Page 1
i
SYNTHESIS OF BISHOMO-INOSITOL DERIVATIVES
A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF NATURAL AND APPLIED SCIENCES
OF MIDDLE EAST TECHNICAL UNIVERSITY
BY
MERVE GÖKÇEN BEKARLAR
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR
THE DEGREE OF MASTER OF SCIENCE IN
CHEMISTRY
SEPTEMBER 2011
Page 2
ii
Approval of the thesis:
SYNTHESIS OF BISHOMO-INOSITOL DERIVATIVES
submitted by MERVE GÖKÇEN BEKARLAR in partial fulfillment of the requirements for the degree of Master of Sciences in Chemistry Department, Middle East Technical University by,
Prof. Dr. Canan ÖZGEN ____________________ Dean, Graduate School of Natural and Applied Sciences Prof. Dr. İlker ÖZKAN ____________________ Head of Department, Chemistry Dept., METU Prof. Dr. Metin BALCI Supervisor, Chemistry Dept., METU ____________________ Examining Committee Members: Prof. Dr. Cihangir TANYELİ ____________________ Chemistry Dept., METU Prof. Dr. Metin BALCI ____________________ Chemistry Dept., METU Prof. Dr. Aliye ALAYLI ALTUNDAŞ ____________________ Chemistry Dept., Gazi University Assist. Prof. Dr. Raşit ÇALIŞKAN ____________________ Chemistry Dept., Süleyman Demirel University Assist. Prof. Dr. Gani KOZA ____________________ Chemistry Dept., Ahi Evran University
Date: 14.09.2011
Page 3
iii
I hereby declare that all information in this document has been obtained and presented in accordance with academic rules and ethical conduct. I also declare that, as required by these rules and conduct, I have fully cited and referenced all material and results that are not original to this work.
Name, Last name : Merve Gökçen Bekarlar
Signature :
Page 4
iv
ABSTRACT
SYNTHESIS OF BISHOMO-INOSITOL DERIVATIVES
Bekarlar, Merve Gökçen
M.Sc., Department of Chemistry
Supervisor: Prof. Dr. Metin Balcı
September 2011, 128 pages
Inositols belong to an important class of biologically active compounds, named as
cyclitols (cyclic polyols), which have attributed high interest in the past decade due
to the glycosidase inhbition property that affects many biological processes.
Initially, 1,3,3a,7a-tetrahydro-2-benzofuran (69) was synthesized as a key compound
to synthesize bishomo-inositol derivatives. Then photooxygenation and
manganese(III) acetate oxidation reactions of this key compound were studied in
order to obtain isomeric inositol derivatives. Moreover, dihydroxylation and acid
catalyzed ring opening reactions were investigated. Finally, new synthetic
approaches for the synthesis of bishomo-inositol derivatives have been developed. In
addition to that whole products were conscientiously purified and characterized and
mechanism for the formation of the products has been discussed.
Keywords: Cyclitol, inositol, bishomo-inositol, photooxygenation, dihydroxylation,
1,3,3a,7a-tetrahydro-2-benzofuran, manganese(III) acetate.
Page 5
v
ÖZ
BİSHOMO-İNOSİTOL TÜREVLERİNİN SENTEZİ
Bekarlar, Merve Gökçen
Yüksek Lisans, Kimya Bölümü
Tez Yöneticisi: Prof. Dr. Metin Balcı
Eylül 2011, 128 sayfa
İnositoller, siklitol adı verilen biyolojik olarak aktif olan önemli bir sınıfa ait olup,
birçok biyolojik olayı etkileyen glykosidaz inhibe edici özelliği nedeniyle son
yıllarda oldukça ilgi çekmiştir.
Öncelikle bishomo-inositol türevlerinin sentezi için 1,3,3a,7a-tetrahidro-2-
benzofuran (69) anahtar molekül olarak sentezlendi. Daha sonra farklı sentez yolları
elde etmek için anahtar molekülün fotooksijenasyon ve mangan(III) asetat
oksidasyon reaksiyonları çalışıldı. Bununla beraber, dihidroksilasyon ve asit
katalizörlüğündeki halka açma reaksiyonları incelendi. Son olarak, bis-homoinositol
türevlerinin sentezi için yeni sentetik yaklaşımlar geliştirildi. Ayrıca bütün ürünler
özenle saflaştırıldı ve karakterize edildi.
Anahtar Kelimeler: Siklitol, inositol, bishomo inositol, 1,3,3a,7a-tetrahidro-2-
benzofuran, fotooksijenasyon, mangan(III) asetat, dihidroksilasyon.
Page 6
vi
ACKNOWLEDGEMENTS
I would like to express my special thanks to my supervisor Prof. Dr. Metin Balcı for
his guidance, patience, supports and encouragements. It was a great chance for me to
be a student of Prof. Balcı.
I also give my thanks to the members of our research group; SYNTHOR especially
to Alper Kılıklı, Berk Müjde, Burak Südemen, Emrah Karahan, Selbi Keskin, Serdal
Kaya for their friendships and helpfulness. I am grateful to Selbi Keskin for her
assistance. I would also like to thank to Arif Baran, Dilem Doğan, Yasemin Altun,
for their helps about the study.
I wish to express my special thanks to my bench mate: Melek Sermin Özer, for her
friendship and endless helps from wherever she is. She was my best suppporter
during my master study and made this time enjoyable for me.
I would like to thank to my friends; Nagehan Keskin, Huriye Erdoğan, Sevinç
Tunçağıl for their invaluable friendship and supports.
My special thanks to Can Nebigil for his encouragement and friendship.
Thanks to all members of METU Chemistry Department.
Finally, my special appreciation and great gratitude is devoted to my family for their
endless love, and encouragement in every moment of my life. Specially, to my sister
Hande Bekarlar for her patience, moral support.
Page 7
vii
TABLE OF CONTENTS
ABSTRACT ................................................................................................................ iv
ÖZ ................................................................................................................................ v
ACKNOWLEDGEMENTS ........................................................................................ vi
TABLE OF CONTENTS ........................................................................................... vii
LIST OF FIGURES ..................................................................................................... x
LIST OF SCHEMES ................................................................................................. xiv
LIST OF ABBREVIATIONS ................................................................................... xvi
CHAPTERS
1. INTRODUCTION.................................................................................................... 1
1.1 Cyclitols ............................................................................................................. 1
1.2 Inositols .............................................................................................................. 1
1.2.1 Biologic functions of inositols .................................................................... 3
1.3 Synthesis of inositol and inositol derivatives ..................................................... 4
1.3.1 Synthesis of myo-inositol ............................................................................ 4
1.3.2 Synthesis of neo-inositol (10) ..................................................................... 5
1.3.3 Synthesis of allo-inositol (11) ..................................................................... 6
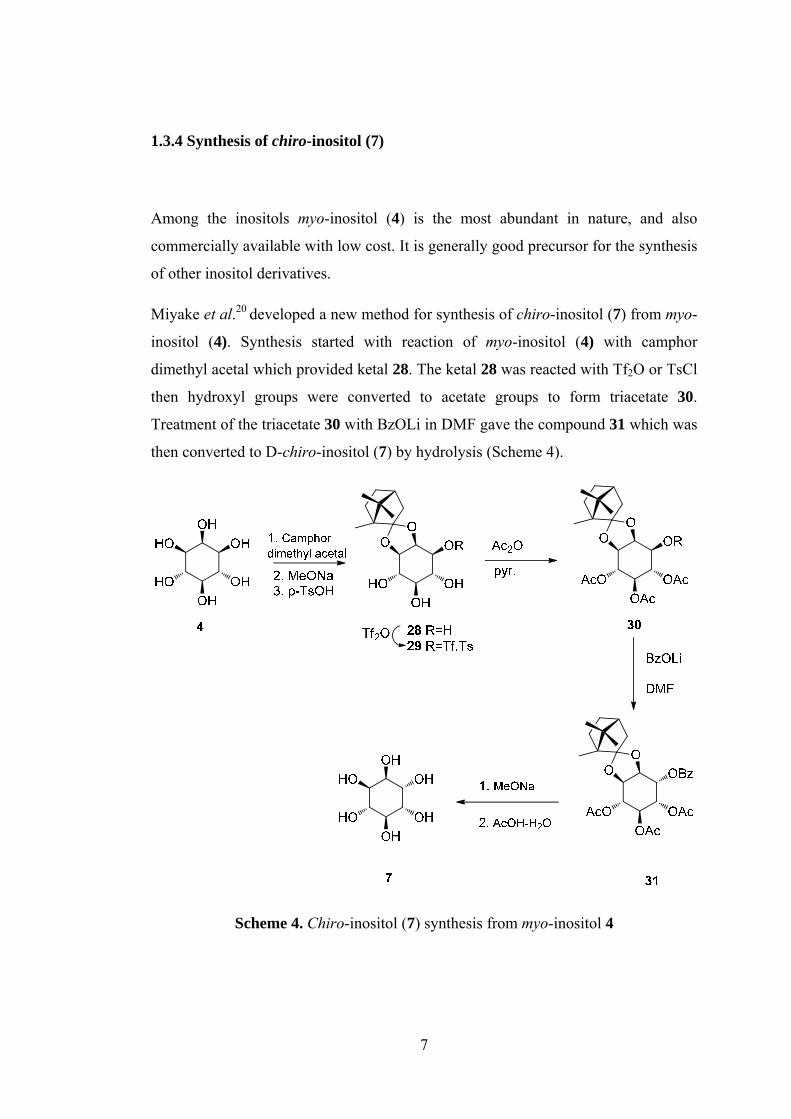
1.3.4 Synthesis of chiro-inositol (7) ..................................................................... 7
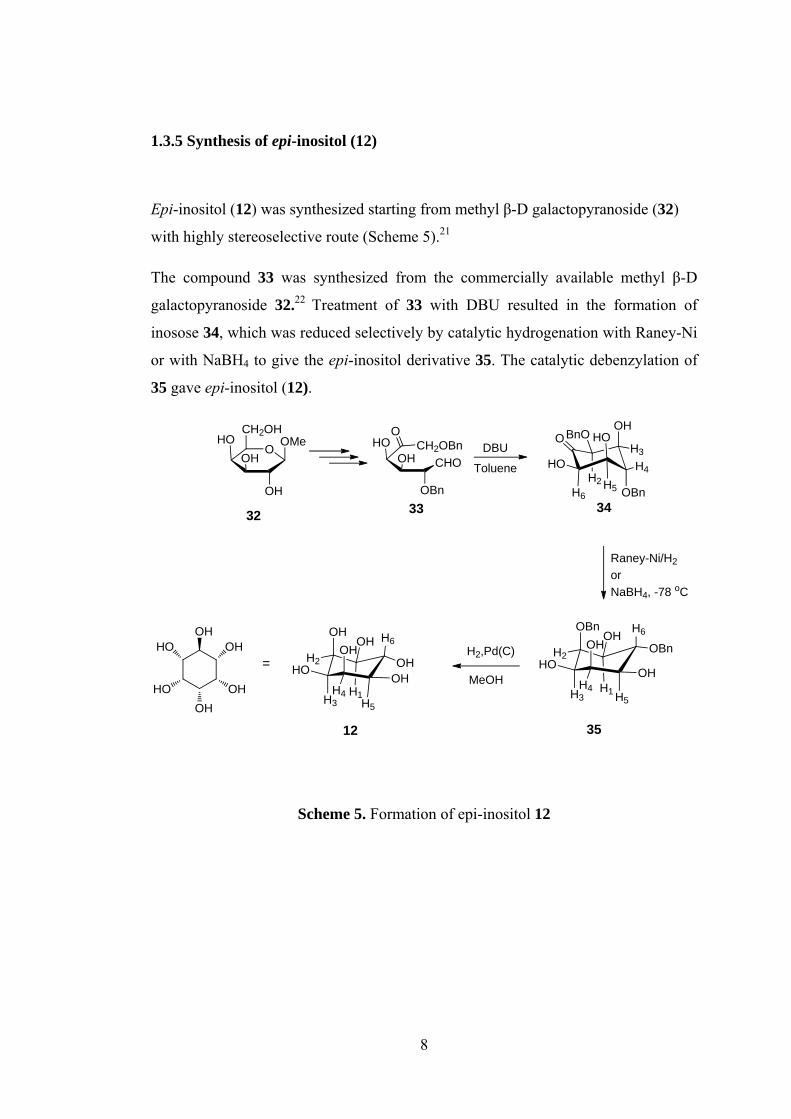
1.3.5 Synthesis of epi-inositol (12) ...................................................................... 8
1.3.6 Synthesis of muco-inositol (6) .................................................................... 9
1.3.7 Synthesis of cis-inositol (9) ......................................................................... 9
1.3.8 Synthesis of scyllo-inositol (5) .................................................................. 11
1.3.9 Synthesis of bicyclic inositols ................................................................... 12
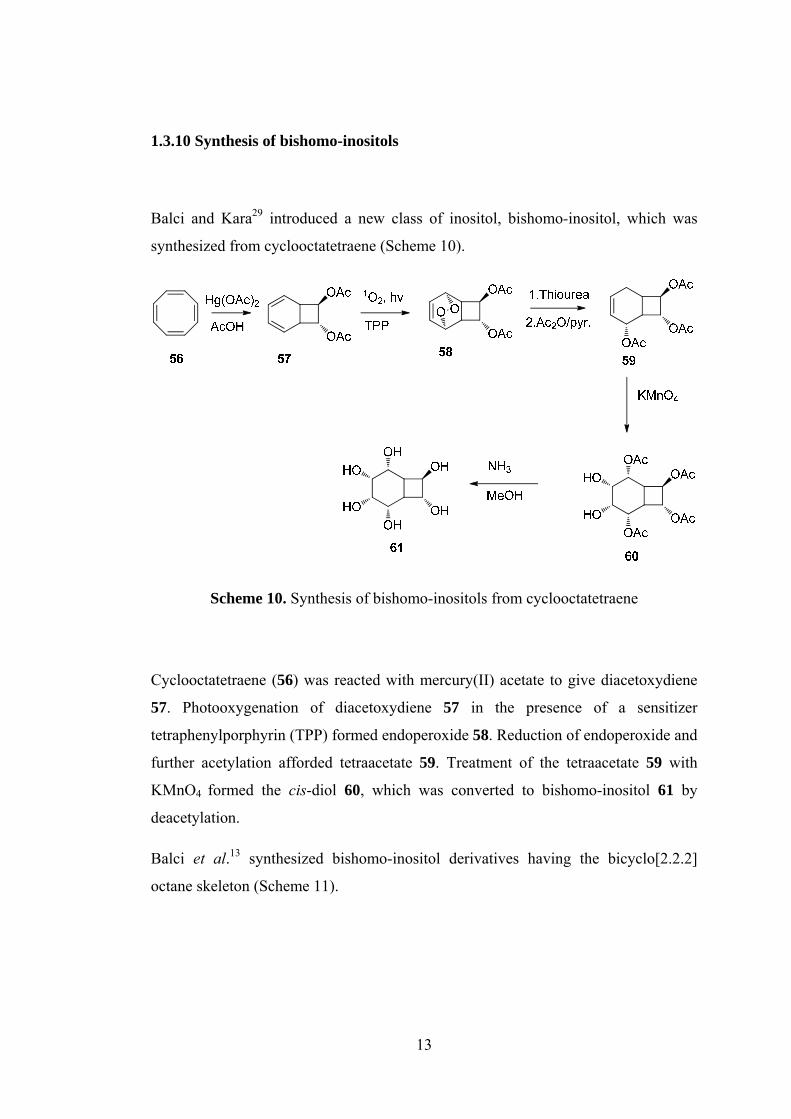
1.3.10 Synthesis of bishomo-inositols ............................................................... 13
