entry Biologic Submission Submissions for SEBs undergo a tailored CDR review. The category 1 requirements are summarized in Table 9. Detailed descriptions of the information that comprise each of the category 1 requirements for an SEB tailored CDR review submission are provided in this section. Where there are specific requirements for a submission filed on a pre-NOC versus post-NOC basis, they are delineated in the descriptions that follow the table.
Table 9: Category 1 Requirements for a Tailored CDR Review: SEB Submission Section Specific items and criteria General Information
• Completed application overview template • Signed cover letter • Completed executive summary template for a submission • Completed SEB submission template • Product monograph
Health Canada Documentation
• NOC or NOC/c • Health Canada clinical reviewers’ report • Table of Clarifaxes
Efficacy, Effectiveness, and Safety Information
• Common Technical Document sections 2.3, 2.5, 2.7.1, 2.7.2, 2.7.3, 2.7.4, 5.2, or statement indicating any section(s) not required for the Health Canada submission
• Reference list and copies of key clinical studies and errata (or statement that there are no errata)
• Table of studies • Reference list and copies of editorial articles (or statement that there are no
editorials) • Literature search strategies • Signed declaration that all known unpublished studies have been disclosed • CONSORT diagrams • Reference list and copies of new data • Reference list and copies of articles for validity of outcome measure
Submission Guidelines for the CADTH Common Drug Review August 2014 25

CADTH Common Drug Review
Table 9: Category 1 Requirements for a Tailored CDR Review: SEB Submission
Section Specific items and criteria Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
Pricing and Distribution Information
• Submitted unit pricing to four decimal places • Method of distribution • Signed commitment to honour the submitted price
Sharing of Information
• Signed letter authorizing unrestricted sharing of information
Pre-NOC Letters • Letter for sending NOC or NOC/c to CADTH a • Letter for finalized category 1 requirements a
CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; SEB = subsequent entry biologic. a This is only a requirement for a submission filed on a pre-NOC basis. a) General Information i. Application Overview Template
• A completed Application Overview Template. ii. Signed Cover Letter
• A signed cover letter (an electronic signature is acceptable) from the applicant, providing the following information: A clear description of the submission being filed (e.g., category 1 requirements for
an SEB submission filed on a pre-NOC basis) If requesting priority review, notification that a priority review is being requested and
the justification for the priority review request (i.e., based on clinical and/or economic criteria) and confirmation that a completed priority review request template has been included
Confirmation that all of the requirements have been provided in the submission The indication(s) to be reviewed under the CDR process The date the NOC or NOC/c was issued for the indication(s) to be reviewed or, in
the case of a submission filed on a pre-NOC basis, the anticipated date the NOC will be issued
The requested listing criteria, if applicable Intention to provide category 2 requirements at least 20 business days before the
targeted Canadian Drug Expert Committee meeting (if not being provided with category 1 requirements)
A statement confirming whether the submitted price is the anticipated or current marketed price, or the confidential price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation, irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer
The names and contact information (email and phone number) for the primary and backup contact(s) that CADTH can contact regarding the submission. The manufacturer may designate the consultant(s) preparing the submission as primary and/or backup contact(s). Any changes in contacts should be communicated to CADTH as soon as possible, by emailing [email protected].
Submission Guidelines for the CADTH Common Drug Review August 2014 26

CADTH Common Drug Review
iii. Executive Summary
• A high-level summary of the submission (five pages maximum excluding reference list), following the Executive Summary Template for a Submission.
iv. Subsequent Entry Biologic Submission Template • A completed Subsequent Entry Biologic Submission Template for submitting the
required clinical and economic information. v. Product Monograph
All submissions filed on a pre-NOC basis: • At the time of filing the submission: copy of the most recent draft product monograph
showing the company, drug brand, and non-proprietary names that correspond to the anticipated NOC.
• As soon as available, send by email to [email protected]: a copy of the draft product monograph initially filed showing, in tracked changes, all
of the clinical and label review changes made up to the time of the product monograph being approved by Health Canada
a copy of the clean and dated product monograph approved by Health Canada. For drugs reviewed by Health Canada’s Therapeutics Products Directorate, the approved product monograph refers to the pristine product monograph to which the Letter of Certification applies.
All submissions filed on a post-NOC basis: • A copy of the most current version of the Health Canada–approved product monograph.
b) Health Canada Documentation i. Health Canada NOC or NOC/c
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating the anticipated
target date for receipt of an NOC or NOC/c for the indication(s) to be reviewed. • A copy of the granted NOC or NOC/c for the indication(s) under review by CDR, dated
and signed by Health Canada, must be sent by email to [email protected] as soon as it is available (i.e., on the day of, or next business day after, receipt from Health Canada).
• If the drug receives an NOC/c for the indication(s) being reviewed by CDR: a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the clinical benefit, including an indication of time frames, must also be provided by email to [email protected] as soon as it is available.
All submissions filed on a post-NOC basis: • A copy of the NOC or NOC/c granted, dated, and signed by Health Canada. The NOC or
NOC/c must be for the indication(s) for which the drug is to be reviewed under the CDR process.
• If the drug in the submission has received an NOC/c for the indication(s) to be reviewed, the manufacturer must provide a copy of the Letter of Undertaking that outlines the confirmatory studies intended to verify the drug’s clinical benefit, including an indication of time frames.
Submission Guidelines for the CADTH Common Drug Review August 2014 27

CADTH Common Drug Review
ii. Health Canada Clinical Reviewers’ Report
All submissions filed on a pre-NOC basis: • At the time of filing the submission: a placeholder document indicating that a copy of
Health Canada’s BSEAR will be provided as soon as it is available. • After NOC or NOC/c is issued: a copy of Health Canada’s BSEAR must be provided by
email to [email protected] as soon as available (i.e., on the day of, or next business day after, receipt from Health Canada). If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
• To avoid delays in providing the report to CADTH, manufacturers are encouraged to request it from Health Canada as soon as they are assured that an NOC or NOC/c will be issued, and forward it immediately upon receipt to CADTH.
All submissions filed on a post-NOC basis: • A copy of Health Canada’s BSEAR. • If the BSEAR is unavailable from Health Canada at the time of filing the submission, a
placeholder document must be included, indicating that a copy of the report will be provided as soon as it is available.
• As soon as available (i.e., on the day of, or next business day after, receipt from Health Canada), a copy of Health Canada’s BSEAR must be provided by email to [email protected]. If the file size exceeds 10 MB, the report must be provided to CADTH on a CD, DVD, or USB flash drive.
iii. Clarifaxes
All submissions filed on a pre-NOC basis: • At time of filing the submission: a summary table of Clarifaxes relating to any clinical
aspects of the Health Canada review of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the time of filing with CADTH. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
• On an ongoing basis up to the point of the NOC or NOC/c being issued, the manufacturer must provide CADTH with revised Clarifax summary tables to reflect any additional Clarifaxes as delineated above. The revised table(s) should be sent to CADTH by email ([email protected]).
All submissions filed on a post-NOC basis: • A summary table of Clarifaxes relating to any clinical aspects of the Health Canada
review of the drug (e.g., clinical studies or product monograph, not chemistry- and manufacturing-related topics) up to the point of the NOC or NOC/c being issued. The date of each Clarifax, topic for clarification, a brief summary of the response, and date of the response must be included.
c) Efficacy, Effectiveness, and Safety Evidence i. Common Technical Document
As shown in Table 10, a copy of selected sections from the Common Technical Document modules. If any of these sections of the Common Technical Document were not a requirement for filing the regulatory submission with Health Canada, a placeholder document with a statement confirming this is required.
Submission Guidelines for the CADTH Common Drug Review August 2014 28

CADTH Common Drug Review
Table 10: Common Technical Document Sectionsa Required for SEB Submissions
Section Title 2.3 Quality Overall Summary 2.5 Clinical Overview 2.7.1 Summary of Biopharmaceutical Studies and Associated Analytical Methods 2.7.2 Summary of Clinical Pharmacology Studies 2.7.3 Summary of Clinical Efficacy 2.7.4 Summary of Clinical Safety 5.2 Tabular Listing of All Clinical Studies
SEB = subsequent entry biologic. a If any of these sections were not a requirement for filing the regulatory submission with Health Canada for the drug to be reviewed through the CADTH Common Drug Review, a placeholder document with a statement confirming this is required.
ii. Clinical Studies
• Copies of published and unpublished studies that address key clinical issues. It is preferred that unpublished data are submitted in manuscript format; however, if
unavailable in manuscript format, the information should be provided in accordance with the CONSORT 2010 Statement Checklist using clearly labelled sections as outlined (i.e., title, abstract, introduction, methods, results, discussion, other information)
Include copies of any errata related to any published studies provided. If there are no errata, a placeholder document with a statement confirming this must be provided
Should an unpublished study submitted as a category 1 requirement become published during the CDR review process, manufacturers must email a copy of the published study to [email protected], indicating that it is the published version of a previously unpublished study included in the category 1 requirements initially submitted
As specified in Appendix 9: Electronic File Structure and Naming Format, the first file in the folder must be a reference list of the articles and errata included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6.
iii. Table of Studies
• A tabulated list of all published and unpublished clinical studies using the Table of Studies Template.
iv. Editorials
• Copies of editorials relating to published clinical studies provided in the submission (i.e., studies listed in the first section of the “table of studies” requirement template) As specified in Appendix 9: Electronic File Structure and Naming Format, the first file
in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6
If there are no editorial articles available, a placeholder document with a statement confirming this must be provided.
v. Search Strategies
• Search strategies used to locate published studies in medical literature databases. All search terms that were used (i.e., MeSH headings and keywords) and the names of
Submission Guidelines for the CADTH Common Drug Review August 2014 29

CADTH Common Drug Review
databases (e.g., MEDLINE, Embase, Cochrane, etc.) that were searched are required. Search results are not required.
vi. Declaration for Disclosure of Known Clinical Studies • A signed declaration that all known unpublished clinical studies have been disclosed
using the Letter Confirming Disclosure of all Known Unpublished Studies template. If CADTH discovers undisclosed unpublished trials through other sources, this may result in the submission being considered at a later CDEC meeting to allow time for the retrieval and review of the trials.
vii. CONSORT Diagrams
• Diagrams following the CONSORT flow diagram reporting standards or similar diagrams that document the flow of patients through trials identified as pivotal trials in Health Canada documentation, as well as any other key trials included in the submission (i.e., studies listed in the first section of the “table of studies” requirement template).
• All information for the four stages of a trial (i.e., enrolment, intervention allocation, follow-up, and analysis) of the CONSORT flow diagram is required. Please consult Appendix 5 and/or the CONSORT web page (http://www.consort-statement.org/consort-statement/flow-diagram) for additional details regarding the structure and content of flow diagrams.
viii. New Data
• Copies of new data, generated since the last date that data were reported in the studies included in the Health Canada submission. Typically, the clinical studies submitted to CDR are the same as those submitted to Health Canada, and sometimes these studies are ongoing, with data collected after submission to Health Canada. The data that become available after the study has been submitted to Health Canada are required. These data will be accepted in a variety of formats, including late draft, Clinical Study Report, synopsis, abstract, or conference proceedings. As specified in Appendix 9: Electronic File Structure and Naming Format, the first file
in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6
If no new data are available, a placeholder document with a statement confirming this must be provided.
ix. Validity of Outcome Measures
• Copies of references supporting the validity of primary outcome measures in clinical studies. As specified in Appendix 9: Electronic File Structure and Naming Format, the first file
in the folder must be a reference list of the articles included in the folder. Guidance on the preparation of reference lists is provided in Appendix 6
If no references are available, a placeholder document is required with a statement confirming that a search was undertaken but no references were located.
d) Disease Prevalence and Incidence
• The prevalence and incidence of the disease(s) or condition(s) for the indication to be reviewed provided for the Canadian population, with a breakdown by participating province, territory, and First Nations populations where available.
Submission Guidelines for the CADTH Common Drug Review August 2014 30

CADTH Common Drug Review
• References must be provided for this document in the following format: in-text citations numbered in their order of appearance a numbered reference list in the Citing Medicine format (Appendix 6).
e) Pricing and Distribution Information i. Submitted Price
• The submitted price for the drug, reported to four decimal places, as follows: price per smallest unit price per smallest dispensable unit for all dosage forms and strengths available in
Canada price for all packaging formats available in Canada.
• The submitted price is the price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation,7 irrespective of the type of recommendation made and whether the Canadian Drug Expert Committee criteria for the listing are the same as the criteria requested by the manufacturer. The submitted price can be an anticipated or current market price in Canada, or a confidential price.
• Only one price (anticipated or current market price, or confidential price) to four decimal places per dispensable unit is to be submitted per drug that is to be reviewed under the CDR process (i.e., only one price for all indications undergoing review by CDR concurrently).
• The submitted price must be used in the pharmacoeconomic evaluation included in the submission and in the BIAs.
ii. Method of Distribution
• Method of distribution to pharmacies (e.g., wholesale, direct, or other arrangements).
iii. Commitment to Honour Submitted Price • A signed commitment to honour the submitted price for all drug plans using
the Commitment to Honour Submitted Price Letter template.
f) Letter Authorizing Unrestricted Sharing of Information • A letter from the holder of the NOC or NOC/c (or from the manufacturer applying for an
NOC in the case of a submission filed on a pre-NOC basis), using the Unrestricted Sharing of Information Letter template, printed on company letterhead and signed by an appropriate senior official, permitting unrestricted sharing of information regarding the drug product under review through the CDR process, between CADTH and: Federal, provincial, territorial governments, including their agencies and departments Patented Medicine Prices Review Board.
7 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 31

CADTH Common Drug Review
g) Additional Letters for Submissions Filed on Pre-NOC Basis
i. Letter for Sending NOC or NOC/c to CADTH • After the NOC or NOC/c has been issued, the manufacturer must provide a signed letter,
using the Letter for Sending NOC or NOC/c to CADTH template, indicating whether or not any wording changes to the Health Canada–approved final product monograph, as compared with the draft product monograph filed in the initial package of category 1 requirements, result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis. The letter should be sent to CADTH by email ([email protected]).
ii. Letter for Finalized Category 1 Requirements
• A signed letter, using the Letter for Finalized Category 1 Requirements template, confirming that all finalized versions of category 1 requirements for the submission filed on a pre-NOC basis have been provided to CADTH. The letter should be sent to CADTH by email ([email protected]).
5.2 Category 2 Requirements Category 2 requirements are used by the drug plans and are not considered as part of the review or recommendation process of CDR. CADTH provides secretariat support to the drug plans by ensuring that category 2 requirements have been filed in accordance with the Submission Guidelines for the CADTH Common Drug Review. When CADTH notifies a manufacturer that category 2 requirements are complete, it indicates that CADTH has confirmed that each of the requirements has been provided by the manufacturer, but it does not imply that the submitted information meets the requirements of the individual drug plans. If any of the drug plans have questions regarding the filed category 2 requirements, they will contact manufacturers directly. One copy of the category 2 requirements8 must be provided to CADTH as a single package in electronic format on a CD, DVD, or USB flash drive, organized as specified in Appendix 9: Electronic File Structure and Naming Format. Category 2 requirements for submissions filed on either a pre-NOC or post-NOC basis must be provided as a single package to CADTH at least 20 business days before the targeted Canadian Drug Expert Committee meeting at which the submission will be considered. Incomplete category 2 requirements will not affect placement of the submission on the targeted Canadian Drug Expert Committee agenda; however, the CDEC Final Recommendation will not be issued until the manufacturer has been notified by CADTH that all category 2 requirements are complete. Category 2 requirements may be submitted concurrently with category 1 requirements, when available. When advised by CADTH that category 2 requirements are complete, manufacturers should provide the drug plans with a copy of the category 2 requirements as described in Appendix 1. No additional copies of category 2 requirements are required by CADTH. The manufacturer is responsible for ensuring that appropriate copyright permissions have been obtained for electronic copies of articles included in category 2 requirements for a submission, to be shared among the drug plans. Category 2 requirements are as follows for all submission types. If priority review status based on the economic criterion was requested, the BIAs, with or without the Certified Product Information
8 If not provided at the same time as category 1 requirements.
Submission Guidelines for the CADTH Common Drug Review August 2014 32

CADTH Common Drug Review
Document, would have been filed as part of the priority review request along with the category 1 requirements for the submission. a) Cover Letter
The following letter is required for all submissions where category 2 requirements were not provided at the same time as category 1 requirements: • A signed cover letter (an electronic signature is acceptable) from the applicant, providing
the following information: a clear description of the submission being filed (e.g., category 2 requirements for a
new drug submission filed on a pre-NOC basis) confirmation that all of the category 2 requirements have been provided.
b) Budget Impact Analyses and Supporting Documentation
• BIAs for all of the following jurisdictions’ drug plans, in accordance with their individual requirements: British Columbia, Alberta, Saskatchewan, Manitoba, Ontario, New Brunswick, Nova Scotia, Prince Edward Island, Newfoundland and Labrador, and the Non-Insured Health Benefits Program. When data specific to Prince Edward Island are unavailable, the BIA for Prince Edward Island is to be based on Nova Scotia data.
• Copies of all supporting documentation used and/or cited in the BIAs. As specified in Appendix 9: Electronic File Structure and Naming Format, the first
file in the folder must be a reference list of the documents included in the folder.
The base unit price used in the BIAs must be the same as the price submitted in the category 1 requirements and must be clearly identified in each BIA. Jurisdiction-specific markups or discounts can then be applied, if applicable.
c) Certified Product Information Document • A completed and approved copy of the Certified Product Information Document (CPID).
In lieu of the CPID, the Master Formula and Final Product Specifications documents are required.
5.3 Additional Information Additional information consists of information that CADTH may require to complete the review. CADTH may request additional information from Health Canada or the manufacturer. Note the manufacturer’s continuing responsibility to advise CADTH of any harms or safety issues that may arise during the time the submission is under review, as detailed in the Harms and Safety Information section below. a) Harms and Safety Information
CADTH may request additional harms and safety information; however, the manufacturer has the responsibility of advising CADTH of all data on harms related to the drug under review (including harms and safety issues that may arise during the time that the submission is under review), and of communiqués (e.g., “Dear Doctor” letters) being prepared to alert health care professionals about safety concerns. Failure to advise CADTH of these issues or of communiqués as soon as they arise may result in CADTH readjusting and extending the usual CDR timelines in order to review this information.
Submission Guidelines for the CADTH Common Drug Review August 2014 33

