jou rn al hom ep age: www.elsev ier .com/ locate /porgcoat
ynthesis and characterization of Schiff base derivative with pyrroleing and electrochromic applications of its oligomer
˙smet Kaya ∗, Ercan Bora, Aysel Aydın anakkale Onsekiz Mart University, Faculty of Sciences and Arts, Department of Chemistry, Polymer Synthesis and Analysis Laboratory,7020 C anakkale, Turkey
r t i c l e i n f o
rticle history:eceived 23 May 2012eceived in revised form 7 November 2013ccepted 11 November 2013vailable online 28 November 2013
a b s t r a c t
In this study, Schiff base monomer, 2-[(1H-pyrrol-2-yl-methylene)amino]phenol (2-PMAP) was obtainedby condensation reaction of 2-aminophenol with pyrrole-2-carbaldehyde. This monomer was oxidizedin an aqueous alkaline medium by NaOCl to obtain the corresponding oligomer (O-2-PMAP). The result-ing compounds were characterized by 1H- and 13C-NMR, FT-IR, UV–vis, TG-DTA and size exclusionchromatography (SEC) techniques. Metal complexes compounds of O-2-PMAP were prepared with var-
ious metal salts, such as Cu (AcO)2·H2O, Zn (AcO)2·2H2O, Cd (AcO)2·2H2O, Pb (AcO)2·3H2O and Co(AcO)2·4H2O. In addition to this, electrochemical copolymerization of O-2-PMAP with thiophene (Th),3,4-ethylenedioxythiophene (EDOT) and pyrrole (Py) were performed onto indium/tin oxide (ITO)-coatedglass plate. The spectral changes of the co-polymeric films were recorded in the range of the differentpotentials. The film stabilities of its EDOT and Py copolymers were determined. Consequently, it was
er fil
hermal degradation realized that the copolym
. Introduction
Recently, Schiff base polymers including active hydroxyl ( OH)nd azomethine ( CH N ) groups have been widely studied [1].hese Schiff base polymers have been used in various fields becausef useful properties such as paramagnetism, semi conductivity,lectrochemical cell and resistance to high energy [2,3]. Theyave been also used to prepare composite thermo-stabilizations,raphite materials, epoxy oligomer and block copolymers, photoesists, and materials [4–6]. Due to the presence of func-ional groups, they also exhibit different properties includingatalytic activity, thermal stability, ion selectivity, conductivitynd anti-microbial properties. Kaya and Aydın have reportedligomer/polymer–metal complexes based on various Schiff baseonomers [7,8]. It is known that the active azomethine and
ydroxyl groups have crucial important for cleaning poisonouseavy metals in industrial waste waters. Therefore, these kindsf products are also important for analytic and environmentalhemistry [9]. Since oligomer/polymer–metal complexes are alsomportant for electrochromic purposes, the electrochromic proper-ies of new Schiff base polymers have been investigated in previous
ork [10].
A new Schiff base oligomer was synthesized via oxidative poly-ondensation in an aqueous alkaline medium by NaOCl as oxidant.
Also, electrocopolymerization reactions of this oligomer were per-formed by thiophene (Th), 3,4-ethylenedioxythiophene (EDOT) andpyrrole (Py) with cyclic voltammetry technique. The structures andcharacterizations of compounds were made various spectral andthermal techniques. Some oligomer–metal complexes compoundswere synthesized by acetate salts of Cu, Zn, Cd, Co and Pb.
2. Experimental
2.1. Materials
2-Aminophenol (Merck), pyrrole-2-carbaldehyde (Merck),pyrrole (Py) (Aldrich), thiophene (Th) (Aldrich), 3,4-ethylenedioxythiophene (EDOT) (Aldrich), boron trifluorideethyl etherate (BF3·EtE) (Fluka), methanol, acetonitrile, acetone,ethyl acetate, chloroform, tetrahydrofurane (THF), dimethyl-formamide (DMF), dimethylsulfoxide (DMSO), KOH, hydrochloricacid (HCl), Cu (AcO)2·H2O, Zn (AcO)2·2H2O, Cd (AcO)2·2H2O,Pb (AcO)2·3H2O and Co (AcO)2·4H2O were supplied by MerckChemical Co. (Germany). They were used as received. Sodiumhypochlorite (NaOCl) (30% aqueous solution) was supplied byPaksoy Chemical Co. (Turkey).
2.2. Characterization
The infrared and ultraviolet–visible spectra were recorded byusing Perkin Elmer FT-IR spectrum one and Perkin Elmer Lambda25, respectively. The FT-IR spectra were obtained by using the ATR
ttachment (4000–550 cm−1). UV–vis spectra of 2-PMAP and O-2-MAP were determined in methanol. 1H- and 13C-NMR spectra of-PMAP and O-2-PMAP were characterized by using Bruker AvancePX-400 and 100.6 MHz, respectively, and they were recorded at5 ◦C by using deuterated DMSO as solvent. TMS was used as inter-al standard. Thermal data were recorded by using Perkin Elmeriamond Thermal Analysis device. TG-DTA measurements wereerformed between 20 and 1000 ◦C (in N2, rate 10 ◦C/min). SECnalysis was carried out at 25 ◦C by using DMF/Methanol (4/1, v/v)s eluent at a flow rate of 0.4 mL/min. The instrument (Shimadzu0AVp series HPLC-SEC system) was calibrated with a mixture ofolystyrene standards (Polymer Laboratories; the peak molecu-
ar weights (Mp) between 162 and 19 880) using GPC softwareor the determination of the number-average molecular weightMn), weight-average molecular weight (Mw) and polydispersityndex (PDI) of the polymer samples. Macherey-Nagel GmbH & Co.100 A and 7.7 nm diameter loading material) 3.3 mm i.d. × 300 mmolumns were used for SEC investigations.