1.4 Singlet Oxygen ................................................................................................. 17
Page 8
viii
1.5 Manganese (III) Acetate Oxidation Reactions ................................................. 20
1.6 Aim of thesis .................................................................................................... 21
2. RESULTS AND DISCUSSION ............................................................................ 23
2.1 Synthesis of 3aR(S),7aS(R)-1,3,3a,7a-tetrahydro-2-benzofuran (69) ............. 23
2.2 Photooxygenation of 3aR(S),7aS(R)-1,3,3a,7a-tetrahydro-2-benzofuran (69) . 24
2.3 Rearrangement of endoperoxide 94 to bisepoxide 100 .................................... 25
2.4 Acid catalyzed ring opening reaction of the bisepoxide 100 ........................... 27
2.4.1. Ring opening reaction of the bisepoxide 100 in the presence of water ... 27
2.4.2. Ring Opening Reaction of the Bisepoxide 100 without Water .................... 29
2.4.3 Manganese (III) Acetate Oxidation Reaction of rel-(3aR,7aS)-1,3,3a,7a-
Tetrahydro-2-benzofuran (69) ............................................................................ 38
2.4.4 Upjohn Dihyroxylation ............................................................................. 43
2.4.5 Reduction of Lactone in Diacetate Isomers 120 & 121 by LiAlH4 .......... 49
3. EXPERIMENTAL ................................................................................................. 50
3.1 General ............................................................................................................. 50
3.2 Synthesis of cis-1,2,3,6,-Tetrahydrophthalyl Alcohol (91).............................. 51
3.3 Synthesis of rel-(1R, 3S)-1,3,3a,4,7,7a-Hexahydro-2-benzofuran (92) ........... 51
3.4 Synthesis of rel-(3aR,5R,6S,7aS)-5,6-Dibromooctahydro-2-benzo furan (94) 52
3.5 Synthesis of rel-(3aR,7aS)-1,3,3a,7a-Tetrahydro-2-benzofuran (69) ............... 53
3.6 Synthesis of rel-(1R,2R,6S,7S)-4,10,11-trioxa-tricyclo[5.2.2.02,6]undec-8-ene
(94) ......................................................................................................................... 53
3.7 Synthesis of rel-(1aR,1bS,2aS,2bS,5aR,5bR)-octahydrobis (oxireno)
isobenzofuran (100) ............................................................................................... 54
3.8 Synthesis of rel-(1R,5S,6R,7S,8S,9S)-3-oxabicyclo[3.3.1]nonane-6,7,8,9-
tetrayl tetraacetate (103) ......................................................................................... 55
3.9 Synthesis of rel-(1R,5S,6R,7S,8S,9S)- 3-oxabicyclo [3.3.1]nonane-6,7,8,9-
tetraol (105) ............................................................................................................ 56
Page 9
ix
3.10 Synthesis of rel-(3aR,4S,5R,6R,7S,7aS)-octahydro-2-benzofuran-4,5,6,7-
tetrayl tetraacetate (102) ......................................................................................... 56
3.11 Manganese (III) Acetate Oxidation Reaction of rel-(3aR,7aS)-1,3,3a,7a-
Tetrahydro-2-benzofuran (69) ................................................................................ 57
3.12 cis-Hydroxylation of rel-(3aR,5aR,8aS,8bR)-1,3,3a,8,8a,8b Hexahydro
benzo[1,2-b:3,4-c’]difuran-7(5aH)-one (115) ........................................................ 59
4. CONCLUSION ...................................................................................................... 61
REFERENCES ........................................................................................................... 63
APPENDIX ................................................................................................................ 65
A. SPECTRAL DATA ........................................................................................... 65
Page 10
x
LIST OF FIGURES
FIGURES
Figure 1. Cyclitol derivatives ....................................................................................... 1
Figure 2. Inositol Stereoisomers ................................................................................. 2
Figure 3. Molecular Orbital Diagram of O2 Molecule ............................................... 18
Figure 4. Electronic States of Molecular Oxygen ...................................................... 19
Figure 5. Formation of Singlet Oxygen with Sensitizer ............................................ 19
Figure 6. Singlet Oxygen Reactions .......................................................................... 20
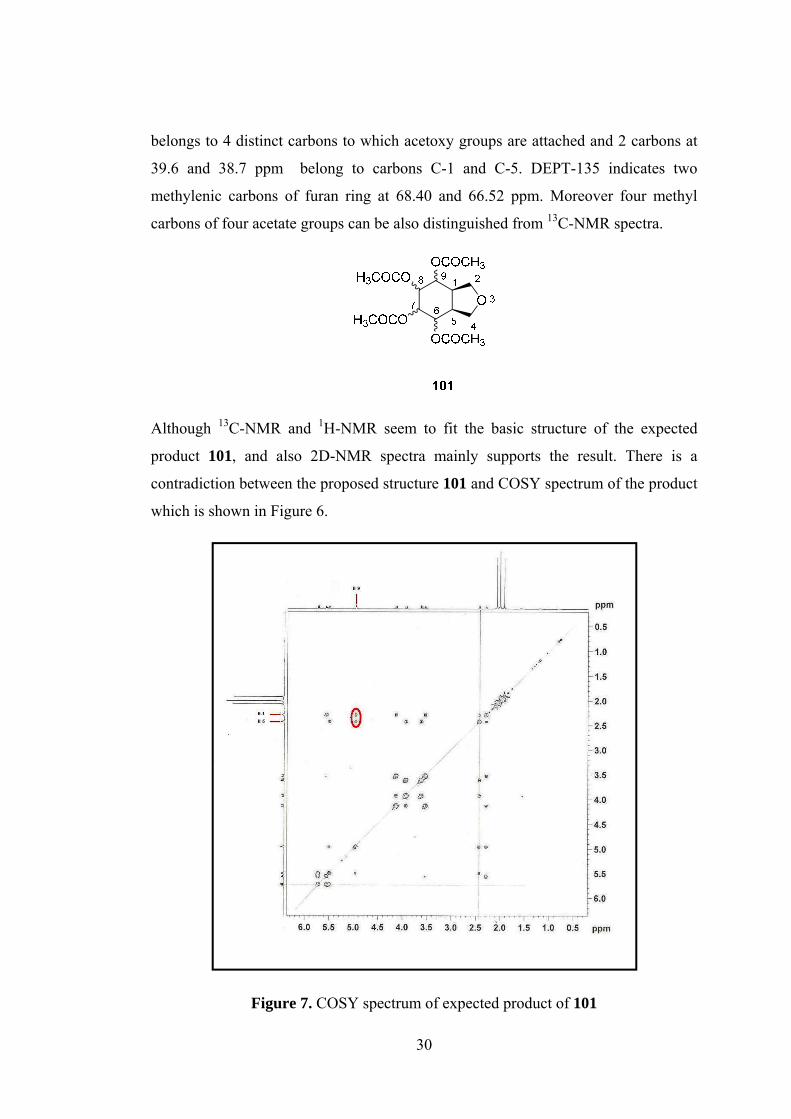
Figure 7. COSY spectrum of expected product of 101 .............................................. 30
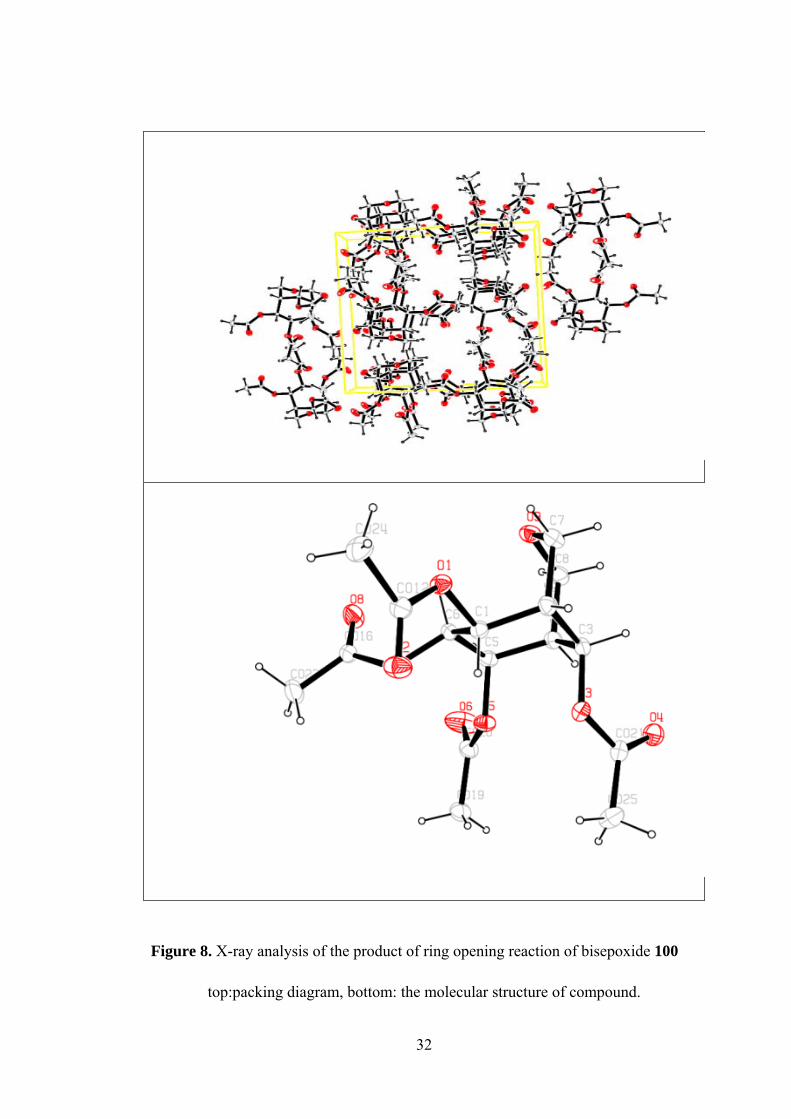
Figure 8. X-ray analysis of the product of ring opening reaction of bisepoxide 100 32
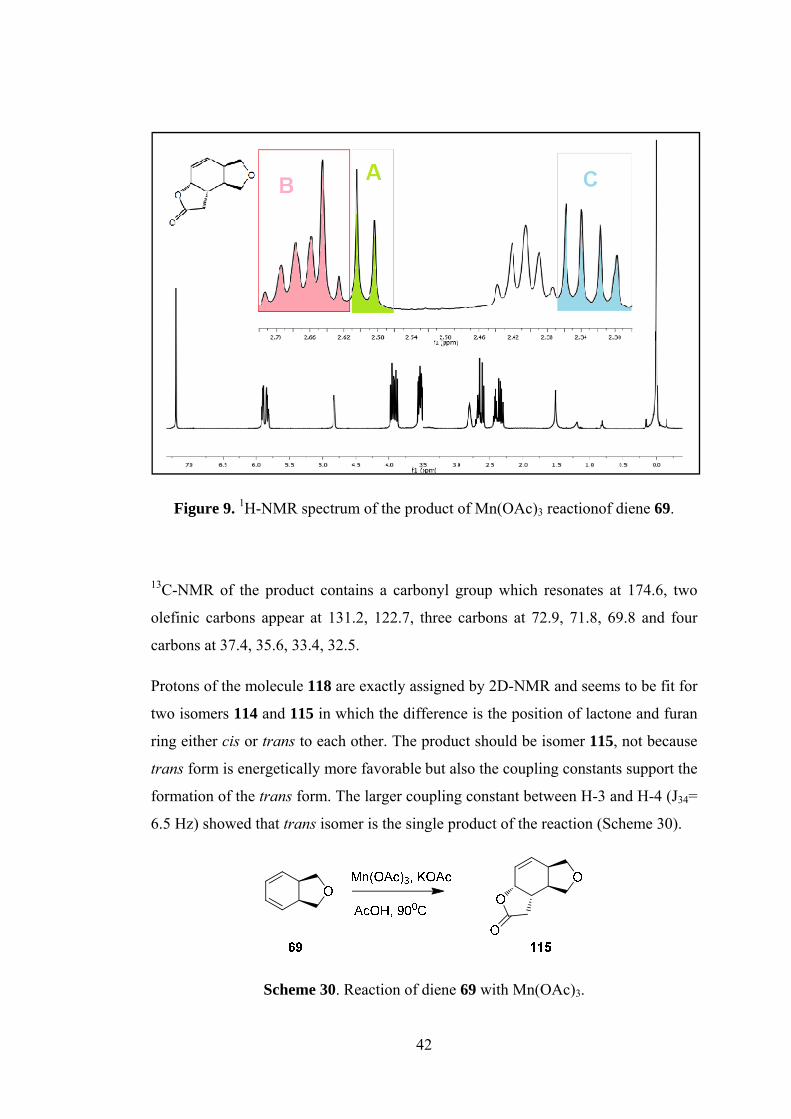
Figure 9. 1H-NMR spectrum of the product of Mn(OAc)3 reactionof diene 69. ....... 42
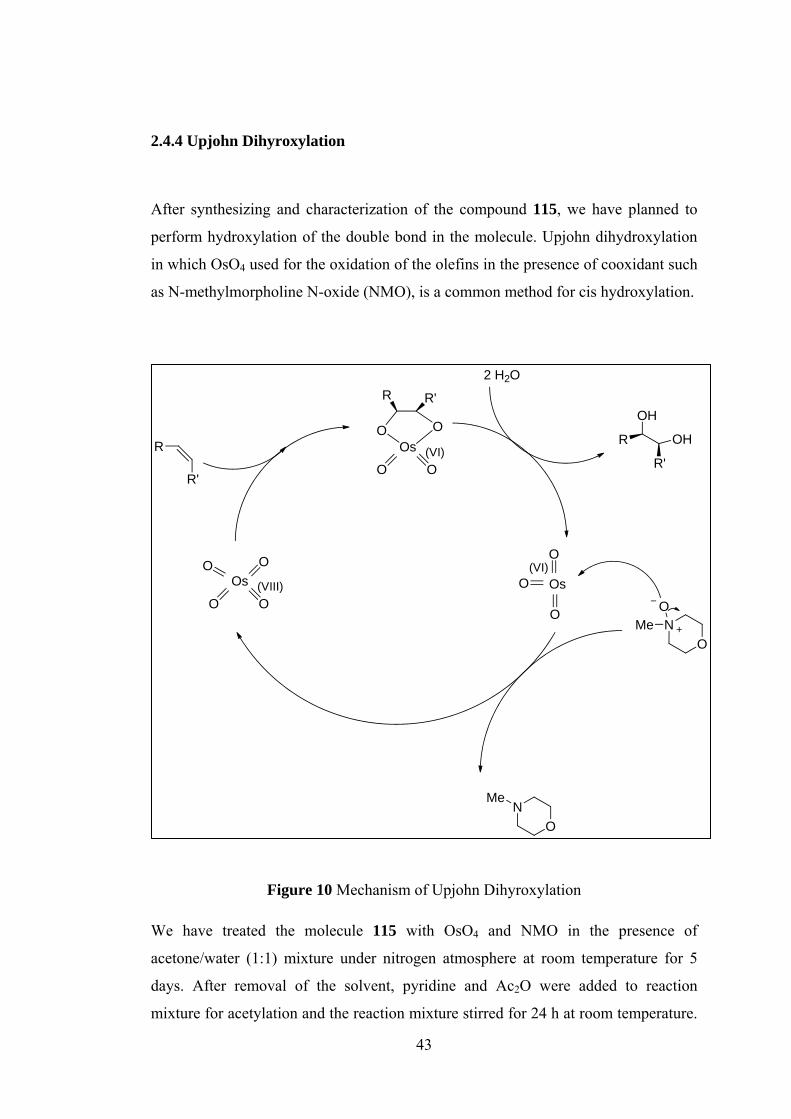
Figure 10 Mechanism of Upjohn Dihyroxylation ...................................................... 43
Figure 11. Optimized geometry of the molecule 115 ................................................ 44
Figure 12. Geometry Optimization of Molecule 120 ................................................. 46
Figure 13. Geometry Optimization of Molecule 121 ................................................. 46
Figure 14. Karplus-Conroy Curve............................................................................. 47
Figure A. 1 1H-NMR spectrum of compound 91 ....................................................... 65
Figure A. 2 13C-NMR spectrum of compound 91 ...................................................... 66
Figure A. 3 IR spectrum of compound 91 ................................................................. 67
Page 11
xi
Figure A. 4 1H-NMR spectrum of compound 92 ....................................................... 68
Figure A. 5 13C-NMR spectrum of compound 92 ...................................................... 69
Figure A. 6 IR spectrum of compound 92 ................................................................. 70
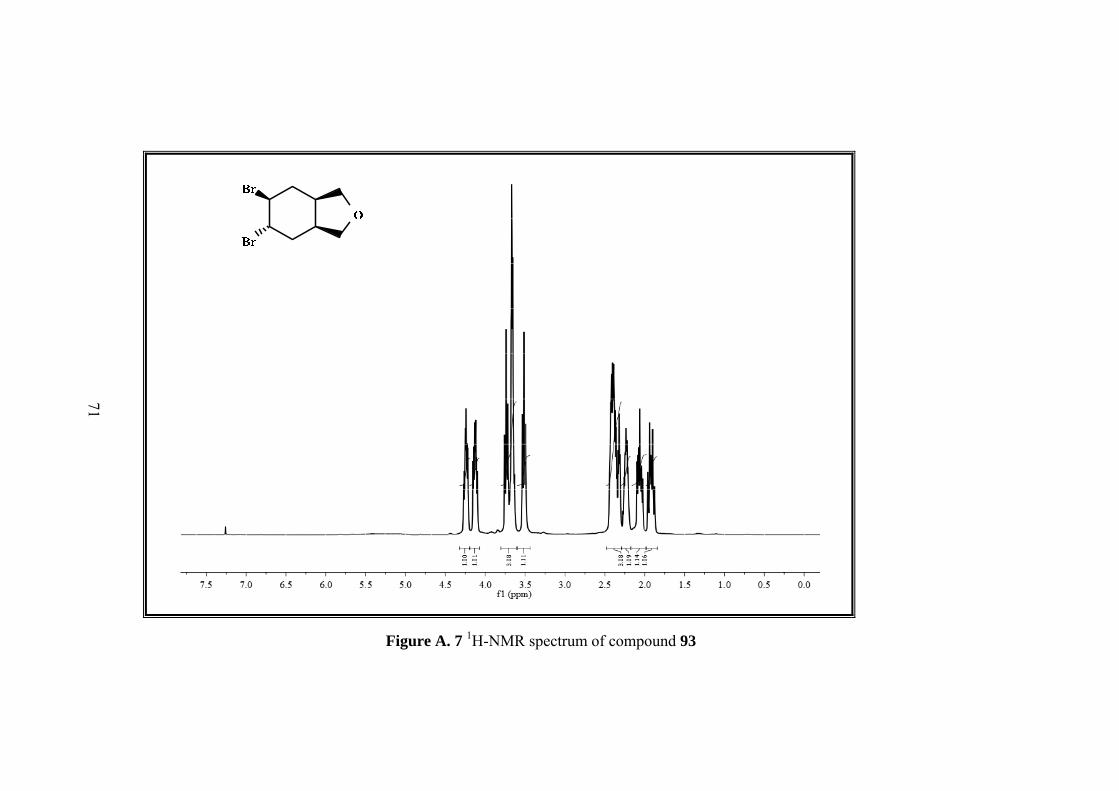
Figure A. 7 1H-NMR spectrum of compound 93 ....................................................... 71
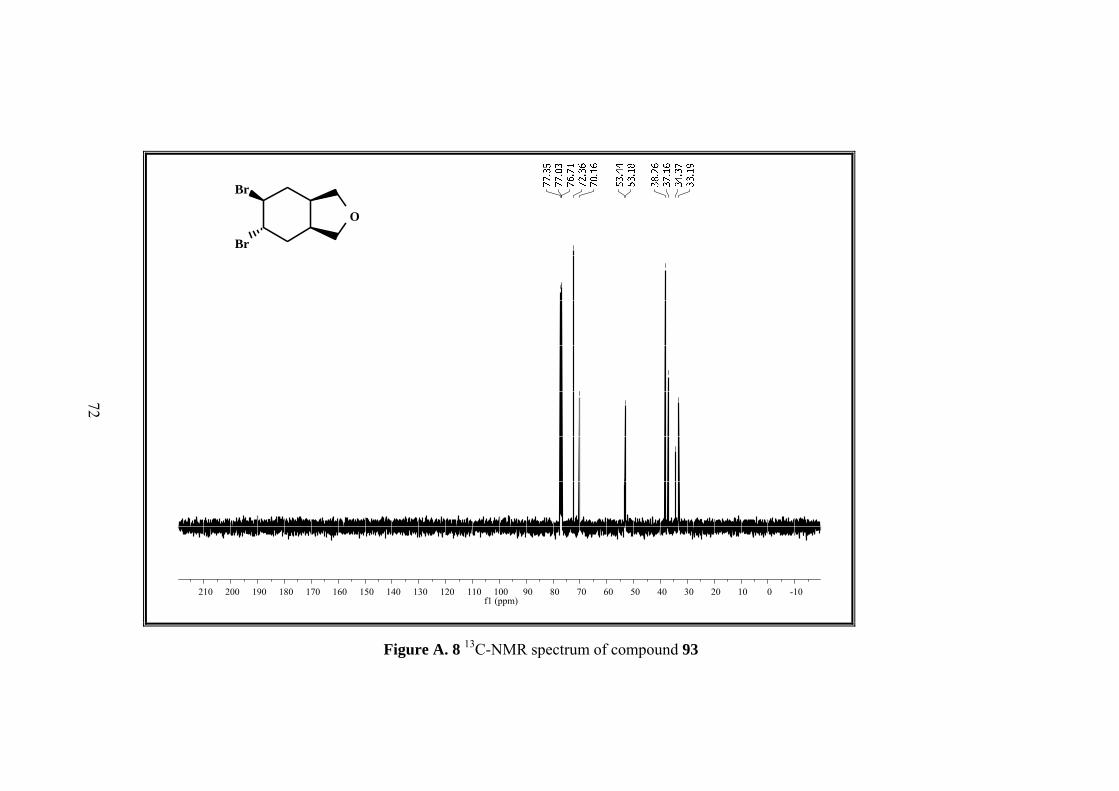
Figure A. 8 13C-NMR spectrum of compound 93 ...................................................... 72
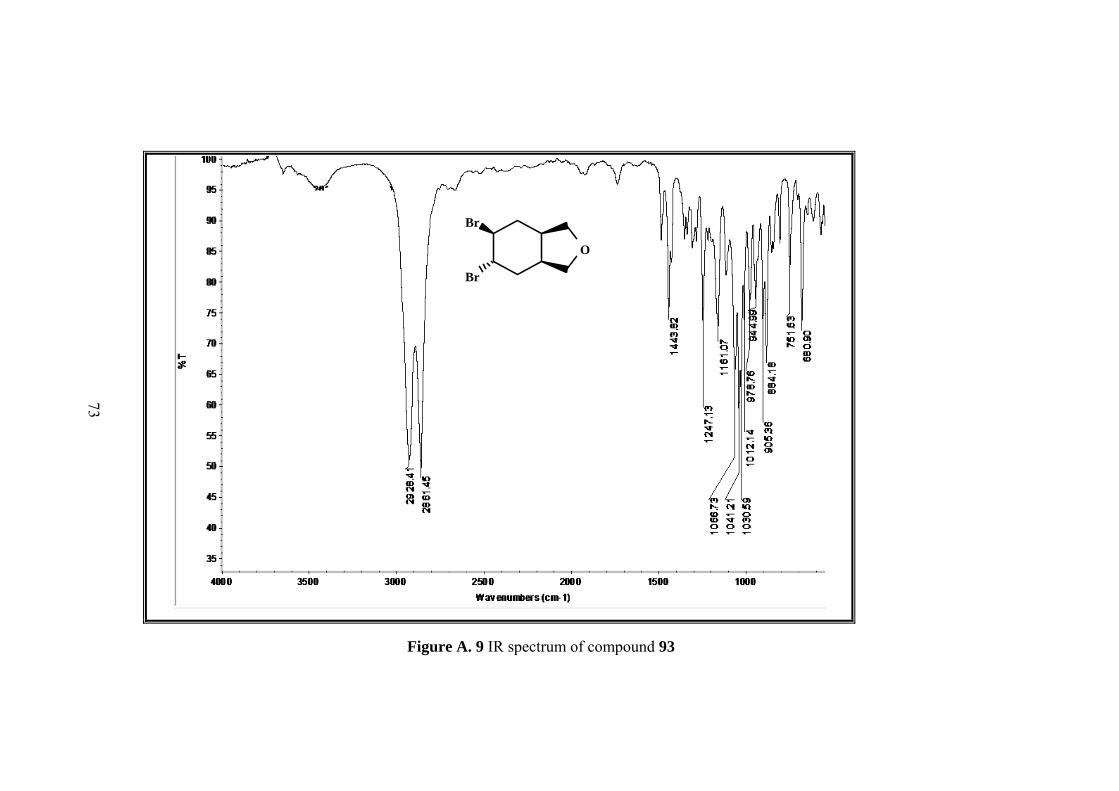
Figure A. 9 IR spectrum of compound 93 ................................................................. 73
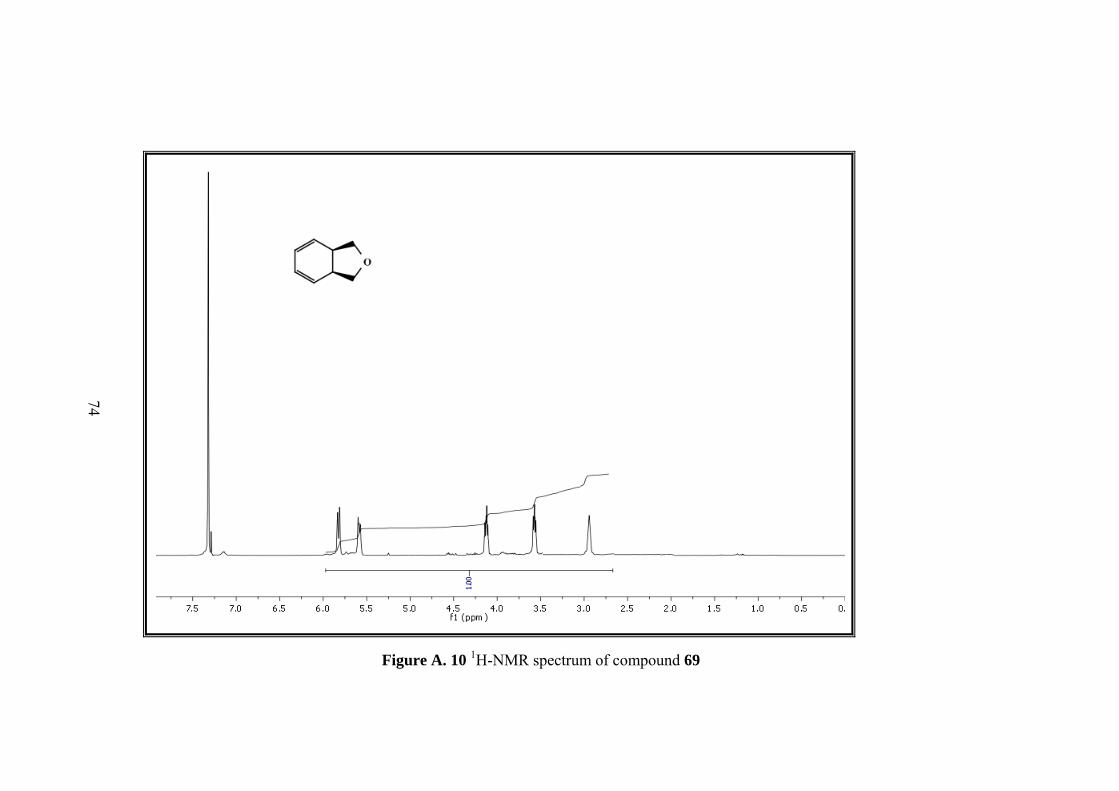
Figure A. 10 1H-NMR spectrum of compound 69 ..................................................... 74
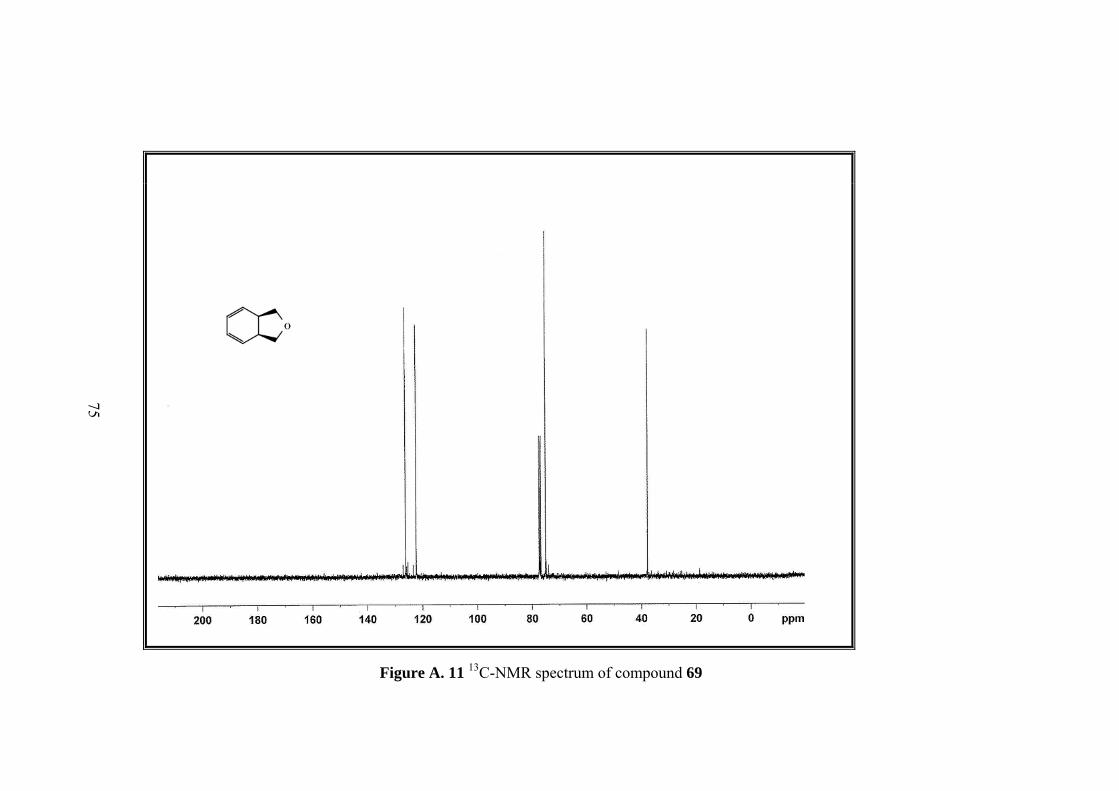
Figure A. 11 13C-NMR spectrum of compound 69 .................................................... 75
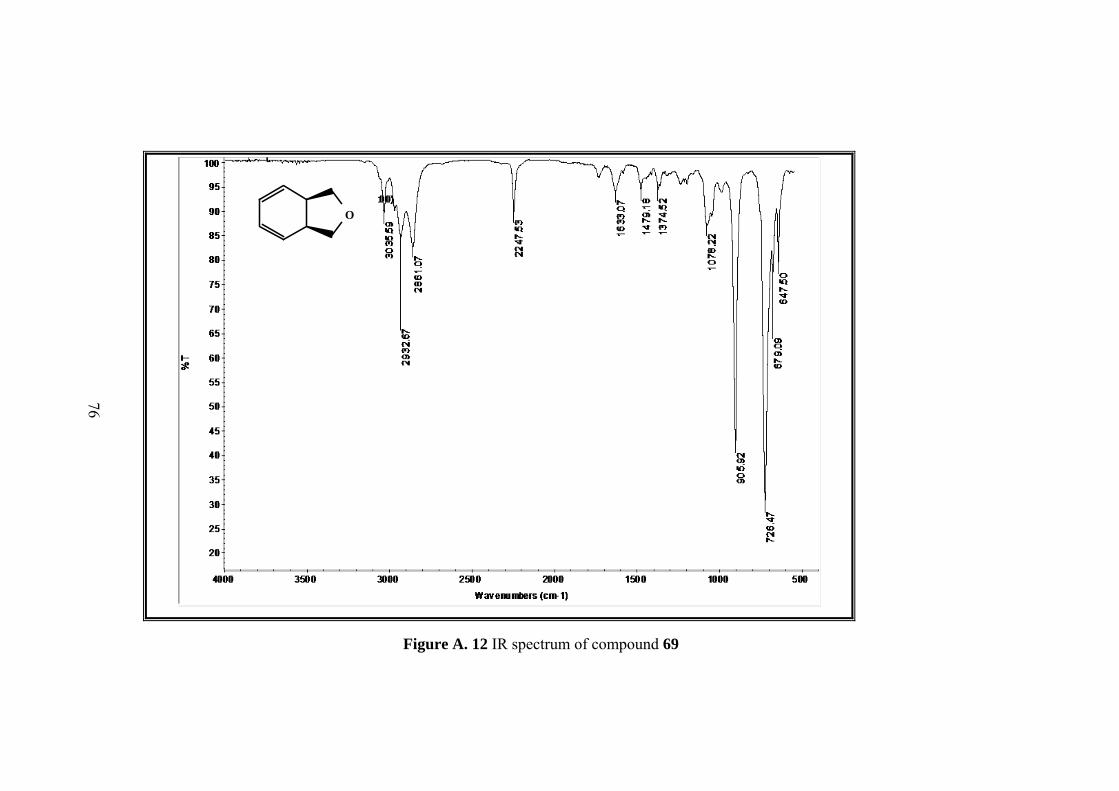
Figure A. 12 IR spectrum of compound 69 ............................................................... 76
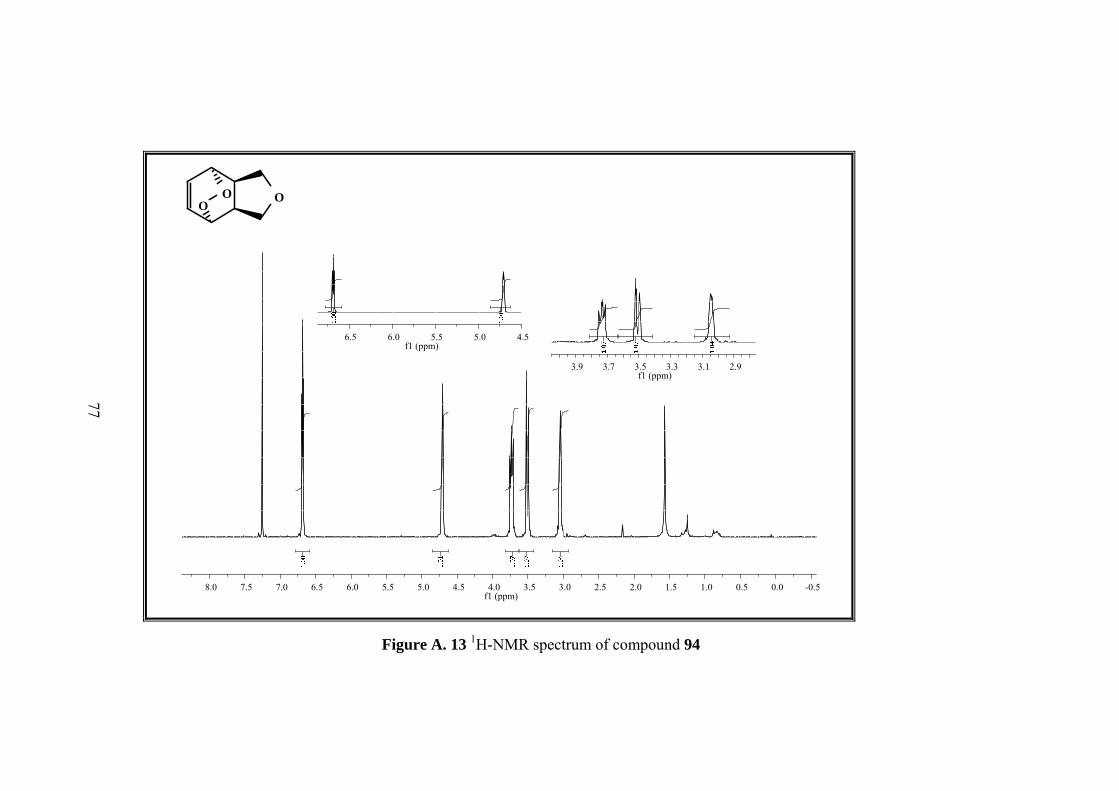
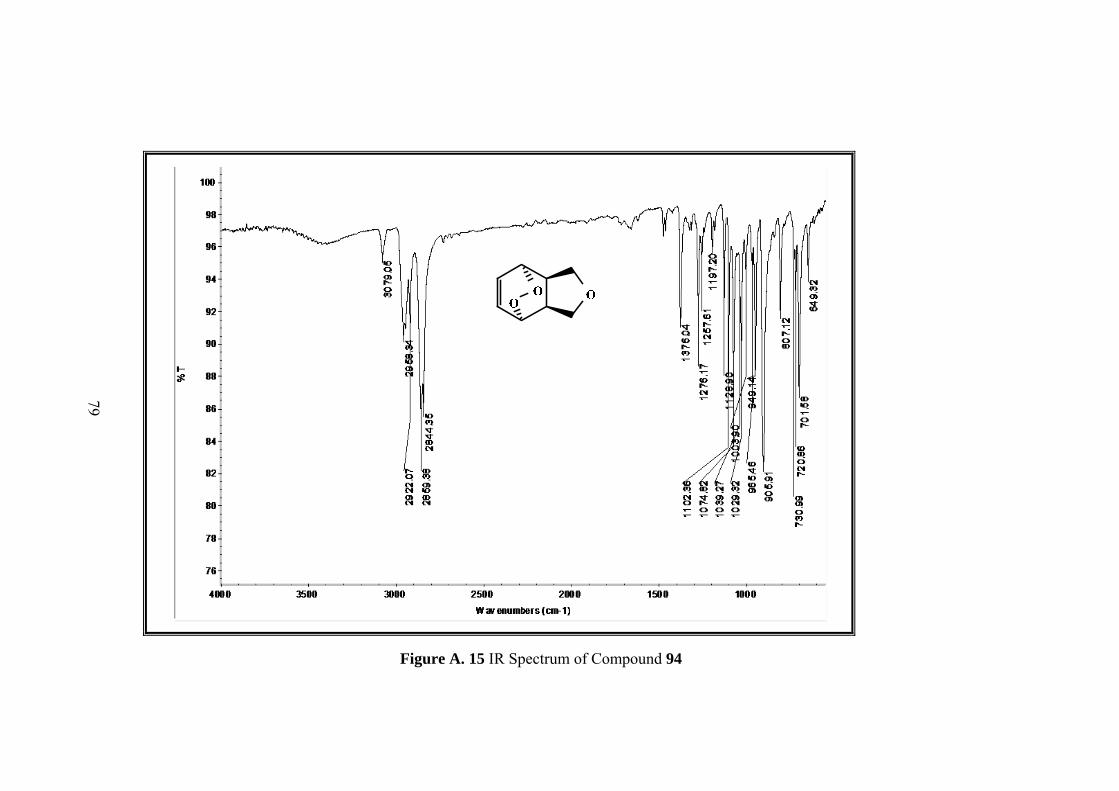
Figure A. 13 1H-NMR spectrum of compound 94 ..................................................... 77
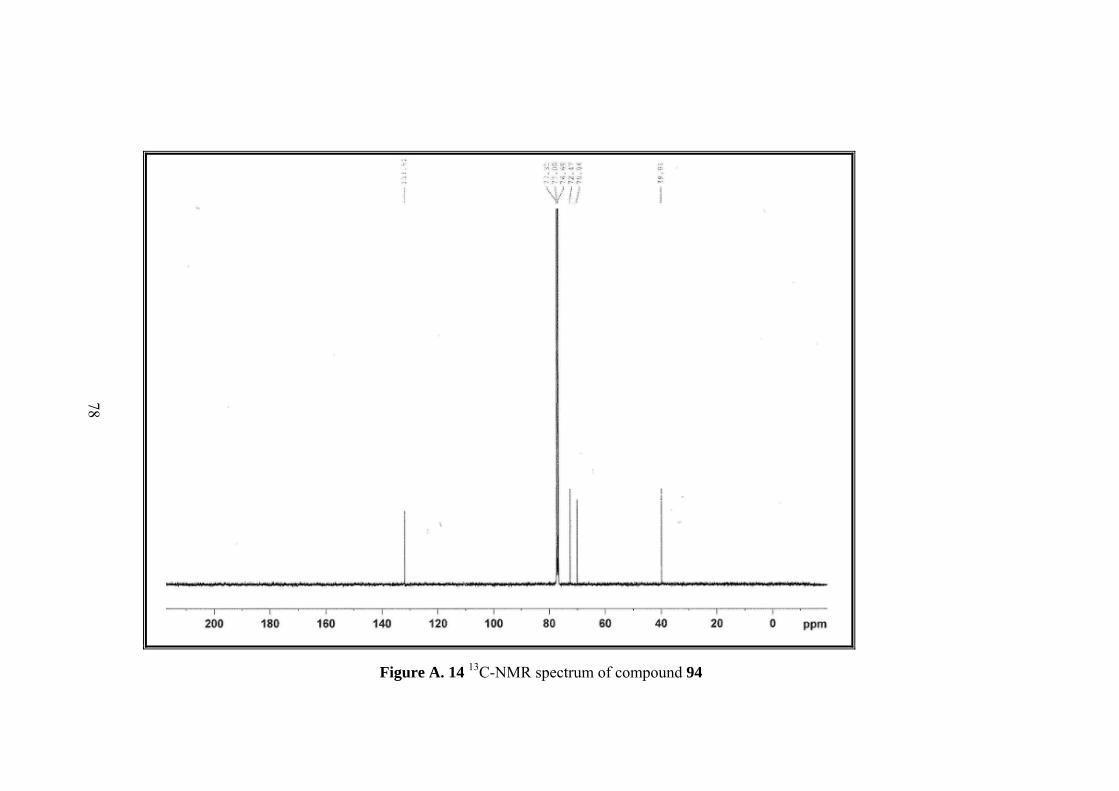
Figure A. 14 13C-NMR spectrum of compound 94 .................................................... 78
Figure A. 15 IR Spectrum of Compound 94 .............................................................. 79
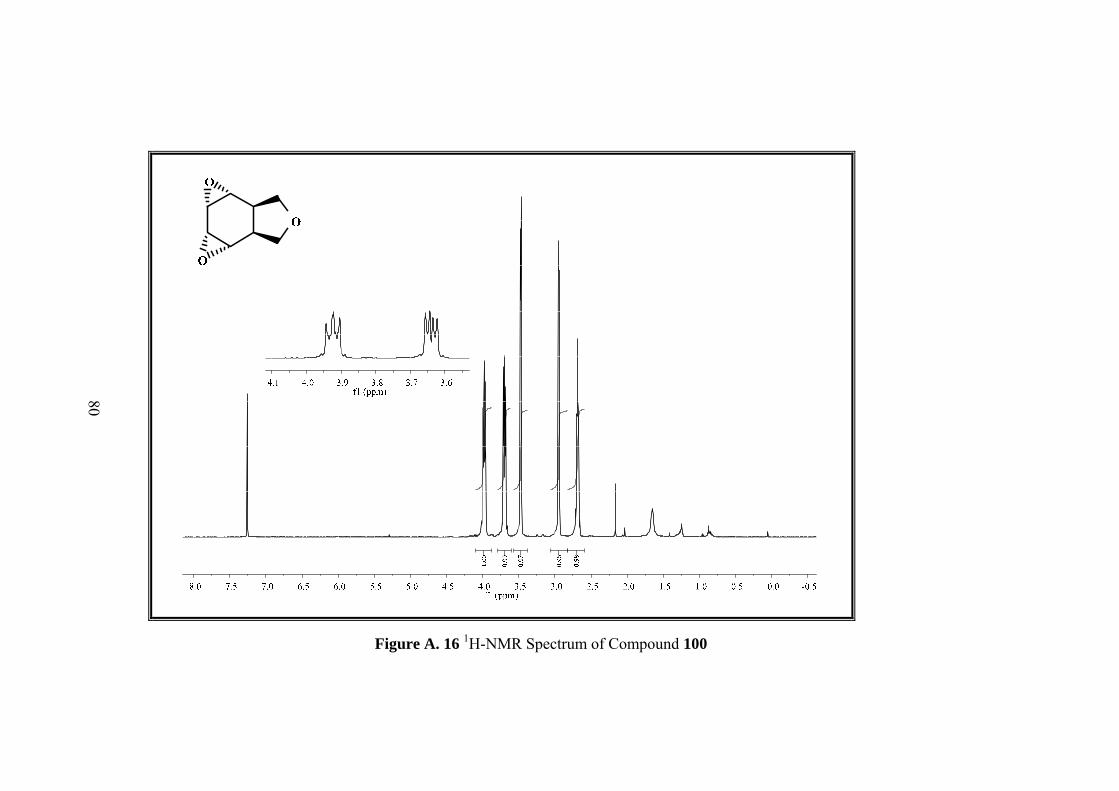
Figure A. 16 1H-NMR Spectrum of Compound 100 ................................................. 80
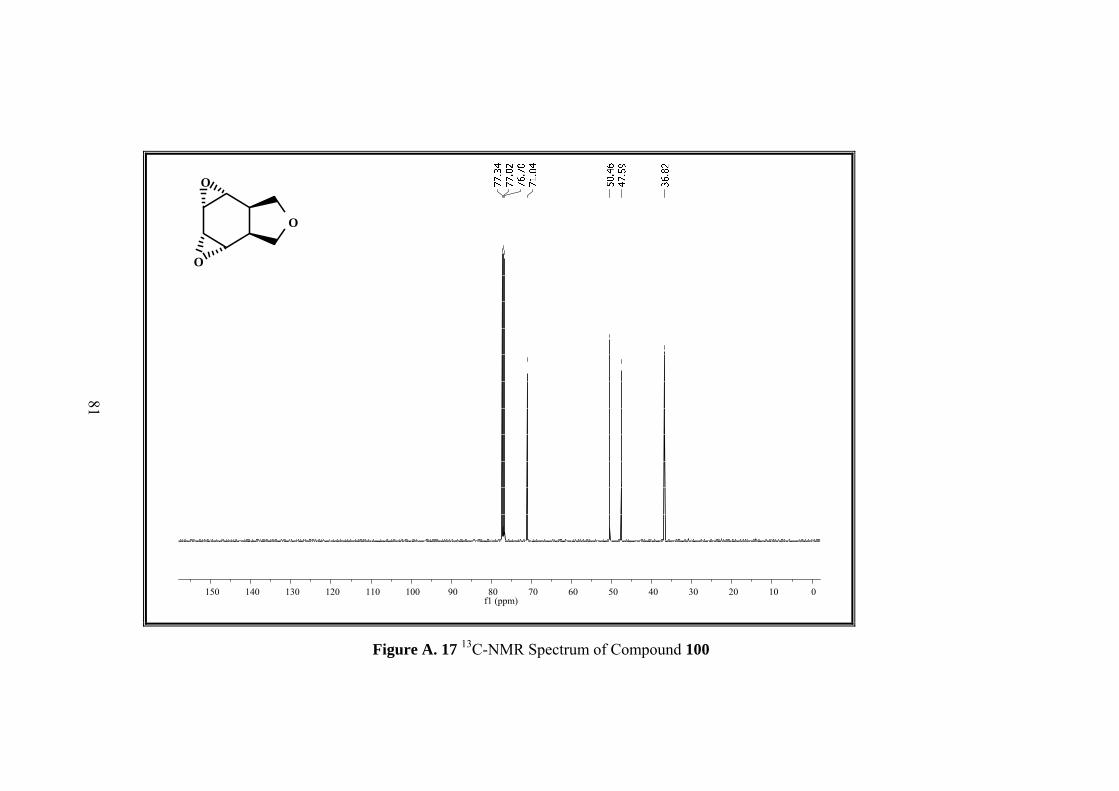
Figure A. 17 13C-NMR Spectrum of Compound 100 ................................................ 81
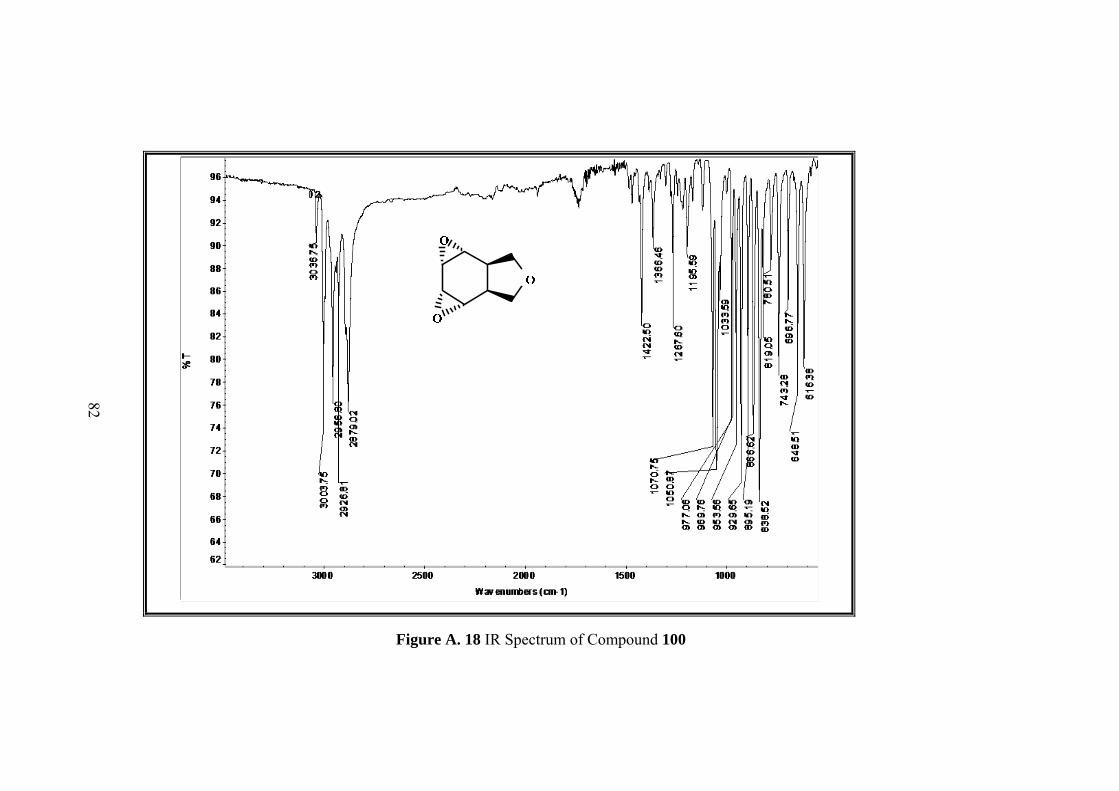
Figure A. 18 IR Spectrum of Compound 100 ............................................................ 82
