Ž . Int. J. Miner. Process. 56 1999 1–30 Surface forces and measuring techniques J.C. Froberg a,b,1 , O.J. Rojas c,2 , P.M. Claesson a, ) ¨ a Laboratory for Chemical Surface Science, Department of Chemistry, Physical Chemistry, Royal Institute of Technology, S-100 44 Stockholm, Sweden b Institute for Surface Chemistry, Box 5607, S-114 86 Stockholm, Sweden c Escuela de Ingenieria Quimica, UniÕersidad de Los Andes, Merida 5101, Venezuela Received 22 March 1998; received in revised form 3 September 1998; accepted 27 November 1998 Keywords: surface forces; double-layer forces; hydrophobic interactions; flotation; mica; surface force apparatus 1. Introduction In this chapter we are concerned with interactions between particles, including air bubbles. Such interactions occurring across aqueous solutions are of special interest Ž since they determine phenomena in mineral processing, including froth flotation Leja, . 1982; Schulze, 1984 . The most important types of colloidal forces will be introduced and discussed together with short descriptions of the most commonly used techniques for studying such interactions. This field is large and subject to extensive research and the account given here is therefore necessarily very concise. The interested reader will find more extended treatments in some of the books and research articles referred to in the text. Some results obtained from surface force measurements will also be provided in order to illustrate what can be learned from such studies. In flotation we are dealing with complex systems comprising, in addition to the minerals to be separated, the gangue or non-mineral containing material, other sus- pended solids, electrolytes, and dissolved and adsorbed polymers. The object of the ) Corresponding author. Institute for Surface Chemistry, Box 5607, S-114 86 Stockholm, Sweden. Fax: q46-8-20-89-98; E-mail: [email protected]1 Fax: q46-8-20-89-98; E-mail: [email protected]. 2 Fax: q58-74-402957; E-mail: [email protected]. 0301-7516r99r$ - see front matter q 1999 Elsevier Science B.V. All rights reserved. Ž . PII: S0301-7516 98 00040-4
Transcript
Ž .Int. J. Miner. Process. 56 1999 1–30
Surface forces and measuring techniques
J.C. Froberg a,b,1, O.J. Rojas c,2, P.M. Claesson a,)¨a Laboratory for Chemical Surface Science, Department of Chemistry, Physical Chemistry, Royal Institute of
Technology, S-100 44 Stockholm, Swedenb Institute for Surface Chemistry, Box 5607, S-114 86 Stockholm, Sweden
c Escuela de Ingenieria Quimica, UniÕersidad de Los Andes, Merida 5101, Venezuela
Received 22 March 1998; received in revised form 3 September 1998; accepted 27 November 1998
In this chapter we are concerned with interactions between particles, including airbubbles. Such interactions occurring across aqueous solutions are of special interest
Žsince they determine phenomena in mineral processing, including froth flotation Leja,.1982; Schulze, 1984 . The most important types of colloidal forces will be introduced
and discussed together with short descriptions of the most commonly used techniquesfor studying such interactions. This field is large and subject to extensive research andthe account given here is therefore necessarily very concise. The interested reader willfind more extended treatments in some of the books and research articles referred to inthe text. Some results obtained from surface force measurements will also be provided inorder to illustrate what can be learned from such studies.
In flotation we are dealing with complex systems comprising, in addition to theminerals to be separated, the gangue or non-mineral containing material, other sus-pended solids, electrolytes, and dissolved and adsorbed polymers. The object of the
) Corresponding author. Institute for Surface Chemistry, Box 5607, S-114 86 Stockholm, Sweden. Fax:q46-8-20-89-98; E-mail: [email protected]
0301-7516r99r$ - see front matter q 1999 Elsevier Science B.V. All rights reserved.Ž .PII: S0301-7516 98 00040-4
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨2
flotation process, to separate unwanted material from the desired product, is usuallyfacilitated by introduction of additives like surfactants and polymers to the alreadycomplex system. These components interact with one another in a variety of ways. This
Žleads to formation of various molecular aggregates micelles, polymer–surfactant com-. Ž .plexes, etc. and to adsorption at air–liquid and solid–liquid interfaces Fig. 1 , which
affect the interactions between the constituents in the flotation system.The study of interaction forces is vital in order to comprehend, control and optimise
the process. However, as will become apparent in the following, interpretations ofmeasured interactions are a difficult matter even in relatively simple model systems. It isadvisable to keep this in mind when applying results obtained on model systems to areal situation where even what might seem to be a minor difference could have a largeeffect on the interactions and thus the efficiency of the flotation process.
The interactions that are important for the flotation process are often divided intoŽ .three regimes Derjaguin and Duhkin, 1960; Schulze, 1984 : The hydrodynamic regime
which is concerned with the motion of particles, aggregates of particles and air bubbles.This is important for the actual separation of the mineral from the undesired material.The second regime is that in which diffusiophoretic forces operate due to lateralmovement of the particle in the electric field emanating from the rising bubble. The thirdand final regime is the subject of this chapter, the long-range interactions of molecularorigin. These interactions are important when the distance between the particles is lessthan a few hundred nanometers. They determine whether the particles aggregate or not,and whether the particles attach to the air bubbles constituting the transporting medium.Another important area deals explicitly with the attachment between the mineral and thecarrier medium. This process is to a large extent governed by the wetting properties ofthe solids, which in turn are related to the short-range interactions. This is dealt with inother chapters in the present volume.
Fig. 1. Illustration of various components present in mineral furnishes.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 3
2. Force measuring techniques
During the last three decades several different techniques have been developed formeasuring interactions between macroscopic solid surfaces, air–water interfaces, andbetween a colloidal particle and a macroscopic surface. Some of these techniques arebriefly described below. The interested reader is referred to the original literature or any
Žof the recent review articles concerned with force measuring techniques Butt et al.,.1995; Israelachvili, 1995; Claesson et al., 1996a,b .
2.1. Interferometric surface forces apparatus
The idea of using optical interferometry to determine the distance between surfaces inŽforce measuring devices can be traced back to Derjaguin and co-workers Derjaguin et
.al., 1954, 1956 . However, the early measurements were not very precise and did notallow measurements to be carried out at shorter separations than about 100 nm. Moreaccurate measurements of force–distance curves in air were later obtained in EnglandŽ .Tabor and Winterton, 1969; Israelachvili and Tabor, 1972 . The technique was later
Žimproved much further, and measurements in liquid media were made possible Israe-.lachvili and Adams, 1978 . Modified versions of the interferometric surface force
Žapparatus, SFA, have been developed in several laboratories Klein, 1983; Parker et al.,.1989; Israelachvili and McGuiggan, 1990 . The basic principles of this instrument are
shown in Fig. 2. The preferred substrate in the SFA is muscovite mica owing to the easewith which one may obtain molecularly smooth and well defined surfaces of thismaterial. The substrate surfaces are silvered on the backside and then glued onto curvedsilica discs, with a cylindrical surface of radius Rf2 cm, using an epoxy resin. Twosuch discs are then mounted in a crossed cylinder geometry which facilitates easyalignment inside the apparatus chamber.
The surface separation is determined using interferometry by introducing white lightinto the optical cavity formed by the silvered back sides of the two mica surfaces. Theemanating light is directed into a spectrometer where a pattern of fringes of equal
Ž .chromatic order FECO, Israelachvili, 1973 is observed which can be analysed to give adistance resolution of 0.1 nm. During the experiment the surface separation is changed
Ž .by means of a motor driven positioning rod 1 nm sensitivity or by applying a voltageto the piezo-electric crystal, on which the upper surface is mounted, which causes aproportional expansion or contraction in steps as small as 0.1 nm. For every change indistance, known from the calibration of the distance controlling device measured at largeseparation where no force is present, the actual displacement is obtained interferometri-cally. The magnitude of the force resulting in a deviation, D x, between the expected andthe actual change in separation can be calculated by applying Hooke’s law using theforce constant, k, of the spring which holds the lower surface.
FskD x 1Ž .
In order to allow comparison between different experiments and with theoreticalpredictions a more suitable quantity is the free energy of interaction per unit area, G,
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨4
Fig. 2. Schematic illustration of the main components of the interferometric surface force apparatus is providedŽ .in A . The stainless steel measuring chamber contains the two interacting surfaces. In a typical experiment,
Ž .one mica surface is glued to a silica disc that is attached to a piezoelectric crystal topmost part . The othersurface, also glued to a silica disc, is mounted on a double cantilever force measuring spring. The surfaces are
w Ž .xoriented in a crossed cylinder configuration see B . White light enters through the window in the bottom ofthe chamber. It had multiple reflections between the silver layers and a standing wave pattern, fringes of equal
Ž . w Ž .xchromatic order FECO , is generated see C . The standing waves exit through the top window and thewavelength and fringe shape are analysed in a spectrometer.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 5
Ž .between flat surfaces which according to the Derjaguin approximation Derjaguin, 1934is related to the force via
Fs2p G 2Ž .
R
This relation holds as long as the distance between the surfaces is much smaller than theradii of the interacting bodies, and while the radii do not become a function of theseparation, i.e., while there is no surface deformation.
