Surface redox chemistry and mechanochemistryof insulating polystyrene nanospheres†
Thomas S. Varley, Martin Rosillo-Lopez, Sandeep Sehmi, Nathan Hollingsworthand Katherine B. Holt*
Cyclic voltammetry (CV) of polystyrene nanospheres was carried out after immobilisation onto boron-doped
diamond electrodes. Although the polystyrene is insulating, a voltammetric response was obtained. This was
attributed to the high surface area of the nanospheres, allowing the redox chemistry of the polystyrene
surface to be probed despite the non-conducting nature of the bulk. The polystyrene redox response was
found to be strongly dependent on prior mechanical agitation. Centrifuged, sonicated and vortexed
polystyrene nanospheres all exhibited significantly higher oxidation currents than the non-agitated
polystyrene. Mechanical treatment by sonication and centrifugation was found to bring about changes to
surface chemistry of the polystyrene spheres, in particular the introduction of oxygen functionalities.
For these samples the CV response is attributed to the presence of surface phenol functionalities. On the
non-agitated and vortex treated polystyrene surfaces X-ray photoelectron spectroscopy revealed an absence
of oxygen functionalities that could explain the redox response. Repetition of the CV experiment in the
presence of a solution spin trap suggests that radical species play a role in the observed response. For the
vortexed sample the increased oxidation currents were attributed to significant surface roughening and
deformation, as revealed by Transmission Electron Microscopy.
1. Introduction
Non-conducting materials, such as wide band-gap polymers,are not typically associated with measurable electrochemistry.However, surface redox chemistry of insulating materials attributedto defect sites, dangling bonds and band-gap states maygive rise to interesting electrochemistry. For nanoparticulatematerials the large surface atom to bulk atom ratio allows the redoxchemistry of surface states and functional groups to dominate overthe insulating response of the bulk, enabling a voltammetricresponse to be obtained. We have previously reported this to bethe case for insulating diamond nanoparticles, where surface stateswithin the band gap are able to exchange electrons with theirsurroundings (solution redox probes, an underlying electrode).1
In this study we report the cyclic voltammetry (CV) response ofelectrode-immobilised polystyrene (PS) nanospheres (Scheme 1).We show here that, despite their insulating nature, PS nanobeadsexhibit a redox response when immobilised on a boron-dopeddiamond (BDD) electrode. In addition, we subject the PS nano-spheres to different forms of mechanical agitation and activation,namely centrifugation, sonication and vortex mixing, prior to
immobilisation onto the electrode. We find that the redoxresponse of the PS is strongly influenced by the nature of theprevious mechanical treatment, indicating that mechanically-induced changes to the PS surface may be detected andquantified electrochemically.
The effect of mechanical force upon the surface chemistry ofinsulating polymers has been previously studied with regardsto understanding mechanical degradation mechanisms2 andthe ‘mechanochemistry’ related to surface radical formation onpolymers.3 Surface radicals (mechanoradicals) are formedvia homolytic bond cleavage of the polymer chain under mechan-ical strain.3,4 Although polymer covalent bonds are very strong(typical C–C bond strength is ca. 350 kJ mol�1) experimental resultsfrom Electron Spin Resonance techniques show that these
radicals can be formed as a result of mechanical work upon thesurface and persist for some period of time.4 There is substan-tial technological relevance to studying this chemistry, asmastication (mechanical breakdown of polymers to achievedesired properties), recycling by pulverisation and lubricationprocesses can all generate mechanoradicals.5
Polymer mechanochemistry, well-established in the tribology andpolymer literature, has recently been re-examined by researchersengaged in study of electrostatic charge accumulation.6–11 Electro-static charging (tribocharging, contact electrification) is the separa-tion of charge that occurs when two surfaces are contacted togetherand then separated.6 Although an everyday phenomenon that hasbeen well studied, a unified theory explaining the mechanism ofcharge accumulation by wide band-gap polymers has not yet beenachieved. The exact nature of the charge is not firmly established,with both electrons7 and ions8 suggested as charge carriers. Transferof material fragments from one surface to another resulting in bothpositively and negatively charged ‘mosaics’ has also been observed.9
Regardless of the nature of the charge, the role of surface radicalshas been shown to be important,10 with recent studies showingcharge and radical co-localisation on polymer surfaces after con-tact.11 Moreover, treatment of a polymer surface with a radical spintrap molecule resulted in no charge accumulation, confirming theco-dependence of the two.11
Our primary motivation in this study was to establish whethersurface redox chemistry for otherwise insulating materials couldbe observed using voltammetry, through the use of high surfacearea nanomaterials. However, given the proposed importance ofsurface radicals in charge accumulation and electrostatic char-ging10,11 we additionally probed the redox response of PS nano-spheres after surface activation by mechanical means, todetermine whether surface mechanoradicals or other reactivespecies thus formed showed different redox activity that could beused in their characterisation.