.3. Syntheses of the compounds
.3.1. Synthesis of 2-[(1H-pyrrol-2-yl methylene)amino]phenol2-PMAP)
2-[(1H-pyrrol-2-yl methylene)amino]phenol was prepared byhe condensation reaction of 2-aminophenol (0.109 g, 0.001 mol)ith pyrrole-2-carbaldehyde (0.095 g, 0.001 mol) in methanol
15 mL). The mixture was refluxed for 3 h at 70 ◦C (Scheme 1). Therecipitated 2-PMAP was filtered, recrystallized from methanol andried in a vacuum oven at 60 ◦C [8] (yield 78%). The precipitatedolid product was yellow.
For 2-PMAP. UV–vis (�max): 204, 238, 304 and 350 nm. FT-IRcm−1): � (O H) 3279 s, � (N H) 3334 s, � (C-H Phenyl) 3034 m,
.3.2. Synthesis of oligo-2-[(1H-pyrrol-2-ylethylene)amino]phenol with NaOCl in aqueous alkaline medium
O-2-PMAP was synthesized through oxidative polyconden-ation of 2-PMAP in aqueous alkaline medium by using NaOCl30%, 1 mL) as oxidant. 2-PMAP (0.186 g, 0.001 mol) was dissolvedn an aqueous solution of KOH (10%, 0.001 mol) and placed into a0-mL three-necked round-bottom flask. It was fitted with a con-
enser, thermometer, stirrer and an addition funnel containingaOCl. After heating, NaOCl was added drop-by-drop over about0 min (Scheme 2). The reaction mixture was cooled to room tem-erature, and then HCl (0.001 mol, 37%) was added into the reaction
Scheme 2. Synthesis of O-2-PMAP.
oatings 77 (2014) 463– 472
medium for neutralization. For the separation of mineral salts andunreacted monomers, the mixture was filtered and washed withhot water (3 × 25 mL) and then the oligomer was dried in a vacuumoven at 60 ◦C [10].
2.3.3. Syntheses of oligo-2-[(1H-pyrrol-2-ylmethylene)amino]phenol-metal complexes
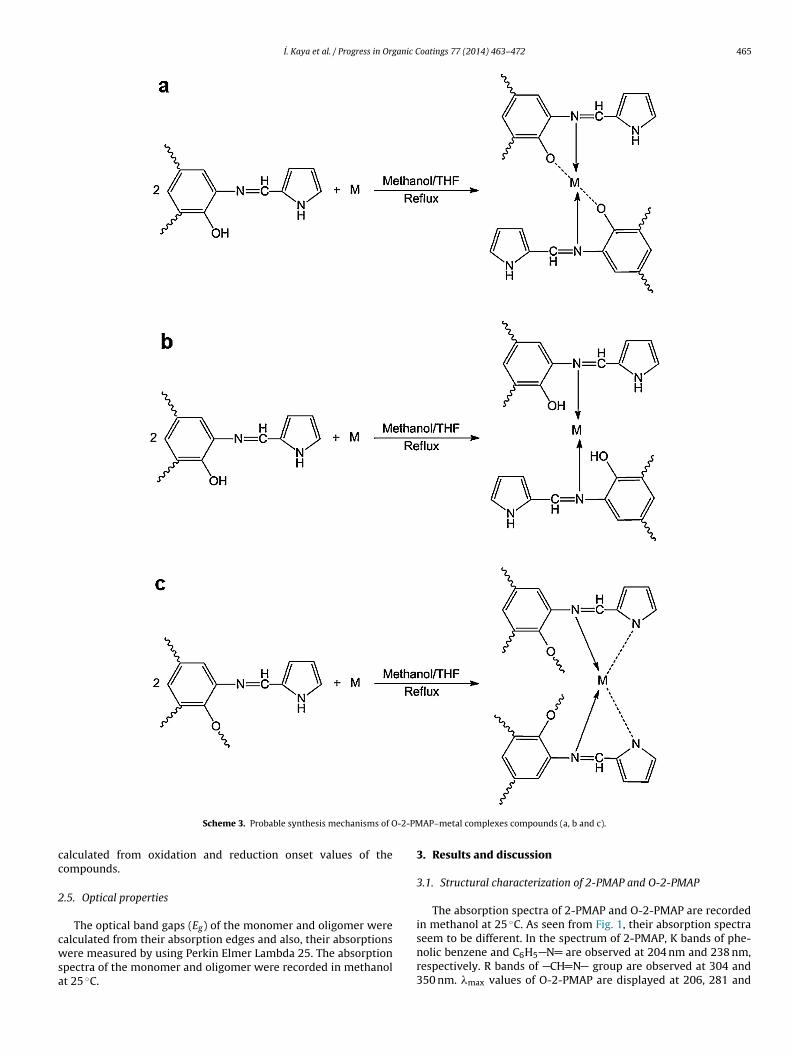
Solutions of Cu (AcO)2·H2O, Zn (AcO)2·2H2O, Cd (AcO)2·2H2O, Pb(AcO)2·3H2O and Co (AcO)2·4H2O (0.001 mol) in methanol (10 mL)were separately added to a solution of O-2-PMAP (0.002 mol/unit)in THF (20 mL). The mixture was stirred and heated at 70 ◦C for 5 h(Scheme 3). The precipitated products were filtered, washed withcold MeOH/THF (1/1, v/v) and then they were dried in a vacuumoven at 60 ◦C [7,8]. Yields of O-2-PMAP-Cd, O-2-PMAP-Pb, O-2-PMAP-Co, O-2-PMAP-Zn, O-2-PMAP-Cu were found as 97%, 19%,73%, 85%, and 94%, respectively. The combination of the metal com-plexes with oligomer units could be shown for different positionsas given in Scheme 3 [11].