2.2. Non-interferometric techniques
Many different types of non-interferometric surface force techniques have beenŽdeveloped during the years see e.g., Derjaguin et al., 1978; Peschel et al., 1982; Ducker
.et al., 1991; Parker, 1994 . Perhaps the most popular of these techniques is the AtomicŽ .Force Microscope AFM which apart from being used for imaging also can be applied
Žto force measurements between a colloidal sized particle and a surface Ducker et al.,.1992; Butt et al., 1995; Larson et al., 1997 or between an air bubble and a surface
Ž .Butt, 1994; Ducker et al., 1994; Fielden et al., 1996 . By using a colloidal probe,Ž .obtained by gluing a spherical particle of radius 5–50 mm onto the cantilever tip, a
known geometry is obtained. A technique in some respects similar to the AFM-colloidalprobe technique is the so-called MASIF, which uses two spherical surfaces, most often
Ž .glass Rf1–2 mm , to measure the interaction forces. Both these techniques depend onelectronic determination of the surface separation and spring deflection, rather thaninterferometry. The zero for the surface separation is set at the hard wall encountered in
Table 1Comparison of the three surface force instruments
SFA MASIF AFM colloidal probe
Mode of operation step-wise or continuous continuous approach continuous approachapproach
Requirement of substrate thin sheets of uniform molecularly smooth colloidal particlethickness, molecularly preferably hard preferably hardsmooth, transparent, well defined geometry well defined geometrypreferably hard
Typical time for one force curve 10–40 min 0.1–2 min 1–2 sTypical number of data points 50–100 1000–10 000 a few hundredTypical radius 1–2 cm 0.1–0.2 cm 5–50 mm
y7 y8 y10Typical force sensitivity 10 N 2=10 N 1=10 NTypical sensitivity in force 5–10 mNrm 10–20 mNrm 2–20 mNrmnormalised by radiusMeasures absolute distance YES not without use of NOand layer thickness interferometryMeasures local radius and YES not without use of NOsurface deformation interferometryMeasures refractive index and YES not without use of NOadsorbed amount interferometry
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨6
each force run when the surfaces have come into contact, assuming that such a hard wallŽ .is reached Schillen et al., 1997 . This procedure is different from the situation in the´
SFA where the zero separation is set at the beginning of each experiment when the twosurfaces are brought into a strong adhesive contact in air. The contact is considered to beunchanged during the course of the experiment and any change in smallest separationcan be attributed to an adsorbed layer. This constitutes a difference between thetechniques, thus conclusions about layer thicknesses cannot readily be inferred fromAFM-colloidal probe and MASIF experiments. There are on the other hand a widervariety of surfaces which can be employed, since there is no requirement that thesurfaces are transparent. Furthermore, one can take advantage of the fact that measure-ments can be performed much faster using these techniques. Some essential features ofthe SFA, MASIF and AFM-colloidal probe techniques are summarised in Table 1.
2.3. Thin film balance
Various types of thin film balance techniques can be used for measuring dynamic andstatic interactions between two air–liquid interfaces, or between an air–liquid interface
Žand a solid surface Scheludko, 1967; Exerowa and Scheludko, 1971; Bergeron et al.,.1993 . A schematic drawing of the porous frit version of this instrument is shown in Fig.
3. It consists of a gas tight measuring cell. The solution under investigation is placed in
Fig. 3. A schematic illustration of the main components of the thin film balance. A macroscopic foam film isformed in a hole drilled in a porous glass frit. The surfactant solution is contained in the frit, in the glasscapillary and at the bottom of the closed cell. The film thickness is determined using interferometry. Thereflected light is viewed by a video camera and the intensity of a selected wavelength is measured with a
Ž .photomultiplier tube PMT . The pressure in the measuring cell is varied by means of a syringe pump andmeasured by a pressure transducer.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 7
the bottom of the cell and it is also contained in a porous glass frit with a capillary tubefused to one of its sides. The foam film is formed in a small hole drilled in the frit. In
Ž .the flat portion of the film the disjoining pressure P equals the capillary pressure,Ž .which in turn equals the difference in the pressure in the gas phase P and in the liquidg
Ž .phase Pl
2g cos uPsP yP sP yP q yD r gh 3Ž .g l g r r
Ž .where P is the reference pressure above the capillary often atmospheric pressure , gr
the surface tension of the liquid film, u the contact angle between the liquid and thecapillary wall, D r the difference in density between the solution and the surroundingair, h is the liquid rise in the capillary and g the gravitational constant. By varying thegas pressure in the measuring cell the disjoining pressure can be changed, and this, ofcourse, changes the equilibrium film thickness. In this way the repulsive forces can bedetermined, commonly referred to as the disjoining pressure isotherm in thin filmbalance literature.
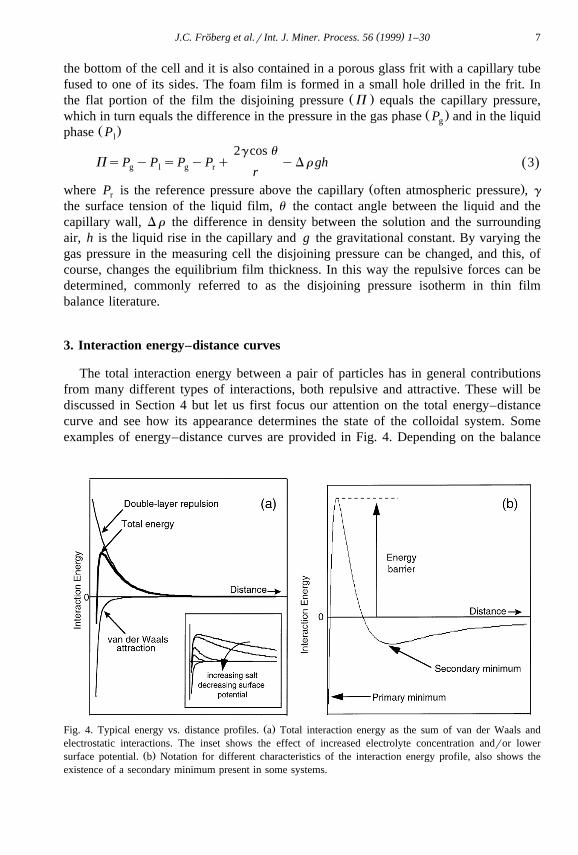
3. Interaction energy–distance curves
The total interaction energy between a pair of particles has in general contributionsfrom many different types of interactions, both repulsive and attractive. These will bediscussed in Section 4 but let us first focus our attention on the total energy–distancecurve and see how its appearance determines the state of the colloidal system. Someexamples of energy–distance curves are provided in Fig. 4. Depending on the balance
Ž .Fig. 4. Typical energy vs. distance profiles. a Total interaction energy as the sum of van der Waals andelectrostatic interactions. The inset shows the effect of increased electrolyte concentration andror lower
Ž .surface potential. b Notation for different characteristics of the interaction energy profile, also shows theexistence of a secondary minimum present in some systems.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨8
between the repulsive and attractive components in the particular system, one of severalsituations is possible. When the interaction is repulsive at all separations the particleswill always repel each other and the colloidal system will remain in the dispersed state,which is also the thermodynamically stable state.
On the other hand when the interaction is purely attractive, except at very shortseparations, the two particles come close together into an energy minimum and anaggregate is formed. The colloidal system is then unstable and will rapidly coagulateŽ .also called irreversible flocculation . In most cases the situation is more complicatedand the total energy of interaction may be repulsive or attractive depending on theinterparticle separation. For instance, the outermost and innermost part of the energy–distance curve may be attractive whereas it may be repulsive at intermediate separationsŽ .Fig. 4 .
The maximum in the energy distance curve is referred to as the energy barrier, andwhen it is small, i.e., comparable to the Brownian energy due to the thermal motion
Ž .k T , the particles may approach each other close enough to reach the primary or innerB
energy minimum. The depth of this minimum is often large compared to k T and theB
colloidal system thus becomes coagulated. On the other hand, when the energy barrier islarge compared to k T the particles cannot reach the primary minimum but they preferB
Ž .to be in the secondary or outer minimum. The colloidal system becomes reversiblyflocculated when the depth of the minimum is comparable to k T. The floc structureB
can in this case easily be broken, for example, by stirring or shearing. If the secondaryminimum is absent or small compared to k T the system remains in a dispersedB
metastable state. One says that the colloidal system is kinetically stabilised since thelarge energy barrier slows down the coagulation rate.
A colloidal system may thus be unstable, thermodynamically stable or kineticallystabilised depending on the interactions. These are in turn determined by the propertiesof the particles; such as surface charge and dielectric properties, and the properties of themedium; pH, ionic strength, and concentration of additives such as surfactants, poly-mers, and polyelectrolytes. It is for the purpose of controlling the interactions in theflotation process that most additives are used.
4. The interaction energy contributions
A better understanding of how the interactions can be controlled can be obtained byconsidering the most important contributions to the total energy–distance curve, themolecular origin of these contributions, and how the strength of the different interactioncontributions can be regulated by additives. This section will start by considering thealways present van der Waals interaction, and the electrostatic double-layer interactionpresent between charged surfaces immersed in electrolyte solutions. These contributionsare considered in the DLVO theory, which is the most widely used framework forunderstanding colloidal stability in the absence of adsorbed polymers. The name stemsfrom the four scientist who developed the theory in the 1940s: Derjaguin and Landau in
ŽRussia and independently Verwey and Overbeek in the Netherlands see Faraday Disc.Ž . .18 1954 for an account of the development . We will furthermore briefly describe
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 9
non-DLVO interactions such as solvation, hydrophobic, and steric interactions. ThereŽare several books which treat the subject extensively Israelachvili, 1991; Evans and
.Wennerstrom, 1994 and the reader is encouraged to consult these for a more extensive¨discussion.
4.1. Õan der Waals interactions
Van der Waals interactions exist between molecules and between particles. The mostimportant molecular contributions are due to interactions between permanent rotating
Ž .dipoles Keesom interactions , between a permanent rotating dipole and an inducedŽ . Ždipole Debye interactions , and between two induced dipoles London or dispersion
.interactions . The dispersion contribution originates from the motion of electrons aroundthe nuclei, and it therefore exists also between non-polar molecules and is alwayspresent. Except for highly polar molecules it can be shown that the dispersion contribu-tion accounts for nearly all the van der Waals interaction. The interaction energy isinversely proportional to the separation between the molecules to the power of six.