2. Experimental methodsPreparation of PS nanosphere suspensions
Sulfate-terminated PS beads of diameter 800 nm were purchasedfrom Sigma-Aldrich (LB7, 10% solids in aqueous suspension). Thesuspension also contains 0.1–0.5% sodium dodecyl sulfate surfac-tant and 0.2% inorganic salts (potassium sulfate and sodiumbicarbonate). To remove these extraneous agents the PS beadswere cleaned by membrane dialysis against water. The membranewas a pre-treated standard grade regenerated cellulose dialysismembrane (Spectrum Laboratories), with a molecular weight cutoff of 3500 Da and a flat width of 45 mm. Membranes were receivedin an aqueous solution containing sodium azide preservative(0.05%), which was removed by dialysis against distilled water for2 h. Dialysis of the PS solution (diluted tenfold from the commer-cial stock solution) was subsequently performed against distilledwater for 48 h, changing the water every 12 h. The resulting cleanedsuspension was 1% PS solids in deionised water and was stored ina glass container throughout. Alternatively the PS nanosphereswere separated from their native solution by centrifugation (with
three cleaning and rinsing steps) followed by re-suspension indeionised water to give a final solution of 1% PS solids. Thesesamples were both centrifuged and stored in polypropylene cen-trifuge tubes (Eppendorf 1.5 mL Flex tube microcentrifuge tubes);according to the manufacturers these tubes are ‘‘manufacturedwithout the use of slip agents, plasticizers and biocides-substancesthat have been shown to leach from plastic consumables into thesample and negatively affect bio-assay results’’.12
For some experiments the PS suspensions were subjected tomechanical agitation, either by sonication for 1 hour in an ultrasonicbath or by vortex mixing. Sonication was performed using a FisherScientific Ultrasonic bath (FB 15049) filled with deionised water. PSsuspensions were held in place via a clamp and positioned such thatas much of the polypropylene centrifuge tube was submerged aspossible. PS solutions were vortex mixed within their polypropylenecentrifuge tubes using a Vortex Genie 2 (Scientific IndustriesBohemia, N.Y., USA) on setting 3. Control experiments were alsocarried out by vortexing samples in a glass container, to ensure thatmaterial transfer from the plastic container walls was not respon-sible for the redox activity (see ESI†).
To further ensure that plasticizers or impurities did not leachfrom container walls and contaminate the suspension, thecomposition of the aqueous supernatant after storage andmechanical treatment was determined by Nuclear MagneticResonance (NMR). The PS nanosphere surface chemistry andmorphology were also investigated, before and after mechanicaltreatment, using Transmission Electron Microscopy (TEM) andX-ray Photoelectron Spectroscopy (XPS). See ESI† for details.
Preparation and electrochemistry of PS-modified electrodes
Prior to modification with PS, the boron-doped diamond (BDD)working electrode was polished with 0.05 mm alumina slurry, rinsedand cycled repeatedly between 0 and 1 V vs. Ag/AgCl in the relevantbuffer solution until a stable background was achieved. The PSsuspensions were drop-coated onto a boron-doped diamond (BDD)electrode to form an adherent film with average coverage (5� 1) �105 beads mm�2. Reproducibility of surface coverage of PS wasevaluated by inspection of each modified electrode under an opticalmicroscope and analysis of the resulting image using imagingsoftware (see ESI† for details). The surface coverage of PS was thesame regardless of suspension preparation method. CV experi-ments with the electrode-immobilised PS was carried out with astandard three electrode cell in 0.2 M phosphate buffer solutions(PBS) containing different proportions of K2HPO4 and KH2PO4 toadjust the pH. All solutions were made up with 18 MO cmdeionised water. Spin trap experiments were performed using thesame three electrode cell, but using 0.2 M PBS containing 10 mMN-tert-butyl-a-phenylnitrone (PBN) as the electrolyte solution.
3. ResultsCV response of electrode-immobilised PS nanospheres cleanedby centrifugation
In contrast to the featureless BDD background response, forthe PS-modified electrode oxidation currents begin to flow
at ca. 0.45 V, rising to a distinct peak centred at ca. 0.7 V (Fig. 1).There is no corresponding reduction peak on the reverse scan,indicating the oxidation process is irreversible. However asmall reduction peak at about 0.25 V is noted on the reversescan and on the second scan a corresponding oxidation peak isobserved at 0.28 V. This reversible couple centred at ca. 0.26 V isstable to repeated cycling and results from the oxidation processat 0.7 V (Fig. 1 inset, ESI†). The peak currents exhibit a propor-tional relationship with voltammetric scan rate, confirming asurface-confined species. On the second and subsequent scansthe oxidation peak at 0.7 V is no longer present, indicating thatthe species responsible is not regenerated following its oxidationduring the first cycle. The reproducibility of the CV response wasdetermined by evaluating the charge passed under the 0.7 V and0.28 V oxidation peaks for ten repetitions with a freshly modifiedelectrode (ESI†). It was determined that (28 � 5) nC of chargeare passed during the oxidation process centred at 0.7 V, while(2.5 � 0.5) nC is passed during the oxidation process at 0.28 V.Inspection of the electrode under a microscope showed thatcoverage of PS was unchanged after the CV scans had been carriedout. This observation indicates that PS remains immobilised on theelectrode throughout the experiment.