Figure A. 19 DEPT-135 spectrum of compound 100 ................................................ 83
Figure A. 20 COSY spectrum of compound 100 ....................................................... 84
Figure A. 21 HSQC spectrum of compound 100 ....................................................... 85
Figure A. 22 HMBC spectrum of compound 100 ...................................................... 86
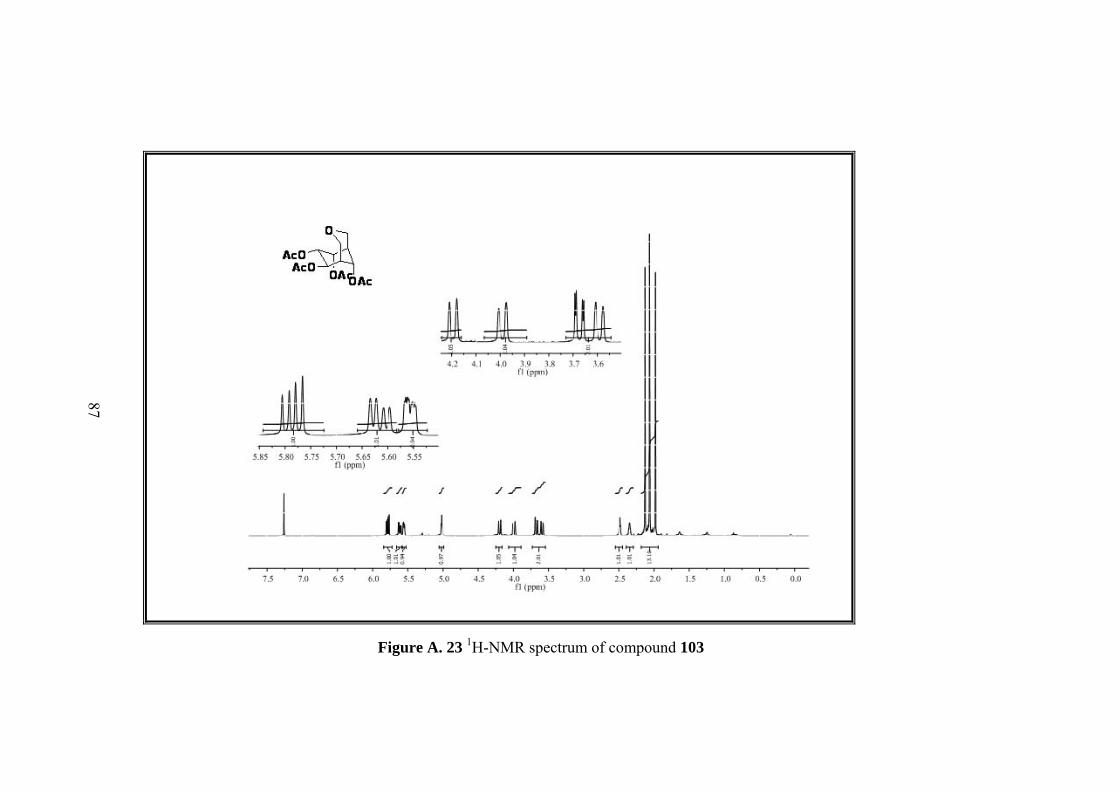
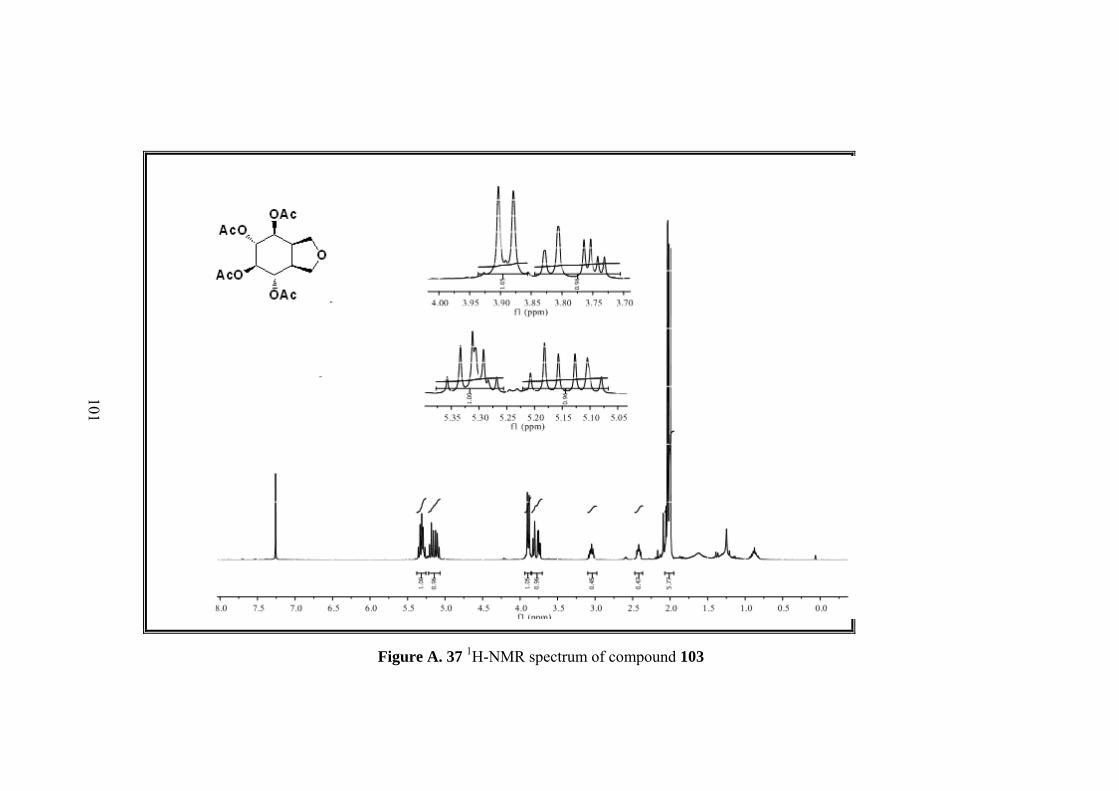
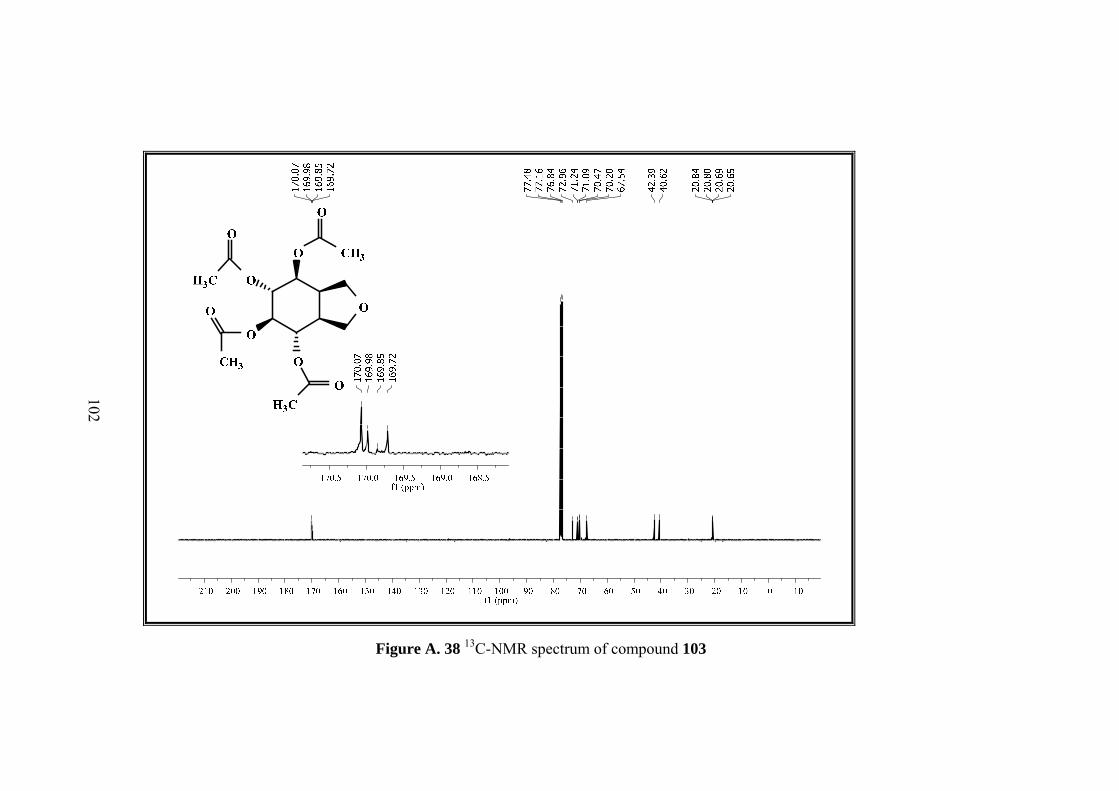
Figure A. 23 1H-NMR spectrum of compound 103 ................................................... 87
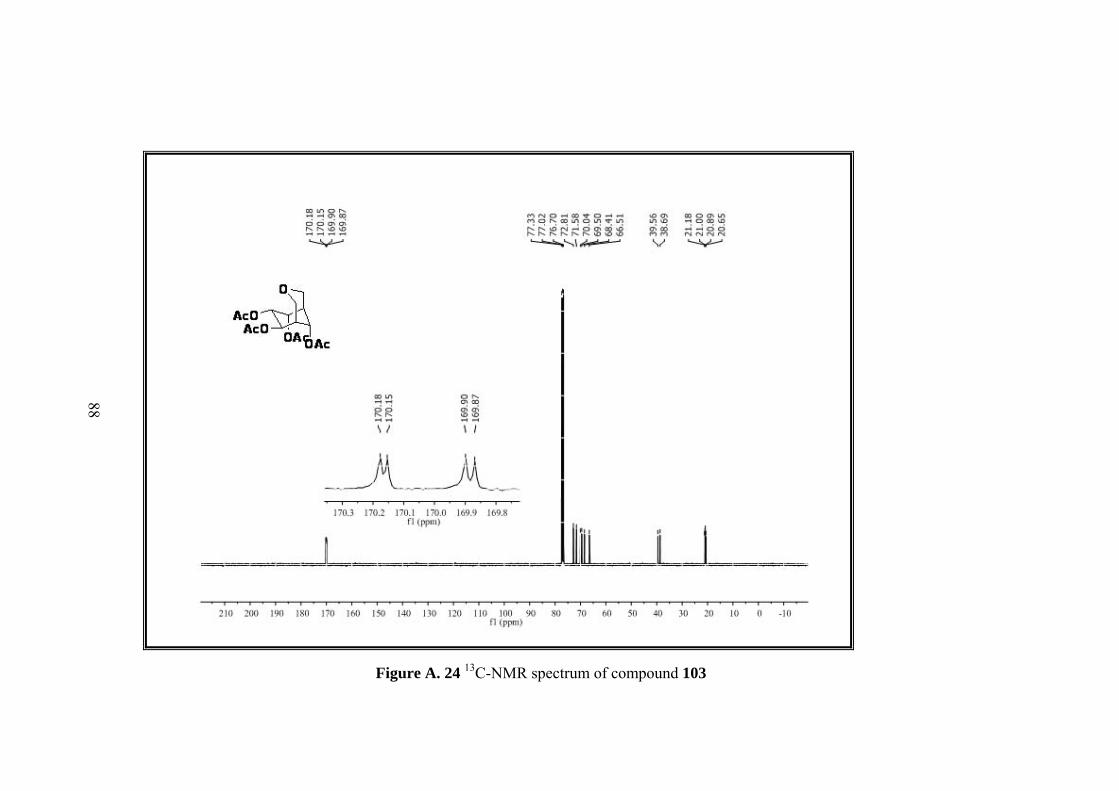
Figure A. 24 13C-NMR spectrum of compound 103 .................................................. 88
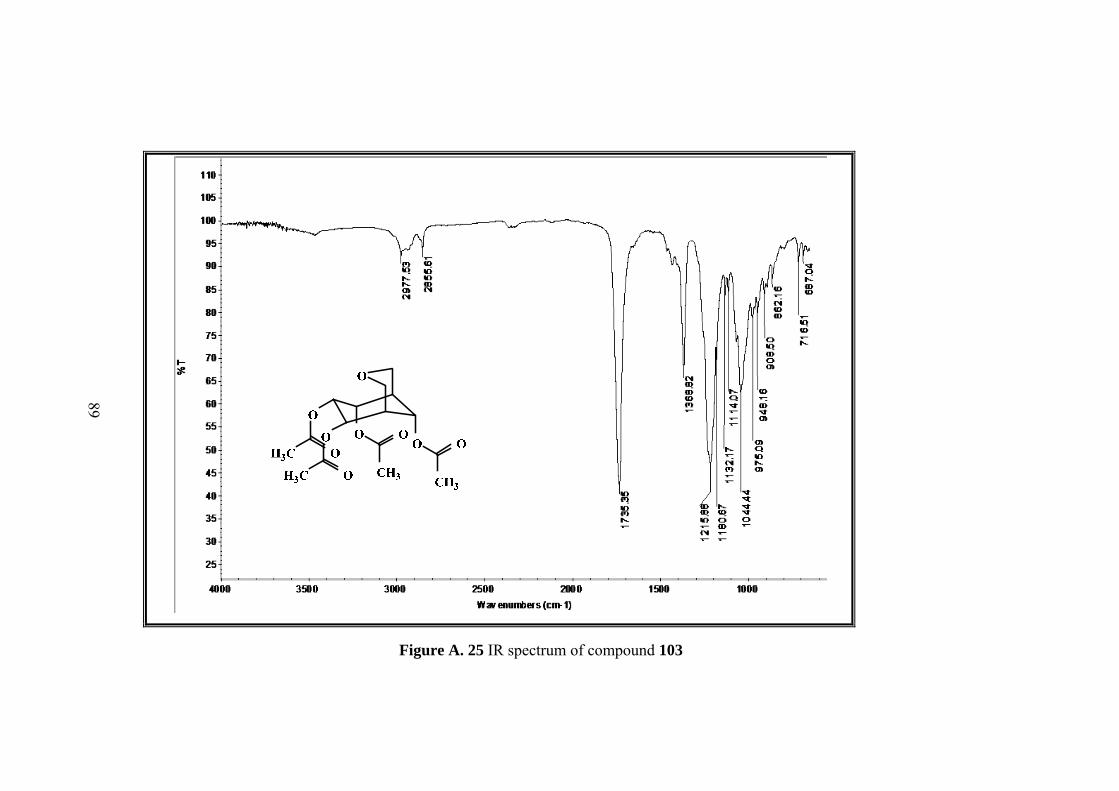
Figure A. 25 IR spectrum of compound 103 ............................................................. 89
Page 12
xii
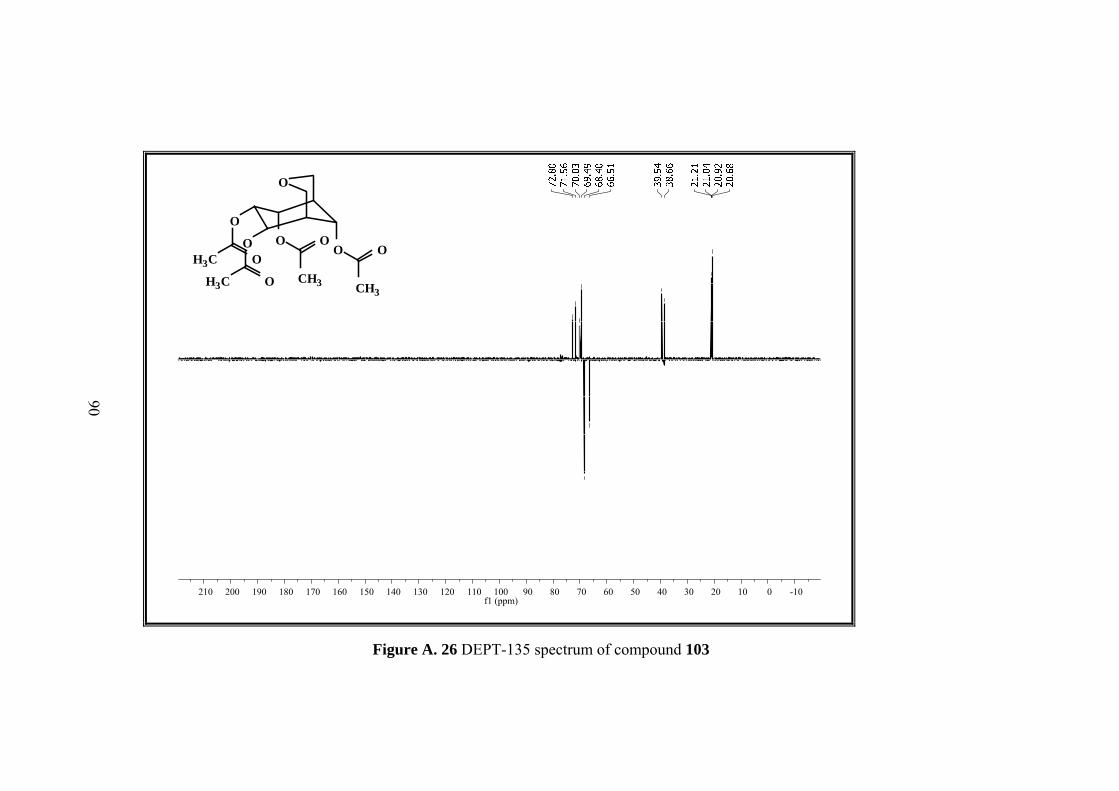
Figure A. 26 DEPT-135 spectrum of compound 103 ................................................ 90
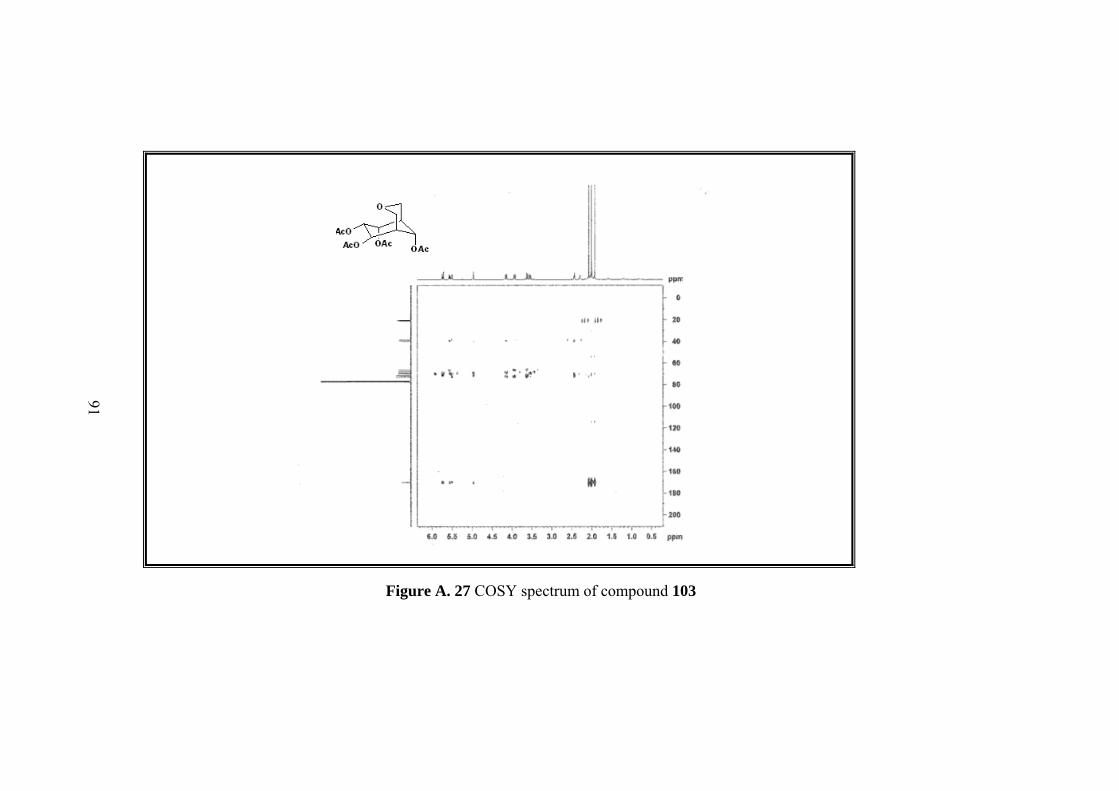
Figure A. 27 COSY spectrum of compound 103 ....................................................... 91
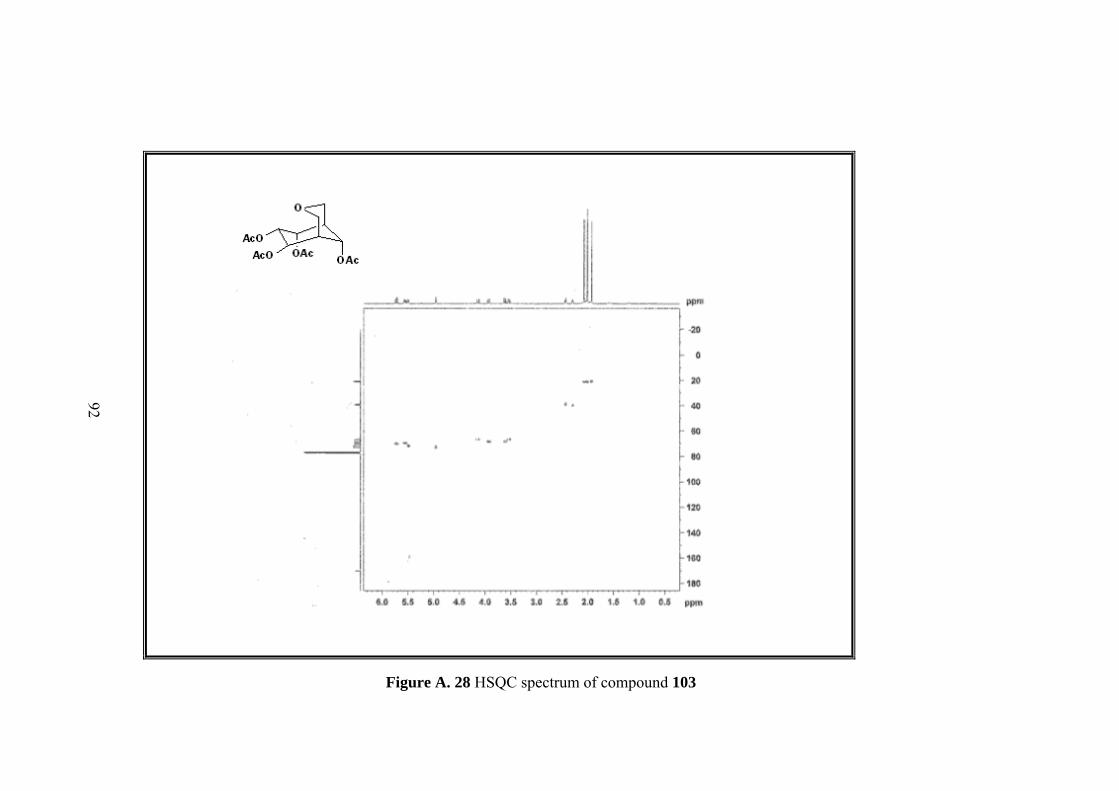
Figure A. 28 HSQC spectrum of compound 103 ....................................................... 92
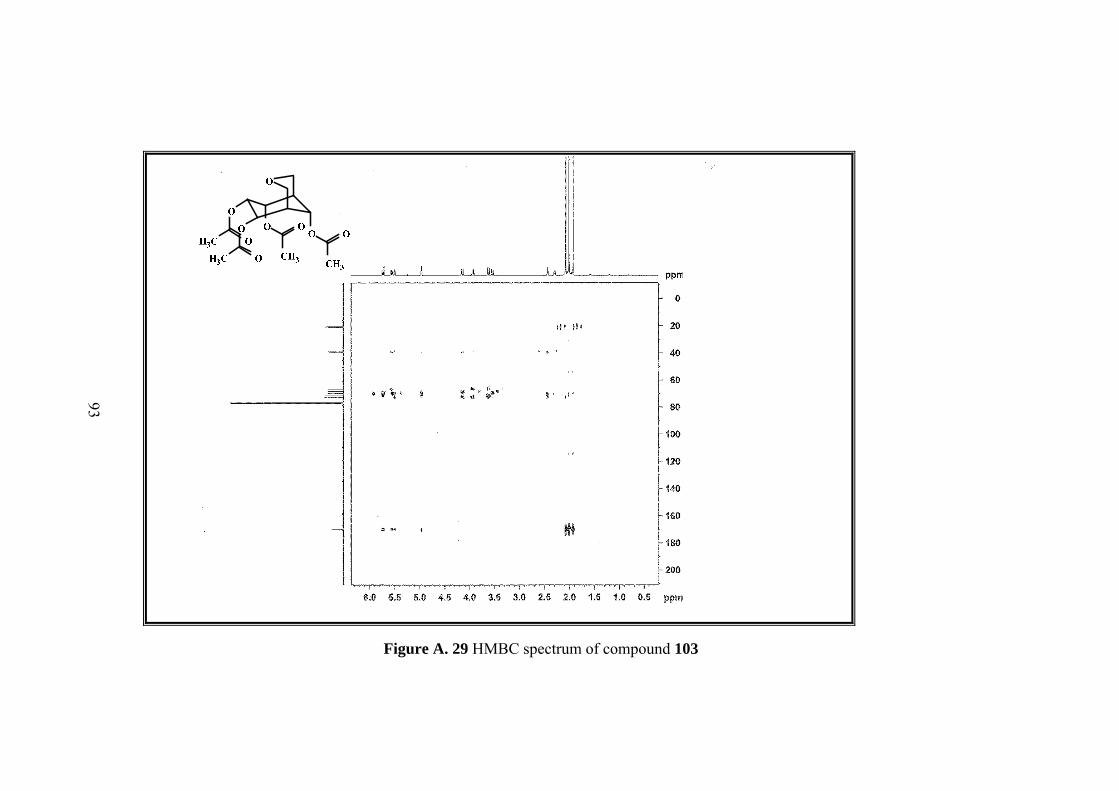
Figure A. 29 HMBC spectrum of compound 103 ...................................................... 93
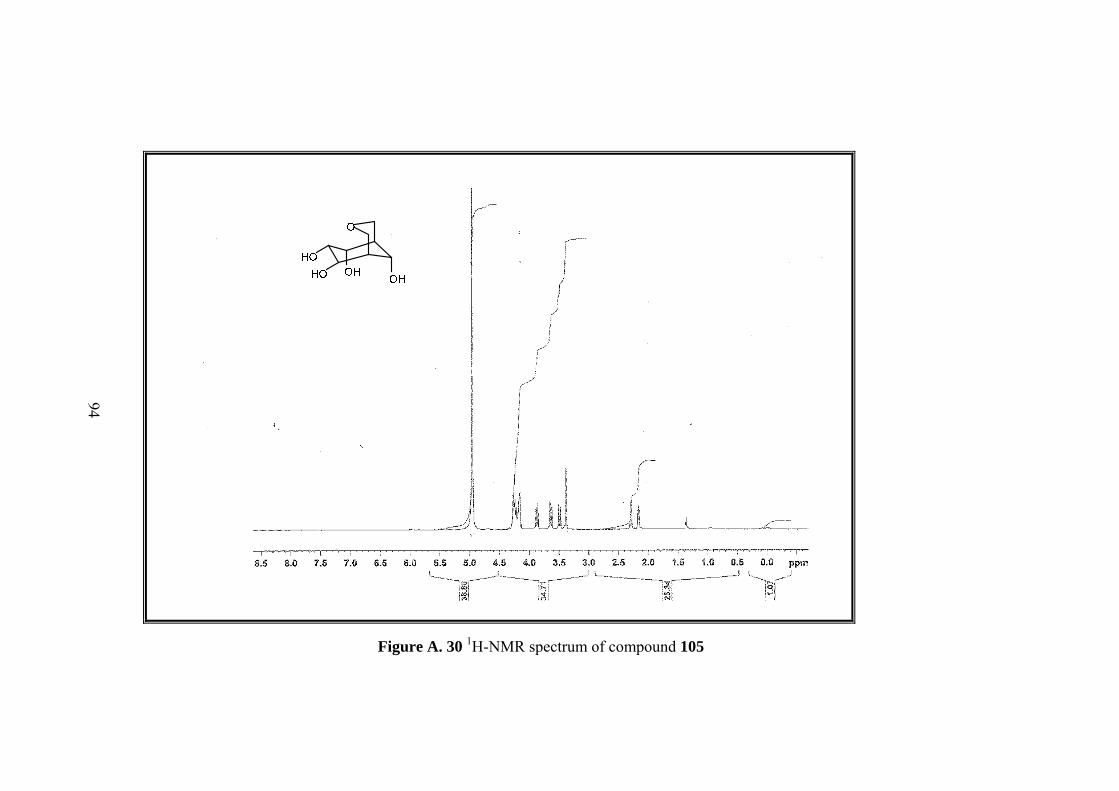
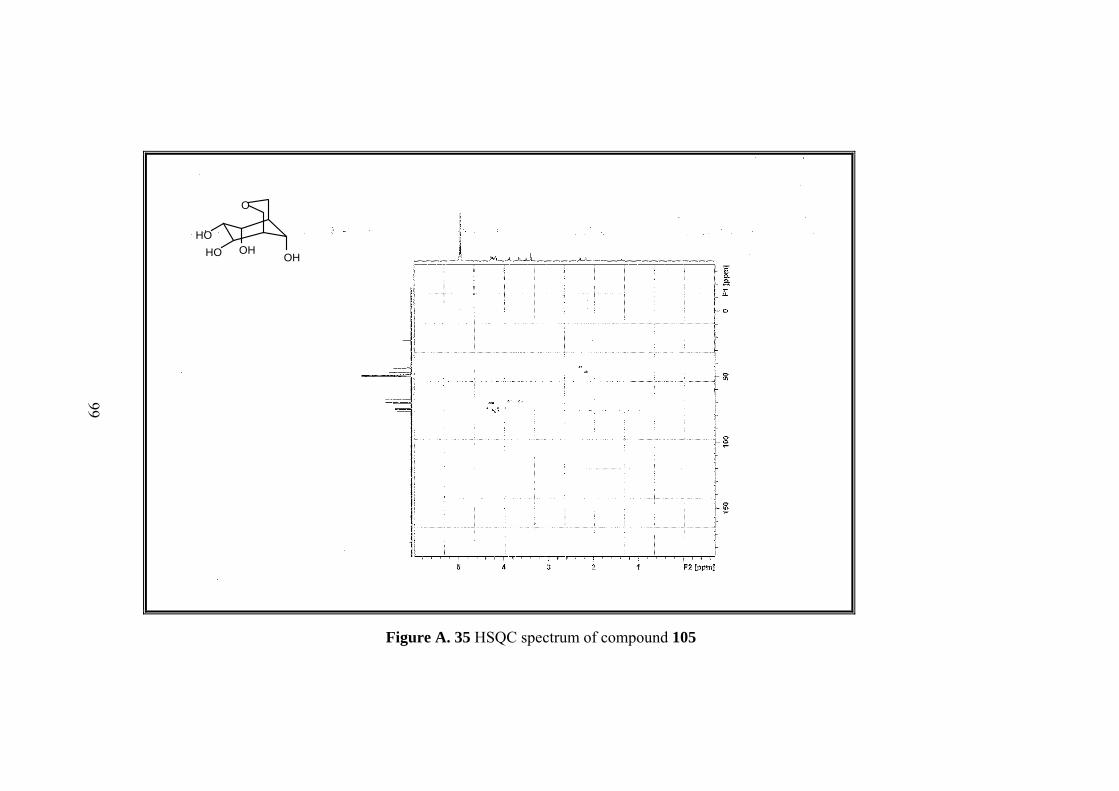
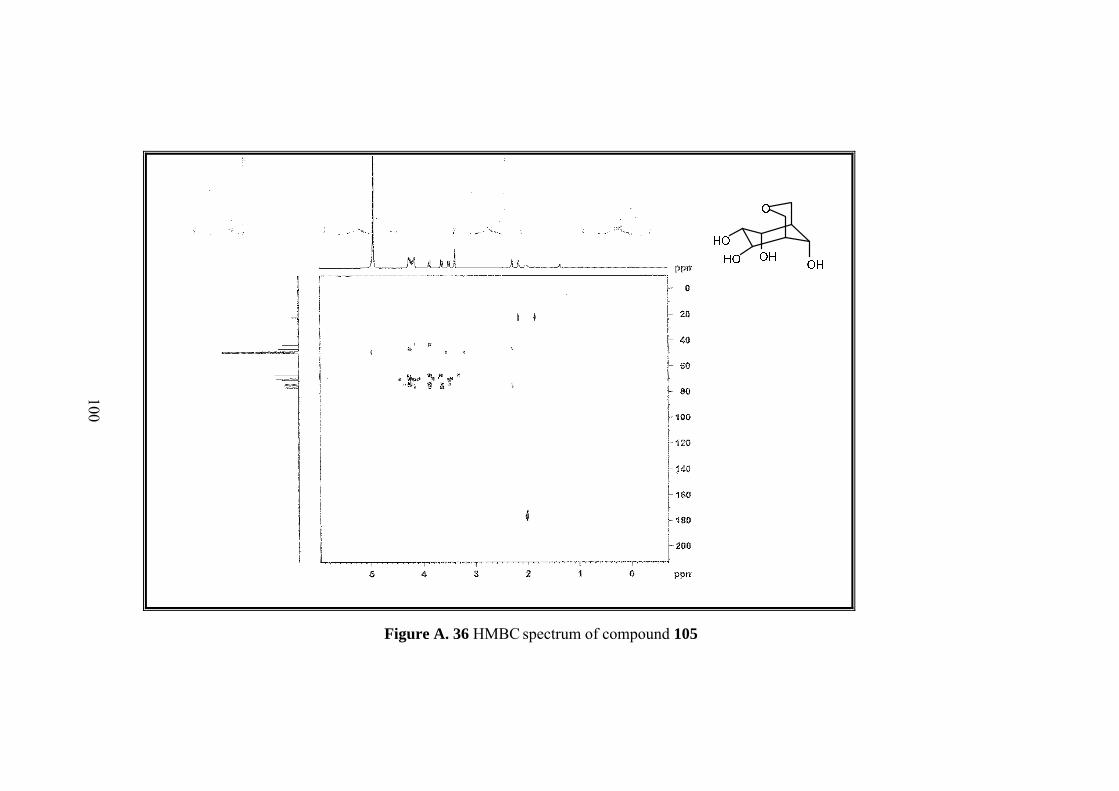
Figure A. 30 1H-NMR spectrum of compound 105 ................................................... 94
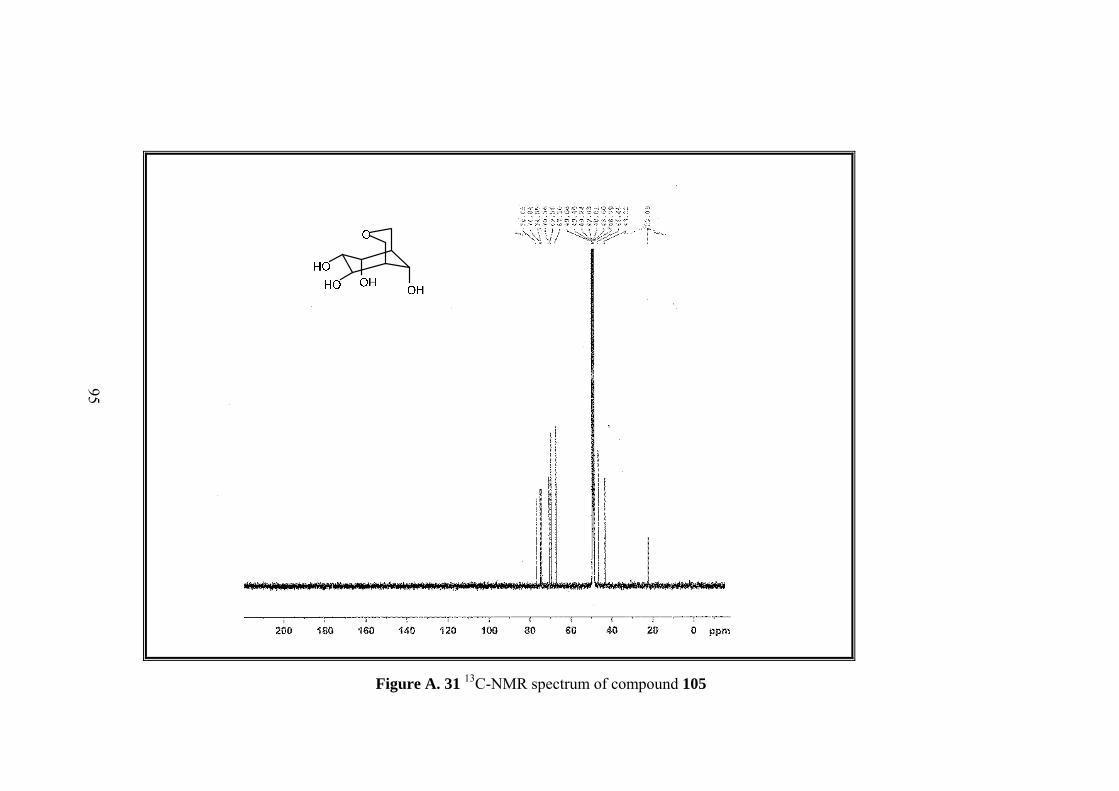
Figure A. 31 13C-NMR spectrum of compound 105 .................................................. 95
Figure A. 32 IR spectrum of compound 105 ............................................................. 96
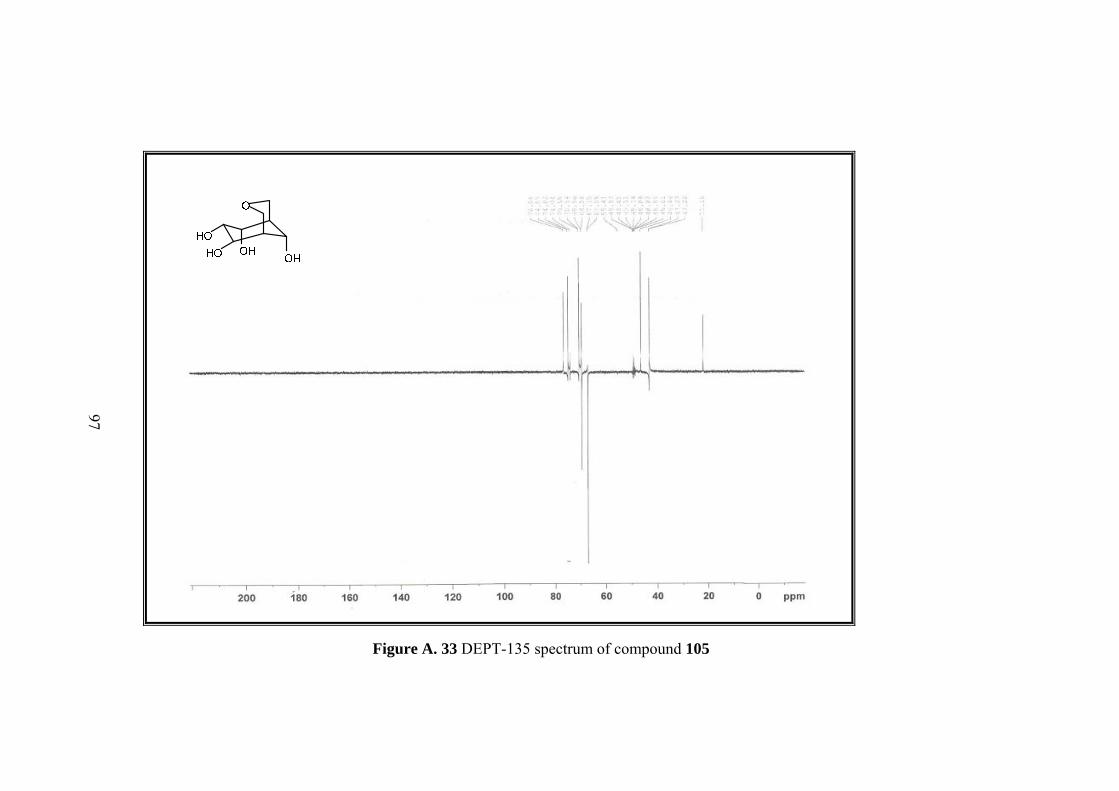
Figure A. 33 DEPT-135 spectrum of compound 105 ................................................ 97
Figure A. 34 COSY spectrum of compound 105 ....................................................... 98
Figure A. 35 HSQC spectrum of compound 105 ....................................................... 99
Figure A. 36 HMBC spectrum of compound 105 .................................................... 100
Figure A. 37 1H-NMR spectrum of compound 103 ................................................. 101
Figure A. 38 13C-NMR spectrum of compound 103 ................................................ 102
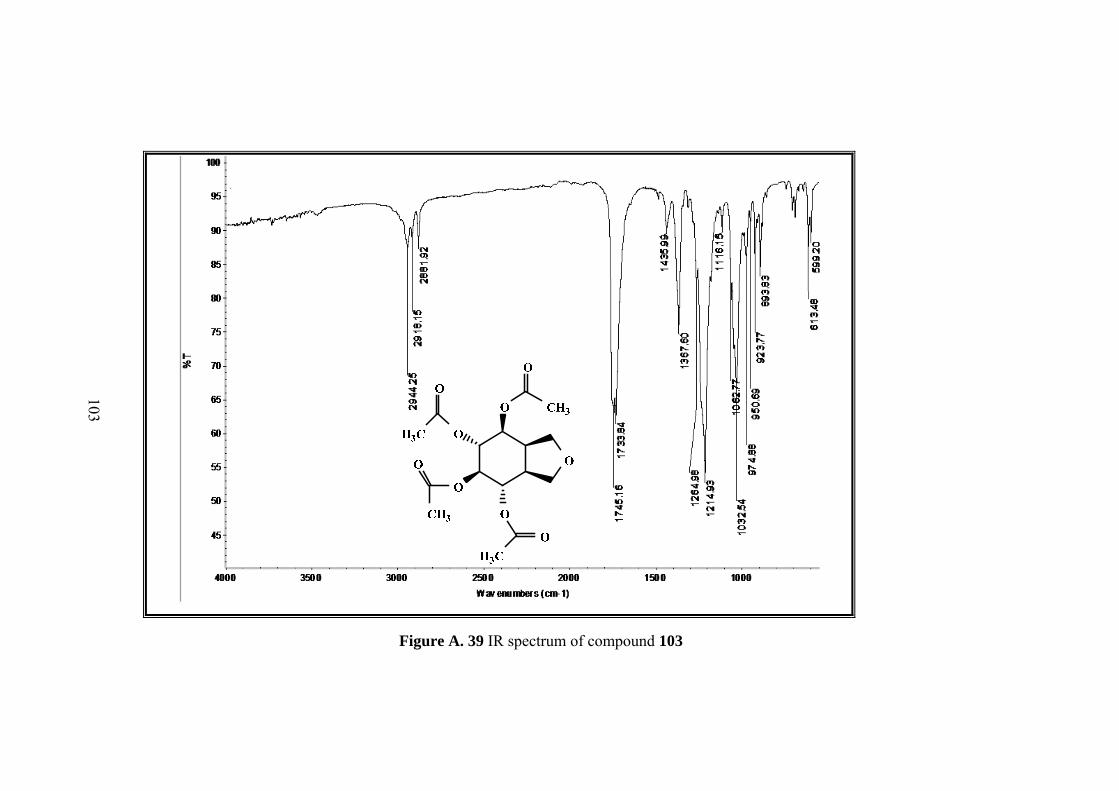
Figure A. 39 IR spectrum of compound 103 ........................................................... 103
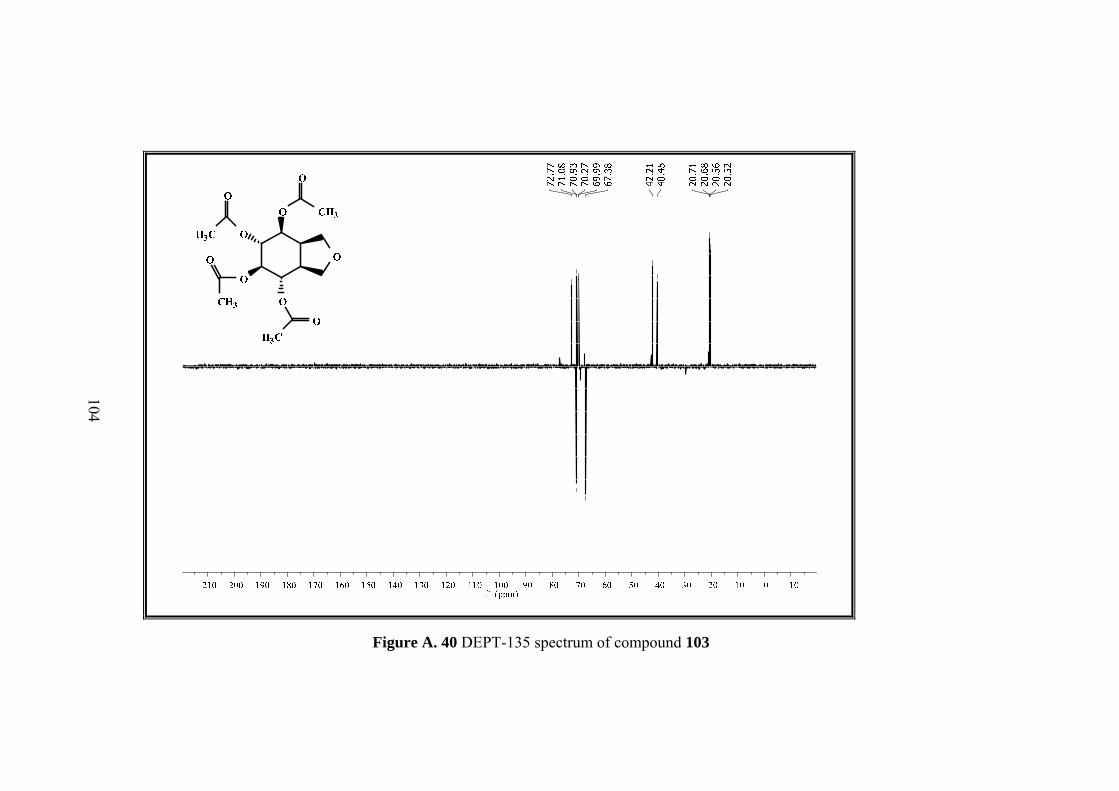
Figure A. 40 DEPT-135 spectrum of compound 103 .............................................. 104
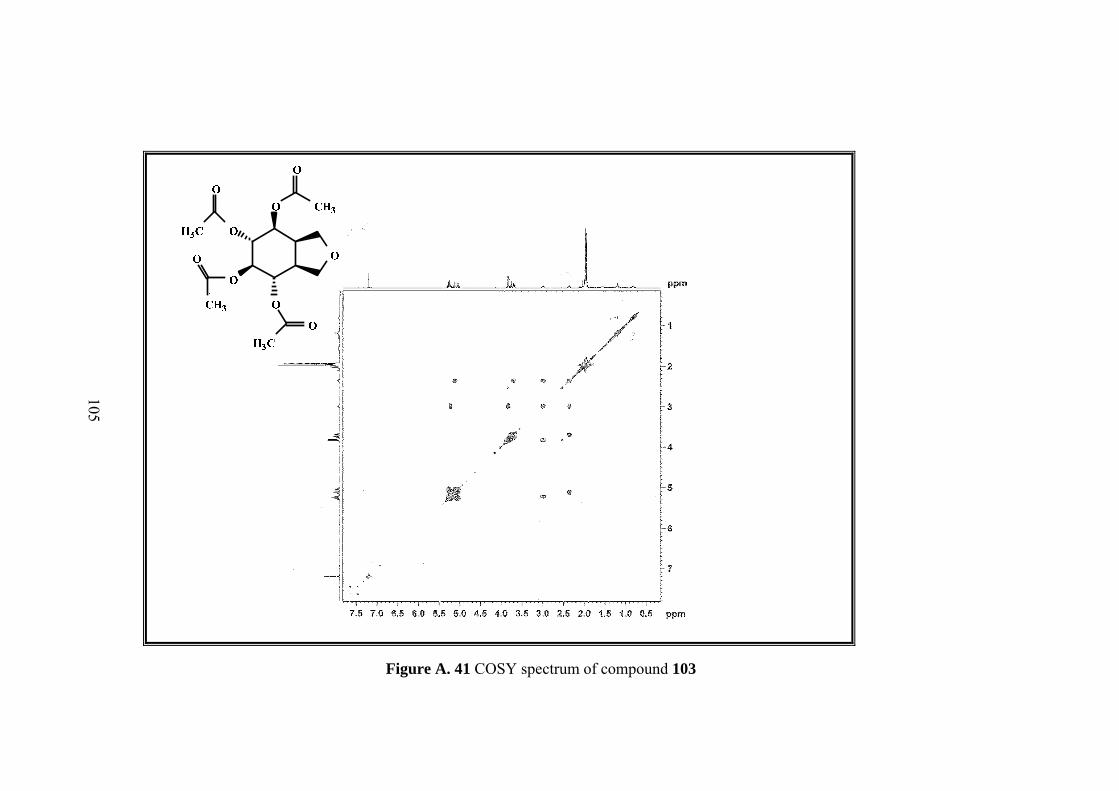
Figure A. 41 COSY spectrum of compound 103 ..................................................... 105
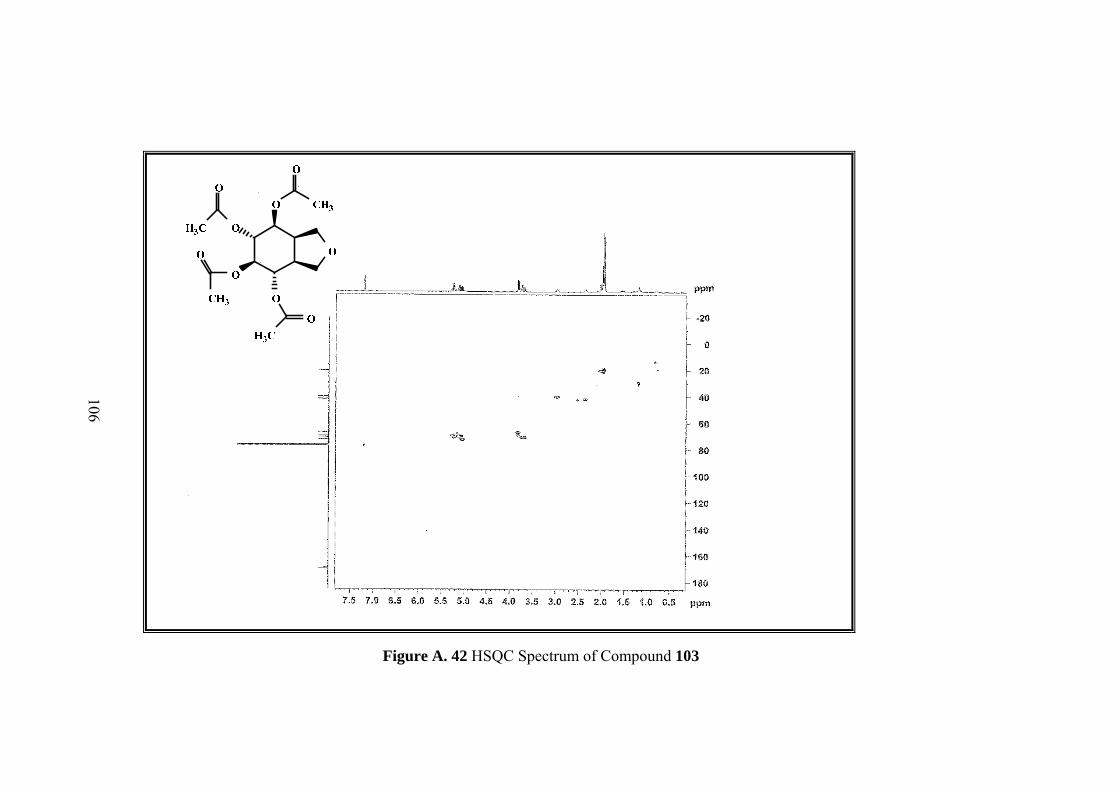
Figure A. 42 HSQC Spectrum of Compound 103 ................................................... 106
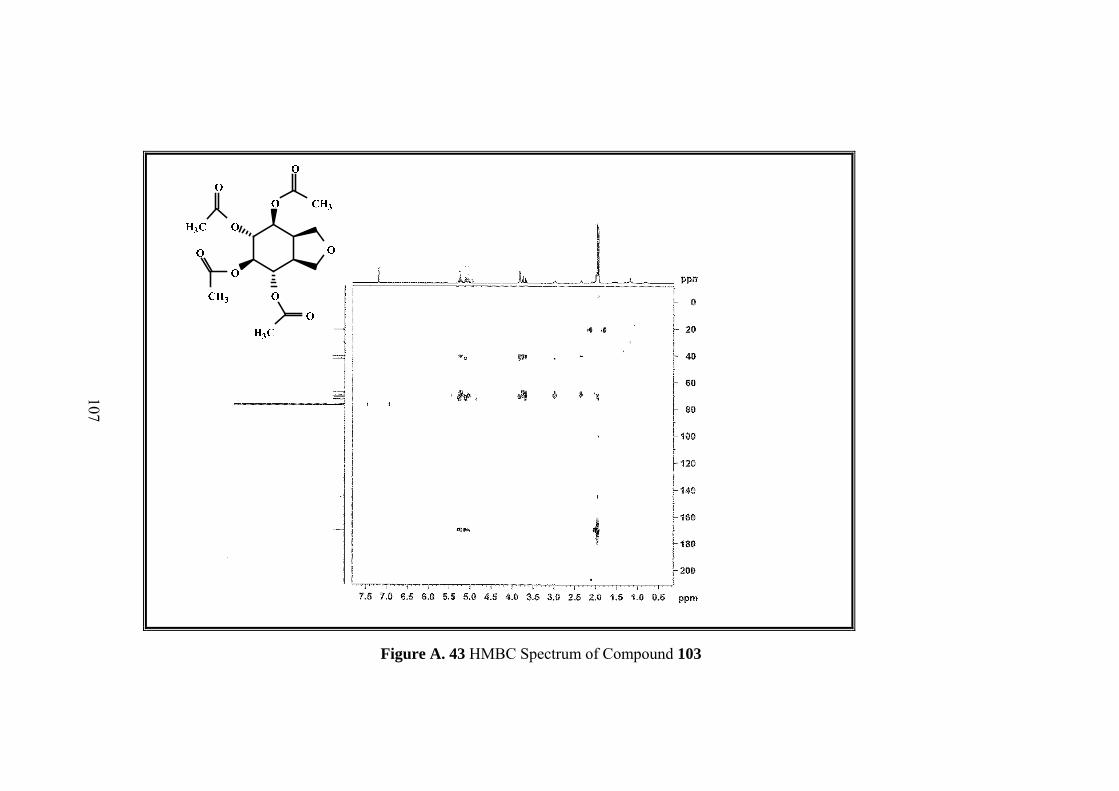
Figure A. 43 HMBC Spectrum of Compound 103 .................................................. 107
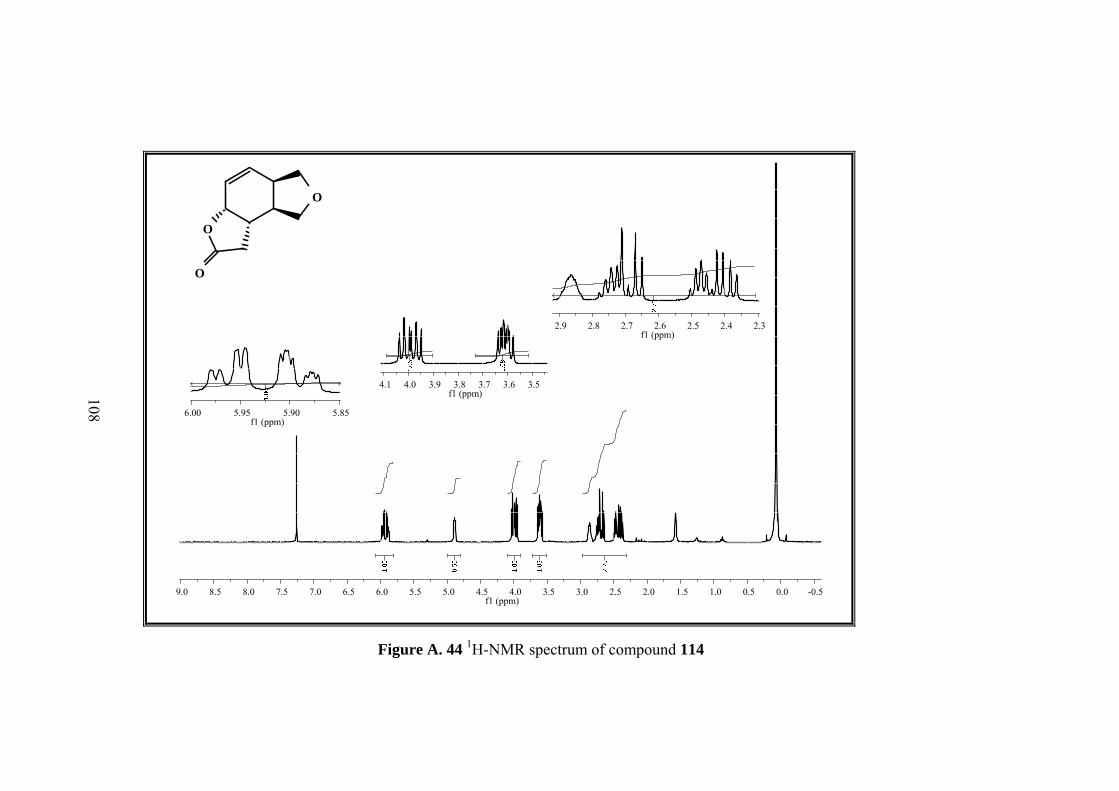
Figure A. 44 1H-NMR spectrum of compound 114 ................................................. 108
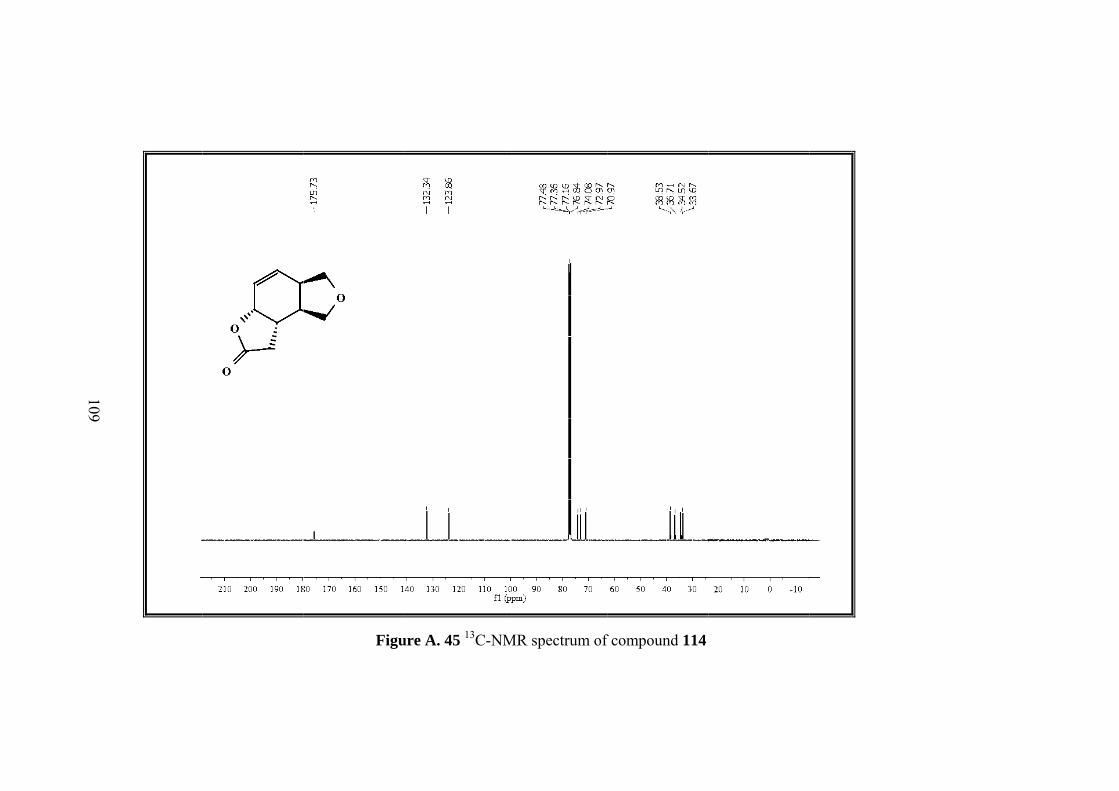
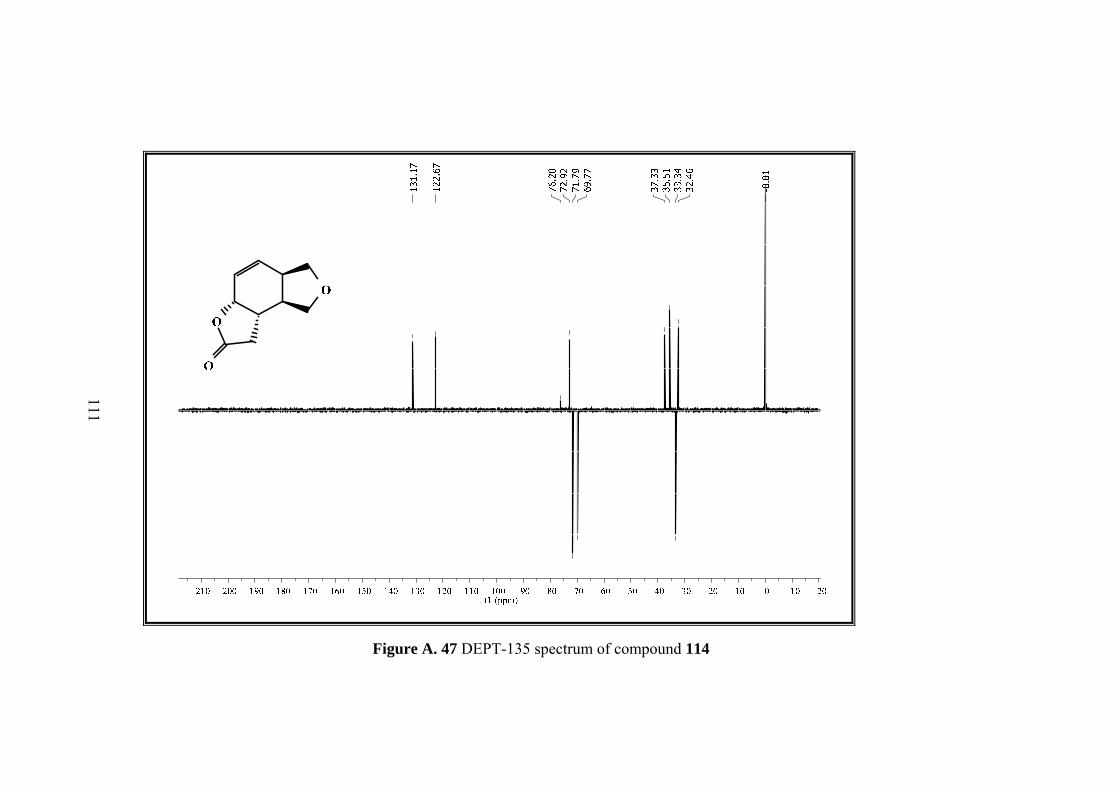
Figure A. 45 13C-NMR spectrum of compound 114 ................................................ 109
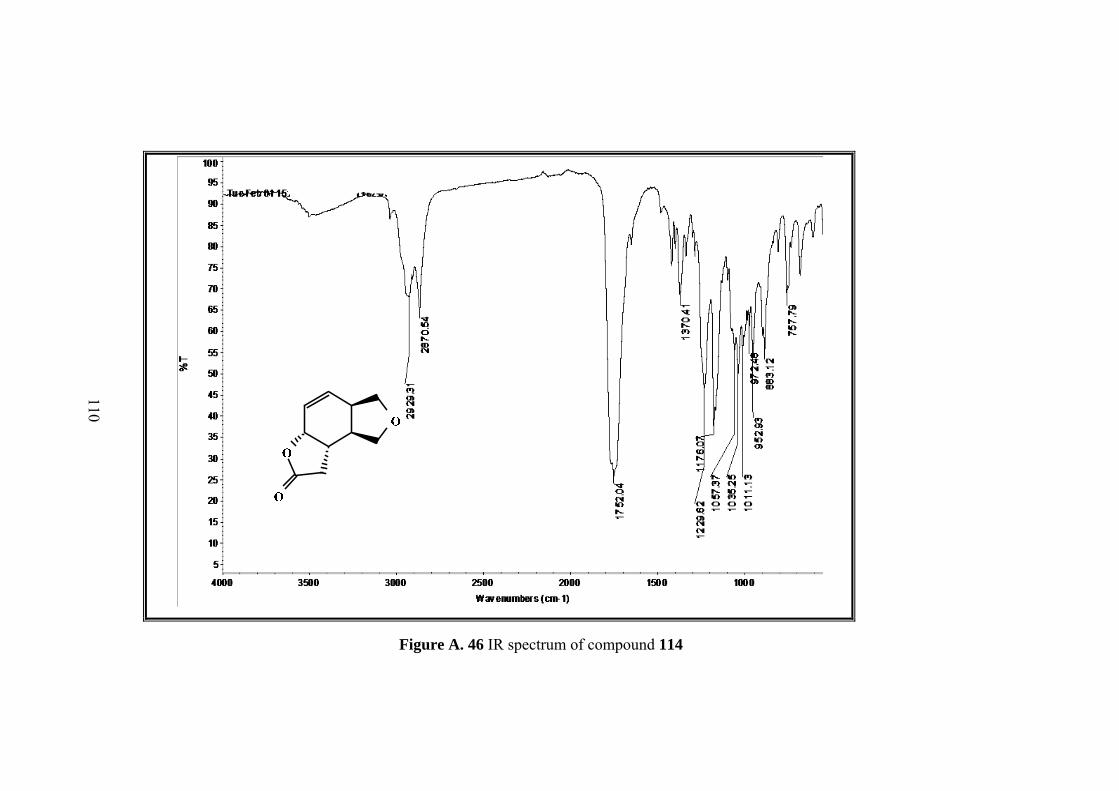
Figure A. 46 IR spectrum of compound 114 ........................................................... 110
Figure A. 47 DEPT-135 spectrum of compound 114 .............................................. 111
Page 13
xiii
Figure A. 48 COSY spectrum of compound 114 ..................................................... 112
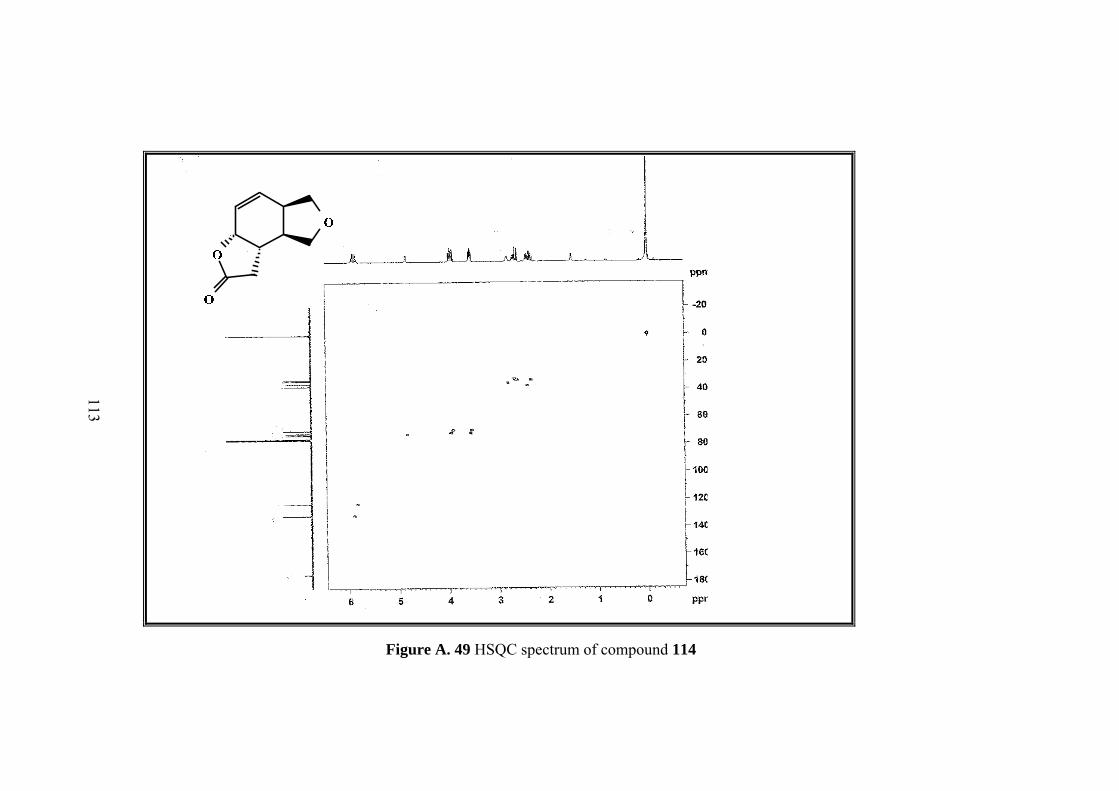
Figure A. 49 HSQC spectrum of compound 114 ..................................................... 113
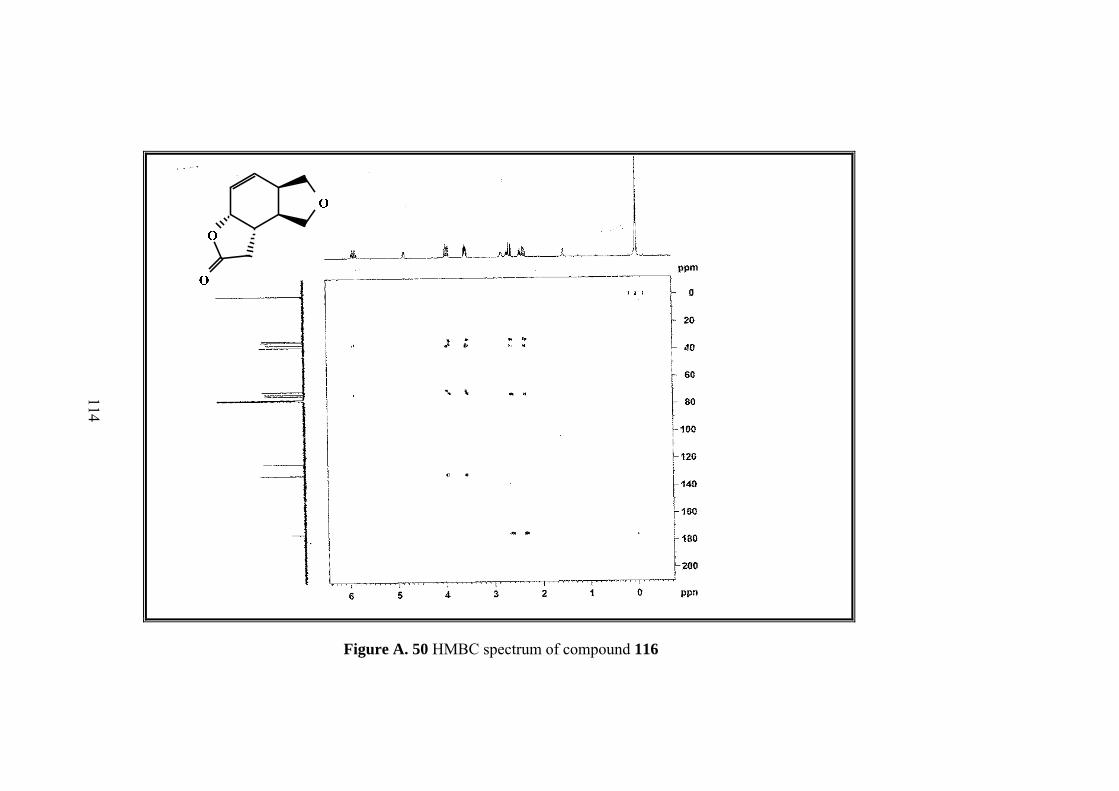
Figure A. 50 HMBC spectrum of compound 116 .................................................... 114
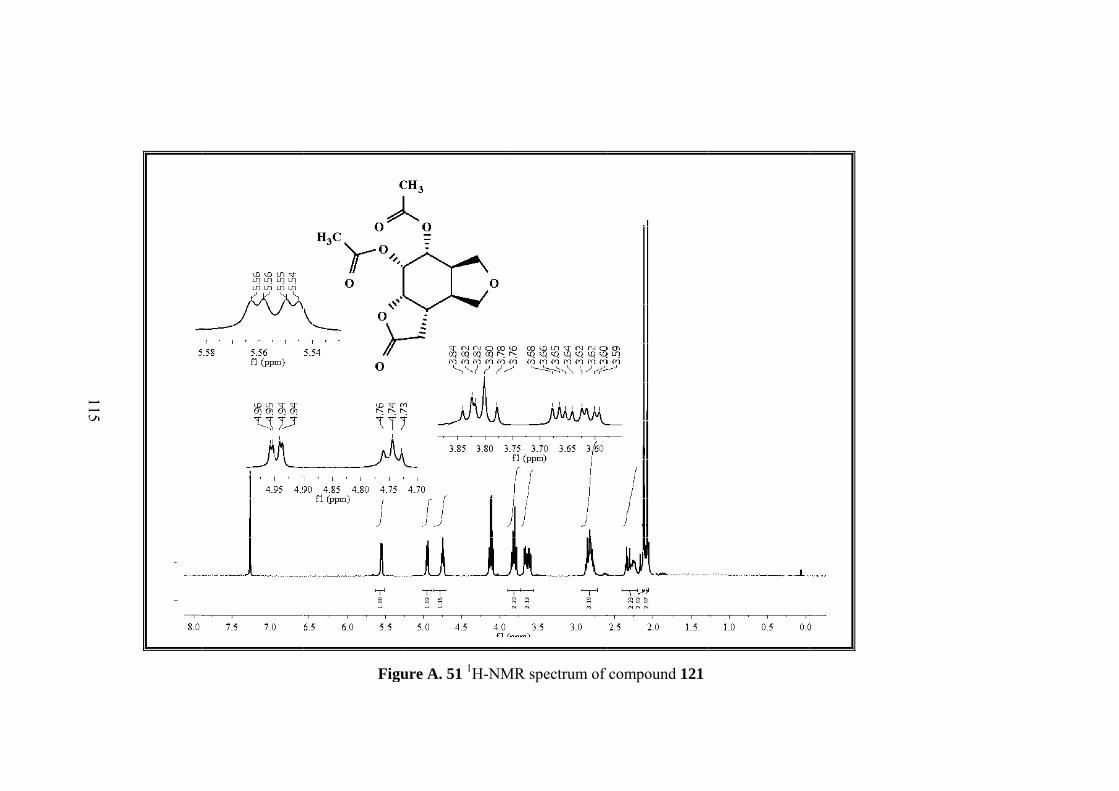
Figure A. 51 1H-NMR spectrum of compound 121 ................................................. 115
Figure A. 52 13C-NMR spectrum of compound 121 ................................................ 116
Figure A. 53 IR spectrum of compound 121 ........................................................... 117
Figure A. 54 DEPT spectrum of compound 121 ..................................................... 118
Figure A. 55 COSY spectrum of compound 121 ..................................................... 119
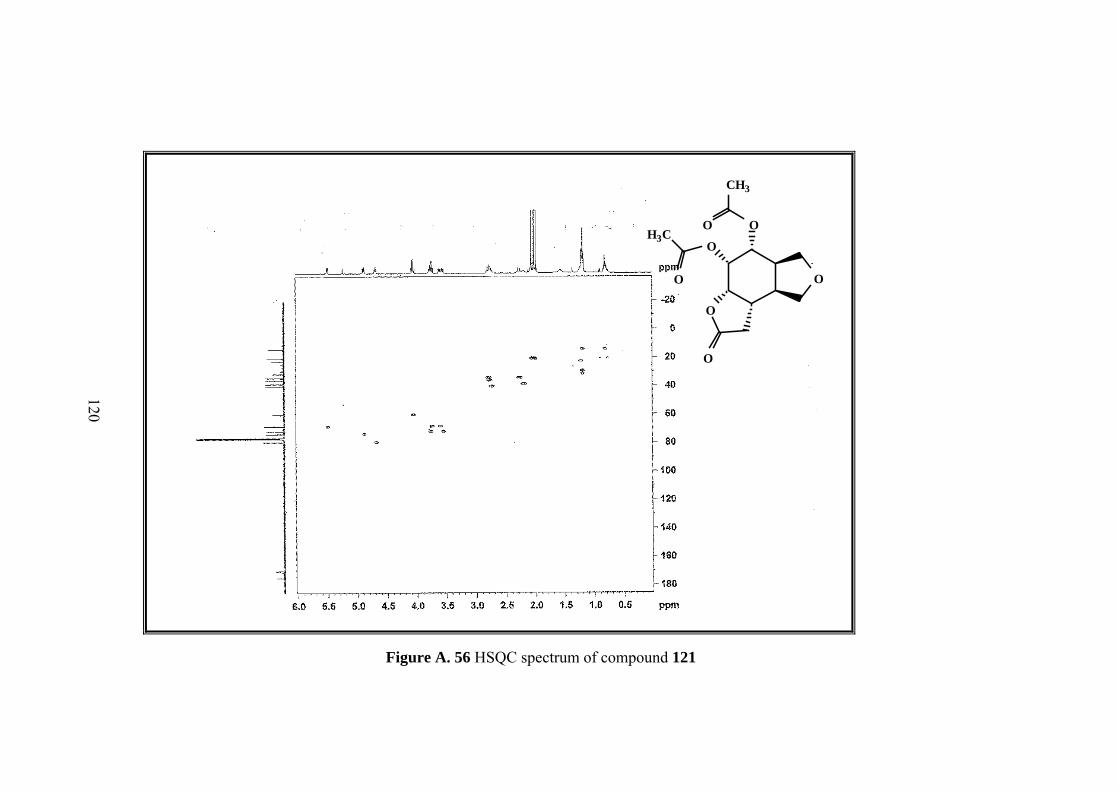
Figure A. 56 HSQC spectrum of compound 121 ..................................................... 120
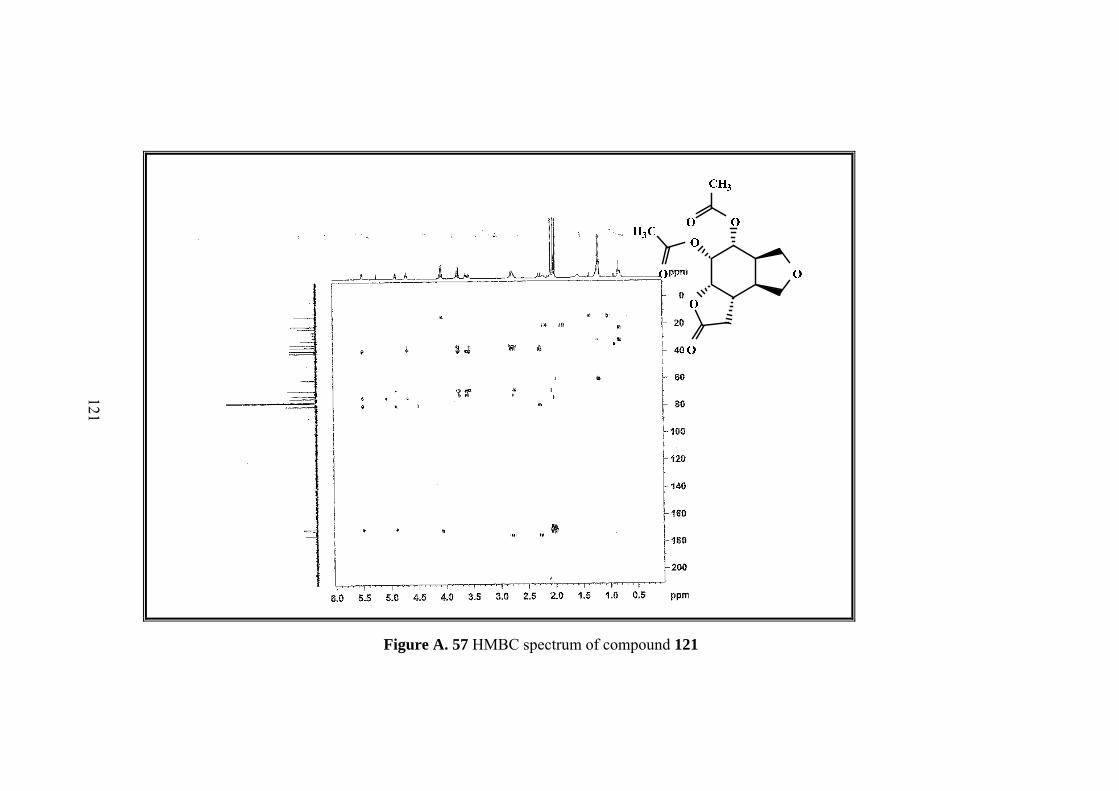
Figure A. 57 HMBC spectrum of compound 121 .................................................... 121
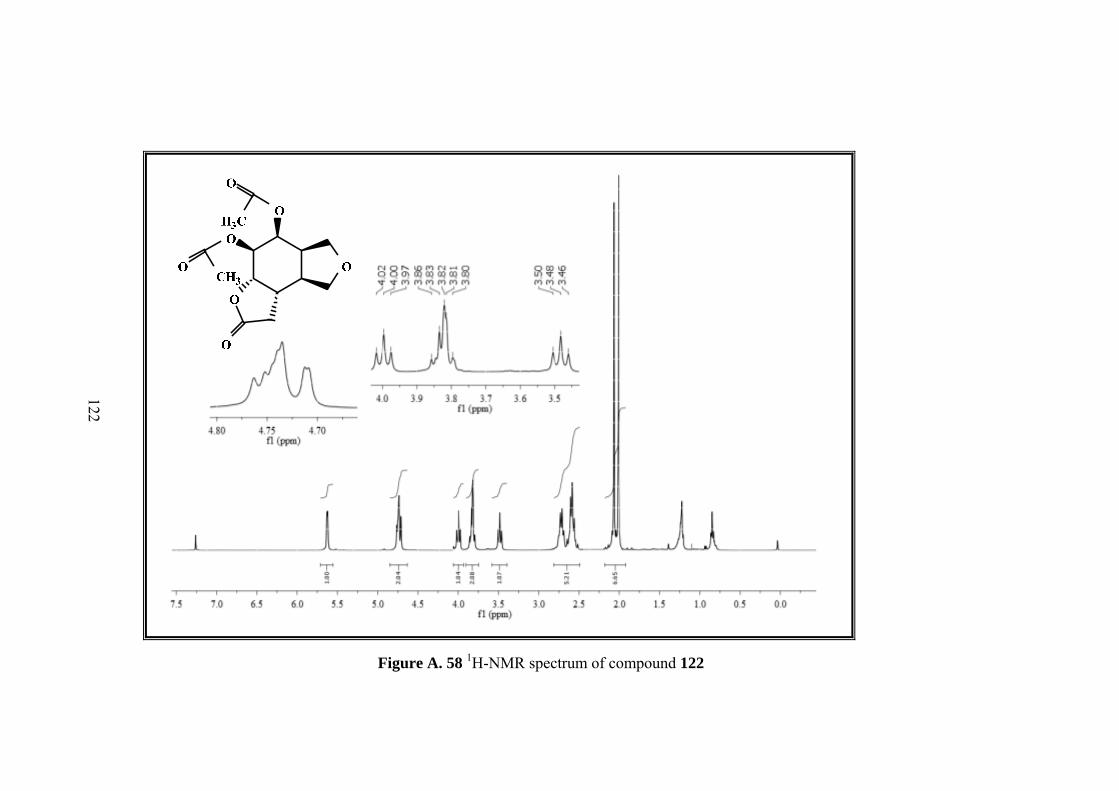
Figure A. 58 1H-NMR spectrum of compound 122 ................................................. 122
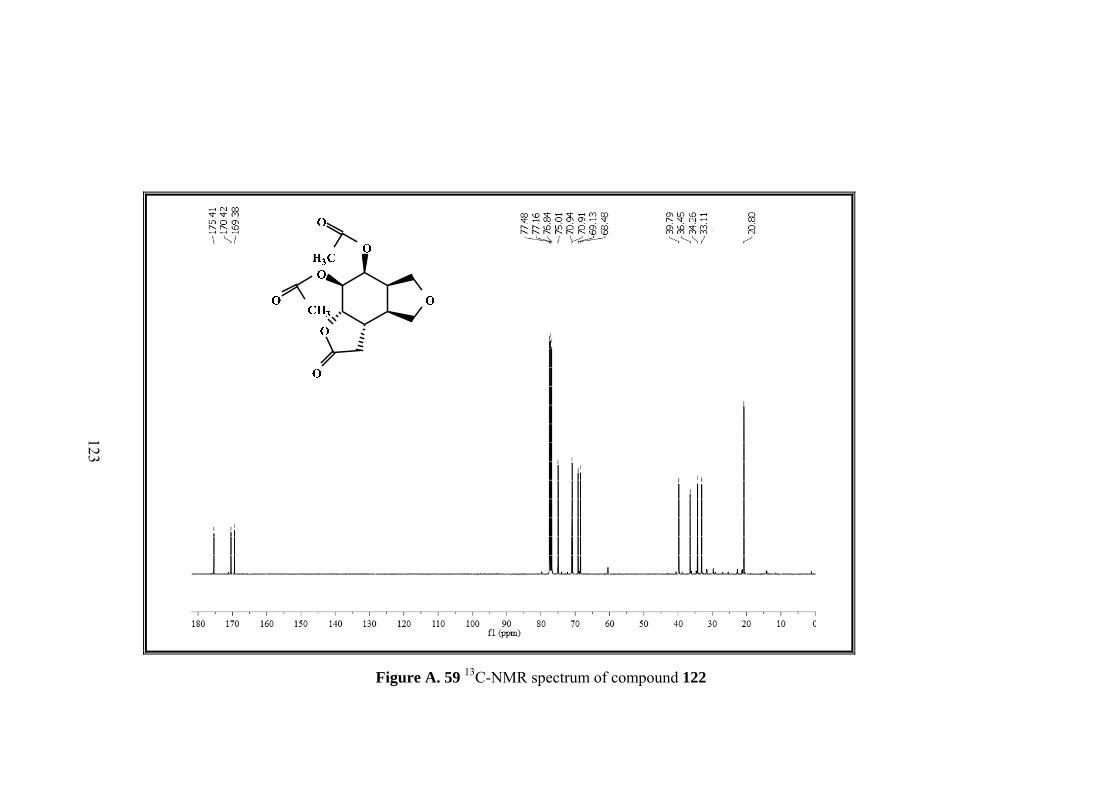
Figure A. 59 13C-NMR spectrum of compound 122 ................................................ 123
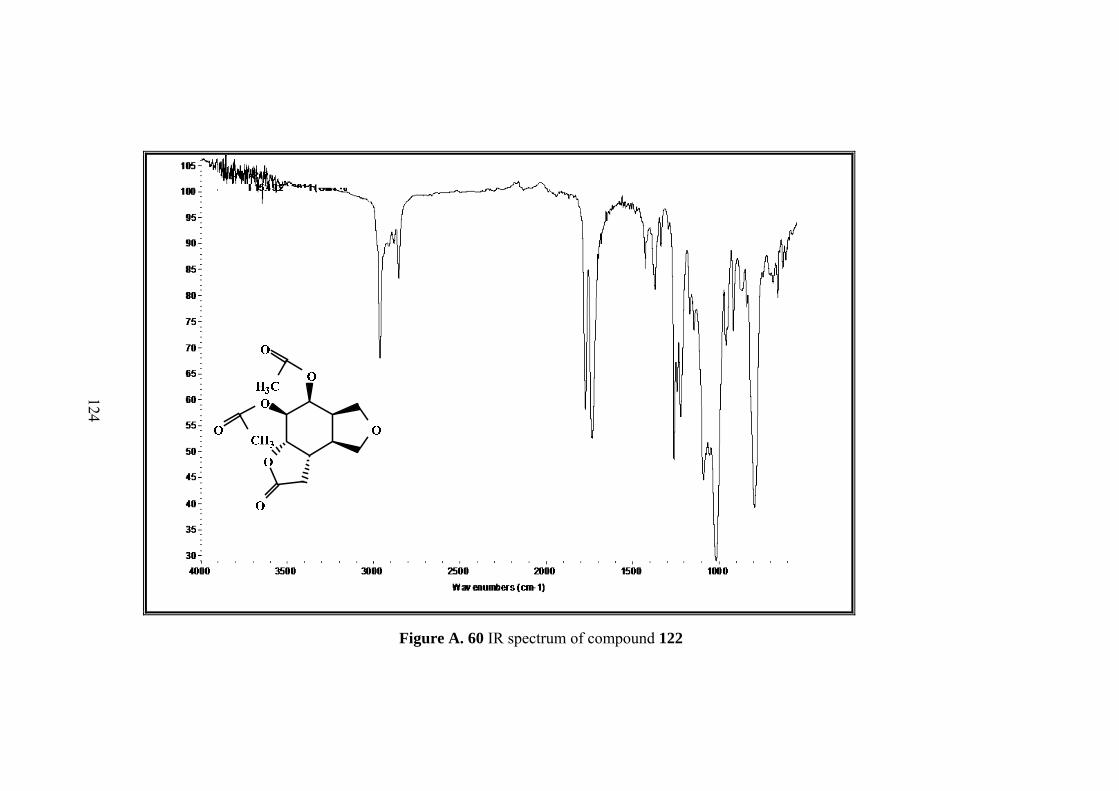
Figure A. 60 IR spectrum of compound 122 ........................................................... 124
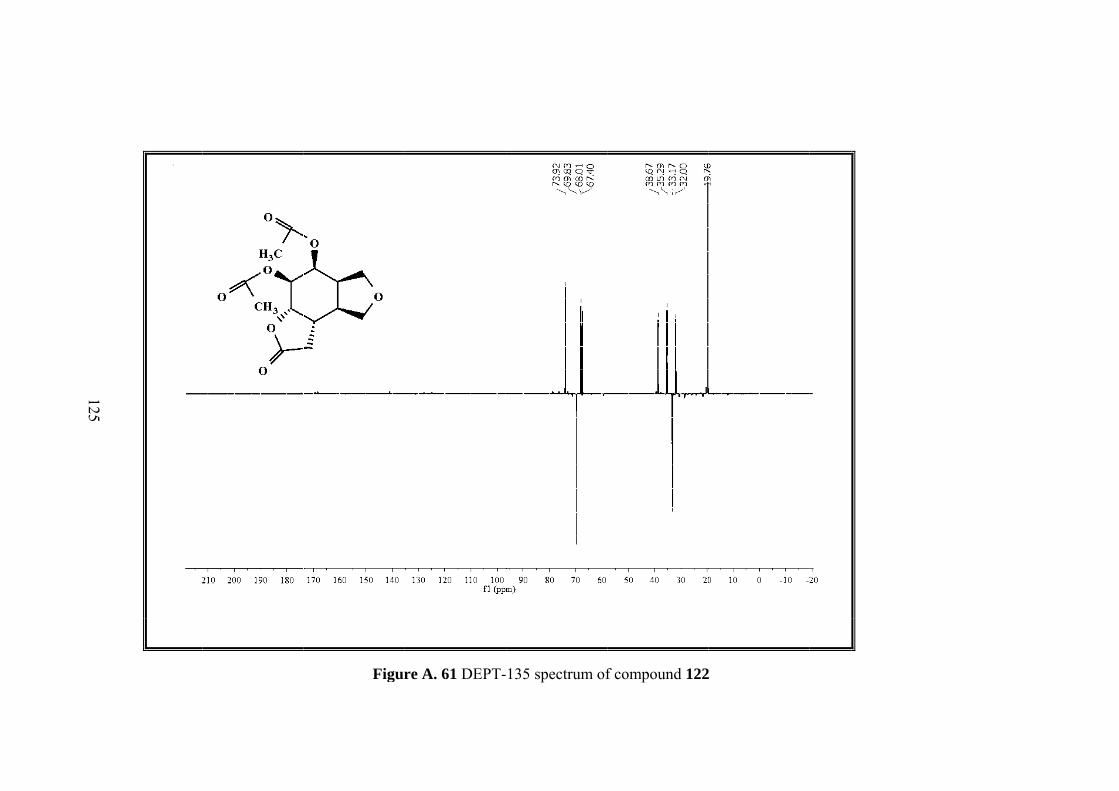
Figure A. 61 DEPT-135 spectrum of compound 122 .............................................. 125
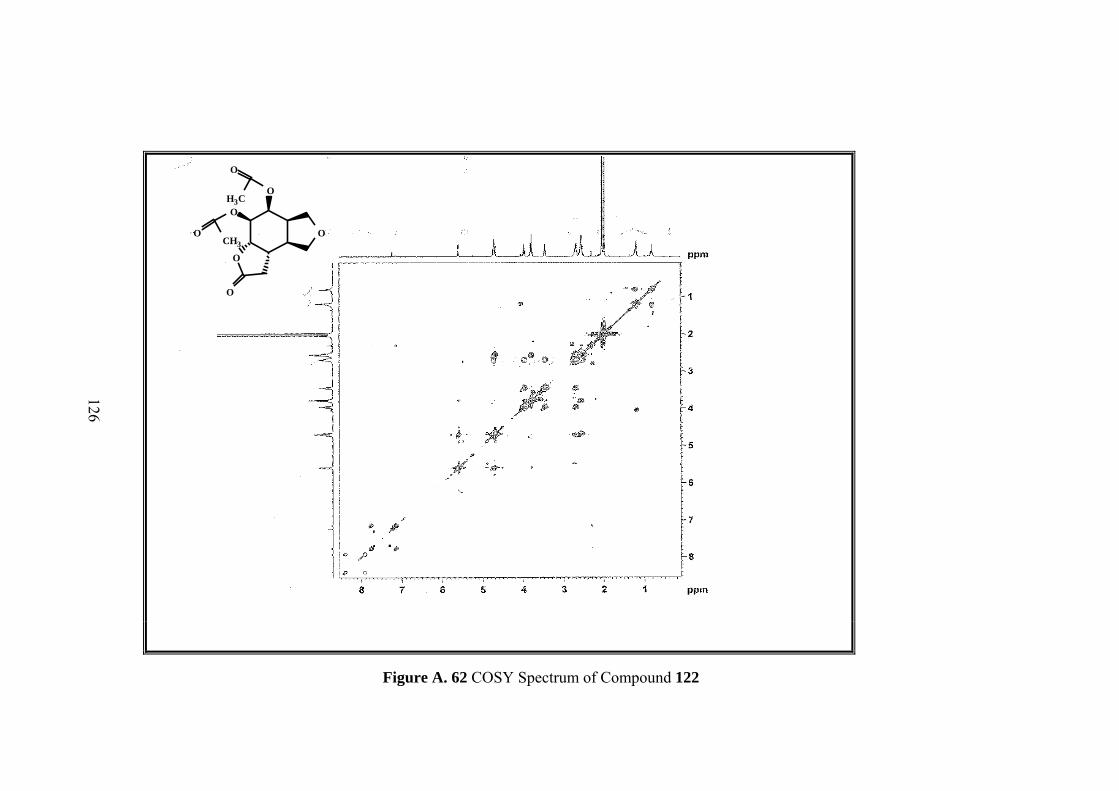
Figure A. 62 COSY Spectrum of Compound 122 ................................................... 126
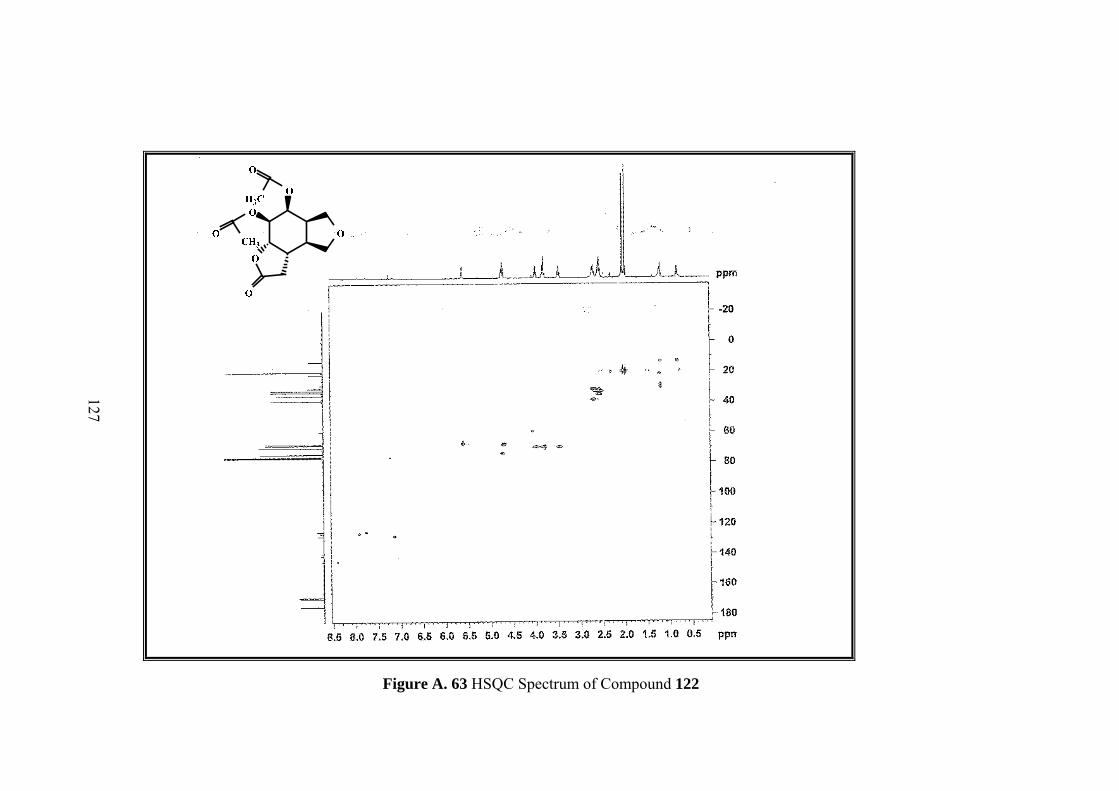
Figure A. 63 HSQC Spectrum of Compound 122 ................................................... 127
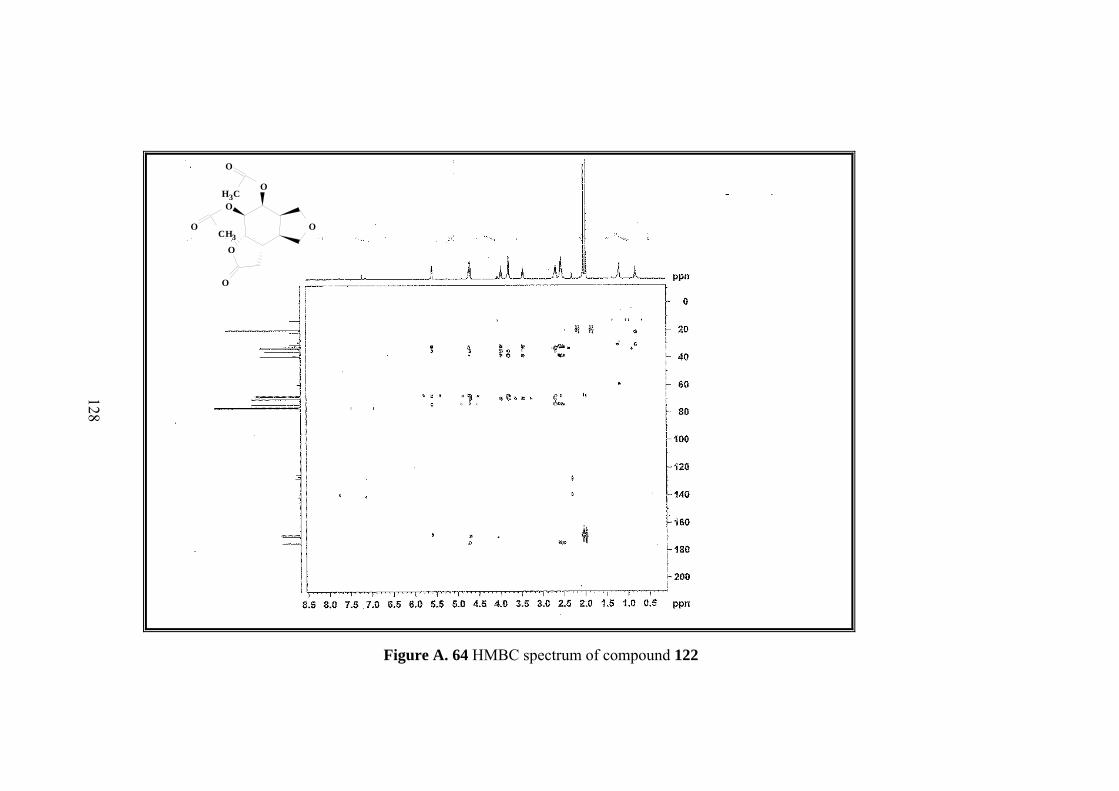
Figure A. 64 HMBC spectrum of compound 122 .................................................... 128
Page 14
xiv
LIST OF SCHEMES
SCHEMES
Scheme 1. The first total synthesis of inositol ............................................................. 4
Scheme 2. Synthesis of neo-inositol 10 ....................................................................... 5
Scheme 3. Formation of allo-inositol 11 ..................................................................... 6
Scheme 4. Chiro-inositol (7) synthesis from myo-inositol 4 ....................................... 7
Scheme 5. Formation of epi-inositol 12 ....................................................................... 8
Scheme 6. Synthesis of muco-inositol 6 ...................................................................... 9
Scheme 7. Synthesis of cis-inositol 9 ......................................................................... 10
Scheme 8. Formation of scyllo-inositol 5 .................................................................. 11
Scheme 9. Synthesis of chiro-inositol derivative 55 from napthalene....................... 12
Scheme 10. Synthesis of bishomo-inositols from cyclooctatetraene ......................... 13
Scheme 11. Synthesis of inositol derivatives having bicyclo[2.2.2]octane skeleton . 14
Scheme 12. Synthesis of bishomo-myo-inositol 74 ................................................... 15
Scheme 13. Bishomo-inositol derivatives from 1,3,3a,7a-tetrahydro-2-benzofuran . 16
Scheme 14. Synthesis of hydroxy-skipped homo-inositol 81 .................................... 17
Scheme 15. Formation of the γ-lactones ................................................................... 20
Scheme 16. Reactions of benzonorbornadiene with Mn(OAc)3 ................................ 21
Scheme 17. Synthesis of benzofuran derivative 92 ................................................... 23
