1 The Application of Empirical Methods of 13 C NMR Chemical Shift Prediction as a Filter for Determining Possible Relative Stereochemistry. A short title: The Application of Empirical NMR Prediction to Determine Stereochemistry Mikhail E. Elyashberg + , Kirill A. Blinov + and Antony J.Williams * . + Advanced Chemistry Development, Moscow Department, 6 Akademik Bakulev Street, Moscow 117513, Russian Federation, ChemZoo Inc., 904 Tamaras Circle, Wake Forest, North Carolina 27587 Abstract: The reliable determination of stereocenters contained within chemical structures usually requires utilization of NMR data, chemical derivatization, molecular modeling, quantum- mechanical calculations and, if available, X-ray analysis. In this article we show that the number of stereoisomers which need to be thoroughly verified can be significantly reduced by the application of NMR chemical shift calculation to the full stereoisomer set of possibilities using a fragmental approach based on HOSE codes. The applicability of this suggested method is illustrated using experimental data published for a series of complex chemical structures. Keywords: NMR, 1 H, 13 C, chemical shift prediction, stereochemistry. Introduction
Transcript
1
The Application of Empirical Methods of 13C NMR Chemical Shift Prediction as a
Filter for Determining Possible Relative Stereochemistry.
A short title:
The Application of Empirical NMR Prediction to Determine Stereochemistry
Mikhail E. Elyashberg+, Kirill A. Blinov+ and Antony J.Williams*.
+Advanced Chemistry Development, Moscow Department, 6 Akademik Bakulev Street,
Moscow 117513, Russian Federation,
ChemZoo Inc., 904 Tamaras Circle, Wake Forest, North Carolina 27587
Abstract:
The reliable determination of stereocenters contained within chemical structures usually
requires utilization of NMR data, chemical derivatization, molecular modeling, quantum-
mechanical calculations and, if available, X-ray analysis. In this article we show that the
number of stereoisomers which need to be thoroughly verified can be significantly
reduced by the application of NMR chemical shift calculation to the full stereoisomer set
of possibilities using a fragmental approach based on HOSE codes. The applicability of
this suggested method is illustrated using experimental data published for a series of
complex chemical structures.
Keywords:
NMR, 1H, 13C, chemical shift prediction, stereochemistry.
Introduction
2
A number of different methods of NMR chemical shift prediction have been applied
to the process of molecular structure elucidation and validation. Empirical methods are
attractive since they are fast enough and fully automatic. The fastest NMR spectra
calculations are provided using an incremental approach and offer a computational speed
of 6,000-10,000 chemical shifts per second on a normal desktop computer (circa 2007)
and provides an average chemical shift deviation for carbon NMR of 1.8 ppm[1,2].
Spectral prediction utilizing artificial neural networks provide similar speed and accuracy
performance[1,2]. The third most popular empirical method is slower and is based on the
application of a database containing reference structures with assigned 13C or 1H
chemical shifts. The target and reference structures are described by means of HOSE
codes[3] and this allows prediction of the chemical shift of an atom from the target
structure using the chemical shifts of the reference structures as the basis. In the
ACD/NMR predictor[4], the prediction algorithms use a library containing 185,000
structures with NMR chemical shifts assigned to carbon and hydrogen atoms. If
information regarding the relative stereochemistry of a given atom ai and its environment
is known then these data are also coded into the reference structures. To predict the
chemical shift of an atom ai in the target structure its HOSE code is compared with the
codes of the corresponding atoms in reference structures. As a result of statistical
processing of the chemical shifts assigned to all “atom-twins” detected in the reference
structures, the chemical shift of an atom from the target structure is predicted. A strategy
based on combining all mentioned methods was suggested[5,6]. It allows selection of the
most probable structure from the output file of expert system developed for the molecular
structure elucidation.
3
At the same time a series of articles have been published espousing the value of ab-
initio quantum mechanical (QM) approaches for NMR chemical shift calculations (for
instance,[7-12]) and, most frequently, the GIAO option of the DFT method[13] has been
employed for the calculation of 1H and 13C chemical shifts. It was shown that DFT based
methods can be applied for the selection of a preferable structural hypothesis by means of
comparing the predicted chemical shifts with those determined experimentally. This
approach was also an efficient tool for evaluating the different conformers of flexible
molecules as well as the elucidation of the most probable stereoisomers[13-17].
