Page 1
Accepted Manuscript
Synthesis and anti-parasitic activity of a novel quinolinone-chalcone series
Marina Roussaki, Belinda Hall, Sofia Costa Lima, Anabela Cordeiro da Silva,
Shane Wilkinson, Anastasia Detsi
PII: S0960-894X(13)01121-9
DOI: http://dx.doi.org/10.1016/j.bmcl.2013.09.047
Reference: BMCL 20899
To appear in: Bioorganic & Medicinal Chemistry Letters
Received Date: 6 August 2013
Revised Date: 13 September 2013
Accepted Date: 16 September 2013
Please cite this article as: Roussaki, M., Hall, B., Lima, S.C., da Silva, A.C., Wilkinson, S., Detsi, A., Synthesis and
anti-parasitic activity of a novel quinolinone-chalcone series, Bioorganic & Medicinal Chemistry Letters (2013),
doi: http://dx.doi.org/10.1016/j.bmcl.2013.09.047
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Page 2
1
Synthesis and anti-parasitic activity of a novel quinolinone-
chalcone series
Marina Roussaki,a Belinda Hall,b Sofia Costa Lima,c Anabela Cordeiro da Silva,c,d
Shane Wilkinson,b Anastasia Detsia*
a Laboratory of Organic Chemistry, School of Chemical Engineering, National
Technical University of Athens, Zografou Campus 15780 Athens, Greece
bSchool of Biological and Chemical Sciences, Queen Mary University of London,
England
cParasite Disease Group, IBMC-Institute for Molecular and Cell Biology, University of
Porto, Rua do Campo Alegre, 823, 4150-180, Porto, Portugal
dDepartment of Biological Sciences, Faculty of Pharmacy, University of Porto, Rua de
Jorge Viterbo Ferreiera, 228, 4050-313 Porto, Portugal
* Corresponding author
e-mail: [email protected]
Tel.: +302107724126
Fax: +302107723072
Abstract
A series of novel quinolinone-chalcone hybrids and analogues were designed,
synthesized and their biological activity against the mammalian stages of
Trypanosoma brucei and Leishmania infantum evaluated. Promising molecular
scaffolds with significant microbicidal activity and low cytotoxicity were identified.
Quinolinone-chalcone 10 exhibited anti-parasitic properties against both organisms,
being the most potent anti-L.infantum agent of the entire series (IC50 value of 1.3 ±
0.1 μM). Compounds 4 and 11 showed potency toward the intracellular, amastigote
stage of L. infantum (IC50 values of 2.1 ± 0.6 and 3.1 ± 1.05 μM, respectively).
Promising trypanocidal compounds include 5 and 10 (IC50 values of 2.6 ± 0.1 and 3.3
± 0.1 μM, respectively) as well as 6 and 9 (both having IC50 values of <5 μM).
Chemical modifications on the quinolinone-chalcone scaffold were performed on
Page 3
2
selected compounds in order to investigate the influence of these structural features
on antiparasitic activity.
Keywords
Quinolinone-chalcones, antiparasitic activity, T. brucei, L. infantum, cytotoxicity
Diseases caused by protozoan parasites belonging to the genera Trypanosoma
and Leishmania are responsible for high mortality and morbidity each year,
particularly in low-income countries.1 With no immediate prospect of a vaccine, drugs
are the only option to treat these infections. The use of current therapies is
controversial as many are toxic, some are mutagenic while clinical resistance is on
the rise. Additionally, many require medical supervision while being administered
which all contribute to the cost per treatment. Against this backdrop, there is an
urgent requirement for new, safe, cheap drugs. However, as drug development is
expensive, the development of new chemotherapies targeting trypanosomatid
parasites is perceived as not being commercially attractive. As a result, trypanosomal
and leishmanial diseases are said to be neglected due to the lack of funds available
to combat these pathologies.
Human African trypanosomiasis (HAT) is prevalent in sub-Saharan Africa with an
estimated 50 million people living in areas deemed at risk.2 This life-threatening
zoonosis is caused by Trypanosoma brucei, which is transmitted to humans by the
blood feeding habits of its insect vector, the tsetse fly.3,4 Due to vector control
programmes coupled with improved public health surveillance, the number of
infections have fallen dramatically over the last 15 years from a peak estimated
between 400,000 - 500,000 in 1998 to less than 30,000 cases in 2009.5,6 However,
previous epidemics have shown that unless such measures are maintained, HAT can
readily flare up causing a rapid increase in prevalence following political and
socioeconomic disorder, potentially resulting in the deaths of tens of thousands of
people.6,7
Leishmaniasis is endemic in many tropical and sub-tropical regions, with
approximately 350 million people living in “at risk” areas. Estimates indicate that 12
million people currently suffer from leishmaniasis, with about 2 million new cases and
50,000 deaths occurring each year.2,8 More than 20 human infectious Leishmania
species are responsible for a complex set of pathologies that range from a self
Page 4
3
limiting cutaneous form to a potentially fatal visceral disease. The parasite is
transmitted through the hematophagous feeding habits of pregnant, female sandflies
belonging to the subfamily Phlebotominae. The main transmission cycle is zoonotic
although anthroponotic routes are now common for some species, especially in an
urban context. However, due to intravenous drug usage/needle sharing, transfusion
with infected blood and other blood-borne contact coupled with global warming and
the spread of the insect vector, leishmaniasis is beginning to emerge as a problem in
non-endemic regions.