CADTH Common Drug Review
b) Clinical Study Reports and Periodic Safety Update Reports
CADTH may request complete copies or sections of Clinical Study Reports and Periodic Safety Update Reports from the manufacturer. These documents should be provided in searchable electronic format (i.e., PDF or Microsoft Word).
6. REQUESTING PRIORITY REVIEW STATUS Manufacturers may request CDR priority review status at the time of filing a submission or resubmission. For details regarding the procedure and assessment process for priority review, please consult the Procedure for the CADTH Common Drug Review. a) A submission or resubmission may be granted priority review status based on clinical criteria
if all of the following criteria are demonstrated: • The drug is indicated or anticipated to be indicated for an immediately life-threatening or
other serious disease. • The drug addresses an unmet medical need. • The drug offers substantial improvement in clinically important outcome measures of
efficacy and effectiveness, when compared with other appropriate comparators. b) A submission or resubmission may be granted priority review status based on economic
criteria if the following criterion is demonstrated: • For the drug under review, the projected combined cost savings for the drug plans is an
average of at least $7.5 million per year for the first three years the product is marketed in Canada, when compared with appropriate comparators.
6.1 Priority Review Requests Based on Clinical Criteria Manufacturers requesting priority review status based on clinical criteria must submit the following information at the time of filing the submission or resubmission:
a) The signed cover letter from the applicant must clearly state that priority review status is being requested based on clinical criteria and that a completed priority review application template has been provided in the submission or resubmission.
b) A completed Priority Review Application Template.
c) Relevant information provided by the manufacturer regarding improvements in safety, tolerability, and quality of life would also be considered during the assessment. Manufacturers may provide details regarding these outcomes in the executive summary.
d) All other category 1 and category 2 requirements must be provided in accordance with the appropriate section of the Submission Guidelines for the CADTH Common Drug Review.
6.2 Priority Review Requests Based on Economic Criteria Manufacturers requesting priority review status based on economic criteria must provide the following information at the time of filing the submission or resubmission:
a) The signed cover letter from the applicant must clearly state that priority review status is being requested based on the economic criterion and that a completed priority review application template has been provided in the submission or resubmission.
Submission Guidelines for the CADTH Common Drug Review August 2014 34

CADTH Common Drug Review
b) A completed Priority Review Application Template providing a clear rationale and analysis to
support the claim of meeting the cost-savings criterion.
c) BIAs must be provided for all of the following jurisdictions’ drug plans, in accordance with their requirements: British Columbia, Alberta, Saskatchewan, Manitoba, Ontario, New Brunswick, Nova Scotia, Prince Edward Island, Newfoundland and Labrador, and the Non-Insured Health Benefits Program. When data specific to Prince Edward Island are unavailable, the BIA for Prince Edward Island is to be based on Nova Scotia data.
In addition, a pan-Canadian BIA (all drug plans9) with an explanation of the BIA and assumptions on which it is based is required.
d) Budge impact models (Excel spreadsheets) for all of the above jurisdictions’ drug plans, as well as a pan-Canadian budget impact model (Excel spreadsheet) showing how the pan-Canadian cost savings are derived, including a summary table of the analysis for all drug plans.
e) Copies of all supporting documentation used and/or cited in the BIAs. • As specified in Appendix 9: Electronic File Structure and Naming Format Appendix 9:
Electronic File Structure and Naming Format, the first file in the folder must be a reference list of the documents included in the folder.
All other category 1 and category 2 requirements must be provided in accordance with the appropriate sections of the Submission Guidelines for the CADTH Common Drug Review. Note that: • If a priority review based on economic criteria is requested for a submission, the only
outstanding category 2 requirement will be the CPID which, if available, can be filed along with the category 1 requirements.
• If priority review status based on the economic criterion is requested for a resubmission, there are no further category 2 requirements to file.
7. RESUBMISSION REQUIREMENTS A manufacturer may file a resubmission for a new drug, a drug with a new indication, a new combination product, a new combination product (funded components or CADTH-designated tailored CDR review), or an SEB that has previously been reviewed through the CDR process and for which a CDEC Final Recommendation has been issued by CADTH. To be eligible for a resubmission, the applicant must submit new information that was not previously provided in the initial submission or a previous resubmission(s). The new information must consist of one or both of the following: • new clinical information in support of improved efficacy or safety • new cost information that significantly affects the cost-effectiveness of the drug. New information in support of improved efficacy must be from a randomized controlled trial, whereas case-control or cohort studies will be accepted when the new information is in support of improved safety, if randomized controlled trials are unavailable.
9 The pan-Canadian BIA must encompass only the drug plans that participate in the CADTH Common Drug Review process.
Submission Guidelines for the CADTH Common Drug Review August 2014 35

CADTH Common Drug Review
New information in support of cost information must be supported by a new pharmacoeconomic evaluation and new BIAs. Table 11 summarizes the supporting information that must be filed for all resubmissions, according to the type of new information providing the rationale for filing.
Table 11: Summary of New Information Required for Resubmissions Type of New Information Supporting Information That Must be Filed
New clinical information supporting improved efficacy
• New RCT(s) • New pharmacoeconomic evaluation • New BIAs (category 2 requirement, unless economics-based priority
review status requested) New clinical information supporting improved safety
• New case-control or cohort study/studies, if RCT(s) unavailable • New pharmacoeconomic evaluation • New BIAs (category 2 requirement, unless economics-based priority
review status requested) New cost information that significantly affects the cost-effectiveness of the drug
• New pharmacoeconomic evaluation • New BIAs (category 2 requirement, unless economics-based priority
review status requested)
BIAs = budget impact analysis; RCT = randomized controlled trial. The CDR resubmission requirements are grouped into the following categories: category 1 requirements, category 2 requirements, and priority review request requirements. A brief description of these requirements is provided in Table 12 and detailed descriptions are provided in subsequent sections and also in section 6 if priority review status is requested. For all resubmissions, the clinical and pharmacoeconomic information provided in the category 1 and category 2 requirements should focus on the indication(s) to be reviewed under the CDR process, unless otherwise specified.10
10 An exception to this is the “table of studies” requirement.
Submission Guidelines for the CADTH Common Drug Review August 2014 36

CADTH Common Drug Review
Table 12: Resubmission Requirement Categories
Requirement Category
Function in the CDR Process Due
Category 1 Used by the CDR review team and CDEC for the review and recommendation process.
At the time of filing the application.
Category 2 Used by the drug plans and are not considered as part of the CDR review or recommendation process. CADTH provides secretariat support to the drug plans by ensuring that category 2 requirements have been filed in accordance with the Submission Guidelines for the CADTH Common Drug Review.
≥ 20 business days before the targeted CDEC meeting.
Priority review request Used by CADTH, CDEC, and the drug plans for determining whether a resubmission should be granted priority review status.
At the time of filing the resubmission.
Additional information Additional information that CADTH may require for completion of the review (e.g., Clinical Study Reports).
As soon as possible following a request by CADTH, to avoid delays in the review process.
CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review. To expedite the screening of resubmissions for completeness and to help with the efficient use of documents, manufacturers must organize all resubmission information in the order prescribed in the category 1 and category 2 requirements below and follow the electronic file folder format in Appendix 9: Electronic File Structure and Naming Format . The resubmission checklists used by CADTH for screening category 1 and category 2 requirements can be found in Appendix 8: Checklists for Preparing CADTH COMMON DRUG REVIEW Applications . These checklists may assist manufacturers in ensuring that all requirements have been included in the resubmission. 7.1 Category 1 Requirements
Category 1 requirements are used by the review team and the Canadian Drug Expert Committee for the review and recommendation process. One copy of all category 1 requirements must be filed with CADTH as a single resubmission package in electronic format on CDs, DVDs, or USB flash drives and accepted by CADTH before the review can proceed. When the category 1 requirements have been screened and accepted for review by CADTH, the manufacturer and the drug plans are informed and steps commence to determine the order and timing for initiating the review. The manufacturer is responsible for ensuring that appropriate copyright permissions have been obtained for electronic copies of articles included in category 1 requirements of a resubmission, to be shared among the CDR review team, the Canadian Drug Expert Committee, and the drug plans for the review of the resubmission. If the resubmission is for a new combination product (funded components or CADTH-designated tailored review), contact CADTH for the resubmission requirements.
Submission Guidelines for the CADTH Common Drug Review August 2014 37

CADTH Common Drug Review
a) General Information i. Application Overview Template
• A completed Application Overview Template. ii. Signed Cover Letter
• A signed cover letter (an electronic signature is acceptable) from the applicant, providing the following information: A clear description of the resubmission being filed and the rationale for the
resubmission (e.g., a resubmission based on new clinical information supporting improved efficacy)
A statement confirming that the information on which the resubmission is based is new (i.e., that it was not included in the initial submission or a previous resubmission) and stating the anticipated change or outcome
If requesting priority review, notification that a priority review is being requested and the justification for the priority review request (i.e., based on clinical and/or economic criteria) and confirmation that a completed priority review request template has been included
Confirmation that all of the requirements have been provided in the resubmission The indication(s) to be reviewed under the CDR process The requested listing criteria, if applicable Intention to provide category 2 requirements at least 20 business days before the
targeted Canadian Drug Expert Committee meeting (if not filing a resubmission based on new cost information)
A statement confirming whether the submitted price is the anticipated or current marketed price, or the confidential price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation, irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer
The names and contact information (email and phone number) for the primary and backup contact(s) that CADTH can contact regarding the resubmission. The manufacturer may designate the consultant(s) preparing the resubmission as primary and/or backup contact(s). Any changes in contacts should be communicated to CADTH as soon as possible, by emailing [email protected].
iii. Executive Summary • A high-level summary of the resubmission (five pages maximum excluding reference
list), following the Executive Summary Template for a Resubmission.
iv. Product Monograph • The copy of the most current version of the Health Canada–approved product
monograph. b) New and Updated Efficacy and/or Safety Evidence i. New Clinical Information
• A reference list (see Appendix 6) of all new clinical information included in the resubmission that was not provided in the initial submission, or a previous resubmission.
Submission Guidelines for the CADTH Common Drug Review August 2014 38

CADTH Common Drug Review
Provide separate reference lists for each category of new information (e.g., new clinical
studies, new editorials, errata, etc.). See the checklist in Appendix 8: Checklists for Preparing CADTH COMMON DRUG REVIEW Applications.
• Copies of all new clinical information included in each of the above lists. The following are examples of new clinical information that, if applicable, must be included in the reference list and of which copies are to be included in the resubmission: • new randomized controlled trial(s) supporting the improved efficacy of the drug • new case-control or cohort studies supporting the improved safety of the drug • new editorial articles and errata relating to published clinical studies provided in both
the initial submission and any previous resubmission(s) (or if there are no editorials or errata, a placeholder document with a statement confirming this)
• new indirect comparison or network meta-analysis, if applicable • for a drug that has been issued an NOC/c: documentation describing the status of, and
most recent analysis results (final or interim) of the confirmatory studies listed in the Letter of Undertaking.
ii. CONSORT Diagrams
• Diagrams following the CONSORT flow diagram reporting standards or similar diagrams that document the flow of patients through new key trials included in the resubmission.
• All information for the four stages of a trial (i.e., enrolment, intervention allocation, follow-up, and analysis) of the CONSORT flow diagram is required. Please consult Appendix 5 and/or the CONSORT web page (http://www.consort-statement.org/consort-statement/flow-diagram) for additional details regarding the structure and content of flow diagrams.
iii. Updated Table of Studies
• An updated tabulated list of all published and unpublished clinical studies using the Table of Studies Template.
iv. Search Strategies
• Search strategies used to locate published studies in medical literature databases. All search terms that were used (i.e., MeSH headings and keywords) and the names of databases (e.g., MEDLINE, Embase, Cochrane, etc.) that were searched are required. Search results are not required.
v. Declaration for Disclosure of Known Clinical Studies
• A signed declaration that all known unpublished clinical studies have been disclosed using the Letter Confirming Disclosure of all Known Unpublished Studies template. If CADTH discovers undisclosed unpublished trials through other sources, this may result in the resubmission being considered at a later Canadian Drug Expert Committee meeting to allow time for the retrieval and review of the trials.
c) New and Updated Economic and Epidemiologic Information i. Pharmacoeconomic Evaluation
• An appropriate pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR. If there are subgroups
Submission Guidelines for the CADTH Common Drug Review August 2014 39

CADTH Common Drug Review
that may benefit from the drug or specific reimbursement criteria requested by the manufacturer, additional analyses should be provided. For example, if a manufacturer is requesting the review of one of several Health Canada–approved indications, the base-case analysis for the pharmacoeconomic evaluation must include the full population for the indication to be reviewed. In addition, if the applicant decides to make a specific listing request for one or more subgroups of the patient population that may benefit from the drug, additional analyses for each subgroup must also be provided in the pharmacoeconomic submission.
• Only one type of pharmacoeconomic analysis is to be submitted, and the type of analysis must be in accordance with section 9 of this document as well as CADTH’s Guidelines for the Economic Evaluation of Health Technologies: Canada. Should the applicant decide to submit more than one pharmacoeconomic analysis, they must clearly indicate which of the analyses submitted should be reviewed. The submission will not be accepted by CADTH for review until the applicant confirms which submitted economic analysis is to be reviewed.
• Only the price submitted to CDR is to be used in all base-case pharmacoeconomic evaluations.
ii. The Economic Model
• Three separate CDs, DVDs, or USB flash drives containing copies of the economic model in unlocked and fully executable format provided only in the manufacturer’s initial resubmission sent for screening of acceptability. No additional copies are required by CADTH once the resubmission is accepted for review by CADTH.
• The submitted model must be used as the basis for the economic evaluation. • The preferred economic model software platforms are Excel, TreeAge, or Arena. Before using other specialized program software, manufacturers must contact
CADTH in advance to ensure that they meet CADTH’s requirements and to receive direction on how the model and software should be provided as part of the resubmission information. Manufacturers are expected to provide CADTH with the necessary requirements to run the model (i.e., licences, laptop with model, and software), which will be returned to the manufacturer at the end of the review process at the manufacturer’s expense.
• The economic model must be provided in its entirety, meaning CADTH must have full access to the programming code and be able to fully execute the model based on modifications to parameters of interest. The CDR review team must be able to vary individual parameters, view the calculations, and run the model to generate results. The type of information that CDR requires for its examination of the model and the preferred format for receiving it are described in Table 13.
• Where the clinical inputs are based on an ITC, the full technical report of the ITC must be provided as part of the filed material.
• If statistical analyses of data sets are included in the model, the manufacturer must provide a description of the data sources and analyses conducted, and results from the analyses.
• Copies of any supporting materials that are used as part of the modelling exercise must be provided.
• Documentation detailing the methods used in the modelling exercise and basic user information must be included.
Submission Guidelines for the CADTH Common Drug Review August 2014 40

CADTH Common Drug Review
Table 13: CADTH Common Drug Review Pharmacoeconomic
Model Information Requirements Information Elements Format Basis for the pharmacoeconomic model
A modela that is unlocked and fully executable. The user should be able to specify inputs, view calculations, run analyses, and have full access to the programming code.
Media CD, DVD, USB flash drive or laptop (see software requirements below regarding when laptop is required).
Software requirements
The submitted model must be in one of the following formats: Excel, TreeAge, or Arena. Where manufacturers are using specialized program software, they must contact CADTH in advance to ensure that the programs meet CADTH’s requirements and to receive direction on how the model and software should be provided in the resubmission. Where the model platform is agreed upon, the manufacturers must provide CADTH with the necessary requirements to run the model (i.e., licences, laptop with model, and software), which will be returned to the manufacturer at the end of the review process at the manufacturer’s expense.
Basic user guide to the model Electronic format Model documentation (manuscripts or a summary of the model report may be provided)
Electronic format
Description of the statistical analyses included in the model (e.g., data sources, methods, and results)
Electronic format
Copies of any supporting materials that are used as part of the modelling exercise
Electronic format
a The pharmacoeconomic model will be examined by internal and external CDR review team members. The pharmacoeconomic model will not be released by CADTH to any third parties.
iii. Number of Patients Accessing a New Drug
The following information is required only for a new drug resubmission, or a new combination product resubmission if one of the components is a “new drug” (as defined in section 2.1.1): • For the indication(s) to be reviewed through CDR, the number of patients in Canada
currently accessing the new drug to within 20 business days of filing the resubmission. This information must include the number of patients accessing the drug through mechanisms such as: compassionate use participation in a clinical trial.
• Use the Number of Patients Accessing New Drug template for providing this information.
iv. Disease Prevalence and Incidence • The prevalence and incidence of the disease(s) or condition(s) for the indication(s) to
be reviewed provided for the Canadian population, with a breakdown by participating
Submission Guidelines for the CADTH Common Drug Review August 2014 41