In our previous report[18] we have shown that empirical methods of NMR chemical
shift prediction can be successfully used at the selection stage of structural hypotheses
which are verified further with application of molecular geometry optimization and QM
chemical shift prediction. In this regard we hypothesize that empirical methods can help
in preliminary selection of a set of the most probable stereoisomers for their subsequent
verification by additional experimental techniques and QM chemical shift prediction.
This may be possible since the stereocenters of structures included into the ACD/CNMR
database and stereochemistry is taken into account by the NMR chemical shift prediction
algorithms. The incremental and neural nets based algorithms of chemical shift prediction
also use the stereochemistry information related to the atoms included into 3-6-membered
cycles[2]. It was interesting to know whether this information can be useful for
stereochemistry determination.
We have tested our hypothesis using a series of examples. We have used examples
from recent literature (2007-8) for novel structures for which relative stereochemistry
was reported. These structures are deliberately absent from the ACD/CNMR database.
4
The application of empirical methods of 13C NMR chemical shift prediction is shown to
allow the selection of a set of the most probable stereoisomers and always includes the
genuine stereoconfiguration.
RESULTS AND DISCUSSION.
Fattorusso et al[15] utilized DFT chemical shift computation to confirm the most
probable stereoisomer of artarborol, 1, a rare nor-caryophyllane derivative, isolated by
the authors[15] and structurally characterized by both 1D and 2D NMR spectroscopic
methods.
1
2
3
4
5
6
7
89
O10
11
12
OH13
CH314
CH315
CH316
H17
H18
H19
1
To select the most probable stereoisomer the authors[15] carried out a series of
investigations. Structure 1 contains five stereogenic carbons (numbered 1-5 on structure
1) with four of them at junctions between the 9-membered ring and the small ring cycles,
while both cis- and trans- junctions of rings adjacent to the nine-membered core are
possible in natural caryophyllanes.
A combination of 2D ROESY experiments with Mosher’s modified method[19]
was used to assess the absolute configuration of C-2 (R) and allowed the authors[15] to
reduce the total number of possible stereoisomers to the following four (Figure 1):
5
1
23
4
5
6
7
89
O10
11
12
OH13
CH314
CH315
CH316
H17
H18
H19
1
23
4
5
6
7
8
9
O10
11
12
OH13
CH314
CH315
CH316
H17
H18
H19
1
23
4
5
6
7
89
O10
11
12
OH13
CH314
CH315
CH316
H17
H18
H19
1
23
4
5
6
7
89
O10
11
12
OH13
CH314
CH315
CH316
H17
H18
H19
A B C D
Figure 1. The four candidate stereoisomer structures of artarborol.
Further selection was made by analyzing the scalar coupling constants and additional
spatial couplings across the entire molecule for which all candidate structures were
subjected to a conformational search. As a result, structures B and D were rejected at the
first step, structure C was then excluded and finally stereoconfiguration A was assigned
to artarborol. To support this stereochemical assignment each conformation of the
stereoisomers A and C were fully optimized by the authors[15], and the NMR chemical
shifts were calculated using the GIAO option of the MPW1PW91/6-31G(d,p) DFT
method[20]. A Boltzmann-weighted average of the 13C NMR chemical shifts for all carbon
atoms in the low-energy conformers was calculated for each configuration, using the ab-
initio standard free energies as weighting factors[21]. The total processing time for each
molecule was approximately 60 h (PC Pentium IV). A comparison of calculated chemical
shifts with those determined experimentally for structures A and C showed that
deviations were smaller for structure A thereby confirming the validity of the solution.
Selection of the most probable stereoisomer was attained as a result of a
comprehensive experimental and theoretical investigation of the compound and its
conceivable 3D models. We investigated what results would be obtained if the problem
6
is solved using 1D and 2D NMR spectra and the empirical chemical shift prediction
methods implemented into the expert system Structure Elucidator[5,6,22].
To perform this analysis structure 1 was input into the system and all carbon and
hydrogen atoms were supplied with chemical shifts in accordance with the author’s
assignment. Then all 25=32 streoisomers were generated by the program and depicted
using conventional designations for stereobonds. 1H and 13C chemical shifts were
calculated for the complete stereoisomer set using the fragment-based approach within
the Structure Elucidator program. In addition, 13C NMR chemical shifts were calculated
using both neural net (N) and incremental (I) approaches.