Nature has provided an inspiration for the design and synthesis of many
compounds, providing novel scaffolds that have been exploited in drug development.
Two groups of molecules that have shown potential in this regard are the chalcones
and quinolinones. Chalcones are open chain flavonoids, comprised of two aryl rings
(rings A and B) linked by an α,β-unsaturated carbonyl moiety (Figure 1). They are
synthesized predominantly by plants and are important precursors in a number of
biochemical pathways ranging from flower pigmentation to biological defence against
phytopathogens and insects.9
(Figure 1)
Due to their distribution in edible plants and to their powerful and diverse
biological activities, chalcones have been intensively investigated as scaffolds in the
development of anti-cancer,10 antioxidant,11,12 anti-angiogenic,13 and anti-
inflammatory14,15 agents. Several natural and synthetic chalcones have been shown
to possess anti-parasitic activities such as licochalcone A (Figure 2), a natural
prenylated chalcone isolated from the Chinese licorice, identified as a potent anti-
leishmanial agent which acts by inhibiting fumarate reductase in L. major and L.
donovani,16,17 and hybrid scaffolds incorporating the chalcone moiety, which are
emerging as potent lead structures targeting Leishmania and Trypanosoma
species.18-20
The quinolinone moiety (Figure 1) is a common structural motif found in numerous
quinolinone alkaloid natural products.21-23 Quinolinone analogues possess important
biological properties such as antioxidant,24,25 anti-inflammatory25,26 and enzyme-
inhibitory activity.27 As with the chalcones, several quinolinone-based compounds
have emerged as anti-parasitic lead structures, notably the natural products
dictyolomide A and B (Figure 2), which possess leishmanicidal activity,28 and 3-
substituted-quinolinone derivatives that target both L. donovani and T. brucei. 29
Page 5
4
(Figure 2)
As a continuation of our studies toward the synthesis and biological evaluation of
chalcone12 and quinolinone24,25 derivatives, here we present the synthesis of novel
hybrid compounds encompassing the 4-hydroxy-2-quinolinone heterocyclic system
and the chalcone moiety in one molecular scaffold in order to investigate the potential
synergistic effect of these two pharmacophores against the protozoan parasites T.
brucei and L. infantum. Our approach substitutes ring A of the chalcone moiety with
the 4-hydroxy-2-quinolinone structure. The literature concerning the synthesis and
biological evaluation of this type of compounds is scarce and, to our knowledge, this
is the first time that their anti-parasitic activities against T. brucei and L. infantum
have been reported.
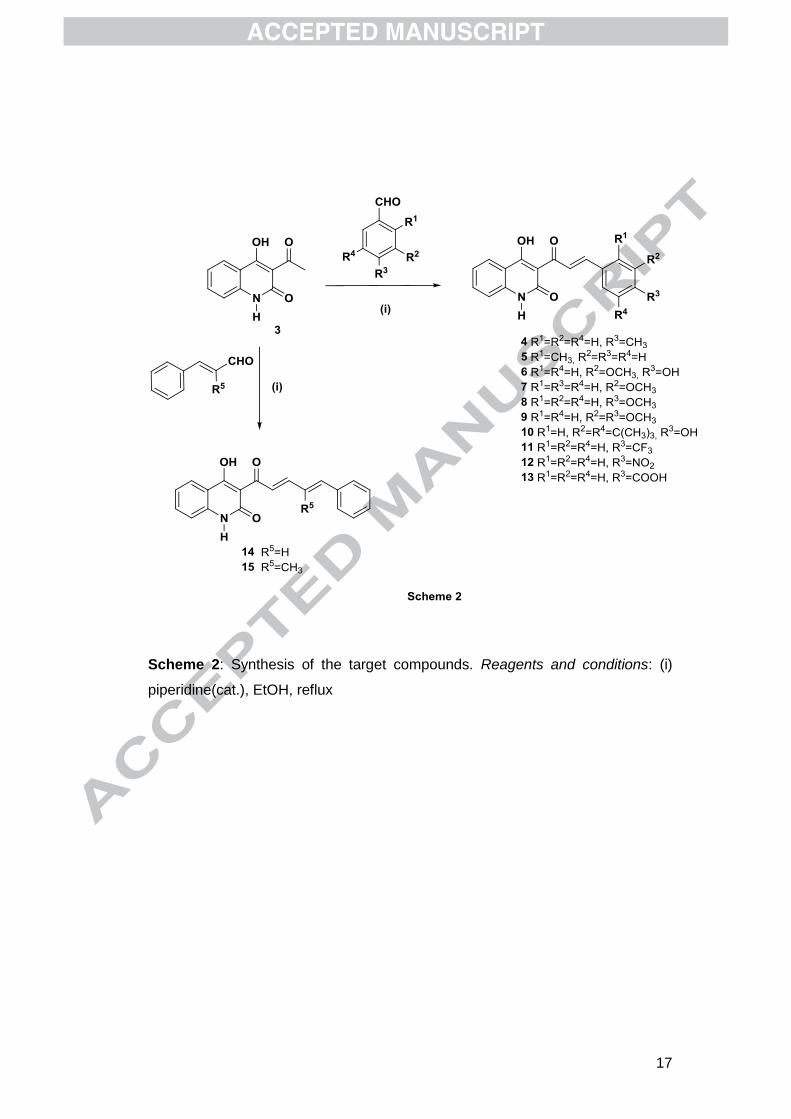
The synthesis of the desired compounds was accomplished via an aldol
condensation reaction between 3-acetyl-4-hydroxy-2-quinolinones and variably