CADTH Common Drug Review
province, territory, and Frist Nations populations where available. References must be provided for this document.
• All references in the following format: in-text citations numbered in their order of appearance a numbered reference list in the Citing Medicine format (Appendix 6).
d) Pricing and Distribution Information i. Submitted Price
• The submitted price for the drug, reported to four decimal places, as follows: price per smallest unit price per smallest dispensable unit for all dosage forms and strengths available in
Canada price for all packaging formats available in Canada.
• The submitted price is the price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation,11 irrespective of the type of recommendation made and whether the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer. The submitted price can be an anticipated or current market price in Canada, or a confidential price.
• Only one price (anticipated or current market price, or confidential price) to four decimal places per dispensable unit is to be submitted per drug that is to be reviewed under the CDR process (i.e., only one price for all indications undergoing review by CDR concurrently).
• The submitted price must be used in the pharmacoeconomic evaluation included in the resubmission and in the BIAs.
ii. Method of Distribution
• Method of distribution to pharmacies (e.g., wholesale, direct, or other arrangements).
iii. Commitment to Honour Submitted Price • A signed commitment to honour the submitted price for all drug plans using
the Commitment to Honour Submitted Price Letter template.
e) Letter Authorizing Unrestricted Sharing of Information • A letter from the holder of the NOC or NOC/c, using the Unrestricted Sharing of
Information Letter template, printed on company letterhead and signed by an appropriate senior official, permitting unrestricted sharing of information regarding the drug product under review through the CDR process, between CADTH and: Federal, provincial, territorial governments, including their agencies and
departments Patented Medicine Prices Review Board.
f) List of Decisions by Drug Plans
• A summary of the benefit status of the drug product for all drug plans at the time of the resubmission, including all criteria for coverage, if applicable.
11 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 42

CADTH Common Drug Review
7.2 Category 2 Requirements Category 2 requirements are used by the drug plans and are not considered as part of the review or recommendation process of CDR. CADTH provides secretariat support to the drug plans by ensuring that category 2 requirements have been filed in accordance with the Submission Guidelines for the CADTH Common Drug Review. When CADTH notifies a manufacturer that category 2 requirements are complete, it indicates that CADTH has confirmed that each of the requirements has been provided by the manufacturer, but it does not imply that the submitted information meets the requirements of the individual drug plans. If any of the drug plans have questions regarding the filed category 2 requirements, they will contact manufacturers directly. One copy of the category 2 requirements12 must be provided to CADTH as a single package in electronic format on a CD, DVD, or USB flash drive, organized as specified in Appendix 9: Electronic File Structure and Naming Format. Category 2 requirements for resubmissions must be provided as a single package to CADTH at least 20 business days before the targeted Canadian Drug Expert Committee meeting at which the resubmission will be considered. Incomplete category 2 requirements will not affect placement of the resubmission on the targeted Canadian Drug Expert Committee agenda; however, the CDEC Final Recommendation will not be issued until the manufacturer has been notified by CADTH that all category 2 requirements are complete. Category 2 requirements may be submitted concurrently with category 1 requirements, when available. When advised by CADTH that category 2 requirements are complete, manufacturers should provide the drug plans with a copy of the category 2 requirements as described in Appendix 1. No additional copies of category 2 requirements are required by CADTH. The manufacturer is responsible for ensuring that appropriate copyright permissions have been obtained for electronic copies of articles included in category 2 requirements of a resubmission, to be shared among the drug plans. Category 2 requirements for resubmissions are as follows. Note that if a request for priority review status has been filed for a resubmission, there will be no category 2 requirements to file. a) Cover Letter
The following letter is required for all resubmissions where category 2 requirements were not provided at the same time as category 1 requirements: • A signed cover letter (an electronic signature is acceptable) from the applicant,
providing the following information: a clear description of the resubmission being filed (e.g., category 2 requirements for
a resubmission based on new cost information) confirmation that all of the category 2 requirements have been provided.
b) Budget Impact Analyses and Supporting Documentation
The following information is required for all resubmissions, unless a priority review based on the economic criterion was requested at the time of filing category 1 requirements:
12 If not provided at the same time as category 1 requirements.
Submission Guidelines for the CADTH Common Drug Review August 2014 43

CADTH Common Drug Review
• BIAs for all of the following jurisdictions’ drug plans, in accordance with their individual
requirements: British Columbia, Alberta, Saskatchewan, Manitoba, Ontario, New Brunswick, Nova Scotia, Prince Edward Island, Newfoundland and Labrador, and Non-Insured Health Benefits Program. When data specific to Prince Edward Island are unavailable, the BIA for Prince Edward Island is to be based on Nova Scotia data.
• Copies of all supporting documentation used and/or cited in the BIAs. As specified in Appendix 9: Electronic File Structure and Naming Format, the first
file in the folder must be a reference list of the documents included in the folder.
The base unit price used in the BIAs must be the same as the price submitted in the category 1 requirements and must be clearly identified in each BIA. Jurisdiction-specific markups or discounts can then be applied, if applicable.
7.3 Additional Information Additional information consists of information that CADTH may require to complete the review. CADTH may request additional information from Health Canada or the manufacturer. Note the manufacturer’s continuing responsibility to advise CADTH of any harms or safety issues that may arise during the time the resubmission is under review, as detailed in the Harms and Safety Information section below. a) Harms and Safety Information
CADTH may request additional harms and safety information; however, the manufacturer has the responsibility of advising CADTH of all data on harms related to the drug under review (including harms and safety issues that may arise during the time that the resubmission is under review), and of communiqués (e.g., “Dear Doctor” letters) being prepared to alert health care professionals about safety concerns. Failure to advise CADTH of these issues or of communiqués as soon as they arise may result in CADTH readjusting and extending the usual CDR timelines in order to review this information.
b) Clinical Study Reports and Periodic Safety Update Reports CADTH may request complete copies or sections of Clinical Study Reports and Periodic Safety Update Reports from the manufacturer. These documents should be provided in searchable electronic format (i.e., PDF or Microsoft Word).
Submission Guidelines for the CADTH Common Drug Review August 2014 44

CADTH Common Drug Review
8. RESUBMISSION BASED ON A REDUCED PRICE DURING EMBARGO PERIOD
8.1 Eligibility A resubmission based on a reduced price during the embargo period can be made on the following grounds: • The Canadian Drug Expert Committee recommendation is “Do not list at the submitted
price”; OR • The Canadian Drug Expert Committee recommendation is “List with clinical criteria and/or
conditions” where there is a condition of a reduced price in comparison with the submitted price, or cost/cost-effectiveness has been identified as a reason for the recommendation, or cost/cost-effectiveness has been identified as a factor in the “Of Note” section of the
recommendation. • In the case of any resubmission based on a reduced price during the embargo period, the
only new information that will be accepted and reviewed is the reduced price and pharmacoeconomic analyses based on the new price. New clinical information will not be considered.
• CADTH will determine the date of the targeted Canadian Drug Expert Committee meeting for such a resubmission based on the amount of pharmacoeconomic information related to the price reduction and the effort required for review.
• See the Procedure for the CADTH Common Drug Review for additional details. 8.2 Requirements The requirement for a resubmission based on a reduced price during the embargo period is as follows: a) Signed Cover Letter A signed cover letter (an electronic signature is acceptable) from a senior company official to CADTH and copied to all drug plans that: • states what the reduced price is as price, reported to four decimal places, per smallest unit;
smallest dispensable unit for all dosage forms and strengths available in Canada; and for all packaging formats available in Canada (only one reduced price per unit is to be submitted)
• indicates if the reduced price is a confidential price • guarantees that the reduced price is the price per unit that must not be exceeded for any of
the drug plans following release of a CDEC Final Recommendation,13 irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer.
13 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 45

CADTH Common Drug Review
9. GUIDELINES FOR THE TYPE OF ECONOMIC ANALYSIS TO BE SUBMITTED
An appropriate pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR is required for all submissions for new drugs, drugs with a new indication, new combination products and resubmissions. If there are subgroups that may benefit from the drug or specific reimbursement criteria requested by the manufacturer, additional analyses should be provided. For example, if a manufacturer is requesting the review one of several Health Canada–approved indications, the base-case analysis for the pharmacoeconomic evaluation must include the full population for the indication to be reviewed. In addition, if a specific listing request is made for a subpopulation or if there are subgroups of the population that may benefit from the drug, additional analyses for each subgroup or subpopulation must also be provided in the pharmacoeconomic submission. This section provides guidance for the type of economic analysis to submit to CDR. Users of this document should also refer to Economic and Epidemiologic Information in sections 5.1.1d) for submissions and section 7.1c) for resubmissions. For methodological details, see the CADTH document Guidelines for the Economic Evaluation of Health Technologies: Canada. Based on the type of drug being submitted and its expected place in therapy, specific guidance is provided in the following information. It should be noted that where a manufacturer is suggesting clinical benefit (in terms of efficacy or harms with their drug), an economic model must be submitted.
Submission Guidelines for the CADTH Common Drug Review August 2014 46

CADTH Common Drug Review
Figure 1: Summary of the Guidelines for the Type of Economic Analysis to Submit
CDR = CADTH Common Drug Review; Mfr = manufacturer.
9.1 The Drug is the First Available for Treatment of the Disorder or Disease, or the First to be Listed by Drug Plans
Drugs that fall under this category include, but are not limited to: • drugs indicated for the treatment of diseases or disorders for which there are currently no
drugs approved in Canada or drugs that establish a new therapeutic class for the treatment of a disease or disorder
• drugs that consist of new molecules • drugs with new mechanisms of action.
Submission Guidelines for the CADTH Common Drug Review August 2014 47

CADTH Common Drug Review
Table 14: Guidelinesa for the Economic Analyses of Drugs
That Are the First Available Treatment Primary analysisb Cost-effectiveness or cost-utility analysis Acceptable pharmacoeconomic outcomes
• Cost per LYG • Cost per QALY • Cost per event avoided
Comparator Standard or current care Details of cost estimates
Price comparison table and health care cost tables
LYG = life-year gained; QALY = quality-adjusted life-year. a See also subsections “Primary Type of Analysis” and “Comparator” below. b Economic model required in all instances.
The preferred clinical outcomes for CDR are the final clinical outcomes. If final and intermediate outcomes are available, the submitted analysis should be based on final clinical outcomes. Where possible, the manufacturer should provide clinical evidence detailing the implications of the submitted treatment on clinical outcomes. Where this information is not available, surrogate outcomes shown to be valid surrogates for final clinical outcomes may be used. If data are not available to support the relationship between surrogate and final clinical outcomes, a cost-consequence should be provided.
a) Primary Type of Analysis The primary type of analysis should be presented as a cost-utility analysis or cost-effectiveness analysis, reporting: • cost per life-year gained or cost per quality-adjusted life-year (QALY) gained • cost per clinical event avoided (only for “non-subjective clinical outcome measures” when
extrapolation to life-years or QALYs is inappropriate). b) Comparator In all cases, the new therapy should be compared with the accepted therapy (existing practice), where the accepted treatment would be the single most prevalent clinical practice (if there is one that is dominant). Where generic versions of the accepted therapies exist, the price of the generic drug should be used. All other reasonable alternative therapies should at least be discussed in the report. See the CADTH document Guidelines for the Economic Evaluation of Health Technologies: Canada for further guidance. c) Requirements for Cost Data Companies should submit a price comparison table (Table 16) and cost tables (Table 17 and Table 18) outlining all appropriate costs, and identifying sources and assumptions for the costs included in each category. 9.1.1 Suggested Content for Submission Manufacturers should include the following information for review by CDR when submitting an economic evaluation in support of a drug for which similar drugs are not currently available: • description of the study treatment • description of, and justification of, comparator(s) (i.e., reflects current management) • description of indication or treatment population • perspective
Submission Guidelines for the CADTH Common Drug Review August 2014 48

CADTH Common Drug Review
• time horizon and justification • discount rates and justification (if applicable) • target audience • type of economic analysis and justification • appropriate and clear description of research methodology • clear description of data sources: effectiveness, safety, cost and resource use, and other
data • description of all assumptions used in the analysis • identification and definition of the key cost drivers • report of total and incremental costs and effects • report of cost breakdown of total costs • description, justification, and comprehensive reporting of sensitivity analyses (if applicable) • discussion of limitations • discussion of equity considerations • discussion of transferability of results across different jurisdictions. For more detailed information on reporting structure, companies should refer to CADTH’s Guidelines for the Economic Evaluation of Health Technologies: Canada. 9.2 The Drug Is Not the First Available Treatment for the Disorder or Disease Drugs that fall under this category include drugs that largely duplicate the action of existing drugs (i.e., drugs in an established class with a similar mechanism of action and therapeutic use) and combination products (for which all constituent drugs are funded). Table 15 shows the analysis required.
Table 15: Guidelines for Economic Analyses for Drugs That Are Not the First Available Treatment
Is the drug one in an established class (i.e., drugs with same mechanism of action and therapeutic use)
No Follow process for first available treatment (section 9.1)
Yes Trial(s) versus other available treatments?
Yes Results from trial(s) show no difference in efficacy and safety?
Yes Complete cost tables (section 9.3)
No Follow process for first available treatment (section 9.1)
No Manufacturer conducted an indirect comparison where similar clinical benefits (efficacy and safety) of drug compared with appropriate comparators are demonstrated?
Yes Complete cost tables (section 9.3)
No Follow process for first available treatment (section 9.1)
Submission Guidelines for the CADTH Common Drug Review August 2014 49

CADTH Common Drug Review
If other available treatments are listed as benefits by any of the drug plans in Canada, the manufacturer must indicate whether a randomized trial has been conducted comparing the new therapy with either the first available drug or other available drugs for the disorder or disease. 9.2.1 Head-to-Head Trials Have Been Conducted Versus Other Available Drugs Note: Other available drugs for the disorder or disease that are listed as benefits by any of the drug plans Do the results of the trial(s) show no difference in safety and efficacy? “No difference in safety and efficacy” is defined as the lack of any statistically significant differences between the intervention and alternatives. This decision should be based on a high-quality clinical assessment of the intervention. Beyond treatment effects, the submitted drug may differ from alternatives in terms of compliance or convenience of use (e.g., due to less frequent drug administration). These differences should only be considered as relevant for claiming clinical benefits if they have been linked to changes in clinically meaningful outcomes. (Note: Inappropriate assumptions of clinical equivalence compared with existing available drugs for treatment of this disease or disorder may compromise the ability to fully review the submitted drug.) a) Results from the head-to-head trial(s) show no difference in safety and
efficacy (non-inferiority or non-superiority) Companies should submit a price comparison table (Table 16) and cost tables (Table 17 and Table 18) outlining all appropriate costs, identifying source, and assumptions for the costs included in each category.
b) Results from the head-to-head trial show difference(s) in safety and/or efficacy Follow the submission process for first available drug (section 9.1).
9.2.2 No Head-to-head Trial(s) Have Been Conducted Versus Another Available Drug Note: Other available drugs for the treatment or management of the disorder or disease that are listed as benefits by any of the drug plans a) Manufacturer assumes drug provides similar clinical benefits versus other
drugs in class based on the results of an indirect comparison Companies should submit a price comparison table (Table 16) and cost tables (Table 17 and Table 18) outlining all appropriate costs, identifying sources, and assumptions for the costs included in each category.
b) Manufacturer assumes drug provides different clinical benefits versus other drugs in class based on the results of an indirect comparison Where the manufacturer is assuming different clinical benefit (efficacy or effectiveness or safety) of the submitted drug compared with other drugs in the class, based on an indirect comparison, the evidence to support these claims should be provided in detail. Reference to other studies that may provide information on clinical benefits other than efficacy and safety,
Submission Guidelines for the CADTH Common Drug Review August 2014 50

CADTH Common Drug Review
or post-marketing data that provide information on longer-term safety should be supplied as required.
Follow the submission process for first available drug (section 9.1).
9.2.3 Suggested Content for Submission Manufacturers should include the following information for a review by CDR when submitting an economic evaluation in support of a drug for which similar drugs are currently available: • description of the study treatment • description of, and justification of, comparator(s) • description of indication or treatment population • perspective • time horizon considered in cost table(s) and justification • discount rates and justification (if considering time horizons beyond one year) • justification for approach to economic submission [cost table(s)] — this may include
reference to specific studies or sections in the submission binder • clear description of data sources: effectiveness, safety, cost and resource use, and other data • appropriate and clear description of research methodology (if applicable) • description of all assumptions used in the analysis • cost table(s) (Table 16, Table 17, and Table 18 in section 9.3) • description, justification, and reporting of sensitivity analyses (if applicable) • discussion of limitations.
9.3 Suggested Format of Cost Information Suggested approaches for reporting cost data are provided in Table 16, Table 17, and Table 18. Manufacturers are strongly urged to consider all potentially relevant costs that may be applicable to their drug; the cost components listed in the sample tables by no means reflect a comprehensive list of all possible costs. In addition, manufacturers may provide tables that are more specific to their submitted drug. The following tables are examples intended to assist those submitting economic information.
Submission Guidelines for the CADTH Common Drug Review August 2014 51

CADTH Common Drug Review
Table 16: Price Comparison Table
Column 1
Column 2
Column 3
Column 4
Column 5
Column 6
Column 7
Column 8
Column 9
Drug
Strength
Form
List Price
Per Unit (Specify)
($)
Average Daily Use
Average Daily Drug Cost Per
Patient
Typical Annual Drug Costa
Range of Plausible
Costs
Extra columns
may include: Daily Pill Burden,
Frequency of Use
Submitted Drug
XX mg Tablet or
Caplet
$Y Z mg daily, twice daily, etc.
$A Consider actual market use of drug
Consider other
patient weights, possible wastage
scenarios
Comparator 1
Comparator 2
Comparator x
(previous standard of
care)
a Where the typical duration of treatment is less than one year, the typical cost for a full course of treatment should be considered. Note: Data sources and assumptions must be clearly stated by the manufacturer. As necessary, it is recommended that the company include sections detailing the calculations resulting in the value reported in each of these cells. Included within this table should be a detailed comparison of cost for the submitted drug with all other available drugs for the treatment of the disease or disorder (if applicable) and with any previous drugs that were considered standard of care. A description of the submitted drug and relevant comparators must be provided in columns 1 through 3. The list price of the treatment per unit and the intended dosing and use should be described in columns 4 to 6. In addition, the typical price of each drug, as used in clinical practice, should also be addressed in this table. “Typical Annual Drug Cost” should account for the actual doses used, wastage (where partial vials cannot be reused), etc. The components included in this figure should be clearly detailed, and details of how this number was derived should be provided. These details should be provided in a footnote to this table or, if necessary, in an appendix to the pharmacoeconomic submission. Factors that may affect the cost of the treatment should be detailed in column 8, “Range of Plausible Costs.” These factors might include patient weight, possible wastage, and treatment resistance. Additional columns may be included if necessary; justification must be provided for the inclusion of additional information. For example (column 9), where complex dosing or treatment regimens are being considered, the number of pills per day, or the frequency of use may be relevant.
Submission Guidelines for the CADTH Common Drug Review August 2014 52

