1 SYNTHETIC METHODOLOGY DEVELOPMENT TOWARD BUILDING BLOCKS BEARING PENTAFLUOROSULFANYL (SF 5 ) GROUPS AND gem- DIFLUOROCYCLOPROPYL MOIETIES By ZHAOYUN ZHENG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2012
Transcript
1
SYNTHETIC METHODOLOGY DEVELOPMENT TOWARD BUILDING BLOCKS BEARING PENTAFLUOROSULFANYL (SF5) GROUPS AND gem-
DIFLUOROCYCLOPROPYL MOIETIES
By
ZHAOYUN ZHENG
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
1.2.1 Applications of the SF5 Group in Medicinal Chemistry ............................ 16
1.2.2 Applications of the SF5 Group in Agrochemistry ...................................... 18 1.2.3 Applications of the SF5 group in Functional Materials ............................. 19
1.3 Synthesis of Pentafluorosulfanyl Substituted Aromatic Rings ........................... 20 1.3.1 Synthesis of SF5-Benzene ....................................................................... 20 1.3.2 The Synthesis of SF5-Furan .................................................................... 23
1.3.3 Synthesis of SF5-Naphthalene................................................................. 24 1.3.4 Synthesis of SF5-Pyrazole and –Triazole ................................................ 25
2 THE PREPARATION OF PENTAFLUOROSULFANYL PYRROLE AND THIOPHENE THROUGH 1,3-DIPOLAR CYCLOADDITION ................................... 26
2.1 Initial Investigations of Synthetic Methods toward SF5-bearing Heterocycles ... 26 2.2 Preparation of SF5-pyrrole Carboxylic Acid Esters ............................................ 30
3.2 The design of the reaction ................................................................................ 57 3.3 Results .............................................................................................................. 60 3.4 Discussion ........................................................................................................ 64 3.5 Conclusion ........................................................................................................ 66 3.6 Experimental section ......................................................................................... 66
4 FACILE PREPARATION OF SF5-CONTAINING POLYMERS BY RING-OPENING METATHESIS POLYMERIZATION (ROMP) AND PRODUCT CHARACTERIZATION ........................................................................................... 71
4.1 Introduction ....................................................................................................... 71 4.2 Results and Discussion ..................................................................................... 72
1-4 SF5-substituted analogs of triflualin .................................................................... 18
1-5 The various applications of SF5 groups in functional materials ........................... 19
1-6 Early preparation method for SF5-benzene ........................................................ 20
1-7 First practical route to prepare SF5-benzene ...................................................... 21
1-8 Preparation of SF5-benzene from SF5Cl gas ...................................................... 21
1-9 Practical preparation of SF5-benzene developed by Umemoto .......................... 22
1-10 Preparation of SF5-furan through retro-Diels-Alder-reaction ............................... 23
1-11 Preparation of SF5-furan through Diels-Alder- and retro-Diels-Alder-reaction .... 24
1-12 Preparation of SF5-naphthalene ......................................................................... 24
1-13 Preparation of SF5-pyrazole and –triazole by 1,3-dipolar cycloaddition .............. 25
2-1 The first attempt synthetic route for SF5-pyrrole ................................................. 26
2-2 Second synthetic route to SF5-Heterocycles catalyzed by palladium ................. 27
2-3 Proposed mechanism for the synthesis of SF5-heterocycles .............................. 29
2-5 The preparation of SF5-Heterocycles based on cycloaddition chemistry ............ 30
2-6 Preparation CF3-pyrrole from azomethine ylide .................................................. 30
2-7 1,3-Dipolar cycloaddition approach to SF5-heterocycles .................................... 32
2-8 Removal of t-butyl group catalyzed by triflic acid ................................................ 32
2-9 Mechanism for the regioselective cycloaddition chemistry ................................. 33
2-10 Proton NMR of 2-8b ........................................................................................... 34
2-11 Proton NMR of 2-9b ........................................................................................... 34
9
2-12 Proton NMR of 2-10 ............................................................................................ 35
2-13 19F-NMR spectrum of compounds 2-10 and 2-9b ............................................... 35
2-14 The high reactivity of azomethine ylide building block 2-11 ................................ 36
2-15 Removal of TIPS group from 2-14f ..................................................................... 39
2-16 Removal of the benzyl group from dihydropyrrole .............................................. 40
2-18 Construction of thiophene through thiocarbonyl ylide ......................................... 41
2-19 1H-NMR of 2-20d ................................................................................................ 42
2-20 1H-NMR of 2-21d ................................................................................................ 43
2-21 19F-NMR of 2-20d and 2-21d .............................................................................. 43
3-1 The reactivity of TFDA and its reaction mechanism ........................................... 54
3-2 Reaction of TFDA with α,β-unsaturated ketones ................................................ 55
3-3 Friedel-Crafts reaction of difluorocyclopropanecarbonyl chloride ....................... 56
3-4 Attempt to prepare substituted difluorocyclopropane ketones ............................ 57
3-5 Preparation of difluorocyclopropenyl ketone and its properties .......................... 57
3-6 Reactivity difluorocyclopropyl ketone with HBr in Ionic Liquid. ........................... 58
3-7 Proposed tautomerization mechanism for β-difluoro enols/enolates .................. 58
3-8 Synthetic approach to substituted difluorocyclopropyl ketones .......................... 59
3-9 Application of HEH as hydride donor .................................................................. 59
3-10 Reactions of difluorocyclopropene with HEH, catalyzed by Brӧnsted acid ......... 63
3-11 Proposed mechanism for the catalytic reduction of difluorocyclopropenyl ketones ............................................................................................................... 64
3-12 Kinetic control reaction ....................................................................................... 65
Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
SYNTHETIC METHODOLOGY DEVELOPMENT TOWARD BUILDING BLOCKS
BEARING PENTAFLUOROSULFANYL (SF5) GROUPS AND gem-DIFLUOROCYCLOPROPYL MOIETIES
By
Zhaoyun Zheng
December 2012
Chair: William R. Dolbier, Jr. Major: Chemistry
Pyrrole and thiophene derivatives bearing a pentafluorosulfanyl (SF5) group were
unknown. Utilizing cycloaddition reactions of azomethine ylide with SF5-alkynes, a
series of SF5-pyrrole carboxylic acid esters were prepared in good yield. Further smooth
processes of SF5-alkynes with N-Benzyl-N-(methoxymethyl)-N-(trimethylsilylmethyl)-
amine initiated by triflic acid demonstrated that 1,3-dipolar cycloadditions are a general
approach to construct heterocyclic compounds containing the SF5 group. These
reactions were subsequently extended to prepare SF5-thiophene derivatives.