substituted benzaldehydes. The required starting material, 3-acetyl-4-hydroxy-2(1H)-
quinolinone (3), was synthesized using our previously developed methodology which
includes C-acylation of ethyl acetoacetate by 2-methyl-3,1-benzoxazin-4-one (1) and
subsequent cyclization of the C-acylation product 2 in basic conditions (Scheme 1).30
(Scheme 1)
The methodologies reported in the literature for the synthesis of quinolinone-
chalcone hybrids are limited: Kalechits et al. have prepared a series of quinolinone-
chalcone analogues by refluxing 3-acetyl-4-hydroxy-2(1H)-quinolinone with various
benzaldehydes in pyridine using drops of piperidine as catalyst.31 In 2006 a
microwave-assisted synthesis of this type of compounds in EtONa/EtOH was
reported32 whereas the most recent methodology (2011) involves a solvent-free
protocol using silica sulfate catalyst which efficiently performs the crossed-aldol
reaction required.33
In the present study, various conditions were investigated in order to synthesize
the target quinolinone-chalcone derivatives. Optimization of the reaction conditions
was performed using 3-acetyl 4-hydroxyquinolin-2(1H)-one (3) and 4-methyl-
benzaldehyde as starting materials and carrying out the reaction using ethanol or
pyridine as solvents and aqueous KOH or piperidine as bases (Table 1).
(Table 1)
Page 6
5
Optimum results were obtained using catalytic amounts of piperidine in EtOH
(Scheme 2). Using these conditions, the reaction is completed in 1–2 h (monitored by
TLC). Acidification with aqueous HCl (10%), followed by filtration of the precipitating
solid, results to the isolation of the desired products in satisfactory yields (40–60%)
and purity, as confirmed by NMR spectroscopy.
(Scheme 2)
Twelve compounds were synthesized in total, bearing either a variety of electron –
donating (CH3, OCH3, C(CH3)3, OH) or electron – withdrawing substituents (NO2,
CF3, COOH) on ring B of the chalcone framework (compounds 4-13) or an extended
conjugated system (compounds 14 and 15).
In order to further investigate the structure – activity relationship of the series of
quinolinone-chalcone hybrids, we decided to modify two of the most characteristic
structural features of the quinolinone-chalcone framework, namely the amide moiety
and the α,β-unsaturated carbonyl system. In this context, modification of the amide
hydrogen of the heterocyclic ring of compounds 5 and 9 by alkylation was initially
undertaken (Scheme 3). The required starting materials, N-ethyl and N-benzyl-3-
acetyl-4-hydroxy-2-quinolinones 18 and 19, were synthesized via reductive alkylation
of methyl anthranilate by the appropriate aldehyde, acylation of the secondary amine
using 2,2,6-trimethyl-1,3-dioxan-4-one as an acylating agent and cyclization of the
crude product under basic conditions.34 The final alkylated quinolinone-chalcones 20-
23 were prepared via a crossed aldol coupling reaction, according to the initial
method (Scheme 3).