Effect of mechanical agitation on voltammetric response of PSnanospheres
When the PS nanospheres were cleaned by dialysis, rather thancentrifugation, the form of the CV was unchanged from that inFig. 1, but the magnitude of the oxidation currents was notice-ably decreased, with charge passed under the 0.7 V oxidationpeak being (13 � 3) nC. Larger currents for the centrifugedbeads could be attributed to mechanical ‘activation’ of the PSsurface by shear forces. To investigate the effect of mechanicalforce on the beads, the dialysed suspension was subject to
deliberate mechanical agitation, either by sonication or vortexmixing, prior to immobilisation onto the electrode. The form ofthe resulting CV for both methods was similar to Fig. 1 buthigher oxidation currents were observed. Fig. 2a shows the meancharge obtained from integration of the 0.7 V oxidation peak forall samples, where a statistically significant difference betweenthe data sets can be observed. In particular the vortexed PSshows greatly enhanced oxidation response, approximately tentimes that of the dialysed samples.
Time and oxygen dependence of voltammetric response formechanically agitated PS
The time-dependence of the enhanced redox response was inves-tigated over a period of 14 days. The PS sample was vortex mixedand every day thereafter three repeat CVs were carried out with thesame sample. The charge for the 0.7 V oxidation peak remainedsignificantly higher than for the dialysed, centrifuged and sonicatedbeads, indicating a permanent change to the PS surface (Fig. 2b).Similarly PS suspensions having undergone centrifugation or soni-cation maintained their enhanced oxidation currents compared tothe dialysed samples, although some decline in electrochemicalactivity was noted over a period of several months.
The effect of dissolved oxygen present during the mechanicalagitation process was explored by sonicating a deoxygenated PSsuspension (previously cleaned by dialysis) prior to drop-coatingonto the electrode. Sonication in the absence of oxygen gives rise totwo peaks associated with oxidation of the PS layer, at ca. 0.63 Vand ca. 0.77 V, in contrast to the single peak at 0.7 V observed aftersonication in the presence of oxygen (Fig. 2c). Additionally thereduction peak at ca. 0.2 V on the reverse peak is much lessprominent for the PS sonicated in the deoxygenated solution.
XPS analysis of the PS surface chemistry after mechanicaltreatment
The carbon 1s region spectrum for the dialysed PS (Fig. 3a) canbe fit with three peaks, corresponding to sp3 carbon–carbonbonding of the backbone (284.9 eV) and sp2 carbon–carbonbonding of the phenyl ring (284.3 eV and 291.1 eV).13 There are noobvious peaks in the region 296–289 eV that could be attributed tocarbon bonded to oxygen.14 Oxygen is present on the surface ofthe PS, as indicated by the oxygen 1s spectrum (ESI†) but this canbe attributed to the sulfate terminating groups.15 The centrifugedPS C 1s spectrum (Fig. 3b) can be fit with an additional two peaks:at 286.6 eV for carbon bound to oxygen via a single bond and at288.8 eV corresponding to carbon bound to more than one oxygen(or engaged in double bonding with one oxygen).14 The sonicatedPS (Fig. 3c) shows a greater degree of oxidation than the centri-fuged sample, with more prominent peaks observed in theregions for C–O (286.6 eV) and CQO (288.8 eV) bonding. Incontrast, the vortexed PS (Fig. 3d) shows similar oxygen content tothe dialysed PS, with no evidence from the C 1s spectrum thatcarbon is engaged in bonding with oxygen.
Additional information about the nature of the carbon–oxygen bonding is obtained from the valence band XPS spectrafor the samples (Fig. 4).14,16 An increase in intensity of thepeak(s) at ca. 25 eV is noted for the centrifuged and sonicated
Fig. 1 CV in 0.2 M pH 6 PBS of centrifuged PS immobilised on BDDelectrode. Red: 1st scan; blue: 2nd scan; black: background response ofunmodified BDD. Scan rate 0.02 V s�1.
samples, concomitant with a decrease in intensity of the peaksat 17 and 9 eV. The peaks at 25 eV are attributed to electronswith oxygen 2p character, i.e. originating from O(2p)–H(1s) andO(2p)–C(2p) molecular orbitals.14 The presence of these peaksfor the centrifuged and sonicated PS is consistent with the carbon–oxygen bonding in the core level spectra and increased oxygencontent for these samples. Valence band peaks at 17 and 9 eV areassigned to electrons of C 2s character, associated with the phenylring of PS.13 The loss of intensity of these peaks for the oxidisedsonicated and centrifuged samples therefore indicates a change incarbon bonding in the phenyl ring, perhaps through insertion ofsingly or doubly bonded oxygen. The sample found to contain thehighest oxygen content was PS sonicated in a deoxygenatedsolution. As noted above, the electrochemical response was differ-ent for this material also; however the increased oxygen content issomewhat surprising given the lack of dissolved oxygen available toreact with the PS surface.
TEM images of the PS samples after mechanical treatment
Dialysed, sonicated and vortexed PS nanospheres were exam-ined with TEM to observe changes in morphology associatedwith the mechanical treatment. Fig. 5a shows the dialysed PSbeads are smooth and spherical with no evidence of surfacedeformation. The sonicated PS beads (Fig. 5b) are still sphericalbut the surface is roughened and black patches of transferredmaterial are observable on the surface. In the regions wherethe sonicated beads touch, ‘strings’ of material can be seenextending between the beads, where material is shared. Underthe heating effect of the TEM beam, occasionally the contactedbeads are seen to move apart and as they do so they leaveirregular dark ‘scars’ on the surface where the beads weretouching and the surface has been roughened and materialtransferred (see ESI†). The dark patches on the beads havetherefore been attributed to prior collisions between thesebeads during the sonication treatment.