The van der Waals interactions between two macroscopic particles can, as a firstapproximation, be calculated by summing the interactions between all molecular pairs
Ž .between the two bodies. In practise this is done by an integration Hamaker, 1937 andthe result is that the distance dependence of the van der Waals interaction betweenmacroscopic surfaces is much weaker than that between molecules, i.e., the interaction ismore long-ranged.
The expression for two spherical particles of radius R and R separated in Õacuum1 2Ž .by a distance D is Hamaker, 1937
2A b b a qabqaV sy q q2ln 4Ž .A 2 2 2ž /12 a qabqa a qabqaqb a qabqaqb
where asDr2 R , bsR rR , and A is the material dependent Hamaker constant.2 1 2Ž .Only the first term in Eq. 1 needs to be considered when the radii of the spheres are
much larger than the surface separation. When this is the case and when the sphereshave the same radii, R, the expression above simplifies to
ARV sy 5Ž .A 12 D
The Hamaker method is conceptually easy but unfortunately not very accurate. Itneglects many-body interactions, entropic contributions and retardation. It is also notvery easy to accurately account for the presence of the medium separating the twointeracting particles.
All these effects are considered in an alternative treatment due to Lifshitz et al.Ž .Dzyaloshinskii et al., 1961 . Here the interacting particles and the intervening mediumare treated as continuous phases and the interaction is viewed as originating frominterference between fluctuating electromagnetic fields extending beyond the surface ofthe particles. The expressions for the interaction energy between bodies of a specificshape have the same functional form as in the Hamaker treatment, but the equivalent to
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨10
the Hamaker constant is a function of the frequency dependent bulk dielectric propertiesof the continuous phases. Rather accurate approximate equations to the Lifshitz expres-sion are often used for calculating the Hamaker constant. The material constants neededin order to perform the calculations are the static dielectric constants, ´ , and refractivei
indices, n , of the different phases i. Thus the non-retarded Hamaker constant for twoi
macroscopic bodies 1 and 2 interacting over a medium 3, is approximately given byŽ .Israelachvili, 1991
2 2 2 2 2 2 2 2' ( ( ( (8 2 n qn n qn n qn q n qnž /1 3 2 3 1 3 2 3
where n is the main electronic absorption frequency in the UV region, here assumed toe
be the same for all three media, k the Boltzmann constant, h Planck’s constant, and TB
the absolute temperature.Implicit in the discussion so far has been that the Hamaker constant will not be
affected by the separation between the particles, i.e., the distance over which the electricfield has to travel to affect the other body. However, this is not correct when theseparation is so large that the time needed for the electric field emanating from one bodyto travel to the other body and being reflected back again becomes comparable to theperiod of the fluctuations that generate the field. Thus the phase correlation between thefields emanating from the two bodies will be lost and the interaction will be weakened.This phenomenon, called retardation, is important at separations larger than about 5 nm,and can be taken care of within the framework of the Lifshitz theory, e.g., as described
Ž .in the book by Mahanty and Ninham 1976 .Ž . Ž .It can easily be seen from Eqs. 5 and 6 that the van der Waals interaction is
always attractive between identical entities, and it is always attractive in vacuum evenwhen the two interacting bodies are not the same. However, when the materialproperties of the bodies are different the interaction may become repulsive in a medium.This is for instance the case for an air bubble interacting with a mineral particle or ahydrophobised mineral particle across water. Fig. 5 shows the non-retarded van derWaals interaction for a few different combinations of phases interacting across water.The figure displays that the van der Waals force can be both attractive and repulsive,with the latter being the case whenever the dielectric properties of the interveningmedium falls between those of the interacting phases.
The van der Waals interaction is altered by the presence of an adsorbed filmwhenever its dielectric properties are different to those of the particles. The non-retardedvan der Waals force between spheres of material 1 and 1X, each coated with a thin layerof material 2 and 2X of thickness L and LX, respectively, interacting across medium 3 can
Ž .be evaluated using the expression Ninham and Parsegian, 1970X X XR A A A A A232 132 1 32 1 21 eff
W D sy y y q syŽ . X X12 D DqL DqL DqLqL 12 DŽ . Ž . Ž .7Ž .
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 11
Fig. 5. Calculated van der Waals forces between two spheres for different combinations of silica, water, air,Ž .and hydrocarbon octane . The different curves correspond to the following combinations and Hamaker
Ž . y2 0— - — air–water–silica Asy0.8=10 J. Note how a negative Hamaker constant results in a repulsivevan der Waals force.
where A is the Hamaker constant for bodies of material m and o interacting inm noŽ .medium n, which can be calculated according to Eq. 6 .
The effective Hamaker constant will consequently be dependent on the separation D:At large separations it will be dominated by the contribution from the material in theparticles, while the influence of the properties of the adsorbed layer will becomeapparent as the separation between the surfaces decreases. The effect of an adsorbedlayer is indicated in Fig. 6a where the dependence of the effective Hamaker constant isshown as a function of the normalised surface separation. The case of a quartz surfaceinteracting with an air bubble in water when bearing identical adsorbed hydrocarbon
Ž .films, calculated according to Eq. 7 , is shown by the solid curve, and the case of twoquartz surfaces with adsorbed hydrocarbon films in water is shown by the dashed curve.For both, the Hamaker constant at small separations will be dominated by the adsorbedlayers while at larger separations it is determined by the properties of the underlyingmedia. In Fig. 6b, the resulting van der Waals forces between two spherical particles aregiven as a function of surface separation for the two systems, the change from a
Ž .repulsion to an attraction at 1 nm for the asymmetric system solid curve reflects thechange in sign of the Hamaker constant whereas the force for the symmetric filmŽ .dashed curve remains attractive at all separations.
4.2. The electrostatic double-layer interaction
It has been shown that the electrostatic interactions between particles and bubblesgreatly affects the flotation of minerals. The flotation performance is, for example,
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨12
Ž . Ž .Fig. 6. a The effective Hamaker constant calculated using Eq. 7 , as a function of normalized surfaceseparation. The dashed curve is for two quartz surfaces coated with a hydrocarbon layer of thickness T s1nm. At short separations the effective Hamaker constant is equal to that between the adsorbed hydrocarbon
Ž y2 0 .layers As0.5=10 J , whereas at large separations it approaches that between uncoated quartz surfacesŽ y2 0 .A s0.83=10 J . The solid curve is for the asymmetric system of a quartz particle interacting with an air
Ž .bubble when both carry an adsorbed layer of hydrocarbon T s1 nm . Again at small separations the adsorbedlayers dominate the Hamaker constant, whereas at longer ranges it approaches the value for a quartz particle
Ž .interacting with air, which in this case leads to a change in sign. b Calculated van der Waals forces for theŽ .two systems in a . It is worth noting that the change in sign of the Hamaker constant for the asymmetric
Ž .system solid line leads to a situation where the force is repulsive at long range while turning attractive atsmall separations, approximately 1 nm. The purely repulsive van der Waals force is for silica interacting withair across water without any adsorbed layers.
Ž .closely correlated to the range of the double-layer force Paulson and Pugh, 1996 . Thisinteraction, therefore, deserves an extensive study if one aims to control and optimisethe mineral flotation process.
A solidraqueous solution interface is most often charged due to one of severalmechanisms; acid–base equilibria as in the case of silanol groups on silica, desorption oflattice ions as in the case of clays and minerals such as mica, or adsorption of ionicsurfactants and polyelectrolytes. The surface charge is exactly balanced by the netcharge in the solution outside the surface. In the region immediately outside the surface,say below 1 nm from the surface, the ions may, in addition to electrostatic forces,experience other interactions with the surface. These ions are said to be adsorbed andthey build up the so called Stern layer. Outside the Stern layer, in the diffuse layer, theions are only affected by electrostatic forces and in this region it is straightforward, in a
Ž .mean field approach such as the Poisson Boltzmann PB model, to calculate the ionconcentration away from the surface. The extension of the diffuse layer will depend onthe surface charge density, ion concentration and valency. A central quantity in thisrespect is the Debye length which describes the rate with which the mean potentialdecays away from the surface. The Debye-length decreases with increasing ionicstrength but it is independent of the surface charge density and surface potential.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 13
An electrostatic double-layer force arises when two surfaces come close enough fortheir diffuse layers to start to overlap. Before discussing how this force arises it isvaluable to consider the ion distribution in the diffuse layer outside a flat surface ascalculated in the non-linear PB-approximation. The classical text on this subject is the
Ž .book by Verwey and Overbeek 1948 , ‘Theory of the Stability of Lyophobic Colloids’,Ž .more recent treatments are given in the books by Israelachvili 1991 , and Evans and
Ž .Wennerstrom 1994 .¨Ž .The concentration number density of ions of type i in the diffuse layer a distance x
Ž .from the surface, r x , will at equilibrium be related to the bulk concentration r andi i,`Ž .the mean potential difference between the bulk solution and layer x, C x , according to
the Boltzmann relation
r x sr eyz i eC Ž x .r k BT 8Ž . Ž .i i ,`
where e is the elementary charge and z the valency of the ion. The potential is relatedi
to the charge density at x via the Poisson equation
z er x sy´´ d2Crd x 2 9Ž . Ž .Ž .Ý i i 0i
where ´ is the permittivity of vacuum and ´ is the dielectric constant of the medium.0
Combining these two relations one obtains the Poisson–Boltzmann equation
d2Crd x 2 sy z er r´´ eyz i eC Ž x .r k BT 10Ž . Ž .Ý i i ,` 0i
This non-linear second order differential equation in C can be solved analytically. Thefirst integration gives a relation between the surface potential, C , and the surface0
charge density, s ,0
1r2yz eC r k Ti 0 Bs s 2k T´´ r e y1 11Ž . Ž .Ý0 B 0 i ,`
i
A second integration gives an expression for the variation of the mean potential withthe distance from the surface and takes the form
zeC x zeCŽ . 0 yk xtanh s tanh e 12Ž .ž /ž /4k T 4k TB B
Ž . Ž .for a symmetrical electrolyte. For small potentials -25 mV Eq. 12 reduces to theDebye–Huckel relation¨
C x sC eyk x 13Ž . Ž .0
Ž .where the quantity 1rk is the Debye screening length mentioned above. It is generallygiven for an electrolyte with bulk number density r of species i byi,`
1r2
´´ k T0 B1rks 14Ž .2� 0z e rŽ .Ý i i ,`
i
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨14
Clearly, the Debye-length and the electrostatic potential outside the surface bothdecrease rapidly with increasing ionic strength.