A novel Brӧnsted acid catalyzed synthetic method for preparation of substituted
gem-difluorocyclopropenyl ketones was designed based on the known tautomerization
mechanism of β-difluoro enols. The reaction, using Hantzsch ester (HEH) as a hydride
transfer reagent, proved to be a general route for preparation of substituted gem-
difluoro-cyclopropyl ketones in high yield. The reaction unexpectedly proceeded to give
largely cis product. Based upon the proposed mechanism, the diastereoselectivity could
be improved by using a more bulky Brӧnsted acid under optimized conditions.
14
A facile method was established for preparing polymers with SF5 group directly
attached to the backbone through ring opening metathesis polymerization (ROMP) of
SF5-substituted cyclooctene followed by hydrogenation. The microstructure of these
novel polymers were well characterized by 1H-NMR, GPC and 19F-NMR. TGA and DSC
experiments showed that the unsaturated polymers and their hydrogenated derivatives
have similar thermal profiles. While P3 and P4 have better thermal stabilities than P1
and P2, the latter pair exhibit higher glass transition temperatures.
15
CHAPTER 1 AN INTRODUCTION TO THE SYNTHESIS OF PENTAFLUOROSULFANYL(SF5)-
CONTAINING AROMATIC COMPOUNDS
1.1 Introduction
Because of its small size, high electronegativity and its ability to form hydrogen
bonds, the fluorine atom can dramatically affect the chemical and physical properties of
organic compounds.1 For example: fluorinated macromolecules usually exhibit low
surface energy, low dielectric constants and high chemical stability;2 fluorinated small
bioactive molecules often display amazingly enhanced biophysical activity by a
combination of factors such as increasing metabolic stability and binding affinity, and
altering lipophilicity and acidity.3 Therefore, fluorine chemistry has found wide
applications in the chemical world ranging from materials to medicine. This importance
has significantly accelerated the synthetic methodology development towards selective
and efficient fluorination of organic molecules as well as development of fluorine-
containing building blocks during the last century.
Extensive investigation has been applied toward the incorporation of a single
fluorine atom into organic molecules, with increasing interest shown in the last several
decades for the exploration of synthetic methods of perfluoroalkylation. The goal is to be
able to fine tune the chemical, physical or biological properties of target molecules by
incorporating various numbers of fluorine atoms.4 Among them, the trifluoromethyl (CF3)
group has proved to be a very important substituent as numerous compounds bearing
the trifluoromethyl moiety have become of great interest in the pharmaceutical
community. Thus considerable attention has been devoted to the synthetic development
of trifluoromethylation.5,6 The pentafluorosulfanyl (SF5) group, first introduced into
organic molecules a half-century ago, has been found to be an interesting substituent
16
that mimics the trifluoromethyl group with regard to electronic and steric factors. Hence,
in recent years SF5 chemistry has become one of the fast-growing fields in fluorine
chemistry after a long period of hibernation.
1.2 Applications of Pentafluorosulfanyl Chemistry
The pentafluorosulfanyl group has been regarded as an alternative to the
trifluoromethyl group, with previous investigations revealing that the SF5 group has a
much higher electronegativity than the CF3 moiety (3.65 vs 3.36), and the steric
demand of the SF5 group approaches that of the t-butyl group. Examination of the
stability of the SF5 group to hydrolysis demonstrated that the SF5 group has higher
hydrolysis stability than the CF3 substituent. All these differences indicate that
substitution of the SF5 group for CF3 may have profound effect on bioactivity.7
1.2.1 Applications of the SF5 Group in Medicinal Chemistry
Figure 1-1. SF5-substituted analogs of fluoxetine, fenfluramine, and norfenfluramine
The clinical agents fluoxetine (1-1), fenfluramine (1-2a) and norfenfluramine (1-
2b), which all bear a trifluoromethyl group, were widely used as serotonin (5-
hydroxytryptamine, 5-HT) inhibitors in the 1970s. In order to search analogs with higher
bioactivity and to test the influence of the SF5 group on bioactive molecules, Welch and
coworkers prepared SF5-substituted analogs for 5-HT inhibitors (Figure 1-1) and
evaluated their bioactivity.8 The examination showed that the SF5 substituent could
17
improve these inhibitors’ selectivity toward 5-hydroxytryptamine receptors. Among them,
compound 1-4b could lead to dramatically increased potency against 5-HT2b, 5-HT2c
and 5-HT6 receptors.
Figure 1-2. SF5- and CF3-substituted analogs of mefloquine
Mefloquine (1-5) is a clinically efficient treatment for malaria, which is a global
health problem with millions of casualties per year. However, its undesirable
neuropsychiatric side-effects such as anxiety, depression, seizure, and the emergence
of drug resistance led scientists to look for a better candidate. The Wipf group
synthesized two sets of mefloquine analogs with SF5 or CF3 substituted at the 6- or 7-
position (Figure 1-2), and they evaluated their bioactivities against parasites and
toxicities against mammalian cells.9 The results revealed the SF5 substituted compound
1-6b to have better bioactivity and selectivity than the CF3 substituted substance 1-6a or
mefloquine, while compound 1-7b was almost equivalent to CF3 analog 1-7a and
mefloquine.