Scheme 18. Synthesis of key compound 69 ............................................................... 24
Page 15
xv
Scheme 19. Photooxygenation reaction of 69 ............................................................ 24
Scheme 20. CoTPP-catalyzed rearrangement of endoperoxide 95 ............................ 25
Scheme 21. Mechanism of CoTPP catalyzed rearrangement of endoperoxide 98 .... 26
Scheme 22. Synthesis of compound 100.................................................................... 26
Scheme 23. Ring opening reaction of 100 ................................................................. 27
Scheme 24. Expected product of the ring opening reaction of bisepoxide 100 ......... 29
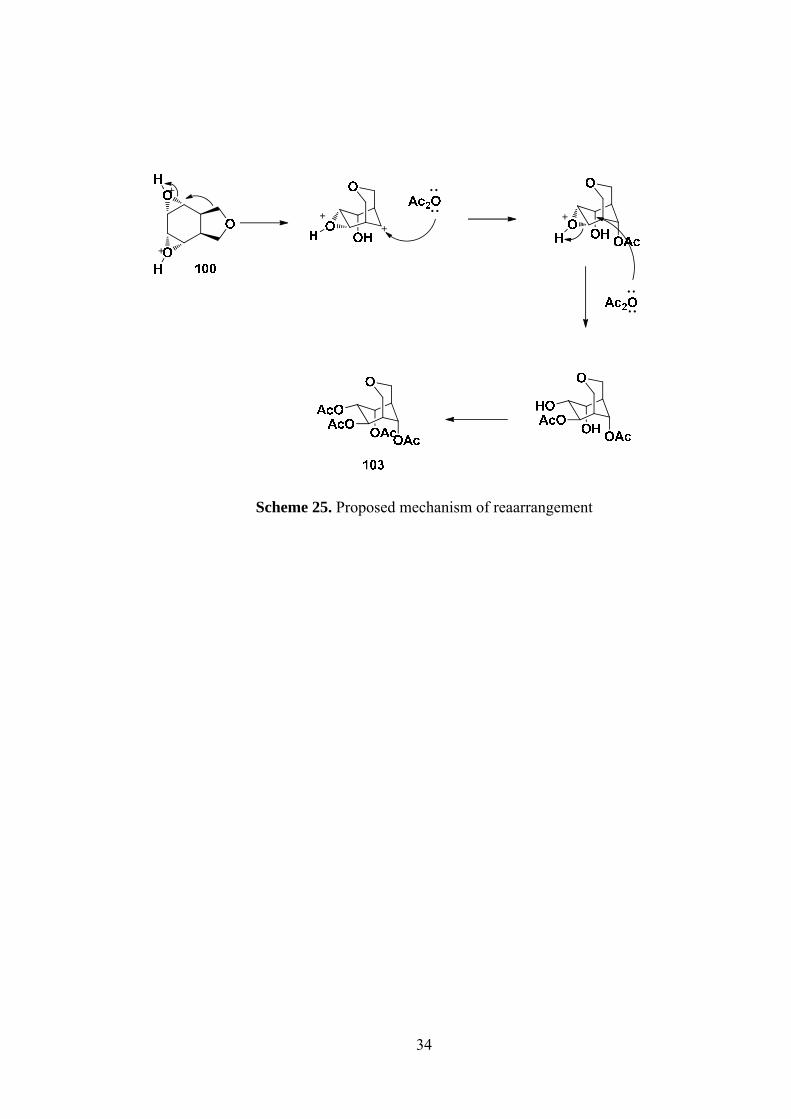
Scheme 25. Proposed mechanism of reaarrangement ................................................ 34
Scheme 26. Acetolysis reaction of tetraacetate 103 ................................................... 37
Scheme 27. Hydrolysis of the molecule 103 .............................................................. 38
Scheme 28. The mechanism of the reaction of olefins with Mn(OAc)3 in acetic acid
.................................................................................................................................... 39
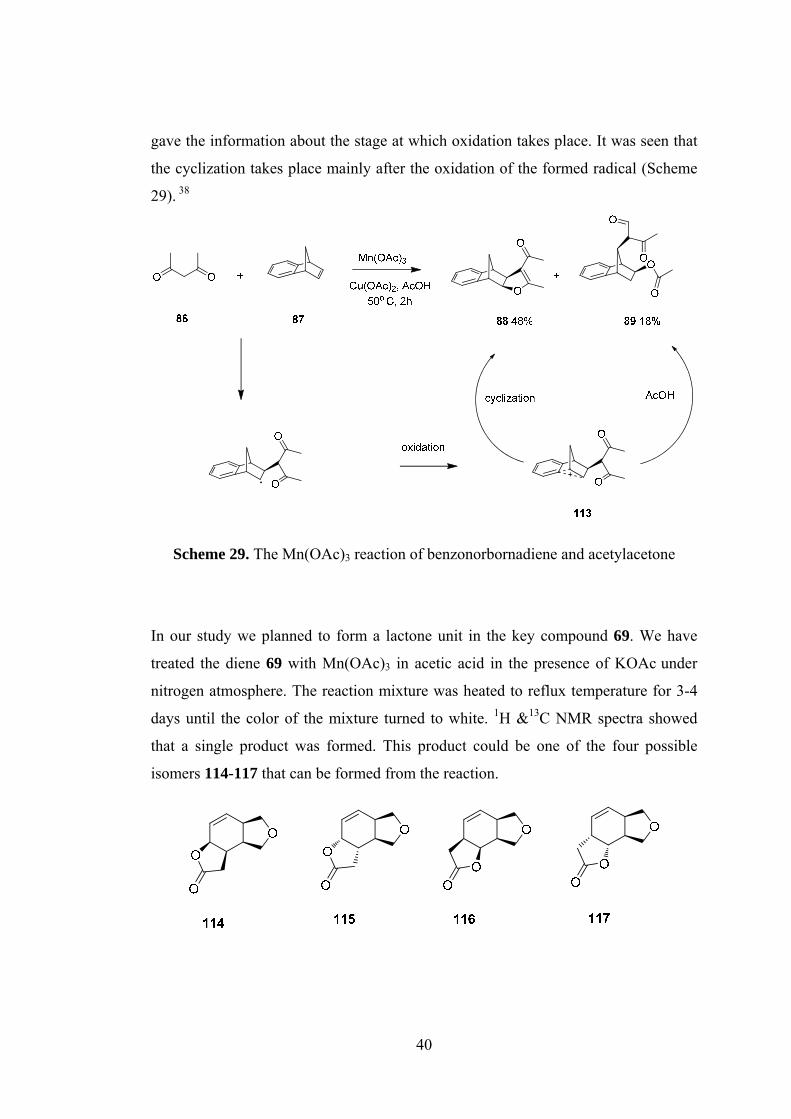
Scheme 29. The Mn(OAc)3 reaction of benzonorbornadiene and acetylacetone ...... 40
Scheme 30. Reaction of diene 69 with Mn(OAc)3. .................................................... 42
Scheme 31. Upjohn dihyroxylation of molecule 115 ................................................ 44
Scheme 32. Dihydroxylation of compound 115 ........................................................ 45
Scheme 33. Acetolysis and hydrolysis reactions of the compounds 120 and 121 .... 49
Scheme 34. Synthesis of tetraacetate 102 .................................................................. 61
Scheme 35. Synthesis of tetraacetate 103 .................................................................. 62
Scheme 36. Synthesis of diacetate molecules 120& 121 ........................................... 62
Page 16
xvi
LIST OF ABBREVIATIONS
DMP: 2,2 Dimethoxypropane
PTSA: Paratoluenesulfonic acid
DBH: 1,3-Dibromo-5,-dimethylhydantoin
AIBN: Azobisisobutyronitrile
NMO: N-methylmorpholine N-oxide
TsCl: Toluenesulfonyl chloride
Tf2O: Trifluoromethanesulfonic anhydride
DMF: Dimethylformamide
DBU: 1,8 Diazobicycloundec-7-ene
mCPBA: metachloroperoxybenzoic acid
TFA: Trifluoroacetic acid
NBS: n-Bromosuccinimide
NMR: Nuclear Magnetic Resonance
DEPT: Distortionless enhancement by polarization transfer
HMBC: Heteronuclear multi-bond coherence
COSY: Correlation spectroscopy
HSQC: Heteronuclear single quantum coherence
ppm: Parts per million
TBAF: Tetra-n-butylammonium floride
Page 17
1
CHAPTER 1
INTRODUCTION
1.1 Cyclitols
Cyclitols are cycloalkanes, which are highly substituted with hydroxy functional
groups. This class of compounds has received high interest due to the fact that many
biologically important molecules and natural products contain a polyhydroxylated
carbocycle. Among the numbers of cyclitols the most common ones are conduritols
(1), quercitols (2) and inositols (3).
Figure 1. Cyclitol derivatives
1.2 Inositols
Inositols are cyclohexanehexols. The first cyclohexanehexol was isolated by Scherer
in 1850. He named the cyclohexanehexol as inositol since ‘inos is the Greek name of
the muscle and he isolated it from the meat extract which is a muscular tissue.1 The
name was used for the other isomers as they were discovered. There are nine possible
stereoisomers for inositol. The naturally occurring isomers are myo- 4, D-chiro- 7,
Page 18
2
scyllo- 5, muco- 6, neo-inositol 10 and the unnatural synthetic isomers are cis- 9, L-
chiro 8, epi- 12 and allo-inositol 11 (Figure 1).
Figure 2. Inositol Stereoisomers
The most abundant isomer of inositols is the myo-inositol (4). It is found in
eukaryotic cells in the form of phosphate derivative. Myo-inositol which is also
commercially available, found in many foods such as beans, nuts, cantaloupe,
orange.2 Myo-inositol is a convenient precursor for the synthesis of the other inositol
derivatives.
Scyllo-inositol (5), was isolated from the organs of fish3, also present in plants like
linden, coconut pulm.4 D-chiro-inositol (7) is not abundant in most diets, they are
less widely found in seeds and plants and in significant quantities in buckwheat
farinetta.5 Neo-inositol (10) phosphates have been isolated from some mammalian
Page 19
3
tissue.6 Muco-inositol (6) is an another naturally occuring inositol is present in
honey.7
The inositols are crystalline substances with high-melting points and low solubility.
Generally they are stable to heat, acids, and alkalis.3
1.2.1 Biologic functions of inositols
Inositols have remarkable biological functions such as glycosidase inhibition,
intercellular communication, phosphate storage etc. In recent years inositols and
phosphate derivatives of inositols, which have significant role in various cellular
functions which are cell migration, cell differentiation, cell growth and endocytosis,
have been studied and new derivatives have been discovered.8
Inositol is also an effective and safe option in the treatment of panic disorder,
obsessive-compulsive disorder (OCD), bulimia nervosa, binge eating and/or
depression.9
Some inositol derivatives such as chiro- (7), muco- (6) and epi-inositol (12) have
been reported to help the action of insulin, which induces glucose uptake involving
the translocation of glucose transporter 4 (GLUT4) to the plasma membrane
particularly in muscle tissues.10
Scyllo-inositol (5) has shown promise as a potential therapeutic for Alzheimer’s
disease, scyllo-inositol derivatives inhibit or disrupt Ab peptide aggregation as
atherapeutic strategy to treat AD.11
Phosphate derivative of D-myo–inositol (4) act as a messenger molecule that used in
intercellular reactions as controlling the intercellular Ca2+ concentration.12
Bishomo-inositols shows inhibition of α-glycosidase which is important in the
treatment of diabetes and HIV infection.13, 14
Page 20
4
1.3 Synthesis of inositol and inositol derivatives
The first synthesis of inositol was performed from benzene by Wieland and Wishart
in 1914.15 They synthesized inositol by hydrogenation reaction of benzenehexol 15
(Scheme 1).16
Scheme 1. The first total synthesis of inositol
Due to biological importance of the inositols different synthetic pathways have been
developed during the years. Inositols have been synthesized from aromatic
compounds, tetrahydrobenzoquinones, sugars, cyclohexene and its derivatives, chiral
acids, norbornenes, naturally occurring inositols, and others.
1.3.1 Synthesis of myo-inositol
Of the nine possible stereoisomers, myo-inositol is generally the most abundant. It is
widespread in all higher organisms and most microorganisms and plays very
important roles in cellular signal transduction.12
Myo-inositol is a crystalline compound with a sweet taste which was first isolated by
Scherer in 1849. Myo-inositol was synthesized from D-glucose in several steps.15
Different synthetic pathways for myo-inositol and their phosphates derivatives have
been developed during the years because of the important biological functions.
Page 21
5
1.3.2 Synthesis of neo-inositol (10)
Hudlicky et al.17 developed a method for the synthesis of different inositol
derivatives including neo-inositol from bromobenzene.
Synthesis of neo-inositol 10 started with reaction of bromobenzene with toluene
dioxygenase which provided the diol 17. The protection reaction of diol and reaction
with 1,3-dibromo-5,-dimethylhydantoin (DBH) gave 18. Epoxide formation with
KOH, followed by the ring opening reaction at elevated temperatures afforded 20.
Debromination of 20 and cis hydroxylation by OsO4 formed molecule 21 which is