To calculate the double layer force per unit area, D P, between two flat surfaces westart by considering the pressure on a flat surface immersed in an electrolyte solution
Ž .which for a surface with surface charge s and with an ion density of r 0 at the0 i
surface, can be written as
s 20
Psk T r 0 y 15Ž . Ž .ÝB iž /2´´ k T0 Bi
This expression states that a force can only act on a surface by way of transfer ofmomentum, the first term, or through the normal component of the Maxwell stress
Ž . Ž .tensor, second term. The contact value theorem, Eq. 15 , Israelachvili, 1991 is exactŽ .in a continuum approach. However, the true values of s and r 0 are generally not0
known with sufficient precision for an accurate calculation of the small differenceŽ .between the two large terms in Eq. 15 constituting the origin to the pressure. Within
the Poisson–Boltzmann approximation one can derive an expression for the pressure onŽ .a flat surface by inserting Eq. 8
s 20yz eC r k Ti 0 BPsk T r e y 16Ž .ÝB i ,`ž /2´´ k T0 Bi
In order to obtain the net pressure on the flat surface one has to subtract the osmoticpressure of the bulk solution to obtain
s 20yz eC r k Ti 0 BD Psk T r e y yk T r 17Ž .Ý ÝB i ,` B i ,`ž /2´´ k T0 Bi i
Ž .For an isolated surface one finds by applying Eq. 11 that the net pressure is zero,showing the internal consistency of the PB-equation.
One can, in the Poisson–Boltzmann treatment, generalise the contact value theorem,Ž .Eq. 15 , to any plane between two interacting surfaces. The net normal pressure
between two flat surfaces separated a distance D can in this case be written2s xŽ .DŽ .yz eC x rk Ti D BD P D sP D yP ` sk T r e y1 y 18Ž . Ž . Ž . Ž . Ž .ÝB i ,` 2´´ k T0 Bi
Ž . Ž .where x takes values between 0 and D, and C x and s x , respectively are theD D
surface potential and surface charge density at x when the surfaces are a distance Dapart. The net normal pressure is of course equal at all points between the surfaces whenmechanical equilibrium is established, but will vary with the surface separation. An
Ž .indefinite integration of Eq. 10 yields2dC x 2k TŽ .D B Ž .yz eC x rk Ti D Bs r e qC D 19Ž . Ž .Ž .Ý i ,`ž /d x ´´0 i
Ž . Ž .where C is a integration constant. Now, by combining Eqs. 18 and 19 with
dC xŽ .Ds sy´´ 20Ž .D 0 d x
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 15
we arrive at a simple expression for the pressure in the normal direction for flat surfacesseparated a distance D
D P D syk T r C D q1 21Ž . Ž . Ž .Ž .ÝB i ,`i
C is thus a function of the surface separation D, taking the value of y1 for an isolatedsurface to give a net pressure of zero. For values of C-y1 the net pressure will berepulsive while for values larger than y1 it will be attractive. The different types ofpossible surface charging mechanisms; constant potential, constant charge, or regulating
Ž .surfaces, will give rise to different boundary conditions for the integration of Eq. 19necessary to find the surface separation corresponding to a given combination of the s0Ž .or c values for the two surfaces and the C value. The result is an expression for the0
surface separation in terms of incomplete elliptic integrals of the first kind. Within thistreatment it is possible to arrive at an expression for the free energy of interaction perunit area as
D DX X X XDGsy D P D d D sk T r C D q1 d D 22Ž . Ž . Ž .Ž .ÝH HB i ,`
` `i
When the value of C as a function of the surface separation has been determined thefree energy of interaction can be evaluated, this time in terms of incomplete ellipticintegrals of the second kind. It is then straightforward to obtain the free energy ofinteraction for spheres as
V DŽ . Dspheres X Xsy DG D d D 23Ž . Ž .Hp R `
The calculations show that when two surfaces of equal surface potential approach eachother, the double layer interaction will be purely repulsive at all separations, independentof whether the interaction takes place under constant potential or constant charge.
Ž .Devereux and de Bruyn 1963 show in their treatment of interaction between dissimilarsurfaces at constant potential, that when the signs of the surface potentials are equal thesurfaces will repel each other at large separations while at shorter distances theinteraction turn attractive. When the potential of the two surfaces have opposite sign theinteraction will be purely attractive. The condition of constant potential for unequalsurfaces does, however, lead to the unrealistic conclusion that the magnitude of thesurface charge density of one of the surfaces approaches infinitely large positive values,while that of the other approaches infinitely large negative values.
The interaction between dissimilar surfaces at constant charge will, on the other hand,be purely repulsive for surfaces with the same sign as shown by Bell and PetersonŽ .1972 . However, when the surface charges have opposite signs on the two surfaces, theinteraction turns out to be attractive at long range while turning repulsive at shorterrange. The exception to this is when the two surfaces have equal but opposite surfacecharge which leads to a purely attractive force. Fig. 7 shows calculated interaction forcesfor two spherical surfaces, when the apparent potentials of the isolated surfaces are 50and y30 mV both for the case of constant charge and constant potential. The interaction
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨16
Fig. 7. Calculated double-layer forces for unequal spheres, with potentials 50 and y30 mV at largeŽ . Ž .separations, interacting at constant charge dashed line and constant potential solid line in a 1-mM 1:1
electrolyte.
is at long range attractive for both boundary conditions while it becomes repulsive atshort range for the case of constant charge.
Finally, when the surfaces show a regulation of the surface charge density charac-terised by the dissociation constant of the chargeable surface groups, a rather complexvariation of the double layer force with surface separation emerges. An account of the
Ž .interaction between dissimilar, regulating surfaces was presented by Chan et al. 1976using both an approximative graphical method giving the main features of the interac-tion curve, as well as an exact Poisson–Boltzmann treatment. They showed that the signof the double layer force may change as often as three times when two such surfacesapproach each other.
To calculate the double-layer force between flat surfaces exactly in the non-linearPB-approximation is thus possible but rather complicated. For spheres which are large
Ž .compared to the range of the force one can use the Derjaguin approximation, Eq. 2 , toobtain accurate expressions for the double-layer interaction. It is, however, sometimesuseful to find simple analytical expressions for the double-layer force. The simplest oneis obtained for small surface potentials in the weak overlap approximation, which isvalid at large separations. For the pressure and energy of interaction between twoidentical flat surfaces one obtains
P D s2´´ k 2C 2eyk D 24Ž . Ž .0 0
and
V D s2´´ kC 2eyk D 25Ž . Ž .0 0
Using the Derjaguin approximation, one obtains for two spheres of the same radii
F DŽ .2 yk Ds2´´ kC e 26Ž .0 0
p R
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 17
and
V DŽ .2 yk Ds2´´ C e 27Ž .0 0
p R
Hence, for this case the double-layer force at large separations decays exponentiallywith surface separation and the decay-length is equal to the Debye-length. It turns outthat this conclusion is valid also when the non-linear PB-approximation is solvedexactly. Thus, the range of the double-layer force decreases rapidly when the ionicstrength is increased.
Several assumptions are inherent in the rather primitive model of the Poisson–Boltz-mann treatment. The mean-field description of the double-layer breaks down at smallenough separations where several effects will become important: The finite size of theions will lead to an increased repulsion resulting from an excluded volume effect. Ioncorrelations are neglected, which has been shown to have a substantial effect on thedouble layer interaction, even turning what in the PB-model is a repulsive interactioninto an attraction, which, for example, is the case for highly charged surfaces insolutions of divalent ions. Furthermore, the description of the surfaces as having auniform charge will also lead to errors, since at small separation the discreteness of the
Žsurface charge will further invalidate the mean-field treatment Guldbrand et al., 1984;.Kjellander and Marcelja, 1984; Attard et al., 1988; Kjellander, 1996 .
4.3. DLVO theory
The DLVO theory considers the total interaction to be due to additive contributionsfrom the van der Waals interaction and the double layer interaction. As stated above, thevan der Waals interaction is attractive whereas the double-layer interaction is repulsivebetween identical surfaces. The van der Waals contribution predominates at smallinterparticle distances and the double-layer contribution gives rise to an energy barrier atlarger separations as illustrated in Fig. 4a. The insert to Fig. 4a illustrates that increasingthe ionic strength or lowering the surface potential results in a lower energy maximum.Fig. 4b shows the situation for a system with a strong van der Waals attraction and a lowdouble-layer repulsion which results in the existence of a secondary minimum atrelatively large interparticle distances. Clearly, we can see that by just considering theDLVO-forces one may obtain all the types of energy–distance curves that are requiredfor having kinetically stable, weakly flocculated or coagulated systems. Further, byincreasing the ionic strength one may go from a kinetically stable colloidal dispersion toone which is coagulated.