Vortex treatment leads to even more dramatic deformation inmorphology (Fig. 5c); here thick strings and lumps of polymermaterial can be seen extending between the beads, almost stickingthem together. Many of the beads show significant deformationand loss of their smooth spherical morphology. In addition someamorphous grey matter can be seen occasionally in the images,attributed to polystyrene material that has been pulled loose fromthe surface of the beads during impact.
Dialysed PS: origin of the redox response
The phenyl rings of PS do not undergo direct oxidation in thepotential range of this experiment, as benzene and tolueneare oxidised above 2 V vs. Ag/AgCl at BDD.17 The terminatingsulphate groups of the dialysed PS beads likewise oxidise athigher potentials (42.5 V vs. Ag/AgCl).18 Substituted phenolsundergo irreversible oxidation at 0.7 V (vs. Ag/AgCl) and above19,20
resulting in generation of redox couples in the region 0.0–0.4 V onsubsequent scans. This behaviour is remarkably similar to thatobserved for the electrode-immobilised PS. However for thedialysed PS the presence of surface phenol moieties is inconsis-tent with the XPS data, which shows that little if any carbon is
Fig. 2 (a) Average charge passed under 0.7 V oxidation peak for differentPS preparation methods; (b) time dependence of charge passed under0.7 V oxidation peak for vortexed PS; (c) CVs in 0.2 M pH 6 PBS (scan rate0.02 V s�1) comparing response of electrode-immobilised PS after priorsonication in air-saturated (black) and deoxygenated (red) solutions.
engaged in C–O bonding (Fig. 3a and 4); it is therefore impor-tant to consider possible origins for voltammetric response ofthe dialysed polystyrene.
While adsorption of impurities onto the electrode, ratherthan the PS, may be responsible for the voltammetric response,we have taken great precautions to prevent this and have beenunable to identify such a contaminant. The dialysed PS is storedexclusively in glass containers and the only brief contact with plasticis with a pipette tip during drop-coating. We ensure that all plasticused in our experiments is free from plasticizers and other possiblecontaminants (see Experimental). The electrode is carefully cleanedand a background response obtained in the absence of PS beforeevery experiment. The same drop-coating procedure (volume, dryingtime, environment) is used for all samples and yet we see asignificant difference between the data sets (Fig. 2a) so it seemsunlikely that an airborne contaminant or a process taking placeduring deposition is responsible for the response. The supernatantis tested for dissolved redox species using NMR (see ESI†) and wehave identified no species that could be responsible for the redoxresponse. There is no detectable solid material other than PS beadsobserved in multiple TEM images. Other control experiments arereported in the ESI.† There may be an as yet unidentified contam-ination source that is responsible for the redox peaks, however in itsapparent absence other explanations for the redox activity of theimmobilised PS layer must be considered.
One explanation for the discrepancy between the observedelectrochemistry and the absence of surface functionalities couldbe the difference in environment between the solution phaseelectrochemistry experiments and the ultra-high vacuum of the
Fig. 3 Carbon 1s region XPS spectra for PS (a) cleaned by dialysis; (b) cleaned by centrifugation; (c) subject to 1 hour sonication; (d) subject to 1 hourvortex mixing.
Fig. 4 Valence band XPS spectra for polystyrene cleaned by dialysis(black); cleaned by centrifugation (blue); subject to 1 hour sonication inair-saturated solution (pink); subject to 1 hour sonication in deoxygenatedsolution (green) and subject to 1 hour vortex mixing (red).
XPS experiments and the reorganisation of the polymer surface inresponse to the environment. It has been shown that PS surfaces arehighly dynamic and reorganise in response to changes in hydro-phobicity/hydrophilicity of the interface conditions.21,22 Whenexposed to water, polar groups are more likely to be pulled fromthe bulk to the surface to interact with the water and reduce the freeenergy at the interface.21 Conversely in a hydrophobic environment(e.g. in a nitrogen atmosphere) oxygen-containing polar groupsmigrate from the surface to the bulk of the polymer.22 If the UHVenvironment can be considered as hydrophobic, oxygen functional-ities, such as phenols, may move into the bulk and so not be soreadily detected with XPS. Another explanation could be presence ofpersistent and long lived radicals on the PS surface, where theunpaired electron is delocalised about the phenyl ring. This speciesis likely to be amenable to further oxidation at relatively mildpotentials with a possible product being a phenolic species. Sucha species would not readily be detected with XPS.