The average deviations of the predicted chemical shifts relative to the
experimental shifts (dA = fragmental approach, dN = NN approach and dI = incremental
approach) were calculated for each of 32 stereoisomers and all stereoisomers were ranked
in ascending order of the 13C deviation values. Since the chemical shifts are insensitive to
the absolute configuration of a stereoisomer and its inverse partner the reduced ranked
stereoisomer set was finally represented as a sequence of 16 stereoisomer pairs, each pair
having equal deviations. Figure 2 shows the first 8 out of 16 “unique” stereoisomers
ranked in ascending order of the average deviations calculated for 13C NMR spectrum.
The remaining stereoisomers are characterized by 13C average deviations dA(13C) falling
in the range between 2.49 and 2.90 ppm.
Figure 2 shows that the correct stereoisomer was distinguished both by its 13C and
1H average deviations. Our experiences in the field of computer-aided structure
elucidation have shown [22] that the dA(1H) deviation is a less reliable criterion compared
with dC and it is usually only used for additional confirmation of the most probable
7
structural isomer[5,6,22]. The difference between the deviations dA(13C) found for the
second and first ranked structures is not large (0.2 ppm), but this value is frequently
observed in the structure elucidation process when the “best structure” is selected[22] . It is
worthy to note that in the stereoisomers 3, 4, 6 and 9, atoms H-17 and H-19 are situated
on opposite sides of the macrocycle and are unlikely to be close enough in space to show
a ROESY coupling. Since the authors[15] made the final choice between structures A and
C on the basis of comparison of differences between experimental and calculated 13C
chemical shifts of all carbon atoms we also compared these values (see Figure 3).
O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 1.773 (v.11.01)
dI(13C): 2.791
dN(13C): 2.738
dA(1H): 0.289 (v.11.01)
1 (ID:29)
A O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 1.959 (v.11.01)
dI(13C): 2.893
dN(13C): 2.817
dA(1H): 0.313 (v.11.01)
2 (ID:4)O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 1.969 (v.11.01)
dI(13C): 2.893
dN(13C): 2.817
dA(1H): 0.312 (v.11.01)
3 (ID:13)O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 1.982 (v.11.01)
dI(13C): 2.893
dN(13C): 2.817
dA(1H): 0.313 (v.11.01)
4 (ID:24)
O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 1.998 (v.11.01)
dI(13C): 2.791
dN(13C): 2.738
dA(1H): 0.293 (v.11.01)
5 (ID:8)
DO
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 2.092 (v.11.01)
dI(13C): 2.791
dN(13C): 2.738