CADTH Common Drug Review
Table 17 details the direct health care costs associated with the submitted drug. For each category, the costs provided should be reported as mean total cost per patient (i.e., taking into account the frequency of an event and its cost). Data sources and assumptions must be clearly stated by the manufacturer. As necessary, it is recommended that the company include sections detailing the calculations resulting in the data for each of these cells. Relevant cost items should be detailed in the rows, under the appropriate heading — examples have been provided of possible cost items. Other comparators may be included in this table; include columns as necessary.
Table 17: Direct Health Care Costs
Category Submitted Drug
Comparator(s) Time Framea
Data Sources A B Other
Drug — List priceb Drug — Typical costb Costs incurred during treatment Administration of drug (specify clinic time, outpatient visit, supplies, where relevant)
Monitoring Other costs induced through use of the drug (specify treatment of adverse events or complications, concomitant drugs, etc.)
Costs that may be affected by treatment (specify surgery, in-hospital stay)
Costs incurred beyond treatment
Medical costs (specify cost of treating disease, complications with treatment)
Total costs a Specify time frame: monthly, annual, or lifetime, etc. b As detailed in Table 16. Costs Incurred During Treatment The costs to be included under this subheading are those incurred by individuals during, for instance, the course of a clinical trial. These might include the cost of administering the drug, such as clinic time, visits to the physician, and supplies. Induced costs of treatment may include items such as additional time required to monitor patients receiving treatment, lab tests, treatment-related complications (which may include the need for additional drugs, outpatient visits, hospitalizations, etc.). Costs that may be affected by the use of a drug for a condition should also be included in this subsection, which might include the treatment of disease-related complications, hospitalizations, and emergency room visits. Where the costs have not been collected alongside the clinical trial, it should be indicated that the costs were derived by the economic model, and specific sources and assumptions should be stated in the last column.
Submission Guidelines for the CADTH Common Drug Review August 2014 53

CADTH Common Drug Review
Costs Incurred Beyond Treatment Based on extrapolation beyond the clinical trial, these are the expected costs for patients receiving the submitted drug and appropriate comparators during the specified, extended time horizon. This would include the costs of complications caused or prevented by the administration of the new drug. These costs tend to be more important when performing full economic evaluations.
Table 18: Non–Health Care Resources and Costs Category Submitted
Drug Comparator(s) Time
Framea Data
Sources A B Other Patient’s time (total) Treatment time Lost productivity Caregiver time Out-of-pocket costs (total)
(Specify individual items: travel expenses, child care, modifications to home)
a Specify time frame: monthly, annual, or lifetime, etc. Table 18 details the non–health care resources and costs associated with the submitted treatment compared with the appropriate comparators. Where non–health care costs are relevant in the economic evaluation, this table must be completed. The manufacturer must clearly state data sources and assumptions. As necessary, it is recommended that the company include sections detailing the calculations resulting in each of these cells. Where assumptions or modelling have been conducted to determine the estimates for this table, the validation process for these estimates must be described and results of the validation reported. Cells should be completed with respect to the expected cost for each item. The costs are derived by the expected number of hours spent multiplied by the wage or cost per hour. These data should be reported separately, and the sources of this information should be presented clearly. Patient’s time should be reported in terms of the time spent receiving medical care and the lost productive time due to the disease or disability, multiplied by the mean wage or expected cost per hour. Patient and caregiver time must be converted to costs. The values used for this calculation must be provided. Calculations should be provided if the estimate is not transparent. The patient’s, or caregiver’s, out-of-pocket costs should be detailed. These items might include travel expenses, child care, or modifications to the home.
Submission Guidelines for the CADTH Common Drug Review August 2014 54

CADTH Common Drug Review
APPENDIX 1: DRUG PLANS PARTICIPATING IN THE CADTH COMMON DRUG REVIEW After receiving confirmation from CADTH that category 1 requirements for a submission or resubmission are accepted for review, manufacturers are encouraged to immediately send copies of the requirements, in the quantity and format specified by each of the drug plans, as outlined in Table 19. Similarly, when CADTH has notified manufacturers that category 2 requirements are complete, copies should immediately be sent to the drug plans in accordance with Table 19. Reference to a “copy of requirements” in the table refers to copies of all of the category 1 requirements and/or category 2 requirements. For submissions filed on a pre-Notice of Compliance (NOC) basis, copies of the finalized category 1 requirements accepted by CADTH must also be sent to the drug plans. When sending copies of the category 1 and/or category 2 requirements to each of the drug plans (including any of the federal drug plans for which preparation of budget impact analyses [BIAs] is not required [e.g., Department of National Defence]), each plan is to be sent copies of all of the BIAs prepared in accordance with the submission or resubmission requirements. For example, in addition to the BIA for British Columbia, British Columbia’s drug plan must also be sent copies of the BIAs prepared for all of the other drug plans as submitted to CADTH in accordance with the Submission Guidelines for the CADTH Common Drug Review. HIV/AIDS Drug Funding Submissions — British Columbia and Alberta British Columbia and Alberta have separate review and reimbursement processes for HIV/AIDS drugs. Funding submissions for these drugs in British Columbia should also be sent to the British Columbia Centre for Excellence in HIV/AIDS. For Alberta, submissions should be sent to Alberta Health (contact information as provided in Table 19). Any questions should be directed to these organizations.
Table 19: Contact Information and Requirements for CDR-Participating Drug Plans
Jurisdictiona Contact/Send CDR Documents to: What to Send British Columbia
Director, Formulary Management Pharmaceutical Services Division Ministry of Health 3-2 1515 Blanshard Street Victoria, BC V8W 3C8 T: 250 952 1183
• 1 copy of requirementsb in hard copy
• 1 copy of requirementsb on CD or DVD
Alberta Director, Professional and Industry Relations Pharmaceutical and Supplementary Health Benefits Alberta Health 10025 Jasper Avenue 11th Floor, ATB Place North Edmonton, AB T5J 1S6 T: 780 427 2653
3 copies of requirementsb on CDs or DVDs
Submission Guidelines for the CADTH Common Drug Review August 2014 55

CADTH Common Drug Review
Table 19: Contact Information and Requirements for CDR-Participating Drug Plans
Jurisdictiona Contact/Send CDR Documents to: What to Send Saskatchewan Director, Pharmaceutical Services
Drug Plan and Extended Benefits Branch Saskatchewan Health 3475 Albert Street, 2nd Floor East Regina, SK S4S 6X6 T: 306 787 3305
• 1 copy of requirementsb on CD or DVD
Manitoba Kathy McDonald Secretary Drug Standards and Therapeutics Committee 1014-300 Carlton Street Winnipeg, MB R3B 3M9 T: 204 786 7317
• 1 copy of requirementsb in hard copy
• 1 copy of requirementsb on CD or DVD
Ontario Director, Drug Program Services Branch Ontario Public Drug Programs Ministry of Health and Long-Term Care 5700 Yonge Street, 3rd Floor Toronto, ON M2M 4K5 T: 416 327 8109
• 3 copies of requirementsb in hard copy
• pharmacoeconomic and BIA unlocked models on CD or DVD
• optional: 1 copy of requirementsc on CD or DVD
New Brunswick Executive Director Pharmaceutical Services Department of Health P.O. Box 5100 520 King Street, 6th Floor Fredericton, NB E3B 5G8 T: 506 453 3884
1 copy of requirementsb on CD or DVD
Nova Scotia Manager Insured Pharmaceutical Programs Nova Scotia Department of Health and Wellness Barrington Tower 1894 Barrington Street, 3rd Floor PO Box 488 Halifax, NS B3J 2A8 T: 902 424 1596
1 copy of requirementsb on CD
Prince Edward Island
Pharmacy Consultant Health System Planning and Development Department of Health and Wellness P.O. Box 2000, 20 Fitzroy Street Charlottetown, PE C1A 7N8 T: 902 368 4907
1 copy of requirementsb on CD or DVD
Submission Guidelines for the CADTH Common Drug Review August 2014 56

CADTH Common Drug Review
Table 19: Contact Information and Requirements for CDR-Participating Drug Plans
Jurisdictiona Contact/Send CDR Documents to: What to Send Newfoundland and Labrador
Director Pharmaceutical Services Division Department of Health and Community Services 57 Margaret’s Place St. John’s, NL A1C 3Z3 T: 709 729 6507
1 copy of requirementsb on CD or DVD
Northwest Territories
Manager Health Services Administration Territorial Services Department of Health and Social Services Bag Service #9 Inuvik, NT X0E 0T0 T: 867 777 7404
1 copy of the following requirement information on a USB flash drive: • Product monograph • Notice of compliance • Submitted price
Yukon Territory
Manager, Extended Benefits and Pharmaceutical Services Insured Health and Hearing Services (H-2) Government of Yukon 4th Floor, 204 Lambert Street (Box 2703) Whitehorse, YT Y1A 2C6 T: 867 667 5628
1 copy of requirementsb on CD or DVD
Nunavut Territory
No copies
Non-Insured Health Benefits
Manager, Pharmacy Policy Development Division Non-Insured Health Benefits First Nations and Inuit Health Branch Health Canada 200 Eglantine Driveway Postal Locator 1902A Ottawa, ON K1A 0K9 T: 613 957 7674
1 copy of requirementsb on CD or DVD
Department of National Defence
CF Drug Benefit Plan Manager Pharmacy Policy and Standards Department of National Defence 1745 Alta Vista Drive, Room 206 Ottawa, ON K1A 0K6 T: 613 945 6600 ext. 3360
1 copy of requirementsb on CD or DVD
BIA = budget impact analysis; CDR = CADTH Common Drug Review; NOC = Notice of Compliance. a For CDR-participating federal drug plans not listed in the table, manufacturers should contact the plans directly if requested to so do by
the plans. b “Copy of requirements” refers to all of the category 1 requirements accepted for review by CADTH and/or category 2 requirements for
which notification of being complete has been received from CADTH, as applicable. For submissions filed on a pre-NOC basis, copies of the finalized category 1 requirements accepted by CADTH must also be sent to the drug plans.
Submission Guidelines for the CADTH Common Drug Review August 2014 57

CADTH Common Drug Review
APPENDIX 2: KEY DEFINITIONS The following are high-level definitions for key terms used in the Procedure for the CADTH Common Drug Review and Submission Guidelines for the CADTH Common Drug Review documents. Readers should consult the appropriate sections of the documents for more detailed context as it relates to some terms.
Active Substance — a therapeutic substance that has pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease (see new active substance).
Additional Information — any information that is requested from the manufacturer by CADTH in addition to the category 1 requirements that is required to complete the review of the submission or resubmission, or to clarify information related to the submission or resubmission.
Applicant — a person, corporation, or entity eligible to file an application for a CDR submission or resubmission. The applicant could be a manufacturer, a supplier, a corporation, or entity recruited by the manufacturer or the supplier.
Application — written documentation filed by an applicant to have a drug reviewed through the CDR process.
Appropriate Comparator — typically a drug listed by one or more drug plans for the indication under review. However, the choice of appropriate comparator(s) in reviews by CDR is made on a case-by-case basis.
Budget Impact Analysis (BIA) — a forecast of the impact of listing a drug on the drug plans’ expenditures.
Business Day — any day (other than a Saturday, Sunday, statutory holiday, or company holiday) on which the CADTH office in Ottawa (Ontario, Canada) is open for business during regular business hours. Please refer to the CADTH website “Contact Us” section for a current listing of CADTH business days.
Common Drug Review (CDR) Process — a single technology drug review process by which CADTH conducts an objective, rigorous, evidence-based, health technology assessment of the relative therapeutic merits and cost-effectiveness of drugs, incorporating patient group–submitted input.
Common Drug Review (CDR) Queuing — queuing is a delay in the initiation of the review of a CDR submission or resubmission.
Common Drug Review (CDR) Review Team — a team assembled by CADTH to undertake the review of a submission or resubmission, or to prepare a report in response to a request for advice. The CDR review team may include CADTH staff, contracted reviewers, and external experts with appropriate qualifications and expertise.
Canadian Drug Expert Committee (CDEC) — an appointed, national, independent advisory committee to CADTH that makes drug-related recommendations and provides drug-related advice through the CDR and Therapeutic Review processes. The Canadian Drug Expert Committee is composed of individuals with expertise in drug therapy, drug evaluation and drug
Submission Guidelines for the CADTH Common Drug Review August 2014 58

CADTH Common Drug Review
utilization, and public members to bring a lay perspective. The Canadian Drug Expert Committee replaced the Canadian Expert Drug Advisory Committee (CEDAC) in September 2011.
Canadian Drug Expert Committee (CDEC) Brief — a compilation of the materials regarding a drug under review by CDR, prepared by CADTH staff for the members of the Canadian Drug Expert Committee. The Canadian Drug Expert Committee brief includes patient group input, CDR review report(s), manufacturer’s comments on the CDR review report(s) and the CDR review team’s responses, and the manufacturer’s executive summary.
Canadian Drug Expert Committee (CDEC) Final Recommendation — provides guidance to the drug plans participating in CDR to make a funding decision regarding the drug under review. Canadian Drug Expert Committee Final Recommendations are non-binding to the drug plans. Each drug plan makes its own drug-listing decisions based on the Canadian Drug Expert Committee Final Recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Canadian Drug Expert Committee (CDEC) Record of Advice — the advice document issued by the Canadian Drug Expert Committee in response, in cases where a request for advice has not resulted in a revised Canadian Drug Expert Committee recommendation Canadian Expert Drug Advisory Committee (CEDAC) — CEDAC was replaced by the Canadian Drug Expert Committee (CDEC) in September 2011. CEDAC was a CADTH advisory body composed of individuals with expertise in drug therapy and drug evaluation and public members. For drugs reviewed through the CDR process, CEDAC made formulary listing recommendations for use by the participating federal, provincial, and territorial publicly funded drug plans. Confidentiality Guidelines — refer to the CADTH Common Drug Review Confidentiality Guidelines document (Appendix 4).
Confidential Price — a price per unit that is submitted in confidence, as part of the CDR submission requirements and to which the provisions of the CADTH Common Drug Review Confidentiality Guidelines apply.
Conflict of Interest Guidelines — the conflict of interest guidelines adopted by CADTH.
Date of Acceptance for Review — the date on which CADTH has confirmed with the applicant that the key requirements for initiating the review process for a submission or resubmission (i.e., category 1 requirements as delineated in the Submission Guidelines for the CADTH Common Drug Review) have been met.
Date of Filing — the date on which a submission or resubmission is received by CADTH’s reception desk.
Date of Initiation of a Review — the date on which CDR review team kicks off a review. Drug — an active substance considered to be a drug under the Canadian Food and Drugs Act and Food and Drug Regulations that has been granted (or will be granted in the case of a submission filed on a pre-Notice of Compliance [NOC] basis), a Health Canada NOC or Notice of Compliance with conditions (NOC/c), and is approved for human use.
Submission Guidelines for the CADTH Common Drug Review August 2014 59

CADTH Common Drug Review
Drug Plans — the federal, provincial, and territorial drug plans participating in CDR.
Embargo Period — refers to the period of time following the issuance of an embargoed Canadian Drug Expert Committee recommendation, during which the embargoed Canadian Drug Expert Committee recommendation is neither acted on by drug plans nor is publicly available. During this period, the manufacturer may submit a request for reconsideration or a resubmission based on a reduced price, or the drug plans may submit a request for clarification.
Embargoed Canadian Drug Expert Committee (CDEC) Recommendation — an evidence-based recommendation issued by CADTH. The embargoed Canadian Drug Expert Committee recommendation is released to the manufacturer and drug plans only, and is not publicly available. The manufacturer must maintain the confidentiality of this document.
External Expert — an individual with appropriate qualifications and expertise required for some aspect of the review of the submission or resubmission, and whose services are obtained on a contract basis, as required.
Formulary — a list of drugs covered as benefits, as determined by each federal, provincial, and territorial drug plan.
Formulary Working Group (FWG) — a working group of the CADTH Drug Policy Advisory Committee (DPAC). The FWG is composed of representatives from the federal, provincial, and territorial drug plans. FWG provides advice to CADTH on pharmaceutical issues and helps with the effective jurisdictional sharing of pharmaceutical information. FWG members are observers at Canadian Drug Expert Committee meetings.
Generic Drugs — copies of Canadian reference products (i.e., Health Canada–approved brand name drugs) that demonstrate bioequivalence on the basis of pharmaceutical equivalence (i.e., they contain identical amounts of the identical active medicinal ingredients, in comparable dosage forms, but do not necessarily contain the same non-medicinal ingredients as the Canadian reference product, and the conditions of use fall with those of the Canadian reference product) and bioavailability characteristics, where applicable, with the Canadian reference product. Generic drugs are not reviewed through the CDR process.
New Active Substance — a therapeutic substance that has never before been approved for marketing in Canada in any form. It may be: • a chemical or biological substance not previously approved for sale in Canada as a drug • an isomer, derivative, or salt of a chemical substance previously approved for sale as a drug
in Canada but differing in properties regarding safety and efficacy.
New Combination Product — consists of two or more drugs that have not been previously marketed in Canada in that combination. It may consist of either two or more new drugs, two or more previously marketed drugs, or a combination of new drug(s) and previously marketed drug(s). Combination products (funded components), a category of new combination products, contain components that are already funded by drug plans and are eligible for a tailored review by CDR and for modified submission requirements.
New Drug — a therapeutic substance that has never before been approved for marketing in any form, regardless of when the Notice of Compliance or Notice of Compliance with conditions was issued. It may be: • a chemical or biological substance not previously approved for sale in Canada as a drug
Submission Guidelines for the CADTH Common Drug Review August 2014 60