18
Figure 1-3. Trypanothione reductase inhibitors
Although numerous SF5-containing analogs of bioactive molecules have been
synthesized, the Diederich group was the first to study the structure-activity relationship
on the molecule’s target level for SF5-bearing derivatives.10 They chose flavoenzyme
trypanothione reductase, which is found in parasites, as a target for the design of SF5-
containing inhibitors. Based on the diphenyl amine core structure, they synthesized
three sets of analogs bearing the SF5 moiety (Figure 1-3). Interestingly, bioactivity tests
showed that all the compounds (1-8b, 9b, 10b) with a SF5 substituent exhibited the low
cytotoxicity as well as good membrane permeability.
1.2.2 Applications of the SF5 Group in Agrochemistry
Figure 1-4. SF5-Substituted analogs of triflualin
Triflualin (1-11a), a widely used herbicide for pre-emergence control of grass, was
one of the annual best sellers in the US. When the Welch group simply modified its
structure by adding a SF5 group, they obtained an amazing result from the herbicidal
activity evaluation.11 In a post-emergence test, 1-11b exhibited almost twice the potency
19
as triflualin while having the same general spectrum of activity. Even more surprisingly,
in pre-emergence screening, 1-11b was approximately 5-fold more potent against
quackgrass and crabgrass. Therefore, 1-11b is a very promising candidate for further
exploration.
1.2.3 Applications of the SF5 group in Functional Materials
Liquid crystals (LC) as display materials have been extensively used in common
electronic devices such as PCs, notebooks, and cell phones. Due to its high polarity and
lipophilicity, the SF5 group was found to significantly improve the properties of LC
materials. When scientists from the Merck corporation prepared various SF5-substituted
LC materials based on the structure of widely-used fluorinated LC molecules (1-12),
they discovered that all these materials had considerably enhanced dielectric anisotropy
and lower birefringence, which are two of the most important parameters for the design
of LC materials.12
Figure 1-5 The various applications of SF5 groups in functional materials
Due to the multiple unique properties of the SF5 group, the Shreeve group found it
to be an excellent motif for the design of energetic materials (1-13). Generally, the SF5-
incorporated compound had high density, good thermal stability and enhanced
detonation performance.13
20
Taking advantage of its high lipophilicity, Gard and researchers from 3M prepared
various SF5-containing surfactants.14 These materials normally exhibited lower surface
tension and better performance than their CF3 analogs (1-14).
1.3 Synthesis of Pentafluorosulfanyl Substituted Aromatic Rings
Fluorinated aromatic compounds are widely used in chemical, pharmaceutical and
agrochemical industries. Thus, there are good reasons to establish practical synthetic
methods to construct SF5-substituted aromatic compounds, which may have great
potential for applications such as those mentioned above. Although the first preparation
of SF5-benzene originated in the 1960s, only in the last decade have several
breakthroughs occurred. In the following sections, a concise introduction of the
synthesis of aromatic rings with an SF5 group directly attached is presented.
1.3.1 Synthesis of SF5-Benzene
Figure 1-6 Early preparation method for SF5-benzene
Because of its significant potential importance, SF5-benzene building blocks
attracted much attention from fluorine chemists working in the field of SF5 chemistry.
Even though Sheppard’s pioneering work on the preparation of SF5-benzene was
reported almost a half-century ago, this molecule remained a challenge to fluorine
scientists for decades because all of the synthetic procedures developed during this
21
period required either harsh reaction conditions or expensive reagents while giving
poor yields (Figure 1-6).15,16,17,18
Figure 1-7 First practical route to prepare SF5-benzene
The first practical and scalable synthetic method was reported by Bowen and
Philip (Figure 1-7).19 Inspired by the previous work, they still used nitro-substituted aryl
disulfides as starting materials (1-16a, b). Diluted F2 gas was creatively employed as a
fluorinating reagent, and the desired product was obtained in reasonable yield at low
temperature. Though the F2 gas was very toxic, corrosive and relatively expensive, this
method was commercialized to facilitate research in other areas during the subsequent
years due to the mild reaction conditions and easy work-up procedure. It is also worth
mentioning that in this article, they investigated the properties of the SF5 group as well.
The investigation revealed that generally the SF5 group could survive in various reaction
conditions such as hydrogenations, coupling reactions, acid-base reactions, and it also
exhibited higher stability than CF3 analogs in a hydrolysis test.
Figure 1-8. Preparation of SF5-benzene from SF5Cl gas
Pentafluorosulfanyl chloride (SF5Cl) gas, one of the few commercially available
SF5 reagents, was used for several decades for construction of SF5-containing building
22
blocks. However, due to its low boiling point (-21oC), normally the reaction required the
use of autoclave and high temperature. In 2002, the Dolbier group discovered that Et3B
was an excellent radical initiator for addition reactions of SF5Cl to alkene and alkyne
substrates.7 This new procedure could be carried out in common glassware at low
temperature with high yield. Based on this creative invention, they designed a novel
route to prepare SF5-benzene.20 Starting from easily available reagent 1,4-
cyclohexadiene, the dichloride substitute intermediate 1-18 was obtained in quantitative
yield through a classical radical process. When this product was submitted to standard
SF5Cl addition conditions initiated by Et3B, followed by simple elimination, the target
molecule was attained with >70% yield over the three steps. Although this method was
quite straight forward, the relatively high price of SF5Cl gas has limited its use.