dA(1H): 0.293 (v.11.01)
6 (ID:20)
C O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 2.358 (v.11.01)
dI(13C): 3.643
dN(13C): 3.306
dA(1H): 0.313 (v.11.01)
7 (ID:12)O
OH
CH3
CH3
CH3H
H
H
H
H
H
H
H
H
H
H
dA(13C): 2.364 (v.11.01)
dI(13C): 2.893
dN(13C): 2.817
dA(1H): 0.309 (v.11.01)
8 (ID:33)
Figure 2. The first 8 out of 16 stereoisomers ranked in ascending order of the average
deviation dA (13C).
8
-10
-8
-6
-4
-2
0
2
4
6
1 3 5 7 9 11 13
Atom number
Ch
em
ical sh
ift
dif
fere
nce, p
pm
A
C
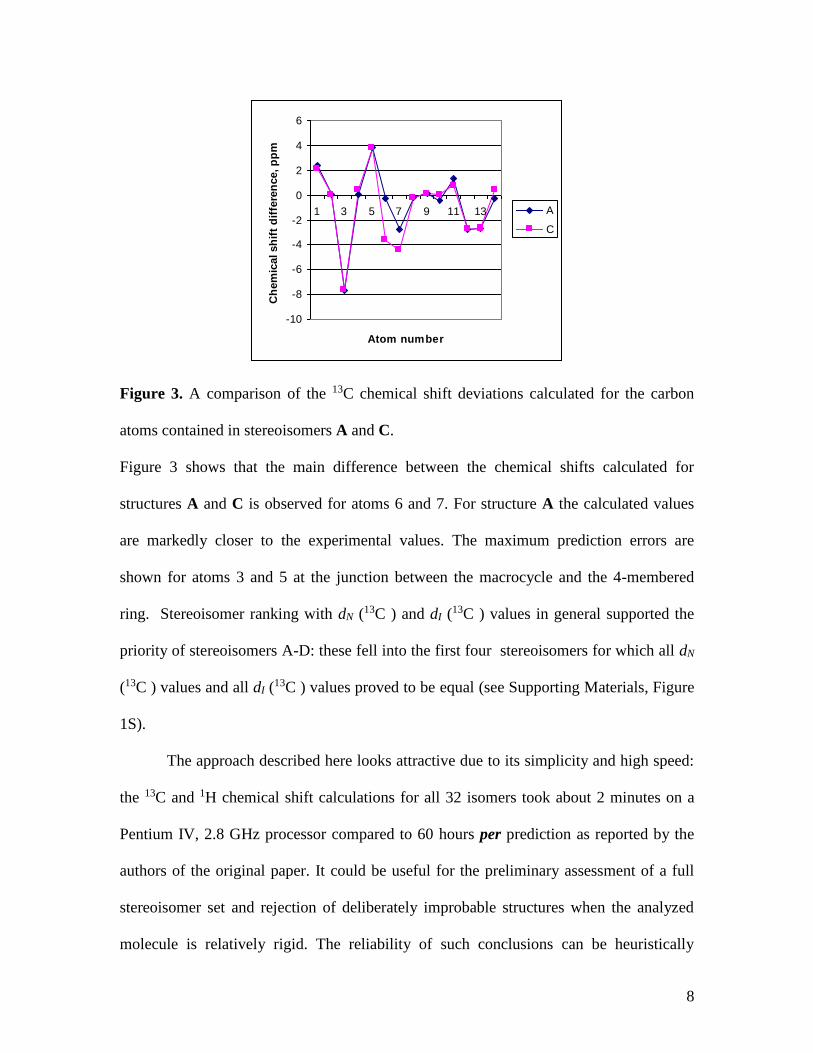
Figure 3. A comparison of the 13C chemical shift deviations calculated for the carbon
atoms contained in stereoisomers A and C.
Figure 3 shows that the main difference between the chemical shifts calculated for
structures A and C is observed for atoms 6 and 7. For structure A the calculated values
are markedly closer to the experimental values. The maximum prediction errors are
shown for atoms 3 and 5 at the junction between the macrocycle and the 4-membered
ring. Stereoisomer ranking with dN (13C ) and dI (13C ) values in general supported the
priority of stereoisomers A-D: these fell into the first four stereoisomers for which all dN
(13C ) values and all dI (13C ) values proved to be equal (see Supporting Materials, Figure
1S).
The approach described here looks attractive due to its simplicity and high speed:
the 13C and 1H chemical shift calculations for all 32 isomers took about 2 minutes on a
Pentium IV, 2.8 GHz processor compared to 60 hours per prediction as reported by the
authors of the original paper. It could be useful for the preliminary assessment of a full
stereoisomer set and rejection of deliberately improbable structures when the analyzed
molecule is relatively rigid. The reliability of such conclusions can be heuristically
9
evaluated by visual comparison of the reference structures used for chemical shift
prediction with the target structure. For instance, a series of structures containing the ring
framework of artarborol were shown by the program when examining the chemical shift
prediction protocol. It should be emphasized that the artarborol molecule (a new
compound) was absent from the library of structures included with the ACD/NMR
prediction program. Reference structure 2 is the most similar structure to the artarborol
structure under investigation:
40.1029.45
24.4551.50
44.2543.75
27.60
63.65
59.80
34.6039.50
O
CH316.90
66.40
HH
CH321.55
CH329.85
OH
H
2
We demonstrated that removing structure 2 from the database did not influence the
results: the deviation characteristic for the best stereoisomer was only slightly increased
from 1.773 to 1.799 ppm.