Some calculated DLVO force curves across an aqueous 1 mM 1:1 electrolyte areshown in Fig. 8a and b. The forces calculated in Fig. 8a are for the case of two solidsurfaces where the van der Waals interaction is characterised by a non-retarded Hamaker
y20 Ž .constant of 0.8=10 J . The double-layer force is calculated using constant chargeboundary conditions. The upper curve is for the case where both surfaces have a surface
Ž .potential at large separations of 50 mV, and here we obtain the classical behaviourwith a repulsion at large separations and an attraction at short distances. The lower curveis for the case where the surface potentials are unequal, 50 and y30 mV, respectively.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨18
Ž .Fig. 8. Calculated DLVO-forces solid lines for interacting spheres in 1-mM 1:1 electrolyte, dashed linesŽ . Ž .represent the electrostatic double layer force only constant charge . a Two solid surfaces with a non-retarded
y2 0 Ž .Hamaker constant of 0.8=10 J, upper curves are for equal potentials 50 mV while the lower are forŽ . Ž .unequal spheres 50 and y30 mV . b One solid surface interacting with an air bubble resulting in a
non-retarted Hamaker constant y0.8=10y20 J and therefore repulsive van der Waals forces, upper and lowerŽ .curves correspond to potentials as given in a .
In this case the total interaction is purely attractive, even though the double-layerŽ .contribution turns repulsive at short separations dashed line . Some experimental data
for the case of two mica surfaces interacting across aqueous KBr solutions are shown inFig. 9. In 0.1-mM KBr, the long-range force is dominated by a double-layer force,
Ž . Ž .Fig. 9. Experimental data for mica interacting across a 0.1-mM Kbr solution I and a 10-mM KBr ' . Thesolid lines are best fit for the DLVO theory, showing that an extra hydration repulsion is present at high ionicstrengths.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 19
whereas the short range force is attractive due to the van der Waals interaction. Thisbehaviour is as expected from DLVO theory. In the 10 mM KBr solution the long-rangeforce is still dominated by a double layer force. However, in this case no attraction isseen at short separations, which can be explained by the appearance of a short-range
Ž .repulsive hydration force not considered in the DLVO theory see below . The data inŽFig. 8b are calculated for the case of a repulsive van der Waals force with a Hamaker
y20 Ž ..constant equal to y0.8=10 J , as for the case of an air bubble interacting withquartz across water. The upper curve, which is purely repulsive, is for the case of bothpotentials being 50 mV. When the potentials instead are unequal, 50 and y30 mV, thelong-range force is attractive whereas the short range force is repulsive. It is seen thatthe repulsive van der Waals interaction prevents the air-bubble from attaching to thequartz particle. To make the quartz particle hydrophobic does not change the fact thatthe van der Waals force is repulsive, even though the Hamaker constant for hydrocar-
y20 Ž .bon–water–air is considerably less negative, y0.2=10 J . However, if also the airbubble carries an adsorbed surfactant layer, the van der Waals force at short separationsbecomes attractive as shown in Fig. 6b. The attachment of air bubbles to hydrophobisedmineral particles are often alternatively explained in terms of an additional attraction dueto hydrophobic interactions.
4.4. Hydrophobic interactions
For the solid minerals to float, their surfaces must be made hydrophobic, that is,wetted only partially by water. In a series of papers Yoon and co-workers haveadvocated that the additional attractive force needed to explain why air bubbles doattach to hydrophobised mineral particles is a long-range hydrophobic interactionŽRabinovich et al., 1993a,b; Yotsumoto and Yoon, 1993a,b; Rabinovich and Yoon,1994; Yoon and Ravishankar, 1994; Yoon and Mao, 1996; Yoon and Ravishankar,
.1996a,b; Ravishankar and Yoon, 1997; Yoon et al., 1997 . At this point it is worthwhileto consider what evidence there are for such a long-range attraction between hydropho-bic surfaces and how it may be related to the hydrophobic interaction between moleculesthat drives the self-assembly of surfactants in aqueous and other hydrogen bonding
Žsolutions Ramadan et al., 1983; Evans and Ninham, 1986; Bergenstahl and Stenius,˚1987; Beesley et al., 1988; Warnheim and Jonsson, 1988; Jonstromer et al., 1990;¨ ¨ ¨
.Warnheim et al., 1990 . Self-assembly of surfactants has been much discussed, for¨Ž .instance by Tanford in his famous book about the hydrophobic effect Tanford, 1980 .
One conclusion was that the hydrophobic effect was short-range and proportional to thesize of the non-polar molecule. The comparison of the temperature dependence of thechanges in free energy, entropy and enthalpy of micelle formation in water and
Ž .hydrazine Ramadan et al., 1983 has provided a good understanding for the associationprocess of surfactants.
One can, artificially but pedagogically, divide the free energy change for theassociation process into two main contributions; one that is due to transfer of non-polar
Ž .groups from the polar solvent and their confinement in the micelle D H and DS .tr tr
Here both the enthalpy contribution and the entropy contribution are negative and theŽ .resulting free energy DG sD H yTDS is also negative. This is the dominanttr tr tr
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨20
contribution to the solvophobic effect in non-structured hydrogen bonding liquids suchŽ . Žas hydrazine and water at high temperatures well above 1008C Evans and Ninham,
.1986 . Thus, in this case the process is enthalpy driven. In water at temperatures wellbelow 1008C there is also a second contribution due to changes in the water structureitself which accompany the removal of non-polar substances from the aqueous environ-
Ž . Ž .ment. Here both the enthalpic D H and entropic DS contributions are large andstr str
positive. In this case one may write, even though the two contributions are, of course,not independent of each other, DGsD H qD H yTDS yTDS , and the totaltr str tr str
enthalpy and entropy change is dominated by the structural contribution. Hence, one cansay that at low temperature the micellisation process in water is entropy driven and thatit is due to the release of water clathrates around the hydrocarbon chains. However, themost remarkable finding is that despite the fact that both D H and DS vary stronglystr str
with temperature, the free energy change accompanying micelle formation is rathertemperature insensitive. Hence, DG sD H yTDS is close to zero at all tempera-str str str
tures and for this reason it has been suggested that it is misleading to view theŽhydrophobic effect as being due to structural changes in the solvent Evans and Ninham,
.1986 .It is thus clear that there is a short-range attractive hydrophobic interaction between
non-polar molecules in aqueous solutions. The question then naturally arises if there is asimilar attraction between non-polar surfaces in water, and in that case which distance
Ž .dependence it has. The first measurements by Israelachvili and Pashley 1982 indicatedthat an additional attraction in excess of the van der Waals force indeed exists betweenhydrophobic surfaces, and they reported an exponential distance dependence of thisinteraction with a decay constant of 1 nm. Later more strongly hydrophobic surfaceswere studied and a decay length of the force of 1.4 nm was reported by Pashley et al.Ž .1985 . Later experiments showed evidence for much more long-ranged forces that werebetter fitted to a two step exponential function
V sV 0 eyD rl1 qV 0 eyD rl2 28Ž .hph hph1 hph2
with one decay constant of 1–3 nm dominating for separations up to 10 nm and one ofaround 5–15 nm dominating at larger separations. Hence, it is clear that there do existan additional long-range attraction between hydrophobic surfaces, but it is not clear if itshould be regarded as a hydrophobic attraction akin to the hydrophobic effect betweenmolecules. From the high interfacial tension between hydrocarbon and water it is evidentthat the short-range interaction between hydrocarbon surfaces immersed in aqueoussolutions should be strongly attractive. However, there is no theory that convincinglyexplain why the effect should extend out to 100 nm.
Measurements of forces between different types of hydrophobic surfaces agree onthat there is an additional attraction in excess of the van der Waals interaction. However,they do not agree on the range of the attractive force. It is of the order of 10 nm for
Ž . Žslightly hydrophobic surfaces Herder, 1990 , for plasma polymers Parker et al.,. Ž .1994b , polymerised Langmuir–Blodgett layers Wood and Sharma, 1994 and some
Ž .silanated surfaces Parker and Claesson, 1994 , whereas it is of the order of 100 nm forŽ .non-polymerised Langmuir–Blodgett deposited films Claesson and Christenson, 1988
Žand some other silanated surfaces Parker et al., 1994a; Rabinovich and Derjaguin,
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 21
.1988 . It is clear that at least some of the data describing the most long-range attractiveŽforces can be rationalised in terms of cavity formation between the surfaces Parker et
.al., 1994a , whereas the interpretation of the data obtained for Langmuir–Blodgett layersŽ .are complicated by the limited stability of such films Eriksson et al., 1997 . The
Ž .literature on the subject was recently reviewed by Hato 1996 , who also studied doublechained surfactants with tails terminated by methyl- or hydroxide groups. He found thatthe most hydrophobic surface showed the least long-range attraction. However, theattraction in the shorter distance regime, D-5–20 nm increased with increasing surfacehydrophobicity. Hence, the more long-range part of the measured force can not beviewed as a hydrophobic interaction. Further support for this view is that the range of
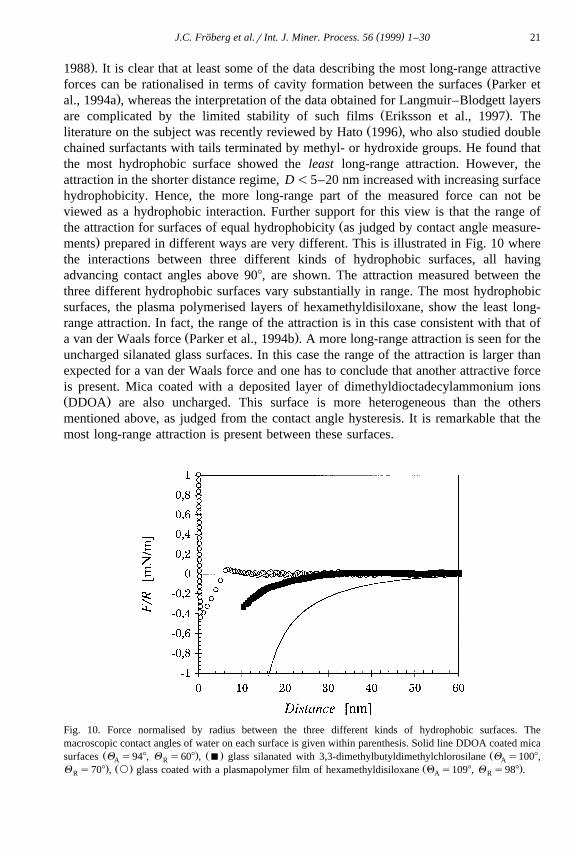
Žthe attraction for surfaces of equal hydrophobicity as judged by contact angle measure-.ments prepared in different ways are very different. This is illustrated in Fig. 10 where
the interactions between three different kinds of hydrophobic surfaces, all havingadvancing contact angles above 908, are shown. The attraction measured between thethree different hydrophobic surfaces vary substantially in range. The most hydrophobicsurfaces, the plasma polymerised layers of hexamethyldisiloxane, show the least long-range attraction. In fact, the range of the attraction is in this case consistent with that of
Ž .a van der Waals force Parker et al., 1994b . A more long-range attraction is seen for theuncharged silanated glass surfaces. In this case the range of the attraction is larger thanexpected for a van der Waals force and one has to conclude that another attractive forceis present. Mica coated with a deposited layer of dimethyldioctadecylammonium ionsŽ .DDOA are also uncharged. This surface is more heterogeneous than the othersmentioned above, as judged from the contact angle hysteresis. It is remarkable that themost long-range attraction is present between these surfaces.