Figure 1-9. Practical preparation of SF5-benzene developed by Umemoto
A major milestone for preparation of SF5-aromatics was established by Umemoto
and his coworkers (Figure 1-9). In 2012, they reported an innovative construction of the
SF5 group through a novel intermediate bearing the SF4Cl group.21 Starting from
commercially available phenyl disulfide or thiol, the SF4Cl group was assembled by
bubbling chlorine gas into a dry potassium fluoride solution and then stirring overnight at
RT. This intermediate 1-19 was not very stable. Following simple filtration and
evaporation of the solvent, it was further treated with SbF3/SbCl5 in CH2Cl2, and the
desired product 1-20 was obtained via a clean transformation. This procedure, which
has been scaled up by the author’s company, exhibited great substrate scope for
23
preparing SF5-benzene and its derivatives. Since all of the reagents are relatively cheap
and commercially available, and the procedure is readily scaled-up, this invention
should significantly benefit the whole chemical community.
1.3.2 The Synthesis of SF5-Furan
Figure 1-10. Preparation of SF5-furan through retro-Diels-Alder-reaction
With the successful preparation of SF5-benzene from SF5Cl and their continuing
interest in SF5-substituted heterocyclic compounds, the Dolbier group designed a new
route to synthesize SF5-furan based on the process of the retro-Diels-Alder reaction
(Figure 1-10).22 Utilizing the previous Et3B initiated conditions, SF5Cl was smoothly
introduced to the easily prepared starting material 1-21, which is the Diels-Alder adduct
of furan and acrylonitrile. The mixture of two regioisomers 1-22a and 1-22b was treated
with strong base LiOH in DMSO to provide the clean elimination products 1-23a and 1-
23b. At high temperature, they underwent retro-Diels-Alder reaction to give the target
molecule 1-24 with decent yield. Currently, this is the first and only reported preparation
method to construct SF5-furan. However, the utilization of the expensive SF5Cl gas and
the narrow substrate scope limited its wide application.
24
Figure 1-11. Preparation of SF5-furan through Diels-Alder- and retro-Diels-Alder-
reaction
In the same article, they reported an alternative method to prepare SF5-furan in
one pot based on a cascade mechanism (Figure 1-11). Starting material 4-
phenyloxazole is an easily prepared building block for facile construction of furan and its
derivatives. Therefore, they treated SF5-substituted alkyne with oxazole at high
temperature, and after an overnight reaction, the desired product was obtained in high
yield after column purification. The reaction was believed to proceed through a Diels-
Alder mechanism with the generation of an unstable adduct 1-25, which underwent a
Diels-Alder reaction to result in the target molecule. This method has relatively broader
substrate scope as SF5-substituted alkynes can be prepared from terminal alkynes
through addition-elimination steps.
1.3.3 Synthesis of SF5-Naphthalene
Figure 1-12. Preparation of SF5-naphthalene
25
With the achievement of SF5-furan through Diels-Alder reactions, the Dolbier
group continued to build SF5-naphthalene by such methodology (Figure 1-12).23 Initial
addition of SF5Cl to benzobarralene and subsequent base-catalyzed elimination of HCl
led to the key intermediate 1-29 in high yield. The ethylene bridge of 1-29 was smoothly
eliminated by heating with the commercially available reagent 3,6-bis-(2-pyridyl)-1,2,3,4-
tetrazine (1-30), and the target molecule was obtained in high yield through this
sequence of reactions.
1.3.4 Synthesis of SF5-Pyrazole and –Triazole
Figure 1-13. Preparation of SF5-pyrazole and –triazole by 1,3-dipolar cycloaddition
In 1964, researchers from Dupont reported the first example of construction of
SF5-bearing heterocycles based on 1,3-dipolar cycloaddition (Figure 1-13).17 Simply
adding SF5-acetylene to diazomethane at 0oC, a mixture of regioisomers (1-32a, 1-32b)
with a ratio of 60:40 was readily obtained. In 2007, Shreeve and her coworkers utilized
the same method to prepare SF5-containing energetic materials.13 Starting from bulky
TIPS substituted SF5-acetylene, only one regioisomer (1-33) as product was obtained in
quantitative yield. They also extended this reaction to prepare various SF5-triazoles (1-
34) as high performance materials using ‘click chemistry’.
26
CHAPTER 2 THE PREPARATION OF PENTAFLUOROSULFANYL PYRROLE AND THIOPHENE
THROUGH 1,3-DIPOLAR CYCLOADDITION
2.1 Initial Investigations of Synthetic Methods toward SF5-bearing Heterocycles
Investigations of pentafluorosulfanyl (SF5) chemistry in Dr. Dolbier’s lab were
initiated by Dr. Samia Ait-Mohand in 2002.7 Her great invention provided a practical
method to add SF5Cl to alkene and alkyne substates without utilizing an autoclave
reactor and high temperature. The reactions were usually carried out at low temperature
(-30oC) in ordinary glassware, initiated by catalytic amount of Et3B (0.1eq.), and
generated the desired products in high yield in a short time (2 hours). Based on this
significant discovery, in subsequent research, Dr. Sergeeva established the earlier-
mentioned approach to SF5-benzene,20 and Dr. Mitani prepared the first furans bearing
an SF5 group through retro-Diels-Alder chemistry.22 With the considerable continued
interest in SF5-containing heterocyclic compounds because of their great potential for
application and commercial value, my challenge was to investigate the preparation
methods for pyrroles and thiophenes bearing an SF5 group, which had never been
made before.
Figure 2-1. The first attempted synthetic route to SF5-pyrrole
With the commercially available SF5Cl gas in hand and inspired by the previous
methods, we designed a short synthetic route for SF5-pyrrole (Figure 2-1). Starting from
the purchased 2,5-dihydropyrrole (2-1a), SF5Cl would be first incorporated into the five-
27
membered ring using the standard Et3B method, followed by elimination of HCl and
oxidation steps, the desired product would be generated using a concise approach.