then converted to neo-inositol 10 with hydrochloric acid (Scheme 2).
BrOH
OH
Br
O
O
Me
Me
HO
Br
Br
O
O
OMe
Me
Br
O
O
Me
Me
HO
OH
Br
O
O
Me
Me
HO
OH
OH
HO
OH
OH
HO
OH
OH
HO
t.dioxygenase DMP,PTSA
DBH,H2O
KOH
DME
reflux
1.Bu3SnH/AIBN
2.OsO4,NMO
16 17 18 19
202110
HCl
MeOH
Scheme 2. Synthesis of neo-inositol 10
Page 22
6
1.3.3 Synthesis of allo-inositol (11)
Altenbach et al.18 introduced new synthetic pathways for inositols including
conduritols as intermediates starting from benzoquinone. Allo-inositol 11 is one of
them which has been synthesized via conduritol-E.
Dibromodiacetate 23 which was synthesized from p-benzoquinone in several steps,19
was converted to dibromodiol isomers 24 and 25 with an enzymatic reaction using
pig pancreas lipase (PPL). Conduritol E derivative 26 was obtained from the reaction
of one of these dibromo isomers with sodium acetate and aqueous acetic acid in 10
days. Then cis-hydroxylation with RuCl3 and NaIO4 provided the molecule 27, which
was then converted to allo-inositol (11) (Scheme 3).
Scheme 3. Formation of allo-inositol 11
Other inositol derivatives; neo-, epi-, chiro- and scyllo-inositol were also synthesized
from p-benzoquinone via corresponding conduritols as intermediates. Neo-inositol
(10) was obtained from conduritol E, chiro-inositol (8) from conduritol B and epi-
inositol (12) from conduritol C.17
Page 23
7
1.3.4 Synthesis of chiro-inositol (7)
Among the inositols myo-inositol (4) is the most abundant in nature, and also
commercially available with low cost. It is generally good precursor for the synthesis
of other inositol derivatives.
Miyake et al.20 developed a new method for synthesis of chiro-inositol (7) from myo-
inositol (4). Synthesis started with reaction of myo-inositol (4) with camphor
dimethyl acetal which provided ketal 28. The ketal 28 was reacted with Tf2O or TsCl
then hydroxyl groups were converted to acetate groups to form triacetate 30.
Treatment of the triacetate 30 with BzOLi in DMF gave the compound 31 which was
then converted to D-chiro-inositol (7) by hydrolysis (Scheme 4).
Scheme 4. Chiro-inositol (7) synthesis from myo-inositol 4
Page 24
8
1.3.5 Synthesis of epi-inositol (12)
Epi-inositol (12) was synthesized starting from methyl β-D galactopyranoside (32)
with highly stereoselective route (Scheme 5).21
The compound 33 was synthesized from the commercially available methyl β-D
galactopyranoside 32.22 Treatment of 33 with DBU resulted in the formation of
inosose 34, which was reduced selectively by catalytic hydrogenation with Raney-Ni
or with NaBH4 to give the epi-inositol derivative 35. The catalytic debenzylation of
35 gave epi-inositol (12).
O
CH2OHHO
OH
OMe
OH CHO
CH2OBnO
HO
OBn
OHDBU
Toluene
OBnH5
HO
HO
H6
BnO
H2
H3
OH
H4
H3H4
OH
OH
H5
H6
OBnOH
H1
H2
OBn
HO
O
Raney-Ni/H2
or
NaBH4, -78 oC
H3H4
OH
OH
H5
H6
OH
OH
H1
H2
OH
HO
H2,Pd(C)
MeOH
32 33 34
3512
OHOHHO
OH
OHHO
=
Scheme 5. Formation of epi-inositol 12
Page 25
9
1.3.6 Synthesis of muco-inositol (6)
Hudlicky et al.17 developed a synthetic pathway for muco-inositol (6) via the
microbial oxidation of bromobenzene. The starting compound epoxide 36 was
synthesized from the diene diol 17,23 which was obtained from bromobenzene as
shown in Scheme 2. Synthesis started with epoxide opening and followed by
reduction to form diol 37. Oxidation of the diol 37 with m-CPBA gave epoxide
isomers 38 and 39. Hydrolysis of the diastereomers formed myo- and muco-inositol
mixture. Muco-inositol (6) was seperated by crystallization (Scheme 6).
Scheme 6. Synthesis of muco-inositol 6
1.3.7 Synthesis of cis-inositol (9)
Takahashi et al.24 synthesized inositol diastereoisomers from PdCl2-catalyzed
Ferrier-II carbocyclization of methyl 6-O-acetyl-5-enopyranosides. cis-Inositol was
synthesized from methyl 6-O-acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranoside 40
which was prepared from methyl glucoside. The β-hydroxyketone 41 was obtained
from corresponding glucopyranoside via Ferrier-II carbocyclization reactions.25
Page 26
10
Reduction of the β-hydroxyketone 41 with NaBH4 gave the molecule 42 as a single
diastereoisomer. Removal of the benzyl groups with H2 in the presence of Pd(OH)2
resulted in the formation of pentaol 43. The regioselective protection of the pentaol
with acetone under acidic conditions provided 44. The inversion of the free hydroxyl
group of compound 44 was succeeded by the reaction of the corresponding triflate 45
with CF3COOCs and 18-crown-6 followed by saponification. Further deprotection
under acidic conditions formed the cis-inositol (9) (Scheme 7).
Scheme 7. Synthesis of cis-inositol 9
Page 27
11
1.3.8 Synthesis of scyllo-inositol (5)
Synthetic pathway for the scyllo-inositol (5) was developed starting from myo-
inositol via its orthoformate.26 The benzoylation of myo-inositol 1,3,5,-orthoformate
followed by the tosylation of the remaining hydroxyl groups to form the compound
47. Removal of the benzoyl group gave the alcohol 47 which was further submitted
to Swern oxidation reaction. Resulted compound 48 treated with the sodium
borohydride for the reduction of carbonyl group. Methanolysis of the tosylates and
then acetylation formed scyllo-inositol 1,3,5,-orthoformate 50 as a triacetate.
Cleavage of the orthoformate yielded scyllo-inositol 5 (Scheme 8).
Scheme 8. Formation of scyllo-inositol 5
Page 28
12
1.3.9 Synthesis of bicyclic inositols
Aromatic compounds were not used as a precursor only for the synthesis of inositols
but also for the inositol analogues. Mehta et al.27 presented the polycylitols as a new
term, which might be biologically active like conduritols and carbosugars. They
proposed bicyclic inositols that have potential for biological activity.6 They have also
synthesized conjoined and annulated inositols.
Naphthalene, anthracene and indene are the precursor for the synthesis of this new
class.
Scheme 9. Synthesis of chiro-inositol derivative 55 from napthalene
Bicyclic inositols were synthesized from napthalene by Mehta and Ramesh28
(Scheme 9). Birch reduction of naphtalene gave isotetralin 51 which was converted
to 52 in 3 steps. The reaction of 52 with NBS for allylic bromination, followed by
dehydrobromination reaction provided the diene 53. Hydroxylation of diene unit in
53 gave the tetrol 54. Hydrolysis of tetrol formed the annulated chiro-inositol 55.
Page 29
13
1.3.10 Synthesis of bishomo-inositols
Balci and Kara29 introduced a new class of inositol, bishomo-inositol, which was
synthesized from cyclooctatetraene (Scheme 10).
Scheme 10. Synthesis of bishomo-inositols from cyclooctatetraene
Cyclooctatetraene (56) was reacted with mercury(II) acetate to give diacetoxydiene
57. Photooxygenation of diacetoxydiene 57 in the presence of a sensitizer
tetraphenylporphyrin (TPP) formed endoperoxide 58. Reduction of endoperoxide and
further acetylation afforded tetraacetate 59. Treatment of the tetraacetate 59 with
KMnO4 formed the cis-diol 60, which was converted to bishomo-inositol 61 by
deacetylation.
Balci et al.13 synthesized bishomo-inositol derivatives having the bicyclo[2.2.2]
octane skeleton (Scheme 11).
Page 30
14
O
O O
OO
OO
O
O
OO
O
O
O
O+
OAcOAc
OAc
OAc
OAcOAc
OAc
OAc
AcO
AcO
OHOH
OH
OH
HO
HO
1. OsO4/ NMO
2. Ac2O/pyr.
NH3
MeOH
1. HCl(g),MeOH2. K2CO3,MeOH3. Ac2O,pyr.
62 63 64
65 66 67
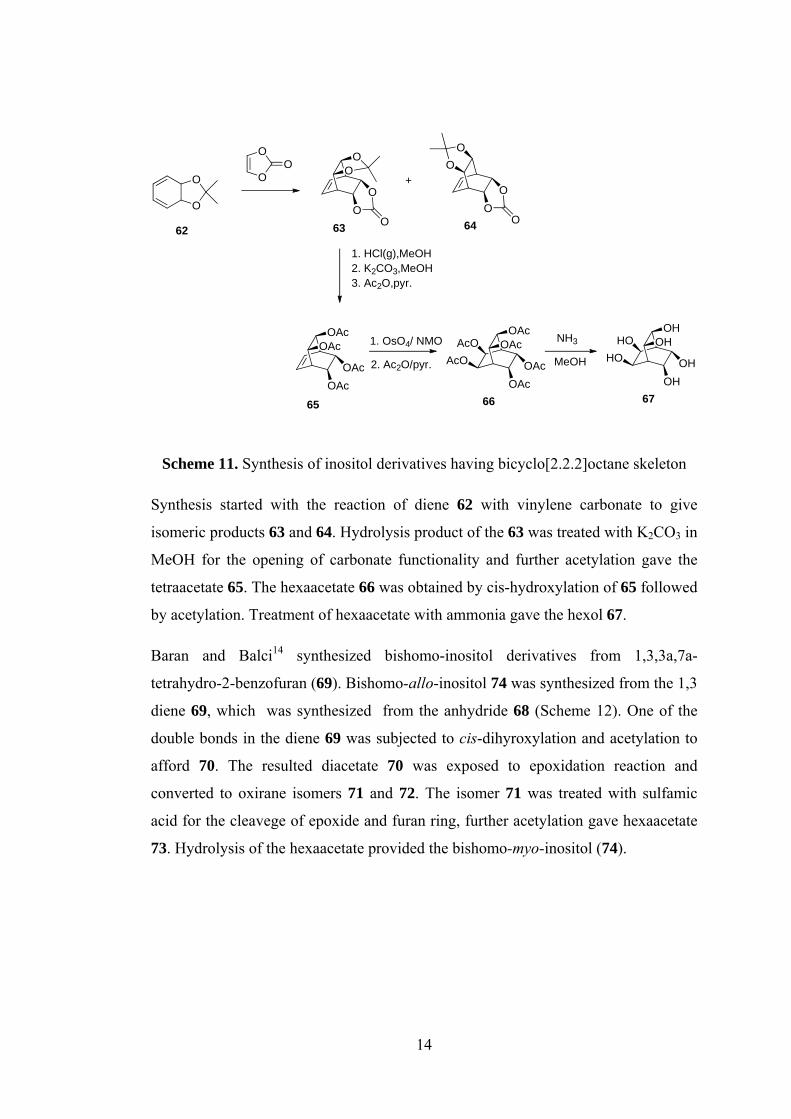
Scheme 11. Synthesis of inositol derivatives having bicyclo[2.2.2]octane skeleton
Synthesis started with the reaction of diene 62 with vinylene carbonate to give
isomeric products 63 and 64. Hydrolysis product of the 63 was treated with K2CO3 in
MeOH for the opening of carbonate functionality and further acetylation gave the
tetraacetate 65. The hexaacetate 66 was obtained by cis-hydroxylation of 65 followed
by acetylation. Treatment of hexaacetate with ammonia gave the hexol 67.