Fig. 10. Force normalised by radius between the three different kinds of hydrophobic surfaces. Themacroscopic contact angles of water on each surface is given within parenthesis. Solid line DDOA coated mica
Ž . Ž . Žsurfaces Q s948, Q s608 , B glass silanated with 3,3-dimethylbutyldimethylchlorosilane Q s1008,A R A. Ž . Ž .Q s708 , ` glass coated with a plasmapolymer film of hexamethyldisiloxane Q s1098, Q s988 .R A R
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨22
Hence, from the examples above it is clear that the contact angle does not correlatewith the range of the attraction. Instead the structure of the hydrophobic surface is veryimportant for the range of the attractive force. Interestingly, this also influences howeasily air-bubbles nucleate on the surfaces and the lateral mobility of the surfacemolecules. Both these phenomena have in some theories been suggested to be importantparameters that determine the range and magnitude of the attractive force between
Ž .hydrophobic surfaces Parker et al., 1994a; Yaminsky et al., 1996 .The subject of ‘hydrophobic interactions’ between macroscopic surfaces and their
range is still controversial and opposing views and interpretations can be found in theliterature. It would go to far to discuss this controversial subject any further here, but inthe remainder of this chapter we will take a pragmatic view. We will use theexperimental finding that an additional attraction does exist between macroscopichydrophobic surfaces, but we will avoid giving this attraction a molecular interpretation.When one adds an exponential attraction to the DLVO-forces, as in the extended DLVOtheory of Yoon et al., and calculate the forces between a hydrophobised mineral particleand an air-bubble under the same conditions as in Fig. 8b, one sees that the short-rangeinteraction now is attractive and the air bubble will be attached to the surface once the
Ž .force barrier has been overcome Fig. 11 .
4.5. Structural or solÕation forces
When the DLVO-forces were discussed it was implicitly assumed that the liquidbetween the surfaces was a structureless continuum, characterised by its bulk dielectricproperties. However, at short separations this is not a good approximation and themolecular nature of the liquid has to be considered. The presence of the surface will
Fig. 11. The solid lines show the expected force between spheres after adding a hydrophobic attraction of theŽ . Ž .form FrR mNrm sy150 exp y Drl , where ls1.5 nm, to the DLVO-forces in Fig. 8b. The Hamaker
y2 0 Ž .constant for the quartz–water–air bubble system is y0.8=10 J, the surface potentials where in a equalŽ .on both surfaces, 50 mV, while in b they where 50 and y30 mV. The long dashed curves correspond to
double-layer forces only, whereas the short dashed ones are obtained after adding the van der Waals force.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 23
modify the packing of liquid molecules outside it, and the density of the liquid will varywith the separation from the surface in an oscillatory fashion approaching the bulk valuea few molecular layers away from the surface. For spherical molecules the periodicity ofthe oscillations is close to the diameter of the molecule. When two particles comesufficiently close to each other their solvent layers start to overlap. This results in achange in liquid density next to the surface that also varies in an oscillatory fashion withparticle separation. This change in liquid density is the cause of the oscillating structural
Žforce observed between smooth solid surfaces immersed in various liquids Christenson,.1988; Israelachvili, 1991 .
The situation in water is, because of its hydrogen bonding properties, more compli-cated than in non-polar liquids. Measurements of the forces acting between micasurfaces in electrolyte solutions have shown that once the salt concentration is highenough, typically above 1–10 mM depending on the electrolyte, a short-range repulsiveforce decaying exponentially with a decay constant of about 1 nm dominates the
Ž .interaction Pashley, 1981a,b; Pashley and Israelachvili, 1984 . One example of such aforce curve is provided in Fig. 9. This force, known as a hydration force, is in the caseof mica presumably due to dehydration of adsorbed cations. It has been given atheoretical interpretation by Gruen and Marcelja in terms of a surface induced polarisa-
Ž .tion of the liquid water Gruen and Marcelja, 1983 . We note that high precisionmeasurements have shown that at short separations an oscillatory force profile is
Žsuperimposed on the exponentially decaying hydration force Israelachvili and Pashley,.1983 indicating, not surprisingly, that normal packing constraints as well as specific
surface–water interactions are influencing the short-range forces between hydrophilicsurfaces in aqueous solutions. It has been convincingly demonstrated that hydration
Ž .forces may affect the stability of colloidal dispersions Healy et al., 1978 but it has toour knowledge not yet been shown that hydration forces are important in the flotationprocess.
4.6. Interactions due to the presence of polymers
High-molecular-weight polymers, used as depressants and flocculating agents com-prises an important component in mineral processing. Organic regulating agents, repre-
Ž .sented by natural polymers or their derivatives , such as modified cellulose, starch, andŽ .tannins are also commonly used Leja, 1982 . The presence of polymers in solution
andror on particle surfaces affects the interparticle forces in a way that depends onwhether they adsorb to the particle surface irreversibly, reversibly or not at all. Otherfactors which determine the sign, range and magnitude of polymer induced forces are,
Že.g., solventrpolymer interactions, surface coverage and polymer concentration Fleer et.al., 1993 . The different types of forces which may be generated by polymers are
discussed briefly below.
4.6.1. Forces between surfaces carrying irreÕersibly adsorbed polymersModels describing the interaction between particles carrying irreversibly adsorbed
Ž .flexible polymers have been developed by de Gennes 1979 and by Scheutjens andŽ .Fleer Fleer et al., 1993 . These theories are applicable when the polymer adsorp-
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨24
tionrdesorption rate is slow compared with how fast the polymer coated interfacesapproach each other. Under such circumstances the total amount of polymer on thesurfaces is independent of the surface separation and the system is not in trueequilibrium with bulk solution. However, it is often the case that the speed of approachis sufficiently slow for the irreversibly adsorbed polymers to adopt the most favourableconformation for each surface separation. Hence, there is an equilibrium within thelayer. This condition is referred to as quasi-equilibrium or restricted equilibrium.
Non-ionic polymers may give rise to an attractive bridging interaction when polymersegments belonging to the same molecule are bound to two surfaces, whereas polyelec-trolytes may generate bridging forces once segments from the same macromolecule are
Ž .close to both surfaces Dahlgren et al., 1993 . Bridging is important when the surfacecoverage is low. This situation may arise when the polymer concentration is low orwhen the time for adsorption is short. In such cases it is possible for the polymer toadopt a larger number of favourable conformations when the surfaces are at intermediatedistances from each other compared to when they are far apart, resulting in an increasein entropy and this is the molecular mechanism behind the attraction. However, at smallenough separations the number of available polymer conformations decreases againwhich results in a repulsion. The range of the attraction is determined by the length ofthe polymer tails extending from the surface.
As the surface coverage increases the effect of bridging polymers becomes lessimportant, whereas force contributions arising from interactions between polymers
Ž .adsorbed onto different surfaces become dominant. Such contributions are: i anosmotic contribution including changes in ideal entropy of mixing and changes in
Ž .solvation of the polymer segments, and ii an elastic contribution due to a reduction ofthe number of possible conformations of the polymer chains at smaller surface separa-tions.
Under restricted equilibrium conditions, the second contribution always becomesincreasingly repulsive as the surface separation is decreased, and it dominates at smallenough separations. The osmotic contribution may be either repulsive or attractivedepending on the solvent quality. It is unfavourable for polymer chains to interpenetratein good solvents, whereas such a process is favourable in poor solvents. The solventquality, which is a measure of the strength of the interaction between individualsegments compared to those between segment and solvent, is usually expressed by thex-parameter. The x-parameter is larger than 0.5 in good and less than 0.5 in poorsolvents. Under poor solvency conditions the outer part of the interaction betweenpolymer coated surfaces is thus expected to be attractive.
The most effective steric stabilisers are often diblock copolymers where one of theblocks, the anchor block, is strongly adsorbed to the surface whereas the other block, thebuoy, experiences good solvency conditions and thus extends far out into the solution.