An alternate explanation for the observed redox response isthat ‘phenol-type’ functionalities are formed on the electrode-immobilised PS surface in situ through reaction with electrogener-ated hydroxyl radicals. It is well-established that hydroxyl radicalsare generated at BDD electrodes at oxidative potentials as a result ofwater discharge23 however usually at more positive potentials thanused in this study. However it has been suggested that non-diamond inclusions (e.g. sp2 impurities) in BDD films may supporttheir formation within the potential range of this study24 and thatelectrogenerated OH plays a role in the oxidation mechanism ofsulphides and disulphides at BDD at potentials well below that ofwater discharge.25 The CV of the unmodified BDD (Fig. 1) showsincreases in current at potentials of 0.6 V and above, which mayindicate water oxidation to form hydroxyl radicals at active sites onthe BDD surface. These hydroxyl radicals would then react imme-diately with the PS immobilised at the electrode interface. Theoxidation of benzene to phenol at different OH radical generatingmetal oxide electrodes has been reported to take place at similarapplied potentials to those used in this study.26
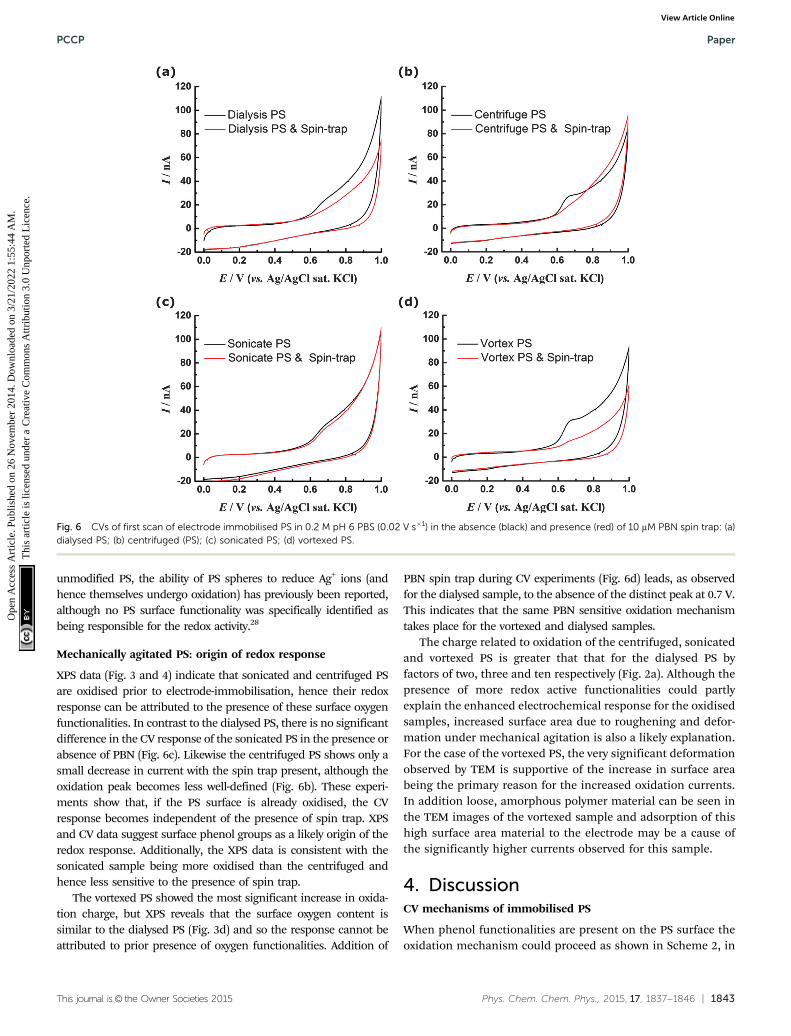
To test this proposed mechanism the CV experiments wererepeated with addition of 10 mM N-tert-butyl-a-phenylnitrone (PBN)to the solution (Fig. 6a). PBN is a radical spin trap molecule, whichcompetitively reacts with the OH radicals as they are generated27
preventing them from reacting with the PS surface. Without PBN theoxidation peak is observed at ca. 0.7 V, however when the spin trap ispresent the oxidation currents are significantly diminished. Thisappears to support a hypothesis that OH radicals generated at theBDD electrode are necessary to observe a redox response for dialysedPS nanospheres and indicates that although PS itself is not redoxactive within the potential range investigated, redox active phenolicgroups may be generated and then detected in situ at the BDDelectrode. However it is also possible that addition of PBN has adifferent effect, for example it may react directly with the surface ofthe PS if carbon-centred radicals are present. This would preventtheir further oxidation and hence suppress the voltammetric peak.Additionally the PBN may adsorb to the surface of the PS, preventingreorganisation and the migration of polar oxygen groups from withinthe bulk to the surface where they can be oxidised. Although wecannot definitively determine the origin of the redox response of the
Fig. 5 TEM images of (a) dialysed PS; (b) PS sonicated for 1 hour; (c) PSvortexed for 1 hour.
unmodified PS, the ability of PS spheres to reduce Ag+ ions (andhence themselves undergo oxidation) has previously been reported,although no PS surface functionality was specifically identified asbeing responsible for the redox activity.28
Mechanically agitated PS: origin of redox response
XPS data (Fig. 3 and 4) indicate that sonicated and centrifuged PSare oxidised prior to electrode-immobilisation, hence their redoxresponse can be attributed to the presence of these surface oxygenfunctionalities. In contrast to the dialysed PS, there is no significantdifference in the CV response of the sonicated PS in the presence orabsence of PBN (Fig. 6c). Likewise the centrifuged PS shows only asmall decrease in current with the spin trap present, although theoxidation peak becomes less well-defined (Fig. 6b). These experi-ments show that, if the PS surface is already oxidised, the CVresponse becomes independent of the presence of spin trap. XPSand CV data suggest surface phenol groups as a likely origin of theredox response. Additionally, the XPS data is consistent with thesonicated sample being more oxidised than the centrifuged andhence less sensitive to the presence of spin trap.