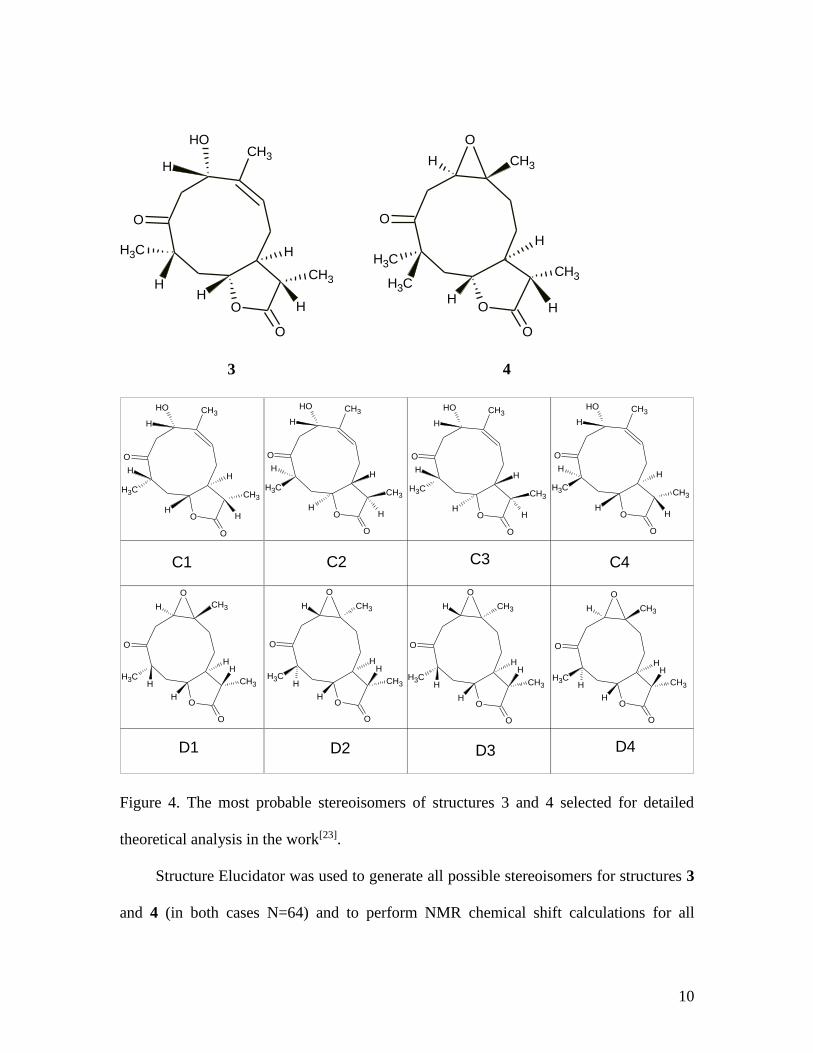
The described approach was also applied to two new ketopelenolides 3 and 4 which
were separated and scrutinized by the same research group[23]. The stereochemistry
shown in structures 3 and 4 was determined by authors[23] as a result of conformational
analysis and QM based 13C chemical shift calculation of the most probable stereoisomers.
The calculations were performed in groups of four for each structure (C1-C4 for structure
3 and D1-D4 for structure 4, see Figure 4). It has been shown that C1 corresponds to
stereoisomer 3 and D1 – to stereoisomer 4.
10
OHCH3
O
CH3
O
O
CH3
H
HH
H
H
O
O
CH3
O
O
CH3
CH3H
CH3H
H
H
3 4
OH CH3
O
CH3
O
O
CH3
H
H
H
H
H
C1
OH CH3
O
CH3
O
O
CH3
H
H
H
H
H
C2
OH CH3
O
CH3
O
O
CH3
H
H
H
H
H
C3
OH CH3
O
CH3
O
O
CH3
H
H
H
H
H
C4
O
O
CH3
O
O
CH3
CH3H
H
H
HH
D1
O
O
CH3
O
O
CH3
CH3H
H
H
HH
D2
O
O
CH3
O
O
CH3
CH3H
H
H
HH
D3
O
O
CH3
O
O
CH3
CH3H
H
H
HH
D4
Figure 4. The most probable stereoisomers of structures 3 and 4 selected for detailed
theoretical analysis in the work[23].
Structure Elucidator was used to generate all possible stereoisomers for structures 3
and 4 (in both cases N=64) and to perform NMR chemical shift calculations for all
11
stereoisomers using empirical methods. 13C chemical shift prediction using the
fragmental method placed stereoisomer C2 in first position in the ranked file and the
genuine stereoisomer C1 at the second position with a difference between deviations of
0.01 ppm. At the same time ranking stereoisomers using dN(13C) values brought
stereoisomers C1-C4 to the 1-4 positions with equal dN(13C) and dI(13C) values for all of
the stereoisomers (see Supporting Materials, Figure 2S). For structure 4 the stereoisomers
were ranked by dA(13C) values in the following order: 1st – D1, 2nd – D2, 3rd – D3, 5th –
D4 (see Supporting Materials, Figure 3S). The correct stereoisomer was placed in first
position and the other most probable stereoisomers selected in[23] were distinguished by
the program as also deserving attention.