Ž .One example of such a block copolymer is B E , where the butylene block B anchors8 41
the polymer to hydrophobic surfaces or to air–water interfaces whereas the ethyleneŽ .oxide block E extends into the solution. The forces measured between two hydropho-
bised mica surfaces coated with a layer of B E are shown in Fig. 12. Here, we see that8 41
adsorption of the block copolymer completely masks the attractive van der Waals andhydrophobic forces that act between uncoated hydrophobic surfaces, and instead a
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 25
Fig. 12. Force normalised by radius as a function of absolute surface separation between hydrophobised micasurfaces across a 0.1-mM KBr solution containing 50 ppm B E .8 41
purely repulsive steric force is encountered. The functional form of the measuredŽinteraction agrees well with forces calculated using lattice mean field theory Schillen et´
.al., 1997 .Ž .The force disjoining pressure acting between two air–water interfaces in a single
foam film stabilised by B E were measured employing the thin film balance tech-8 41
nique. Just as was the case for hydrophobic solid surfaces coated with this polymer, theŽ .interaction is completely dominated by a steric force Fig. 13 . The range of the force is
Ž .about the same, 22–23 nm, at all B E concentrations 20–300 ppm . This indicates8 41
that the length of the longest tails is the same at all polymer concentrations. However, as
Fig. 13. Disjoining pressure isotherms for single foam films stabilised by the diblock copolymer B E . In8 41
addition to the polymer, the solutions also contained 10 mM KBr. The diblock copolymer concentrations wereŽ . Ž . Ž . Ž .20 ppm l , 50 ppm e , 100 ppm ' , and 300 ppm ^ . The measurements were carried out with the thin
film balance technique.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨26
the polymer concentration is increased the steric interaction at smaller distances be-comes larger, indicating that as the adsorbed amount increases it becomes increasinglyunfavourable to compress the adsorbed layers. Another feature that is evident from thedata displayed in Fig. 13 is that one may distinguish three regimes in the disjoiningpressure isotherm. In the outermost regime the film thickness decreases rapidly withincreasing pressure. However, once the disjoining pressure has reached about 2 kPa, theisotherm becomes considerably steeper. The film thickness at this transition is observed
Žat larger separations for more concentrated polymer solutions from 18.5 nm in the 20.ppm solution to 21 nm in the 300 ppm B E solution . The reason for this change in8 41
character of the interaction is not clear. At very high pressures the slope of the disjoiningpressure isotherm decreases again. We propose that this is due to a reduction in theadsorbed amount at high pressures. This depletion of the polymer from the interfaceeventually leads to rupture of the foam film at high pressures.
4.6.2. Forces between surfaces carrying reÕersibly adsorbed polymersŽ .The theoretical models developed by de Gennes 1987 and Scheutjens and Fleer
Ž .Fleer et al., 1993 both predict that under full equilibrium conditions, i.e., whenpolymers are free to desorb as the particles approach each other, purely attractive forces
Ž .are generated by homopolymers de Gennes, 1982 . The reason for this is that, as somepolymers desorb from the surfaces, a greater fraction of the remaining polymers willattach to both surfaces and a bridging attraction will pull them towards each other.Hence, there cannot be any steric stabilisation by adsorbed homopolymers under fullequilibrium conditions, but the reason that homopolymers can be used as stericstabilisers is that, due to a very slow desorption, they are retained on the surface duringa collision event and the interactions are thus better described by theories for interac-tions between surfaces carrying irreversibly adsorbed polymers.
4.6.3. Forces due to non-adsorbing polymersThe concentration of polymer segments of non-adsorbing polymers is for entropic
reasons lower close to a surface than in bulk solution. An attractive osmotic force isgenerated when the polymer depleted layer extends all the way between the surfaces.The magnitude of this so-called depletion attraction increases strongly with the polymerconcentration. In concentrated systems the magnitude of the force is sufficient to cause
Ž .flocculation Feigin and Napper, 1980; Fleer et al., 1993 .
5. Final remarks
It can be said that the interactions between like interfaces are fairly well understood,and the mathematical treatment allows for fairly easy calculations of the interactionspresent. However, systems more apt to flotation most often involves interfaces which are
Ž .different, for instance with respect to their surface charge or potential , their ability todeform under load, and whose affinity for the additives present in most applied systemsdiffer. Another question not dwelled upon here is the fact that presence of divalent andother ions of higher valency will necessitate further assumptions in the treatment of the
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 27
double-layer interactions. Yet another complication is the vast presence of hydrophobicmolecules which imply that hydrophobic interactions should be taken into account, andtoday there is no widespread agreement on how to do this. It therefore becomesnecessary to invoke more approximate relations for asymmetric systems which facilitatecalculations for estimates of the interactions present, methods which might have lesspredictive power. It is nevertheless essential to obtain an assessment of the magnitudeand range of the interaction forces to be expected in a certain system, in order to be ableto predict its behaviour and make proper adjustments to the formulations used. Thedevelopment of new techniques such as the AFM and Thin Film Balance, also makes itpossible to experimentally determine the interaction profile for air-bubble–particlesystems. Increased understanding of the interactions in such systems may provide a newmeans to enhance the efficiency of certain processes by removing constituents ratherthan adding a few more, which in turn can turn out to be a more efficient way to obtainan ecologically acceptable and therefore economical process.
References
Attard, P., Mitchell, D.J., Ninham, B.W., 1988. Beyond Poisson–Boltzmann: images and correlations in theŽ .electric double layer: II. Symmetric electrolyte. J. Chem. Phys. 89 7 , 4358–4367.
Beesley, A.H., Evans, D.F., Laughlin, R.G., 1988. Evidence for the essential role of hydrogen bonding inŽ .promoting amphiphilic self-assembly: measurements in 3-methylsydnone. J. Phys. Chem. 92 3 , 791–793.
Bell, G.M., Peterson, G.C., 1972. Calculation of the electric double-layer force between unlike spheres. J.Ž .Colloid Interface Sci. 41 3 , 542–566.
Bergenstahl, B., Stenius, P., 1987. Phase diagrams of dioleoylphosphatidylcholine with formamide, methylfor-˚mamide and dimethylformamide. J. Phys. Chem. 91, 5944–5948.
Bergeron, V., Fagan, M.E., Radke, C.J., 1993. Generalized entering coefficients: a criterion for foam stabilityagainst oil in porous media. Langmuir 9, 1704–1713.
Butt, H.-J., 1994. A technique for measuring the force between a colloidal particle in water and a bubble. J.Colloid Interface Sci. 166, 109–117.
Butt, H.-J., Jaschke, M., Ducker, W., 1995. Measuring surfaces forces in aqueous electrolyte solution with theatomic force microscope. Bioelectrochem. Bioenerg. 38, 191–201.
Chan, D.Y.C., Healy, T.W., White, L.R., 1976. Electrical double layer interactions under regulation by surfaceionization equilibria—dissimilar amphoteric surfaces. J. Chem. Soc., Faraday Trans. 1 72, 2844–2865.
Christenson, H.K., 1988. Non-DLVO forces between surfaces—solvation, hydration, and capillary effects. J.Dispersion Sci. Technol. 9, 171.
Claesson, P.M., Christenson, H.K., 1988. Very long range attractive forces between uncharged hydrocarbonand fluorocarbon surfaces in water. J. Phys. Chem. 92, 1650.
Dahlgren, M.A.G. et al., 1993. Salt effects on the interaction between adsorbed cationic polyelectrolyte layersŽ .—theory and experiment. J. Phys. Chem. 97 45 , 11769.
de Gennes, P.-G., 1979. Scaling Concepts in Polymer Physics. Cornell Univ. Press, Ithaca.de Gennes, P.G., 1982. Polymers at an interface: 2. Interaction between two plates carrying adsorbed polymer
layers. Macromolecules 15, 492–500.de Gennes, P.G., 1987. Polymers at an interface. A simplified view. Adv. Colloid Interface Sci. 27, 189.Derjaguin, B., 1934. Untersuchungen uber die Reibung und Adhasion, IV. Kolloid Zeitung 69, 155–164.¨ ¨Derjaguin, B.V., Duhkin, S.S., 1960. Theory of flocculation of small and medium size particles. Trans. Inst.
Min. Metal. 70, 221.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨28
Derjaguin, B.V., Abrikosova, I.I., Lifshitz, E.M., 1956. Direct measurement of molecular attraction betweenŽ .solids separated by a narrow gap. Q. Rev. 10 3 , 295–329.
Derjaguin, B.V., Titiyevskaya, A.S., Abrikossova, I.I., Malkina, A.D., 1954. Investigations of the forces ofinteraction of surfaces in different media and their application to the problem of colloidal stability. Discus.Faraday Soc. 18, 24–41.
Derjaguin, B.V., Rabinovich, Y.I., Churaev, N.V., 1978. Direct measurement of molecular forces. Nature 272,313–318.
Devereux, O.F., de Bruyn, P.L., 1963. Interaction of Plane-Parallel Double Layers. MIT Press, MA.Ducker, W.A., Pashley, R.M., Senden, T.J., 1991. Direct measurements of colloidal forces using an atomic
force microscope. Nature 353, 239.Ducker, W.A., Senden, T.J., Pashley, R.M., 1992. Measurement of forces in liquids using a force microscope.
Langmuir 8, 1831–1836.Ducker, W.A., Xu, Z., Israelachvili, J.N., 1994. Measurements of hydrophobic and DLVO forces in
bubble–surface interactions in aqueous solutions. Langmuir 10, 3279–3289.Dzyaloshinskii, I.E., Lifshitz, E.M., Pitaevskii, L.P., 1961. The general theory of van der Waals forces. Adv.
Phys. 10, 165–209.Eriksson, L.G.T., Claesson, P.M., Ohnishi, S., Hato, M., 1997. Stability of dimethyldioctadecylammonium
bromide Langmuir–Blodgett films on mica in aqueous salt solutions-implications for surface forcemeasurements. Thin Solid Films 300, 240–255.
Evans, D.F., Ninham, B.W., 1986. Molecular forces in the self-organization of amphiphiles. J. Phys. Chem. 90Ž .2 , 226–234.
Evans, D.F., Wennerstrom, H., 1994. The Colloidal Domain Where Physics, Chemistry, Biology, and¨Technology Meet. VCH Publisher, New York.