However, the first step reaction did not occur as we expected. In the beginning we
thought it might be due to the presence of a proton on the dihydropyrrole nitrogen, but
even with the subsequent change of hydrogen to a phenyl group (2-1b) the reaction still
did not occur. Since there have been few reported examples of SF5Cl addition into
internal alkenes compared to reported addition to terminal alkenes, we realized that
steric hindrance could play a key role in this situation, as SF5 group is as bulky as a t-
butyl group.
Figure 2-2. Second synthetic route to SF5-heterocycles catalyzed by palladium
28
After the failure of the first attempt, we considered that SF5Cl might not be a good
starting material for direct construction of heterocycles, due to the narrow substrate
scope of its addition reactions. Compared to SF5Cl gas, SF5-substituted alkynes should
be better building blocks for several reasons. First, numerous chemical transformations
based on alkynes have been established; secondly, the one-step construction of
heterocycles from alkynes catalyzed by transition-metals have been well explored in
recent decades; thirdly, many successful synthetic precedents for preparation of CF3-
heterocycles from CF3-substituted alkynes have been reported, probably correlating
with SF5 chemistry; lastly, SF5-alkynes were readily prepared from SF5Cl based on the
previously developed method.
Therefore we designed a second route towards various SF5-containing
heterocyclic compounds based on SF5-alkyne building blocks. Konno and his coworkers
had demonstrated a general method to prepare CF3-containing benzoheterocycles
catalyzed by palladium (Figure 2-2),24,25 and we expected to obtain at least one of the
desired compounds from those diverse transformations. However we did not achieve
any positive results except recycling the starting materials when SF5- alkyne was mixed
with the aromatic iodide (2-3a, b, c). From the proposed mechanism, we rationalized
that the problem may be due to steric hindrance, which prevents the addition of the
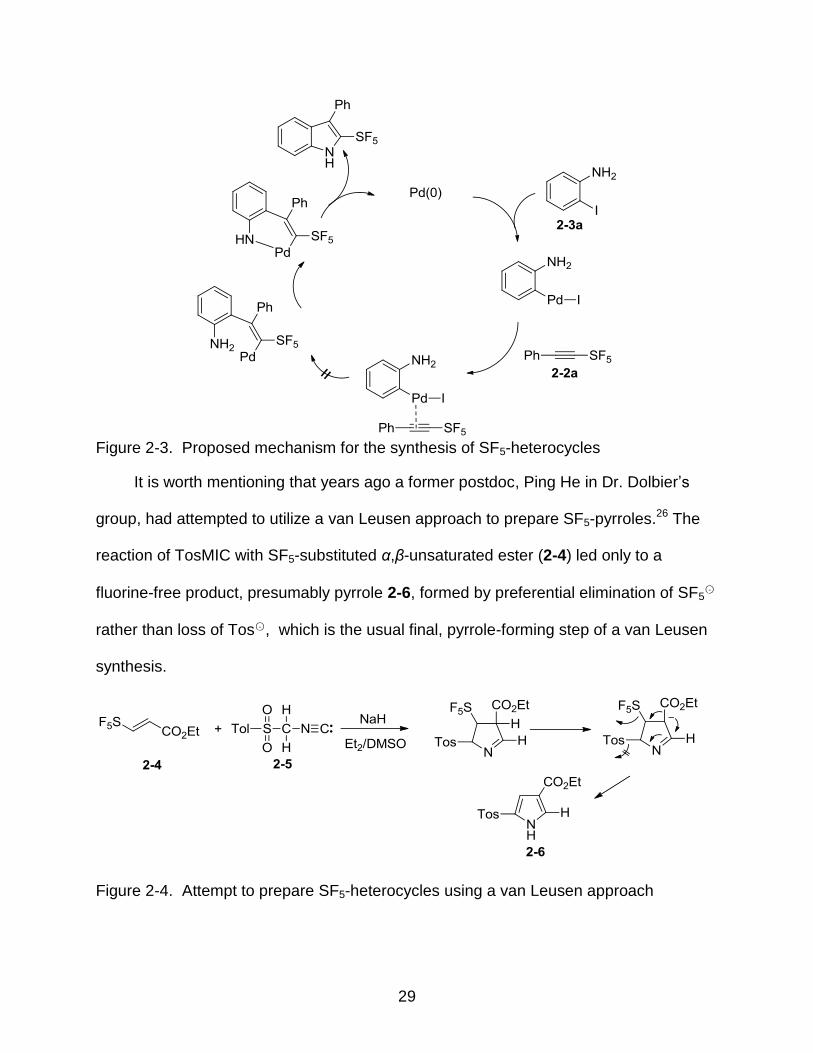
aromatic palladium intermediate into the alkyne substrate (Figure 2-3).
29
Figure 2-3. Proposed mechanism for the synthesis of SF5-heterocycles
It is worth mentioning that years ago a former postdoc, Ping He in Dr. Dolbier’s
group, had attempted to utilize a van Leusen approach to prepare SF5-pyrroles.26 The
reaction of TosMIC with SF5-substituted α,β-unsaturated ester (2-4) led only to a
fluorine-free product, presumably pyrrole 2-6, formed by preferential elimination of SF5○-
rather than loss of Tos○- , which is the usual final, pyrrole-forming step of a van Leusen
synthesis.
Figure 2-4. Attempt to prepare SF5-heterocycles using a van Leusen approach
30
2.2 Preparation of SF5-pyrrole Carboxylic Acid Esters
2.2.1 Introduction
Figure 2-5. The preparation of SF5-heterocycles based on cycloaddition chemistry
After all attempts mentioned above to generate the desired SF5-heterocyclic
compounds failed, we carefully examine the possible reasons via a literature review.