(Scheme 3)
Moreover, pyrazoline analogues of compounds 5 and 9 were synthesized, by
refluxing the corresponding chalcone with phenyl hydrazine or hydrazine hydrate in
glacial acetic acid solution35 (Scheme 4). The final pyrazoline analogues 24 and 25
were obtained after filtration of the solid product formed and the pure compounds
were collected after trituration with methanol in 40% yield.
(Scheme 4)
Page 7
6
All the 1H NMR spectra of the synthesized chalcones showed the characteristic
signals of the vinylic protons α and β at 6.5-8 ppm, with coupling constants J=12-18
Hz, indicating the E-geometric configuration for all compounds, and a single peak at
17-18 ppm assigned to the enolic proton of position 4 which is involved in a strong
hydrogen bond with the carbonyl oxygen.
The anti-parasitic activities of the compounds synthesized in this work were
evaluated in vitro against bloodstream form Trypanosoma brucei and axenically
cultured intracellular Leishmania infantum amastigotes (Table 2).
(Table 2)
Out of twelve quinolinone-chalcone analogues screened, five were shown to have
growth inhibitory activity against T. brucei. The most potent of these was compound 5
(IC50 = 2.6 ± 0.1 μM) followed by compounds 10, 6, 9 and 7 (decreasing potency),
with all five trypanocidal structures containing electron-donating substituents on ring
B of the chalcone motif. Analysis of data suggests that the position and number of
these groupings all contribute to a compound’s anti-parasitic property. For the most
effective trypanocidal agent, compound 5, the electron-donating methyl substituent is
located at the 2-position on ring B while its isomer, compound 4 that contains the
methyl grouping at the 4-position, had no effect on trypanosomal growth at
concentration up to 10 μM. Similarly, compound 7, which contains a single methoxy
at the 3-position on ring B is trypanocidal (IC50 value of 6.5 ± 0.1 μΜ) while the
isomeric chalcone 8, that contains a 4-methoxy group, had no effect on parasite
growth. The presence of two electron-donating substituents was shown to lead to a
slight increase in trypanocidal activity: compound 9 which contains two methoxy
groups at the 3- and 4- positions on ring B generated an IC50 value of 4.9 ± 0.2 μΜ
while an IC50 value of 4.9 ± 0.1 μΜ was displayed by compound 6 that contains a
methoxy and hydroxyl at the 3- and 4-positions, respectively. The remaining
trypanocidal chalcone, compound 10, possesses an ortho-(di-tert-butyl)-phenol
substitution. Despite being the second most potent anti-trypanosomal structure of the
chalcone series (IC50 value of 3.3 ± 0.1 μΜ), it displayed high cytotoxicity (IC50 value
26 μΜ).
For compounds 5 and 9, analogues containing an alkyl substituent on the amidic
nitrogen present on the heterocyclic ring of the quinolinone moiety (compounds 20-
Page 8
7
23) were synthesized. N-ethyl-analogues 20 and 21 did not exhibit growth inhibitory
properties against bloodstream form T. brucei. Compounds 22 and 23, the N-benzyl
analogues of chalcones 5 and 9, respectively, showed lower activity against T. brucei
than the non-alkylated compounds, albeit higher than the N-ethyl analogues. These
results suggest that the hydrogen of the heterocyclic amide group is important in the
mechanism of action of these compounds. Likewise, alterations to the quinolinone-
chalcone structure at other sites, by incorporating electron withdrawing groups
(COOH, CF3 and NO2) on ring B of the chalcone (compounds 11, 12, and 13) or
extending the conjugation system between the quinolinone and chalcone motifs
(compounds 14 and 15), had no effect on trypanosomal growth at concentration up to
10 μM.
Modifying the α,β-unsaturated carbonyl system by synthesizing the pyrazoline
analogues 24 and 25, resulted to a remarkable increase in antiparasitic activity
against bloodstream form T. brucei: both compounds exhibited IC50 lower than the
reference drug nifurtimox thus are the most active against T. brucei parasites among
all the 18 compounds tested in this work. Pyrazoline 24 (IC50 value of 1.46 ± 0.1) can
be regarded as a promising lead antiparasitic compound as it is also not cytotoxic
against THP1 cells.