For preliminary evaluation of the generality of the described approach we
repeated the work using the structures of natural products belonging to a number of
different classes, i.e. steroids, alkaloids, terpenes, cembranoids, etc. A set of such
structures whose relative stereochemistry was recently described in a series of
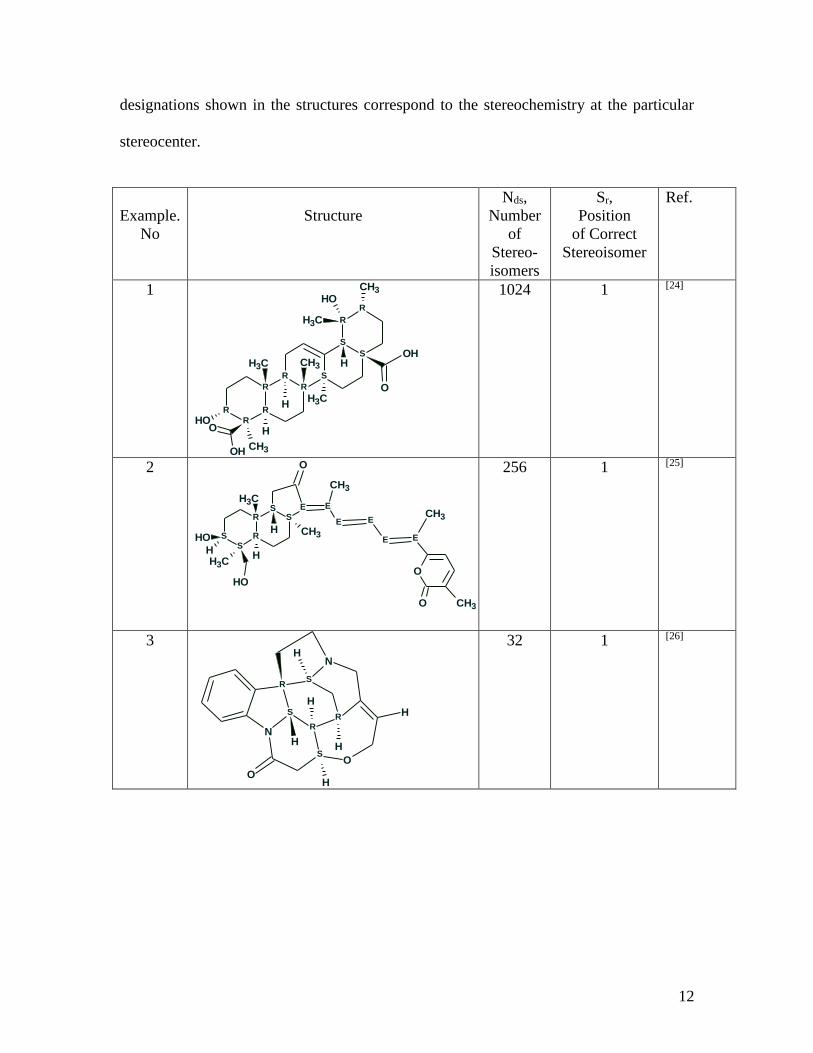
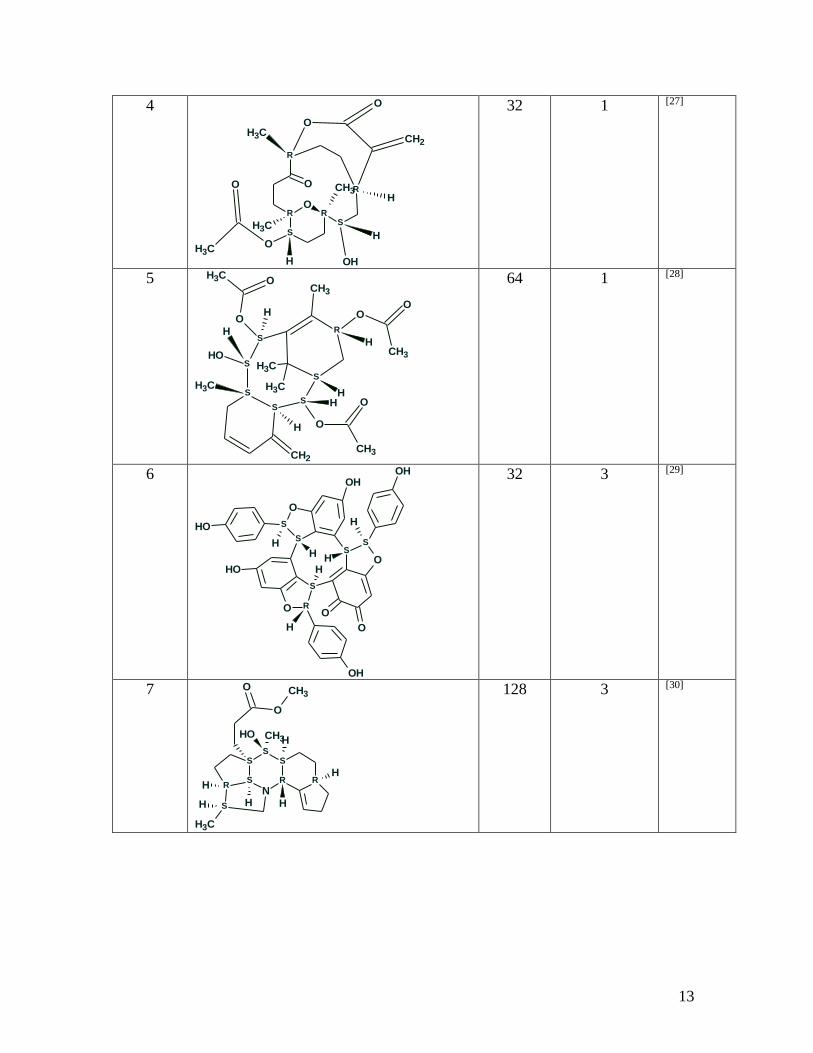
publications was chosen (see Table 1).
Table 1. Examples of structures for which sets of preferable stereoisomers were selected
using empirical methods of 13C NMR chemical shift prediction. The R and S
12
designations shown in the structures correspond to the stereochemistry at the particular
stereocenter.
Example.
No
Structure
Nds,
Number
of
Stereo-
isomers
Sr,
Position
of Correct
Stereoisomer
Ref.
1
R
R
R
R
R
R S
S
S
R
R
OH
CH3
CH3
OH
O
CH3
CH3
OH
OH
O
CH3 CH3
H
H
H
1024 1 [24]
2
R
RS
S
SS E
OH
CH3
OH
CH3E
E
CH3
E
E E
CH3
O
O CH3
O
CH3
H
H
H
256 1 [25]
3
RR
S
S
S
R
N
N
O
O
H
H
H
H
H
H
32 1 [26]
13
4
OR R
S
R
OR
S
OCH3
O
CH3
CH3
CH3
OH
O
O
CH2
H
H
H
32 1 [27]
5 CH3
CH3
CH3
CH3
CH3
CH3
CH3
S
S
S
R
S
S
S
CH2
OO
O
O
O
O
OH
H
H
H
H
H
H
64 1 [28]
6
S
S
S S
S
R
OH
OH
OHOH
OH
O
O
O
O
O
HH
H
H
H
H
32 3 [29]
7
S
R
S
N
S
S RR
S
CH3
O
O
CH3
OH CH3
H H
H
H
H
H
128 3 [30]
14
8
R
R
S
R
R
R
S
S
O
OH
OH
OH
CH3
CH3
R
CH3
R
OO
R
*
CH3
OH
CH3
H
H
HH
H
H
H
2048 3 [31]
9
R
R
SS
S
S R
S
RR
R
CH3
OH
O
O
CH3O
O
CH3
CH3
O
O
O
CH3
CH3
CH3
O
CH3
O
CH3
O
CH3
HH
H
H H
H
H
H
H
1024 3 [32]
10
CH3
CH3
CH3
CH3
CH3
CH3
CH3CH3
CH2
R
R
S
S
R
RS
EE
SS
O
O
O
O
O
O
O
O
O
O
OH
OH
HH
H
H
H
H
H
512 3 [28]
11
R
S
R
S
RS
NCH3
CH3
CH3
CH3S N
CH3CH3
CH3
H
H
H
64 3 [33]
15
12
O
S RS
R
CH3
OHCH2
S
RO
CH3
O
CH3 O
O
CH2
HH
H
H
32 4 [27]
13
SS
S
S
SZ
Z
S
O
R
R
S
CH2
O
CH3
O
O
CH3
O
CH3
OCH3
O
CH3
OCH3
OH
O
CH3
OH
HH
H
H
H
H
256 8 [34]
14
R
R
S
R
R
R
S
RS
OH
OHCH3
OHCH3
OH
CH3
O
OH
HH
H
H
H
256 12 [35]
All selected structures were supplied with assigned experimental 1H and 13C NMR
chemical shifts. Three similar structures borrowed from earlier publications (of 2003 and
2004) were temporarily removed from the database during our research. For each
molecule a full set of N possible stereoisomers was generated and the 13C NMR chemical