When carefully analyzing the previous cases for preparing SF5-aromatic compounds,
we rationalized that concerted cycloaddition chemistry could be one possible method as
furan, pyrazole and triazole had all been generated by cycloaddition reactions starting
from SF5-alkynes (Figure 2-5).22,13
Figure 2-6. Preparation CF3-pyrrole from azomethine ylide
The 1,3-dipolar cycloaddition of azomethine ylide, generated from thermal opening
of 2-7, to alkynes or alkenes has been demonstrated to be a general approach to
construct pyrrolines or pyrrolidines, respectively. La Porta and co-workers successfully
implemented this method to prepare trifluoromethyl pyrroles starting from cycloaddition
31
of CF3-alkyne with azomethine ylide and following with DDQ oxidation.27 Therefore, we
expected this to be a feasible way to our desired product.
2.2.2 Result and Discussion
Table 2-1. Reaction of SF5-alkyne with azomethine ylide
2-7(equiv) 5h 20h 40%
1 -- 32% 37%
3 40% 100% --
When the readily prepared aziridine 2-7 was heated with equimolar amount of
SF5-alkyne 2-2b in xylene, to our delight, a clean chemical transformation occurred as
shown by 19F-NMR, even though in 20h it only gave moderate conversion and
prolonging of the reaction time did not help. By increasing the amount of 2-2b to 3
equiv, the reaction rate was dramatically accelerated and full conversion could be
achieved overnight (Table 2-1). The isolated yield of 2-8b was 60% and its structure
was fully characterized. Smooth DDQ (2equiv) oxidation provided the target molecule 2-
9b in quantitative yield.
32
Figure 2-7. 1,3-Dipolar cycloaddition approach to SF5-heterocycles
To explore the reaction’s limitations, more substrates were prepared and
submitted to the reaction. In order to simplify the procedure, the crude pyrroline
intermediate was treated with DDQ directly after removing the solvent without isolation.
Generally the one pot reaction showed good substrate scope with moderate to good
yields (Figure 2-7).
Figure 2-8. Removal of t-butyl group catalyzed by triflic acid
To demonstrate the full scope of this method, the removal of t-butyl group was
accomplished by utilizing the reported method. While heating 2-9b in CH2Cl2 with a
catalytic amount triflic acid for 2h, the unprotected pyrrole 2-10 was obtained in an
unoptimized yield of 72% (Figure 2-8).
33
Figure 2-9. Mechanism for the regioselective cycloaddition chemistry
Notably, the present procedure gave exclusively one regioisomer based on SF5-
alkyne compared to the regioisomeric mixture (~75:25) obtained from CF3-alkyne.27 The
mechanistic analysis clearly explained the regioselective chemistry (Figure 2-9). As for
the CF3 substrate, the reaction was dominated by the electronic effect while the steric
effect led to the minor product, as the strain was greatly released between ester group
and CF3 part. When it came to the SF5 substrate, the electronic and steric effects both
preferred the same regioselectivity. This result also agreed well with the structural
properties of CF3 and SF5 groups as both are strong electron-withdrawing groups but
have considerably different size.
34
2.2.3 Structure Characterization
Figure 2-10. Proton NMR of 2-8b
Figure 2-11. Proton NMR of 2-9b
35
Figure 2-12. Proton NMR of 2-10
Figure 2-13. 19F-NMR spectrum of compounds 2-10 and 2-9b
In Figure 2-10, the appearance of the aromatic proton signals (7.23, 7.34 ppm),
the sharp t-butyl (0.98 ppm) and methyl peak (3.72 ppm) clearly indicate the formation
of a dihydropyrrole ring by the cycloaddition reaction. After oxidation with DDQ, the
signal of CH2 (3.92, 4.10 ppm) and CH (4.49 ppm) groups in compound 2-8b
disappeared, and a new peak in the aromatic region emerged by integration, which
proved the success of aromatization. In Figure 2-12, the characteristic t-butyl signal
36
(1.68 ppm) of compound 2-9b completely disappeared and a new broad singlet peak at
9.34 ppm appeared when 2-9b was treated with catalytic triflic acid, which confirmed the
removal of the protecting group.
19F-NMR is also a powerful tool to monitor the reaction with the advantage of little
interference from solvents and other substrates when compared to proton NMR. The
SF5 group gives 19F-NMR signals with the characteristic AB4 system. Therefore, the
doublet peaks around 74.30 ppm in 2-9b that moved to 73.56 ppm in 2-10 clearly
demonstrate the cleavage of the N-C bond (Figure 2-13).
2.3 Preparation of SF5-pyrrole
2.3.1 Introduction
Based on the above successful preparation of SF5-pyrrole carboxylic acid ester
where SF5-alkynes acted as dipolarophiles, we wondered if the cycloaddition reaction
could be a good general approach to SF5-pyrrole. If so, a wide variety of SF5-pyrrole
structures could be built for potential medicinal applications. Therefore, another
azomethine ylide building block 2-11 was selected to examine this hypothesis.
Figure 2-14. The high reactivity of azomethine ylide building block 2-11
N-Benzyl-N-(methoxymethyl)-N-(trimethylsilylmethyl)amine 2-11 was first
recognized as an azomethine ylide synthon by Hosomi and coworkers in 1984,28 and
later on its properties and reactivity were fully investigated by Padwa etc.29 All studies
demonstrated that this compound had unique advantages over others: First, 2-11 is
37
readily prepared and is already commercially available; secondly, the reaction condition
is adjustable as it could be initiated by either catalytic amount of H+ or an F- source; and
thirdly, it has been widely used to build bioactive molecules or natural products because
of its extensive substrate scope; 30,31 last but not the least, it has excellent reactivity as
even under mild condition the dearomization occurred when it interacted with
dinitrobenzene (Figure 2-14).32 Hence, there are good reasons to believe that 2-11
could also react with SF5-alkynes.