When the quinolinone-chalcone hybrid series were screened against the
intracellular amastigote stage of L. infantum, eleven structures (compounds 4-15, 20
and 21) were shown to affect parasite growth. The most potent of these were
compounds 10, 4 and 11 which yielded IC50 values of 1.3 ± 0.1, 2.1 ± 0.6 and 3.1 ±
1.0 µM, respectively. Intriguingly, the structure activity relationships observed using
this compounds series against L. infantum were markedly distinct from those noted
when targeting T. brucei, with many of the differences being directly opposite. For
example, moving the methyl substituent from 4-position (compound 4) to 2-position
(compound 5) or the methoxy group from the 4-position (compound 8) to 3-position
(compound 7) resulted in a significant reduction in leishmancidal activity in contrast to
the situation noted for T. brucei (Table 2). Moreover, the electronic nature of the
substituents does not seem to play a very important role in the anti-leishmanial
activity as it did for T. brucei: compound 11 bearing an electron-withdrawing
trifluoromethyl group (a methyl group isostere) at the 4-position of ring B is the third
more active agent of the series against L. infantum with the NO2 and COOH
containing derivatives (compounds 12 and 13) having an affect against L. infantum
amastigotes. Why these conflicting SAR differences arise is unclear. This could
Page 9
8
reflect that these parasites reside at different locations within the mammalian host: T.
brucei is an extracellular pathogen found in the hosts’ bloodstream whereas L.
infantum can invade and grow in host macrophages, residing within an acidic
compartment. Alternatively, this could simply be due to the concentration ranges
used in the screens: many leishmanicidal compounds have IC50 values >10 µM, the
maximum level used against T. brucei.
N-alkylation of the heterocyclic amide group resulted in compounds with lower or
slightly better antileishmanial activity as compared to the non-alkylated analogues,
indicating that the N-H group is probably crucial for anti-leishmanial activity as it was
also in the case of anti-trypanosomal activity. Pyrazoline 24 showed lower activity
against L. infantum in comparison with the chalcone analogue 9 whereas pyrazoline
25, an analogue of chalcone 5, was the most active anti-leishmanial agent among all
the compounds tested exhibiting IC50 value of 0.71 μM. It is worth noting that
pyrazoline 25 showed the best antiparasitic activity against both parasites.
To evaluate their effects on mammalian cells, cytotoxicity assays were performed
with all compounds against differentiated THP-1 macrophages (Table 2). Of the most
potent agents (IC50 values <10 μM), the compounds 4, 5, 7, 9, 11 and 24 had a no
growth-inhibitory effect on THP-1 cells at concentrations at 30 M (5, 7 and 9) or 50
M (4, 11 and 24) whereas compounds 6 and 10 had IC50 values of around 20 μM.
Exact IC50 cytotoxicity data could not be determined due to the issues relating to
compound solubility in DMSO and other common solvents. Comparison of potency
against the parasite and mammalian lines indicates that pyrazoline 24 and chalcone
5 are the most effective trypanocidal agents while chalcone analogues 4 and 11 are
interesting lead structures targeting L. infantum.
Studies concerning the actual mechanism of action of chalcones against
kinetoplastids are still underway by several research groups. For instance, Chen et
al.17 have shown that chalcones active against Leishmania major and L. Donovani,
inhibit the action of fumarate reductase (FRD), one of the enzymes of the parasite
respiratory chain which is not present in mammalian cells, and suggested that this
enzyme might be the specific target for the chalcones. However, fumarate reductase
activity does not appear to be essential in the bloodstream form of T. brucei, so other
mechanisms must be involved in cytotoxicity towards these parasites.36 Recently,
Page 10
9
several chalcones have been shown to induce production of ROS and trigger
apoptosis in cancer cells37 and that this kind of pathway is present in kinetoplastids.38
The role of the quinolinone moiety is unclear at this stage and no reports
concerning the antiparasitic activity of quinolinone-chalcone hybrids or the
corresponding pyrazoline analogues are available, therefore we can only speculate
that our compounds could follow either of the above mentioned mechanisms. These
studies could be the subject of further investigation.
The emphasis of this work was the synthesis of novel hybrid compounds,
encompassing the quinolinone and the chalcone moiety in one molecular scaffold. A
series of these analogues, bearing a wide variety of substituents were synthesized
and their in vitro anti-parasitic activity against bloodstream form T. brucei and the
intracellular L. infantum amastigotes, as well as their cytotoxicity against mammalian
cells, were evaluated. Several compounds elicited growth inhibitory activity against
one or both parasites, promoting further investigations into deciphering any structure
activity relationships. Of this series, the most potent structure against T. brucei was
compound 5 (IC50 value of 2.6 ± 0.1 μM) followed by compounds 6 and 9 (IC50 values
of <5 μM), while compounds 4 and 11 showed potential as leishmanidal agents (IC50
values of 2.1 ± 0.6 and 3.1 ± 1.0 μM, respectively). None of these lead structures
displayed cytotoxicity towards the mammalian THP-1 cell line. In contrast chalcone
10, although active against both parasites (and the most potent against L. infantum),
did display toxicity toward mammalian cells. We were able to show that substituents
on ring B of the chalcone motif and the amidic nitrogen present on the heterocyclic
ring of the quinolinone moiety are important determinates in mediating the biological
activities of these compounds. These features can be incorporated into the next
generation of quinolinone-chalcone hydrid scaffolds with the goal of developing new,
safe, cost effective treatments targeting these neglected tropical diseases. Structural
modification of the α,β-unsaturated carbonyl system via transformation to the
corresponding pyrazoline analogues, resulted to compounds 24 and 25 with
remarkable in vitro anti-parasitic activity against bloodstream form T. brucei. Although
pyrazoline 25 exhibited the best activity against both parasites and is the most potent
among all the compounds tested in this work, its high cytotoxicity againt mammalian
cells prohibits its further consideration as a lead compound. On the other hand,
pyrazoline 24 should be considered as a promising lead trypanocidal agent as it
possesses high antiparasitic activity and is not cytotoxic. To our knowledge the
pyrazoline scaffold, although extensively studied for other biological acitivities, has
Page 11
10
not been yet explored as a structural feature for the development of antiparasitic
agents.