CADTH Common Drug Review
• an isomer, derivative, or salt of a chemical substance previously approved for sale as a drug
in Canada but differing in properties regarding safety and efficacy.
New Indication — a disease condition for which the use of a particular drug has not previously been approved by Health Canada.
New Information — new clinical information and/or new cost information that was not part of an originally filed submission or resubmission.
Notice of Compliance (NOC) — authorization issued by Health Canada to market a drug in Canada when regulatory requirements for the safety, efficacy, and quality are met.
Notice of Compliance with conditions (NOC/c) — authorization issued by Health Canada to market a drug under the Notice of Compliance with conditions policy. This indicates that the sponsor has agreed to undertake additional studies to confirm the clinical benefit of the product.
Patient Group — an organized group of patients or caregivers in Canada.
Patient Group–Submitted Input — information, submitted by a patient group, that describes the experiences and perspectives of patients living with the condition for which a drug in a CDR submission or resubmission is indicated and the impact of drug therapy on the lives of those with that illness or condition.
Post-Notice of Compliance (NOC) — the timing of filing a CDR submission after Health Canada has granted a Notice of Compliance (NOC) or Notice of Compliance with conditions (NOC/c) for the indication(s) to be reviewed under the CDR process.
Pre-Notice of Compliance (NOC) — the timing of filing a CDR submission before Health Canada has granted a Notice of Compliance (NOC) or Notice of Compliance with conditions (NOC/c) for the indication(s) to be reviewed under the CDR process, and for which the anticipated date of NOC or NOC/c is within 90 calendar days of the submission being filed.
Priority Review — a preferred status in the review queue for drugs meeting the CDR priority review criteria. All steps in the CDR process are completed and timelines are not truncated.
Queuing — see CDR Queuing
Reasons for Recommendation — these represent the key considerations and rationale used by the Canadian Drug Expert Committee in formulating the recommendation.
Request for Advice — a written request made by drug plans for Canadian Drug Expert Committee advice regarding a previous CEDAC or CDEC Final Recommendation. A request for advice can result in a revised Canadian Drug Expert Committee recommendation or a CDEC Record of Advice.
Request for Clarification — a written request from drug plans for clarification of an embargoed Canadian Drug Expert Committee recommendation.
Request for Reconsideration — a written request from a manufacturer for an embargoed Canadian Drug Expert Committee recommendation to be reconsidered.
Submission Guidelines for the CADTH Common Drug Review August 2014 61

CADTH Common Drug Review
Request for Withdrawal — a written request by an applicant to withdraw a submission or resubmission from the review process. These may be filed any time before the CDEC Final Recommendation has been issued.
Resubmission — an application filed to review a previous submission or resubmission for the same indication(s) on the basis of new information after a CDEC Final Recommendation has been issued.
Review Team — see CDR Review Team
Standard Common Drug Review (CDR) Review — consists of the CDR review team conducting a systematic review of clinical evidence provided by the manufacturer along with studies identified through its independent, systematic literature search, and an appraisal of the manufacturer-provided pharmacoeconomic evaluation.
Stopped Review — the cessation of the review of a submission or resubmission under the CDR process before all steps of the review process are completed. Work on a stopped submission or resubmission does not resume.
Submission — an application filed for an initial review of a drug under the CDR process for a specific indication(s), for any of the following CDR–eligible drug submission types: new drug, drug with a new indication, new combination product, new combination product (funded components) or a subsequent entry biologic.
Submission Guidelines — the guidelines adopted by CADTH that outline how submissions and resubmissions from manufacturers must be prepared and submitted.
Submission or Resubmission Requirements — information that is required by CADTH to review a submission or resubmission through the CDR process.
Submission Status Report — a document posted on the CADTH website for every CDR submission, resubmission, or request for advice filed with CADTH that provides key targeted time frames and the status of the review. The report is generally updated on a weekly basis.
Submitted Price — the price per unit that is submitted to CDR and that must not be exceeded for any of the drug plans following release of a CDEC Final Recommendation,14 irrespective of the type of recommendation made and whether or not the Canadian Drug Expert Committee criteria for listing are the same as the criteria requested by the manufacturer.
Subsequent Entry Biologic (SEB) — a biologic drug (i.e., a drug derived from living sources versus a chemically synthesized drug), demonstrating a high degree of similarity to an already authorized biologic drug (i.e., a “reference product” that has been authorized in Canada, or in some circumstances can be an authorized non-Canadian biologic from a jurisdiction that has an established relationship with Health Canada). Similarity between an SEB and the reference product is established in accordance with Health Canada’s Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs), for the authorized indications.
14 A CDEC Final Recommendation is non-binding on the drug plans. Each of the drug plans subsequently makes its own drug-listing decisions based on the Canadian Drug Expert Committee recommendation in addition to other factors, including the plan’s mandate, jurisdictional priorities, and financial resources.
Submission Guidelines for the CADTH Common Drug Review August 2014 62

CADTH Common Drug Review
Suspended Review — refers to the temporary cessation of the review of a submission or resubmission under the CDR process. This occurs if questions or issues arise outside of the regular review process or if the CDR review team is unable to perform a thorough assessment of the submission or resubmission due to incomplete or non-transparent information. Once the issue is resolved, the review proceeds from the point at which it was suspended. The applicant is not required to file a submission or resubmission to re-initiate the review.
Tailored Common Drug Review (CDR) Review — consists of the CDR review team conducting an appraisal of the clinical evidence and pharmacoeconomic evaluation filed by the manufacturer using a CADTH-provided review template that is specific to the type of drug product to be reviewed.
Therapeutic Review — a review of publicly available evidence regarding a single drug, a therapeutic category of drugs, or a pharmacologic class of drugs. The scope and depth of the review are determined by jurisdictional needs. An important characteristic of a therapeutic review is that it is conducted to coincide with a CDR submission review, and thus, informs the CDR submission review and listing recommendation and informs drug plan decisions.
Submission Guidelines for the CADTH Common Drug Review August 2014 63

CADTH Common Drug Review
APPENDIX 3: CADTH CONTACT AND CADTH COMMON DRUG REVIEW APPLICATION FILING INFORMATION Table 20: How and Where to Direct CDR-Related Inquiries or CDR Applications Type of Inquiry or CADTH CDR Application
How and Where to Direct
• General CDR inquiries • CDR process or procedure-
related inquiries
In writing to: Email: [email protected] Fax: 613-226-5392 Mail15: Central Intake CADTH 600-865 Carling Avenue Ottawa, ON K1S 5S8
Filing CDR applications for a submission or resubmission
By registered mail, courier, or in person to: Central Intake CADTH 600-865 Carling Avenue Ottawa, ON K1S 5S8
Inquiries regarding a CDR application for which the review has been initiated
By email to:
• the designated submission coordinator contact provided by CADTH in the category 1 requirement acceptance letter
CDR = CADTH Common Drug Review.
Table 21: Delivery Times Means of Delivery When Considered to Have Been Delivered By courier, registered mail, regular mail, in person
• On the day of receipt by CADTH’s reception desk
Email or fax • On the day of transmittal if sent during CADTH business hours • On the business day that normal business hours next occur, if sent
outside of CADTH business hours For CADTH business hours and holiday schedule, please check the CADTH website, www.cadth.ca, under “Contact Us.”
15 If the party sending a CADTH Common Drug Review–related inquiry or other correspondence, or filing a CADTH Common Drug Review application knows, or should reasonably know, of any disruption or difficulty with the postal system that might affect the delivery of mail, any such inquiry, correspondence, or CADTH Common Drug Review application should not be mailed, and should instead be delivered to CADTH by electronic means, if applicable (e.g., resubmissions based on a reduced price during the embargo period may be submitted by email; CADTH Common Drug Review applications cannot be filed electronically), by courier, or in-person delivery.
Submission Guidelines for the CADTH Common Drug Review August 2014 64

CADTH Common Drug Review
APPENDIX 4: CADTH COMMON DRUG REVIEW CONFIDENTIALITY GUIDELINES These guidelines are intended to ensure the confidential information obtained for the purposes of CDR is protected and handled in a consistent manner by CADTH. By filing a submission or supplying other information to CADTH for the CDR process, a manufacturer consents to these guidelines and agrees to be bound by the terms and conditions herein. Confidential Information Manufacturer’s must clearly and obviously label or mark any confidential information as such. Only information labelled in this manner will be considered as confidential, and CADTH will only be bound by the terms of these guidelines for labelled information. Confidential information includes any non-public scientific, technical, or commercial information about a manufacturer’s business or a manufacturer’s product received through the exchange of information as part of the CDR process, but which does not include information that:
a) was already in the possession of CADTH, external reviewer(s) assigned to review the submission or resubmission, Canadian Drug Expert Committee members, external experts (when contracted to provide specific information in relation to the submission or resubmission), federal, provincial, territorial (F/P/T) governments (including their agencies and departments) or the Patented Medicine Prices Review Board (PMPRB); without restriction as to its use or disclosure
b) is or becomes available to the general public other than as a result of a breach of the procedures contained herein (information available to the general public includes but is not limited to published articles, drug prices, and product monographs)
c) a third party (who is not under any obligation as to confidentiality or non-disclosure) rightfully discloses to CADTH, external reviewer(s) assigned to review the submission or resubmission, Canadian Drug Expert Committee members, external experts (when contracted to provide specific information in relation to the submission or resubmission), F/P/T governments (including their agencies and departments), or PMPRB; without restriction as to its use or disclosure.
Confidential information also includes information about a manufacturer’s product that is provided to CADTH by Health Canada, with authorization from the manufacturer. Handling Confidential Information
1. Responsibilities of CADTH a) CADTH will use reasonable care to prevent the unauthorized use, disclosure, publication, or
dissemination of confidential information that is labelled confidential and included in and related to submissions and resubmissions
b) CADTH will not disclose confidential information in and related to a submission or resubmission to any third party except as permitted by these guidelines, or as required by law or by order of a legally qualified court or tribunal
Submission Guidelines for the CADTH Common Drug Review August 2014 65

CADTH Common Drug Review
c) CADTH will use the submission or resubmission and confidential information solely for the
purpose of carrying out its responsibilities with respect to CDR
d) CADTH shall utilize secure filing and storage, websites, and tracking processes for handling submissions or resubmissions and confidential information.
2. Release of Manufacturer’s Information a) CADTH may release a manufacturer’s submission or resubmission, including confidential
information, to the following “authorized recipients”: • CADTH staff • CDR review team members (including contractors and clinical experts) • Canadian Drug Expert Committee members • F/P/T government representatives (including their agencies and departments) • PMPRB representatives.
b) While CADTH is an independent not-for-profit organization and is therefore not subject to access to information legislation, some of the authorized recipients listed above have their own confidentiality procedures and are subject to freedom of information and access to information legislation over which CADTH has no control. By filing a submission or resubmission, manufacturers consent to their information, including confidential information, being exchanged with F/P/T governments (including their agencies and departments) and the PMPRB by signing a template letter provided in the Submission Guidelines for the CADTH Common Drug Review.
c) CADTH staff members are required, as a condition of employment, to comply with CADTH’s confidentiality requirements, Code of Conduct, and Conflict of Interest Guidelines.
d) All CDR review team members, Canadian Drug Expert Committee members, and external experts must abide by the confidentiality clauses contained in their Code of Conduct and/or Conflict of Interest Guidelines and/or contracts.
e) CADTH’s Drug Policy Advisory Committee (DPAC) Formulary Working Group (FWG) members are required to sign a non-disclosure agreement requiring them to comply with these confidentiality guidelines.
3. Documents Shared with Authorized Recipients a) The documents which CADTH may share with the authorized recipients include:
• Manufacturer’s submission or resubmission • Redacted and unredacted CDR review report(s)16 • Manufacturer’s comments about CDR review report(s) • CDR review team’s responses to manufacturer’s comments • Embargoed Canadian Drug Expert Committee recommendation • CDEC Final Recommendation • CDEC Record of Advice • CDEC brief
16 The term “CDR review report(s)” in this document refers to the CADTH Common Drug Review Clinical Review Report and the Common Drug Review Pharmacoeconomic Report typically prepared for a standard CDR review, and/or the combined CADTH Common Drug Review Clinical and Pharmacoeconomic Review Report prepared for a tailored CDR review, and/or the Common Drug Review Request for Advice report prepared in response to a request for advice.
Submission Guidelines for the CADTH Common Drug Review August 2014 66

CADTH Common Drug Review
• CDEC reconsideration brief.
b) CADTH provides the following documents to the submitting manufacturer. The manufacturer
shall maintain the confidentiality of these documents. • CDR review report(s) • Embargoed Canadian Drug Expert Committee recommendation • Canadian Drug Expert Committee recommendation on reconsideration • Technical version of the CDEC Final Recommendation (until posted on CADTH website) • Plain language version of the CDEC Final Recommendation (until posted on CADTH
website) • Response to request for clarification (if applicable).
c) The documents which CADTH may post on its website include:
• Tracking document indicating the status of a CDR review, including a submission filed on a pre-NOC basis.
• CDEC Final Recommendation with confidential information redacted (both the technical version and the plain language version).
• CDR review report(s) with confidential information redacted, as per the current Procedure for the CADTH Common Drug Review.
• CDEC Record of Advice.
4. Making Reference to Confidential Information in Public CADTH Common Drug Review Documents
CADTH may use confidential information supplied by the manufacturer in the preparation of the CDR review report(s), Canadian Drug Expert Committee recommendations, CDR Request for Advice reports, and CDEC Record of Advice documents. Before these documents are posted in the public domain, the manufacturer will be asked to identify any confidential information for redaction. The following principles and provisions shall apply:
The following provisions will apply to any confidential information which the manufacturer requests to be redacted from the CDR review report(s), CDEC Final Recommendation, or the CDEC Record of Advice: • CADTH will redact the confidential information using redaction software and will indicate that
the manufacturer requested that the confidential information be redacted, pursuant to the CADTH Common Drug Review Confidentiality Guidelines. CADTH may provide a general description of the type of information (e.g., confidential
price, unpublished study results) that was redacted. • Confidential submitted prices will be redacted; however, the outputs of economic models
(e.g., incremental cost-effectiveness ratios) are not considered confidential and will not be redacted.
• In the case of a disagreement expressed by the manufacturer regarding redactions made in the CDR review report(s), CADTH may require additional time to resolve the disagreement in consultation with the manufacturer. This additional time could delay publication of the CDR review report(s); however, any such delays will not affect the timelines for issuing the CDEC Final Recommendation.
Submission Guidelines for the CADTH Common Drug Review August 2014 67

CADTH Common Drug Review
5. Archiving of Documents Containing Confidential Information a) CADTH may retain two master copies of electronic documents, including documents
containing confidential information associated with the review of a drug, for as long as there may be a need to consult them, as follows: • one copy is kept securely on a CADTH server • one copy is kept on the manufacturer-provided media (CDs, DVDs or USB flash drives)
in secure storage. b) One complete set of all hard copy documents,17 including documents containing confidential
information associated with the review of a drug, is kept on file by CADTH in secure storage for as long as there may be a need to consult them.
c) CADTH will determine at its sole discretion if there is a need to consult this information.
d) CADTH staff undertakes regular reviews of archived material. Any material that CADTH determines to be no longer required is disposed of, as described in section 6.
6. Disposal of Documents Containing Confidential Information a) CADTH disposes of extra hard copies17 of the submission or resubmission.
b) CADTH disposes of electronic documents on any manufacturer-provided media (CDs, DVDs, USB flash drives), other than the copies which are retained as per paragraph 5 a) of these guidelines.
c) At the completion of a review, reviewers are directed to delete all electronic documentation that was provided during the review, which includes confidential information that may have been stored on the hard drive of a computer or in emails.
17 If applicable. CADTH discontinued the requirement for hard copies of category 1 requirements for all CDR submissions received on or after October 9, 2013.
Submission Guidelines for the CADTH Common Drug Review August 2014 68

CADTH Common Drug Review
APPENDIX 5: EXAMPLE OF CONSORT DIAGRAM This is an example of the type of information that is required for the CDR process. It can be provided in a different format as long as all of the information shown in the flowchart below is provided.
Figure 2: Example of CONSORT Flowchart
Flow Diagram of the progress through the phases of a randomized trial (i.e., enrolment, intervention allocation, follow-up, and data analysis)
For Reference: Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. PloS Med [Internet]. 2010 [cited 2014 Jul17];7(3):e1000251. Available from: http://www.plosmedicine.org/article/info%3Adoi%2F10.1371%2Fjournal.pmed.1000251
Assessed for eligibility (n = ...)
Excluded (n = ...) • Not meeting inclusion criteria
(n = ...) • Declined to participate (n = ...) • Other reasons (n = ...)
Allocated to intervention (n = ...) • Received allocated intervention
(n = ...) • Did not receive allocated intervention
(give reasons) (n = ...)
Lost to follow up (n = ...) (give reasons)
Discontinued intervention (n = ...) (give reasons)
Analyzed (n = ...) • Excluded from analysis
(give reasons) (n = ...)
Randomized (n = ...)
Ana
lysi
s
Fo
llow
-up
Allo
catio
n
E
nrol
lmen
t
Allocated to intervention (n = ...) • Received allocated intervention
(n = ...) • Did not receive allocated intervention
(give reasons) (n = ...)
Lost to follow up (n = ...) (give reasons)
Discontinued intervention (n = ...) (give reasons)
Analyzed (n = ...) • Excluded from analysis
(give reasons) (n = ...)
Submission Guidelines for the CADTH Common Drug Review August 2014 69