The vortexed PS showed the most significant increase in oxida-tion charge, but XPS reveals that the surface oxygen content issimilar to the dialysed PS (Fig. 3d) and so the response cannot beattributed to prior presence of oxygen functionalities. Addition of
PBN spin trap during CV experiments (Fig. 6d) leads, as observedfor the dialysed sample, to the absence of the distinct peak at 0.7 V.This indicates that the same PBN sensitive oxidation mechanismtakes place for the vortexed and dialysed samples.
The charge related to oxidation of the centrifuged, sonicatedand vortexed PS is greater that that for the dialysed PS byfactors of two, three and ten respectively (Fig. 2a). Although thepresence of more redox active functionalities could partlyexplain the enhanced electrochemical response for the oxidisedsamples, increased surface area due to roughening and defor-mation under mechanical agitation is also a likely explanation.For the case of the vortexed PS, the very significant deformationobserved by TEM is supportive of the increase in surface areabeing the primary reason for the increased oxidation currents.In addition loose, amorphous polymer material can be seen inthe TEM images of the vortexed sample and adsorption of thishigh surface area material to the electrode may be a cause ofthe significantly higher currents observed for this sample.
4. DiscussionCV mechanisms of immobilised PS
When phenol functionalities are present on the PS surface theoxidation mechanism could proceed as shown in Scheme 2, in
Fig. 6 CVs of first scan of electrode immobilised PS in 0.2 M pH 6 PBS (0.02 V s�1) in the absence (black) and presence (red) of 10 mM PBN spin trap: (a)dialysed PS; (b) centrifuged (PS); (c) sonicated PS; (d) vortexed PS.
an overall four electron/four proton oxidation at ca. 0.7 V withthe final product being a quinone. Assuming uniform mono-layer coverage of 5 � 105 beads mm�2 and transfer of ca. 13 nCof charge for the dialysed PS, the number of electrons trans-ferred during the oxidation process at 0.7 V is of the order of104 per bead (for detailed calculations see ESI†). Assumingsmooth spherical particles, this translates very roughly to 3 or4 electrons per styrene unit (i.e. per C6H5–C2H4-moiety) on thesurface of the sphere and within tunnelling distance of theelectrode, consistent with the mechanism in Scheme 2.
The generated quinone species then undergoes a reversiblereduction to the hydroquinone at potentials consistent with thesmall redox couple observed for oxidised PS at ca. 0.26 V. Thenoted charge discrepancy between the oxidation peaks at 0.7 Vand 0.28 V for all samples (for the dialysed PS (28 � 5) nCand (2.5 � 0.5) nC respectively) indicates that this is not theonly reaction pathway and other redox inactive products mustbe formed.
A proposed mechanism to explain the generation of pheno-lic species at a previously unoxidised PS surface is proposed inScheme 3, here electrogenerated OH radicals abstract a hydrogenatom from the polymer backbone to produce a tertiary carbon
radical. If dissolved oxygen is present a peroxy radical may beformed and this may be a terminating step. Alternatively theradical may be delocalised around the aromatic ring resulting inphenolic species after reaction with water or dissolved oxygen. Inaddition direct addition of OH radical to the aromatic ring willresult in formation of phenolic moieties. The rate constants forOH reaction via H abstraction or by aromatic ring addition arecomparable (of the order 109 M�1 s�1 in aqueous solution)29
hence the two processes take place in competition. The phenolsgenerated via either route then undergo immediate oxidationat ca. 0.7 V, via the mechanism shown in Scheme 2, therebyexplaining why a voltammetric response consistent with thepresence of phenol is observed, despite no phenol functionalitybeing present at the PS surface at the start of the experiment.
Polymer mechanochemistry: changes to the PS surface inducedby mechanical forces
The XPS and electrochemical results show clearly that changes toPS surface chemistry are induced by different mechanical agita-tion techniques. The vortexed sample does not show any changesto surface termination as a result of the mechanical forcesimposed, which is surprising given the degree of morphological
Scheme 2 Suggested mechanism for oxidation of phenol surface groups, as potential origin of voltammetric response of electrode-immobilised PS.
Scheme 3 Proposed mechanism for generation of redox-active phenolic groups on the PS surface through reaction with hydroxyl radicals.
deformation observed. This is perhaps a consequence of the typesof forces experienced by the PS nanospheres. During vortexmixing the main forces are likely to be compressive, rather thanshear forces. While this leads to substantial changes in surfacemorphology as the beads impact against each other, the collisionsmay have insufficient energy to allow homolytic cleavage of thepolymer backbone.