Ackowledgements
This collaboration has been promoted by the COST Action CM0801 “New drugs for
neglected diseases”.
Supplementary data
Supplementary data associated with this article can be found, in the online version,
at……..
References
1. “Sustaining the drive to overcome the global impact of neglected tropical
diseases”, Second WHO report on neglected tropical diseases, Crompton D.
WT, Ed., 2013, WHO reference number: WHO/HTM/NTD/2013.1
2. Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gurtler, R.E.; McKerrow, J.; Reed,
S.; Tarleton, R., J. Clin. Invest. 2008, 118, 1301.
3. Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H.,
Antimicrob. Agents Chemoth. 2010, 54, 2893.
4. Kennedy, P.G.E., J. Neurol. 2006, 253, 411.
5. Simarro, P. P.; Jannin, J.; Cattand, P., PLoS Med. 2008, 5, 55.
6. Brun, R.; Blum, J.; Chappuis, F.; Burri, C., Lancet 2010, 375, 148.
7. Steverding, D., Parasites and Vectors 2008, 12, 1, 3.
8. Croft, S. L.; Coombs, G. H., Trends in Parasitol. 2003, 19, 502.
9. Batovska, D.I.; Todorova, I., Curr. Clin. Pharmac. 2010, 5, 1.
10. Roman, B. I.; Heugebaert, T. S. A.; Bracke, M. E.; Stevens, C. V. Curr. Med.
Chem. 2013, 20, 186.
11. Shenvi, S.; Kumar, K.; Hatti, K. S.; Rijesh, K.; Diwakar, L.; Reddy, G. C. Eur. J.
Med. Chem. 2013, 62, 435.
12. Detsi A.; Majdalani M.; Kontogiorgis C.A.; Hadjipavlou-Litina D.; Kefalas P.,
Bioorg. Med. Chem. 2009, 17, 8073.
13. Mojzis, J.; Varinska, L.; Mojzisova, G.; Kostova, I.; Mirossay, L., Pharmacol.
Res. 2008, 57, 259.
14. Nowakowska, Z., Eur. J. Med. Chem. 2007, 42, 125.
15. Bano, S.; Javed, K.; Ahmada, S.; Rathish, I.G.; Singh, S.; Chaitanya, M.;
Arunasree, K.M.; Alam, M.S. Eur. J. Med. Chem. 2013, 65, 51.
Page 12
11
16. Kayser, O.; Kiderlen, A.F.; Croft, S.L., Parasitol. Res. 2003, 90, S55.
17. Chen, M.; Zhai, L.; Christensen, S. B.r; Theander, T. G.; Kharazmi, A.
Antimicrob. Agents Chemoth. 2001, 45, 2023.
18. Ryczak, J.; Papini, M.; Lader, A.; Nasereddin, A.; Kopelyanskiy, D.; Preu, L.;
Jaffe, C.L.; Kunick, C. Eur. J. Med. Chem. 2013, 64, 396.
19. Tyagi, V.; Khan, S.; Shivahare, R.; Srivastava, K.; Gupta, S.; Kidwai, S.;
Srivastava, K.; Puri, S. K.; Chauhan, P. M. S. Bioorg. Med. Chem. Lett. 2013,
23, 291.
20. Qiao, Z.; Wang, Q.; Zhang, F.; Wang, Z.; Bowling, T.; Nare, B.; Jacobs, R.T.;
Zhang, J.; Ding, D.; Liu, Y., J. Med. Chem. 2012, 55, 3553.
21. He, J.; Lion,U.; Sattler,I.; Gollmick, F.A.; Grabley,S.; Cai, J.; Meiners, M.;
Schunke, H.; Schaumann, K.; Dechert,U.; Krohn, M. J. Nat. Prod. 2005, 68,
1397.