CADTH Common Drug Review
APPENDIX 6: PREPARING LISTS OF REFERENCES These instructions may be used to assist applicants in preparing reference lists in the Citing Medicine format18 for the following CDRcategory 1 or 2 requirements:
Category 1 Requirements Category 2 Requirements • Executive Summary • Clinical Studies and Errata
• BIA Supporting Documentation
• Editorials • New data after NDS • Validity of Outcomes
BIA = budget impact analysis; NDS = new drug submission. Preparing the List of References: • Use 11-point Arial font. • Provide a numbered list of all references included in the folder (e.g., Clinical Studies) using
the Citing Medicine format. Example: 1. Klarenbach S, Cameron C, Singh S, Ur E. Cost-effectiveness of second-line
antihyperglycemic therapy in patients with type 2 diabetes mellitus inadequately controlled on metformin. CMAJ [Internet]. 2011 Oct 3 [cited 2011 Nov 8];183(16):E1213-E1220. Available from: http://www.cmaj.ca/content/183/16/E1213.long
2. Cameron C, Coyle D, Ur E, Klarenbach S. Cost-effectiveness of self-monitoring of blood glucose in patients with type 2 diabetes mellitus managed without insulin. CMAJ [Internet]. 2010 Jan 12 [cited 2010 Jan 15];182(1):28-34. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2802601
• Save the reference list as a PDF or Microsoft Word file (e.g., doc or docx) with the following
file naming structure (please note the initial underscore in the file name): _List of References
Numbering the Files in Each Folder • Prefix each of the file names in the folder using the numbers used in the reference list.
Example: 1 - Klarenbach 2011.pdf 2 - Cameron 2010.pdf
18 The following website provides further information on the Citing Medicine format for various types of citations: http://www.nlm.nih.gov/pubs/formats/recommendedformats.html (accessed August 27, 2014).
Submission Guidelines for the CADTH Common Drug Review August 2014 70

CADTH Common Drug Review
APPENDIX 7: LIST OF TEMPLATES Pre-submission Phase • CADTH Common Drug Review Voluntary Pipeline Notification Template • Pre-submission Meeting Request Form • CADTH Common Drug Review Mandatory Notification Submission Template • CADTH Common Drug Review Mandatory Notification Resubmission Template • New Combination Product Considerations Form Templates for Category 1 Requirements • Application Overview Template • Executive Summary Template for a Submission • Executive Summary Template for Resubmission • Table of Studies Template • Letter Confirming Disclosure of all Known Unpublished Studies • Number of Patients Accessing New Drug • Commitment to Honour Submitted Price Letter • Unrestricted Sharing of Information Letter • Letter for Sending NOC or NOC/c to CADTH • Letter for Finalized Category 1 Requirements
Tailored CDR Review Templates • Subsequent Entry Biologic Submission Template • New Combination Product Submission Template
Priority Review Request • Priority Review Application Template
Submission Guidelines for the CADTH Common Drug Review August 2014 71

CADTH Common Drug Review
APPENDIX 8: CHECKLISTS FOR PREPARING CADTH COMMON DRUG REVIEW APPLICATIONS Manufacturers may use the checklists used by CADTH, as provided in this appendix, to help ensure that all submission or resubmission requirements for a CDR application have been included. Category 1 Requirements
A. New drug, drug with a new Indication, or new combination product submission filed on a pre-NOC basis
B. New drug, drug with a new indication, or new combination product submission filed on a post-NOC basis
C. New combination product (funded components or CADTH-designated tailored CDR review) filed on a pre-NOC basis
D. New combination product (funded components or CADTH-designated tailored CDR review) submission filed on a post-NOC basis
E. Subsequent entry biologic submission filed on a pre-NOC basis F. Subsequent entry biologic submission filed on a post-NOC basis G. All resubmissions H. Priority review applications Category 2 Requirements I. All submissions J. All resubmissions
CDR = CADTH Common Drug Review; NOC = Notice of Compliance. Various hyperlinked templates (e.g., for letters, tailored CDR reviews, tables) are provided throughout the Submission Guidelines for the CADTH Common Drug Review and are to be used when filing a CDR application for a submission or resubmission. These templates are also available on the CADTH website.
Submission Guidelines for the CADTH Common Drug Review August 2014 72

CADTH Common Drug Review
A. Category 1 Requirements for a Standard CDR Review: New Drug, Drug with a
New Indication, or New Combination Product Submission Filed on a Pre-NOC Basis
Table 22: Category 1 Requirements for a Standard CDR Review
Submission Filed on a Pre-NOC Basis Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter • Clear description of submission filed ☐
• Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Anticipated date of NOC for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20
business days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the anticipated or current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is
included ☐
Executive Summary • Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Product Monograph At the time of filing: • A copy of the most recent draft product monograph ☐ After NOC or NOC/c is issued: • Draft product monograph with tracked clinical and label review
changes up to time of Health Canada approval ☐
• Clean and dated version of Health Canada–approved product monograph
☐
Health Canada Documentation NOC At the time of filing:
• A placeholder document indicating the anticipated NOC date for the indications(s) to be reviewed by CDR
☐
After NOC or NOC/c is issued: • Copy of NOC or NOC/c granted for the indication(s) under review ☐ • Letter of Undertaking (only if NOC/c granted) ☐
Health Canada Reviewers’ Report
At the time of filing: • A placeholder document indicating that a copy of Health
Canada’s PSEA or BSEAR, as applicable, will be provided as ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 73

CADTH Common Drug Review
Table 22: Category 1 Requirements for a Standard CDR Review
Submission Filed on a Pre-NOC Basis Requirement Specific Items and Criteria Included
soon as it is available After NOC or NOC/c is issued: • A copy of Health Canada’s PSEA or BSEAR, as applicable, as
soon as it is available ☐
Clarifaxes At time of filing: • Summary table of any clinical Clarifaxes up to time of filing ☐ Ongoing basis until NOC or NOC/c is issued: • Revised clinical Clarifax summary table(s) ☐
Efficacy, Effectiveness, and Safety Information Common Technical Document
• Section 2.5 ☐ • Section 2.7.1 ☐ • Section 2.7.3 ☐ • Section 2.7.4 ☐ • Section 5.2 • Or a placeholder document for any section(s) not required by
Health Canada for the regulatory submission, if applicable
☐ ☐
Clinical Studies and Errata
• Reference list of key clinical issues studies (published and unpublished) and any errata
☐
• Copies of studies addressing key clinical issues • Copies of any errata (or placeholder document stating none
found)
☐ ☐
Table of Studies • Completed table of studies template ☐ Editorials • Reference list of editorial articles (or placeholder document
stating none found) • Copies of editorial articles
☐ ☐
Search Strategies • Literature search strategies • Included all search terms and names of databases searched
☐
Disclosure of Studies • Signed declaration that all known unpublished studies have been disclosed (used the provided letter template)
☐
CONSORT Diagrams • For pivotal trials in Health Canada submission ☐ • For other key trials included in the submission (as per first
section of the “table of studies” template) ☐
New Data • Reference list of new data (or statement that none available) ☐ • Copies of new data available ☐
Validity of Outcome Measures
• Reference list (or statement that none available) ☐ • Copies of validity of outcome measure references available ☐
Economic and Epidemiologic Information Pharmacoeconomic Evaluation
• Pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR
☐
Economic Model • Three separate CDs, DVDs, or USB flash drives, each with a copy of the unlocked and fully executable economic model
☐
• Required economic model supporting documentation ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 74

CADTH Common Drug Review
Table 22: Category 1 Requirements for a Standard CDR Review
Submission Filed on a Pre-NOC Basis Requirement Specific Items and Criteria Included Number of Patients Accessing a New Drug
• Number of patients accessing the new drug up to within 20 business days of filing the submission. (Note: this requirement is only for a new drug submission or a new combination product submission if one of the components is a new drug.) Used the provided template.
☐
Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced ☐ Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used the provided letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
Additional Letters for Submissions Filed on Pre-NOC Basis Letter for Sending NOC or NOC/c to CADTH
After NOC or NOC/c is issued: • A signed letter indicating whether any wording changes to the
Health Canada–approved final product monograph result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis (used the provided letter template)
☐
Letter for Finalized Category 1 Requirements
After NOC or NOC/c is issued: • A signed letter confirming that all finalized versions of category 1
requirements for the submission have been provided to CADTH (used the provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; PSEA = Pharmaceutical Safety and Efficacy Assessment.
Submission Guidelines for the CADTH Common Drug Review August 2014 75

CADTH Common Drug Review
B. Category 1 Requirements for Standard CDR Review: New Drug, Drug with a
New Indication, or New Combination Product Submission Filed on a Post-NOC Basis
Table 23: Category 1 Requirements for a Standard CDR Review
Filed on a Post-NOC Basis Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter • Clear description of submission filed ☐
• Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Date of NOC or NOC/c for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐
If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is
included ☐
Executive Summary • Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Product Monograph • A copy of the most current version of the Health Canada–approved product monograph
☐
Health Canada Documentation NOC • Copy of NOC or NOC/c granted for the indication(s) to be
reviewed ☐
• Letter of Undertaking (only if NOC/c granted) ☐ Health Canada Reviewers’ Report
• A copy of Health Canada’s PSEA or BSEAR, as applicable, at time of filing, or placeholder document indicating it will be provided as soon as it is available
☐
Clarifaxes • Summary table of any clinical Clarifaxes up to the time of NOC or NOC/c being issued
☐
Efficacy, Effectiveness, and Safety Information Common Technical Document
• Section 2.5 ☐ • Section 2.7.1 ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 76

CADTH Common Drug Review
Table 23: Category 1 Requirements for a Standard CDR Review
Filed on a Post-NOC Basis Requirement Specific Items and Criteria Included
• Section 2.7.3 ☐ • Section 2.7.4 ☐ • Section 5.2 • Or a placeholder document for any section(s) not required by
Health Canada for the regulatory submission, if applicable
☐
Clinical Studies and Errata
• Reference list of key clinical issues studies (published and unpublished) and any errata
☐
• Copies of studies addressing key clinical issues • Copies of any errata (or placeholder document stating none found)
☐ ☐
Table of Studies • Completed table of studies template ☐ Editorials • Reference list of editorial articles (or placeholder document
stating none found) • Copies of editorial articles
☐ ☐
Search Strategies • Literature search strategies • Included all search terms and names of databases searched
☐
Disclosure of Studies
• Signed declaration that all known unpublished studies have been disclosed (used the provided letter template)
☐
CONSORT Diagrams
• For pivotal trials in Health Canada submission ☐ • For other key trials included in the submission (as per first section
of the “table of studies” template) ☐
New Data • Reference list of new data (or statement that none available) ☐ • Copies of new data available ☐
Validity of Outcome Measures
• Reference list (or statement that none available) ☐ • Copies of validity of outcome measure references available ☐
Economic and Epidemiologic Information Pharmacoeconomic Evaluation
• Pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR
☐
Economic Model • Three separate CDs, DVDs, or USB flash drives, each with a copy of the unlocked and fully executable economic model
☐
• Required economic model supporting documentation ☐ Number of Patients Accessing a New Drug
• Number of patients accessing the new drug up to within 20 business days of filing the submission. (Note: this requirement is only for a new drug submission or a new combination product submission if one of the components is a new drug.) Used the provided template
☐
Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced
☐
Submission Guidelines for the CADTH Common Drug Review August 2014 77

CADTH Common Drug Review
Table 23: Category 1 Requirements for a Standard CDR Review
Filed on a Post-NOC Basis Requirement Specific Items and Criteria Included Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used the provided letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; PSEA = Pharmaceutical Safety and Efficacy Assessment.
Submission Guidelines for the CADTH Common Drug Review August 2014 78

CADTH Common Drug Review
C. Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated for Tailored CDR Review) Filed on a Pre-NOC Basis
Table 24: Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Pre-NOC Basis
Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter
• Clear description of submission filed ☐ • Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Anticipated date of NOC for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the anticipated or current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is
included ☐
Executive Summary
• Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Tailored CDR Review Template
• Completed new combination product submission template ☐
Product Monograph
At the time of filing: • A copy of the most recent draft product monograph ☐ After NOC or NOC/c is issued: • Draft product monograph with tracked clinical and label review
changes up to time of Health Canada approval ☐
• Clean and dated version of Health Canada–approved product monograph
☐
Health Canada Documentation NOC At the time of filing:
• A placeholder document indicating the anticipated NOC date for the indications(s) to be reviewed by CDR
☐
After NOC or NOC/c is issued: • Copy of NOC or NOC/c granted for the indication(s) under
review ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 79

CADTH Common Drug Review
Table 24: Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Pre-NOC Basis
Requirement Specific Items and Criteria Included • Letter of Undertaking (only if NOC/c granted) ☐
Health Canada Reviewers’ Report
At the time of filing: • A document stating that a copy of Health Canada’s PSEA or
BSEAR, as applicable, will be provided as soon as it is available ☐
After NOC or NOC/c is issued: • A copy of Health Canada’s PSEA or BSEAR, as applicable, as
soon as it is available ☐
Clarifaxes At time of filing: • Summary table of any clinical Clarifaxes up to time of filing ☐ Ongoing basis until NOC or NOC/c is issued: • Revised clinical Clarifax summary table(s) ☐
Clinical Study Clinical Study • Copy of one clinical study using the new combination product, not
the individual components; this can be a pharmacokinetic study ☐
Disease Prevalence and Incidence Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced ☐ Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used the provided letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
Additional Letters for Submissions Filed on Pre-NOC Basis Letter for Sending NOC or NOC/c to CADTH
After NOC or NOC/c is issued: • A signed letter indicating whether any wording changes to the
Health Canada-approved final product monograph result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis (used the provided letter template)
☐
Letter for Finalized Category 1 Requirements
After NOC or NOC/c is issued: • A signed letter confirming that all finalized versions of category 1
requirements for the submission have been provided to CADTH (used the provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; PSEA = Pharmaceutical Safety and Efficacy Assessment.
Submission Guidelines for the CADTH Common Drug Review August 2014 80

CADTH Common Drug Review
D. Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Post-NOC Basis
Table 25: Category 1 Requirements for a Tailored CDR Review: New Combination Product (Funded Components or CADTH-Designated Tailored CDR Review)
Submission Filed on a Post-NOC Basis Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter
• Clear description of submission filed ☐ • Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Date of NOC or NOC/c for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is
included ☐
Executive Summary
• Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Tailored CDR Review Template
• Completed new combination product submission template ☐
Product Monograph
• A copy of the most current version of the Health Canada-approved product monograph
☐
Health Canada Documentation NOC • Copy of NOC or NOC/c granted for the indication(s) to be reviewed ☐
• Letter of Undertaking (only if NOC/c granted) ☐ Health Canada Reviewers’ Report
• A copy of Health Canada’s PSEA or BSEAR, as applicable, at time of filing, or placeholder document indicating it will be provided as soon as it is available
☐
Clarifaxes • Summary table of any clinical Clarifaxes up to the time of NOC for NOC/c being issued
☐
Clinical Study Clinical Study • Copy of one clinical study using the new combination product, not
the individual components; this can be a pharmacokinetic study
☐
Submission Guidelines for the CADTH Common Drug Review August 2014 81
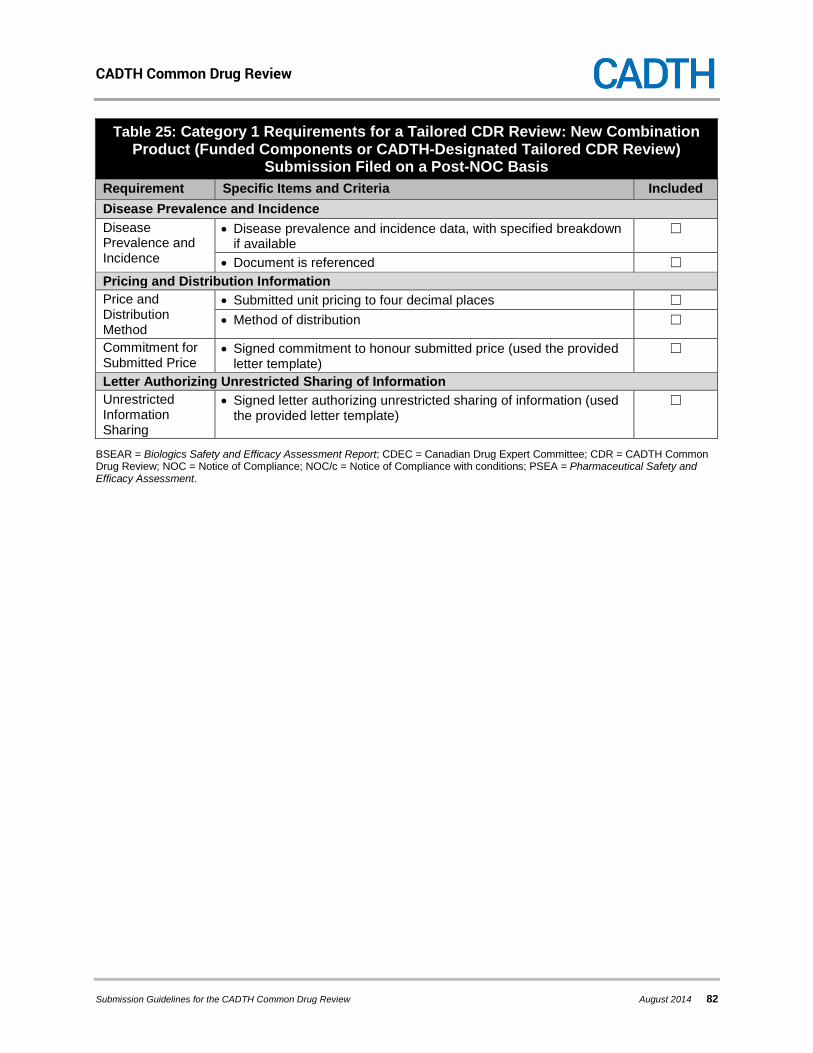
CADTH Common Drug Review
Table 25: Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated Tailored CDR Review) Submission Filed on a Post-NOC Basis
Requirement Specific Items and Criteria Included Disease Prevalence and Incidence Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced ☐ Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used the provided letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; PSEA = Pharmaceutical Safety and Efficacy Assessment.
Submission Guidelines for the CADTH Common Drug Review August 2014 82