2.3.4. Electrochemical copolymerization reactions of O-2-PMAPwith Th, EDOT and Py
Electrochemical measurements were carried out with a CHI660C Electrochemical Analyzer (CH Instruments, Texas, USA) at apotential scan rate of 0.25 V/s. The electroactivity of the compoundswere determined from the oxidation–reduction peak potentials.Electrocopolymerization reactions were separately carried out inthe presence of Th, EDOT and Py. The system consists of a CV cellcontaining indium/tin oxide (ITO)-coated glass plate as the work-ing electrode, platinum wire as the counter electrode, and Ag wireas the reference electrode. The measurements were performed inLiClO4 (0.1 M)/acetonitrile (AN:BF3·EtE) (10/1, v/v) solvent mix-ture at room temperature [12]. Electrocopolymerization reactionswere carried out as follows: 15 mg of O-2-PMAP was dissolved in10 mL 0.1 M LiClO4/AN:BF3·EtE (10/1, v/v) and placed into a CVcell. One drop of Th or EDOT or Py was added into the cell foreach solution. Electrocopolymerization reactions were separatelyrun by repeated electrochemical scanning of the solutions betweendifferent potentials for each compound. Scan rate and cycle num-ber were calibrated to 0.25 V/s and 20 for all the measurements,respectively.
2.4. Electrochemical properties
Cyclic voltammetry (CV) measurements were performed bya CHI 660C Electrochemical Analyzer at a potential scan rateof 0.25 V/s. All the experiments were carried out in a drybox under Ar atmosphere at room temperature. The electro-chemical potential of Ag was calibrated with respect to theferrocene/ferrocenium (Fc/Fc+) couple. The half-wave potential(E1/2) of (Fc/Fc+) measured in 0.1 M tetrabutylammoniumhexafluo-rophosphate (TBAPF6) acetonitrile solution is 0.39 V vs. Ag wire or
0.38 V vs. supporting calomel electrolyte (SCE). The voltammetrymeasurements were carried out for the monomer and oligomer inacetonitrile and DMSO/acetonitrile mixture, respectively [13,14].The electrochemical HOMO and LUMO energy gaps (Eg) were
I. Kaya et al. / Progress in Organic Coatings 77 (2014) 463– 472 465
-2-PM
cc
2
cwsa
Scheme 3. Probable synthesis mechanisms of O
alculated from oxidation and reduction onset values of theompounds.
.5. Optical properties
The optical band gaps (Eg) of the monomer and oligomer were
alculated from their absorption edges and also, their absorptionsere measured by using Perkin Elmer Lambda 25. The absorption
pectra of the monomer and oligomer were recorded in methanolt 25 ◦C.
AP–metal complexes compounds (a, b and c).
3. Results and discussion
3.1. Structural characterization of 2-PMAP and O-2-PMAP
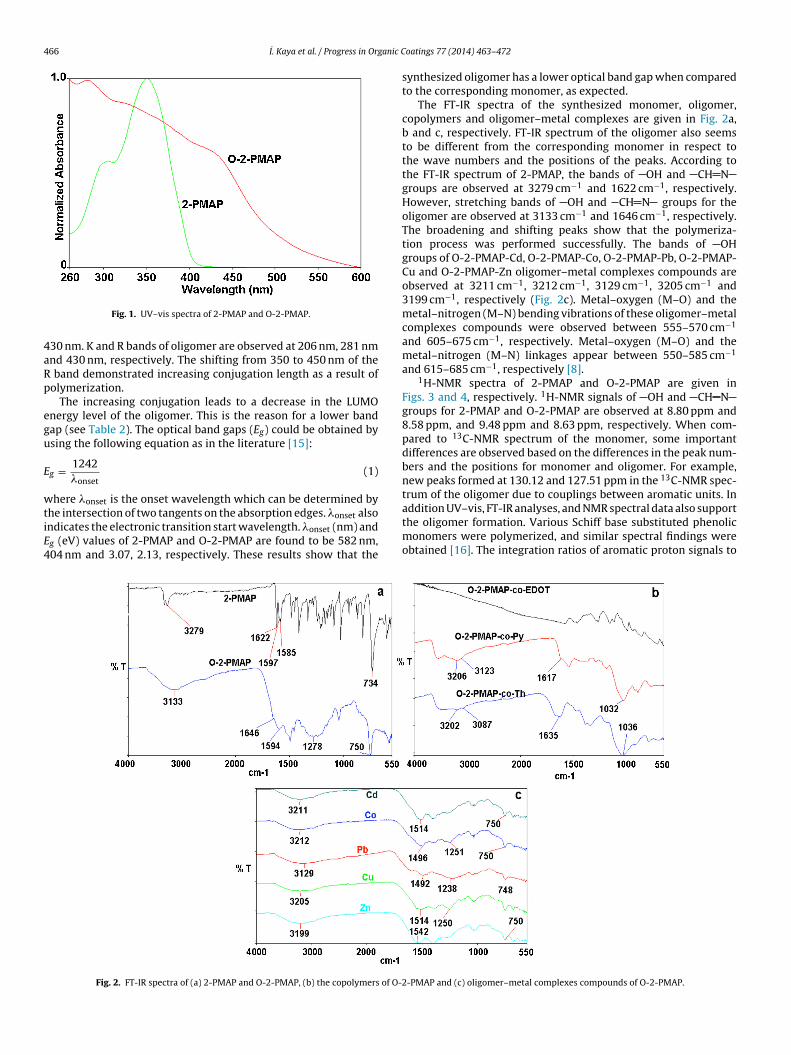
The absorption spectra of 2-PMAP and O-2-PMAP are recordedin methanol at 25 ◦C. As seen from Fig. 1, their absorption spectra
seem to be different. In the spectrum of 2-PMAP, K bands of phe-nolic benzene and C6H5 N are observed at 204 nm and 238 nm,respectively. R bands of CH N group are observed at 304 and350 nm. �max values of O-2-PMAP are displayed at 206, 281 and
466 I. Kaya et al. / Progress in Organic C
4aRp
egu
E
wtiE4
Fig. 1. UV–vis spectra of 2-PMAP and O-2-PMAP.