Baran and Balci14 synthesized bishomo-inositol derivatives from 1,3,3a,7a-
tetrahydro-2-benzofuran (69). Bishomo-allo-inositol 74 was synthesized from the 1,3
diene 69, which was synthesized from the anhydride 68 (Scheme 12). One of the
double bonds in the diene 69 was subjected to cis-dihyroxylation and acetylation to
afford 70. The resulted diacetate 70 was exposed to epoxidation reaction and
converted to oxirane isomers 71 and 72. The isomer 71 was treated with sulfamic
acid for the cleavege of epoxide and furan ring, further acetylation gave hexaacetate
73. Hydrolysis of the hexaacetate provided the bishomo-myo-inositol (74).
Page 31
15
Scheme 12. Synthesis of bishomo-myo-inositol 74
Baran and Balci14 synthesized several inositol analogs starting from 1,3,3a,7a-
tetrahydro-2-benzofuran (69) via different pathways (Scheme 13). Bishomo-allo-
inositols 76, 77 and bishomo-chiro-inositol 75 were also synthesized and their α-
glycosidase inhibition activities were determined.
Page 32
16
Scheme 13. Bishomo-inositol derivatives from 1,3,3a,7a-tetrahydro-2-benzofuran
Recently, Mahapatra and Nanda30 synthesized hydroxy-skipped homo-inositol 81
analogs from enantiomers of 5-hydroxymethyl-2-cyclohexenone (Scheme 14).
The synthesis started with the protection of the free hydroxyl group of (S)-5-
hydroxymethyl-2-cyclohexenone 78 by tert-butyl diphenylchlorosilane (TBDPS-Cl).
Hydroxymethylation of TBDPS ether resulted in the formation of 79. Asymmetric
dihydroxylation of the compound 79 with AD-mix-α gave diol 80. The reduction
reaction with NaBH4 followed by deprotection provided the inositol 81.
Page 33
17
Scheme 14. Synthesis of hydroxy-skipped homo-inositol 81
1.4 Singlet Oxygen
Singlet oxygen, which was first identified in 1924, is an important and reactive
species in chemical reactions. Not only the chemistry of singlet oxygen but also its
biological functions especially in certain blood diseases, in cancer-inducing
mechanism and in metabolic hydroxylation raise the study of singlet oxygen. Beside
its biological role, reactions and applications of singlet oxygen have also been
searched.31 The reactivity of singlet oxygen can be understood by the molecular
orbital diagram as illustrated in Figure 2.
Page 34
18
Figure 3. Molecular Orbital Diagram of O2 Molecule
In the ground state, the molecular oxygen has two unpaired electrons with parallel
spin at π*2p orbitals and is shown as triplet oxygen 3O2. Because of the parallel spin
of unpaired electrons, ground state oxygen molecule is paramagnetic and biradical.
Moreover parallel electron spins are not allowed for the direct access of paired
electrons and enable the single-electron step reactions.
The O2 molecule can absorb energy in order to change its electron configuration to
one of the singlet configurations. The two lowest electronic states of molecular
oxygen known as singlet oxygen have electrons with antiparallel spins (Figure 3).
Page 35
19
Figure 4. Electronic States of Molecular Oxygen
In both forms of singlet oxygen the spin restriction is removed and so the oxidizing
ability is great. Life time of the singlet oxygen is very short with respect to triplet
oxygen. The lower energy state, 1∆ is much longer lived than the other energy states.
Photosensitization reactions are the general method for the formation of singlet
oxygen in laboratory. The reactions are performed under light in the presence of a
sensitizer. Excitation of the sensitizer with light, continue with intersystem crossing
(ISC) and transfer of the energy to an adjacent oxygen molecule generating the
singlet oxygen and ground state sensitizer back (Figure 4). Generally dye molecules
are used as sensitizers such as methylene blue, rose bengal and porphyrins.
Figure 5. Formation of Singlet Oxygen with Sensitizer
There are different types of reactions of singlet oxygen with unsaturated molecules.
The three most common reaction types of singlet oxygen with olefins are the ene
Page 36
20
reaction, Diels-Alder reaction, and the [2+2] cycloaddition reaction (Figure 5). As a
result of ene reaction of singlet oxygen with olefins hydroperoxides are mainly
formed. On the other hand, the reaction of 1O2 with dienes and/or electron rich
olefins generate bicyclic endoperoxides and [2+2] cycloaddition reaction provides
1,2-dioxetane All three types of singlet oxygen reactions have been utilized in
organic synthesis for the regiospecific and stereospecific oxidation of olefins.
1.5 Manganese (III) Acetate Oxidation Reactions
Mn(OAc)3, which is an oxo-centered coordination complex, is a versatile oxiding
agents for the synthesis of many important organic compounds. Heiba, Dessau,
Bush, and Finkbeiner have demonstrated in 1968 that reactions of olefins with
Mn(OAc)3 in acetic acid at reflux resulting the γ-lactones32(Scheme 15).
Scheme 15. Formation of the γ-lactones
Figure 6. Singlet Oxygen Reactions
Page 37
21
This was the basis for the oxidatively initiate free radical reactions of Mn(OAc)3
which has been further developed and used in synthetic chemistry till today with its
different mechanistic pathways34.
Balci and co workers also studied Mn(OAc)3 reactions. Mn(OAc)3 treated with
benzonorbornadiene and its derivatives. The mechanism of the reaction with the
affect of the co-oxidant Cu(OAc)2 were investigated.38
Scheme 16. Reactions of benzonorbornadiene with Mn(OAc)3
1.6 Aim of thesis
Due to their potential for biological activities, developing new synthetic pathways for
the inositol derivatives is important. Various methodologies have been developed for
the synthesis of inositols and their derivatives. The synthesis of inositols was
achieved either via the transformation of other cyclitols, generally the myo-inositol,
or via appropriate substrates. Balci and co-workers formulated strategy of synthesis
based on the photooxygenation of the appropriate dienes.
Page 38
22
Baran and Balci14 achieved the stereoselective synthesis of isomeric bishomo-inositol
derivatives starting from the diene 69 and introduced the complex stereochemistry by
combination of photooxygenation, epoxidation, and cis hydroxylation reactions. The
α-glycosidase inhibitory activities were also investigated and it was found that some
of the isomers showed α-glycosidase inhibitions.
In this work, we are interested in synthesis of bishomo-inositol derivatives from
1,3,3a,7a-tetrahydro-2-benzofuran 69 via different pathways. The key compound 69
is synthesized in several steps. Photooxygenation reaction of diene 69, followed by
formation of bisepoxide and acid catalyzed ring opening reactions of this epoxide
were the planned steps to perform. In the second part of the study we decided to
perform manganese (III) oxidation of the diene 69 and further cis hydroxylation
reactions.
All products will be purified and characterized also the formation mechanism of the
products would be studied.
Page 39
23
CHAPTER 2
RESULTS AND DISCUSSION
In order to synthesize different bishomo-inositol derivatives we have chosen
1,3,3a,7a-tetrahydro-2-benzofuran (69) as a key compound. Using two different
pathways starting from the key compound 69, different bishomo-inositol derivatives
were synthesized. Firstly, we started to synthesize the key compound 69 from cis-
1,2,3,6-tetrahydrophytalic anhydride (90) in several steps for further reactions.
2.1 Synthesis of 3aR(S),7aS(R)-1,3,3a,7a-tetrahydro-2-benzofuran (69)
The commercially available cis-1,2,3,6-tetrahydrophytalic anhydride (90) was
reduced to corresponding alcohol 91 by LiAlH4 in THF at 0 oC. Treatment of the
resulted alcohol 91 with p-toluenesulfonyl chloride in pyridine gave the benzofuran
derivative 92. The resulted benzofuran derivative was purified by vacuum distillation
to give colorless liquid 92. The 1H NMR and 13C NMR spectra of the compounds 91
and 92 are in agreement with those reported in the literature.14
.
Scheme 17. Synthesis of benzofuran derivative 92
Page 40
24
The compound 92 was submitted to bromination reaction to form addition product
93. Elimination of this dibromide 93 with DBU in benzene provided the key
compound, rel-(3aR,7aS)-1,3,3a,7a-tetrahydro-2-benzofuran (69).
Scheme 18. Synthesis of key compound 69
2.2 Photooxygenation of 3aR(S),7aS(R)-1,3,3a,7a-tetrahydro-2-benzofuran (69)
After synthesizing the key compound 69, we decided to follow two different
pathways to synthesize different bishomo-inositol derivatives. One of these pathways
started with singlet oxygen reaction. Singlet oxygen reaction was performed with
oxygen gas in the presence of a sensitizer and a light source. The key compound 69
and the sensitizer, tetraphenylporphyrine (TPP), were dissolved in CH2Cl2. The
mixture was irradiated with a projection lamp (500 W) for 12 h at room temperature
while oxygen was passed through the solution. After the reaction was completed,
singlet oxygen addition product 94 was obtained in 81 % yield.
Scheme 19. Photooxygenation reaction of 69
Formation of single isomer 94 was proved by 1H and 13C spectra. 1H and 13C spectra
were in agreement with the reported in the literature.14 Although, dissymmetric diene
unit in the key compound 69 seems to be open to attack for both sides, the structure
of the single product 94 showed that one side was favored. Baran and Balci
Page 41
25
explained this favor as a result of the repulsive interaction between the nonbonded
electrons of oxygen in the tetrahydrofuran ring and on the singlet oxygen molecule.14
2.3 Rearrangement of endoperoxide 94 to bisepoxide 100
After the characterization of endoperoxide 94, the next step was treatment of 94 with
cobalt meso tetraphenylporphine (CoTPP) complex to get the rearranged product of
endoperoxide 94. Balci and coworkers39 studied the reactions of various unsaturated
bicyclic endoperoxides with CoTPP. Although diepoxides were major products, it
was seen that open-chain epoxy aldehydes were also formed (Scheme 20). From the
various product distribution with different endoperoxides, Balci et al.39 explained the
mechanism as both diepoxides and epoxy ketones formed from the oxygen-oxygen
diradicals. However, ring closures of these radicals are in competition with the 1,2-
hydrogen shift required to yield an epoxy ketone and the difference in activation
energy causes the epoxide formation to be favored.
Scheme 20. CoTPP-catalyzed rearrangement of endoperoxide 95
Adam et al.35 performed the CoTPP-catalyzed rearrangement of keto endoperoxide
98 which afford bisepoxide 99 as a major product.Decomposition reaction of the keto
endoperoxide 98 in the presence of CoTPP involves radical pathways. The possible
mechanism for the formation of bisepoxide 99 from the keto endoperoxide 98 was
given by Adam et al.35 (Scheme 21).
Page 42
26
Scheme 21. Mechanism of CoTPP catalyzed rearrangement of endoperoxide 98
The CoTPP complex, synthesized from TPP, was dissolved in CH2Cl2 and was
added dropwise to a solution of endoperoxide 94 in an ice-bath. When addition was
completed, reaction mixture was stirred for additional 2 h at room temperature.
Crude product was submitted to column chromatography eluting with hexane/EtOAc
(2:3) (Scheme 22).
Scheme 22. Synthesis of compound 100
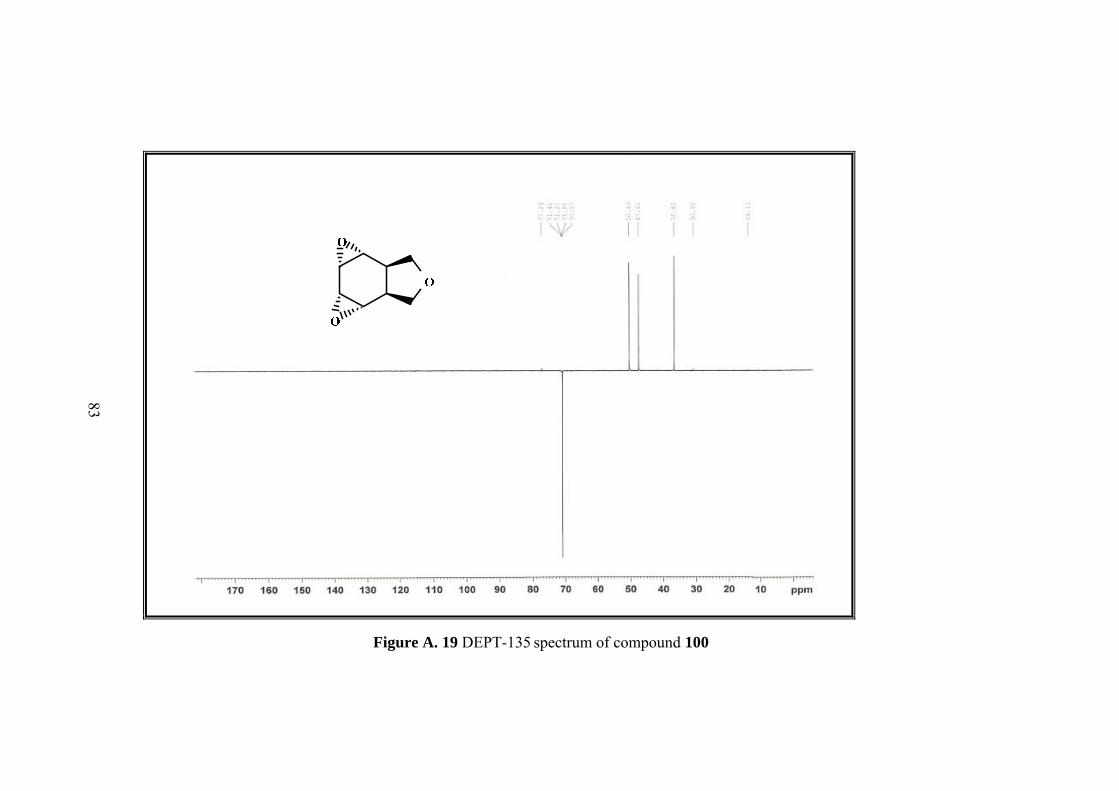
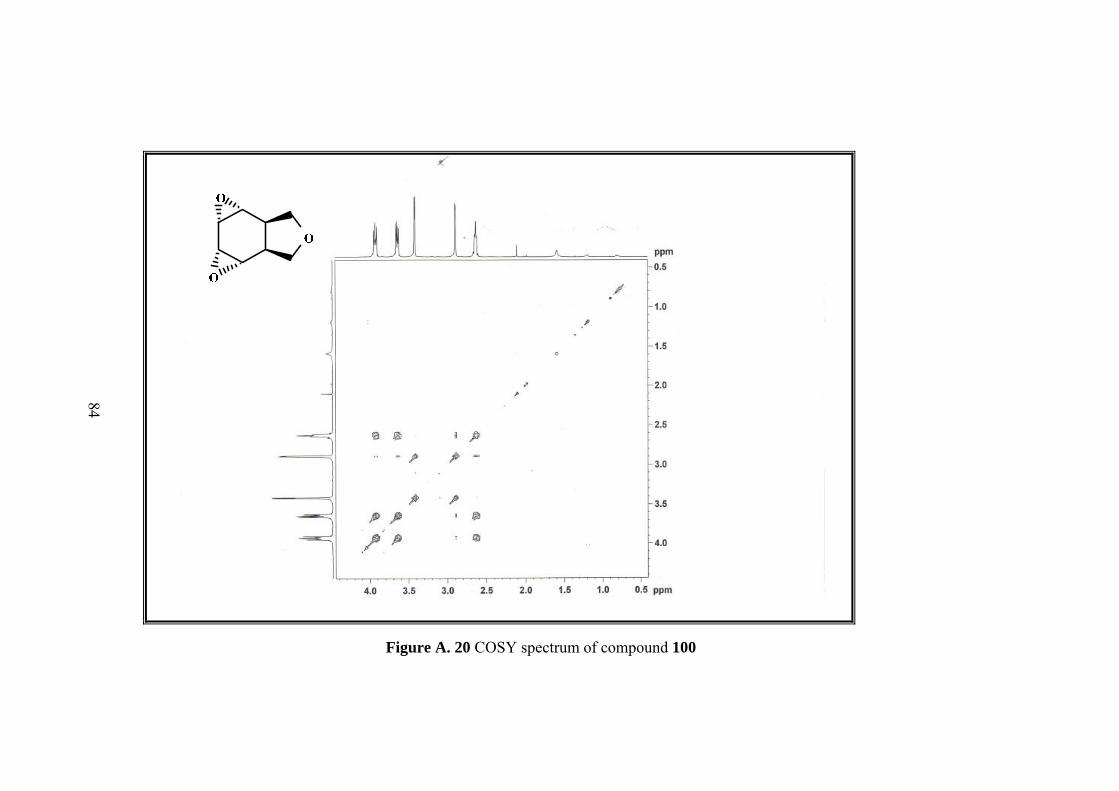
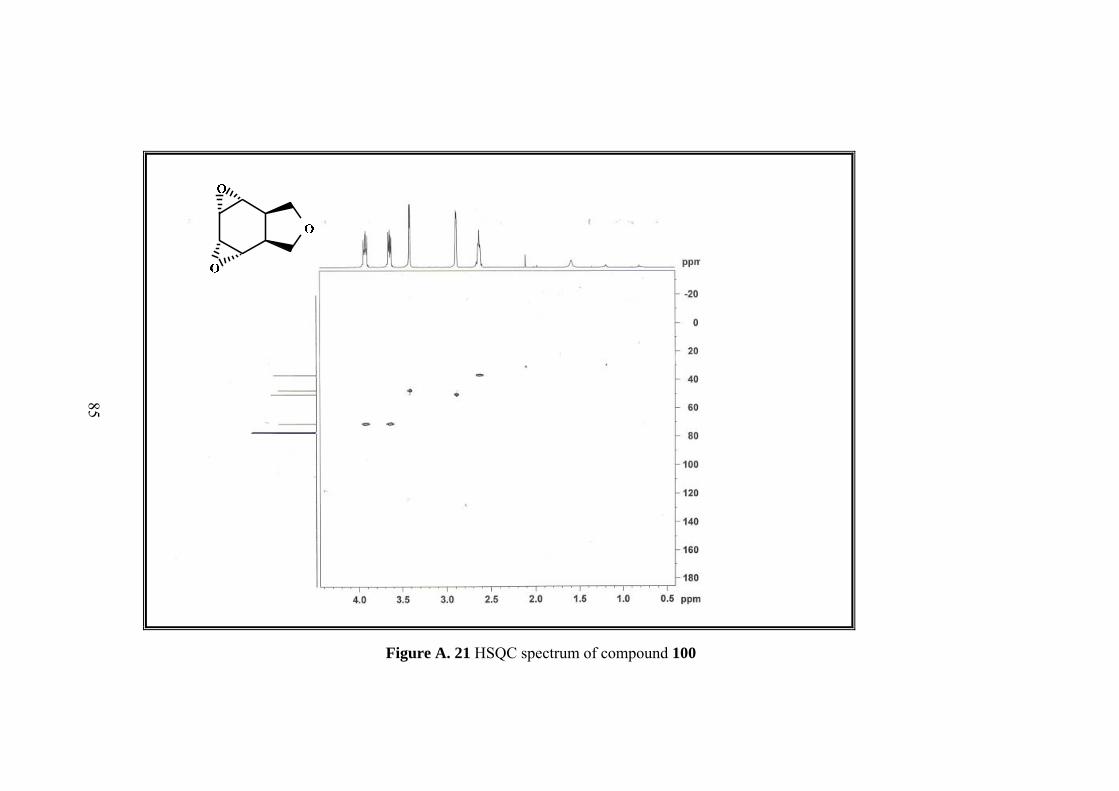
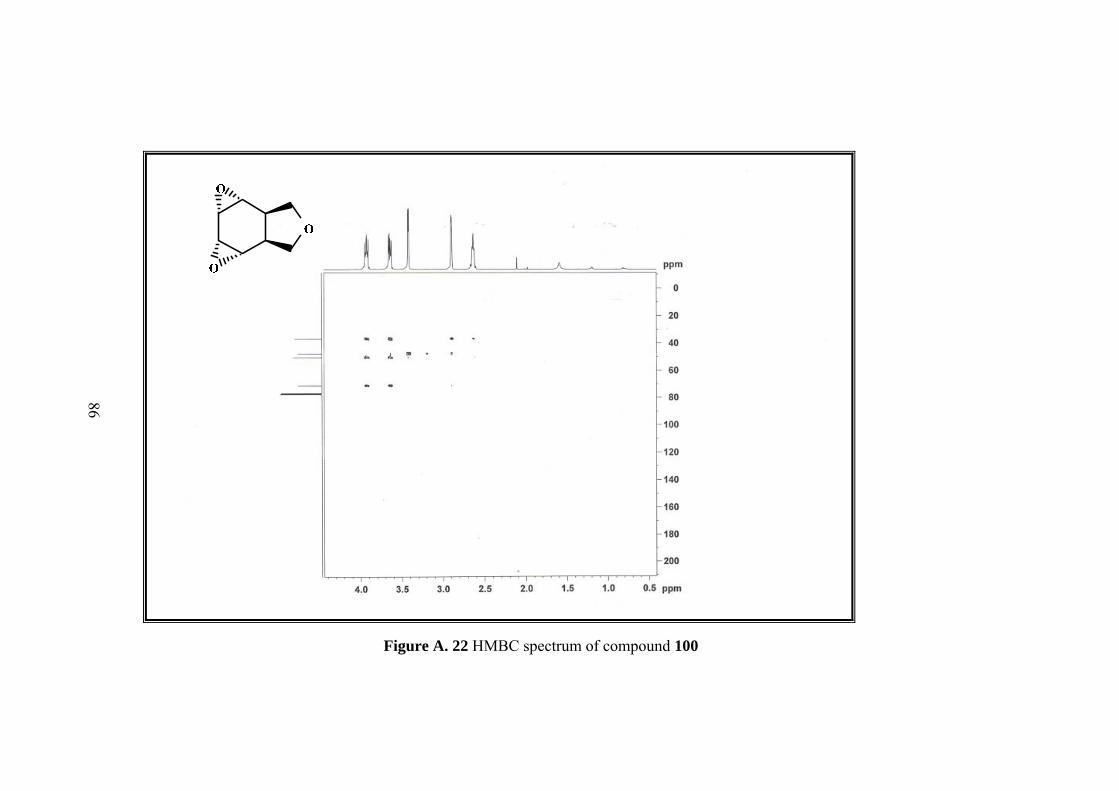
Resulting single product was characterized by 1D&2D NMR. Five sets of signals in 1H-NMR and four sets of singlets in 13C-NMR of the compound 100 indicated the
formation of symmetrical compound. Moreover both 1H-NMR and 13C-NMR spectra
showed that the signals in the olefinic region were disappeared.
In 1H-NMR spectrum methylenic protons of the furan ring resonate as an AB-system
between 3.94 to 3.62 ppm. Four epoxide protons gave two symmetrical signals which
resonate as doublet at 3.41 ppm and multiplet at 2.63 ppm. The remaining methine
protons appear at 2.97 as multiplet.
13C NMR also shows the symmetry with four signals at 126.2, 122.3, 75.0, 37.8 ppm.
Page 43
27
2.4 Acid catalyzed ring opening reaction of the bisepoxide 100
There are different routes for the opening of the epoxide rings. A wide range of
nucleophiles can attack to epoxide ring under both acidic and basic conditions
resulting in ring opening. Generally in basic medium ring opening reactions followed
SN2 mechanism whereas in acidic medium depending on the type of the epoxide both
SN2 and SN1 mechanism can proceed.36
In our study we preferred an acid catalyzed ring opening reaction for the bisepoxide
100. To synthesize the tetraacetate 101, the bisepoxide was reacted with H2SO4
under two different conditions; in the presence and absence of water.
2.4.1. Ring opening reaction of the bisepoxide 100 in the presence of water
In order to open the epoxide rings in compound 100, we decided to perform acid
catalyzed ring opening reaction with H2SO4 in the presence of water followed by
acetylation of the formed tetrol by Ac2O in pyridine. Due to the presence of two
epoxide functionalities and a tetrahydrofuran ring, formation of different
configurational isomers was expected.
Scheme 23. Ring opening reaction of 100
From the spectral data it was determined that three isomers were formed with the
ratio 67 %, 16.5%, 16.5%. The mixture was submitted to column chromatography on
silica gel and as the major product rel-(3aR,4S,5R,6R,7S,7aS)-4,6,7-
tris(acetyloxy)octahydro-2-benzofuran-5-yl acetate (102), was obtained.
Page 44
28
The structure of the major compound was characterized by spectroscopic methods
such as 1D- and 2D-NMR.
In 1H-NMR spectrum four acetoxy protons resonate between 5.29 to 5.02 ppm.
Methylene protons of the furan ring resonate as two distinct AB-system in the region
3.83 to 3.67 ppm. The two methine protons in the intersection of the cyclohexane and
furan ring resonate as multiplets at 2.98 and 2.35 ppm. The remaining protons of
acetate groups resonate between 1.97-1.93 ppm as singlets.
13C-NMR consist of sixteen lines containing four carbonyl group at 169.9, 169.8,
169.7, 169.6 ppm, four acetoxy carbon at 72.8, 71.1, 70.93, 70.3 ppm, two
methylenic carbons at 70.0, 67.4 ppm. Methyl and methine carbon resonances appear
at 42.2, 40.5, 20.7, 20.68, 20.57, 20.53 ppm.
Assignment of the product was done by using 2D-NMR especially the correlations in
HMBC&COSY. To determine the exact configuration of the acetate groups, the
corresponding coupling constants were measured between the relevant protons. The
acetoxy proton H-5 in 102 resonates as doublet of doublets with the couplings J45 =
9.5 Hz, J56 = 9.1 Hz, indicating the trans configuration of H-5 with respect to
neighboring protons H-4 & H-6. Beside the proton H-5 acetoxy proton H-6 also
gives coupling with H-7 with the coupling constant J76 = 10 Hz, which shows the
trans configuration of H-6 to H-7. In addition to that H-7 resonates as triplet with a
coupling constant 10 Hz, which indicated its trans position to neighboring protons.
As a result, configuration of H-7 is cis to furan ring. However for H-4 the situation is
reverse; coupling constant between H-4 and H-3a is relatively smaller, J43a = 6.0 Hz,
which shows us H-4 is trans to furan ring.
Page 45
29
2.4.2. Ring Opening Reaction of the Bisepoxide 100 without Water
In order to open the bisepoxide, we have treated compound 100 with acetic
anhydride in the presence of H2SO4. Although we expected the formation of
tetraacetate isomers from the nucleophilic substitution reaction of acetic anhydride,
spectral data confirmed the formation of single isomer.
Scheme 24. Expected product of the ring opening reaction of bisepoxide 100
The 1H-NMR and 13C-NMR spectra of the resulted compound, indicate that there is
an unsymmetrical compound which involves 13 different proton signals and 15 line
carbon atoms.
Assignment of the protons and carbons were done with the combination of 1D&2D-
NMR. In addition to the chemical shifts, correlations, which were shown by HMBC
and COSY spectra, were important to label the protons and carbons.
In 1H-NMR, the presence of 10 different proton signals beside the methyl protons at
2.08-1.92 ppm, shows that bisepoxide was opened in an unsymmetrical way. Four
different protons in low field belong to methine protons of cyclohexane ring which
are adjacent to acetate groups. Acetoxy protons resonate at 5.74, 5.56 ppm as doublet
as doublets, at 5.50 ppm as doublet of doublets of doublets and at 4.96 ppm as broad
triplet. Methylenic protons of the furan ring (H-2,2’ and H-4,4’) resonate as 2
different AB systems between 4.14 and 3.54 ppm. Two signals at 2.43 and 2.29 ppm
belong to methine protons H-5 and H-1 of the compound. Six methyl protons of
acetate groups resonate at 2.08 (6H), 2.01(3H) and 1.92(3H) ppm.
13C-NMR spectra with DEPT-90 and DEPT-135 also support the formation of single
unsymmetric product. It is seen that four different carbonyl carbons exist in the
compound. DEPT-90 shows that the carbon resonances between 72.8 and 69.5 ppm
Page 46
30
belongs to 4 distinct carbons to which acetoxy groups are attached and 2 carbons at
39.6 and 38.7 ppm belong to carbons C-1 and C-5. DEPT-135 indicates two
methylenic carbons of furan ring at 68.40 and 66.52 ppm. Moreover four methyl
carbons of four acetate groups can be also distinguished from 13C-NMR spectra.
Although 13C-NMR and 1H-NMR seem to fit the basic structure of the expected
product 101, and also 2D-NMR spectra mainly supports the result. There is a
contradiction between the proposed structure 101 and COSY spectrum of the product
which is shown in Figure 6.
Figure 7. COSY spectrum of expected product of 101
Page 47
31
The correlation of methine protons H-1 and H-5 with neighboring methine protons of
cyclohexane ring H-9 and H-6 is an expected result since these protons interact with
each other over 3 or 4 bonds. However, it is also expected that each proton would
give different correlation by different intensities with the same proton. In COSY
spectrum of the product (Figure 6) it is seen that the correlation between H-1& H-9
and H-5&H-9 have approximately same intensities which means the couplings of the
protons are likely to be same which is not possible for the predicted structure 101.
To be sure from the structure we decided to take X-Ray analysis from the crystals.
The X-ray analysis of structure, as shown in Figure 7, confirmed that the structure of
product is not the expected one.
Page 48
32
Figure 8. X-ray analysis of the product of ring opening reaction of bisepoxide 100
top:packing diagram, bottom: the molecular structure of compound.
Page 49
33
X-ray analysis showed that a rearranged product was formed. Actually no change
was expected for furan ring in the bisepoxide 100 in the reaction, however X-ray
results indicated the existence of an arrangement occurred during the reaction
involving the furan ring. This rearrangement explains the doubtful correlation of the
protons. With the exact configuration of the product 103, it is reasonable for H-1 and
H-5 to have the correlations with H-9 in same intensities.
After exact structure was found, we have proposed a mechanism for this unexpected
product 103. The reaction that takes place would be a kind of neighboring group
participation which is a well-known mechanism results from the interaction of sigma
bond or pi bond electrons or lone pair electrons in an atom. Neighboring group
participation also called anchimeric assistance occurred when the nucleophilicity of
atom or bond within the molecule is compatible with the nucleophilicity of the
substance in the medium.37 In our case an alkyl substitution seems to be placed. In
the acidic medium, bisepoxide oxygen was protonated, with the substitution of furan
ring which is antiperiplanar to epoxide ring a secondary carbocation formed which is
further attacked by acetic anhydride. Second epoxide ring was opened by direct
nucleophilic attack of acetic anhydride (Scheme 25).
Page 50
34
Scheme 25. Proposed mechanism of reaarrangement
Page 51
37
2.4.2.1. Acetolysis Reaction of rel-(1R,5S,6R,7S,8S,9S)-3-oxabicyclo[3.3.1]
nonane-6,7,8,9-tetrayl tetraacetate (103)
After synthesizing and characterizing the compound 103, we planned to treat it with
sulfamic acid which is used for acetolysis of furan ring. Although, the rearrangement
product 103 has a tetrahydropyran instead of tetrahydrofuran ring, we applied same
procedure in acetolysis of furan ring. We have dissolved the compound 103 in a
mixture of acetic anhyride and acetic acid (1:1) and added sulfamic acid at room
temperature in catalytic amount. After the reflux for 24 hours, reaction was stopped.
The product of the reaction characterized by NMR and it was shown that there was
no change in the starting material 103. Since the reaction failed, reaction was
repeated with increasing amount of catalytic sulfamic acid. However, reaction was
not succeeded. We assume that tetrahydropyran is more stable than the furan ring to
ring opening reaction.
Scheme 26. Acetolysis reaction of tetraacetate 103
Page 52
38
2.4.2.2 Hydrolysis Reaction of rel-(1R,5S,6R,7S,8S,9S)-
3oxabicyclo[3.3.1]nonane-6,7,8,9-tetrayl tetraacetate (103)
Rearranged tetraacetate molecule 103 was dissolved in absolute methanol. While dry
NH3(g) was passed through solution, the mixture was stirred for 4 h. Evaporation of
solvent and formed acetamide gave tetrol 105.
Scheme 27. Hydrolysis of the molecule 103
Full assignment of the tetrol molecule 105 was made by 1D&2D NMR. Four
methine protons (H-6,7,8,9) neighboring hydroxyl groups and one of the methylenic
protons on the tetrahydropyran ring (H-2) resonate at the same region between 4.27-
4.15 ppm. Other metylenic protons (H-4,4’) gave an AB system; A part resonate at
3.86 as doublet and B part at 3.63 ppm as doublet of doublets. H-2’ resonates at 3.5
ppm as a broad doublet. Methine protons H-5 and H-1 resonate as broad singlet at
2.29 and 2.16 ppm.
2.4.3 Manganese (III) Acetate Oxidation Reaction of rel-(3aR,7aS)-1,3,3a,7a-
Tetrahydro-2-benzofuran (69)
The second route, that we have planned for the synthesis of different inositol
derivatives from the key compound rel-(3aR,7aS)-1,3,3a,7a-tetrahydro-2-benzofuran,
started with manganese acetate oxidation reaction. Manganese(III) acetate is an
powerful one electron oxidant which mediated free radical reactions for a new bond
formation.
Heiba and Dessau proposed the mechanism of the reaction of olefins with Mn(OAc)3
in acetic acid at reflux, in which the oxidation of the radical 107 to the cation 112
Page 53
39
continue with cyclization to the tetrahydrofuran derivative and then loss of a proton
to give 111 (route A).32 On the other hand, Fristad et al.33 have proposed an
alternative route B, where the formed radical 107 undergoes first a cyclization
reaction followed by oxidation (Scheme 28).
Scheme 28. The mechanism of the reaction of olefins with Mn(OAc)3 in acetic acid
Beside these conflictions in the mechanism, Balci and co workers38 investigated the
mechanism of Mn(OAc)3 reactions with benzonorbornadiene derivatives. The
Mn(OAc)3 reactions of benzonorbornadiene and its hetero derivatives;
oxabenzonorbornadiene, azabenzonorbornadiene with dimedone and acetylacetone
were performed in the presence and absence of an co-oxidant Cu(OAc)2. In order to
decide the stage at which oxidation takes place (route A or B in scheme 28), the
structure of the products and possible intermediates investigated.
It is well-known that a radical type of intermediate does not undergo rapid
rearrangement whereas a cation in a benzonorbornyl system has great tendency to
form rearranged products. Therefore, the structure of the formed products would
Page 54
40
gave the information about the stage at which oxidation takes place. It was seen that
the cyclization takes place mainly after the oxidation of the formed radical (Scheme
29). 38
Scheme 29. The Mn(OAc)3 reaction of benzonorbornadiene and acetylacetone
In our study we planned to form a lactone unit in the key compound 69. We have
treated the diene 69 with Mn(OAc)3 in acetic acid in the presence of KOAc under
nitrogen atmosphere. The reaction mixture was heated to reflux temperature for 3-4
days until the color of the mixture turned to white. 1H &13C NMR spectra showed
that a single product was formed. This product could be one of the four possible
isomers 114-117 that can be formed from the reaction.
Page 55
41
From the both 1H NMR & 13C NMR spectra, it was seen that symmetry in the diene
molecule 69 was distorted. There are 12 different protons and 10 different carbon
signals for the product. The appearance of carbonyl peak at 174.6 is a signal for the
formation of lactone unit. The four carbons in the olefinic region were reduced to
two; which shows the lactone formation from one of the double bond in diene
molecule 69. From the COSY spectrum we can easily eliminate the two isomers 116
and 117, which are the constitutional isomers of 114 and 115, due to presence of
correlation of H-12 with H-3 instead of H-2. In order to decide which configurational
isomer 114 or 115 is the product, correlation between the double bond proton and
alkoxy proton with the coupling constants were investigated.
In the 1H-NMR two olefinic protons resonate at 5.90 and 5.83 as doublet of doublets
of doublets and a methine proton resonance was observed at 4.82 ppm, which should
belong to the proton of the carbon at the junction of the lactone unit. Deshielding
would be due to oxygen atom. Since the product is not symmetric anymore,
methylenic protons of the furan ring resonate as two different AB systems. Beside
AB systems, an ABC system belongs to the methylenic protons of lactone unit and
neighboring methine proton. One of the methylenic proton in lactone ring H-12
resonates at 2.67 forms B part of ABC system, methine proton H-3 resonate at 2.60
and gives A part of ABC system and other metylenic proton H-12’ resonates at 2.33
and gives C part of ABC system.
Page 56
42
Figure 9. 1H-NMR spectrum of the product of Mn(OAc)3 reactionof diene 69.
13C-NMR of the product contains a carbonyl group which resonates at 174.6, two
olefinic carbons appear at 131.2, 122.7, three carbons at 72.9, 71.8, 69.8 and four
carbons at 37.4, 35.6, 33.4, 32.5.
Protons of the molecule 118 are exactly assigned by 2D-NMR and seems to be fit for
two isomers 114 and 115 in which the difference is the position of lactone and furan
ring either cis or trans to each other. The product should be isomer 115, not because
trans form is energetically more favorable but also the coupling constants support the
formation of the trans form. The larger coupling constant between H-3 and H-4 (J34=
6.5 Hz) showed that trans isomer is the single product of the reaction (Scheme 30).
Scheme 30. Reaction of diene 69 with Mn(OAc)3.