In contrast the centrifuged PS is likely to experience sig-nificant shear force, due to the direction in which the force isimposed relative to the surface, resulting in homolytic bondbreakage, forming mechanoradical species.2–4 The sonicatedsamples will also experience shear force as the PS spheres rubagainst other, but additionally may be exposed to cavitationevents.30 For both sonicated and centrifuged PS incorporationof oxygen functionality into the PS is observed, presumablydue to mechanoradical formation (through cleavage of thebackbone) followed by reaction with dissolved oxygen or withwater.3 Cavitation during ultrasound treatment may alsogenerate hydroxyl radicals that can react with the PS.30
The role of dissolved oxygen in the aqueous mechanochem-istry of PS is clear from the different electrochemical response(Fig. 2c) and XPS spectra (Fig. 4) obtained for PS sonicated inair-saturated and argon-saturated solutions. Homolytic bondcleavage results in formation of alkyl radicals on the PS back-bone, and these react rapidly with dissolved oxygen, resultingin unstable peroxy radicals3,4 that rapidly decompose to alco-hols and other functionalities.31 In the absence of oxygen, thealkyl radical is more long-lived, resulting in propagation ofchain reactions and radical delocalisation around the phenylring. Reaction of the resulting species with water or hydroxylradicals produced through cavitation may lead to a wider rangeof oxidised species than obtained in the presence in oxygen,where formation of the peroxy radical is a terminating step.31
This may explain the surprising result that, according to theXPS analysis, PS sonicated in the absence of oxygen has ahigher oxygen content than that sonicated with oxygen present.The difference in CV response for the two PS samples mayprovide insight into the identity of the redox active functional-ities present and is the subject of further investigation.
Relevance to electrostatic charging of insulating materials
Insulating polymers such as Teflon, PMMA, nylon and poly-ethylene have been shown to perform redox chemistrywhen immersed in solution after electrostatic charging.7 Theseobservations suggested that electron transfer between polymersurfaces could be responsible for the observed charge separa-tion that takes place when two materials are rubbed together.However, the feasibility of electron transfer as a mechanism forelectrostatic charging has been questioned due to the highenergy required to transfer an electron from the HOMO of onewide band-gap insulator to the LUMO of another.8a In response, theconcept of a ‘cryptoelectron’ was invoked to explain how electrons ofmid band-gap energy may be available for transfer. ‘Cryptoelectrons’were defined as ‘electrons residing in a material whatever theirsource (e.g. surface states, impurities, bulk defects) with energiessignificantly different from those expected from molecular states (or
bands) in the material’.7c Recently at least some of the chemistryattributed to the transfer of ‘cryptoelectrons’ has been associatedwith the presence of surface mechanoradicals.10
In the present study the mechanical activation of the PSnanospheres has been carried out in solution, rather thanrubbing together two materials in an ambient environment,making a direct comparison to previous studies inappropriate.However we do see evidence for chemical modification of thePS surface after mechanical agitation in solution, where theshear forces and interfacial environment between two polymersurfaces may be comparable to that achieved by rubbingtogether two polymers in humid air. The surface modificationswe observe are consistent with radical formation.3 The electro-chemical response of the resulting surface species showsthat after mechanical treatment surface groups and states areavailable in the energy gap between the frontier molecularorbitals of the polystyrene and could potentially facilitateelectron transfer between polymer surfaces if the energies ofthe surface states was matched.
5. Conclusions
This relatively simple CV study has revealed an unexpected degree ofinsight into the reactivity and mechanochemistry of the PS surfacein solution. Further work is planned to compare and contrast thebehaviour of other polymers under similar conditions. One of themotivations of this study was to determine whether the redoxchemistry of insulating polymers could be measured using voltam-metric techniques. A complex, pH dependent CV response haspreviously been obtained for electrode-immobilised undoped (non-conductive) diamond nanoparticles.1 The response was attributed tothe presence of surface functionalities (alcohols, quinones etc.) thatcould undergo direct electron transfer with the electrode and withdissolved redox species. The present study confirms that, if redoxactive surface functionalities are present, a CV response can alsobe obtained for polymer nanoparticles such as PS, despite theinsulating nature of the bulk. This presents a limitless opportunityfor further investigation of the surface chemistry of insulatingmaterials, suggesting that many materials at the nanoscale mayexhibit an electrochemical response.
Acknowledgements
EPSRC Grant no EP/J0100061 is acknowledged for funding. TheElectrostatics Discharge (ESD) Association is thanked for theaward of a University Research Grant.
References
1 (a) K. B. Holt, Phys. Chem. Chem. Phys., 2010, 12, 2048;(b) K. B. Holt, D. J. Caruana and E. J. Millan-Barrios, J. Am.Chem. Soc., 2009, 131, 11272.
2 (a) T. Q. Nguyen and H. M. Kausch, Macromolecules, 1990,23, 5137; (b) J. A. Odell, A. Keller and A. J. Muller, ColloidPolym. Sci., 1992, 270, 307.
3 (a) M. K. Beyer and H. Clausen-Schaumann, Chem. Rev.,2005, 105, 2921; (b) G. Ayrey, C. G. Moore and W. F. Watson,J. Polym. Sci., 1956, 19, 1; (c) S. N. Zhurkov andV. E. Korsukov, J. Polym. Sci., Polym. Phys., 1974, 12, 385.