30 nm. K and R bands of oligomer are observed at 206 nm, 281 nmnd 430 nm, respectively. The shifting from 350 to 450 nm of the
band demonstrated increasing conjugation length as a result ofolymerization.
The increasing conjugation leads to a decrease in the LUMOnergy level of the oligomer. This is the reason for a lower bandap (see Table 2). The optical band gaps (Eg) could be obtained bysing the following equation as in the literature [15]:
g = 1242�onset
(1)
here �onset is the onset wavelength which can be determined by
he intersection of two tangents on the absorption edges. �onset alsondicates the electronic transition start wavelength. �onset (nm) andg (eV) values of 2-PMAP and O-2-PMAP are found to be 582 nm,04 nm and 3.07, 2.13, respectively. These results show that the
Fig. 2. FT-IR spectra of (a) 2-PMAP and O-2-PMAP, (b) the copolymers of O-
oatings 77 (2014) 463– 472
synthesized oligomer has a lower optical band gap when comparedto the corresponding monomer, as expected.
The FT-IR spectra of the synthesized monomer, oligomer,copolymers and oligomer–metal complexes are given in Fig. 2a,b and c, respectively. FT-IR spectrum of the oligomer also seemsto be different from the corresponding monomer in respect tothe wave numbers and the positions of the peaks. According tothe FT-IR spectrum of 2-PMAP, the bands of OH and CH Ngroups are observed at 3279 cm−1 and 1622 cm−1, respectively.However, stretching bands of OH and CH N groups for theoligomer are observed at 3133 cm−1 and 1646 cm−1, respectively.The broadening and shifting peaks show that the polymeriza-tion process was performed successfully. The bands of OHgroups of O-2-PMAP-Cd, O-2-PMAP-Co, O-2-PMAP-Pb, O-2-PMAP-Cu and O-2-PMAP-Zn oligomer–metal complexes compounds areobserved at 3211 cm−1, 3212 cm−1, 3129 cm−1, 3205 cm−1 and3199 cm−1, respectively (Fig. 2c). Metal–oxygen (M–O) and themetal–nitrogen (M–N) bending vibrations of these oligomer–metalcomplexes compounds were observed between 555–570 cm−1
and 605–675 cm−1, respectively. Metal–oxygen (M–O) and themetal–nitrogen (M–N) linkages appear between 550–585 cm−1
and 615–685 cm−1, respectively [8].1H-NMR spectra of 2-PMAP and O-2-PMAP are given in
Figs. 3 and 4, respectively. 1H-NMR signals of OH and CH Ngroups for 2-PMAP and O-2-PMAP are observed at 8.80 ppm and8.58 ppm, and 9.48 ppm and 8.63 ppm, respectively. When com-pared to 13C-NMR spectrum of the monomer, some importantdifferences are observed based on the differences in the peak num-bers and the positions for monomer and oligomer. For example,new peaks formed at 130.12 and 127.51 ppm in the 13C-NMR spec-trum of the oligomer due to couplings between aromatic units. In
addition UV–vis, FT-IR analyses, and NMR spectral data also supportthe oligomer formation. Various Schiff base substituted phenolicmonomers were polymerized, and similar spectral findings wereobtained [16]. The integration ratios of aromatic proton signals to
2-PMAP and (c) oligomer–metal complexes compounds of O-2-PMAP.
I. Kaya et al. / Progress in Organic Coatings 77 (2014) 463– 472 467
pbipc
sm(r2(pcssp
Fig. 3. 1H-NMR spectrum of 2-PMAP.
OH proton signals indicate that the polymerization process takeslace with both C O C and C C coupling mechanisms [17]. Theroadening in proton signals also indicates the presence of repeat-
ng aromatic units with the different chemical surrounding. Thus,olymerization proceeds by two mechanisms: C C and C O Couplings [18].
The reaction mechanism on the coupling selectivity wastudied by Kaya and co-workers and three possible reactionechanisms for the C C coupling selectivity were proposed
Scheme 4): (i) coupling of Schiff base substituted free phenoxyadicals resulting from one-electron-oxidation of 2-[(1H-pyrrol--yl methylene)amino]phenol and other Schiff base monomers,ii) coupling of phenoxy radicals coordinated to each other orthoosition by using oxidants such as NaOCl, H2O2 and air and (iii)
oupling through phenoxonium anion formed by using KOH. Regio-electivity of coupling was found in the polymerization of somepecific phenols and Schiff base monomers. The possible couplingositions of O-2-PMAP are given in Scheme 4 [19].
Fig. 4. 1H-NMR spectrum of O-2-PMAP.
Scheme 4. The C C and C O C coupling structure of O-2-PMAP.
3.2. Solubility and SEC analysis
O-2-PMAP is a dark brown color with powder form. While it iscompletely soluble in organic solvents such as DMF, DMSO and THF,it is insoluble in ethyl acetate, and it is partly soluble in acetone andchloroform. While 2-PMAP is partly soluble in DMSO, DMF, THF, andethyl acetate, it is insoluble in acetone and chloroform.