Page 57
43
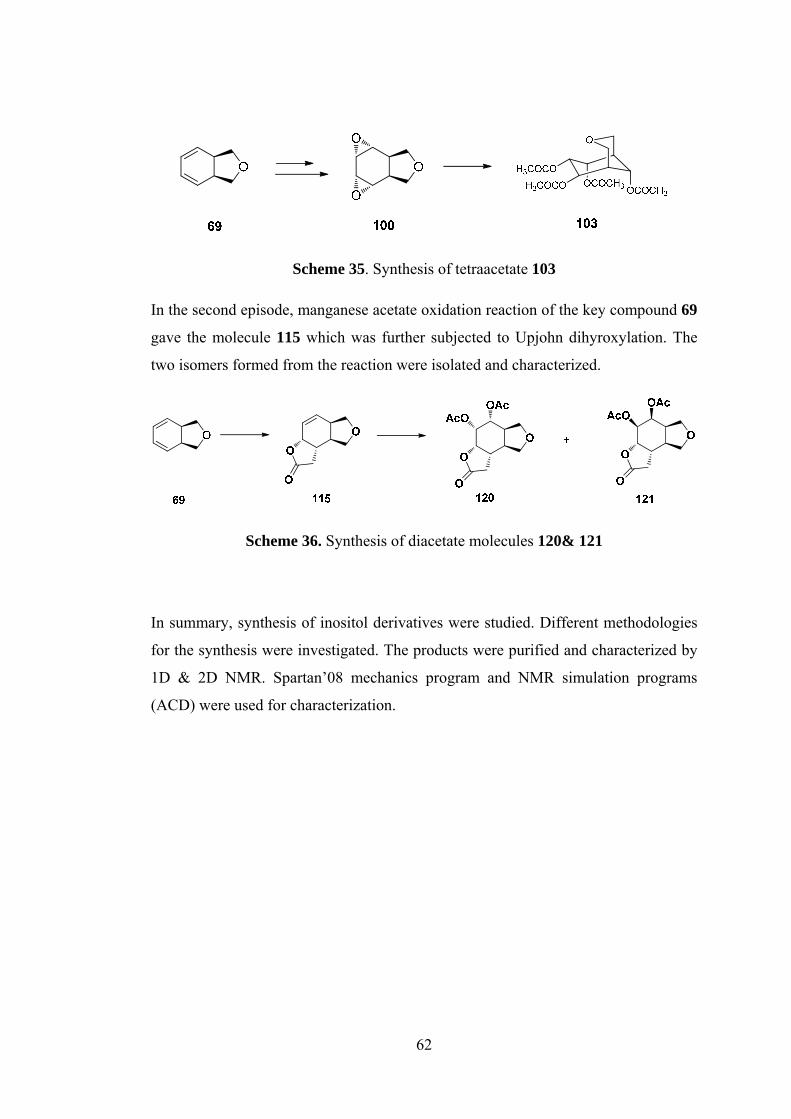
2.4.4 Upjohn Dihyroxylation
After synthesizing and characterization of the compound 115, we have planned to
perform hydroxylation of the double bond in the molecule. Upjohn dihydroxylation
in which OsO4 used for the oxidation of the olefins in the presence of cooxidant such
as N-methylmorpholine N-oxide (NMO), is a common method for cis hydroxylation.
Figure 10 Mechanism of Upjohn Dihyroxylation
We have treated the molecule 115 with OsO4 and NMO in the presence of
acetone/water (1:1) mixture under nitrogen atmosphere at room temperature for 5
days. After removal of the solvent, pyridine and Ac2O were added to reaction
mixture for acetylation and the reaction mixture stirred for 24 h at room temperature.
Os
OO
OO
R
R'
(VIII)
Os
OO
OO
(VI)
R R'
N
O
MeO
N
O
Me
2 H2O
R
R'
OH
OH
Os
O
O
O
(VI)
Page 58
44
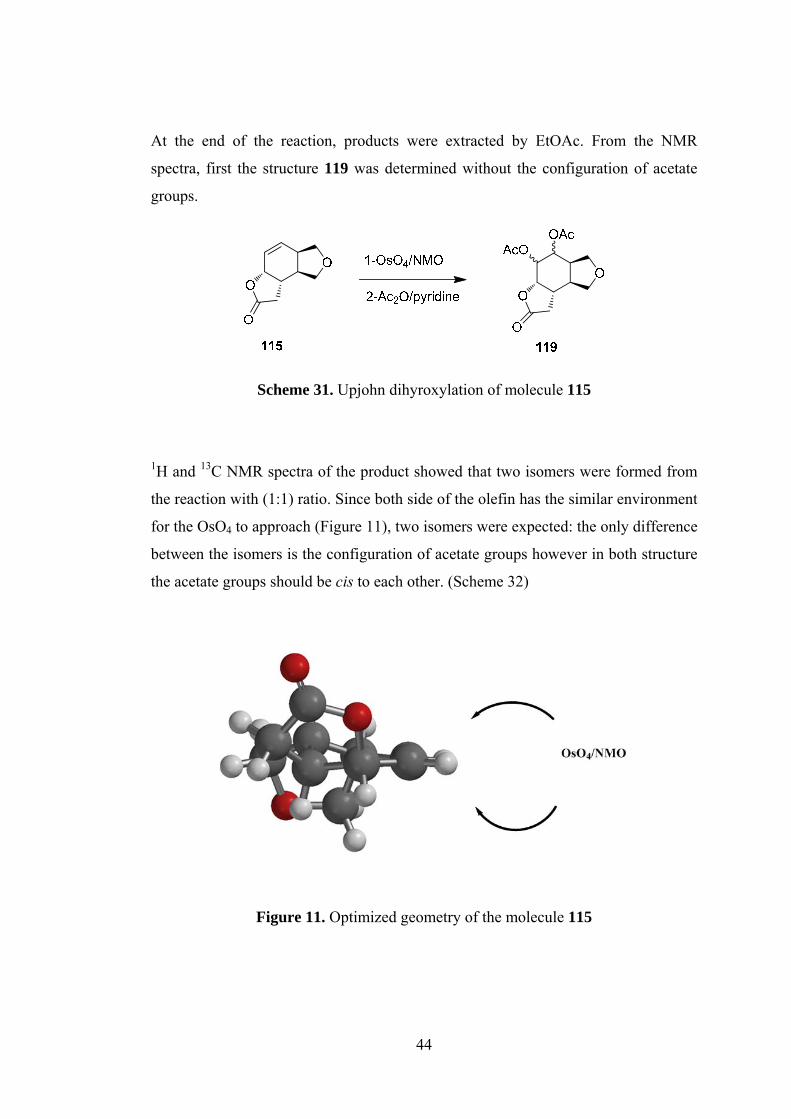
At the end of the reaction, products were extracted by EtOAc. From the NMR
spectra, first the structure 119 was determined without the configuration of acetate
groups.
Scheme 31. Upjohn dihyroxylation of molecule 115
1H and 13C NMR spectra of the product showed that two isomers were formed from
the reaction with (1:1) ratio. Since both side of the olefin has the similar environment
for the OsO4 to approach (Figure 11), two isomers were expected: the only difference
between the isomers is the configuration of acetate groups however in both structure
the acetate groups should be cis to each other. (Scheme 32)
Figure 11. Optimized geometry of the molecule 115
Page 59
45
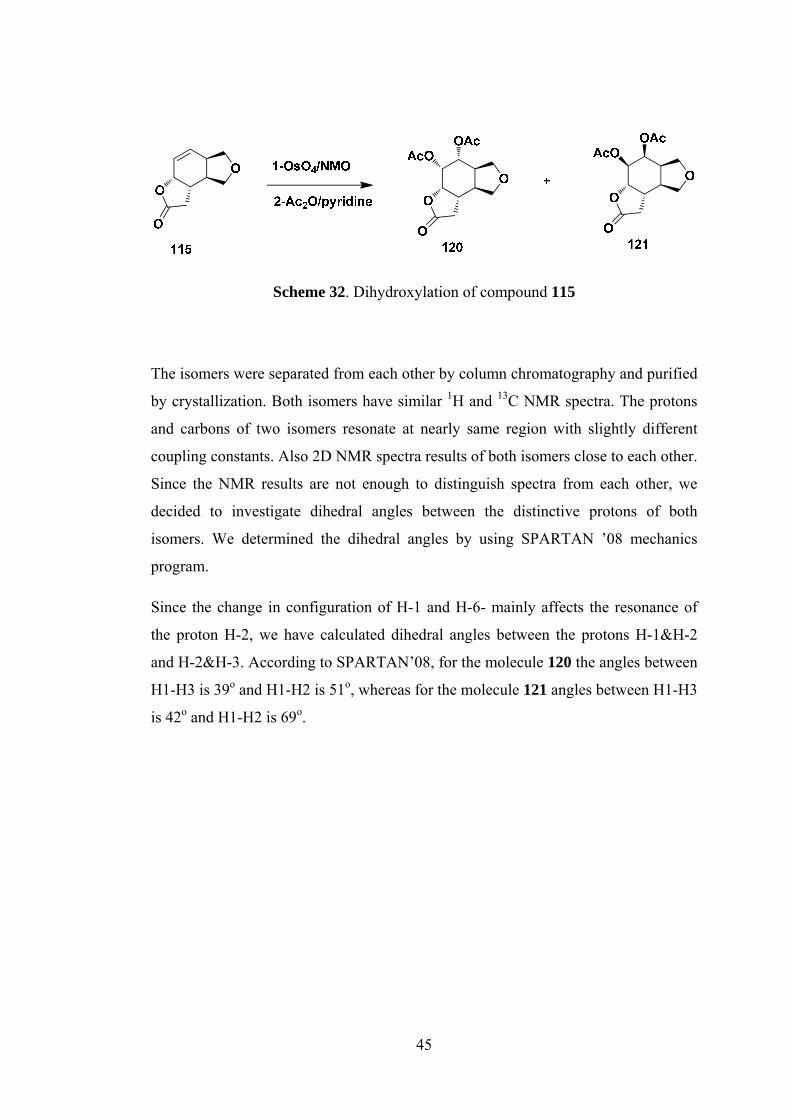
Scheme 32. Dihydroxylation of compound 115
The isomers were separated from each other by column chromatography and purified
by crystallization. Both isomers have similar 1H and 13C NMR spectra. The protons
and carbons of two isomers resonate at nearly same region with slightly different
coupling constants. Also 2D NMR spectra results of both isomers close to each other.
Since the NMR results are not enough to distinguish spectra from each other, we
decided to investigate dihedral angles between the distinctive protons of both
isomers. We determined the dihedral angles by using SPARTAN ’08 mechanics
program.
Since the change in configuration of H-1 and H-6- mainly affects the resonance of
the proton H-2, we have calculated dihedral angles between the protons H-1&H-2
and H-2&H-3. According to SPARTAN’08, for the molecule 120 the angles between
H1-H3 is 39o and H1-H2 is 51o, whereas for the molecule 121 angles between H1-H3
is 42o and H1-H2 is 69o.
Page 60
46
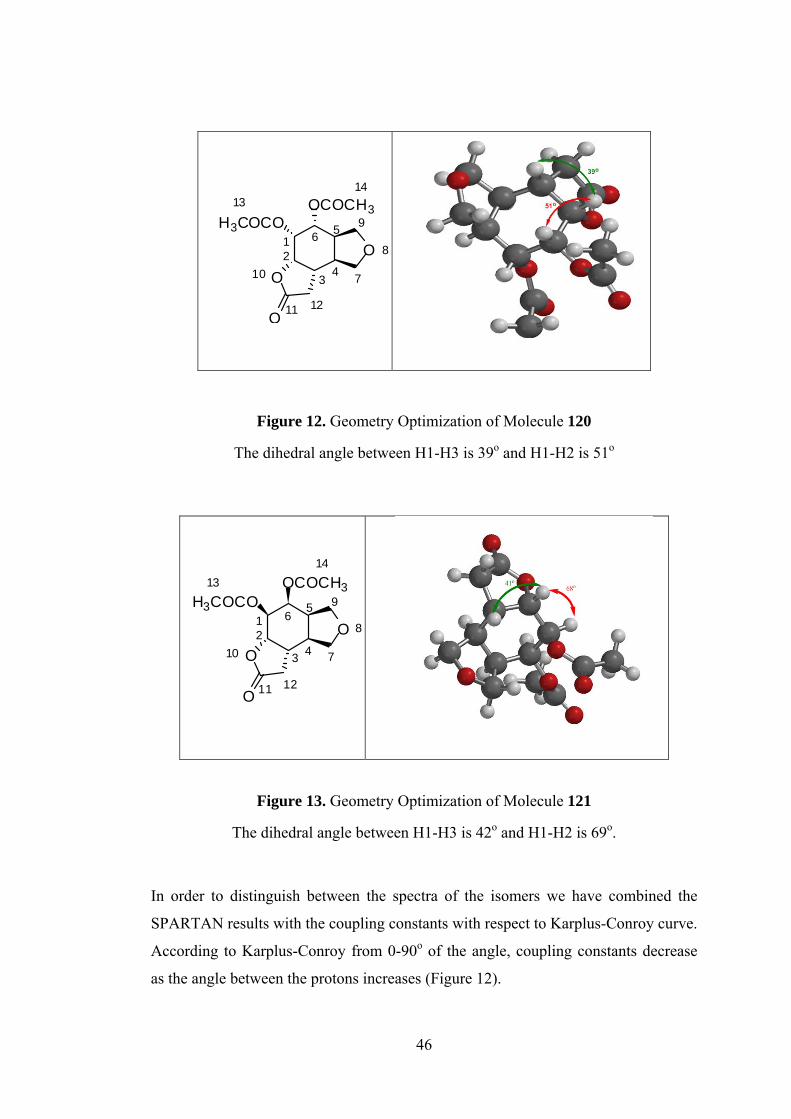
Figure 12. Geometry Optimization of Molecule 120
The dihedral angle between H1-H3 is 39o and H1-H2 is 51o
Figure 13. Geometry Optimization of Molecule 121
The dihedral angle between H1-H3 is 42o and H1-H2 is 69o.
In order to distinguish between the spectra of the isomers we have combined the
SPARTAN results with the coupling constants with respect to Karplus-Conroy curve.
According to Karplus-Conroy from 0-90o of the angle, coupling constants decrease
as the angle between the protons increases (Figure 12).
12
34
56
7
O 8
9
O10
11 12
OCOCH3H3COCO
O
1314
13
14
12
34
56
7
O 8
9
O10
11 12
OCOCH3H3COCO
O
Page 61
47
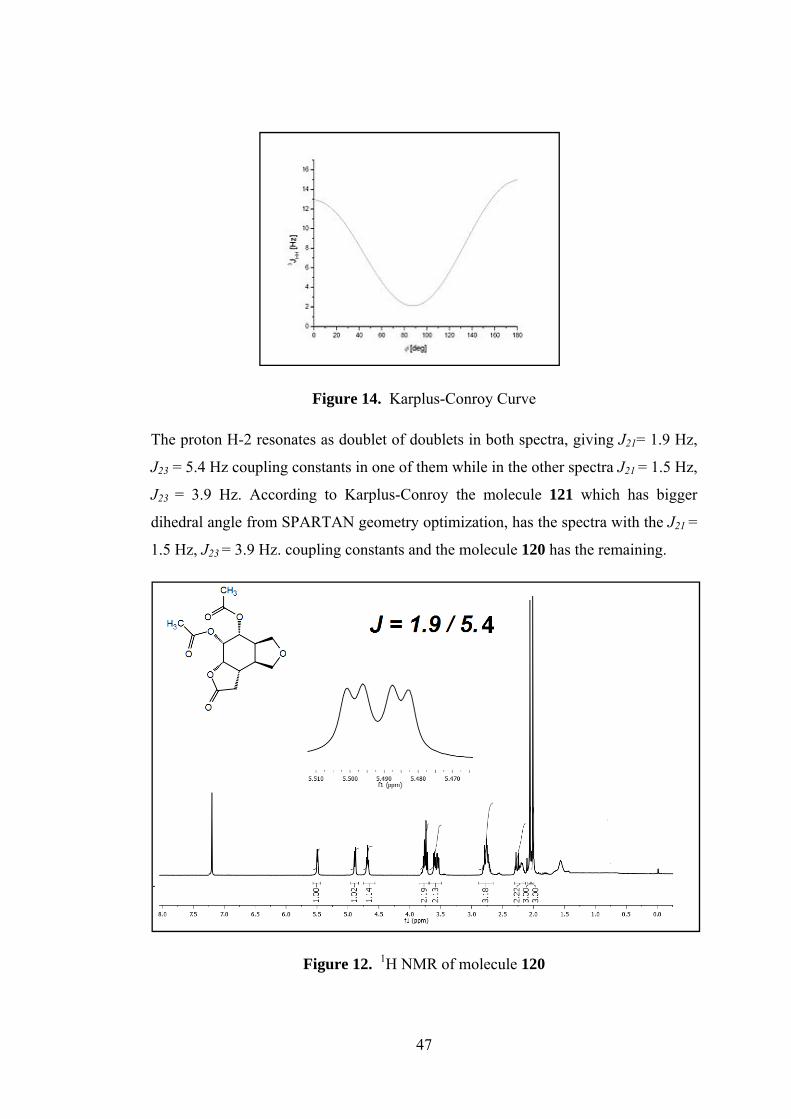
Figure 14. Karplus-Conroy Curve
The proton H-2 resonates as doublet of doublets in both spectra, giving J21= 1.9 Hz,
J23 = 5.4 Hz coupling constants in one of them while in the other spectra J21 = 1.5 Hz,
J23 = 3.9 Hz. According to Karplus-Conroy the molecule 121 which has bigger
dihedral angle from SPARTAN geometry optimization, has the spectra with the J21 =
1.5 Hz, J23 = 3.9 Hz. coupling constants and the molecule 120 has the remaining.
Figure 12. 1H NMR of molecule 120
Page 62
48
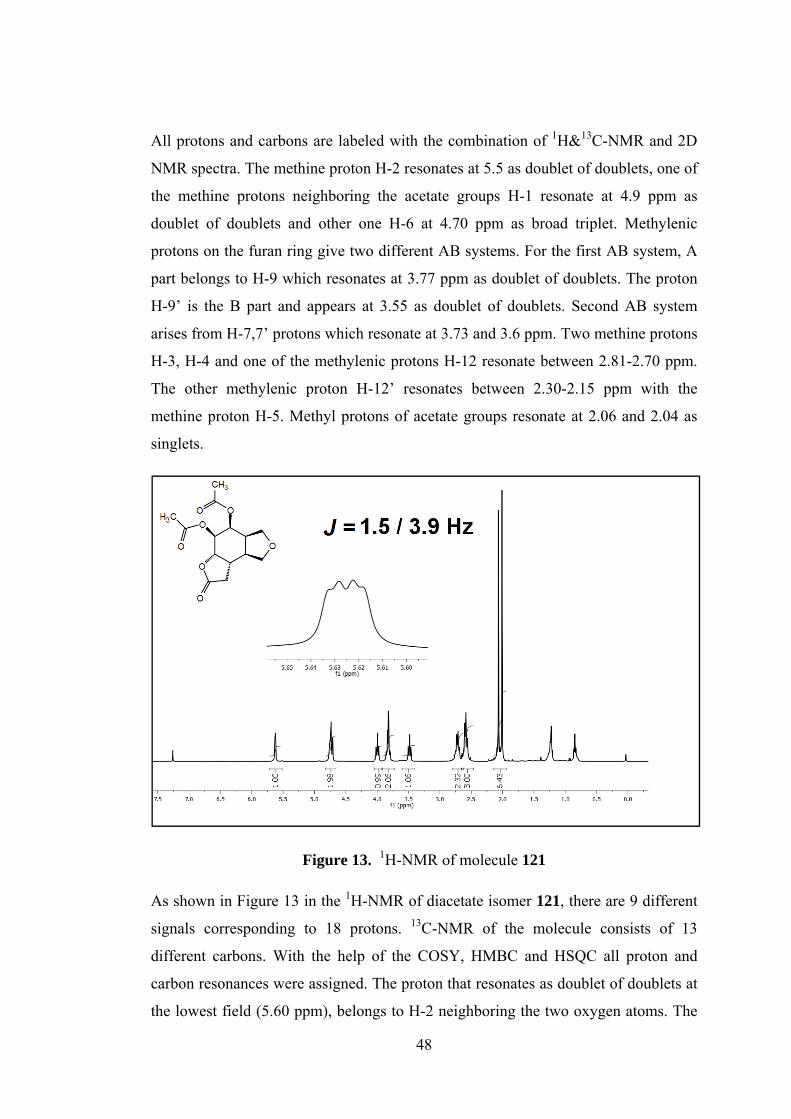
All protons and carbons are labeled with the combination of 1H&13C-NMR and 2D
NMR spectra. The methine proton H-2 resonates at 5.5 as doublet of doublets, one of
the methine protons neighboring the acetate groups H-1 resonate at 4.9 ppm as
doublet of doublets and other one H-6 at 4.70 ppm as broad triplet. Methylenic
protons on the furan ring give two different AB systems. For the first AB system, A
part belongs to H-9 which resonates at 3.77 ppm as doublet of doublets. The proton
H-9’ is the B part and appears at 3.55 as doublet of doublets. Second AB system
arises from H-7,7’ protons which resonate at 3.73 and 3.6 ppm. Two methine protons
H-3, H-4 and one of the methylenic protons H-12 resonate between 2.81-2.70 ppm.
The other methylenic proton H-12’ resonates between 2.30-2.15 ppm with the
methine proton H-5. Methyl protons of acetate groups resonate at 2.06 and 2.04 as
singlets.
Figure 13. 1H-NMR of molecule 121
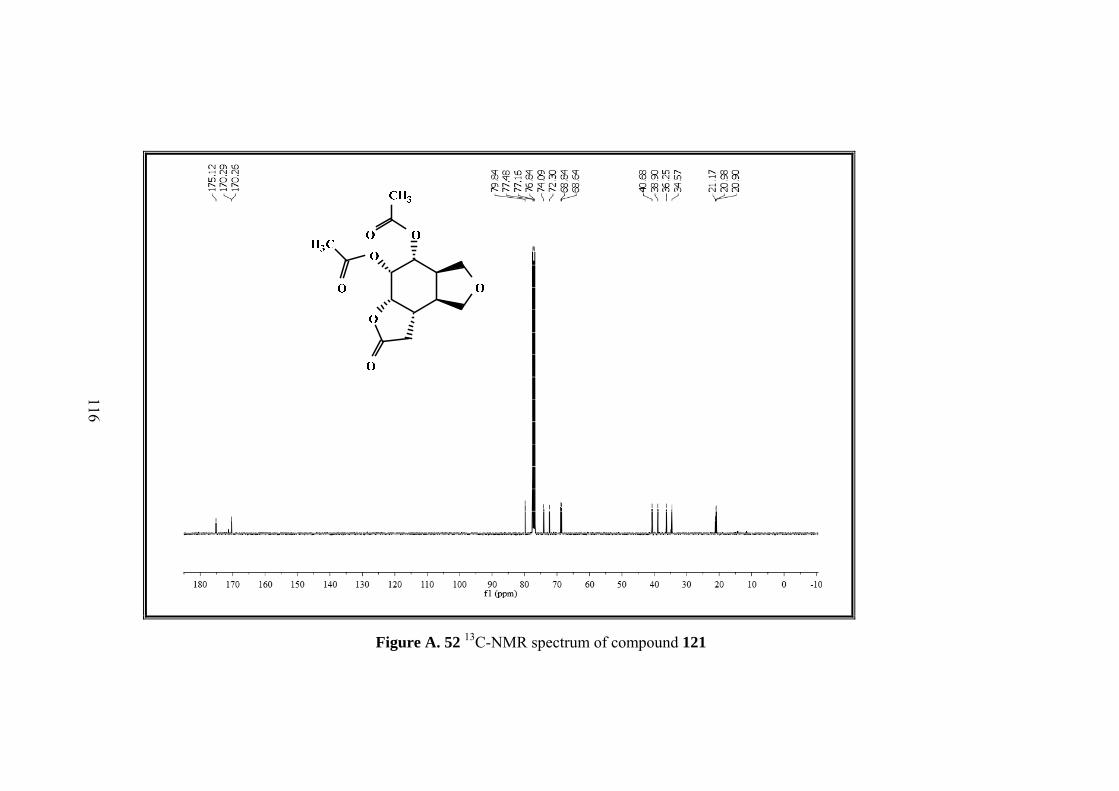
As shown in Figure 13 in the 1H-NMR of diacetate isomer 121, there are 9 different
signals corresponding to 18 protons. 13C-NMR of the molecule consists of 13
different carbons. With the help of the COSY, HMBC and HSQC all proton and
carbon resonances were assigned. The proton that resonates as doublet of doublets at
the lowest field (5.60 ppm), belongs to H-2 neighboring the two oxygen atoms. The
Page 63
49
multiplet at 4.7 ppm belongs to different methine protons H-1 and H-6. The
methylenic protons of the furan ring give 3 different signals: H-9 at 3.96 as triplet, H-
9’, H-7 between 3.82-3.75 ppm as multiplet and H-7’ at 3.45 ppm as triplet. Two
methine protons H-3 and H-4 at the intersections of the rings resonate at 2.7 ppm as
multiplets whereas H-5 resonates at the same region with the methylenic protons H-
12 and H-12’ between 2.60-2.51 ppm. Methyl protons of the acetate groups resonate
as singlets at 2.03 and 1.97 ppm.
2.4.5 Reduction of Lactone in Diacetate Isomers 120 & 121 by LiAlH4
After characterization and isolation of diacetate isomers 121 & 122 we planned to
perform the reduction of the lactone unit in the isomers followed by acetolysis and
hydrolysis reactions to get the corresponding inositol derivatives. However we could
not complete because of the time limitation.
Scheme 33. Acetolysis and hydrolysis reactions of the compounds 120 and 121
Page 64
50
CHAPTER 3
EXPERIMENTAL
3.1 General
Nuclear magnetic resonance (1H-NMR and 13C-NMR) spectra were recorded on a
Bruker Instrument Avance Series-Spectrospin DPX-400 Ultrashield instrument in
MeOD and CDCl3 with TMS as internal reference. Chemical shifts (δ) were
expressed in units parts per million (ppm). Spin multiplicities were specified as
singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets
(ddd) triplet (t) and multiplet (m) and coupling constants (J) were reported in Hertz
(Hz).
Infrared spectra were recorded on a Matson 1000 FT-IR spectrometer and Vertex 70
series FT-IR spectrometer. Band positions were reported in reciprocal centimeters
(cm-1).
Elemental analysis was performed at Atatürk University.
Column chromatographic separations were performed by using Fluka Silica Gel 60
plates with a particle size of 0.063–0.200 mm. Thin layer chromatography (TLC)
was performed by using 0.25 mm silica gel plates purchased from Fluka.
Compounds were named by using ChemDraw Ultra 11.0.
Solvents were purified as reported in the literature.37
Page 65
51
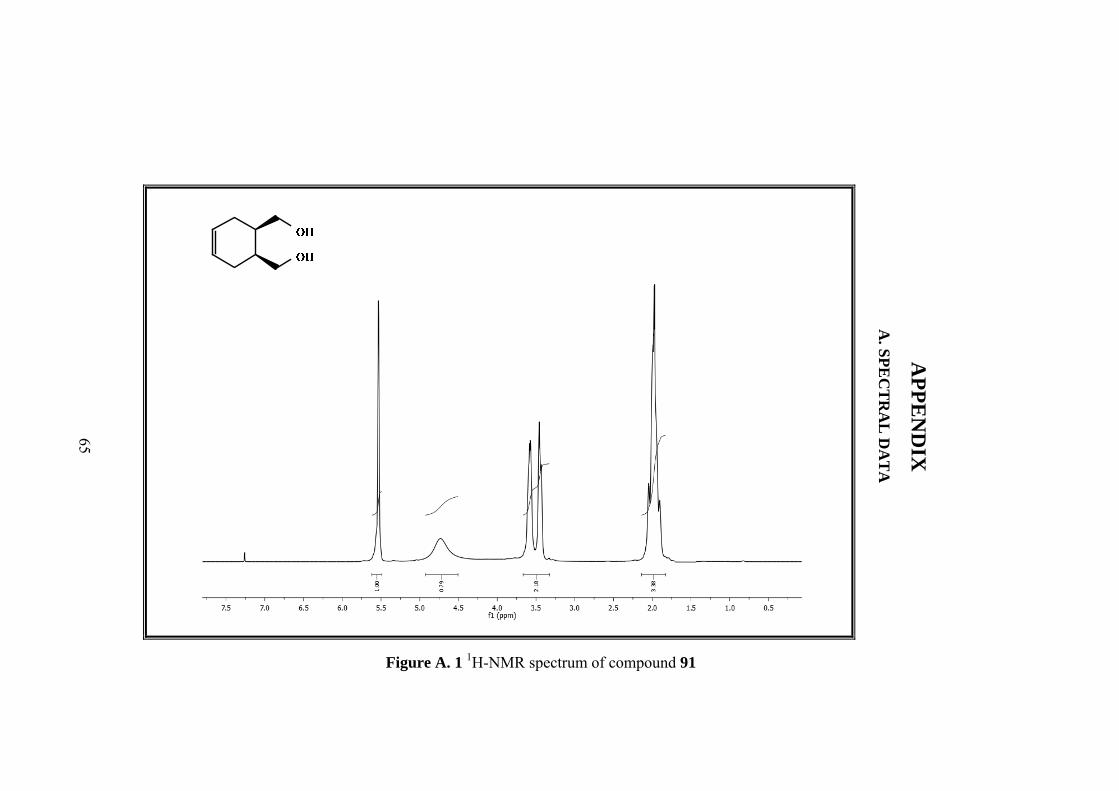
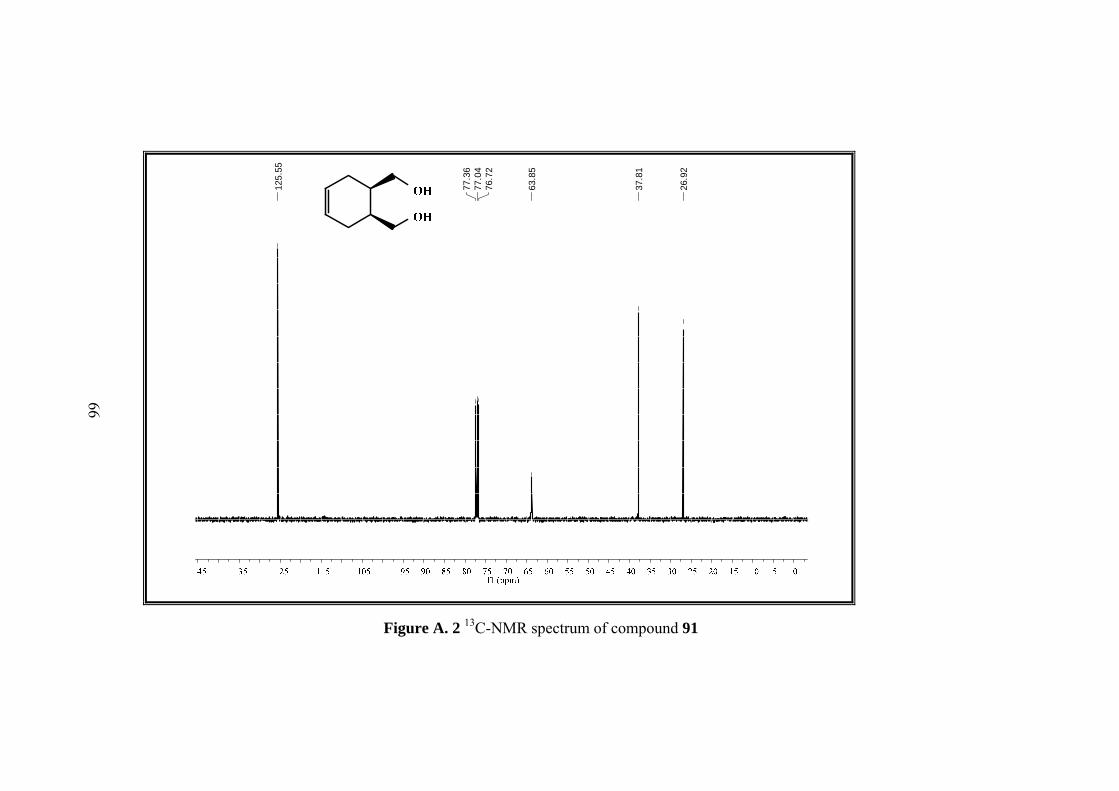
3.2 Synthesis of cis-1,2,3,6,-Tetrahydrophthalyl Alcohol (91)
The cis-1,2,3,6-tetrahydrophytalic anhydride (30 g, 0.20 mol) was dissolved in 158
mL of dry THF, and then the resulting solution was added dropwise to a stirring
mixture of LiAlH4 (8.4 g,0.22 mol) in 50 mL THF in an ice-bath. After the addition,
the mixture was refluxed for 24 h. Then saturated Na2SO4 solution was added to
hydrolyze till the mixture turned to white. When the mixture was cooled to room
temperature, it was suction-filtered and washed with MeOH. The filtrate was
cextracted with EtOAc and the extracts were dried over MgSO4. After the removal of
the solvent, colorless diol 91 was obtained. (24.16 g, 85% yield)
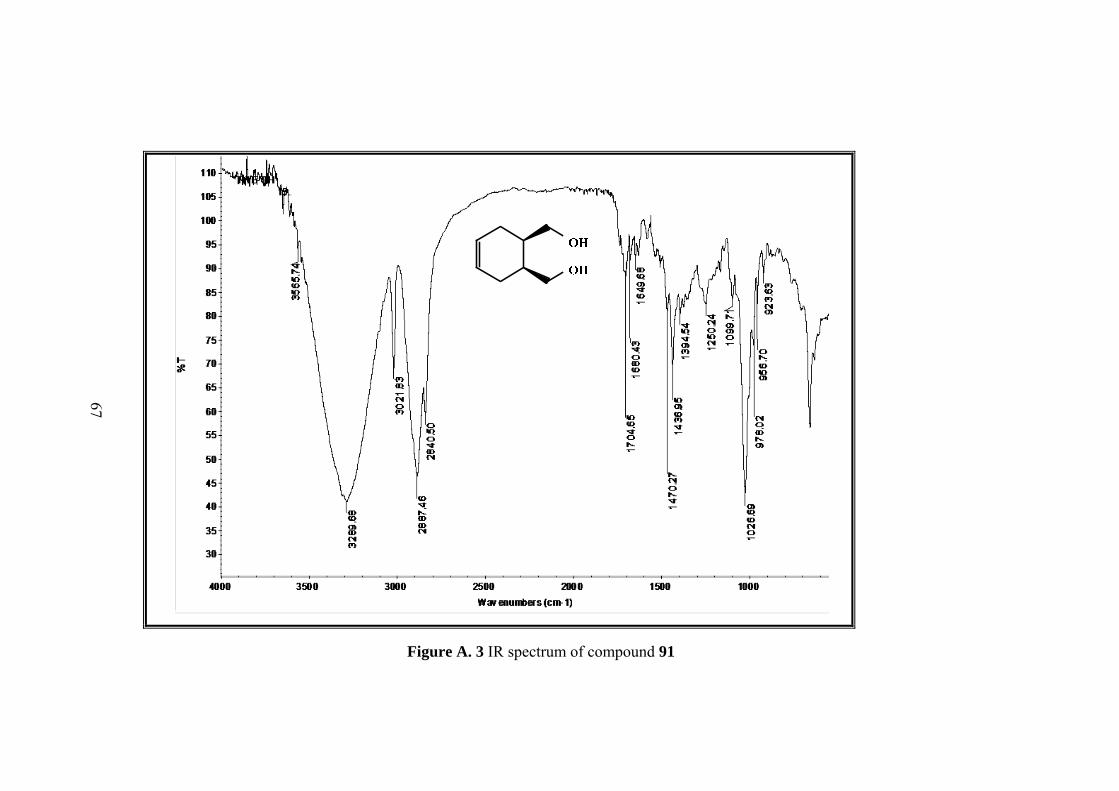
1H NMR (400 MHz, CDCl3) δ: 5.56 (s, 2H, H-4,H-5) 3.61 (m,
2H, H-7,H-9) 3.48 (m, 2H,H-7’,H-9’), 2.07-1.93 (m, 6H, H-1,H-
2,H-3,H-3’,H-6,H-6’). 13C-NMR (100 MHz, CDCl3) : 125.6, 63.9, 37.8, 26.9.
IR (ATR): 3565, 3289, 3021, 2887, 2840, 1704, 1680, 1649, 1470, 1436, 1250,
1099, 1016, 978, 946, 923.