shifts of Nds differing stereoisomers (Nds =N/2, N=2n, n – number of stereocenters) were
calculated by all three mentioned algorithms. A stereoisomer file was ranked in the same
way as in the artarborol case – in descending order of dA(13C) values, and the position of
16
the correct stereoisomer, as determined in the corresponding article, was detected in the
ranked file. The result of each computational experiment was characterized by an Sr value
where Sr is the number of stereoisomers for which the deviations dA(13C) are less than or
equal to the deviation calculated for the right stereoisomer. For instance, Sr =1 means that
the right stereoisomer was ranked the first in the file with deviation dA1(13C), and dA1(
13C)
< dA2(13C), where dA2(
13C) is the deviation calculated for the stereoisomer ranked in
second position. The notation Sr =4 means that the correct stereoisomer is among the first
four stereoisomers in the ranked file.
Table 1 shows that our suggested approach can indeed be used for selecting a set
of the most probable stereoisomers from all possible members of the family. Even for
rather complex structures the preferable stereoisomer was ranked early in the set.
Stereoisomer ranking using dN(13C) is not as effective as dA(13C) but nevertheless in this
case the right stereoisomer most frequently fell into the set of the first 8 ranked
stereoisomers. Consequently, the neural net approach can be used for preliminary ranking
the stereoisomer file for subsequent spectrum prediction based on fragmental method as
is common in Structure Elucidator system[6]. When NOESY/ROESY data were available
from the corresponding articles, application of these data to structures presented in top
sets (Sr =3-12) allowed us to conclude that the right stereoisomer is the preferred one
algorithmically also. Examples of the several top ranked sets of stereoisomers are
presented in the Supporting Materials.
Computational Details.
17
All calculations were performed using ACD/NMR predictor Version 11.00. A personal
computer equipped with a 2.8 GHz Intel processor and 2Gb of RAM and running the
Windows2000 operating system was used. All computer programs are an integral part of
the Structure Elucidator expert system. Other than supplying a set of structures,