According to the SEC analysis, three fractions are observed in thechromatogram of O-2-PMAP. At the first fraction, Mn, Mw, PDI and% values of O-2-PMAP are found to be 6261 g mol−1, 6794 g mol−1,1.085 and 40%. At the second fraction, Mn, Mw, PDI and % values ofO-2-PMAP are found to be 4497 g mol−1, 4022 g mol−1, 1.118 and50%. At the third fraction, Mn, Mw, PDI and % values of O-2-PMAPare found to be 925 g mol−1, 762 g mol−1, 1.214 and 10%. Therefore,the use of NaOCl as oxidant is important to obtain a more monodis-persed polymerization product because its PDI value is close to1.
3.3. Thermal analyses of 2-PMAP, O-2-PMAP andO-2-PMAP–metal complexes
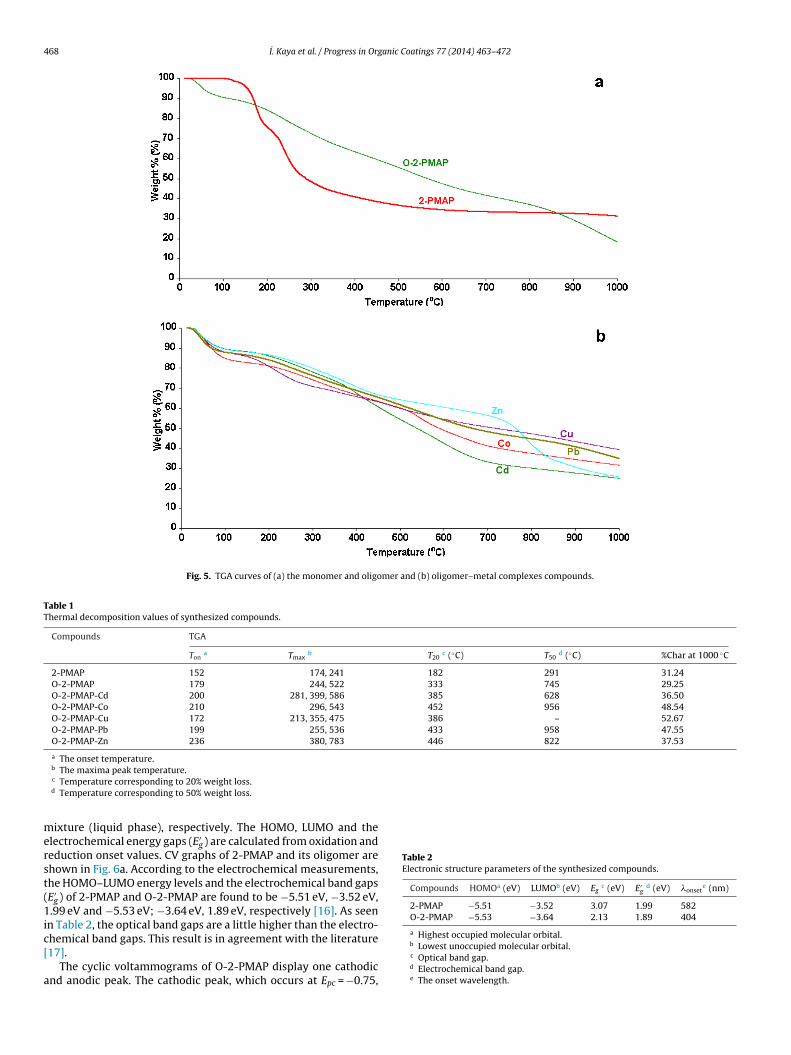
TGA curves of the monomer, oligomer and oligomer–metal com-plexes compounds are given in Fig. 5a and b, respectively. Theother thermal data related to the synthesized compounds are listedin Table 1. As seen in Table 1, the initial degradation tempera-tures of 2-PMAP and O-2-PMAP are found to be 152 ◦C and 179 ◦C,respectively. According to DTA data of 2-PMAP, it has an endother-mic peak at 127 ◦C. TGA analysis indicates that O-2-PMAP is morethermally stable than its corresponding monomer, as expected.Due to long conjugated band systems, the oligomer exhibits ahigher thermal resistance than its corresponding monomer. It isalso revealed that 20% and 50% weight losses of the oligomer arehigher than its monomer. The onset temperatures of the oligomer-metal complexes compounds are listed in Table 1 and their onsettemperatures were between 172 and 236 ◦C (see Fig. 5b). Tmax val-ues of all the synthesized compounds are given in Table 1.
3.4. Electrochemical properties of 2-PMAP and O-2-PMAP
The cyclic voltammetry measurements of 2-PMAP and O-2-PMAP were carried out in acetonitrile and acetonitrile–DMSO
468 I. Kaya et al. / Progress in Organic Coatings 77 (2014) 463– 472
Fig. 5. TGA curves of (a) the monomer and oligomer and (b) oligomer–metal complexes compounds.
Table 1Thermal decomposition values of synthesized compounds.
The maxima peak temperature.c Temperature corresponding to 20% weight loss.d Temperature corresponding to 50% weight loss.
ixture (liquid phase), respectively. The HOMO, LUMO and thelectrochemical energy gaps (E′
g) are calculated from oxidation andeduction onset values. CV graphs of 2-PMAP and its oligomer arehown in Fig. 6a. According to the electrochemical measurements,he HOMO–LUMO energy levels and the electrochemical band gapsE′
g) of 2-PMAP and O-2-PMAP are found to be −5.51 eV, −3.52 eV,.99 eV and −5.53 eV; −3.64 eV, 1.89 eV, respectively [16]. As seen
n Table 2, the optical band gaps are a little higher than the electro-
hemical band gaps. This result is in agreement with the literature17].