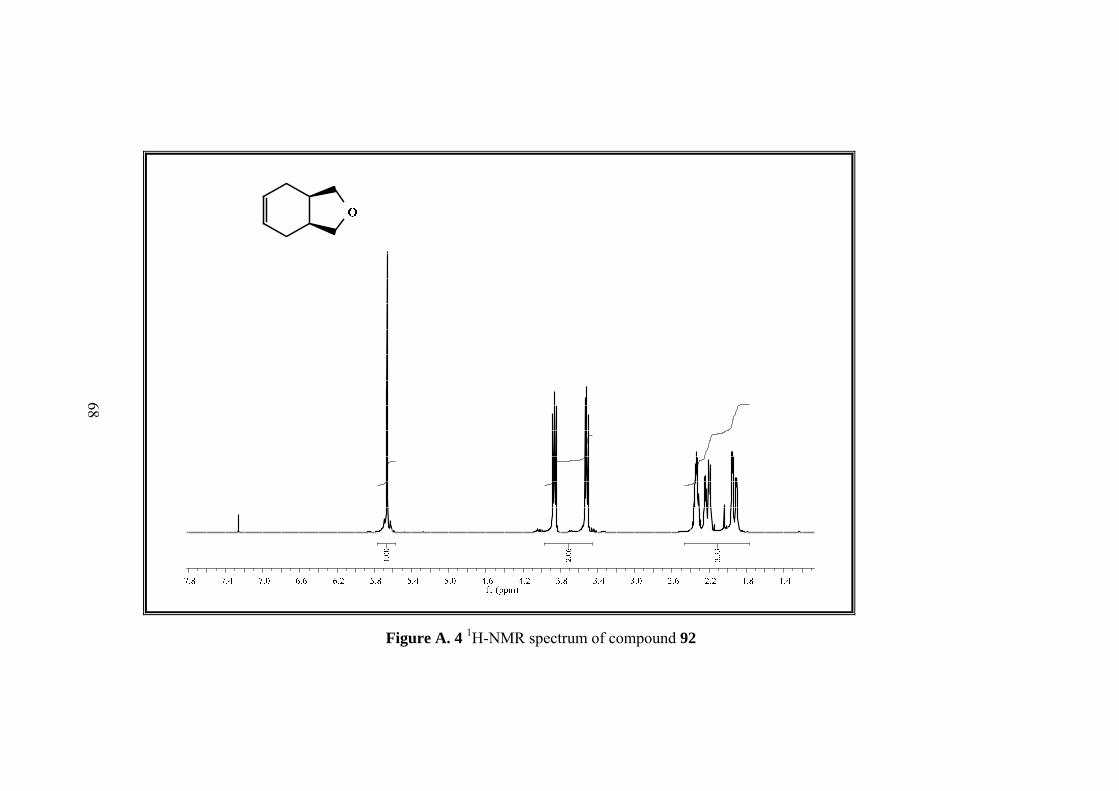
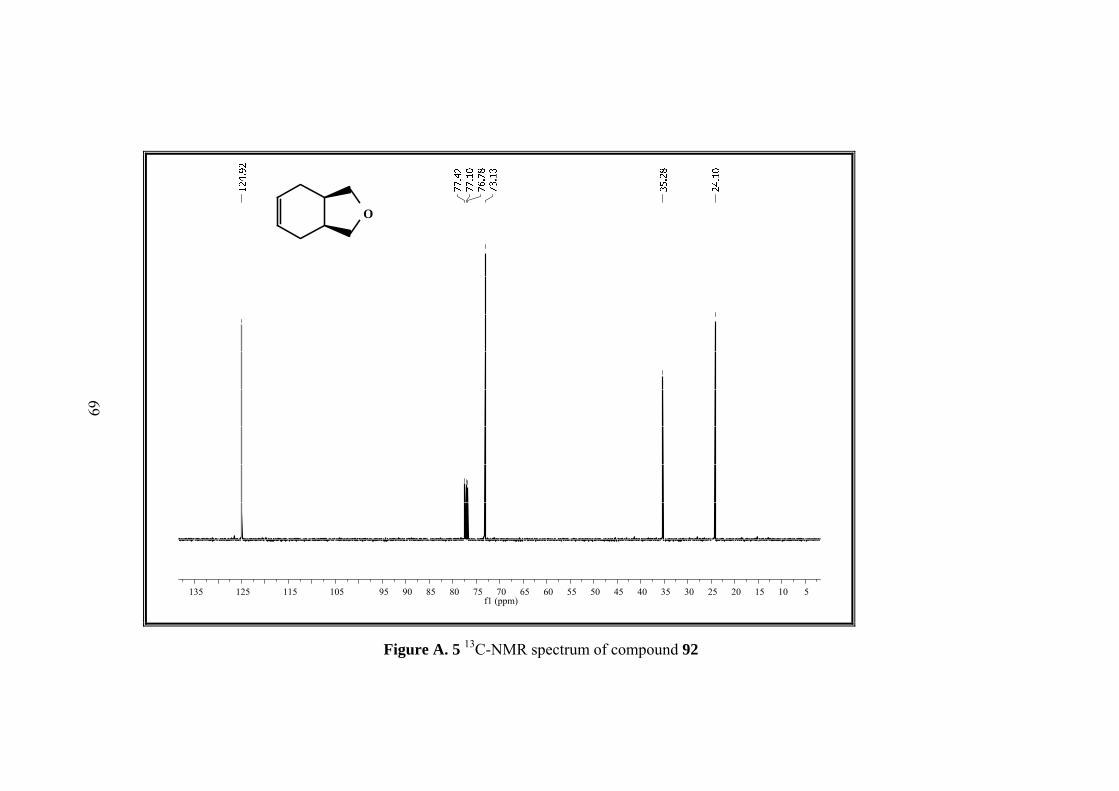
3.3 Synthesis of rel-(1R, 3S)-1,3,3a,4,7,7a-Hexahydro-2-benzofuran (92)
The tosyl chloride (44.82g, 0.24 mol) was dissolved in 64 mL pyridine, and added
dropwise to the refluxing solution of diol 91 (28g, 0.19 mol) in 45 mL pyridine.
After addition was completed, the mixture was refluxed for additional 4 h. When the
mixture was cooled to room temperature, it was poured into H2SO4 ice bath for
neutralization of the pyridine. The mixture was extracted with water and Et2O and
extracts dried over MgSO4. After solvent was evaporated, the crude product
submitted to vacuum distillation for purification resulting in colorless rel-(1R,3S)-
1,3,3a,4,7,7a-hexahydro-2-benzofuran (92).(15.1g, 62%).
5
43
2
16
9
OH10
OH 8
7
Page 66
52
1H-NMR (400MHz,CDCl3) δ: 5.63 (s, 2H, H-5, H-6), 3.82 (dd, A
part of AB-system, J = 7.8Hz and 6.3 Hz, 2H, H-1,H-3), 3.47 (dd,
B part of AB-system, J = 7.8Hz and 5.8 Hz, 2H, H-1’, H-3’), 2.29
(m, 2H, H-3a, H-7a), 2.20-2.13 (m, 2H, H-4, H-7), 1.91-1.85 (m, 2H, H-4’,H-7’) 13C-NMR (100 MHz, CDCl3) 124.9, 73.1, 35.3, 24.1
IR (ATR): 3026, 2970, 2928, 2860, 2842, 2244, 1437, 1088, 1052, 1007, 968, 941,
907, 882, 728.
3.4 Synthesis of rel-(3aR,5R,6S,7aS)-5,6-Dibromooctahydro-2-benzo furan (94)
A solution of bromine (19.0 g, 0.12 mol) in 65 mL of CH2Cl2 was added dropwise to
a magnetically stirred solution of furan 92 (15.0 g, 0.12 mol) in 70 mL of CH2Cl2 at
0 °C. After the mixture was stirred for an additional 2 h at room temperature, the
reaction was stopped. The excess bromine was quenched with saturated Na2S2O5
solution; organic layer was taken and dried over Mg2SO4. The solvent was removed.
Crystallization of the residue from CH2Cl2 at 0 °C gave pure white crystals 93 (24.5
g, 72%). Mp: 58 – 60 oC.
1HNMR (400MHz, CDCl3) δ 4.42-4.39 (m, 1H, H-6), 4.23-4.19
(m, 1H, H-5), 3.88-3.75 (m, 3H, H-3, H-3, H-1’), 3.69-3.58 (m,
1H, H-1), 2.61-2.46 (m, 3H,H-4,H-7,H-7’), 2.42-2.36 (m,1H,H-
4’, 2.34-2.28 (m, 1H), 2.12-1.98 (m, 1H)
13C NMR (100 MHz, CDCl3) δ 72.4, 70.2, 53.4, 38.3, 37.2, 34.4, 33.2
IR (ATR) 2928, 2861, 1443, 1247, 1141, 1056, 1031, 1030, 1002, 978, 944, 905,
884, 751, 680.
5
67
7a
3a4
1
O 2
3
5
67
7a
3a4
1
O 2
3Br8
Br9
Page 67
53
3.5 Synthesis of rel-(3aR,7aS)-1,3,3a,7a-Tetrahydro-2-benzofuran (69)
To a stirred solution of dibromide 93 (14.0 g, 0.08 mol) in 50 mL benzene was added
a solution of 1,8-diazabicyclo[5.4.0]undec-7-ene (19.0 g, 0.123 mol) in 20 mL of
benzene at room temperature. The reaction mixture was refluxed for 24 h and then
cooled to room temperature. The mixture suction filtered, resulted solid washed with
ether. The filtrate washed with saturated aqueous sodium bicarbonate and dried over
Mg2SO4. After the removal of the solvent, vacuum distillation was performed to
give the colorless liquid 69. (6.24 g, 64 %)
1H-NMR (400 MHz, CDCl3) δ 5.83-5.80 (m, 2H, H-4,H-7), 5.60-5.57
(m, 2H, H-5,H-6), 4.15-4.10 (m, 2H, H-1, H-3), 3.59-3.55 (m, 2H, H-
1’, H-3’), 2.94 (bs, 2H, H-3a, H-7a).
13C NMR (100 MHz, CDCl3) δ 126.2, 122.3, 75.0, 37.8
IR (ATR) 3035, 2932, 2861, 2247, 1633, 1479, 1374, 1078, 905, 726, 679, 674
3.6 Synthesis of rel-(1R,2R,6S,7S)-4,10,11-trioxa-tricyclo[5.2.2.02,6]undec-8-ene
(94)
The cyclohexadiene derivative 69 (5.0g, 0.04 mmol) and 100 mg of
tetraphenylporphyrine (TPP) were dissolved in 250 mL of CH2Cl2. The mixture was
irradiated with a projection lamp (500 W) while oxygen was passed through the
solution. After 12 h at room temperature, the reaction was stopped and solvent was
removed. The resulting crude product was purified by column chromatography on
silica gel (70g) eluting with hexane/CH2Cl2 (7:3). Eluted solvent was evaporated.
Crystallization of the residue from hexane/CH2Cl2 (7:3) gave 4.9 g of pure
endoperoxide 94 (81%) as colorless crystals. Mp: 123-126 oC.
5
67
7a
3a4
1
O 2
3
Page 68
54
1H-NMR (400 MHz, CDCl3) δ 6.62 (quasi t, A-part of AA′XX′-
system, 2H, H-8, H-9), 4.65 (m, X-part of AA′XX′-system, 2H, H-1,
H-7), 3.69-3.64 (m, 2H, H-3, H-5), 3.45 (dd, J 33′(55′) = 9.3 and
J23′(65′) = 2.6 Hz, 2H, H-3’, H-5’), 2.97 (m, 2H, H-2, H-6)
13C NMR (100 MHz, CDCl3) δ 131.9, 72.5, 70.0, 39.9
IR (ATR) 3079, 2948, 2922, 2859, 2844, 1376, 1276, 1258, 1197, 1129, 1101,
1074, 1039, 1019, 1075, 1030, 1019, 965, 949, 905
3.7 Synthesis of rel-(1aR,1bS,2aS,2bS,5aR,5bR)-octahydrobis (oxireno)
isobenzofuran (100)
To a stirring solution of endoperoxide 94 (2.2g, 0.014mol) in 30 mL CH2Cl2 was
added the solution of 0.058 g cobalt tetraphenylporphyrine (CoTPP) in 25 mL of
CH2Cl2 at 0oC. After the addition was complete, the mixture was stirred for 2 h at
room temperature. Removal of the solvent gave the crude product which was
submitted to column chromatography on silica gel (50g) eluting with hexane/EtOAc
(2:3).The resulting pure bisepoxide 100 was recrystallized from chloroform (1.84g,
84%). Mp: 73-75 oC.
1H-NMR (400 MHz, CDCl3) δ 3.94-3.90 (m, 2H, H-9, H-3), 3.66-
3.62 (m, 2H, H-3’, H-5’), 3.41 (d, J = 2.7, 2H, H-1a, H-1b), 2.66-
1.60 (m, 2H, H-2a, H-5b), 2.97 (m, 2H, H-2b, H-5a)
13C NMR (100 MHz, CDCl3) δ 71.0, 50.5, 47.6, 30.9
IR (ATR) 3037, 3004, 2947, 2927, 2879, 1423, 1366, 1268, 1196, 1071, 1051,
1034,977, 970, 944,930, 894, 839
8
91
2
67
3
O 4
5
O 11O10
23
55a5b
1a
2a
O2b
1b
O1
O
4
Page 69
55
3.8 Synthesis of rel-(1R,5S,6R,7S,8S,9S)-3-oxabicyclo[3.3.1]nonane-6,7,8,9-
tetrayl tetraacetate (103)
The bisepoxide (300mg, 1.94 mmol) was 100 dissolved in 6 mL of acetic anhydride.
A solution of H2SO4 (6-7 drops) in 3 mL acetic anhydride, was added dropwise to
bisepoxide solution. The reaction mixture stirred for 24 h at room temperature. When
reaction was completed, the reaction mixture was washed with saturated NaHCO3
solution. Organic phase was extracted with EtOAc (3 x 20 mL) and dried over
Mg2SO4. The solvent was evaporated, and the residue (54% was submitted to column
chromatography on silica gel (85g). Elution with hexane/EtOAc (3:1) gave single
product 3-oxabicyclo [3.3.1]nonane-6,7,8,9-tetrayl tetraacetate 103. Recrystallization
of tetraacetate from hexane/ EtOAc (1:4) gave colorless crystals. Mp: 164-166 oC.
1H-NMR (400 MHz, CDCl3) δ 5.75-5.71 (dd, J78 = 10.3
Hz, J76 = 5.4 Hz, 1H, H-7), 5.58-5.54 (dd, J78 = 10.3Hz,
J81= 4.6Hz, 1H, H-8), 5.51-5.49 (ddd, J76 = 5.4Hz, J65 =
2.3 Hz, J69 = 0.9Hz, 1H, H-6), 4.96 (bt, J = 2.7 Hz, 1H, H-9), 4.14 (bd, A-part of AB
system, J22’ = 11.4 Hz, 1H, H-2), 3.94 (d, A-part of AB system, J44’ = 12.1Hz, 1H,
H-4), 3.62 (dd, B-part of AB system, J4’5 = 2.2 Hz, J44’ = 12.1 Hz, 1H, H-4’), 3.54
(d, B-part of AB system, J22’= 11.4 Hz, 1H, H-2’), 2.43 (m, 1H, H-5), 2.29 (m, 1H,
H-1), 2.08 (s, 6H, COCH3), 2.01 (s, 3H, COCH3), 1.92 (s, 3H, COCH3)
13C NMR (100 MHz, CDCl3) δ 170.2, 170.1, 169.9, 169.87, 72.8, 71.6, 69.5, 68.4,
66.5, 39.6, 38.7, 21.2, 21, 20.9, 20.7
IR (ATR) 2978, 2856, 1735, 1369, 1216, 1171, 1132, 1114, 1035, 975, 948, 909,
862
Anal. Calcd for C16H22O9: C, 53.63; H, 6.19 Found: C, 53.55; H, 6.07
79
18
O3
OCOCH3
6 5H3COCO
H3COCOOCOCH3
4
2
Page 70
56
3.9 Synthesis of rel-(1R,5S,6R,7S,8S,9S)- 3-oxabicyclo [3.3.1]nonane-6,7,8,9-
tetraol (105)
Tetraacetate 103 (100 mg, 0.28 mmol) was dissolved in 10 mL methanol. The
solution was stirred at room temperature for 24 h while NH3(g) passed through the
solution. After the evaporation of the solvent and the removal of the acetamide which
was formed during the reaction, tetrol was obtained (46 mg, 86%).
1H-NMR (400 MHz, MeOD) δ 4.27-4.15 (m, 5H, H-2, H-6, H-7,
H-8, H-9), 3.88-3.85 (d, A-part of AB system, J44’=11.8 Hz, 1H,
H-4) 3.65-3.62 (dd, B-part of AB system, J44’ = 11.8 Hz, J4’5 = 2.2
Hz, 1H, H-4’), 3.51-3.48 (bd, B-part of AB system, J22’ =11.6 Hz, 1H, H-2’), 2.29
(bs, 1H, H-5), 2.16 (bs, 1H, H-1)
13C-NMR (100 MHz, MeOD) δ 76.64, 74.84, 74.05, 70.56, 69.56, 66.94, 46.46,
43.22
IR (ATR) 3264, 3356, 2921, 2856, 1659, 1394, 1259, 1142, 1127, 1101, 1059, 970
3.10 Synthesis of rel-(3aR,4S,5R,6R,7S,7aS)-octahydro-2-benzofuran-4,5,6,7-
tetrayl tetraacetate (102)
The bisepoxide (2 g, 0.013 mol) 100 dissolved in 10 mL of water. After the addition
of H2SO4 (5 drops) was added to the solution, the reaction mixture stirred for 24 h at
room temperature. The solvent was evaporated, and the residue dissolved in 6 mL of
pyridine, then 9 mL of acetic anhydride was added to the solution. The mixture
stirred at room temperature for 24 h. When the reaction was completed, the reaction
mixture was poured into HCl-ice bath. The aq. phase was extracted with EtOAc (3 x
50 mL) and saturated NaHCO3 solution. The organic phase was dried over Mg2SO4.
7
9
56
O3
OH8 1
HO
HO OH
2
4
Page 71
57
65
43a
7a7
3
O 2
1
O
O
O
O
O
O
O
O
Evaporation of the solvent gave the mixture of three isomers, the mixture was
submitted to column chromatography on silica gel (120 g). Elution with
hexane/EtOAc (4:1) gave as major product rel-(3aR,4S,5R,6R,7S,7aS)- octahydro-2-
benzofuran-4,5,6,7-tetrayl tetraacetate (102) with 67% ratio, which was
recrystallized from hexane/EtOAc (1:4) resulting white crystals.
Mp: 133-135 oC.
1H-NMR (400 MHz, CDCl3) δ 5.29-5.25 (dd, J45 = 9.5Hz, J56
= 9.1Hz, 1H, H-5), 5.24-5.21(dd, J45 = 9.5 Hz, J43a = 6.0Hz,
1H, H-4), 5.14-5.09 (t, J77a = J76 = 10Hz, 1H, H-7), 5.06-5.02
(dd, J67 = 10 Hz, J56 = 9.1Hz, 1H, H-6) 3.83 (d, J33’= 9.7 Hz,
2H, H-3, H-3’) 3.75(d, A part of AB system, J11’=8.9 Hz,1H,
H-1), 3.70-3.67(dd, B part of AB system, J1’7a = 4.4Hz,
J11’=8.9 Hz, 1H, H-1’), 3.02-2.94 (m, 1H, H-3a), 2.38-2.33 (m, 1H, H-7a), 1.97 (s,
6H, COOCH3), 1.94(s, 3H, COOCH3),1.93 (s, 3H,COOCH3)
13C-NMR (100 MHz, CDCl3) δ 169.9, 169.8, 169.7, 169.6 72.8, 71.1, 70.93, 70.3,
70.0, 67.4, 42.2, 40.5, 20.7, 20.68, 20.57, 20.53
IR (ATR) 2944, 2918, 2882, 1745, 1734, 1456, 1367, 1265, 1215, 1114, 1053, 1033,
975, 940, 923, 894
Anal. Calcd for C16H22O9: C, 53.63; H, 6.19 Found: C, 53.5; H, 5.94
3.11 Manganese (III) Acetate Oxidation Reaction of rel-(3aR,7aS)-1,3,3a,7a-
Tetrahydro-2-benzofuran (69)
Manganese (III) acetate, potassium acetate and diene 69 (2g, 0.016 mol) were
dissolved in 300 mL acetic acid. The mixture was refluxed at constant temperature
(90 oC) under nitrogen atmosphere until it turned to white. When the reaction was
completed, the mixture was washed with CH2Cl2 (2 x 100mL), water (2 x 100mL)
Page 72
58
and saturated NaHCO3 solution (2 x 50 mL). The solution was dried over MgSO4
and purified by column chromatography on silica gel (70 g) eluting with hexane /
EtOAc (7:3). After removal of the solvent, residue (1.8g, 61%) was recrystallized
from EtOAc resulting rel-(3aR,5aR,8aS,8bR)-1,3,3a,8,8a,8b-hexahydrobenzo[1,2-
b:3,4-c’]difuran-7(5aH)-one (115). Mp: 142-144 oC.
1H-NMR (400 MHz, CDCl3) δ 5.91-5.88 (ddd, J45 = 10.2 Hz,
J43a = 3.7 Hz, J45a = 0.7 Hz, 1H, H-4), 5.84-5.80 (ddd, J54 =
10.2 Hz, J55a = 3.1 Hz, J53a = 1.9 Hz, 1H, H-5) 4.82 (m, 1H,
H-5a), 3.97-3.93 (dd, A part of AB system, J33a = 7.6 Hz, J33’
= 8.5 Hz, 1H, H-3), 3.92-3.88 (dd, A part of AB system, J11’=
8.4Hz, J18b= 7.4 Hz, 1H, H-1), 3.57-3.55 (dd, B part of AB system, J3’3a = 4.9 Hz,
J33’ = 8.5 Hz, 1H, H-3’), 3.55-3.51 (dd, B part of AB system, J11’= 8.4Hz, J1’8b = 6.4
Hz, 1H, H-1’), 2.8 (m, 1H, H-3a), 2.71-2.63 (B part of ABC system, Jac = 7.4 Hz,
1H, H-8a), 2.61-2.58 (A part of ABC system, Jbc = 8.1 Hz, 1H, H-8), 2.41 (p, J = 6.5
Hz , 1H, H-8b), 2.36-2.30 (C part of ABC system, Jab = 16.5 Hz, 1H, H-8’)
13C-NMR (100 MHz, CDCl3) δ 174.6, 131.2, 122.7, 72.9, 71.8, 69.8, 37.4, 35.6,
33.4, 32.5
IR (ATR) 2929, 2871, 1752, 1370, 1229, 1176, 1057, 1035, 1001, 972, 942, 883,
758
Found HRMS [M+H]+ calcd for C10H12O3 ;Exact mass:180.07864; Found:
180.08054
1
2
345
7
O
8
O
O
6
3a
5a
8a8b
Page 73
59
3.12 cis-Hydroxylation of rel-(3aR,5aR,8aS,8bR)-1,3,3a,8,8a,8b Hexahydro
benzo[1,2-b:3,4-c’]difuran-7(5aH)-one (115)
To 700 mg (0.039 mol) 115 in 14 mL acetone/water (1:1), 1.5g (13.1mmol) NMO
and 3mg (0.012mmol) OsO4 were added at 0oC. The mixture stirred at room
temperature for 72 h under nitrogen atmosphere. The reaction was stopped, and pH
of the solution adjusted to 2 with HCl. After the removal of the solvent, pyridine (7.5
mL) and Ac2O (12 mL) were added and the reaction mixture stirred for 24 h at room
temperature. When the reaction was completed, reaction mixture hydrolyzed by HCl-
ice solution, extracted with EtOAc (2x100 mL) and washed with NaHCO3 (2x100
mL). Evaporation of the organic phase gave the mixture of 2 diacetate isomers with
(1:1) ratio. The residue was submitted to column chromatography with silica gel
(70g). Elution with hexane/EtOAc (7:3) gave as the first isomer rel-
(3aR,4R,5S,5aS,8aS,8bS)-7-oxodecahydrobenzo[1,2-b:3,4-c’]difuran-4,5-diyl
diacetate 120, and as second isomer rel-(3aR,4S,5R,5aS,8aS,8bS)-7-
oxodecahydrobenzo[1,2-b:3,4-c’]difuran-4,5-diyl diacetate 121 separately.
Recrystallization of the products from EtOAc provided white crystals. Mp: 178-180 oC.
1H-NMR (400 MHz, CDCl3) δ 5.50-5.48 (dd, J55a= 1.9
Hz, J5a8a = 5.4Hz, 1H, H-5a), 4.90-4.87 (dd, J55a = 1.9Hz,
J54= 6.5 Hz, 1H, H-5), 4.70-4.67 (bt, J = 6.5 Hz, 1H, H-4),
3.79-3.75 (dd, A part of AB system, J33’ = 9.1 Hz, J33a=
6.7 Hz, H-3), 3.74-3.72 (bd, A part of AB system, J =
9.2Hz, 1H, H-1), 3.62-3.58 (dd, B part of AB system, J11’
= 9.2 Hz, J1’8b = 5.13 Hz, 1H, H-1’), 3.56-3.53 (dd, B part
of AB system, J33’ = 9.1 Hz, J3’3a= 3.4 Hz, 1H, H-3’), 2.81-2.70 (m, 3H, H-8, H-8a,
H-8b), 2.30-2.15 (m, 2H, H-8’, H-3a), 2.06 (s, 3H, H-9), 2.04 (s, 3H, H-10)
13C-NMR (100 MHz, CDCl3) 174.9, 170.14, 170.11, 79.7, 73.9, 72.2, 68.7, 68.5,
40.5, 38.8, 36.1, 34.4, 20.7, 20.8
IR (ATR) 2962, 2853, 1773, 1733, 1368, 1259, 1241, 1222, 1149, 1143, 1088, 1008,
948, 916, 839, 794
5
5a
8a8b
3a4
1
O2
3
O6
7 8
OCOCH3
H3COCO
O
9
10
Page 74
60
Anal. Calcd for C14H18O7: C, 56.37; H, 6.08 Found: C, 56.02; H, 6.15
1H-NMR (400 MHz, CDCl3) δ 5.60-5.58 (dd, J55a = 1.54
Hz, J5a8a = 3.9 Hz, 1H, H-5a), 4.73-4.67 (m, 2H, H-5, H-4
), 3.96 (t, J = 8.5Hz, 1H, H-3), 3.82-3.75 (m, 2H, H-3’,
H-1), 3.45 (t, J = 8.8 Hz, 1H, H-1’), 2.72-2.65 (m, 2H, H-
8a, H-8b) 2.60-2.51 (m, 3H, H-3a, H-8, H-8’), 2.03 (s,
3H, COCH3), 1.97 (s, 3H, COCH3)
13C-NMR (100 MHz, CDCl3) 174.4, 169.4, 168.3, 73.9, 69.88, 69.85, 68.1, 67.4,
38.7, 35.4, 33.2, 32.1, 19.7
IR (ATR) 3004, 1786, 1749, 1711, 1421, 1361, 1220, 1172, 1092, 1019, 904
Found HRMS [M+Na]+ calcd for C14H18O7; Exact mass: 298.10525; Found:
298.10846
9
10
55a
8a8b
3a4
1
O 2
3
O6
7 8
OCOCH3H3COCO
O
Page 75
61
CHAPTER 4
CONCLUSION
The development of various synthetic pathways for the bishomo- inositol derivatives,
which have potential to show biological activity, is important. Different bishomo-
inositol derivatives were synthesized from the same key compound with distict
pathways.
In the first part of the study, key compound 69 was subjected to photooxygenation to
form corresponding endoperoxide 94 which is further converted to epoxide 100.
Acid catalyzed ring opening reaction of bisepoxide was performed in two different
way; in the presence and absence of water. Reactions were followed by acetylation
for the characterization. In the presence of water rel-(3aR,4S,5R,6R,7S,7aS)-
octahydro-2-benzofuran-4,5,6,7-tetrayl tetraacetate (102) was formed as a major
product.
Scheme 34. Synthesis of tetraacetate 102
However, in the absence of water, single product was formed and characterized.
Unexpected rearrangement was occured during the reaction. Formation mechanism
of the rearranged product 103 was discussed.
Page 76
62
Scheme 35. Synthesis of tetraacetate 103
In the second episode, manganese acetate oxidation reaction of the key compound 69
gave the molecule 115 which was further subjected to Upjohn dihyroxylation. The
two isomers formed from the reaction were isolated and characterized.
Scheme 36. Synthesis of diacetate molecules 120& 121
In summary, synthesis of inositol derivatives were studied. Different methodologies
for the synthesis were investigated. The products were purified and characterized by
1D & 2D NMR. Spartan’08 mechanics program and NMR simulation programs
(ACD) were used for characterization.
Page 77
63
REFERENCES
1. Scherer, J. J.Lie. Ann. Chem. 1850, 73, 322.
2. Clements, R. S. Jr.; Darnell, B. Am. J. Clin. Nutr. 1980, 33, 1944.
3. Posternak, T. Cyclitols HERMANN, PARIS 1965, 76.
4. Ichimura, K.; Kohata, K.; Yamaguchi, Y.; Douzono, M.; Ikeda, H.; Koketsu, M.
Biosci. Biotechnol. Biochem. 2000, 64, 865.
5. Obendorf, R.L.; Horbowicz, M.; Dickerman, A.M.; Brenac, P.; Smith, M.E.;
Crop Science 1998, 38, 78.
6. Sherman, W. R.; Goodwin, S. L.; Gunnell, K. D. Biochemistry 1971, 10, 3491.
7. Sanz, M. L.; Sanz, J.; Martínez-Castro, I. Food Chem. 2004, 133.
8. Balci M.; Kılbas, B. Tetrahedron 2011, 67, 2355.
9. a) Levine, J. Am. J. Psychiat. 1994, 152, 792. b) Benjamin, J. Am. J. Psychiat.
1994, 152, 1084. c) Gelber, H. Int. J. Eat. Disord. 2001, 29, 345. d) Palatnik, A.
J. Clin. Psychopharm. 2001, 21, 335. e) Fux, M. Am. J. Psychiat. 1996, 153,
1219.
10. Ikura, K. Animal Cell Technology: Basic & Applied Aspects, Springer Science,
Business Media B.V. 2009, 15, 217.
11. Nitz, M. Bioorgan. Med. Chem. 2008, 16, 7177.
12. a) Berridge, M. J. Nature 1993, 361, 315. b) Streb, H.; Irvine, R. F.; Berridge,
M. J.; Schulz, I. J. Nature 1983, 306, 67.
13. Baran, A.; Günel, A.; Balci. M. J. Org. Chem. 2008, 73, 4371.
14. Baran, A.; Balci. M. J. Org. Chem. 2009, 74, 88.
15. Hudlicky, T.; Duchek, J.; Adams, D. R., Chem. Rev. 2011, 111, 4224.
16. Wieland, H.; Wishart, R. S. Ber. Dtsch. Chem. Ges. 1914, 47, 2082
17. Hudlicky, T.; Restrepo-Sanchez, N.; Kary, P. D.; Jaramillo-Gomez, L. M.
Carbohydr. Res. 2000, 324, 200.
18. Plettenburg,O.; Adelt, S.; Vogel G.; Altenbach, H-J. Tetrahedron-Asymmetr.
2000, 11,1057.
Page 78
64
19. Adelt, S.; Plettenburg,O.; Stricker, R.; Reiser, G.; Altenbach, H.-J.; Vogel, G. J.
Med. Chem. 1999, 42, 1262.
20. Takahashi, Y; Nakyama, H; Katagiri, K; Ito, N; Takita, T; Takeuchi, T; Miyake,
T. Tetrahedron Lett. 2001, 42, 1053.
21. Pistarà, V.; Barili, P.L.; Catelani, G.; Corsaro, A.; D’Andrea, F.; Fisichella, S.
Tetrahedron Lett. 2000, 41, 3253.
22. Barili, P. L.; Berti, G.; Catelani, G.; D’Andrea, F. Gazz. Chim. Ital. 1992, 122,
135.
23. Adelt, S.; Plettenburg, O.; Stricker, R.; Reiser, G.; Altenbach, H. J.; Vogel, G. J.
Med. Chem. 1999, 42, 1262.
24. Takahashi, H.; Kittaka, H.; Ikegami, S. J. Org. Chem. 2001, 66, 2705.
25. a) Lipta´k, A.; Jodal, I.; Nanasi, P. Carbohydr. Res. 1975, 44, 1. b) Pfitzner, K.
E.; Moffat, J. G. J. Am. Chem. Soc. 1963, 85, 3027. c) Takahashi, H.; Kittaka,
H.; Ikegami, S. J. Org. Chem. 2001, 66, 2705.
26. Sarmah, M. P.; Shashidhar, M. S. Carbohydr. Res. 2003, 338, 999.
27. Mehta, G.; Reddy, D. S. Tetrahedron Lett. 1999, 40, 9137.
28. Mehta,G.; Ramesh, S. S. Tetrahedron Lett. 2003, 44, 3105.
29. Kara,Y.; Balci, M. Tetrahedron 2003, 59, 2063.
30. Mahapatra, T.; Nanda, S. Tetrahedron-Asymmetr. 2010, 21, 2199.
31. Balcı, M. Chem. Rev., 1981, 81, 91.
32. a) Bush, J. B.; Finkbeiner, H. J. Am. Chem. Soc. 1968, 90, 5903. b) Heiba, E. I.;
Dessau, R. M.; Koehl, W. J. J. Am. Chem. Soc. 1968, 90, 590.
33. a) Fristad, W. E.; Peterson, J. R. J. Org. Chem. 1985, 50, 10. b) Fristad, W. E.;
Peterson, J. R.; Ernst, A. B. J. Org.Chem. 1985, 50, 3143. c) Fristad, W. E.;
Peterson, J. R.; Ernst, A. B.; Urbi, G. B. Tetrahedron 1986, 42, 3429.
34. Snider, B. Tetrahedron 2009, 65, 10735.
35. Adam, W.; Balci, M.; Kiliç, H. J. Org. Chem. 2000, 65, 5926.
36. Balci, M. Reaksiyon Mekanizmaları, TÜBA, 2008, 112.
37. Laurence, M. Polar Rearrangement, Harwood Oxford University Presss,
1991,7.
38. Caliskan, R.; Ali, M. F.; Sahin, E.; Watson, W. H.; Balci, M. J. Org. Chem. 2007, 72, 3353.
39. Balci, M.; Sacan, H.; Sutbeyaz, Y. J.Org. Chem. 1988, 53, 2312.
Page 79
Figure A. 1 1H-NMR spectrum of compound 91
AP
PE
ND
IX
A. S
PE
CT
RA
L D
AT
A
65
Page 80
Figure A. 2 13C-NMR spectrum of compound 91
26.9
2
37.8
1
63.8
5
76.7
277
.04
77.3
6
125.
55
66
Page 81
Figure A. 3 IR spectrum of compound 91
67
Page 82
Figure A. 4 1H-NMR spectrum of compound 92
68
Page 83
Figure A. 5 13C-NMR spectrum of compound 92
5101520253035404550556065707580859095105115125135f1 (ppm)
O
69
Page 84
Figure A. 6 IR spectrum of compound 92
70
Page 85
Figure A. 7 1H-NMR spectrum of compound 93
71
Page 86
Figure A. 8 13C-NMR spectrum of compound 93
-100102030405060708090100110120130140150160170180190200210f1 (ppm)
O
Br
Br
72
Page 87
Figure A. 9 IR spectrum of compound 93
O
Br
Br
73
Page 88
Figure A. 10 1H-NMR spectrum of compound 69
74
Page 89
Figure A. 11 13C-NMR spectrum of compound 69
75
Page 90
Figure A. 12 IR spectrum of compound 69
O
76
Page 91
Figure A. 13 1H-NMR spectrum of compound 94
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
4.55.05.56.06.5f1 (ppm)
2.93.13.33.53.73.9f1 (ppm)
OOO
77
Page 92
Figure A. 14 13C-NMR spectrum of compound 94
78
Page 93
Figure A. 15 IR Spectrum of Compound 94
79
Page 94
Figure A. 16 1H-NMR Spectrum of Compound 100
80
Page 95
Figure A. 17 13C-NMR Spectrum of Compound 100
0102030405060708090100110120130140150f1 (ppm)
O
O
O
81
Page 96
Figure A. 18 IR Spectrum of Compound 100
82
Page 97
Figure A. 19 DEPT-135 spectrum of compound 100
83
Page 98
Figure A. 20 COSY spectrum of compound 100
84
Page 99
Figure A. 21 HSQC spectrum of compound 100
85
Page 100
Figure A. 22 HMBC spectrum of compound 100
86
Page 101
Figure A. 23 1H-NMR spectrum of compound 103
87
Page 102
Figure A. 24 13C-NMR spectrum of compound 103
88
Page 103
Figure A. 25 IR spectrum of compound 103
89
Page 104
Figure A. 26 DEPT-135 spectrum of compound 103
-100102030405060708090100110120130140150160170180190200210f1 (ppm)
O
O O
CH3
O O
CH3
O
OCH3
O
OCH3
90
Page 105
Figure A. 27 COSY spectrum of compound 103
91
Page 106
Figure A. 28 HSQC spectrum of compound 103
92
Page 107
Figure A. 29 HMBC spectrum of compound 103
93
Page 108
Figure A. 30 1H-NMR spectrum of compound 105
94
Page 109
Figure A. 31 13C-NMR spectrum of compound 105
95
Page 110
Figure A. 32 IR spectrum of compound 105
O
OHOH
OH OH
96
Page 111
Figure A. 33 DEPT-135 spectrum of compound 105
97
Page 112
Figure A. 34 COSY spectrum of compound 105
98
Page 113
Figure A. 35 HSQC spectrum of compound 105
O
OHOH
OH OH
99
Page 114
Figure A. 36 HMBC spectrum of compound 105
100
Page 115
Figure A. 37 1H-NMR spectrum of compound 103
101
Page 116
Figure A. 38 13C-NMR spectrum of compound 103
102
Page 117
Figure A. 39 IR spectrum of compound 103
103
Page 118
Figure A. 40 DEPT-135 spectrum of compound 103
104
Page 119
Figure A. 41 COSY spectrum of compound 103
105
Page 120
Figure A. 42 HSQC Spectrum of Compound 103
106
Page 121
Figure A. 43 HMBC Spectrum of Compound 103
107
Page 122
Figure A. 44 1H-NMR spectrum of compound 114
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
5.855.905.956.00f1 (ppm)
3.53.63.73.83.94.04.1f1 (ppm)
2.32.42.52.62.72.82.9f1 (ppm)
O
O
O
108
Page 123
109
Figuure A. 45 13C-NM
MR spectrum of f compound 114
Page 124
Figure A. 46 IR spectrum of compound 114
110
Page 125
Figure A. 47 DEPT-135 spectrum of compound 114
111
Page 126
Figure A. 48 COSY spectrum of compound 114
112
Page 127
Figure A. 49 HSQC spectrum of compound 114
113
Page 128
114
Figure A. 50 HMBC spectrum of compound 116
Page 129
115
Figuure A. 51 1H-NM
MR spectrum of compound 121
Page 130
Figure A. 52 13C-NMR spectrum of compound 121
116
Page 131
Figure A. 53 IR spectrum of compound 121
117
Page 132
118
Figgure A. 54 DEP
T spectrum of coompound 121
Page 133
Figure A. 55 COSY spectrum of compound 121
119
Page 134
Figure A. 56 HSQC spectrum of compound 121
O
O
O
O
O
CH3OO
CH3
120
Page 135
Figure A. 57 HMBC spectrum of compound 121
121
Page 136
Figure A. 58 1H-NMR spectrum of compound 122
122
Page 137
Figure A. 59 13C-NMR spectrum of compound 122
123
Page 138
Figure A. 60 IR spectrum of compound 122
124
Page 139
125
Figurre A. 61 DEPT-
135 spectrum off compound 1222
Page 140
Figure A. 62 COSY Spectrum of Compound 122
O
O
O
O
OCH3
O
O
CH3
126
Page 141
Figure A. 63 HSQC Spectrum of Compound 122
127
Page 142
Figure A. 64 HMBC spectrum of compound 122
O
O
O
O
OCH3
O
O
CH3
128