2.3.2 Results and Discussion
The initial investigation followed the reported conditions of using an equal amount
of cesium fluoride and 2-11 with acetonitrile as solvent at RT, however, no desired
product was detected by 19F-NMR. The attempt to increase the temperature or switch to
lithium fluoride did not lead to the target either. When 1.0M TBAF solution was
employed, instead of recovering SF5-alkyne, all of the substrate decomposed for
unknown reasons.
Gratefully after switching to an acid catalyzed system, 65% conversion was
obtained with 0.2 eq trifluoroacetic acid applied in CH2Cl2 at RT. As shown in Table 2-2
full conversion was readily achieved by increasing the amount of 2-11 to 4 eq.
Additional optimization reactions demonstrated that only 2.5 eq of azomethine ylide was
required under reflux conditions, with isolated yield as high as 96%.
More SF5-alkynes were prepared according with previous methods in order to test
the reaction scope. In practice the intermediate dihydropyrroles were not separated but
were converted, in situ, to the respective pyrroles directly by treatment with DDQ.
Generally the reaction provided good to excellent yields with wide substrate scope
38
(Table 2-3). Even for the considerably bulky TIPS-substituted SF5-alkyne, it still gave
78% yield.
Table 2-2. Investigation of the reaction of SF5-alkyne with 2-11
7 Ait-Mohand, S.; Dolbier, W. R. D. Jr. Org. Lett. 2002, 17, 3013-3015.
8 Welch. J. T.; Lim, D. S. Bioorg. Med. Chem. 2007, 15, 6659-6666.
9 Wipf, P.; Mo, T.; Geib, S.; Caridha, D.; Dow, G. S.; Gerena, L.; Roncal, N.; Milner, E. E. Org. Biomol. Chem. 2009, 7, 4163-4165.
10 Stump, B.; Eberle, C.; Schweizer, W. B.; Kaiser, M.; Brun, R.; Krauthsiegel, R. L.; Lentz, D.; Diederich, F. ChemBioChem 2009, 10, 79-83.
11 Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. 2007, 32, 255-259.
12 Kirsch, P.; Hahn, A. Eur. J. Org. Chem. 2005, 3095-3100
13 Ye, C.; Gard, G. L.; Winter, R. W.; Syvret, R. G.; Twamley, B.; Sheeve, J. M. Org. Lett. 2007, 9, 3841-3844.
14 Winner, S. W.; Winter, R. W.; Smith, J. A.; Gard, G. L.; Hannah, N. A.; Rananavare, S. B.; Piknova, B.; Hall, S. B. Mendeleev Commun. 2006, 16, 182-184.
15 Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064-3072.
16 Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072-3076.
17 Hoover, F. W.; Coffman, D. D. J. Org. Chem. 1964, 29, 3567-3570.
18 Ou, X.; Janzen, A. F. J. Fluorine Chem. 2000, 101, 279-283.
19 Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399-3408.
20 Sergeeva, T. A.; Dolbier, W. R. Jr. Org. Lett. 2004, 14, 2417-2419.
21 Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461-471.
96
22 Dolbier, W. R. Jr.; Mitani, A.; Xu, W.; Ghiviriga, I. Org. Lett. 2006, 8, 5573-5575.
23 Dolbier, W. R. Jr.; Mitani, A.; Warren, R. Tetrahedron Letters 2007, 48, 1325-1326.
24 Konno, T.; Chae, J.; Ishihara, T.; Yamanaka, H. J. Org. Chem. 2004, 69, 8258-8265.
25 Konno, T.; Chae, J.; Miyabe, T.; Ishihara, T. J. Org. Chem. 2005, 70, 10172-10174.
26 Joule, J. A.; Mills, K. Heterocyclic Chemistry; Blackwell Science: Oxford, 1995.
27 La Porta, P.; Capuzzi, L.; Bettarini, F. Synthesis 1994, 287-290.
33 Olofson, R. A.; Martz, J. T.; Senet, J.-P.; Piteau, M.; Malfroot, T. J. Org. Chem. 1984, 49, 2081-2082.
34 Ye, X.-S.; Wong, H. N. C. J. Org. Chem. 1997, 62, 1940-1954.
35 Hosomi, A.; Matsuyama, Y.; Sakurai, H. J. Chem. Soc., Chem. Commun. 1986, 1073-1074.
36 Hogberg, H.-E.; Karlsson, S.; Org. Lett. 1999, 1, 1667-1669.
37 Isanbor, C.; O'Hagan, D. J. Fluorine Chem. 2006, 127, 303−319
38 Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013−1029
39 Begue, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992−1012.
40 a) Ren, Y.; Lodge, T. P.; Hillmyer, M. A. J. Am. Chem. Soc. 1998, 120, 6830-6831; b) Barnett, C. J.; Huff, B.; Kobierski, M. E.; Letourneau, M.; Wilson, T. M.; c) Nowak, I.; Robins, M. J. J. Org. Chem. 2006, 71, 8876-8883; d) Nowak, I.; Cannon, J. F.; Robins, M. J. J. Org. Chem. 2007, 72, 523-537; e) Nowak, I.; Robins, M. J. J. Org. Chem. 2007, 72, 3319-3325; f) Lenhardt, J. M.; Ogle, J. W.; Ong, M. T.; Choe, R.; Martinez, T.; Craig, S. L. J. Am. Chem. Soc. 2001, 133, 3222-3225.
97
41 a) Tian, F.; Kruger, V.; Bautista, O.; Duan, J.; Li, A.; Dolbier, W. R. Jr.; Chen, Q. Org.