The cyclic voltammograms of O-2-PMAP display one cathodicnd anodic peak. The cathodic peak, which occurs at Epc = −0.75,
a Highest occupied molecular orbital.b Lowest unoccupied molecular orbital.c Optical band gap.d Electrochemical band gap.e The onset wavelength.
I. Kaya et al. / Progress in Organic Coatings 77 (2014) 463– 472 469
F -PMA0
iqrt
ig. 6. Cyclic voltammograms of (a) 2-PMAP and O-2-PMAP and (b) O-2-PMAP, O-2.1 M LiClO4 in AN (scan rate: 0.25 V/s, cycle number: 20 for each compound).
s assigned to the reduction of the azomethine group. Epc value isuite low, because azomethine ( CH N ) group shifts to a loweregion due to the presence of the pyrrole group and it also hashe most important electro activity property [20]. On the other
P-co-Py, O-2-PMAP-co-EDOT and O-2-PMAP-co-Th, respectively, in the presence of
hand, the observed anodic peaks in the structure of the oligomer ataround Epa = +1.14 V might correspond to oxidation of OH groups[21]. Similar to 2-PMAP, there are two irreversible peaks, suchas one cathodic and one anodic peak. The cathodic peak which
470 I. Kaya et al. / Progress in Organic Coatings 77 (2014) 463– 472
of O-2
o(a[Ftmag
aEE
Fp
Scheme 5. Electropolymerization reaction
ccurs at Epc = −0.91 is assigned to the reduction of the azomethine CH N ) group. On the other hand, the anodic peaks that occurst Epa = +1.78 V might correspond to the oxidation of OH groups1]. Cyclic voltammograms of 2-PMAP and O-2-PMAP are given inig. 6a. By introduction of appropriate substituents into the struc-ure of the polymer like 3,4-ethylenedioxy groups into the pyrrole
oieties, a significant improvement in the stability and the cyclicbility could be expected by emergence of narrower and lower bandap characteristics [22].
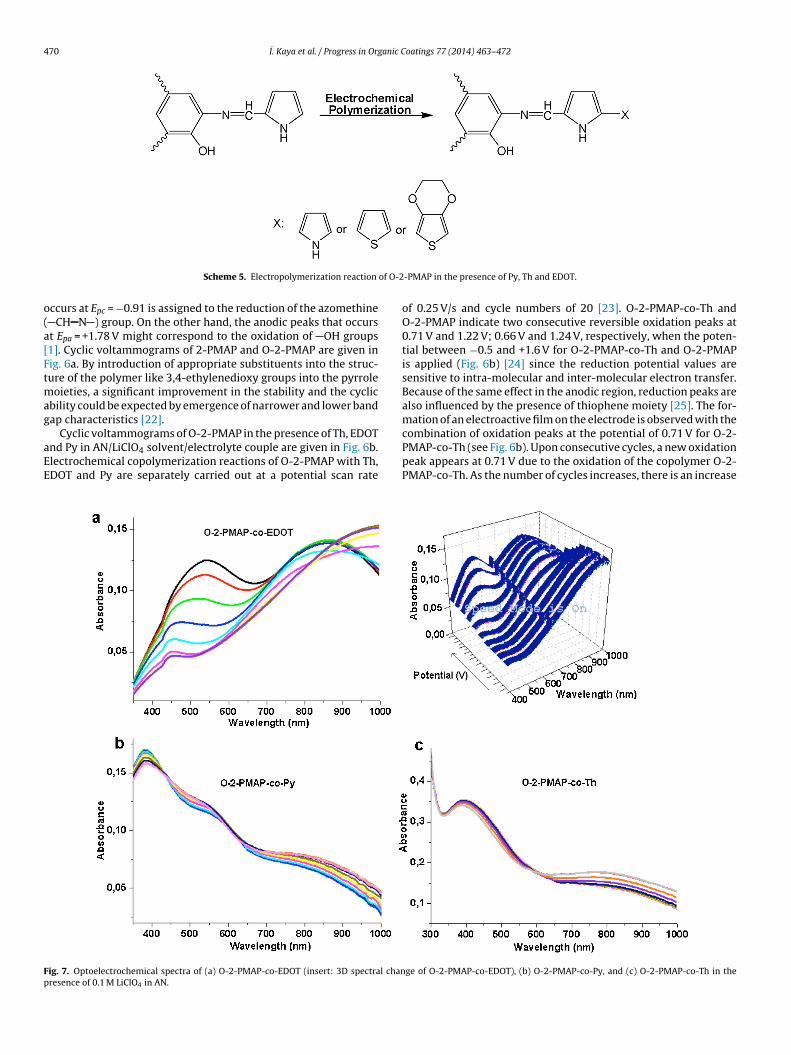
Cyclic voltammograms of O-2-PMAP in the presence of Th, EDOTnd Py in AN/LiClO4 solvent/electrolyte couple are given in Fig. 6b.lectrochemical copolymerization reactions of O-2-PMAP with Th,DOT and Py are separately carried out at a potential scan rate
ig. 7. Optoelectrochemical spectra of (a) O-2-PMAP-co-EDOT (insert: 3D spectral chanresence of 0.1 M LiClO4 in AN.