22. Nakashima, K.; Oyama, M.; Ito, T.; Akao, Y.; Witono, J.R.; Darnaedi, D.;
Tanaka, T.; Murata, J.; Iinuma, M. Tetrahedron 2012, 68, 2421.
23. Gao, H.; Zhang, L.; Zhu, T.; Gu, Q.; Li, D. Chem. Pharm. Bull. 2012, 60, 1458.
24. Angeleska, S.; Kefalas, P.; Detsi, A. Tetrahedron Lett. 2013, 54, 2325.
25. Detsi A.; Bouloumbasi D.; Prousis K.C.; Koufaki M.; Athanasellis G.; Melagraki
G.; Afantitis A.; Igglessi-Markopoulou O.; Kontogiorgis C.; Hadjipavlou-Litina
D.J., J. Med. Chem. 2007, 50, 2450-2458.
26. Jonsson, S.; Andersson, G.; Fex, T.; Fristedt, T.; Hedlund, G.; Jansson, K.;
Abramo, L.; Fritzson, I.; Pekarski, O.; Runstrom, A.; Sandin, H.; Thuvesson, I.;
Bjork, A. J. Med. Chem. 2004, 47, 2075.
27. Larsson, E.A.; Jansson, A.; Ng, F.M.; Then, S.W.; Panicker,R.; Liu, B.;
Sangthongpitag, K.; Pendharkar, V.; Tai, S.J.; Hill,J.; Dan,C.; Ho, S.Y.; Cheong,
W.W.; Poulsen,A.; Blanchard, S.; Lin, G.R.; Alam,J.; Keller,T.H.; Nordlund, P. J.
Med. Chem. 2013, 56, 4497.
28. Lavaud C.; Massiot G.; Vasquez C.; Moretti C.; Sauvain M.; Balderrama L.,
Phytochemistry 1995, 40, 317.
29. Audisio D.; Messaoudi S.; Cojean S.; Peyrat J.F.; Brion J.D.; Bories C.; Huteau
F.; Loiseau P.M.; Alami M., Eur. J. Med. Chem. 2012; 52; 44-50.
30. Detsi A.; Bardakos V.; Markopoulos J.; Igglessi-Markopoulou O., J. Chem. Soc.
Perkin Trans. 1996, 1, 2909.
31. Kalechits G.V.; Olkhovik V.K.; Kalosha I.I.; Skakovskii E.D.; Pap A.A.; Zenyuk
A.A.; Matveenko Y.V., Russ. J. Gen. Chem. 2001, 71, 1257.
32. Sarveswari, S.; Raja, T. K., Ind. J. Heterocyclic Chem., 2006, 16, 171.
33. Bhupathi, Raja S.; Devi, B. Rama; Dubey, P. K. Asian J. Chem. 2011, 23, 4215.
Page 13
12
34. Boyle R.G.; Imogai H.J.; Cherry M.; Humphries A.J; Navarro E.F.; Owen D.R.;
Dales N.A.; Lamarche M.; Cullis C., Gould A.E. WO 2005/028474 A2
35. Sivakumar P.M.; Ganesan S.; Veluchamy P.; Doble M., Chem. Biol. Drug. Des.,
2010, 76, 407.
36. Alsford, S.; Turner, D. J.; Obado, S. O.; Sanchez-Flores, A.; Glover, L.;
Berriman, M.; Hertz-Fowler, C.; Horn, D. Genome Res., 2011, 21, 915.
37. Wu, W.; Ye, H.; Wan, L.; Han, X.; Wang, G.; Hu, J.; Tang, M.; Duan, X.; Fan, Y.;
He, S.; Huang, L.; Pei, H.; Wang, X.; Li, X.; Xie, C.; Zhang, R.; Yuan, Z.; Mao,
Y.; Wei, Y.; Chen, L., Carcinogenesis, 2013, 34, 1636.
38. Piacenza, L.; Irigoin, F.; Alvarez, M. N.; Peluffo, G.; Taylor, M. C.; Kelly, J. M.;
Wilkinson, S. R.; Radi, R., Biochem. J., 2007, 403, 323.
Page 14
13
Captions of Figures, Schemes and Tables
Figure 1. General structure of the chalcone and quinolinone molecular scaffolds.
Figure 2. Quinolinone and chalcone analogues with antiparasitic activity
Scheme 1. Synthesis of the starting 3-acetyl-4-hydroxy-2(1H)-quinolinone (3).
Reagents and conditions: (i) (CH3CO)2O, 130oC, 2h; (ii) CH3COCH2COOEt, t-BuOK,
t-BuOH, r.t.; (iii) aq. Na2CO3/NaOH, r.t.