stereoisomer generation and NMR chemical shift calculation requires no intervention
from the chemist and are performed fully automatically.
Conclusions.
The possibility of applying empirical methods of 13C NMR chemical shift prediction for
selection of a set of the most probable stereoisomers related to a given chemical structure
has been shown for a series of examples. Application of this approach to the elucidation
of the preferred stereoisomer of artarborol has been considered in more detail. We
selected the most probable stereoisomer of artarborol using a simple and fast empirical
method of chemical shift prediction based on HOSE codes. We suggest that it is worth
employing this approach for the preliminary evaluation of all possible stereoisomers
generated by the expert system Structure Elucidator. We expect that this approach will
show general utility when the analyzed structure is relatively rigid and the reference
structures used for chemical shift prediction contain large common fragments with stereo
assignments. This approach can markedly reduce the number of stereoisomers that should
be thoroughly investigated on the basis of NOE correlations, coupling constant values
and quantum-mechanical calculations to finally establish the preferable stereoisomer. The
method can be enhanced by utilizing the methodology suggested in our work[36] and vice
versa: if a starting stereoisomer fed as input to the genetic algorithm for prediction and is
18
close to the right one the genetic algorithm will complete the calculations in a shorter
time.
To continue to develop an optimal strategy and deduce further practical
recommendations it is necessary to investigate a larger set of diverse structures. In this
way we can further refine our methods of NMR chemical shift prediction and make them
more sensitive to relative stereochemistry. For this aim a statistically relevant collection
of material must be accumulated and generalized. This work is in progress, and results
will be presented in our next publication.
References
[1] Blinov KA, Smurnyy YD, Elyashberg ME, Churanova TS, Kvasha M, Steinbeck
C, Lefebvre BE, Williams AJ. J. Chem. Inf. Model. 2008; 48: 550.
[2] Smurnyy YD, Blinov KA, Churanova TS, Elyashberg ME, Williams AJ. J. Chem.