-PMAP in the presence of Py, Th and EDOT.
of 0.25 V/s and cycle numbers of 20 [23]. O-2-PMAP-co-Th andO-2-PMAP indicate two consecutive reversible oxidation peaks at0.71 V and 1.22 V; 0.66 V and 1.24 V, respectively, when the poten-tial between −0.5 and +1.6 V for O-2-PMAP-co-Th and O-2-PMAPis applied (Fig. 6b) [24] since the reduction potential values aresensitive to intra-molecular and inter-molecular electron transfer.Because of the same effect in the anodic region, reduction peaks arealso influenced by the presence of thiophene moiety [25]. The for-mation of an electroactive film on the electrode is observed with the
combination of oxidation peaks at the potential of 0.71 V for O-2-PMAP-co-Th (see Fig. 6b). Upon consecutive cycles, a new oxidationpeak appears at 0.71 V due to the oxidation of the copolymer O-2-PMAP-co-Th. As the number of cycles increases, there is an increase
ge of O-2-PMAP-co-EDOT), (b) O-2-PMAP-co-Py, and (c) O-2-PMAP-co-Th in the
anic Coatings 77 (2014) 463– 472 471
irc
tAtACTigePispaepmiha
3
ie(ivwAi(oP5ttienpcoama
3
casca−sofcs
I. Kaya et al. / Progress in Org
n the intensity of the current. An increase observed in the activeegion of the working electrode owing to the electroactive polymeroating on the electrode [26].
Electrochemical copolymerization reactions of O-2-PMAP inhe presence of active co-monomer agents like EDOT and Py inN/LiClO4 solvent/electrolyte mixture were also tried at room
emperature by adding one drop of EDOT or Py into the CV cell.dditionally, CV studies were performed in order to investigate theV behavior of the copolymers of O-2-PMAP with EDOT and Py.here are three different copolymers containing electropolymer-zable pure thiophene, phenolic benzene and pyrrole substitutedroup in the structure of O-2-PMAP. Due to the electron attractiveffect of the thiophene ring on the azomethine group of O-2-MAP, it increases the oxidation potential. As a result, thiophenes expected to be the first oxidized species and the polymerizationtarts by forming the polaron structure (radical cation) of thio-hene. Moreover, the obtained polymers could have random orlternate co-polymeric structures. Scheme 5 generally shows howlectro copolymerization reaction of O-2-PMAP takes place in theresence of Th, Py and EDOT [27]. As seen in the cyclic voltam-ograms of O-2-PMAP, it can be said that the peak at around 1.24 V
s related to OH groups (see Fig. 6b) [28]. On the other hand, theydroxyl vibration signals for these copolymers are observed atround 3200 cm−1 (see Fig. 2b).
.5. Spectroelectrochemical properties
Spectroelectrochemistry is the best way to examine the changesn optical properties of a polymer. It provides information on thelectronic properties of conjugated polymers, such as band gapEg) and the intergap states. To investigate spectroelectrochem-cal properties of the polymers, both UV–vis spectra and cyclicoltammetry are used [29]. Spectroelectrochemical measurementsere performed between various potentials for each copolymer inN/LiClO4 (0.1 M) solution. Fig. 7 shows the spectroelectrochem-
cal spectra of O-2-PMAP copolymers with (a) EDOT, (b) Py andc) Th at different potentials. As seen in Fig. 7, the �max valuesf the �–�* transitions for O-2-PMAP-co-EDOT, O-2-PMAP-co-y and O-2-PMAP-co-Th at 0 V (neutral states) are found to be00 nm, 385 nm, 550 nm and 850 nm, and 390 nm, respectively. Ashe copolymer becomes oxidized, the intensities of the �–�* transi-ions decrease while the charge carrier bands at about 800–900 nmncrease due to polaron formation [30]. Although P(O-2-PMAP) waslectropolymerizable, its spectroelectrochemical properties couldot be investigated since its film could not be electrochemicallyrepared. It can be said that O-2-PMAP may be solved in a CVell due to the macromolecular structure. Active hydroxyl groupsf polymer which form a hydrogen bond in acetonitrile lead to
decrease in the film ability. Therefore, spectroelectrochemicaleasurements of the electroactive copolymer on ITO could not be
In order to carry out the spectroelectrochemical measurements,yclic voltammetry coupled with optical spectroscopy was usednd a repeated scan mode was selected. Fig. 8 shows electrochromicwitching measurements of O-2-PMAP-co-EDOT and O-2-PMAP-o-Py. Potential scan ranges for O-2-PMAP-co-EDOT (see Fig. 8a)nd O-2-PMAP-co-Py (see Fig. 8b) shown in different colors are0.4 V and +1.2 V; −0.6 V and +1.4 V, respectively, at a chosen
can rate of 0.2 V/s. Measurements were carried out by using
ne wavelength for O-2-PMAP-co-EDOT and three wavelengthsor O-2-PMAP-co-Py. It is observed that the absorption intensitylearly changes in the range of these wavelengths. During thewitching experiment, the �max values are measured as 540 nm for
Fig. 8. Electrochromic switching measurements of (a) O-2-PMAP-co-EDOT and (b)O-2-PMAP-co-Py.
O-2-PMAP-co-EDOT and 385, 550 and 850 nm for O-2-PMAP-co-Py. The polymer film is deposited on ITO-coated glass slidesunder constant potential conditions, as mentioned above. Theabsorbances are then monitored at �max while the copolymers arebeing switched at different potentials. During this measurement, %transmittance at the wavelength of the maximum contrast is deter-mined by using a UV–vis spectrophotometer. As seen in Fig. 8a,O-2-PMAP-co-EDOT shows high film stability between −0.4 and+1.2 V during the repeated potential scan. The optical contrast ismeasured as the difference between T% values at neutral state andfound to be 15% for 540 nm (see Fig. 8a). Moreover, as seen in Fig. 8b,2-PMAP-co-Py has a good absorption recovery at applied poten-tials which indicates the percentage transmittance change (�T%)at three different wavelengths. Although the co-polymeric film ofO-2-PMAP-co-Th is electrochemically synthesized and its spectro-electrochemical measurement is performed, it can be said that itdoes not have film stability. The stability between the reductionand oxidation states during several scans is the most importantproperty for an electroactive polymer to be useful in construction
of new electrochromic devices [31].