CADTH Common Drug Review
E. Category 1 Requirements for a Tailored CDR Review: Subsequent Entry
Biologic Submission Filed on a Pre-NOC Basis
Table 26: Category 1 Requirements for a Tailored CDR Review: SEB Submission Filed on a Pre-NOC Basis
Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter
• Clear description of submission filed ☐ • Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Anticipated date of NOC for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the anticipated or current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is included ☐
Executive Summary
• Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Tailored CDR Review Template
• Completed SEB submission template
Product Monograph
At the time of filing: • A copy of the most recent draft product monograph ☐ After NOC or NOC/c is issued: • Draft product monograph with tracked clinical and label review
changes up to time of Health Canada approval ☐
• Clean and dated version of Health Canada-approved product monograph
☐
Health Canada Documentation NOC At the time of filing:
• A placeholder document specifying the anticipated NOC date for the indications(s) to be reviewed by CDR
☐
After NOC or NOC/c is issued: • Copy of NOC or NOC/c granted for the indication(s) under review ☐ • Letter of Undertaking (only if NOC/c granted) ☐
Health Canada Reviewers’ Report
At the time of filing: • A placeholder document indicating that a copy of Health Canada’s
BSEAR will be provided as soon as it is available ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 83

CADTH Common Drug Review
Table 26: Category 1 Requirements for a Tailored CDR Review:
SEB Submission Filed on a Pre-NOC Basis Requirement Specific Items and Criteria Included
After NOC or NOC/c is issued: • A copy of Health Canada’s BSEAR as soon as it is available ☐
Clarifaxes At time of filing: • Summary table of any clinical Clarifaxes up to time of filing ☐ Ongoing basis until NOC or NOC/c is issued: • Revised clinical Clarifax summary table(s) ☐
Efficacy, Effectiveness, and Safety Information Common Technical Document
• Section 2.3 ☐ • Section 2.5 ☐ • Section 2.7.1 ☐ • Section 2.7.2 ☐ • Section 2.7.3 ☐ • Section 2.7.4 ☐ • Section 5.2 • Or a placeholder document for any section(s) not required by
Health Canada for the regulatory submission, if applicable
☐ ☐
Clinical Studies and Errata
• Reference list of key clinical issues studies (published and unpublished) and any errata
☐
• Copies of studies addressing key clinical issues • Copies of any errata (or placeholder document stating none found)
☐ ☐
Table of Studies • Completed table of studies (used the provided letter template) ☐ Editorials • Reference list of editorial articles (or placeholder document stating
none found) • Copies of editorial articles
☐ ☐
Search Strategies
• Literature search strategies • Included all search terms and names of databases searched
☐
Disclosure of Studies
• Signed declaration that all known unpublished studies have been disclosed (used the provided letter template)
☐
CONSORT Diagrams
• For pivotal trials in Health Canada submission ☐ • For other key trials included in the submission (as per first section
of the “table of studies” template) ☐
New Data • Reference list of new data (or placeholder document stating none available)
☐
• Copies of new data available ☐ Validity of Outcome Measures
• Reference list (or placeholder document stating none available) ☐ • Copies of validity of outcome measure references available ☐
Disease Prevalence and Incidence Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced
☐
Submission Guidelines for the CADTH Common Drug Review August 2014 84

CADTH Common Drug Review
Table 26: Category 1 Requirements for a Tailored CDR Review:
SEB Submission Filed on a Pre-NOC Basis Requirement Specific Items and Criteria Included Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price • Used the provided letter template
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
Additional Letters for Submissions Filed on Pre-NOC Basis Letter for Sending NOC or NOC/c to CADTH
After NOC or NOC/c is issued: • A signed letter indicating whether any wording changes to the
Health Canada–approved final product monograph result in revisions to the clinical or pharmacoeconomic information filed on a pre-NOC basis (used the provided letter template)
☐
Letter for Finalized Category 1 Requirements
After NOC or NOC/c is issued: • A signed letter confirming that all finalized versions of category 1
requirements for the submission have been provided to CADTH (used the provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; SEB = subsequent entry biologic.
Submission Guidelines for the CADTH Common Drug Review August 2014 85

CADTH Common Drug Review
F. Category 1 Requirements for a Tailored CDR Review: Subsequent Entry
Biologic Submission Filed on a Post-NOC Basis
Table 27: Category 1 Requirements for a Tailored CDR Review: SEB Submission Filed on a Post-NOC Basis
Requirement Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter
• Clear description of submission filed ☐ • Confirmation that all requirements have been included ☐ • The indication(s) to be reviewed ☐ • Date of NOC for indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the anticipated or current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is
included ☐
Executive Summary
• Completed executive summary template for a submission ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Tailored CDR Review Template
• Completed SEB submission template ☐
Product Monograph
• A copy of the most current version of the Health Canada–approved product monograph
☐
Health Canada Documentation NOC • Copy of NOC or NOC/c granted for the indication(s) to be
reviewed ☐
• Letter of Undertaking (only if NOC/c granted) ☐ Health Canada Reviewers’ Report
• A copy of Health Canada’s BSEAR at time of filing, or placeholder document indicating it will be provided as soon as it is available
☐
Clarifaxes • Summary table of any clinical Clarifaxes up to the time of NOC or NOC/c being issued
☐
Efficacy, Effectiveness, and Safety Information Common Technical Document
• Section 2.3 ☐ • Section 2.5 ☐ • Section 2.7.1 ☐ • Section 2.7.2 ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 86

CADTH Common Drug Review
Table 27: Category 1 Requirements for a Tailored CDR Review:
SEB Submission Filed on a Post-NOC Basis Requirement Specific Items and Criteria Included
• Section 2.7.3 ☐ • Section 2.7.4 ☐ • Section 5.2 • Or a placeholder document for any section(s) not required by
Health Canada for the regulatory submission, if applicable
☐ ☐
Clinical Studies and Errata
• Reference list of key clinical issues studies (published and unpublished) and any errata
☐
• Copies of studies addressing key clinical issues • Copies of any errata (or placeholder document stating none found)
☐ ☐
Table of Studies • Completed table of studies template ☐ Editorials • Reference list of editorial articles (or placeholder document stating
none found) • Copies of editorial articles
☐ ☐
Search Strategies
• Literature search strategies • Included all search terms and names of databases searched
☐
Disclosure of Studies
• Signed declaration that all known unpublished studies have been disclosed (used the provided letter template)
☐
CONSORT Diagrams
• For pivotal trials in Health Canada submission ☐ • For other key trials included in the submission (as per first section
of the “table of studies” template) ☐
New Data • Reference list of new data (or statement that none available) ☐ • Copies of new data available ☐
Validity of Outcome Measures
• Reference list (or statement that none available) ☐ • Copies of validity of outcome measure references available ☐
Disease Prevalence and Incidence Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced ☐ Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used provide letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used provided letter template)
☐
BSEAR = Biologics Safety and Efficacy Assessment Report; CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; SEB = subsequent entry biologic.
Submission Guidelines for the CADTH Common Drug Review August 2014 87

CADTH Common Drug Review
G. Category 1 Requirements for all Resubmission Types
Table 28: Category 1 Requirements for a Resubmission Section Specific Items and Criteria Included General Information Overview • Completed application overview template ☐ Signed Cover Letter
• Clear description of resubmission filed and the rationale for resubmission (e.g., resubmission based on new clinical and new cost information)
☐
• Confirmation that the information on which the submission is based is new
☐
• Confirmation that all requirements have been included ☐ • Whether changes to the current product monograph are anticipated ☐ • The indication(s) to be reviewed ☐ • Requested listing criteria, if applicable ☐ • Intention to provide category 2 requirements at least 20 business
days before targeted CDEC meeting (if not filed at the same time as category 1)
☐
• Confirmation of whether submitted price is the current market price, or a confidential price
☐
• Names and contact information for primary and backup contacts ☐ If priority review requested: • Statement that priority review status is being requested ☐ • Justification (i.e., clinical and/or economic criteria) ☐ • Confirmation that a completed priority review application is included ☐
Executive Summary
• Completed executive summary template ☐ • Maximum five pages (excluding references) ☐ • Document referenced with all supporting references ☐
Product Monograph • A copy of the most current version of the Health Canada–approved product monograph
☐
New and Updated Efficacy and/or Safety Information New Clinical Information
• A reference list of all new clinical information included in the resubmission that was not provided in the initial submission, or a previous resubmission
☐
• Copies of all new clinical information included in the above list ☐ CONSORT Diagrams
• For new clinical studies included in the resubmission ☐
Table of Studies • An updated tabulated list of all published and unpublished clinical studies using the provided table of studies template
☐
Search Strategies • Literature search strategies • Included all search terms and names of databases searched
☐
Disclosure of Studies
• Signed declaration that all known unpublished studies have been disclosed (used the provided letter template)
☐
New and Updated Economic and Epidemiologic Information Pharmacoeconomic Evaluation
• Pharmacoeconomic evaluation for the full population identified in the approved Health Canada indication(s) to be reviewed by CDR
☐
Submission Guidelines for the CADTH Common Drug Review August 2014 88

CADTH Common Drug Review
Table 28: Category 1 Requirements for a Resubmission
Section Specific Items and Criteria Included Economic Model • Three separate CDs, DVDs, or USB flash drives, each with a copy of
the unlocked and fully executable economic model ☐
• Required economic model supporting documentation ☐
Number of Patients Accessing a New Drug
• Number of patients accessing the new drug up to within 20 business days of filing the resubmission. (Note: this requirement is only for a new drug resubmission or a new combination product resubmission if one of the components is a new drug.) Used the provided letter template.
☐
Disease Prevalence and Incidence
• Disease prevalence and incidence data, with specified breakdown if available
☐
• Document is referenced ☐ Pricing and Distribution Information Price and Distribution Method
• Submitted unit pricing to four decimal places ☐ • Method of distribution ☐
Commitment for Submitted Price
• Signed commitment to honour submitted price (used the provided letter template)
☐
Letter Authorizing Unrestricted Sharing of Information Unrestricted Information Sharing
• Signed letter authorizing unrestricted sharing of information (used the provided letter template)
☐
Drug Plan Decisions Drug Plan Decisions
• A summary of the benefit status of the drug for all drug plans, including all criteria for coverage, if applicable
☐
CDEC = Canadian Drug Expert Committee; CDR = CADTH Common Drug Review.
Submission Guidelines for the CADTH Common Drug Review August 2014 89
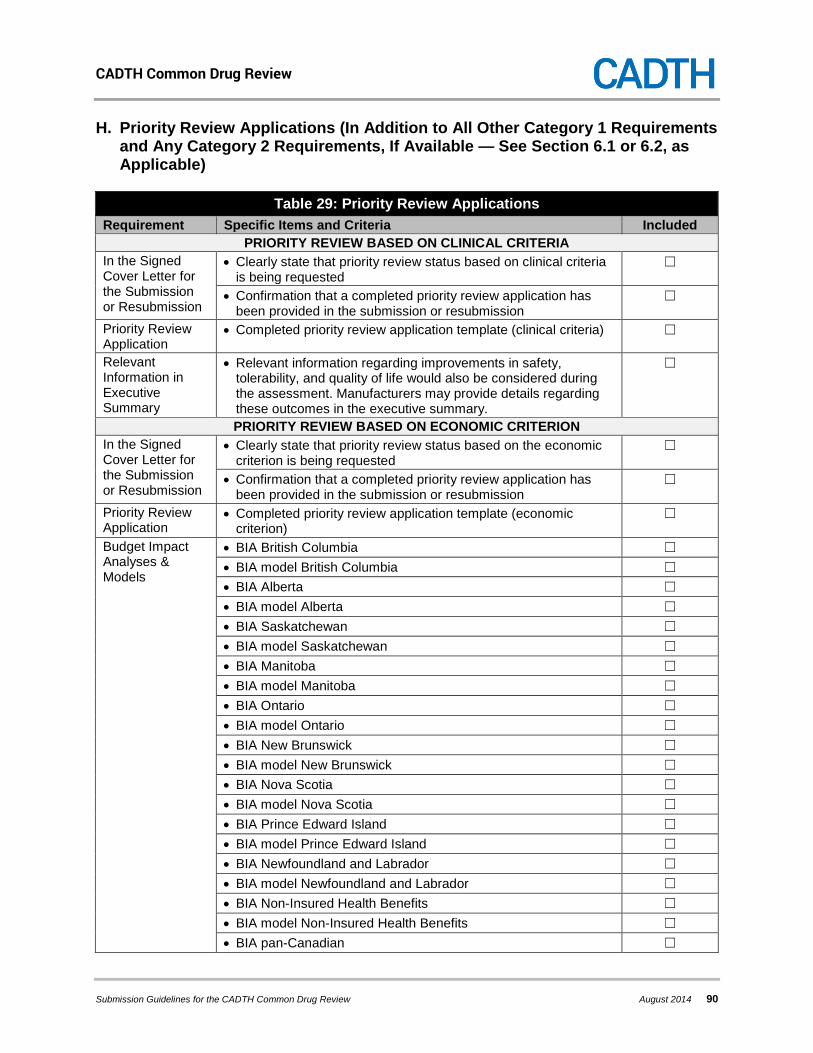
CADTH Common Drug Review
H. Priority Review Applications (In Addition to All Other Category 1 Requirements
and Any Category 2 Requirements, If Available — See Section 6.1 or 6.2, as Applicable)
Table 29: Priority Review Applications
Requirement Specific Items and Criteria Included PRIORITY REVIEW BASED ON CLINICAL CRITERIA
In the Signed Cover Letter for the Submission or Resubmission
• Clearly state that priority review status based on clinical criteria is being requested
☐
• Confirmation that a completed priority review application has been provided in the submission or resubmission
☐
Priority Review Application
• Completed priority review application template (clinical criteria) ☐
Relevant Information in Executive Summary
• Relevant information regarding improvements in safety, tolerability, and quality of life would also be considered during the assessment. Manufacturers may provide details regarding these outcomes in the executive summary.
☐
PRIORITY REVIEW BASED ON ECONOMIC CRITERION In the Signed Cover Letter for the Submission or Resubmission
• Clearly state that priority review status based on the economic criterion is being requested
☐
• Confirmation that a completed priority review application has been provided in the submission or resubmission
☐
Priority Review Application
• Completed priority review application template (economic criterion)
☐
Budget Impact Analyses & Models
• BIA British Columbia ☐ • BIA model British Columbia ☐ • BIA Alberta ☐ • BIA model Alberta ☐ • BIA Saskatchewan ☐ • BIA model Saskatchewan ☐ • BIA Manitoba ☐ • BIA model Manitoba ☐ • BIA Ontario ☐ • BIA model Ontario ☐ • BIA New Brunswick ☐ • BIA model New Brunswick ☐ • BIA Nova Scotia ☐ • BIA model Nova Scotia ☐ • BIA Prince Edward Island ☐ • BIA model Prince Edward Island ☐ • BIA Newfoundland and Labrador ☐ • BIA model Newfoundland and Labrador ☐ • BIA Non-Insured Health Benefits ☐ • BIA model Non-Insured Health Benefits ☐ • BIA pan-Canadian ☐
Submission Guidelines for the CADTH Common Drug Review August 2014 90

CADTH Common Drug Review
Table 29: Priority Review Applications
Requirement Specific Items and Criteria Included • BIA Model pan-Canadian ☐
BIA Supporting Documentation
• Reference list of all supporting documentation used and/or cited in BIAs
☐
• Copies of all supporting documentation used and/or cited in BIAs
☐
BIA = budget impact analysis.
Submission Guidelines for the CADTH Common Drug Review August 2014 91