Scheme 2: Synthesis of the target compounds. Reagents and conditions: (i)
piperidine(cat.), EtOH, reflux
Scheme 3. Synthetic approach to N-alkylated quinolinone-chalcones. Reagents and
conditions: (i) RCHO, H-B(OAc)3Na, CH3COOH, CH2Cl2, r.t.; (ii) 2,2,6-trimethyl-1,3-
dioxan-4-one, toluene, reflux;(iii) EtOH, EtONa, reflux;(iv) piperidine (cat.), EtOH,
reflux
Scheme 4. Synthetic approach to pyrazoline analogues 24, 25. Reagents and
conditions: (i) PhNHNH2 (for compound 24) or H2NNH2.H2O (for compound 25),
CH3COOH, reflux
Table 1. Optimization of reaction conditions for the synthesis of quinolinone-
chalcones
Table 2: Anti-parasitic and cytotoxic activities of compounds 4-15 and 20-25
Page 15
14
Figure 1. General structure of the chalcone and quinolinone molecular scaffolds.
Page 16
15
Figure 2. Chalcone and quinolinone analogues with antiparasitic activity
Page 17
16
Scheme 1. Synthesis of the starting 3-acetyl-4-hydroxy-2(1H)-quinolinone (3).
Reagents and conditions: (i) (CH3CO)2O, 130oC, 2h; (ii) CH3COCH2COOEt,
t-BuOK, t-BuOH, r.t.; (iii) aq. Na2CO3/NaOH, r.t.
Page 18
17
Scheme 2: Synthesis of the target compounds. Reagents and conditions: (i)
piperidine(cat.), EtOH, reflux
Page 19
18
Scheme 3. Synthetic approach to N-alkylated quinolinone-chalcones. Reagents
and conditions: (i) RCHO, H-B(OAc)3Na, CH3COOH, CH2Cl2, r.t.; (ii) 2,2,6-
trimethyl-1,3-dioxan-4-one, toluene, reflux;(iii) EtOH, EtONa, reflux;(iv) piperidine
(cat.), EtOH, reflux
Page 20
19
Scheme 4. Synthetic approach to pyrazoline analogues 24, 25. Reagents and
conditions: (i) PhNHNH2 (for compound 24) or H2NNH2.H2O (for compound 25),
CH3COOH, reflux
Page 21
20
Table 1. Optimization of reaction conditions for the synthesis of quinolinone-chalcone
hybrids
Base Solvent Yield (%)
1eq 1eq KOH(5eq) EtOH 18
1eq 1eq Piperidine(1.2eq) EtOH <10
1eq 1eq Piperidine(1.2eq) Pyridine <10
1eq 1eq Piperidine(cat.) EtOH 49
Page 22
21
Table 2: Anti-parasitic and cytotoxic activities of compounds 4-15 and 20-25
Compound IC50 (µM)
T.brucei L. infantum THP1
4 >10 2.1 ± 0.6 >50a
5 2.6 ± 0.1 >50 >50a
6 4.9 ± 0.1 11.5 ± 1.7 23.3 ± 1.4a
7 6.5 ± 0.1 28.4 ± 3.5 38.5 ± 2.1a
8 >10 12.7 ± 3.0 >50a
9 4.9 ± 0.2 7.5 ± 2.7 >50a
10 3.3 ± 0.1 1.3 ± 0.1 26.2 ± 2.7 a
11 >10 3.1 ± 1.0 >50a
12 >10 26.9 ± 4.6 >50a
13 >10 18.4 ± 6.2 >50a
14 >10 >50 >50a
15 >10 20.0 ± 6.6 >50a
20 >10 24.8 ± 3.9 >50a
21 >10 >50 >50a
22 6.17±0.7 >25 45.7 ± 3.1 a
23 5.68±0.5 >25 28.7 ± 1.7 a
24 1.46±0.1 13.46±0.61 >50a
25 1.43±0.1 0.71±0.07 4.15±0.19
Nifurtimox 2.9 0.3 - >100b
Amphotericin B - 1.2 0.1 23.8 2.3c
a Performed using the MTT assay. b The cytotoxicity of nifurtimox toward
differentiated THP-1 cells was performed using alamarBlue™. c The cytotoxicity
towards Ampotericin B was established through MTT assay.
Page 23
Graphical Abstract
Compound IC50 (μM)
T. brucei L. infantum
4 R1
=R2= R
4=H, R
3=CH3 >10 2.1± 0.6
5 R1=CH3, R
2=R
3=R
4=H 2.6 ± 0.1 >50
10 R1=H, R
2=R
4=C(CH3)3,R
3=OH 3.3 ± 0.1 1.3 ± 0.1
Nifurtimox 2.9 0.3 -
Amphotericin B - 1.2 0.1