Lett. 2000, 2, 563-564; b) Chang, Y.; Cai, C. Chemistry Letters 2005, 34, 1440-1441; c) Fujioka, Y.; Amii. H. Org. Lett. 2008, 10, 769-772; d) Oshiro, K.; Morimoto, Y.; Amii, H. Synthesis 2010, 12, 2080-2084; e) Wang, F.; Luo, T.; Hu, J.; Wang, Y.; Krishnan, H.; Jog, P. V.; Ganesh, S. K.; Prakash, G. K. S.; Olah, G. A. Angew. Chem. Int. Ed. 2011, 50, 7153-7157; f) Wang, F.; Zhang, W.; Zhu, J.; Li, H.; Huang, K.; Hu, J. Chem. Commun. 2011, 47, 2411-2413.
42 Xu, W.; Dolbier, W. R. Jr.; Salazar, J. J. Org. Chem. 2008, 73, 3535-3538.
43 Dolbier, W. R. Jr.; Cornett, E.; Martinez, H.; Xu, W. J. Org. Chem. 2011, 76, 3450-3456.
47 Barbe, G.; Charette, A. B. J. Am. Chem. Soc. 2008, 130, 18-19
48 Akiyama, T. Chem. Rev. 2007, 107, 5744-5758
49 Cox; R. J.; Riston, D. J.; Dane, T. A.; Berge, J.; Charmant, J. P. H.; Kantacha, A. Chem. Commun. 2005, 8, 1037-1039
50 Dolbier, W. R. Jr.; Tian, F.; Duan, J.-X.; Li, A.-R.; Ait-Mohand, S.; Bautista, O.; Buathong, S.; Baker, J. M.; Crawford, J.; Anselme, P.; Cai, X.-H.; Modzelewska, A.; Koroniak, H.; Battiste, M.; Chen, Q.-Y. J. Fluorine Chem. 2004, 125, 459-469
51 a) Bruno, A. Macromolecules 2010, 43, 10163-10184. b) Reisinger, J. J.; Hillmyer, M. A. Prog. Polym. Sci. 2002, 27, 971-1005. c) Hillmyer, M. A.; Lodge, T. P. J. Polym. Sci. Part A: Polym. Chem. 2002, 40, 1-8
52 a) Ameduri, B.; Boutevin, B. Well-Architectured Fluoropolymers: Synthesis, Properties and Applications; Elsevier: Amsterdam, 2004; pp187-230. b) Cais, R. E.; Kometani, J. M. Polymer 1988, 29, 168-172.
53 Lee, J.-K.; Fong, H. H.; Zakhidov, A. A.; McCluskey, G. E.; Taylor, P. G.; Santiago-Berrios, M. ; Abruna, H. D.; Holmes, A. B.; Malliaras, G. G.; Ober, C. K. Macromolecules 2010, 43, 1195-1198.
54 Imbesi, P. M.; Fidge, C.; Raymond, J. E.; Cauet, S. I.; Wooley, K. L. ACS Macro Lett. 2012, 1, 473-477
56 A) Terjeson, R. J.; Gard, G. L. J. Fluorine Chem. 1987, 35, 653-659. B) Gard, G. L.;
Winter, R. ; Nixon, P. G.; Hu, Y.-H.; Holcomb, N. R.; Grainger, D. W.; Castner, D. G. Polym. Prepr. 1998, 39, 962-963. C) Yan, M.; Gard, G.; Mohtasham, J.; Winter, R. W.; Lin, J.; Wamser, C. C. Polym. News 2001, 26, 283-288. D) Winter, R.; Nixon, P. G.; Gard, G. L.; Castner, D. G.; Holcomb, N. R.; Hu, Y.-H; Grainger, D. W. Chem. Mater. 1999, 11, 3044-3049
57 A) Kostov, G.; Ameduri, .; Sergeeva, T.; Dolbier, W. R. Jr.; Winter, R.; Gard, G. L. Macromolecules 2005, 38, 8316-8326. B) Boyer, C.; Ameduri, B.; Boutevin, B.; Dolbier, W. R.; Winter, R.; Gard, G. Macromolecules 2008, 41, 1254-1263.
58 Hillmyer, M.; Laredo, W. R.; Grubbs, R. H. Macromolecules 1995, 28, 6311-6316.
59 Simon, Y. C.; Coughlin, E. B. Journal of polymer Science: Part A: Polymer Chemistry 2010, 48, 2557-2563.
99
BIOGRAPHICAL SKETCH
Zhaoyun Zheng was brought up in Nanjing, People’s Republic of China. He
received his B.S. degree from Nanjing Normal University in 2003 and his M.S. degree
from Nankai University in 2006. After that he worked for a pharmaceutical company in
Tianjin as a synthetic research scientist. In July 2008, he came to University of Florida
and worked in the group of Dr. William R. Dolbier, Jr. as a research scholar. In spring
2010 he enrolled in the PhD program of the Department of Chemistry, University of
Florida, under Dr. Dolbier’s supervision. In the fall of 2012, he received his Ph.D. from
the University of Florida.
Zhaoyun and his wife, Lijuan Yue, have one lovely daughter, Fiona Haoting








































































































![Toward a Methodology for Measuring the Security of ... · PDF fileDownloaded By: [University of Colorado at Denver] At: 18:10 3 October 2007 Toward a Methodology for Measuring the](https://static.documents.pub/doc/80x56/5a9de1797f8b9adb388b5fc0/toward-a-methodology-for-measuring-the-security-of-by-university-of-colorado.jpg)