CADTH Common Drug Review
I. Category 2 Requirements for all Submission Types
Table 30: Category 2 Requirements for all Submission Types Requirement Specific Items and Criteria Included Signed Cover Lettera
• Clear description of submission filed ☐ • Confirmation that all category 2 requirements have been
provided ☐
Budget Impact Analysesb
• British Columbia ☐ • Alberta ☐ • Saskatchewan ☐ • Manitoba ☐ • Ontario ☐ • New Brunswick ☐ • Nova Scotia ☐ • Prince Edward Island ☐ • Newfoundland and Labrador ☐ • Non-Insured Health Benefits ☐
Supporting BIA Documentation
• Reference list of all supporting documentation used and/or cited in BIAs
☐
• Copies of all supporting documentation used and/or cited in BIAs ☐ CPID • Copy of approved CPID ☐
BIA = budget impact analysis; CPID = Certified Product Information Document. a A cover letter is not required if category 2 requirements are being filed at the same time as the category 1 requirements. b BIAs and supporting documentation are not required if they were already provided as part of category 1 requirements for a
submission requesting priority review status based on the economic criterion.
Submission Guidelines for the CADTH Common Drug Review August 2014 92
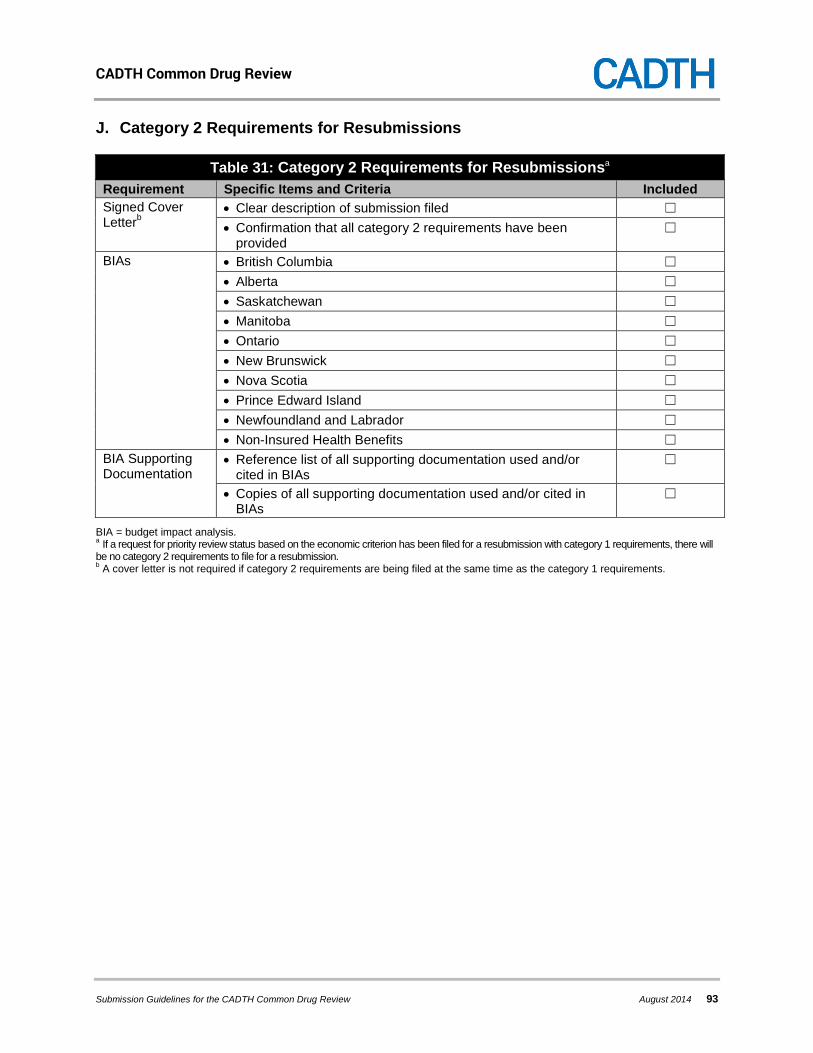
CADTH Common Drug Review
J. Category 2 Requirements for Resubmissions
Table 31: Category 2 Requirements for Resubmissionsa Requirement Specific Items and Criteria Included Signed Cover Letterb
• Clear description of submission filed ☐ • Confirmation that all category 2 requirements have been
provided ☐
BIAs • British Columbia ☐ • Alberta ☐ • Saskatchewan ☐ • Manitoba ☐ • Ontario ☐ • New Brunswick ☐ • Nova Scotia ☐ • Prince Edward Island ☐ • Newfoundland and Labrador ☐ • Non-Insured Health Benefits ☐
BIA Supporting Documentation
• Reference list of all supporting documentation used and/or cited in BIAs
☐
• Copies of all supporting documentation used and/or cited in BIAs
☐
BIA = budget impact analysis. a If a request for priority review status based on the economic criterion has been filed for a resubmission with category 1 requirements, there will be no category 2 requirements to file for a resubmission. b A cover letter is not required if category 2 requirements are being filed at the same time as the category 1 requirements.
Submission Guidelines for the CADTH Common Drug Review August 2014 93

CADTH Common Drug Review
APPENDIX 9: ELECTRONIC FILE STRUCTURE AND NAMING FORMAT
Instructions for Manufacturers Please carefully review the following electronic file structure and naming convention before assembling CDR submission or resubmission requirements. If you have any questions regarding the CDR application process for a submission or resubmission, please email [email protected] with the complete details of your question(s). Filing Category 1 and Category 2 Requirements:
• Documents should be organized on three CDs, DVDs, or USB flash drives, as follows: Category 1 requirements (excluding the economic model and supporting documentation)
on one CD, DVD, or USB flash drive Category 1 economic model and supporting documentation requirement on one CD,
DVD, or USB flash drive (note that three copies of this information on separate media are to be sent to CADTH as part of the category 1 requirements)
Category 2 requirements on one CD, DVD, or USB flash drive (unless being filed at the same time as category 1 requirements, in which case they can be on the same CD, DVD, USB flash drive, if they all fit; if not, provide on separate media).
• File names cannot exceed 64 characters; therefore, manufacturers are asked to use
abbreviations when necessary.
• The media (CDs, DVDs, or USB flash drives) used for filing the requirements must be clearly labelled with the drug brand name, submission date (DD/MM/YYYY), and brief description of contents (e.g., Category 1, Category 2, Category 1 and 2).
• Documents must be provided in PDF or Microsoft Word format, unless otherwise indicated in the requirement descriptions. These files must be unlocked, searchable, and printable. Users must be able to extract information or combine documents.
• Documents must be organized and labelled according to the file structure and naming format provided in this appendix.
• If any extra supporting documents that do not have a designated folder are being submitted at the applicant’s discretion (e.g., an indirect comparison, a Clinical Study Report), these should be appropriately named and filed in a logical location in the file structure.
Providing Additional Information During the Review:
• If CADTH requests additional information during the course of a review under the CDR process, manufacturers can provide the requested information to CADTH by email or on a CD, DVD, or USB flash drive. If the electronic documents are less than a total of 10 MB in size, they can be sent by
email to the designated submission coordinator. If the files exceed 10 MB, the documents must be provided on clearly labelled CDs,
DVDs, or a USB flash drive.
Submission Guidelines for the CADTH Common Drug Review August 2014 94

CADTH Common Drug Review
• The documents must be provided in PDF or Microsoft Word format. These files must be
unlocked, searchable, and printable. Users must be able to extract information or combine documents.
• File names cannot exceed 64 characters; therefore, manufacturers are asked to use abbreviations when necessary.
Submission Guidelines for the CADTH Common Drug Review August 2014 95

CADTH Common Drug Review
A. Category 1 Requirements for a Standard CDR Review: New Drug, Drug with a
New Indication, or New Combination Product Submission The following folder and file structure reflects each of the CDR category 1 requirements for a new drug, drug with a new indication, or new combination product submission and the order in which they are to be provided on the submitted CDs, DVDs, or USB flash drives.
Represents one folder
Represents a PDF or Microsoft Word file (unlocked, searchable, and printable)
CD, DVD, or USB flash drive #1: Brand Name_Date — Category 1
1_General Information 1 - Application Overview 2 - Signed Cover Letter
3 - Executive Summary 4 - Product Monograph
2_Health Canada Documentation 1 - Health Canada Notice of Compliance (NOC) 2 -Letter of Undertaking (Note: only if applicable; adjust following file numbers if necessary) 3 - Health Canada Reviewers’ Report
4 - Table of Clarifaxes
3_Clinical Information
3.1_Common Technical Document 1 - Section 2.5 2 - Section 2.7.1 3 - Section 2.7.3 4 - Section 2.7.4 5 - Section 5.2
3.2_Clinical Studies and Errata _List of Studies and Errata _No Errata (Note: placeholder document, only if applicable) 1 - Trial Name_Author_Year 2 - Trial Name_Author_Year 3 - Trial Name_Author_Year Erratum
3.3_Table of Studies Table of Studies
3.4_Editorials _List of Editorials 1 - Author_Year
3.5_Search Strategies Search Strategies
3.6_Disclosure of Studies
Submission Guidelines for the CADTH Common Drug Review August 2014 96

CADTH Common Drug Review
Signed Disclosure of Studies 3.7_CONSORT Diagrams
1 - CONSORT (Study Name) 2 - CONSORT (Study Name)
3.8_New Data _List of New Data 1 - Trial Name_Author_Year
3.9_Validity of Outcomes _List of References 1 – Author_Year
3.10_Indirect Comparison (Note: not a requirement; may be provided at discretion of the applicant) Indirect Comparison
4_Economic and Epidemiologic
4.1_Economic Information (Note: Include three separate CDs, DVDs or USB flash drives with the economic model and required supporting documentation)
Pharmacoeconomic evaluation
4.2_Epidemiologic Information Number Patients Accessing New Drug (Note: this requirement is only for a new drug submission or a new combination product submission if one of the components is a new drug) Disease Prevalence and Incidence
5_Pricing and Distribution Pricing and Distribution
Commitment for Submitted Price 6_Sharing of Information
Information Sharing Letter If requesting priority review based on clinical criteria:
7_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file) If requesting priority review based on economic criteria:
7_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file)
7.1_Brand Name Budget Impact Analyses (BIAs) and Models 1 - BIA BC 2 - BIA Model BC 3 - BIA AB 4 - BIA Model AB 5 - BIA SK 6 - BIA Model SK 7 - BIA MB 8 - BIA Model MB
Submission Guidelines for the CADTH Common Drug Review August 2014 97

CADTH Common Drug Review
9 - BIA ON 10 - BIA Model ON 11 - BIA NB 12 - BIA Model NB 13 - BIA NS 14 - BIA Model NS 15 - BIA PE 16 - BIA Model PE 17 - BIA NL 18 - BIA Model NL 19 - BIA Non-Insured Health Benefits (NIHB) 20 - BIA Model NIHB 21 - BIA pan-Canadian 22 - BIA Model pan-Canadian
7.2_BIA Supporting Documentation _List of References 1 - Name of document
Submission Guidelines for the CADTH Common Drug Review August 2014 98

CADTH Common Drug Review
B. Category 1 Requirements for a Tailored CDR Review: New Combination
Product (Funded Components or CADTH-Designated for Tailored CDR Review) Submission
The following folder and file structure reflects each of the category 1 requirements for a new combination product (funded components or CADTH-designated for tailored CDR review) and the order in which they are to be provided on a CD, DVD, or USB flash drive.
Represents one folder
Represents a PDF or Microsoft Word file (unlocked, searchable, and printable)
CD, DVD, or USB flash drive #1: Brand Name_Date — Category 1
1_General Information 1 - Application Overview
2 - Signed Cover Letter 3 - Executive Summary
4 - New Combination Submission Template 5 - Product Monograph
2_Health Canada Documentation 1 - Health Canada NOC 2 - Letter of Undertaking (Note: only if applicable; adjust following file numbers if necessary) 3 - Health Canada Reviewers’ Report
4 - Table of Clarifaxes
3_Clinical Information Clinical Study with New Combination
4_Epidemiologic Information Disease Prevalence and Incidence
5_Pricing and Distribution Pricing and Distribution
Commitment for Submitted Price 6_Sharing of Information
Information Sharing Letter If requesting priority review based on clinical criteria:
7_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file) If requesting priority review based on economic criteria:
7_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file)
7.1_Brand Name BIAs and Models 1 - BIA BC
Submission Guidelines for the CADTH Common Drug Review August 2014 99

CADTH Common Drug Review
2 - BIA Model BC 3 - BIA AB 4 - BIA Model AB 5 - BIA SK 6 - BIA Model SK 7 - BIA MB 8 - BIA Model MB 9 - BIA ON 10 - BIA Model ON 11 - BIA NB 12 - BIA Model NB 13 - BIA NS 14 - BIA Model NS 15 - BIA PE 16 - BIA Model PE 17 - BIA NL 18 - BIA Model NL 19 - BIA NIHB 20 - BIA Model NIHB 21 - BIA pan-Canadian 22 - BIA Model pan-Canadian
7.2_BIA Supporting Documentation _List of References 1 - Name of document
Submission Guidelines for the CADTH Common Drug Review August 2014 100

CADTH Common Drug Review
C. Category 1 Requirements for a Tailored CDR Review: Subsequent Entry
Biologic Submission The following folder and file structure reflects each of the category 1 requirements for a subsequent entry biologic (SEB) submission and the order in which they are to be provided on a CD, DVD, or USB flash drive.
Represents one folder
Represents a PDF or Microsoft Word file (unlocked, searchable, and printable)
CD, DVD, or USB flash drive #1: Brand Name_Date — Category 1
1_General Information 1 - Application Overview
2 - Signed Cover Letter 3 - SEB Submission Template
4 - Executive Summary 5 - Product Monograph
2_Health Canada Documentation 1 - Health Canada NOC 2 - Letter of Undertaking (Note: only if applicable; adjust following file numbers if necessary) 3 - Health Canada Reviewers’ Report
4 - Table of Clarifaxes
3_Clinical Information
3.1_Common Technical Document 1 - Section 2.3
2 - Section 2.5 3 - Section 2.7.1
4 - Section 2.7.2 5 - Section 2.7.3 6 - Section 2.7.4 7 - Section 5.2
3.2_Clinical Studies and Errata _List of Articles and Errata _No Errata (Note: placeholder document, only if applicable) 1 - Trial Name_Author_Year 2 - Trial Name_Author_Year 3 - Trial Name_Author_Year Erratum
3.3_Table of Studies Table of Studies
3.4_Editorials _List of Editorials 1 - Author_Year
3.5_Search Strategies Search Strategies
Submission Guidelines for the CADTH Common Drug Review August 2014 101

CADTH Common Drug Review
3.6_Disclosure of Studies Signed Disclosure of Studies 3.7_CONSORT Diagrams
1 - CONSORT (Study Name) 2 - CONSORT (Study Name)
3.8_New Data _List of New Data 1 - Trial Name_Author_Year 2 - Trial Name_Author_Year
3.9_Validity of Outcomes _List of References 1 – Author_Year
4_Disease Prevalence and Incidence Disease Prevalence and Incidence
5_Pricing and Distribution Pricing and Distribution
Commitment for Submitted Price 6_Sharing of Information
Information Sharing Letter If requesting priority review based on economic criteria:
7_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file)
7.1_Brand Name BIAs & Models 1 - BIA BC 2 - BIA Model BC 3 - BIA AB 4 - BIA Model AB 5 - BIA SK 6 - BIA Model SK 7 - BIA MB 8 - BIA Model MB 9 - BIA ON 10 - BIA Model ON 11 - BIA NB 12 - BIA Model NB 13 - BIA NS 14 - BIA Model NS 15 - BIA PE 16 - BIA Model PE 17 - BIA NL 18 - BIA Model NL 19 - BIA NIHB 20 - BIA Model NIHB
Submission Guidelines for the CADTH Common Drug Review August 2014 102

CADTH Common Drug Review
21 - BIA pan-Canadian 22 - BIA Model pan-Canadian
7.2_BIA Supporting Documentation _List of References 1 - Name of document
Submission Guidelines for the CADTH Common Drug Review August 2014 103

CADTH Common Drug Review
D. Category 1 Requirements for All Resubmissions The following folder and file structure reflects each of the category 1 requirements for all CDR resubmissions and the order in which they are to be provided on a CD, DVD, or USB flash drive.
1_General Information 1 - Application Overview 2 - Signed Cover Letter
3 - Executive Summary 4 - Product Monograph
2_New Clinical Information 2.1_New Clinical Studies
_List of New Clinical Studies 1 - Trial Name_Author_Year 2 - Trial Name_Author_Year
2.2_New Editorials and Errata _List of Editorials and Errata _No Editorials or Errata (Note: placeholder document, only if applicable) 1 - Author_Year_Editorial 2 – Trial Name_Author_Year_Erratum 2.3_Other New Clinical Information (Note: add files, as applicable, for any other new clinical information included in the resubmission) _List of Other New Information _Name of New Information
2.4_CONSORT Diagrams 1 - CONSORT (Study Name) 2 - CONSORT (Study Name)
2.5_Updated Table of Studies Table of Studies
2.6_Updated Search Strategies Search Strategies
2.7_Disclosure of Studies Signed Disclosure of Studies
3_Economic and Epidemiologic
3.1_Economic Information (Note: Include three separate CDs, DVDs or USB flash drives with the economic model and required supporting documentation)
Pharmacoeconomic evaluation
3.2_Epidemiologic Information Number Patients Accessing New Drug (Note: this requirement is only for a new drug submission or a new combination product submission if one of the components is a new drug) Disease Prevalence and Incidence
4_Pricing and Distribution Pricing and Distribution
Commitment for Submitted Price
Submission Guidelines for the CADTH Common Drug Review August 2014 104

CADTH Common Drug Review
5_Sharing of Information Information Sharing Letter
6_Drug Plan Decisions Drug Plan Decisions If requesting priority review based on clinical criteria:
8_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file)
If requesting priority review based on economic criteria:
8_Priority Review Request Priority Review Application (Note: must be submitted as a doc or docx file)
8.1_Brand Name BIAs & Models 1 - BIA BC 2 – BIA Model BC 3 - BIA AB 4 – BIA Model AB 5 - BIA SK 6 – BIA Model SK 7 - BIA MB 8 - BIA Model MB 9 - BIA ON 10 - BIA Model ON 11 - BIA NB 12 – BIA Model NB 13 - BIA NS 14 – BIA Model NS 15 - BIA PE 16 – BIA Model PE 17 - BIA NL 18 - BIA Model NL 19 - BIA NIHB 20 - BIA Model NIHB 21 - BIA pan-Canadian 22 - BIA Model pan-Canadian
8.2_BIA Supporting Documentation _List of References 1 - Name of document
Submission Guidelines for the CADTH Common Drug Review August 2014 105

CADTH Common Drug Review
E. Category 2 Requirements for all Submissions and Resubmissions The following folder and file structure reflects each of the category 2 requirements for all CDR submission types and resubmissions, and the order in which they are to be provided on a CD, DVD, or USB flash drive.
Represents one folder
Represents a PDF or Microsoft Word file (unlocked, searchable, and printable)
CD, DVD, or USB flash drive #2: Brand Name_Category 2
1_Cover Letter (Note: only include if category 2 requirements not submitted at the same time as category 1 requirements)
Signed Cover Letter
2_Brand Name BIAs (Note: Do not include if already provided with category 1 requirements for an economic-based priority review application) 2.1_BIAs
1 - BIA BC 2 - BIA AB 3 - BIA SK 4 - BIA MB 5 - BIA ON 6 - BIA NB 7 - BIA NS 8 - BIA PE 9 - BIA NL 10 - BIA NIHB
2.2_BIA Supporting Documentation _List of References 1 - Name of document
3_CPID (Note: The Certified Product Information Document (CPID) is required only for a
submission and not for a resubmission) CPID
Submission Guidelines for the CADTH Common Drug Review August 2014 106

