4 (a) J. Sohma, Prog. Polym. Sci., 1989, 14, 451; (b) M. Sackaguchiand J. Sohma, J. Polym. Sci., Polym. Phys., 1975, 13, 1233.
5 (a) G. Schmidt-Naake, A. Frendel, M. Drache and G. Janke,Chem. Eng. Technol., 2001, 24, 889; (b) A. R. Nesarikar,S. H. Carr, K. Khait and F. M. Mirabella, J. Appl. Polym.Sci., 1997, 63, 1179; (c) J. Klein, Annu. Rev. Mater. Sci., 1996,26, 581.
6 D. J. Lacks and R. M. Sankaran, J. Phys. D: Appl. Phys., 2011,44, 453001.
7 (a) C. Liu and A. J. Bard, Nat. Mater., 2008, 7, 505; (b) C. Liuand A. J. Bard, Chem. Phys. Lett., 2009, 480, 145; (c) C. Liu andA. J. Bard, J. Am. Chem. Soc., 2009, 131, 6397; (d) C. Liu andA. J. Bard, Chem. Phys. Lett., 2010, 485, 231.
8 (a) L. S. McCarty and G. M. Whitesides, Angew. Chem., Int.Ed., 2008, 47, 2188; (b) S. W. Thomas III, S. J. Vella,G. K. Kaufman and G. M. Whitesides, Angew. Chem., Int.Ed., 2008, 47, 6654; (c) A. F. Diaz and R. M Felix-Navarro,J. Electrost., 2004, 62, 277.
9 (a) H. T. Baytekin, A. Z. Patashinski, M. Branicki,B. Baytekin, S. Soh and B. A. Grzybowski, Science, 2011,333, 308; (b) H. T. Baytekin, B. Baytekin, J. T. Incorvati andB. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 1.
10 B. Baytekin, H. T. Baytekin and B. A. Grzybowski, J. Am.Chem. Soc., 2012, 134, 7223.
11 H. T. Baytekin, B. Baytekin, T. M. Hermans, B. Kowalczykand B. A. Grzybowski, Science, 2013, 341, 1368.
13 J. J. Pireaux, J. Riga, R. Caudano, J. J. Verbist, J. Delhalle,S. Delhalle, J. M. Andre and Y. Golillon, Phys. Scr., 1977, 16, 329.
14 E. H. Lock, D. Y. Petrovykh, P. Mack, T. Carney, R. G. White,S. G. Walton and R. F. Fernsler, Langmuir, 2010, 26, 8857.
15 M. M. Nasef, H. Saidi, H. M. Nor and M. O. Yarmo, J. Appl.Polym. Sci., 2000, 76, 336.
16 R. Foerch, G. Beamson and D. Briggs, Surf. Interface Anal.,1991, 17, 842.
17 R. T. S. Oliviera, G. R. Salazar-Banda, M. C. Santos,M. L. Calegaro, D. W. Miwa, S. A. S. Machado andL. A. Avaca, Chemosphere, 2007, 66, 2152.
18 C. Provent, W. Haenni, E. Santoli and P. Rychen, Electrochim.Acta, 2004, 49, 3737.
19 X. Zhu, S. Shi, J. Wei, F. Lv, H. Zhao, J. Kong, Q. Hi and J. Ni,Environ. Sci. Technol., 2007, 41, 6541.
20 T. A. Enache and A. M. Oliveira-Brett, J. Electroanal. Chem.,2011, 655, 9.
21 J. A. Wiles, M. Fialkowski, M. R. Radowski, G. M. Whitesidesand B. A. Grzybowski, J. Phys. Chem. B, 2004, 108, 20296.
22 T. Murakami, S. Kuroda and Z. Osawa, J. Colloid InterfaceSci., 1998, 202, 37.
23 A. Stefanova, S. Ayata, A. Erem, S. Ernst and H. Baltruschat,Electrochim. Acta, 2013, 110, 560.
24 T. A. Enache, A.-M. Chiorcea-Paquim, O. Fatibello-Fihlo andA. M. Oliveira-Brett, Electrochem. Commun., 2009, 11, 1342.
25 C. Terashima, T. N. Rao, B. V. Sarada, Y. Kubota andA. Fujishima, Anal. Chem., 2003, 75, 1564.
26 B. Lee, H. Naito and T. Hibino, Angew. Chem., Int. Ed., 2012,51, 440.
27 (a) Z. Ma, B. Zhao and Z. Yuan, Anal. Chim. Acta, 1999,389, 213; (b) E. G. Janzen, Y. Kotake and R. D. Hinton, FreeRadicals Biol. Med., 1992, 12, 169.
28 L. Johnson and D. A. Walsh, J. Mater. Chem., 2011, 21, 7555.29 (a) L. M. Dorfman, I. A. Taub and D. A. Harter, J. Chem.
Phys., 1964, 41, 2954; (b) M. Anbar, D. Meyerstein andP. Neta, J. Chem. Soc. B, 1966, 742.
30 T. Q. Nguyen, Q. Z. Liang and H. H. Kausch, Polymer, 1997,38, 3783.
31 C. von Sonntag, E. Bothe, P. Ulanski and A. Adhikary,Radiat. Phys. Chem., 1999, 55, 599.