Exerowa, D., Scheludko, A., 1971. Porous plate method for studying microscopic foam and emulsion films.Chim. Phys. 24, 47–50.
Feigin, R.I., Napper, D.H., 1980. Depletion stabilization and depletion flocculation. J. Colloid Interface Sci. 75Ž .2 , 525–541.
Fielden, M.L., Hayes, R.A., Ralston, J., 1996. Surface and capillary forces affecting air bubble–particleŽ .interactions in aqueous electrolyte. Langmuir 12 15 , 3721–3727.
Ž .study. J. Chem. Phys. 80 5 , 2221–2228.Hamaker, H.C., 1937. The London–van der Waals Attraction between spherical particles. Physica IV, 1058.Hato, M., 1996. Attractive forces between surfaces of controlled ‘hydrophobicity’ across water: a possible
range of ‘hydrophobic interactions’ between macroscopic hydrophobic surfaces across water. J. Phys.Chem. 100, 18530–18538.
Healy, T.W., Homola, A., James, R.O., Hunter, R.J., 1978. Coagulation of amphoteric latex colloids:reversibility and specific ion effects. Faraday Discus. Chem. Soc. 65, 156–163.
Herder, P.C., 1990. Forces between hydrophobed MICA surfaces immersed in dodecylammonium chloridesolution. J. Colloid Interface Sci. 134, 336–345.
Israelachvili, J.N., 1973. Thin film studies using multiple-beam interferometry. J. Colloid Interface Sci. 44,259.
Israelachvili, J.N., 1991. Intermolecular and Surface Forces. Academic Press, London.Israelachvili, J.N., 1995. Surface forces and microrheology of molecularly thin liquid films. In: Bhushan, B.
Ž .Ed. , Handbook of MicrorNanotribology. CRC Series, Mechanical and Materials Science. CRC Press,Boca Raton.
Israelachvili, J.N., Adams, G.E., 1978. Measurement of forces between two mica surfaces in aqueousŽ .electrolyte solutions in the range 0–100 nm. J. Chem. Soc., Faraday Trans. 1 74 , 975–1001.
Israelachvili, J.N., McGuiggan, P.M., 1990. Adhesion and short-range forces between surfaces: I. Newapparatus for surface force measurements. J. Mater. Res. 5, 2223–2231.
Israelachvili, J., Pashley, R., 1982. The hydrophobic interaction is long range, decaying exponentially withdistance. Nature 300, 341–342.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨ 29
Israelachvili, J.N., Pashley, R.M., 1983. Molecular layering of water at surfaces and origin of repulsivehydration forces. Nature 306, 249–250.
Israelachvili, J.N., Tabor, D., 1972. The measurement of van der Waals dispersion forces in the range 1.5 to130 nm. Proc. R. Soc. A 331, 19.
Jonstromer, M., Sjoberg, M., Warnheim, T., 1990. Aggregation and solvent interaction in nonionic surfactant¨ ¨ ¨systems with formamide. J. Phys. Chem. 94, 7549–7555.
Kjellander, R., 1996. Ion–ion correlations and effective charges in electrolyte and macroion systems. Berichteder Bunsengesellschaft fur Physikaische Chemie 100, 894–904.¨
Kjellander, R., Marcelja, S., 1984. Correlation and image charge effects in electric double layers. Chem. Phys.Lett. 112, 49–53.
Klein, J., 1983. Forces between mica surfaces bearing adsorbed macromolecules in liquid media. J. Chem.Ž .Soc., Faraday Trans 1 79 , 99.
Larson, I., Drummond, C.J., Chan, D.Y.C., Grieser, F., 1997. Direct force measurements between silica andalumina. Langmuir 13, 2109–2112.
Leja, J., 1982. Surface Chemistry in Froth Flotation. Plenum, New York.Mahanty, J., Ninham, B.W., 1976. Dispersion Forces. Academic Press, London.Ninham, B.W., Parsegian, V.A., 1970. van der Waals forces across triple-layer films. J. Chem. Phys. 52,
4578–4587.Parker, J.L., 1994. Surface force measurements in surfactant systems. Prog. Surface Sci. 47, 205.Parker, J.L., Claesson, P.M., 1994. Forces between hydrophobic silanated glass surfaces. Langmuir 10,
635–639.Parker, J.L., Christenson, H.K., Ninham, B.W., 1989. Device for measuring the force and separation between
two surfaces down to molecular separations. Rev. Sci. Instrum. 60, 3135–3138.Parker, J.L., Claesson, P.M., Attard, P., 1994a. Bubbles, cavities, and the long-ranged attraction between
hydrophobic surfaces. J. Phys. Chem. 98, 8468–8480.Parker, J.L., Claesson, P.M., Wang, J.-H., Yasuda, H.K., 1994b. Surface forces between plasma polymer
films. Langmuir 10, 2766–2773.Pashley, R.M., 1981a. DLVO and hydration forces between mica surfaces in Liq, Naq, Kq and Csq
electrolyte solutions: a correlation of double-layer and hydration forces with surface cation exchangeproperties. J. Colloid Interface Sci. 83, 531.
Pashley, R.M., 1981b. Hydration forces between mica surfaces in aqueous electrolyte solutions. J. ColloidInterface Sci. 80, 153–162.
Pashley, R.M., Israelachvili, J.N., 1984. DLVO and hydration forces between mica surfaces in Mg2q, Ca2q,Sr 2q and Ba2q chloride. J. Colloid Interface Sci. 97, 446.
Pashley, R.M., McGuiggan, P.M., Ninham, B.W., Evans, D.F., 1985. Attractive forces between unchargedhydrophobic surfaces: direct measurements in aqueous solution. Science 229, 1088–1089.
Paulson, O., Pugh, R.J., 1996. Flotation of inherently hydrophobic particles in aqueous solutions of inorganicelectrolytes. Langmuir 12, 4808–4813.
Peschel, G., Belouschek, P., Muller, M.M., Muller, M.R., Konig, R., 1982. The interaction of solid surfaces in¨ ¨ ¨aqueous systems. Colloid Polymer Sci. 260, 444–451.
Rabinovich, Y.I., Derjaguin, B.V., 1988. Interaction of hydrophobized filaments in aqueous electrolytesolutions. Colloids and Surfaces 30, 243–251.
Rabinovich, Y.I., Yoon, R.-H., 1994. Use of atomic force microscope for the measurements of hydrophobicforces between silanated silica plate and glass sphere. Langmuir 10, 1903–1909.
Rabinovich, Y.I., Guzonas, D.A., Yoon, R.-H., 1993a. An infrared investigation of the structure ofŽ .Langmuir–Blodgett films of dimethyldioctadecylammonium bromide DDOAB on silicon in water. J.
Colloid Interface Sci. 155, 221–225.Rabinovich, Y.I., Guzonas, D.A., Yoon, R.-H., 1993b. Role of chain order in the long-range attractive force
between hydrophobic surfaces. Langmuir 9, 1168–1170.Ramadan, M.S., Evans, D.F., Lumry, R., 1983. Why micelles form in water and hydrazine. A reexamination
of the origins of hydrophobicity. J. Phys. Chem. 87, 4538–4543.Ravishankar, S.A., Yoon, R.-H., 1997. Long-range hydrophobic forces in the amine flotation of quartz.
Ž .Minerals and Metallurgical Processing May , 10–17.Scheludko, A., 1967. Thin liquid films. Adv. Colloid Interface Sci. 1, 391–464.
( )J.C. Froberg et al.r Int. J. Miner. Process. 56 1999 1–30¨30
ŽSchillen, K., Claesson, P.M., Malmsten, M., Linse, P., Booth, C., 1997. Properties of poly ethylene´. Ž .oxide –poly butylene oxide diblock copolymers at the interface between hydrophobic surfaces and water.
J. Phys. Chem. B 101, 4238–4252.Schulze, H.J., 1984. Physico-chemical Elementary Processes in Flotation. Developments in Mineral Process-
ing, 4. Elsevier, Amsterdam.Tabor, D., Winterton, R.H.S., 1969. The direct measurement of normal and retarded van der Waals forces.
Proc. R. Soc. A 312, 435.Tanford, C., 1980. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. Wiley, New
York.Verwey, E.J.W., Overbeek, J.T.G., 1948. Theory of the Stability of Lyophobic Colloids. Elsevier, Amsterdam.Warnheim, T., Jonsson, A., 1988. Phase diagrams of alkultrimethylammonium surfactants in some polar¨ ¨
solvents. J. Colloid Interface Sci. 125, 627–633.Warnheim, T., Jonsson, A., Sjoberg, M., 1990. Phase diagrams for cationic surfactant in polar solvent systems.¨ ¨ ¨
Prog. Colloid Polymer Sci. 82, 271–279.Wood, J., Sharma, R., 1994. Preparation of a robust hydrophobic monolayer on mica. Langmuir 10,
hydrophobic monolayers. Langmuir 12, 1936–1943.Yoon, R.-H., Mao, L., 1996. Application of extended DLVO theory: IV. Derivation of flotation rate equation
from first principles. J. Colloid Interface Sci. 181, 613–626.Yoon, R.-H., Ravishankar, S.A., 1994. Application of extended DLVO theory: III. Effect of octanol on the
long-range hydrophobic forces between dodecylammonium-coated mica surfaces. J. Colloid Interface Sci.166, 215–224.
Yoon, R.-H., Ravishankar, S.A., 1996a. Long-range hydrophobic forces between mica surfaces in alkalinedodecylammonium chloride solutions. J. Colloid Interface Sci. 179, 403–411.
Yoon, R.-H., Ravishankar, S.A., 1996b. Long-range hydrophobic forces between mica surfaces in dodecylam-monium chloride solutions in the presence of dodecanol. J. Colloid Interface Sci. 179, 391–402.