The response time of the copolymer films is a very impor-tant parameter in electrochromic applications. The response timeof O-2-PMAP-co-EDOT between −0.4 V and +1.2 V was found to
4 anic C
bP5ten
4
mumpfa6isrfio
A
z
R
[[[[[[
[[[[[
[
[
[
[
[[
[
[
72 I. Kaya et al. / Progress in Org
e 3 s. On the other hand, the response times of O-2-PMAP-co-y in the range of −0.6 V/+1.4 V were 3 s, 6 s, and 8 s at 385 nm,50 nm and 850 nm, respectively. The color change is also an impor-ant parameter for electrochromic materials [32]. The synthesizedlectrochemical copolymers on ITO-glass exhibit red colors at theeutral states and blue colors in the oxidized states.
. Conclusions
Schiff base substituted phenol monomer, 2-[(1H-pyrrol-2-yl-ethylene)amino]phenol (2-PMAP), was converted to oligomer by
sing NaOCl as oxidant in an aqueous alkaline medium. The copoly-ers of O-2-PMAP with Th, Py and EDOT were electrochemically
repared. The reason for the spectral differences was the copolymerormation. As seen in the film stabilities, both O-2-PMAP-co-EDOTnd O-2-PMAP-co-Py co-polymeric films were quite stable for00 s. Although O-2-PMAP-co-Th had electrochromic properties,
t did not have film stability. They could ideally be used in con-truction of novel electrochromic devices due to possessing shortesponse times for the copolymer. The observed band gaps are suf-ciently low to make this polymer highly promising for electronic,ptoelectronic, electroactive and photovoltaic applications.
cknowledgment
The authors would like to thank Government Planning Organi-ation for the financial support (Project No: GPO2010K120710).
eferences
[1] I. Kaya, M. Yıldırım, A. Aydın, D. S enol, React. Funct. Polym. 70 (2010) 815.[2] B.A. Bolto, J. Macromol. Sci. Chem. A 14 (1980) 107.
[
[[[
oatings 77 (2014) 463– 472
[3] U. Cosellato, P.A. Vigato, M. Vidali, J. Coord. Chem. Rev. 23 (1977) 31.[4] E.F. Seriven, Chem. Soc. Rev. 12 (1983) 129.[5] K.S. Sahni, J. Reedijk, Coord. Chem. Rev. 59 (1984) 1.[6] M.N. Patel, S.H. Patil, J. Macromol. Sci. Part A A16 (1981) 1429.[7] I. Kaya, A. Aydın, Chin. J. Polym. Sci. 27 (2009) 465.[8] I. Kaya, A. Aydın, J. Appl. Polym. Sci. 121 (2011) 3028.[9] I. Kaya, M. Gül, Eur. Polym. J. 40 (2004) 2025.10] I. Kaya, M. Yıldırım, A. Aydın, Org. Electron. 12 (2011) 210.11] L.Y. Yang, Q.Q. Chen, G.Q. Yang, J.S. Ma, Tetrahedron 59 (2003) 10037.12] I. Kaya, A. Aydın, Prog. Org. Coat. 73 (2012) 239.13] I. Kaya, A. Aydın, M. Yıldırım, J. Fluoresc. 22 (2012) 495.14] I. Kaya, A. Bilici, J. Macromol. Sci. Pure Appl. Chem. 43 (2006) 719.15] K. Colladet, M. Nicolas, L. Goris, L. Lutsen, D. Vanderzande, Thin Solid Films
451–452 (2004) 7.16] I. Kaya, A. Aydın, E-Polymers 071 (2008) 1.17] I. Kaya, M. Yıldırım, M. Kamacı, Eur. Polym. J. 45 (2009) 1586.18] I. Kaya, A. Bilici, Polimery 52 (2007) 827.19] I. Kaya, A. Bilici, Synth. Met. 156 (2006) 736.20] Y.W. Chen-Yang, J.L. Li, T.L. Wu, W.S. Wang, T.F. Hon, Electrochim. Acta 49 (2004)
24] E. Yıldız, P. C amurlu, C. Tanyeli, I. Akhmedov, L. Toppare, J. Electroanal. Chem.612 (2008) 247.
25] A. Aydın, I. Kaya, Electrochim. Acta 65 (2012) 104.26] M. Jadamiec, M. Lapkowski, M. Matlengiewicz, A. Brembilla, B. Henry, L. Rode-
huser, Electrochim. Acta 52 (2007) 6146.27] S. Tarkuc, E. S ahin, L. Toppare, D. C olak, I. Cianga, Y. Yagcı, Polymer 47 (2006)
2001.28] A. Guenbour, A. Kacemi, A. Benbachir, L. Aries, Prog. Org. Coat. 38 (2000) 121.
29] P. Wagner, P.H. Aubert, L. Lutsen, D. Vanderzande, Electrochem. Commun. 4
(2002) 912.30] P. C amurlu, L. Toppare, J. Macromol. Sci. Pure Appl. Chem. 43 (2006) 449.31] B. Sankaran, J.R. Reynolds, Macromolecules 30 (1997) 2582.32] J. Roncali, Chem. Rev. 97 (1997) 173.